

Abstract
Patient-derived xenografts (PDXs) are resected human tumors engrafted into mice for preclinical studies and therapeutic testing. It has been proposed that the mouse host affects tumor evolution during PDX engraftment and propagation, impacting the accuracy of PDX modeling of human cancer. Here we exhaustively analyze copy number alterations (CNAs) in 1,451 PDX and matched patient tumor (PT) samples from 509 PDX models. CNA inferences based on DNA sequencing and microarray data displayed substantially higher resolution and dynamic range than gene expression-based inferences, and they also showed strong CNA conservation from PTs through late-passage PDXs. CNA recurrence analysis of 130 colorectal and breast PT/PDX-early/PDX-late trios confirmed high-resolution CNA retention. We observed no significant enrichment of cancer-related genes in PDX-specific CNAs across models. Moreover, CNA differences between patient and PDX tumors were comparable to variations in multi-region samples within patients. Our study demonstrates the lack of systematic copy number evolution driven by the PDX mouse host.
Human tumors engrafted into transplant-compliant recipient mice (patient-derived xenografts, PDX) have advantages over prior model systems of human cancer (e.g. genetically engineered mouse models1,2 and cancer cell lines3) for preclinical drug efficacy studies because they allow researchers to directly study human cells and tissues in vivo4-7. Comparisons of genome characteristics and histopathology of primary tumors and xenografts of various cancer types8-14 have demonstrated that the biological properties of patient-derived tumors are largely preserved in xenografts. A growing body of literature supports their use in cancer drug discovery and development15-17.
A caveat to PDX models is that intratumoral evolution can occur during engraftment and passaging18-22. Such evolution could potentially modify treatment response of PDXs with respect to the patient tumors19,23,24, particularly if the evolution were to systematically alter cancer-related genes. Recently, Ben-David et al.23 reported extensive PDX copy number divergence from the patient tumor of origin and across passages, based mainly on large-scale assessment of copy number alterations (CNA) profiles inferred from gene expression microarray data. They raised concerns about genetic evolution in PDXs as a consequence of mouse-specific selective pressures, which could impact the capacity of PDXs to faithfully model patient treatment response. Such results contrast with reports that have observed genomic fidelity of PDX models with respect to the originating patient tumors and from early to late passages by direct DNA measurements in several dozen PDX models8,11,25.
Here we resolve these contradicting observations by systematically evaluating CNA changes and the genes they affect during engraftment and passaging in a large, internationally collected set of PDX models, comparing both RNA and DNA-based approaches. The data collected, as part of the U.S. National Cancer Institute (NCI) PDXNet (PDX Development and Trial Centers Research Network) Consortium and EurOPDX consortium, comprises patient tumor (PT) and PDX samples from >500 models. Our study demonstrates that prior reports of systematic copy number divergence between PTs and PDXs are incorrect, and that there is high retention of copy number during PDX engraftment and passaging. This work also finely enumerates the copy number profiles in hundreds of publicly available models, which will enable researchers to assess the suitability of each for individualized treatment studies.
RESULTS
Catalog of copy number alterations in PDXs.
We have assembled copy number alteration (CNA) profiles of 1,451 unique samples (324 PT and 1,127 PDX samples) corresponding to 509 PDX models contributed by participating centers of the PDXNET, the EurOPDX consortium, and other published datasets11,26 (see Methods, Supplementary Methods, Supplementary Table 1, and Supplementary Fig. 1). We estimated copy number (CN) from five data types: single nucleotide polymorphism (SNP) array, whole-exome sequencing (WES), low-pass whole-genome sequencing (WGS), RNA sequencing (RNA-seq) and gene expression array data, yielding 1,548 tumor datasets including samples assayed on multiple platforms (see Methods, Supplementary Methods, and Supplementary Data 1). Paired-normal DNA, and in some cases paired normal RNA, were also obtained to calibrate WES and RNA-seq tumor samples.
The combined PDX data represent 16 broad tumor types derived from American, European and Asian cancer patients (see Methods), with 64% (n = 324) of the models having their corresponding patient tumors assayed and another 64% (n = 328) having multiple PDX samples of either varying passages (P0-P21) or varying lineages from propagation into distinct mice (Fig. 1a and Supplementary Table 2). The distributions of PT and PDX samples across different tumor types, passages and assay platforms (Fig. 1b and Supplementary Figs. 2-12) show the wide spectrum of this combined dataset, which, to the best of our knowledge, is the most comprehensive copy number profiling of PDXs compiled to date (Supplementary Note 1). Additionally, our data include seven patients with multiple tumors collected either from different relapse time points or different metastatic sites, resulting in multiple PDX models derived from a single patient.
Fig. 1: Patient derived xenograft datasets used for copy number profiling across 16 tumor types.

(a) Numbers of PDX models for each tumor type, with models also having multiple PDX samples or having matched patient tumor samples specified. (b) Distributions of datasets by passage number and assay platform for patient tumors and PDX samples, separated by tumor type. “Late” passages include P18, P19 and P21 samples.
Comparison of CNA profiles from SNP array, WES and gene expression data.
To compare the CNA profiles from different platforms in a controlled fashion, we assembled a dataset with matched measurements across multiple platforms (Supplementary Table 3 and Supplementary Figs. 13-17). Copy number calling has been reported to be noisy for several data types27,28, and we observed that quantitative comparisons between CNA profiles are sensitive to: (i) the thresholds and baselines used to define gains and losses, (ii) the dynamic range of copy number values from each platform, and (iii) the differential impacts of normal cell contamination for different measurements. To control for such systematic biases, we assessed the similarity between two CNA profiles using the Pearson correlation of their log2(CN ratio) values across the genome in 100-kb windows. Regions with discrepant copy number were identified as those with outlier values from the linear regression model (see Methods).
CNAs from WES are consistent with CNAs from SNP array data.
As earlier studies reported that CNA estimates from WES data have more uncertainties than those from SNP arrays29,30, we implemented a WES-based CNA pipeline and validated it against SNP array-based estimates31,32 for matched samples. Copy number gain/loss segments (see Methods) from SNP arrays were of a higher resolution (Fig. 2a; median/mean segment size: 1.49/4.05 Mb for SNP, 4.70/14.6 Mb for WES, P < 2.2 × 10−16) and wider dynamic range (Fig. 2b; range of log2(CN ratio): −8.62-2.84 for SNP, −3.04-1.85 for WES, P < 2.2 × 10−16). The difference in range is apparent in the linear regressions between platforms (Supplementary Fig. 18). These observations take into account the broad factors affecting CNA estimates across platforms, such as the positional distribution of sequencing loci, the sequencing depth of WES, and the superior removal of normal cell contamination by SNP array CNA analysis workflows using SNP allele frequencies33.
Fig. 2: Comparisons of resolution and accuracy for copy number alterations estimated by DNA-based and expression-based methods.

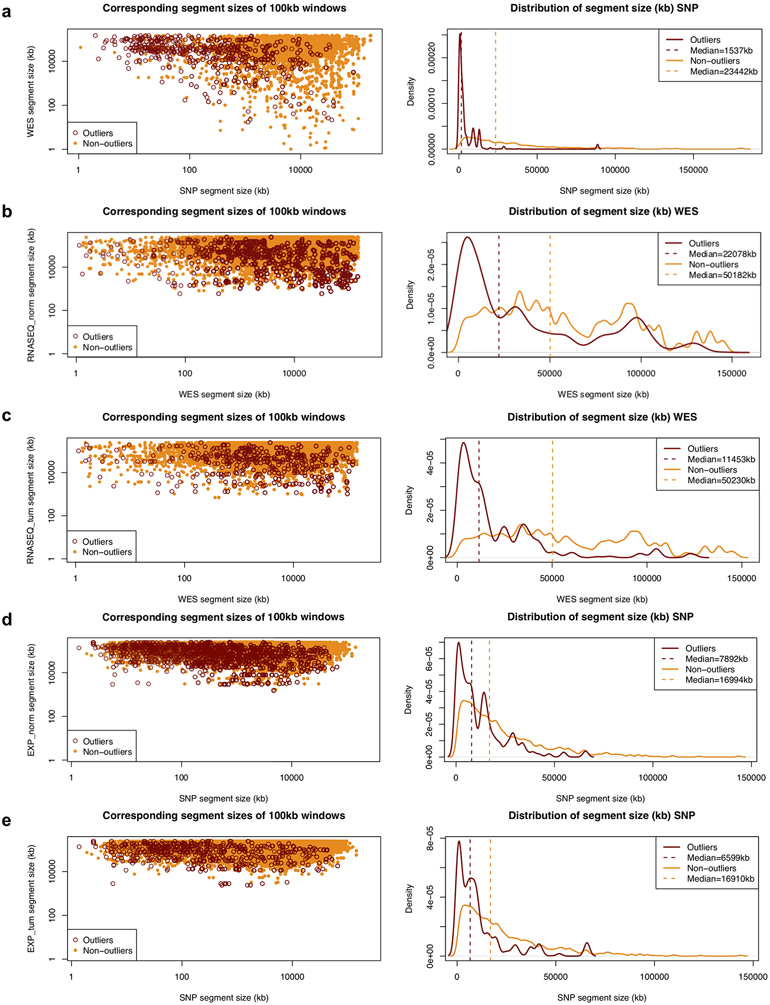
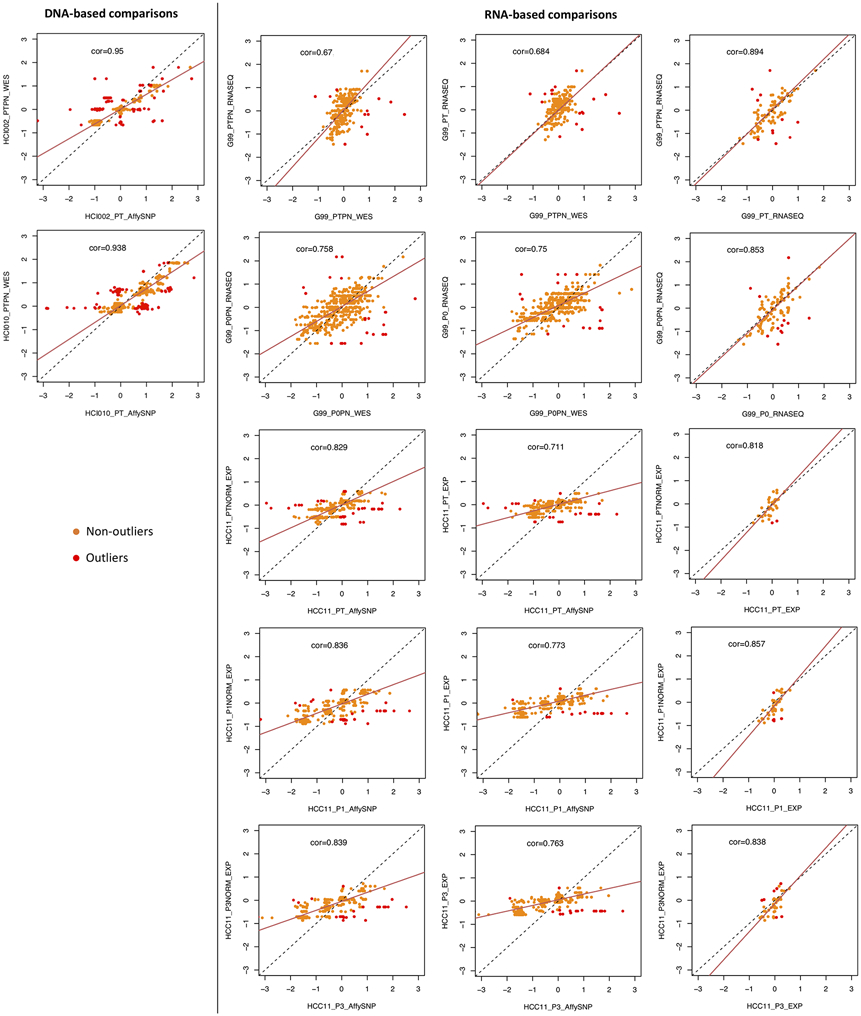
(a) Pairwise comparisons of distributions of segment size (Mb) of CNAs estimated by different measurement platforms in the validation dataset. CNAs are regions with (∣log2(CN ratio)∣ ≥ 0.1). P-values indicate significance of difference between distributions by two-sided Wilcoxon rank sum test. (b) Pairwise comparisons of distributions of log2(CN ratio) of CNA segments. P-values were computed by two-sided Kolmogorov-Smirnov test. (c) Distributions of Pearson correlation coefficient of median-centered log2(CN ratio) in 100-kb windows from CNA segments between pairs of samples estimated by different platforms. Samples with non-aberrant profiles in SNP array and WES data are omitted (5-95% inter-percentile range of log2(CN ratio) < 0.3). P-values were computed by two-sided Wilcoxon rank sum test. In the boxplots, the center line is the median, box limits are the upper and lower quantiles, whiskers extend 1.5 × the interquartile range, dots represent the outliers. (d) Examples of CNA profiles in comparisons of different platforms. Pearson correlation coefficients of CNA segments between pairs of samples are shown on the right. In all the plots, SNP: SNP array, WES: whole-exome sequencing, RNASEQ: RNA sequencing, EXPARR: gene expression array, NORM: normalization by median expression of normal samples, TUM: normalization by median expression of tumor samples, see Supplementary Table 3 for number of samples per group.
