

Abstract
Background
A better understanding of the pathophysiology involving coronary artery calcification (CAC) in patients on hemodialysis (HD) will help to develop new therapies. We sought to identify the differences in metabolomics profiles between patients on HD with and without CAC.
Methods
In this case-control study, nested within a cohort of 568 incident patients on HD, the cases were patients without diabetes with a CAC score >100 (n=51), and controls were patients without diabetes with a CAC score of zero (n=48). We measured 452 serum metabolites in each participant. Metabolites and pathway scores were compared using Mann–Whitney U tests, partial least squares–discriminant analyses, and pathway enrichment analyses.
Results
Compared with controls, cases were older (64±13 versus 42±12 years) and were less likely to be Black (51% versus 94%). We identified three metabolites in bile-acid synthesis (chenodeoxycholic, deoxycholic, and glycolithocholic acids) and one pathway (arginine/proline metabolism). After adjusting for demographics, higher levels of chenodeoxycholic, deoxycholic, and glycolithocholic acids were associated with higher odds of having CAC; comparing the third with the first tertile of each bile acid, the OR was 6.34 (95% CI, 1.12 to 36.06), 6.73 (95% CI, 1.20 to 37.82), and 8.53 (95% CI, 1.50 to 48.49), respectively. These associations were no longer significant after further adjustment for coronary artery disease and medication use. Per 1 unit higher in the first principal component score, arginine/proline metabolism was associated with CAC after adjusting for demographics (OR, 1.83; 95% CI, 1.06 to 3.15), and the association remained significant with additional adjustments for statin use (OR, 1.84; 95% CI, 1.04 to 3.27).
Conclusions
Among patients on HD without diabetes mellitus, chenodeoxycholic, deoxycholic, and glycolithocholic acids may be potential biomarkers for CAC, and arginine/proline metabolism is a plausible mechanism to study for CAC. These findings need to be confirmed in future studies.
Introduction
In patients with ESKD on hemodialysis (HD), coronary artery calcification (CAC) is prevalent and independently predicts the risk of cardiovascular disease, which is the leading cause of death in this population (1–6). The mechanisms of CAC in patients on HD beyond the passive process of calcium-phosphate precipitation are not well understood. In this hypothesis-generating study, we used metabolomic profiling to identify new biomarkers and pathways for CAC among patients on HD. Metabolomic profiling, which studies small molecules such as amino acids in biologic specimens, may provide more insight into the factors involved in cardiovascular calcification. In general, the metabolome is downstream of transcriptional and translational processes and incorporates inputs from diet, environment, and the microbiome—thus, metabolomics reflects a summative assessment of genes, proteins, and exogenous factors (7). In addition, metabolomics has the potential for rapid translational application, because some metabolites may be amenable to therapeutic targeting (8).
Previous studies have examined serum metabolites and cardiovascular mortality in patients on HD (9,10), but none have studied the relationship between serum metabolites and cardiovascular calcification. We performed a case-control study, nested in an existing HD cohort from the Predictors of Arrhythmic and Cardiovascular Risk in ESKD (PACE) study, and compared serum metabolites in patients on HD who were nondiabetic with and without CAC. For metabolites and pathways identified through this analysis, we further explored their relationships with mineral metabolism, calcification inhibitors, and inflammation.
Materials and Methods
Study Design and Population
We conducted a nested, case-control study within PACE, a prospective cohort designed to determine cardiovascular and dialysis-related risk factors in patients on HD (11). PACE was approved by the Johns Hopkins School of Medicine and MedStar Institutional Review Boards. A total of 568 incident patients on HD (i.e., on HD for <6 months) were recruited from 25 freestanding outpatient HD units and two hospital-based outpatient units in Baltimore, Maryland and its surrounding area from 2008 to 2012. CAC was measured at the baseline visit using computed tomography (CT) scanning and was quantified using Agatston score (11). All PACE participants were evaluated for potential inclusion into our metabolomics study. A total of 130 participants who were nondiabetic had serum stored and underwent CAC measurement (Figure 1, Supplemental Methods, Supplemental References; n was used to denote the number of participants, and m for the number of metabolites). We limited our study to participants without diabetes mellitus to avoid potential confounding effects of diabetes-associated metabolic dysregulation (12). Controls (n=48) were participants with a CAC score of zero. Cases (n=51) were patients defined as having a CAC score >100. The remaining 31 participants with CAC scores between zero and 100 were not included in the study. CT is a sensitive method for detecting coronary calcium, with a sensitivity as high as 99% using a cutoff score of zero; however, its specificity is lower, approximately 65% (13,14). By using a cutoff of 100 to define cases, we increased the specificity to 77% (14). We compared the baseline characteristics of study population with unselected participants in Supplemental Table 1. Cases and controls were not matched.