We observed strong agreement between SNP arrays and WES, with significantly higher Pearson correlation coefficients on matched samples than samples of different models (range: 0.913-0.957 for matched samples, 0.0366-0.354 for unmatched samples, P = 1.02 × 10−6), with the exception of two samples that lacked CNA aberrations and were removed (Fig. 2c and Supplementary Figs. 13, 18, and 19). The discordant copy number regions largely correspond to small focal events (average size 1.53 Mb) detectable by SNP arrays but missed by WES (Supplementary Fig. 18 and Extended Data Fig. 1a; see Methods). Hence, CNA profiling by WES is reliable in most regions in this small dataset, with 99% of the genome locations across the samples consistent with the values from SNP arrays (Supplementary Note 2). These PT-based observations are also applicable to PDXs given that mouse DNA is absent in SNP array signal and removed from WES reads34-36.
Low accuracy for gene expression-derived CNA profiles.
To compare the suitability of gene expression for quantifying evolutionary changes in CNA, we adapted the e-karyotyping method23,37,38 for RNA-seq and gene expression array data (Supplementary Figs. 15 and 17; see Methods). Copy number segments calibrated by non-tumor expression were of higher resolution (Fig. 2a; median/mean segment size: 36.0/51.9 Mb for RNASEQ NORM, 48.2/65.3 Mb for RNASEQ TUM, P < 2.2 × 10−16; 62.0/72.4 Mb for EXPARR NORM, 80.1/85.2 Mb for EXPARR TUM, P = 2.20 × 10−7) and wider dynamic range (Fig. 2b; range of log2(CN ratio): −2.07-2.17 for RNASEQ NORM, −1.79-1.81 for RNASEQ TUM, P < 2.2 × 10−16; −1.40-1.89 for EXPARR NORM, −1.13-1.59 for EXPARR TUM, P = 4.09 × 10−7) compared to segments calculated by calibration with tumor samples. These alternative expression calibrations yielded biased gain and loss frequencies (Supplementary Note 3 and Supplementary Fig. 20) and strong variability (Pearson correlation range: 0.218-0.943 for RNASEQ NORM vs. TUM, 0.377-0.869 for EXPARR NORM vs. TUM) in the CNA calls (Fig. 2c and Supplementary Fig. 21). This range of correlations was far greater than was observed in comparisons between the DNA-based methods (P = 9.37 × 10−5 and P = 3.28 × 10−7 relative to SNP vs. WES). This indicates the problematic nature of RNA-based CNA calling with calibration by tumor samples, which has been used when normal samples are not available.
Furthermore, expression-based calling had segmental resolution an order of magnitude worse than the DNA-based methods (Fig. 2a and Supplementary Figs. 14-17; median/mean segment size: 3.45/14.0 Mb for WES, 36.0/51.9 Mb for RNASEQ NORM, P < 2.2 × 10−16; 1.73/ 5.18 Mb for SNP, 62.0/72.4 Mb for EXPARR NORM, P < 2.2 × 10−16). The range of detectable copy number values was also superior for DNA-based methods (Fig. 2b; range of log2(CN ratio): −6.00-5.33 for WES, −2.07-2.17 for RNASEQ NORM, P < 2.2 × 10−16; −9.19-4.65 for SNP, −1.40-1.89 for EXPARR NORM, P < 2.2 × 10−16). In addition, there was a lack of correlation between the expression-based and DNA-based methods (range: 0.0541-0.942 for WES vs. RNASEQ (NORM); 0.00517-0.921 for SNP vs. EXPARR (NORM)) (Fig. 2c and Supplementary Figs. 22 and 23). CNA estimates after tumor-based expression normalization resulted in further discordance with DNA-based copy number results (range: −0.182-0.929, P = 0.0468 for WES vs. RNASEQ (TUM); −0.0274-0.847, P = 2.20 × 10−6 for SNP vs. EXPARR (TUM)). Many focal copy number events detected by DNA-based methods, as well as some larger segments, were missed by the expression-based methods (Extended Data Fig. 1b-e). Representative examples illustrating the superior resolution and accuracy from DNA-based estimates are given in Figure 2d (correlations shown in Extended Data Fig. 2).
Concordance of PDXs with patient tumors and during passaging.
We next adopted a pan-cancer approach to elucidate potential tumor type-independent copy number evolution in PDXs driven by the mouse host. We tracked the similarity of CNA profiles during tumor engraftment and passaging by calculating the Pearson correlation of gene-level copy-number for samples measured on the same platform (see Methods, Extended Data Fig. 3, and Supplementary Figs. 24-60 and 62). All pairs of samples derived from the same PDX model were compared, yielding 501 PT-PDX and 1,257 PDX-PDX pairs (Supplementary Note 4).
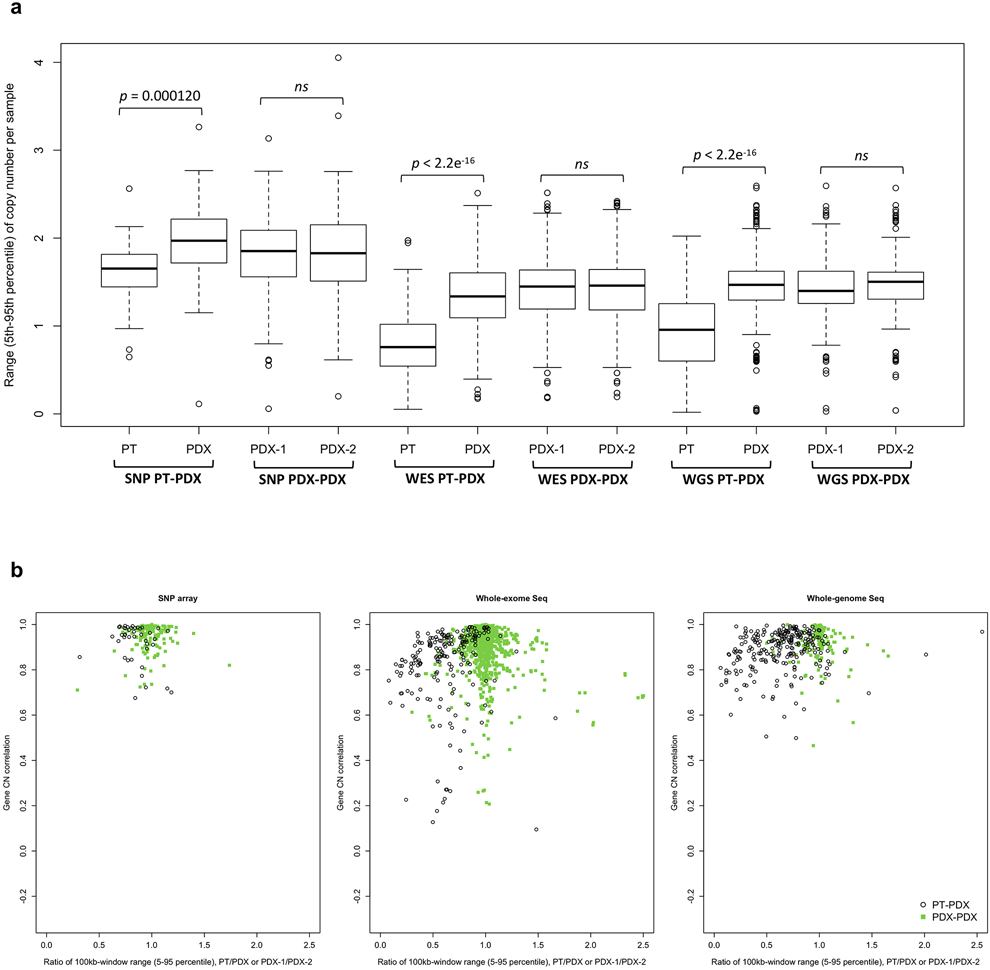
For all DNA-based platforms, we observed strong concordance between matched PT-PDX and PDX-PDX pairs, significantly higher than between different models from the same tumor type and the same center (P < 2.2 × 10−16) (Fig. 3a-c, correlation heatmaps in Supplementary Figs. 24-60). We observed no significant difference in the correlation values between PT-PDX and PDX-PDX pairs for SNP array data (median correlation PT-PDX = 0.950, PDX-PDX = 0.964; P > 0.05), though there were small but statistically significant shifts for WES (PT-PDX = 0.874, PDX-PDX = 0.936; P = 2.31 × 10−16) and WGS data (PT-PDX = 0.914, PDX-PDX = 0.931; P = 0.000299). PT samples have a smaller CNA range than their derived PDXs (median ratio PT/PDX / PDX/PDX: 0.832/0.982, P = 0.000120 for SNP; 0.626/0.996, P < 2.2 × 10−16 for WES; 0.667/1.00, P < 2.2 × 10−16 for WGS; Supplementary Fig. 62b and Extended Data Fig. 4), which can be attributed to stromal DNA in PT samples “diluting” the CNA signal. In PDXs, the human stromal DNA is reduced11,13. The minimal effect for SNP array data confirms this interpretation as human stromal DNA contributions can be removed from SNP arrays based on allele frequencies of germline heterozygous sites, while such contributions to WES and WGS have higher uncertainties. We also performed intra-model comparisons using RNA-based approaches, which showed that the expression-based comparison of CNA profiles between PT and PDXs can lead to the overestimation of copy number changes during engraftment and passage (Supplementary Fig. 63 and Supplementary Note 5).
Fig. 3: Comparisons of copy number alterations from patient tumor to early and late PDX passages.

(a-c) Distributions of Pearson correlation coefficient of gene-based copy number, estimated by (a) SNP array, (b) WES, and (c) WGS, between: PT-PDX samples from the same model; PDX-PDX samples of the same model; samples of different models from a common tumor type and contributing center. P-values were computed by one-sided Wilcoxon rank sum test (ns: not significant, p > 0.05). Number of pairwise correlations are indicated in the legend. (d-f) Distributions of Pearson correlation coefficients of gene-based copy number, estimated by (d) SNP array, (e) WES, and (f) WGS, among patient tumor and PDX passages of the same model. Comparisons relative to PT and P0 are shown (higher passages are shown in Extended Data Fig. 5). In the boxplots, the center line is the median, box limits are the upper and lower quantiles, whiskers extend 1.5 × the interquartile range, dots represent the all data points. (g) Schematic of lineage splitting during passaging and expansion of tumors into multiple mice. This is a simplified illustration for passaging procedures in which different fragments of a tumor are implanted into different mice. (h) Pearson correlation distributions for PDX sample pairs of different lineages and sample pairs within the same lineage: for JAX SNP array, PDMR WES, and EuroPDX WGS datasets. P-values were computed by one-sided Wilcoxon rank sum test. For all boxplots and violin plots, number of pairwise correlations are indicated in the horizontal axis labels.
Late PDX passages maintain CNA profiles similar to early passages.
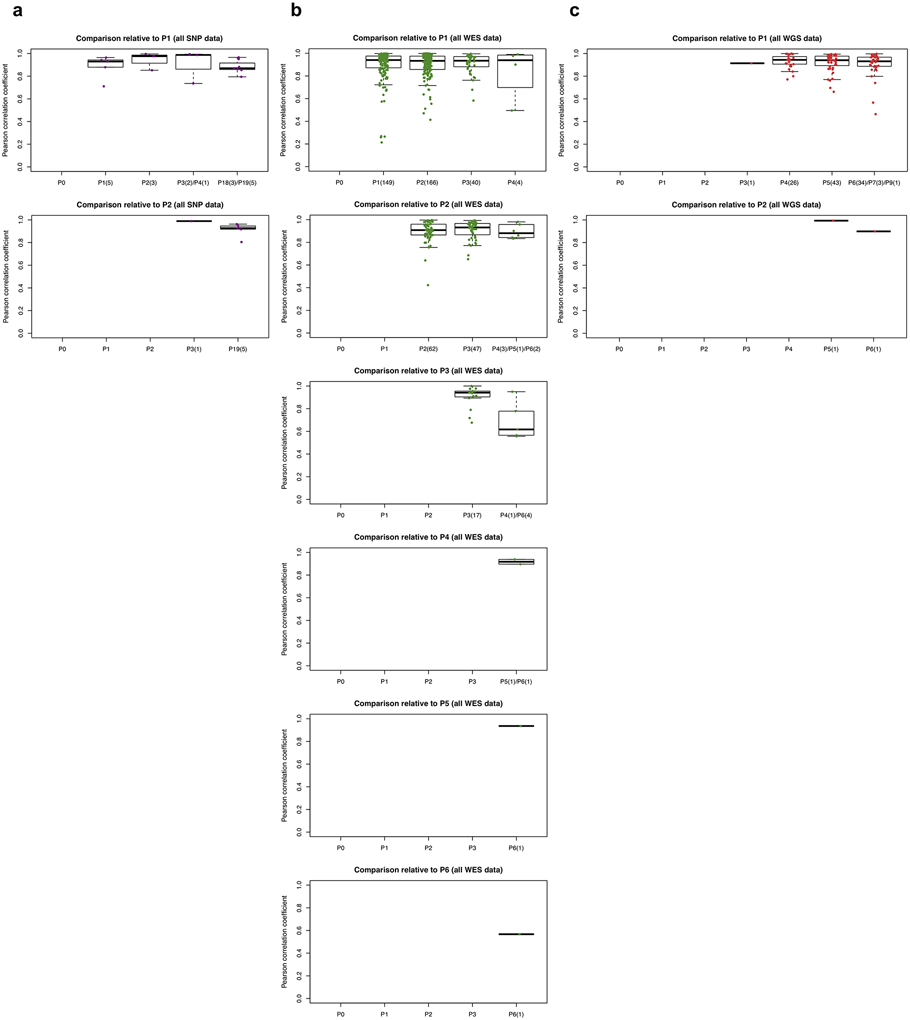
Systematic mouse environment-driven evolution, if present, should reduce CN correlations at each subsequent passage. However, we observed no apparent effect during passaging on the SNP, WES, or WGS platforms (Fig. 3d-f and Extended Data Fig. 5). For example, the SNP data showed no significant difference between passages (Fig. 3d and Extended Data Fig. 5a). For those models having very late passages, there was a small but statistically significant correlation decrease compared to models with earlier passages (P < 8.98 × 10−5, Extended Data Fig. 6b), indicating some copy number changes can occur over long-term passaging (Supplementary Fig. 35). However even at these late passages, the correlations to early passages remained high (median = 0.896). In any given comparison, only a small proportion of the genes were affected by copy number changes (median: 2.72%, range: 1.03-11.9%). Genes that are deleted and subsequently gained in the later passages (top left quadrant of regression plots, Extended Data Fig. 6a) suggest selection of preexisting minor clones as the key mechanism in these regions. For WES and WGS data, more variability in the correlations can be observed (Fig. 3e,f and Extended Data Fig. 5b,c), likely due to a few samples having more stromal contamination or low aberration levels (Supplementary Fig. 62b and Extended Data Fig. 4). However, the lack of downward trend over passaging was also apparent in these sets (Supplementary Note 6).
PDX copy number profiles trace lineages.
We next compared the similarity of engrafted PDXs of the same model with the same passage number. Surprisingly, we discovered that these pairs were not more similar than pairs of PDXs from different passage numbers (Fig. 3d,e, Extended Data Fig. 5, and Supplementary Note 7). Such similarity in correlations suggested that copy number divergence might be associated with effects other than passaging. To further this analysis, we defined, for JAX SNP array and PDMR WES datasets, samples within a lineage as those differing only by consecutive serial passages, while we defined lineages as split when a tumor was divided and propagated into multiple mice (Fig. 3g). For the EurOPDX CRC and BRCA WGS datasets, such lineage splitting was due only to cases with initial engraftment of different fragments of the PT, i.e., PDX samples of different passages were considered as different lineages if they originate from different PT fragments. We observed lower correlation between PDX samples from different lineages compared to within a lineage (Fig. 3h, P = 0.0233 for SNP, P = 0.00119 for WES, P = 0.000232 for WGS), despite a majority of these pairwise comparisons exhibiting high correlation (>0.9) (Supplementary Notes 8 and 9). This suggests that lineage-splitting is often responsible for deviations in CNAs between samples, and that copy number evolution during passaging mainly arises from evolved spatial heterogeneity24.
We further explored whether the stability of copy number during engraftment and passaging is affected by mutations in genes known to impact genome stability (see Methods). Overall, we observed that presence of mutations in such genes does not lead to increased copy number changes during PDX engraftment and passaging (Supplementary Note 10 and Supplementary Fig. 66).
Genes with copy number alterations acquired during engraftment and passaging show no preference for cancer or treatment-related functions.
Next, we investigated which genes tend to undergo copy number changes. Genes with changes during engraftment or during passaging were identified based on a residual threshold with respect to the improved linear regression39 (see Methods; Extended Data Fig. 3). To test for functional biases, we compared CNA-altered genes to gene sets with known cancer- and treatment-related functions40-43 (see Methods). We calculated the proportion of altered genes for sample pairs from each model across all platforms and tumor types. In agreement with the high maintenance of CNA profiles described above, we found the proportion of altered protein-coding genes to be low (median/IQR: 1.90%/ 4.11% PT-PDX, 1.25%/ 3.60% PDX-PDX pairs, Fig. 4a). Only 8.78% of PT-PDX pairs and 4.53% PDX-PDX pairs showed >10% of their protein-coding genes altered. We observed no significant increase (P > 0.1) in alterations among any of the cancer gene sets compared to the background of all protein-coding genes, for either the PT-PDX or PDX-PDX comparisons. This provides evidence that there is no systematic selection for CNAs in oncogenic or treatment-related pathways during engraftment or passaging. We next considered tumor-type-specific effects, focusing on tumor types with larger numbers of models to ensure statistical power. We observed no significant increase in alterations in tumor-type-specific driver gene sets significantly altered in TCGA44-47 compared to the background (P > 0.1) for either PT-PDX or PDX-PDX comparisons (Fig. 4b and Supplementary Note 11).
Fig. 4: Cancer gene sets analysis for copy number altered genes during engraftment and passaging.

(a) Distribution of proportion of altered genes between pairwise PT-PDX or PDX-PDX comparisons of the same model in various gene sets. Protein-coding: protein-coding genes annotated by Ensembl; Oncogenic pathways: genes in oncogenic signaling pathways identified by TCGA; JAX CKB Amp/Del: genes with copy number gain or over-expression / copy number loss or under-expression associated with therapeutic sensitivity or resistance or changes in drug response; Census Amp Del: genes from Cancer Gene Census frequently altered by amplifications or deletions. CNA genes were identified by ∣residual∣ > 0.5 from linear regression model. (b) Distribution of proportion of altered genes between pairwise PT-PDX or PDX-PDX comparisons of the same model in various gene sets within breast cancer, colorectal cancer, lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) models. TCGA Gistic Amp/Del: significantly amplified/deleted genes from TCGA GISTIC analysis for the corresponding tumor type. For all violin plots, P-values were computed by one-sided Wilcoxon rank sum test (ns: not significant, p > 0.1); number of pairwise comparisons are indicated in the plot title, number of genes per gene set is indicated in the horizontal axis labels. (c) Recurrence frequency of protein coding genes with copy number alterations, ∣residual∣ > 1, across all models in PT-PDX and PDX-PDX comparisons. Number of models is indicated in the horizontal axis labels.
Low recurrence of altered genes across models.
We observed a very low recurrent frequency (Fig. 4c, see Methods), with only 12 and 2 genes recurring at > 5% frequency for PT-PDX and PDX-PDX comparisons, respectively (Supplementary Table 4). No gene had a recurrence frequency higher than 8.96% (Supplementary Note 12). None of these recurrent genes overlapped cancer- or treatment-related gene sets, nor did they intersect genes (n = 3) reported by Ben-David et al.23 to have mouse-induced copy number changes associated with drug response in the CCLE48,49 database (Supplementary Note 12).
Absence of CNA shifts in 130 WGS patient tumor, early passage PDX and late passage PDX trios.
We next investigated whether recurrent CNA changes occur in PDXs in a tumor-type specific fashion. To this aim, we analyzed further the WGS-based CNA profiles of large metastatic colorectal (CRC) and breast cancer (BRCA) series, composed of matched trios of PT, PDX at early passage (PDX-early) and PDX at later passage (PDX-late). Genomic Identification of Significant Targets in Cancer (GISTIC)50,51 analysis was applied separately to identify recurrent CNAs in each PT, PDX-early and PDX-late cohorts of CRC and BRCA (see Methods, Supplementary Table 6). As expected, CRCs and BRCAs generated different patterns of significant CNAs, but within each tumor type, GISTIC profiles of the PT, PDX-early, and PDX-late cohorts were virtually indistinguishable (Fig. 5a, Extended Data Fig. 7, and Supplementary Note 13), demonstrating no gross genomic alteration systematically acquired or lost in PDXs.
Fig. 5: Absence of mouse-driven recurrent CNAs during engraftment and propagation of colorectal (CRC) and breast cancer (BRCA) PDXs.

(a) Bar charts representing genome-wide GISTIC G-score for amplifications and deletions in each of the three cohorts of CRC (87 trios) and BRCA (43 trios): PT, PDX-early (P0-P1 for CRC, P0-P2 for BRCA), PDX-late (P2-P7 for CRC, P3-P9 for BRCA). (b-c) Scatter plots comparing gene-level GISTIC G-score between each of the three cohorts, for (b) CRC and (c) BRCA. Bottom-right panels of (b) and (c): scatter plots comparing ΔG-scores from PT to PDX-early and from PDX early to PDX-late. (d-e) Scatter plots comparing GSEA Normalized Enrichment Score (NES) for gene sets between each of the three cohorts, for (d) CRC (e) and BRCA. Bottom-right panels of (d) and (e): scatter plots comparing ΔNES from PT to PDX-early and from PDX-early to PDX-late.
We then carried out gene-level analysis, where each gene was attributed the GISTIC score (G-score) of the respective segment (Supplementary Table 7). In both the CRC and BRCA cohorts, gene-level G-scores of the PTs were highly correlated with the respective PDX-early and PDX-late cohorts (Fig. 5b,c). Moreover, PT versus PDX correlations were comparable to PDX-early versus PDX-late correlations. To search for progressive shifts, we compared the change in G-score (ΔG): (i) from tumor to PDX-early and (ii) from PDX-early to PDX-late. Correlations in these two ΔG values were absent or even slightly negative (bottom-right panels of Fig. 5b,c and Supplementary Note 13). Overall, these results confirmed the absence of systematic CNA shifts in PDXs even under high resolution, gene-level analysis. To evaluate the possibility of systematic copy number evolution at the pathway level in these trios, we performed Gene Set Enrichment Analysis (GSEA)52,53 using G-scores to rank genes in each cohort (see Methods and Supplementary Note 14). For both CRC and BRCA, the Normalized Enrichment Score (NES) profiles for the ~8,000 gene sets of PTs were highly correlated with the respective PDX-early and PDX-late cohorts (Fig. 5d,e). Moreover, PT versus PDX correlations were comparable to PDX-early versus PDX-late correlations. To search for progressive shifts, we calculated for each significant gene set ΔNES values between PT and PDX-early, as well as between early and late PDX. Similar to what was observed for the ΔG-scores, correlations were absent or at most slightly negative (bottom-right panels of Fig. 5d,e), confirming the absence of systematic CNA-based functional shifts in PDXs.
CNA evolution across PDXs is no greater than variation in patient multi-region samples.
As a reference for the treatment relevance of PDX-specific evolution, we compared to levels of copy number variation in multi-region samples of patient tumors. For this we used copy number data from multi-region sampling of non-small-cell lung cancer from the TRACERx Consortium54, performing analogous CNA correlation and gene analyses between multi-region pairs (Supplementary Fig. 69). We observed no significant differences in correlation (P > 0.05) between patient multi-region and lung cancer PT-PDX pairs, while PDX-PDX pairs in fact showed significantly better correlation than the multi-region pairs (P < 0.05, Fig. 6a), consistent across all lung cancer subtypes. Cancer gene set analyses confirmed these results, with multi-region samples showing greater differences than either PT-PDX or PDX-PDX comparisons, across all cancer gene sets considered (P < 0.05; Fig. 6b and Extended Data Fig. 8). These results show that PDX-associated CNA evolution is no greater than what patients experience naturally within their tumors. Our PDX collection also contains a few cases in which the patient tumor was assayed at multiple time points (relapse/metastasis) or multiple metastatic sites, allowing for controlled comparison of intra-patient variation versus PDX evolution (Supplementary Figs. 3, 4, and 7). Despite a lower median in correlations among intra-patient samples, the difference compared to CNA evolution during engraftment (PT-PDX) is not statistically significant (P > 0.05, Fig. 6c). CNA profiles for these samples are shown visually in Figure 6d.