Figure 1. |. We conducted a case-control study nested in the Predictors of Arrhythmic and Cardiovascular Risk in ESKD (PACE) cohort and compared serum metabolites and pathway scores between cases and controls.
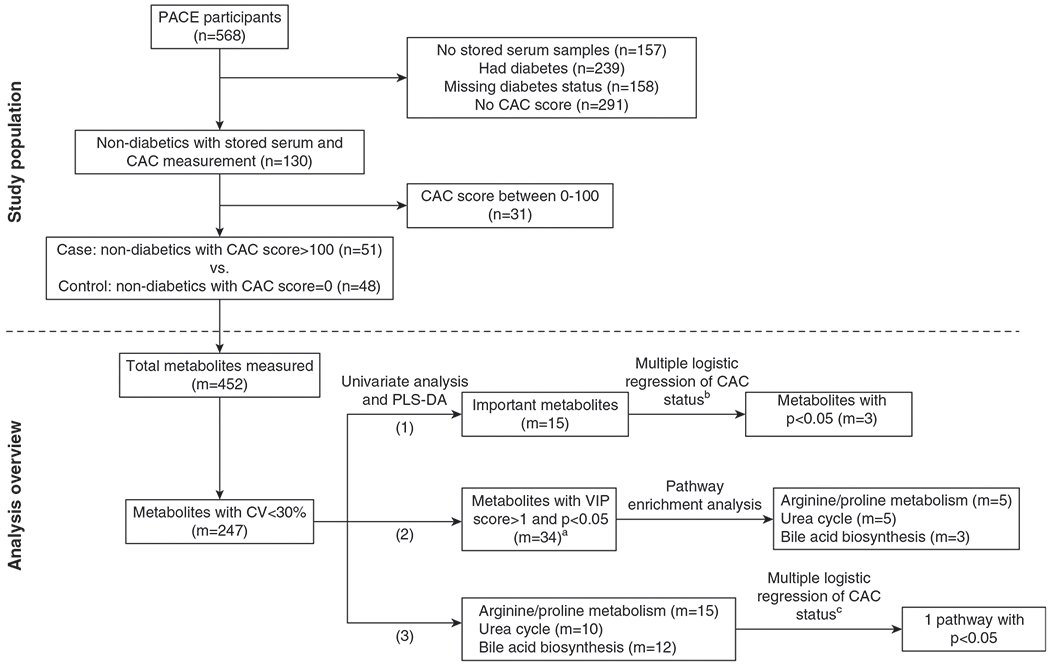
Cases were participants without diabetes with a coronary artery calcification (CAC) score >100 (n=551), whereas controls were those with a CAC score of 0 (n=548). Metabolites (m=5247) and pathway scores were compared between cases and controls. We identified three metabolites and one pathway that remained significant after adjusting for age and race. aThere were 36 metabolites with variable importance in projection (VIP) score greater than one and P value <0.05. Two of these metabolites do not have Human Metabolome Database identification: lumazine (food component) and 3-hydroxybenzaldehyde (chemical), and they were not included in the pathway analysis. bAdjusted for age and race using each metabolite. cAdjusted for age and race using the first principal component of the pathways. CV, coefficient of variation; m, number of metabolites; n, number of participants; PLS-DA, partial least squares–discriminant analyses.