Fig. 6: Comparison of CNA variation during PDX engraftment and passaging to CNA variation among patient multi-region, tumor relapse, and metastasis samples.

(a) Distributions of Pearson correlation coefficients of gene-based copy number for lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), and other lung cancer subtypes, comparing different datasets. TracerX multiregion: multi-region tumor samples of the same patient from TRACERx (92 patient tumors, 295 multi-region samples); PT-PDX samples of the same model; PDX-PDX samples of the same model. P-values were computed by two-sided Wilcoxon rank sum test (ns: not significant, p > 0.05). (b) Distributions of proportion of altered genes between multi-region tumor pairs from TRACERx, and PT-PDX and PDX-PDX pairs for various gene sets for LUAD and LUSC. Gene sets and CNA thresholds are the same as Fig. 4. TCGA Gistic Amp/Del and JAX CKB Amp Del gene sets are shown (other gene sets are shown in Extended Data Fig. 8). P-values were computed by one-sided Wilcoxon rank sum test. Number of genes per gene set are indicated in the plot title. (c) Distributions of Pearson correlation coefficients of gene-based copy number between intra-patient PT (primary/relapse/metastasis) pairs from the same patient and corresponding PT-PDX (derived from the same model; a different PT sample from the same patient generates a different model) pairs for the same set of patients. P-values were computed by two-sided Wilcoxon rank sum test (ns: not significant, p > 0.05). Number of patients and models are indicated in the plot title. For all box plots and violin plots, number of pairwise comparisons are indicated in the horizontal axis labels. In all boxplots, the center line is the median, box limits are the upper and lower quantiles, whiskers extend 1.5 × the interquartile range, dots represent the all data points. (d) CNA profiles of PT and PDX samples from patients with PDX models derived from multiple PT collection (primary/relapse/metastasis).
DISCUSSION
Here we have investigated the evolutionary stability of patient-derived xenografts, an important model system for which there have been prior reports of mouse-induced copy number evolution. To better address this, we assembled the largest collection of CNA profiles of PDX models reported to date, comprising PDX models with multiple passages and their originating patient tumors. Our analysis demonstrated the reliability of copy number estimation by DNA-based measurements over RNA-based inferences, which are substantially inferior in terms of resolution and accuracy (Supplementary Note 15). The importance of DNA measurements is supported by the inconsistent conclusions by two independent studies, Ben-David et al.23,55 and Mer et al.56, on the same PDX expression array dataset by Gao et al.15. Ben-David et al. concluded that drastic copy number changes, driven by mouse-specific selection, often occur within a few passages. On the other hand, Mer et al. reported high similarity between passages of the same PDX model based on direct correlations of gene expression, consistent with our findings in large, independent DNA-based datasets.
The CN shifts inferred by Ben-David et al. are inherently impacted by major technical issues. First, the microarray signal for PT samples is diluted by introgressed human stromal cells, while in PDXs mouse stromal transcripts hybridize only to a fraction of the human probes57. Consequently, PT samples with substantial stromal content would display a reduced signal compared to the corresponding PDX, which can lead to an erroneous inference of systematic increase in aberrations during PDX engraftment when gain/loss regions are directly compared. Second, the mouse host microenvironment can affect the transcriptional profile of the PDX tumor58 and the quantity of mouse stroma can vary across passages. This can result in variability in the expression signal which can be wrongly inferred as CN changes, both from the tumor itself and through cross hybridization of mouse RNA to the human microarray. Although improved concordance in expression between PT and PDX can be achieved with RNA sequencing with the removal of mouse reads59,60, we observed that expression-based copy number inferences still have low resolution and robustness. Hence, many cancer-driving genes, which are found mainly in focal events with a size of 3 Mb or lower61-64, cannot be evaluated for PDX-specific alterations. These issues are further worsened by the lack of tissue-matched normal gene expression profiles for calibration37, which have been only intermittently available but can substantially impact copy number inferences. Because of these considerations, the question of how much PDXs evolve as a consequence of mouse-specific selective pressures cannot be adequately addressed by expression data.
The studies we have presented here take into account the above issues by use of DNA data, as well as by assessing copy number changes by pairwise correlation/residual analysis to control for systematic biases, and they overall confirm the high retention of CNA profiles from PDX engraftment to passaging. We do observe larger deviations between PT-PDX than in PDX-PDX comparisons, though this is likely due to dilution of PT signal by human stromal cells. Interestingly, we found that a major contributor to the differences between PDX samples is lineage-specific drift associated with splitting of tumors into fragments during PDX propagation. This spatial evolution within tumors appears to affect sample comparisons more than time or the number of passages. This suggests that PDX expansion and passaging is the bottleneck of copy number evolution in PDXs, reflecting stochasticity in sampling within spatially heterogenous tumors (Supplementary Note 16).
A challenge for evaluating any model system is that there is no clear threshold for genomic change that determines whether the model will still reflect patient response. Genetic variation among multi-region samples within a patient can shed light on this point54,65-68 since the goal of a successful treatment would be to eradicate all of the multiple regions of the tumor. We found that the copy number differences between PT and PDX are no greater than the variations among multi-region tumor samples or intra-patient samples. Thus, concerns about the genetic stability of the PDX system are likely to be less important than the spatial heterogeneity of solid tumors themselves. This result is consistent with our results on lineage effects during passaging, which indicate that intratumoral spatial evolution is the major reason for genetic drift.
We observed no evidence for systematic mouse environment-induced selection for cancer or treatment-related genes via copy number changes, though individual cases vary (see example in Extended Data Fig. 6c). Moreover, only a small fraction of sample pairs (2.44%, 43 out of 1,758) shows large CNA discordance (see Methods), suggesting that clonal selection out of a complex population is rare. These results indicate that the variations observed in PDXs are mainly due to spontaneous intratumoral evolution rather than murine pressures (Supplementary Note 17).
In summary, our in-depth tracking of CNAs throughout PDX engraftment and passaging confirms that tumors engrafted and passaged in PDX models maintain a high degree of molecular fidelity to the original patient tumors and their suitability for pre-clinical drug testing. At the same time, our study does not rule out that PDXs will evolve in individual trajectories over time, and for therapeutic dosing studies, the best practice is to confirm the existence of expected molecular targets and obtain sequence characterizations in the cohorts used for testing as close to the time of the treatment study as is practical.
METHODS
Experimental details for sample collection, PDX engraftment and passaging, and array or sequencing.
Consolidating tumor types from different datasets.
As the terminology of tumor types/subtypes by the different contributing centers were not consistent, we used the Disease Ontology database69 (http://disease-ontology.org/), cancer types listed in NCI website (https://www.cancer.gov/types) and in TCGA publications70,71 to unify and group the tumor types/subtypes under broader terms as shown in Figure 1 and Supplementary Table 2.
Copy number alteration (CNA) estimation methods.
SNP array.
The estimation of CNA profiles from SNP array were detailed previously34. In short, for Affymetrix Human SNP 6.0 arrays, PennCNV-Affy and Affymetrix Power Tools72 were used to extract the B-allele frequency (BAF) and Log R Ratio (LRR) from the CEL files. Due to the absence of paired-normal samples, the allele-specific signal intensity for each PDX tumor were normalized relative to 300 randomly selected sex-matched Affymetrix Human SNP 6.0 array CEL files obtained from the International HapMap project73. For Illumina Infinium Omni2.5Exome-8 SNP arrays (v1.3 and v1.4 kit), the Illumina GenomeStudio software was used to extract the B-allele frequency (BAF) and Log R Ratio (LRR) from the signal intensity of each probe. The single sample mode of the Illumina GenomeStudio was used, which normalizes the signal intensities of the probes with an Illumina in-house dataset. The single tumor version of ASCAT33 (v2.4.3 for JAX SNP data, v2.5.1 for SIBS SNP data) was used for GC correction, predictions of the heterozygous germline SNPs based on the SNP array platform, and estimation of ploidy, tumor content and allele-specific copy number segments. The resultant copy number segments were annotated with log2 ratio of total copy number relative to predicted ploidy from ASCAT.
Whole-exome sequencing (WES) data.
Aligned bams (see Supplementary Methods) were subset to target region by GATK 4.0.5.1, and SAMTools74 v0.1.18 was used to generate the pileup for each sample. Pileup data were used for CNA estimation as calculated with Sequenza29 v2.1.2. Both tumor and normal data, which utilized the same capture array, were used as input. pileup2seqz and GC-windows (-w 50) modules from sequenza-utils.py utility were used to create the native seqz format file for Sequenza and compute the average GC content in sliding windows from hg38 genome, respectively. We ran the three Sequenza modules with these modified parameters (sequenza.extract: assembly = “hg38”, sequenza.fit: chromosome.list = 1:23, and sequenza.results: chromosome.list = 1:23) to estimate the segments of copy number gains/losses. Finally, segments lacking read counts, in which ≥50% of the segment with zero read coverage, were removed. A reference implementation of this workflow (Supplementary Fig. 71) is developed and deployed in the Cancer Genomics Cloud by Seven Bridges (https://cgc.sbgenomics.com/public/apps#pdxnet/pdx-wf-commit2/wes-cnv-tumor-normal-workflow/, https://cgc.sbgenomics.com/public/apps#pdxnet/pdx-wf-commit2/pdx-wes-cnv-xenome-tumor-normal-workflow/).
Low-pass whole-genome sequencing (WGS) data.
For EuroPDX CRC liver metastasis data, raw copy number profiles for each sample were estimated by QDNAseq75 R package v1.20 by dividing the human reference genome in non-overlapping 50 kb windows and counting the number of reads (see Supplementary Methods) in each bin. Bins in problematic regions were removed76. Read counts were corrected for GC content and mappability by a LOESS regression, median-normalized and log2-transformed. Values below −1,000 in each chromosome were floored to the first value greater than −1,000 in the same chromosome. Raw log2 ratio values were then segmented using the ASCAT33 algorithm implemented in the ASCAT R package v2.0.7. For EuroPDX BRCA tumors, raw copy number profiles were estimated for each sample by dividing the human reference genome in non-overlapping 20-kb windows and counting the number of reads (see Supplementary Methods) in each bin. Only reads with at least mapping quality 37 were considered. Bins within problematic regions (i.e. multimapper regions) were excluded. Downstream analysis to estimate copy number was conducted as described above.
RNA-sequencing (RNA-seq) and gene expression microarray (EXPARR) data.
For expression-based copy number inference, we referred to the previous protocols for e-karyotyping and CGH-Explorer37,38,77,78. For each cancer type, expression values (see Supplementary Methods) of tumor and corresponding normal samples were merged in a single table, and gene identifiers were annotated with chromosomal nucleotide positions. Genes located on sex chromosomes were excluded. Genes which values below 1 TPM (RNA-seq) or probeset log2-values below 6 (microarray) in more than 20% of the analyzed dataset were removed. Remaining gene expression values below the thresholds were respectively raised to 1 TPM or log2-value of 6. In the case of multiple transcripts (RNA-seq) or probesets (microarray) per gene, the one with the highest median value across the entire dataset was selected. According to the e-karyotyping protocol, the sum of squares of the expression values relative to their median expression across all samples was calculated for each gene, and 10% most highly variable genes were removed. For each gene, the median log2 expression value in normal samples was subtracted from the log2 expression value in each tumor sample and subsequently input in CGH-explorer. For tumor-only datasets, the median log2 expression value in the same set of tumor samples was instead subtracted. The preprocessed expression profiles of each sample were individually analyzed using CGH-Explorer (http://heim.ifi.uio.no/bioinf/Projects/CGHExplorer/). CGH-PCF analysis was carried out to call copy number according to parameters previously reported23: least allowed deviation = 0.25; least allowed aberration size = 30; winsorize at quantile = 0.001; penalty = 12; threshold = 0.01.
Statistical methods.
All statistical analysis for data comparison were performed using either one-tailed or two-tailed Wilcoxon rank sum test, two-tailed Kolmogorov–Smirnov test, or one-tailed Wilcoxon signed rank test.
Filtering and gene annotation of copy number segments.
Copy number (CN) segments with log2 copy number ratio estimated from the various platforms were processed in the following steps (Extended Data Fig. 3). Segments <1 kb were filtered based on the definition of CNA79. In addition, SNP array segments had to be covered by >10 probes, with an average probe density of 1 probe per 5 kb. The copy number segments were then binned into 10-kb windows to derive the median log2(CN ratio), which was subsequently used to re-center the copy number segments. Median-centered copy number segments were visualized using IGV80 v2.4.13 and GenVisR81 v1.16.1. Median-centered copy number of genes was calculated by intersecting the genome coordinates of copy number segments with the genome coordinates of genes (Ensembl Genes 93 for human genome assembly GRCh38, Ensembl Genes 96 for human genome assembly GRCh37). In the case where a gene overlaps multiple segments, the most conservative (lowest) estimate of copy number was used to represent the copy number of the entire intact gene.
Comparison of CN gains and losses.
For the comparison of resolution, range of CN values and frequency of gains and losses between different platforms and analysis methods, we defined copy number gain or loss segments as – Gain: log2(CNratio) > 0.1; Loss: log2(CN ratio) < −0.1.
Correlation of CNA profiles.
The overall workflow to compare CNA profiles is shown in Extended Data Figure 3. PDX samples without passage information were omitted in the following downstream analysis. The copy number segments were binned into 100-kb windows or smaller using Bedtools82 v2.26.0, and the variance of log2(CN ratio) and 5-95% inter-percentile range of log2(CN ratio) values across all the bins were calculated as a measure of degree of aberration for each CNA profile. A non-aberrant profile results in a low variance or range. While variance can be biased for CNA profiles with small segments of extreme gains or losses, we preferred the use of 5-95% inter-percentile range of log2(CN ratio) to identify samples with low degree of aberration, such that a narrow range indicates ≥90% of the genome has very low-level gains and losses. The similarity of two CNA profiles is quantified by the Pearson correlation coefficient of log2(CN ratio) of 100-kb windows binned from segments or genes between two samples. Gene-based and segment-based (100-kb windows) correlations were highly similar (data not shown). Using correlation avoided the issue of making copy number gain and loss calls based on thresholds. Sample-based variations in baseline due to median-normalization and range in copy number values could introduce further inconsistencies gain and loss calls between samples. Such variations are further impacted by sample-specific variation in human stromal contamination or sensitivity of copy number detection by different platforms. As median-centering of each CNA profile approximates normalization by the sample ploidy, we confirmed that in general ploidy (estimated from ASCAT analysis of SNP array samples) had no association with the copy number correlation values (Pearson’s product-moment correlation, P > 0.05, cor = 0.0248). One caveat of our approach, however, is that it cannot distinguish genome-wide multiplication of ploidy between samples, as the correlation statistic is invariant to such genome-wide transformations. As such we cannot assess whether ploidy changes occur between samples of a given model.
Comparison of CNA profiles between different platforms.
The copy number segments of each pair of data were intersected and binned into 100-kb windows or smaller using Bedtools. The Pearson correlation coefficient and linear regression model was calculated for the log2(CN ratio) of the windows. Windows with discrepant copy number were identified by outliers of the linear regression model defined by ∣studentized residual∣ > 3. These outlier windows were mapped to their corresponding segments to identify the size of CNA events that were discordant between the different copy number estimation methods. The proportion of the genome discordant CNA was calculated from the summation of the outlier windows.
Identification of genes with CNA between different samples of the same model.
To compare the CNA profiles between different samples (PT or PDX) of the same model, the Pearson correlation coefficient and linear regression model was calculated for the log2(CN ratio) of the genes for each pair of data. Prior to that, deleted genes with log2(CN ratio) < −3 were rescaled to −3 to avoid large shifts in the correlation coefficient and linear regression model due to extremely negative values on the log scale. Extreme outliers of the linear regression model defined by ∣studentized residual∣ > 3 were removed to derive an improved linear regression model39 not biased by few extreme values. Genes with copy number changes between the samples were identified by the difference in log2(CN ratio) relative to the improved linear regression model of ∣standard residual∣ < 0.5. We also removed some samples with low correlation due to sample mislabeling as they displayed high correlation with samples from other models. We also omitted samples with low correlation values (<0.6) which resulted from non-aberrant CNA profiles in genomically stable tumors (5-95% inter-percentile range of log2(CN ratio) < 0.3, Supplementary Fig. 62).
Identification of aberrant sample pairs with highly discordant CNA profiles.
Aberrant CNA profiles were identified based on the 5-95% inter-percentile range of log2(CN ratio) >0.5, for both samples. Sample pairs with Pearson correlation <0.6 were selected as highly discordant CNA profiles between them.
Association of mutations with copy number correlations.
Mutational calls for each WES sample used in this study were obtained using a tumor-normal variant calling workflow developed for patient tumor and PDXs35. Subsequently, genes with either germline and somatic variants that pass through the quality filters (FILTER = PASS or germline) and IMPACT = MODERATE or HIGH by SnpEff (v4.3) annotation are labeled as mutated, and wildtype if otherwise. For SNP array and WGS data, we collected the mutational status (wild-type or mutated) of TP53, BRCA1, and BRCA2 per model where available, which may or may not be obtained from the exact same tumor samples used in this study. For the JAX SNP array dataset, variant calls (tumor-only) were made from various targeted sequencing approaches (TruSeq Amplicon Cancel Panel, JAX Cancer Treatment Profile panel and whole exome). The workflow and filtering criteria to call mutations is described elsewhere34. For the HCI SNP array data, mutations were obtained from whole exome sequencing (unpublished data) and were filtered for frameshift, inframe, missense, and nonsense and splice-site mutations. For BCM SNP array data, mutational status were obtained from clinical samples by immunohistochemistry or Sequenom83 (unpublished data). For WGS data, mutations were obtained from whole exome or targeted panel sequencing84 (unpublished data) and high-quality and likely functional mutations were retained. For each sample pair with copy number correlations, mutational status of TP53 or BRCA was obtained for each individual sample for WES data, while the mutational status was available on a per model basis for SNP and WGS data. BRCA is labeled as mutated when either BRCA1 or BRCA2 is mutated. For mutations in DNA repair genes85 from the WES data, each pair of samples was classified as mutated if any DNA repair gene was reported to be mutated in either sample.
Annotation with gene sets with known cancer or treatment-related functions.
A low copy number change threshold (∣log2(CN ratio) change∣ > 0.5) was selected to include genes with subclonal alterations. Copy number altered genes (∣residual∣ > 0.5) were annotated by various gene sets with cancer or treatment-related functions gathered from various databases and publications (Extended Data Fig. 3):
Genes in 10 oncogenic signaling pathways curated by TCGA and were found to be frequently altered in different cancer types40.
Genes with gain in copy number or expression, or loss in copy number or expression that conferred therapeutic sensitivity, resistance or increase/decrease in drug response from the JAX Clinical Knowledgebase41,42 (JAX-CKB) based on literature curation (https://ckbhome.jax.org/, as of 06-18-2019).
Genes with evidence of promoting oncogenic transformation by amplification or deletion from the Cancer Gene Census43 (COSMIC v89).
Significantly amplified or deleted genes in TCGA cohorts of breast cancer44, colorectal cancer45, lung adenocarcinoma46 and lung squamous cell carcinoma47 by GISTIC analysis, which identified significantly altered genomic driver regions which can be used to differentiate tumor types and subtypes.
Identification of genes with recurrent copy number changes.
A stringent CNA threshold (∣log2(CN ratio) change∣ > 1.0 with respect to linear regression model) was selected to distinguish genes with possible functional impact. Genes with ∣residual∣ > 1.0 with respect to the improved regression linear model (without discriminating gain or loss) were selected for each pairwise comparison between different samples of the same model. Pairwise cases in which genes are deleted in both samples (log2(CN ratio) ≤ −3 ) are omitted. Recurrent frequency for each gene across all models was calculated on a model basis such that genes with copy number between multiple pairs of the same model was counted as once. This avoided the bias towards models with many samples of similar copy number changes between the different pairs.
Drug response analysis using CCLE data.
We developed a pipeline to evaluate gene copy number effects on drug sensitivity86,87 by using the Cancer Cell Line Encyclopedia48,88 (CCLE) cell line genomic and drug response data (CTRP v2). We downloaded the CCLE drug response data from Cancer Therapeutics Response Portal (www.broadinstitute.org/ctrp), and CCLE gene-level CNA and gene expression data from depMap data portal (‘public_19Q1_gene_cn.csv’ and ‘CCLE_depMap_19Q1_TPM.csv’, https://depmap.org/portal/download/). For CCLE drug response data, we used the area-under-concentration-response curve (AUC) sensitivity scores for each cancer cell line and each drug. In total, we collected gene-level log2 copy number ratio data derived from the Affymetrix SNP 6.0 platform from 668 pan-cancer CCLE cell lines, with a total of 545 cancer drugs tested. With the CCLE gene-level CNA and AUC drug sensitivity scores, we performed gene-drug response association analyses for genes with recurrent copy number changes. Pearson correlation p-values between each gene’s log2 (CN ratio) and each drug’s AUC score across all cell lines were calculated, and q-values were calculated by multiple testing Bonferroni correction. Significant gene-CNA and drug associations were kept (q-value < 0.1) to further evaluate gene-expression and drug response associations. If a gene’s expression was also significantly correlated with AUC drug sensitivity scores, particularly in the same direction (either positively or negatively correlated) as the gene-CNA and drug association, that gene would be considered as significantly correlated with drug response based on both its CNA and gene expression.
Genomic Identification of Significant Targets in Cancer (GISTIC) analysis of WGS data.
We carried out GISTIC analysis to identify recurrent CNAs by evaluating the frequency and amplitude of observed events. To obtain perfectly matching and comparable PT–PDX cohorts, for GISTIC analysis, CRC trios in which at least one sample displaying non-aberrant CNA profiles were excluded from the analysis resulting in a total of 87 triplets. The GISTIC51 algorithm (GISTIC 2.0 v6.15.28) was applied on the segmented profiles using the GISTIC GenePattern module (https://cloud.genepattern.org/), with default parameters and genome reference files Human_Hg19.mat for EuroPDX CRC data and hg38.UCSC.add_miR.160920.refgene.mat for EuroPDX BRCA data. For each dataset, GISTIC provides separate results (including segments, G-scores and FDR q-values) separately for recurrent amplifications and recurrent deletions. Deletion G-scores were assigned negative values for visualization. We observed that the G-Score range was systematically lower in PT cohorts, which is likely the result of the dilution of CNA by normal stromal DNA. In contrast, human stromal DNA in PDX samples was lower or negligible. To account for this difference in gene-level G-scores, PDXs at early and late passages were scaled with respect to PT gene-level G-score values using global linear regression, separately for amplification and deletion outputs.
Gene set enrichment analysis (GSEA) of WGS data.
To assess the biological functions associated with the recurrent alterations detected by the GISTIC analysis, we performed GSEAPreranked analysis52,53 (GSEA v3.0) on gene-level GISTIC G-score profiles for both amplifications and deletions. In particular, we applied the algorithm with 1,000 permutations on various gene set collections from the Molecular Signatures Database89,90 (MSigDB v6.2): H (Hallmark), C2 (Curated: CGP chemical and genetic perturbations, CP canonical pathways), C5 (Gene Ontology: BP biological process, MF molecular function, CC cellular component) and C6 (Oncogenic Signatures) composed of 50, 4,762, 5,917, and 189 gene sets, respectively. We also included gene sets with known cancer or treatment-related functions described in an earlier section. We noted that multiple genes with contiguous chromosomal locations, typically in recurrent amplicons, generated spurious enrichment for gene sets which consists of multiple genes of adjacent positions, while very few or none of them had a significant GISTIC G-score. To avoid this confounding issue, we only considered the “leading edge genes”, i.e. those genes with increasing Normalized Enrichment Score (NES) up to its maximum value, that contribute to the GSEA significance for a given gene set. The leading-edge subset can be interpreted as the core that accounts for the gene set’s enrichment signal (http://software.broadinstitute.org/gsea). We included a requirement that the leading edge genes passing the GISTIC G-score significant thresholds based on GISTIC q-value 0.25 (Supplementary Table 8 and Extended Data Fig. 7) make up at least 20% of the gene set. This 20% threshold was chosen as the minimal threshold at which gene sets assembled from TCGA-generated lists of genes with recurrent CNA in CRC or BRCA were identified as significant in GSEA (see Supplementary Table 9). Finally, gene sets with a NES > 1.5 and a FDR q-value < 0.05, which passed the leading edge criteria, were considered significantly enriched in genes affected by recurrent CNAs.