Measurement of Serum Metabolites
Serum was collected on a non-HD day, at the baseline visit after approximately 8 hours of fasting, and stored at −80°C. Using triple quadrupole mass spectrometry (AB Sciex 6500+ QTRAP; Sciex, Concord, Canada) coupled with a Waters (Milford, MA) Ultra Performance Liquid Chromatography system as previously described (15), we performed widely targeted metabolomics (452 metabolites) in stored serum. A pooled quality-control sample was injected six times and used to calculate the coefficients of variation (CVs). A distribution of CVs is shown in Supplemental Figure 1A. Metabolites with a CV <30% (n=247) were included in the analyses, and their pathways are shown in Supplemental Figure 1B. Missing metabolite values (1%) were replaced by half of the minimum positive value in the original dataset.
Measurement of Covariates
Potential confounders included demographics, education level, smoking history, comorbidities, medication use, dialysis access, and dialysis clearance. Participants’ demographic factors (age, sex, and race), education level, smoking history, and medication use were self-reported. Comorbidities, including diabetes mellitus, coronary artery disease (CAD), and hypertension, were adjudicated by a committee of physicians. Dialysis clearance was assessed by singlepool Kt/V. We also measured serum markers of mineral metabolism (calcium, phosphorous, intact parathyroid hormone, C-terminal fibroblast growth factor 23 [FGF-23], and soluble klotho); circulating inhibitors of cardiovascular calcification (osteoprotegerin, dephosphorylated and uncarboxylated matrix glutamate protein [dp-ucMGP], and fetuin-A); secondary calciprotein particle size, and the time of half maximal transformation from primary to secondary calciprotein particle (T50); and high-sensitive C-reactive protein. Details of these measurements are available in Supplemental Methods.
Statistical and Pathway Analyses
Baseline participant characteristics were examined by CAC status using two-sample t test, Mann–Whitney U test, or chi-squared test, as appropriate. Metabolomic data were normalized using auto scaling (centered on the mean and divided by the SD of each metabolite). Figure 1 includes an analysis flowchart to identify significant metabolites and pathways.
First, we compared each metabolite (non-normally distributed) by the CAC status using Mann–Whitney U tests and partial least squares–discriminant analyses (PLS-DA). For Mann–Whitney U tests, we used a P value threshold of <0.005 and a fold-difference threshold of 1.0 to obtain approximately ten metabolites that differentiated cases from controls. The results were presented in a volcano plot. PLS-DA is commonly used to analyze metabolomics datasets because of its ability to analyze data with high colinearity (16). For PLS-DA, we identified the top 15 metabolites on the basis of the variable importance in project (VIP) scores. For the top metabolites (m=15) identified by Mann–Whitney U tests or PLS-DA, we examined the association of each metabolite with CAC using multiple logistic-regression models, adjusting for demographics (age, sex, and race). For the metabolites that had a P value <0.05 after adjusting for demographics (m=3; chenodeoxycholic, deoxycholic, and glycolithocholic acids), we generated fully adjusted models using backward elimination. In addition to individual metabolite (in continuous scale) and demographics, backward elimination selected from the following nine covariates: body mass index; smoking history; education level; single-pool Kt/V; CAD; and medication use, including vitamin D therapy, calcium-based phosphate binder, renin-angiotensin-aldosterone system (RAAS) blockade, and statin. The significance threshold to remove a covariate from the models was 0.1. The backward-elimination approach generated the same set of covariates for chenodeoxycholic, deoxycholic, and glycolithocholic acids, and the final full models were adjusted for demographics; CAD; and use of calcium-base phosphate binder, RAAS blockade, and statin. Additionally, we refitted logistic models using tertiles of the three bile acids, and log-transformed the bile acids to examine their correlations with each other using Pearson correlation.
Second, we performed pathway-enrichment analyses using the Small Molecule Pathway Database. To produce robust results, we used the metabolites that met the following three criteria: a VIP score greater than one, P value <0.05, and available human metabolome database identification (m=34). The top three pathways were identified on the basis of the combination of pathway significance and impact. Pathway significance was assessed using global tests with a false discovery rate–adjusted P value threshold of 0.05, and pathway impact was assessed using topology analysis with relative-betweenness centrality.