DATA AVAILABILITY
Copy number calls from all datasets are available in Supplementary Data 1, and these are used for all figures. Raw sequence data for these calls are a combination of previously described sources (notably the publicly available NCI Patient Derived Models Repository, pdmr.cancer.gov) and newly sequenced data. New sequence data from the PDXNet are being shared as part of the NCI Cancer Moonshot initiative through the Cancer Data Service. For further details, contact the authors. The SNP array data generated by The Jackson Laboratory can be requested via the Mouse Models of Human Cancer Database (tumor.informatics.jax.org). The whole genome sequencing data generated by EurOPDX can be made available by directly contacting the EurOPDX consortium (dataportal.europdx.eu). Other publicly available data used in the analyses include GSE90653, GSE3526, GSE33006 and E-MTAB-1503-3, CCLE cell line genomic and drug response data (CTRP v2), MSigDB v6.2 and TRACERx NSCLC data (DOI: 10.1056/NEJMoa1616288).
CODE AVAILABILITY
We have used well-established computational sequence analysis and statistical analysis techniques, so no code is provided. Full descriptions of all analysis techniques are provided in the Methods. The implementation of the copy number estimation workflow from whole-exome sequencing data is deployed in the cancer genomics cloud at SevenBridges (https://cgc.sbgenomics.com/public/ apps#pdxnet/pdx-wf-commit2/wes-cnv-tumor-normal-workflow/, https://cgc.sbgenomics.com/public/apps#pdxnet/pdx-wf-commit2/ pdx-wes-cnv-xenome-tumor-normal-workflow/).
Extended Data
Extended Data Fig. 1. Comparison of segment sizes between different platforms.
The left panel compares the combined corresponding segment sizes of outlier and non-outliers from the linear regression of the log2(CN ratio) of 100-kb windows binned from copy number segments between matched samples estimated from two different platforms or methods combined. Outliers of the linear regression are identified by studentized residuals > 3 and < −3. a, SNP vs. WES. b, WES vs. RNASEQ (NORM). c, WES vs. RNASEQ (TUM). d, SNP vs. EXPARR (NORM). e, SNP vs. EXPARR (TUM) (see Supplementary Table 3). The right panel compares the distribution of the segment sizes of outliers and non-outliers for the platform or method of higher resolution.
Extended Data Fig. 2. Comparison of copy number between different platforms.
Pearson correlation and linear regression of the log2(CN ratio) of 100-kb windows binned from copy number segments of CNA profiles between matched patient tumor samples estimated from different platforms or analysis methods for examples shown in Figure 2d. Outliers of the linear regression are identified by studentized residuals > 3 and < −3. RNA-seq and expression array samples denoted with “PN” or “NORM” are normalized by the median expression of normal samples.
Extended Data Fig. 3. Analysis workflow to compare CNA between two samples of the same PDX model.

A correlation and robust regression approach to quantify similarity of CNA profiles and identify genes with copy number changes between two samples.
Extended Data Fig. 4. Correlations between PT-PDX and PDX-PDX pairs.
a, The 5-95% inter-percentile range of CNA profiles between PT-PDX or PDX-PDX sample pairs from the same model on different platforms as shown in Figure 3a-c. The 5-95% inter-percentile range of log2(CN ratio) values were calculated across all 100-kb windows per sample. P-values were computed by one-sided Wilcoxon rank sum test (ns: non-significant, P > 0.05). In the boxplots, the center line is the median, box limits are the upper and lower quantiles, whiskers extend 1.5× the interquartile range, and dots represent the outliers. b, Pearson correlation of the samples versus the ratio of 5-95% inter-percentile range between two samples (PT/PDX or PDX-1/PDX-2). Samples pairs with ratio of range much greater or less than 1 (i.e. one sample is much less aberrant than the other) tend to have lower correlations. PDX-1, lower passage PDX; PDX-2, later passage PDX or same passage PDX of different lineage.
Extended Data Fig. 5. Distribution of Pearson correlation coefficients of gene-based copy number.
a-c, Estimated by SNP array (a), WES (b), and WGS (c) between different combinations of patient tumor and PDX passages of the same model. Comparisons relative to passages P1 or later passages (refer to Fig. 3d-f for comparisons with PT and P0). In the boxplots, the center line is the median, box limits are the upper and lower quantiles, whiskers extend 1.5× the interquartile range, and dots represent all data points.
Extended Data Fig. 6. Comparison of CNA between early and very-late passages.
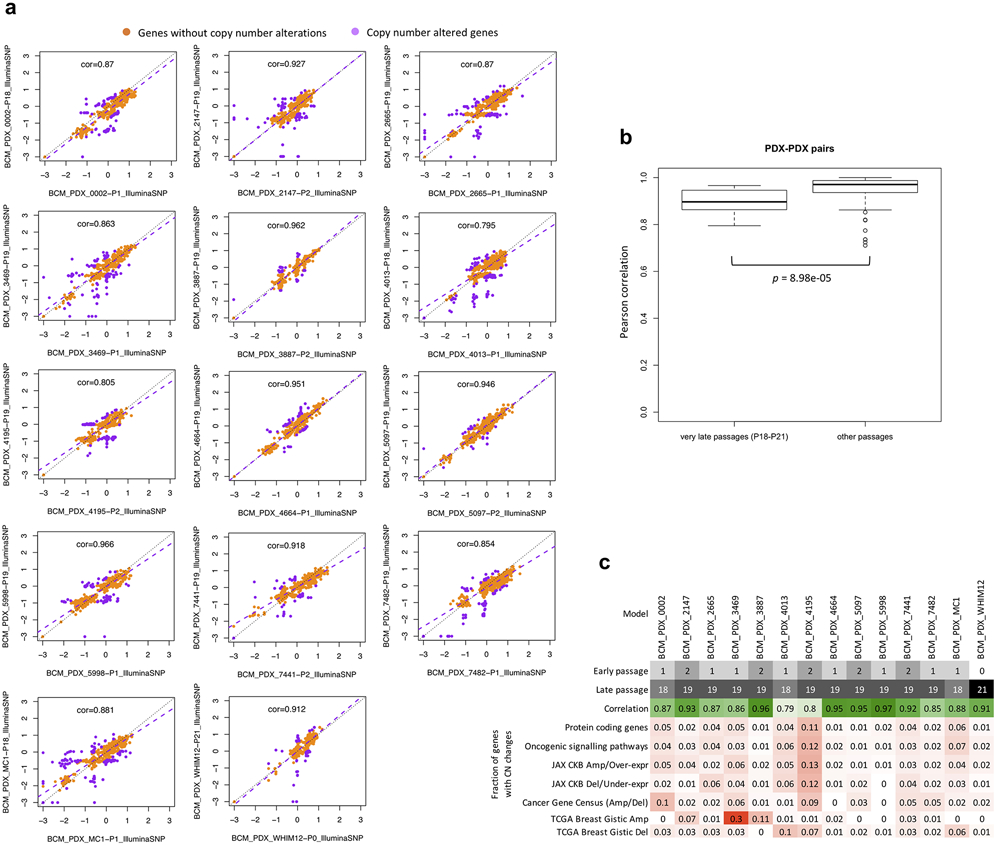
In the BCM SNP array breast cancer dataset. a, Correlation and robust regression of gene-based copy number between early (P0-P2) and very-late passages (P18-P21) of the same model. Genes with copy number changes between the passages are identified by ∣residual∣ > 0.5. Some genes show signs of complete deletion (log2(CN ratio) < −2) but then reappear in later passages. This can only be explained by the early and late passages being dominated by different pre-existing subclones. b, Distribution of Pearson correlation coefficients of gene-based copy number between early and very-late passages of the same model (14 models/pairwise correlations) compared to correlation coefficients between lower passages denoted as “other passages” (< P4). Correlation for “other passages” are based on models from all other non-BCM SNP array datasets (111 pairwise correlations). P-values were computed by one-sided Wilcoxon rank sum test. In all boxplots, the center line is the median, box limits are the upper and lower quantiles, whiskers extend 1.5× the interquartile range, and dots represent outliers. c, Summary of passage numbers, copy number correlation, and fraction of genes of different gene sets with copy number changes (∣residual∣ > 0.5) between passages of each breast cancer model.
Extended Data Fig. 7. GISTIC analysis of recurrent CNAs.

a,b, GISTIC plots showing amplified and deleted regions in the EurOPDX WGS of trios of PTs and derived PDXs, at early and late passages, of colorectal cancer (a, 87 trios) and breast cancer (b, 43 trios). For each GISTIC plot, the top axis reports the G-score and the bottom axis the q-value.
Extended Data Fig. 8. Distribution of proportion of altered genes for lung cancer samples.
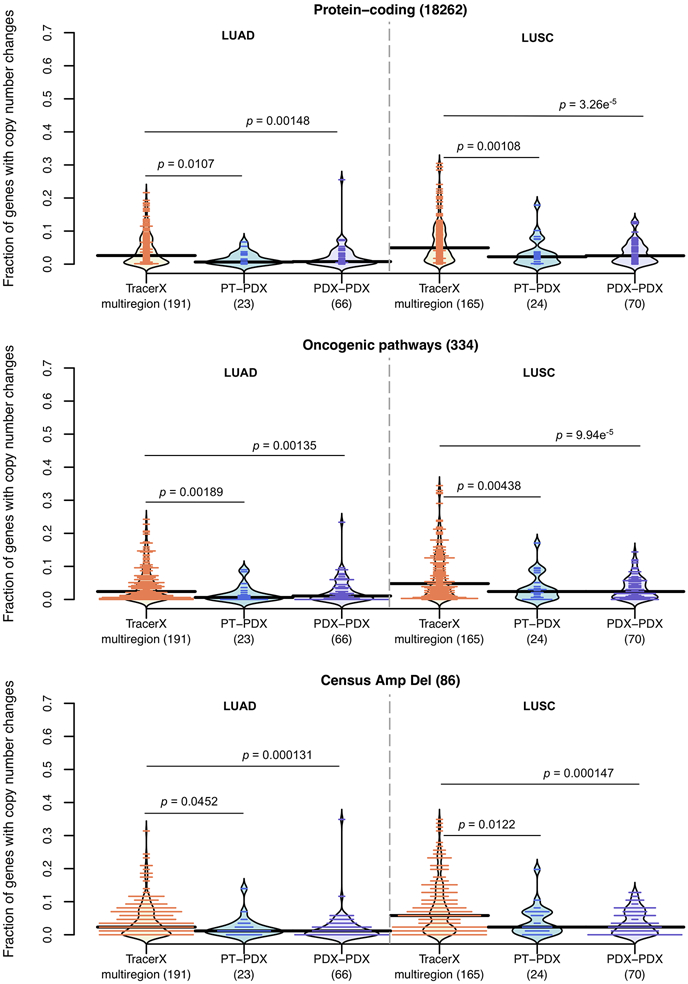
Comparison between multi-region tumor pairs from TRACERx, and PT-PDX and PDX-PDX pairs for various gene sets for LUAD and LUSC. Gene sets and CNA thresholds are the same as Figure 4, other gene sets are shown in Figure 6b. P-values were computed by one-sided Wilcoxon rank sum test. Numbers of genes per gene set are indicated in the plot title, and number of pairwise comparisons are indicated in the horizontal axis labels.