Third, we tested the associations between the top three pathways and CAC status. To do that, we used principal component analysis and metabolites with CV <30% (m=247) to score the top three pathways identified in step 2. The first principal component (PC1) score of each pathway was then used to represent the pathway. We examined the association between the PC1 of each pathway with CAC using multiple logistic regression. Models were first adjusted for demographics, and then, similar to step 1, we generated fully adjusted models using backward elimination, which selected covariates in addition to individual PC1 of each pathway and demographics. The fully adjusted models were adjusted for demographics and statin use for arginine/proline-metabolism and urea-cycle pathways. For the bile-acid synthesis pathway, models were adjusted for demographics; CAD; and use of calcium-based phosphate binder, RAAS blockade, and statin. For secondary analyses, we used Tobit regression to simultaneously model the presence and severity of CAC, and to examine their associations with the key metabolites and pathways identified in step 1 and 2 (17). Because CAC score has a right skewed distribution and zero scores, we log-transformed CAC score plus 1, and used left censoring at zero and bootstrap techniques with 999 repetitions.
Lastly, using Spearman rank correlation, we examined the correlations of key metabolites and/or pathways with the markers of mineral metabolism, inflammation, circulating inhibitors of cardiovascular calcification, and properties of calciprotein particle transformation. MetaboAnalyst 4.0 (McGill University, Montreal, Canada) (18) and STATA 14.1 (StataCorp, College Station, TX) were used for statistical and pathway analyses. A two-sided P value of <0.05 was considered statistically significant in the multiple logistic and Tobit regression models.
Results
Participant Characteristics
As shown in Table 1, the mean age of participants was 53616 years, 38% were women, and 72% were Black. All participants had hypertension and 27% had CAD. Among cases, the median CAC score was 466 (interquartile range, 246–981). Compared with controls, cases were older (64±13 versus 42±12 years), less likely to be Black (51% versus 94%), and more likely to have CAD (45% versus 8%). Cases were also less likely to be taking medications that blocked RAAS (36% versus 58%) and were more likely to be on statin therapy (42% versus 13%).
Table 1.
Baseline participant characteristics by CAC status
| Characteristics | Total (n=99) | Control (CAC Score=0, n=48) | Case (CAC Score >100, n=51) | P Value |
|---|---|---|---|---|
| Age, yr | 53±16 | 42±12 | 64±13 | <0.001 |
| Women, n (%) | 38 (38) | 19 (40) | 19 (37) | 0.81 |
| Black race, n (%) | 71 (72) | 45 (94) | 26 (51) | <0.001 |
| Body mass index, kg/m2 | 27±6 | 28±7 | 27±5 | 0.66 |
| History of smoking, n (%) | 67 (68) | 30 (63) | 37 (73) | 0.29 |
| Education less than grade 11, n (%) | 44 (44) | 24 (50) | 20 (39) | 0.50 |
| Coronary artery disease, n (%) | 27 (27) | 4 (8) | 23 (45) | <0.001 |
| Medication use | ||||
| RAAS blockade, n (%) | 41 (46) | 23 (58) | 18 (36) | 0.04 |
| Calcium-based phosphate binder, n (%) | 37 (37) | 15 (31) | 22 (43) | 0.22 |
| Vitamin D therapy, n (%) | 69 (70) | 34 (71) | 35 (69) | 0.81 |
| Statin use, n (%) | 27 (27) | 6 (13) | 21 (42) | 0.001 |
| Single-pool Kt/V | 1.8±0.3 | 1.8±0.3 | 1.9±0.3 | 0.08 |
| Dialysis access, n (%) | 0.31 | |||
| AVF | 32 (32) | 12 (25) | 20 (39) | |
| AVG | 5 (5) | 3 (6) | 2 (4) | |
| Catheter | 62 (63) | 33 (69) | 29 (57) | |