Supplementary Material
ACKNOWLEDGEMENTS
Support for the PDXNET consortium included funding provided by the NIH to the PDXNet Data Commons and Coordination Center (NCI U24-CA224067), to the PDX Development and Trial Centers (NCI U54-CA224083, NCI U54-CA224070, NCI U54-CA224065, NCI U54-CA224076, NCI U54-CA233223, and NCI U54-CA233306), and to the National Cancer Institute Cancer Genomics Cloud (HHSN261201400008C and HHSN261201500003I). The Jackson Laboratory (JAX) PDX resource data were supported by the National Cancer Institute of the National Institutes of Health under the JAX Cancer Center NCI Grant (Award Number P30CA034196). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The genomic data for JAX PDX tumors used in this work were generated by JAX Genome Technologies and Single Cell Biology Scientific Service. The development of PDX models and generation of data from Seoul National University, in collaboration with The Jackson Laboratory, was supported by the Korean Healthcare Technology R&D project through the Korean Health Industry Development Institute, funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI13C2148). C.L. is supported in part by the operational funds from The First Affiliated Hospital of Xi’an Jiaotong University. C.L. was a distinguished Ewha Womans University Professor supported in part by the Ewha Womans University Research grant of 2018-2019. Sample procurement and next generation sequencing at Huntsman Cancer Institute was performed at the Genomics and Bioinformatics Analysis and Biorepository and Molecular Pathology Shared Resources, respectively, supported by NCI P30CA042014. SNP arrays were performed at the University of Utah Health Sciences Center Genomics Core. We are grateful to Michael P. Klein for assistance with SNP array data. M.H.B. is funded by the National Institutes of Health under Ruth L. Kirschstein National Research Service Award Institutional Training Grant 5T32HG008962-05. M.T.L. is supported by a P30 Cancer Center Support Grant CA125123 and a Core Facility Support Grant from the Cancer Research and Prevention Initiative of Texas RP170691. PDX generation and whole exome sequencing at the University of Texas MD Anderson Cancer Center were supported by the University of Texas MD Anderson Cancer Center Moon Shots Program, Specialized Program of Research Excellence (SPORE) grant CA070907. J.A.R. is supported in part by the National Institutes of Health/National Cancer Institute through The University of Texas MD Anderson Cancer Center’s Cancer Center Support Grant (CCSG) CA-016672 - Lung Program and Shared Core Facilities, Specialized Program of Research Excellence (SPORE) Grant CA-070907, and Lung Cancer Moon Shot Program. The development of PDX models and generation of data from Wistar Institute was supported by National Cancer Institute, National Institutes of Health (NCI R50-CA211199). PDMR data has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health (Contract Number HHSN261200800001E). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. The breast cancer PDX models from Washington University used for this study were developed in part through the support from The Breast Cancer Research Foundation and Fashion Footwear Charitable Foundation of New York, Inc. The pancreatic cancer PDX models from Washington University used in this study were developed with the support of NCI grants P50 CA196510, P30 CA091842 and The Foundation for Barnes-Jewish Hospital’s Cancer Frontier Fund through the Siteman Cancer Center Investment Program. The data for these models were provided by U54 CA224083. Support for the EurOPDX consortium included funding provided by Fondazione AIRC under 5 per Mille 2018 - ID. 21091 program (E.M., A.B., L.T.), AIRC Investigator Grants 18532 (L.T.) and 20697 (A.B.), AIRC/CRUK/FC AECC Accelerator Award 22795 (L.T.), EU H2020 Research and Innovation Programme, grant agreement no. 731105 “EDIReX” (E.M., A.B., L.T., A.T.B., V.S., J.J.), Fondazione Piemontese per la Ricerca sul Cancro-ONLUS 5 per mille Ministero della Salute 2015 (E.M., L.T.), 2014 and 2016 (L.T.), My First AIRC Grant (MFAG) 19047 (C.I.), EU H2020 Research and Innovation Programme, Grant Agreement No. 754923 “COLOSSUS” (A.T.B., D.L., L.T.), European Research Council Consolidator Grant 724748 – BEAT (A.B.), Science Foundation Ireland under grant 13/CDA/2183 ”COLOFORETELL’ (A.T.B), Irish Health Research Board grant ILP-POR-2019-066 (A.T.B.), ISCIII - Miguel Servet program CP14/00228 and GHD-Pink/FERO Foundation grant (V.S.), Netherlands Organization for Scientific Research (NWO) Vici grant 91814643 (J.J.), European Research Council (ERC) Synergy project CombatCancer (J.J.), Oncode Institute (J.J., R.d.B.) and Dutch Cancer Society (J.J., R.d.B.) , NCI grant U24 CA204781 (J.H.C., T.F.M.). The EurOPDX consortium thank C. Saura from the Breast Cancer and Melanoma Group (VHIO) and J. Balmaña from the Hereditary Cancer Genetics Group (VHIO) for providing study samples. We thank D. Krupke from The Jackson Laboratory for assistance with organizing the tumor type information.
CONSORTIA
PDXNET Consortium
Xing Yi Woo1,#,§, Anuj Srivastava1, Zi-Ming Zhao1, Michael W. Lloyd4, Rajesh Patidar7, Li Chen7, Sandra Scherer8, Matthew Bailey8,9, Chieh-Hsiang Yang8, Emilio Cortes-Sanchez8, Yuanxin Xi10, Jing Wang10, Jayamanna Wickramasinghe11, Andrew V. Kossenkov11, Vito Rebecca11, Hua Sun12, R. Jay Mashl12, Sherri R. Davies12, Ryan Jeon13, Christian Frech13, Jelena Randjelovic13, Jacqueline Rosains13, Dennis A. Dean, II13, Brandi Davis-Dusenbery13, Yvonne A. Evrard7, James H. Doroshow21, Alana L. Welm8, Bryan E. Welm8,22, Michael T. Lewis23, Bingliang Fang24, Jack A. Roth24, Funda Meric-Bernstam25, Meenhard Herlyn11, Michael Davies26, Li Ding12, Shunqiang Li12, Ramaswamy Govindan12, Jeffrey A. Moscow27,§, Carol J. Bult4,§, Jeffrey H. Chuang1,§,*, Peter N. Robinson1, Brian J. Sanderson1, Steven B. Neuhauser4, Lacey E. Dobrolecki23, Xiaofeng Zheng10, Mourad Majidi24, Ran Zhang24, Xiaoshan Zhang24, Argun Akcakanat25, Kurt W. Evans25, Timothy A. Yap25, Dali Li25, Erkan Yucan25, Christopher D. Lanier25, Turcin Saridogan25, Bryce P. Kirby25, Min Jin Ha29, Huiqin Chen29, Scott Kopetz30, David G. Menter30, Jianhua Zhang31, Shannon N. Westin32, Michael P. Kim33, Bingbing Dai33, Don L. Gibbons34, Coya Tapia35, Vanessa B. Jensen36, Gao Boning37, John D. Minna37, Hyunsil Park37, Brenda C. Timmons37, Luc Girard37, Dylan Fingerman11, Qin Liu11, Rajasekharan Somasundaram11, Min Xiao11, Vashisht G. Yennu-Nanda26, Michael T. Tetzlaff38, Xiaowei Xu38, Katherine L. Nathanson39, Song Cao12, Feng Chen12, John F. DiPersio12, Kian H. Lim12, Cynthia X. Ma12, Fernanda M. Rodriguez12, Brian A. Van Tine12, Andrea Wang-Gillam12, Michael C. Wendl12, Yige Wu12, Matthew A. Wyczalkowski12, Lijun Yao12, Reyka Jayasinghe12, Rebecca L. Aft40, Ryan C. Fields40, Jingqin Luo12,40, Katherine C. Fuh31, Vicki Chin13, John DiGiovanna13, Jeffrey Grover13, Soner Koc13, Sara Seepo13, Tiffany Wallace42, Chong-Xian Pan43, Moon S. Chen, Jr43, Luis G. Carvajal-Carmona44, Amanda R. Kirane45, May Cho45, David R. Gandara45, Jonathan W. Riess45, Tiffany Le45, Ralph W. deVere White45, Clifford G. Tepper45, Hongyong Zhang46, Nicole B. Coggins46, Paul Lott46, Ana Estrada46, Ted Toal46, Alexa Morales Arana46, Guadalupe Polanco-Echeverry46, Sienna Rocha46, Ai-Hong Ma44, Nicholas Mitsiades47,48, Salma Kaochar47 , Bert W. O’Malley48, Matthew J. Ellis23, Susan G. Hilsenbeck23, Michael Ittmann49
29Department of Biostatistics, The University of Texas M.D. Anderson Cancer Center, Houston, TX, USA.
30Department of Gastrointestinal Medical Oncology, The University of Texas M.D. Anderson Cancer Center, Houston, TX, USA.
31Department of Genomic Medicine, The University of Texas M.D. Anderson Cancer Center, Houston, TX, USA.
32Department of Gynecologic Oncology and Reproductive Medicine, The University of Texas M.D. Anderson Cancer Center, Houston, TX, USA.
33Department of Surgical Oncology, The University of Texas M.D. Anderson Cancer Center, Houston, TX, USA.
34Department of Thoracic/Head & Neck Medical Oncology, The University of Texas M.D. Anderson Cancer Center, Houston, TX, USA.
35Department of Translational Molecular Pathology, The University of Texas M.D. Anderson Cancer Center, Houston, TX, USA.
36Department of Veterinary Medicine & Surgery, The University of Texas M.D. Anderson Cancer Center, Houston, TX, USA.
37Hamon Center For Therapeutic Oncology, UT Southwestern Medical Center, Dallas, TX, USA.
38Department of Pathology and Laboratory Medicine, Hospital of the University of Pennsylvania, Philadelphia, PA, USA.
39Abramson Cancer Center, University of Pennsylvania, Philadelphia, PA, USA.
40Department of Surgery, Washington University School of Medicine in St. Louis, St. Louis, MO, USA.
41Department of Obstetrics and Gynecology, Washington University School of Medicine in St. Louis, St. Louis, MO, USA.
42Center to Reduce Cancer Health Disparities, National Cancer Institute, Bethesda, MD, USA.
43Department of Internal Medicine, Division of Hematology/Oncology, University of California Davis, Sacramento, CA, USA.
44Department of Biochemistry and Molecular Medicine, University of California Davis, Sacramento, CA, USA.
45UC Davis Comprehensive Cancer Center, University of California Davis, Sacramento, CA, USA.
46UC Davis Genome Center, University of California Davis, Sacramento, CA, USA.
47Department of Medicine, Baylor College of Medicine, Houston, TX, USA.
48Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, TX, USA.
49Department of Pathology, Baylor College of Medicine, Houston, TX, USA.
EurOPDX Consortium
Jessica Giordano2,3,#, Roebi de Bruijn5, Francesco Galimi2,3, Andrea Bertotti2,3, Adam Lafferty14, Alice C. O’Farrell14, Elodie Modave15,16, Diether Lambrechts15,16, Petra ter Brugge5, Violeta Serra17, Elisabetta Marangoni18, Rania El Botty18, Claudio Isella2,3,§, Livio Trusolino2,3,§, Annette T. Byrne14,§, Jos Jonkers5,§, Enzo Medico2,3,§,*, Simona Corso2,3, Alessandro Fiori2,3, Silvia Giordano2,3, Marieke van de Ven5, Daniel S. Peeper5, Ian Miller14, Cristina Bernadó17, Beatriz Morancho17, Lorena Ramírez17, Joaquín Arribas17, Héctor G. Palmer17, Alejandro Piris-Gimenez17, Laura Soucek17, Ahmed Dahmani18, Elodie Montaudon18, Fariba Nemati18, Virginie Dangles-Marie18, Didier Decaudin18, Sergio Roman-Roman18, Denis G. Alférez50, Katherine Spence50, Robert B. Clarke50, Mohamed Bentires-Alj51, David K. Chang52, Andrew V. Biankin52, Alejandra Bruna53, Martin O’Reilly53, Carlos Caldas53, Oriol Casanovas54, Eva Gonzalez-Suarez54, Purificacíon Muñoz54, Alberto Villanueva54, Nathalie Conte55, Jeremy Mason55, Ross Thorne55, Terrence F. Meehan55, Helen Parkinson55, Zdenka Dudova56, Ales Křenek56, Dalibor Stuchlík56, Olivier Elemento57, Giorgio Inghirami57, Anna Golebiewska58, Simone P. Niclou58, G. Bea A. Wisman59, Steven de Jong59, Petra Kralova60, Radislav Sedlacek60, Elisa Claeys61, Eleonora Leucci61, Massimiliano Borsani62, Luisa Lanfrancone62, Pier Giuseppe Pelicci62, Gunhild Mari Mælandsmo63, Jens Henrik Norum63, Emilie Vinolo64
50Manchester Breast Centre, Division of Cancer Sciences, University of Manchester, Manchester, UK.
51University Hospital Basel, University of Basel, Basel, Switzerland.
52Institute of Cancer Sciences, University of Glasgow, Glasgow, UK.
53Cancer Research UK Cambridge Institute, Cambridge Cancer Centre, Cambridge, UK.
54Catalan Institute of Oncology, L’Hopistalet de Llobregat, Barcelona, Spain.
55European Bioinformatics Institute, European Molecular Biology Laboratory, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK.
56Institute of Computer Science, MazarykUniversity, Brno, Czech Republic.
57Weill Cornell Medical College, Cornell University, New York, NY, USA.
58NorLux Neuro-Oncology Laboratory, Department of Oncology, Luxembourg Institute of Health, Luxembourg, Luxembourg.
59University Medical Centre Groningen, Groningen, The Netherlands.
60Czech Center for Phenogenomics, Institute of Molecular Genetics, Vestec, Czech Republic.
61TRACE PDX platform, Katholieke Universiteit Leuven, Leuven, Belgium.
62European Institute of Oncology, Milan, Italy.
63Oslo University Hospital, Oslo, Norway.
64seeding science SPRL, Limelette, Belgium.
Footnotes
ETHICS COMPLIANCE
All xenograft studies were completed in accordance with animal research ethics regulations. For details, see Methods and references provided for each contributing group.
COMPETING INTERESTS
A.L.W. and B.E.W. receive a portion of royalties if University of Utah licenses certain PDX models to for-profit entities. M.T.L. is a founder of, and equity stake holder in, Tvardi Therapeutics Inc., a founder of, and limited partner in, StemMed Ltd., and a Manager in StemMed Holdings LLC. He also receives a portion of royalties if Baylor College of Medicine licenses certain PDX models to for-profit entities. J.A.R. serves as a consultant and received stocks from Genprex, Inc., and receives royalties from patents issued. F.M.-B. reports receiving commercial research grants from Novartis, AstraZeneca, Calithera, Aileron, Bayer, Jounce, CytoMx, eFFECTOR, Zymeworks, PUMA Biotechnology, Curis, Millennium, Daiichi Sankyo, Abbvie, Guardant Health, Takeda, Seattle Genetics, and GlaxoSmithKline as well as grants and travel related fees from Taiho, Genentech, Debiopharm Group, and Pfizer. She also served as a consultant to Pieris, Dialectica, Sumitomo Dainippon, Samsung Bioepis, Aduro, OrigiMed, Xencor, The Jackson Laboratory, Zymeworks, Kolon Life Science, and Parexel International, and advisor to Inflection Biosciences, GRAIL, Darwin Health, Spectrum, Mersana, and Seattle Genetics. L.T. reports receiving research grants from Symphogen, Servier, Pfizer, and Merus, and he is in the speakers’ bureau of Eli Lilly, AstraZeneca, and Merck KGaA. J.J. reports receiving funding for collaborative research from Artios Pharma. He also serves as SAB member of Artios Pharma. The other authors declare no competing financial interests.
REFERENCES
- 1.Richmond A & Su Y Mouse xenograft models vs GEM models for human cancer therapeutics. Disease models & mechanisms 1, 78–82 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walrath JC, Hawes JJ, Van Dyke T & Reilly KM Genetically engineered mouse models in cancer research. Advances in cancer research 106, 113–164 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hait WN Anticancer drug development: the grand challenges. Nature Reviews Drug Discovery 9, 253–254 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Shultz LD, Ishikawa F & Greiner DL Humanized mice in translational biomedical research. Nature Reviews Immunology 7, 118–130 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Brehm MA, Shultz LD & Greiner DL Humanized mouse models to study human diseases. Current opinion in endocrinology, diabetes, and obesity 17, 120–125 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hidalgo M et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discovery 4, 998–1013 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Byrne AT et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nature Reviews Cancer 17, 254–268 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Bruna A et al. A biobank of breast cancer explants with preserved intra-tumor heterogeneity to screen anticancer compounds. Cell 167, 260–274.e22 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reyal F et al. Molecular profiling of patient-derived breast cancer xenografts. Breast Cancer Research 14, R11 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Landis MD, Lehmann BD, Pietenpol JA & Chang JC Patient-derived breast tumor xenografts facilitating personalized cancer therapy. Breast Cancer Research 15, 201 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeRose YS et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nature Medicine 17, 1514–1520 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bankert RB et al. Humanized mouse model of ovarian cancer recapitulates patient solid tumor progression, ascites formation, and metastasis. PLOS ONE 6, e24420 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Julien S et al. Characterization of a large panel of patient-derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clinical Cancer Research 18, 5314–5328 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Lee HW et al. Patient-derived xenografts from non-small cell lung cancer brain metastases are valuable translational platforms for the development of personalized targeted therapy. Clin Cancer Res 21, 1172–1182 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Gao H et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nature Medicine 21, 1318–1325 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Hidalgo M et al. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Molecular Cancer Therapeutics 10, 1311–1316 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tentler JJ et al. Patient-derived tumour xenografts as models for oncology drug development. Nature reviews Clinical oncology 9, 338–350 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eirew P et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 518, 422–426 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cho S-Y et al. Unstable genome and transcriptome dynamics during tumor metastasis contribute to therapeutic heterogeneity in colorectal cancers. Clinical Cancer Research 25, 2821–2834 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding L et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 464, 999–1005 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giessler KM et al. Genetic subclone architecture of tumor clone-initiating cells in colorectal cancer. The Journal of Experimental Medicine 214, 2073–2088 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sato K et al. Multiregion genomic analysis of serially transplanted patient-derived xenograft tumors. Cancer Genomics Proteomics 16, 21–27 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ben-David U et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nature Genetics 49, 1567–1575 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim H et al. High-resolution deconstruction of evolution induced by chemotherapy treatments in breast cancer xenografts. Scientific Reports 8, 17937 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li S et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep 4, 1116–1130 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He S et al. PDXliver: a database of liver cancer patient derived xenograft mouse models. BMC Cancer 18, 550 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zare F, Hosny A & Nabavi S Noise cancellation using total variation for copy number variation detection. BMC Bioinformatics 19, 361 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wineinger NE & Tiwari HK The impact of errors in copy number variation detection algorithms on association results. PLOS ONE 7, e32396 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Favero F et al. Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann Oncol 26, 64–70 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Talevich E, Shain AH, Botton T & Bastian BC CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol 12, e1004873 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zack TI et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet 45, 1134–1140 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor AM et al. Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell 33, 676–689.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Loo P et al. Allele-specific copy number analysis of tumors. Proceedings of the National Academy of Sciences 107, 16910–16915 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Woo XY et al. Genomic data analysis workflows for tumors from patient-derived xenografts (PDXs): challenges and guidelines. BMC Medical Genomics 12, 92 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Evrard YA et al. Systematic establishment of robustness and standards in patient-derived xenograft experiments and analysis. Cancer Research 80, 2286–2297 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conway T et al. Xenome--a tool for classifying reads from xenograft samples. Bioinformatics 28, i172–i178 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ben-David U, Mayshar Y & Benvenisty N Virtual karyotyping of pluripotent stem cells on the basis of their global gene expression profiles. Nature Protocols 8, 989–997 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Ben-David U et al. The landscape of chromosomal aberrations in breast cancer mouse models reveals driver-specific routes to tumorigenesis. Nature Communications 7, 12160 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Motulsky HJ & Brown RE Detecting outliers when fitting data with nonlinear regression – a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinformatics 7, 123 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanchez-Vega F et al. Oncogenic signaling pathways in The Cancer Genome Atlas. Cell 173, 321–337.e10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patterson SE et al. The clinical trial landscape in oncology and connectivity of somatic mutational profiles to targeted therapies. Human Genomics 10, 4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patterson SE, Statz CM, Yin T & Mockus SM Utility of the JAX Clinical Knowledgebase in capture and assessment of complex genomic cancer data. npj Precision Oncology 3, 2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sondka Z et al. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nature Reviews Cancer 18, 696–705 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.The Cancer Genome Atlas Network et al. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.The Cancer Genome Atlas Network et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.The Cancer Genome Atlas Research Network et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.The Cancer Genome Atlas Research Network et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barretina J et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ghandi M et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beroukhim R et al. Assessing the significance of chromosomal aberrations in cancer: Methodology and application to glioma. Proceedings of the National Academy of Sciences 104, 20007–20012 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mermel CH et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biology 12, R41 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Subramanian A et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mootha VK et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature Genetics 34, 267–273 (2003). [DOI] [PubMed] [Google Scholar]
- 54.Jamal-Hanjani M et al. Tracking the evolution of non–small-cell lung cancer. New England Journal of Medicine 376, 2109–2121 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Ben-David U, Beroukhim R & Golub TR Genomic evolution of cancer models: perils and opportunities. Nature Reviews Cancer 19, 97–109 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mer AS et al. Integrative pharmacogenomics analysis of patient-derived xenografts. Cancer Research 79, 4539–4550 (2019). [DOI] [PubMed] [Google Scholar]
- 57.Isella C et al. Stromal contribution to the colorectal cancer transcriptome. Nature Genetics 47, 312–319 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Park ES et al. Cross-species hybridization of microarrays for studying tumor transcriptome of brain metastasis. Proceedings of the National Academy of Sciences 108, 17456–17461 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu Y et al. Gene expression differences between matched pairs of ovarian cancer patient tumors and patient-derived xenografts. Scientific Reports 9, 6314 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Isella C et al. Selective analysis of cancer-cell intrinsic transcriptional traits defines novel clinically relevant subtypes of colorectal cancer. Nature Communications 8, 15107 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leary RJ et al. Integrated analysis of homozygous deletions, focal amplifications, and sequence alterations in breast and colorectal cancers. Proceedings of the National Academy of Sciences 105, 16224–16229 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bierkens M et al. Focal aberrations indicate EYA2 and hsa-miR-375 as oncogene and tumor suppressor in cervical carcinogenesis. Genes, Chromosomes and Cancer 52, 56–68 (2013). [DOI] [PubMed] [Google Scholar]
- 63.Krijgsman O, Carvalho B, Meijer GA, Steenbergen RDM & Ylstra B Focal chromosomal copy number aberrations in cancer—Needles in a genome haystack. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1843, 2698–2704 (2014). [DOI] [PubMed] [Google Scholar]
- 64.Bignell GR et al. Signatures of mutation and selection in the cancer genome. Nature 463, 893–898 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.de Bruin EC et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 346, 251–256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gerlinger M et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nature Genetics 46, 225–233 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gerlinger M et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. New England Journal of Medicine 366, 883–892 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rosenthal R et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 567, 479–485 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
METHODS-ONLY REFERENCES
- 69.Schriml LM et al. Human Disease Ontology 2018 update: classification, content and workflow expansion. Nucleic Acids Research 47, D955–D962 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.The Cancer Genome Atlas Network et al. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517, 576–582 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Abeshouse A et al. Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell 171, 950–965.e28 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang K et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 17, 1665–1674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.International HapMap Consortium. The International HapMap Project. Nature 426, 789–796 (2003). [DOI] [PubMed] [Google Scholar]
- 74.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Scheinin I et al. DNA copy number analysis of fresh and formalin-fixed specimens by shallow whole-genome sequencing with identification and exclusion of problematic regions in the genome assembly. Genome research 24, 2022–2032 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Desmedt C et al. Uncovering the genomic heterogeneity of multifocal breast cancer. Journal of Pathology 236, 457–466 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Weissbein U, Schachter M, Egli D & Benvenisty N Analysis of chromosomal aberrations and recombination by allelic bias in RNA-Seq. Nature Communications 7, 12144 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lingjaerde OC, Baumbusch LO, Liestol K, Glad IK & Borresen-Dale AL CGH-Explorer: a program for analysis of array-CGH data. Bioinformatics 21, 821–822 (2005). [DOI] [PubMed] [Google Scholar]
- 79.Redon R et al. Global variation in copy number in the human genome. Nature 444, 444–454 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thorvaldsdottir H, Robinson JT & Mesirov JP Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in Bioinformatics 14, 178–192 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Skidmore ZL et al. GenVisR: Genomic Visualizations in R. Bioinformatics 32, 3012–3014 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang XM et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Research 73, 4885–4897 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Coussy F et al. A large collection of integrated genomically characterized patient-derived xenografts highlighting the heterogeneity of triple-negative breast cancer. International Journal of Cancer 145, 1902–1912 (2019). [DOI] [PubMed] [Google Scholar]
- 85.Riaz N et al. Pan-cancer analysis of bi-allelic alterations in homologous recombination DNA repair genes. Nature Communications 8, 857 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Adams DJ et al. NAMPT is the cellular target of STF-31-like small-molecule probes. ACS Chemical Biology 9, 2247–2254 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Viswanathan VS et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stransky N et al. Pharmacogenomic agreement between two cancer cell line data sets. Nature 528, 84–87 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liberzon A et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liberzon A et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Copy number calls from all datasets are available in Supplementary Data 1, and these are used for all figures. Raw sequence data for these calls are a combination of previously described sources (notably the publicly available NCI Patient Derived Models Repository, pdmr.cancer.gov) and newly sequenced data. New sequence data from the PDXNet are being shared as part of the NCI Cancer Moonshot initiative through the Cancer Data Service. For further details, contact the authors. The SNP array data generated by The Jackson Laboratory can be requested via the Mouse Models of Human Cancer Database (tumor.informatics.jax.org). The whole genome sequencing data generated by EurOPDX can be made available by directly contacting the EurOPDX consortium (dataportal.europdx.eu). Other publicly available data used in the analyses include GSE90653, GSE3526, GSE33006 and E-MTAB-1503-3, CCLE cell line genomic and drug response data (CTRP v2), MSigDB v6.2 and TRACERx NSCLC data (DOI: 10.1056/NEJMoa1616288).