| Markers of mineral metabolism | ||||
| Serum calcium, mg/dl | 8.7±0.7 | 8.7±0.7 | 8.7±0.7 | 0.82 |
| Serum phosphorous, mg/dl | 5.3±1.2 | 5.4±1.4 | 5.2±1.0 | 0.37 |
| Serum magnesium, mEq/L | 1.8±0.2 | 1.8±0.2 | 1.7±0.2 | 0.36 |
| Intact parathyroid hormone, pg/ml | 400 (270–581) | 402 (293–583) | 387 (252–580) | 0.49 |
| FGF-23, RU/ml | 777 (222–1310) | 819 (171–1284) | 754 (316–1500) | 0.53 |
| Soluble klotho, pg/ml | 356 (268–489) | 397 (285–524) | 306 (263–453) | 0.05 |
| Markers of cardiovascular calcification | ||||
| Fetuin-A, mg/L | 533±193 | 547±192 | 519±195 | 0.48 |
| dp-ucMGP, ppm | 1441 (967–2042) | 1258 (783–1800) | 1734 (1101–2437) | 0.02 |
| Osteoprotegerin, pmol/L | 10.1±4.8 | 8.2±3.4 | 11.8±5.2 | <0.001 |
| CPP2 size, nm | 289 (192–394) | 289 (187–394) | 289 (207–394) | 0.97 |
| T50, min | 307 (227–383) | 306 (252–378) | 315 (213–398) | 0.71 |
| C-reactive protein, μg/ml | 4.5 (1.7–11.4) | 3.4 (1.5–8.7) | 5.3 (2.1–17.1) | 0.18 |
If normally distributed, values for continuous variables with normal distribution are provided as mean±SD and were tested with two-sample t test. Otherwise, they are provided as median (interquartile range) and were tested using Mann–Whitney test. Categoric variables are presented as absolute number with percentage and were tested with chi-square test. CAC, coronary artery calcification; RAAS, renin-angiotensin-aldosterone system; AVF, arteriovenous fistula; AVG, arteriovenous graft; FGF-23, fibroblast growth factor23; dp-ucMGP, dephosphorylated and uncarboxylated matrix glutamate protein; CPP2, secondary calciprotein particle; T50, time of half maximal transformation from primary to secondary calciprotein particle.
Identification of Significant Metabolites
Using the Mann–Whitney U test, and a set threshold of fold difference (1.0) and raw P value (0.005), we identified nine metabolites that differed between cases and controls, as shown in the volcano plot (Supplemental Figure 2, Supplemental Table 2). Compared with the controls, bile-acid levels of chenodeoxycholic acid, deoxycholic acid, and glycolithocholic acid were more than two-fold higher in cases (Figure 2). Using PLS-DA, we identified the top 15 metabolites on the basis of VIP scores (Supplemental Figures 3 and 4). The metabolites that were identified by the volcano plot and PLS-DA are summarized in Supplemental Table 2. In addition to the nine metabolites shown in the volcano plot, PLS-DA identified three metabolites involved in arginine/proline metabolism: arginine, ornithine, and succinic acid (Figure 2).
Figure 2. |. Cases and controls had different levels of selected metabolites in arginine and proline metabolism, urea cycle, and bile-acid synthesis.
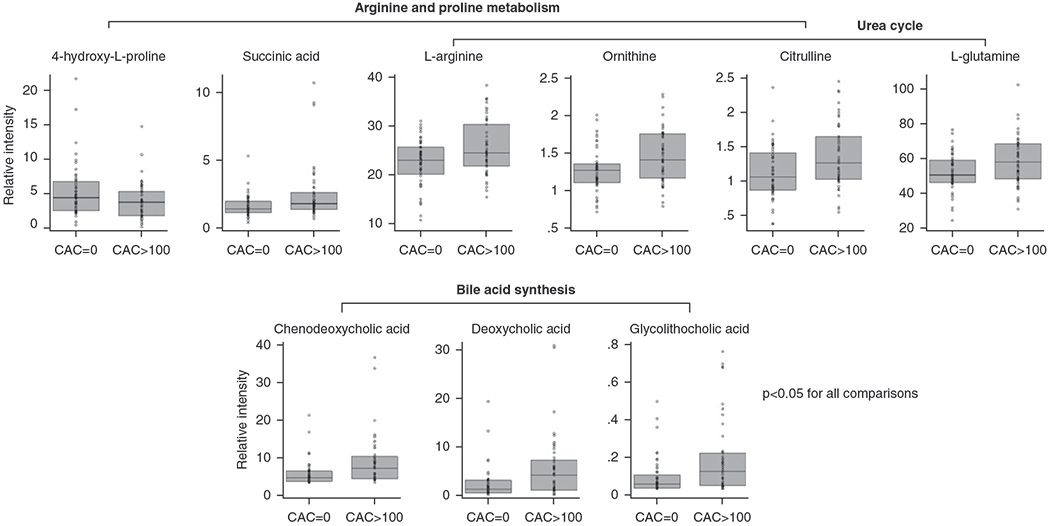
Mann–Whitney U tests were used to compare metabolites between cases (CAC >100) and controls (CAC =0). Metabolites with a P value <0.05 are presented above. l-Arginine (fold difference, 1.14; P=0.03), ornithine (fold difference, 1.17; P=0.008), and citrulline (fold difference, 1.21; P=0.01) are in both arginine and proline metabolism and urea cycle. 4-Hydroxy-l-proline (fold difference, 0.74; P=0.04) and succinic acid (fold difference, 1.44; P=0.005) are only in arginine and proline metabolism, whereas l-glutamine (fold difference, 1.12; P=0.02) is only in the urea-cycle pathway. Chenodeoxycholic acid (fold difference, 2.33; P=0.002), deoxycholic acid (fold difference, 2.32; P=0.001), and glycolithocholic acid (fold difference, 2.44; P=0.004) are in the bile-acid synthesis pathway. Box plots represent median and interquartile range.
After adjusting for demographics, only bile acids remained significantly associated with CAC status (Supplemental Table 2). Higher levels of chenodeoxycholic, deoxycholic, and glycoolithocholic acids were associated with higher odds of CAC. After adjusting for demographics, compared with the participants with bile-acid levels in the first tertile, those in the third tertile had higher odds of CAC with an odds ratio (OR) of 6.34 (95% CI, 1.12 to 36.06; P=0.04), 6.73 (95% CI, 1.20 to 37.82; P=0.03), and 8.53 (95% CI, 1.50 to 48.49; P=0.02) for chenodeoxycholic, deoxycholic, and glycolithocholic acid, respectively (Table 2). However, the associations were no longer significant after additional adjustment for CAD and use of calcium-based phosphate binder, RAAS blockade, and statin.
Table 2.
Logistic regression of CAC status with chenodeoxycholic, deoxycholic, and glycolithocholic acids
| Metabolites | Cases (n) | Unadjusted |
Adjusted for Demographics |
Fully Adjusted |
|||
|---|---|---|---|---|---|---|---|
| OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | ||
| Chenodeoxycholic acid | |||||||
| Continuousa | — | 1.17 (1.04 to 1.33) | 0.01 | 1.18 (1.01 to 1.38) | 0.04 | 1.14 (0.97 to 1.34) | 0.11 |
| Tertile 1 | 12 | Reference | Reference | Reference | |||
| Tertile 2 | 13 | 1.14 (0.42 to 3.08) | 0.80 | 0.84 (0.17 to 4.19) | 0.83 | 0.04 (0.001 to 1.03) | 0.05 |
| Tertile 3 | 26 | 6.5 (2.17 to 19.43) | 0.001 | 6.34 (1.12 to 36.06) | 0.04 | 1.23 (0.11 to 13.26) | 0.87 |
| Deoxycholic acid | |||||||
| Continuousa | — | 1.18 (1.04 to 1.34) | 0.01 | 1.17 (1.00 to 1.37) | 0.049 | 1.14 (0.97 to 1.33) | 0.10 |
| Tertile 1 | 11 | Reference | Reference | Reference | |||
| Tertile 2 | 14 | 1.47 (0.54 to 4.01) | 0.45 | 0.93 (0.19 to 4.61) | 0.93 | 0.05 (0.002 to 1.28) | 0.07 |
| Tertile 3 | 26 | 7.43 (2.46 to 22.42) | <0.001 | 6.73 (1.20 to 37.82) | 0.03 | 1.58 (0.16 to 15.66) | 0.69 |
| Glycolithocholic acid | |||||||
| Continuousa | — | 1.05 (1.01 to 1.09) | 0.01 | 1.07 (1.00 to 1.13) | 0.04 | 1.04 (0.97 to 1.12) | 0.27 |
| Tertile 1 | 11 | Reference | Reference | Reference | |||
| Tertile 2 | 16 | 1.88 (0.70 to 5.09) | 0.21 | 1.60 (0.31 to 8.10) | 0.57 | 1.45 (0.16 to 13.52) | 0.74 |
| Tertile 3 | 24 | 5.33 (1.86 to 15.30) | 0.002 | 8.53 (1.50 to 48.49) | 0.02 | 3.78 (0.32 to 44.84) | 0.29 |
n=33 in each tertile. Demographics included age, sex, and race. Full models were adjusted for demographics; coronary artery disease; and use of calcium-based phosphate binder, renin-angiotensin-aldosterone system blockade, and statin for all three bile acids. CAC, coronary artery calcification; OR, odds ratio.
Units for continuous variables were 1 unit in relative intensity for chenodeoxycholic acid and deoxycholic acid, and 0.01 unit in relative intensity for glycolithocholic acid.
Chenodeoxycholic, deoxycholic, and glycolithocholic acids were correlated with each other (Supplemental Figure 5). For secondary analyses, Tobit regression revealed overall similar observations as the logistic regression (Supplemental Table 3). After adjusting for demographics, the difference in the CAC score of those with a CAC score greater than zero (weighted by the probability of having a CAC score greater than zero) and in the probability of having CAC score greater than zero (weighted by the expected value of CAC score if greater than zero) was 0.15 per 1 unit higher in relative intensity of chenodeoxycholic acid (P=0.02). The association remained significant after further adjustment for CAD and medication use (P=0.03). Deoxycholic and glycolithocholic acids were associated with CAC using Tobit regression after adjusting for demographics, but not in the fully adjusted models.
Identification of Significant Pathways
Pathway-enrichment analyses revealed three top pathways: arginine/proline metabolism, urea cycle, and bile-acid synthesis (Table 3). Metabolites analyzed in these three pathways are shown in Supplemental Table 4. When comparing cases with controls, five metabolites in arginine/proline metabolism, four in urea cycle, and three in bile-acid synthesis had a P value <0.05 (Figure 2). l-Arginine, ornithine, and citrulline were present in both arginine/proline-metabolism and urea-cycle pathways.
Table 3.
Impact and significance of arginine/proline metabolism, urea cycle, and bile-acid synthesis
| Pathway | Metabolites in SMPD Pathway (m) | Metabolites Analyzed (m) | Pathway Impact | FDR-Adjusted P |
|---|---|---|---|---|
| Arginine and proline metabolism | 53 | 15 | 0.21 | 5.8×10−4 |
| Urea cycle | 29 | 10 | 0.23 | 0.002 |
| Bile-acid synthesis | 65 | 12 | 0.01 | 0.005 |
For pathway-enrichment analyses, we used SMPD and included 34 metabolites that had a variable importance in projection score greater than one and P value <0.05. Global tests were used to assess the significance of pathways, and topology analysis with relative betweenness centrality was used to assess pathway impact. SMPD, Small Molecule Pathway Database; FDR, false discovery rate.
We used principal component analysis to generate scores for the three pathways. The PC1 score explained 20% of variance in the arginine/proline metabolism, 24% in urea cycle, and 37% in bile-acid synthesis (Supplemental Figure 6). PC1s of arginine/proline metabolism and urea cycle were associated with CAC status, but only PC1 of arginine/proline metabolism remained significant after adjusting for demographics and statin use, with an adjusted OR of 1.84 (95% CI, 1.04 to 3.27) per 1 unit higher of PC1 score (P=0.04; Table 4). In secondary analyses using Tobit regression, we found that the presence and severity of CAC were associated with the PC1 of both arginine/proline-metabolism (P=0.02) and urea-cycle pathways (P=0.04) in the fully adjusted model, but not with PC1 of bile-acid synthesis (Supplemental Table 3).
Table 4.
Logistic regression of CAC status with arginine/proline metabolism, urea cycle, and bile-acid synthesis
| Pathway | Unadjusteda |
Adjusted for Demographics |
Fully Adjusted |
|||
|---|---|---|---|---|---|---|
| OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | |
| Arginine/proline metabolism | 1.43 (1.06 to 1.92) | 0.02 | 1.83 (1.06 to 3.15) | 0.03 | 1.84 (1.04 to 3.27) | 0.04 |
| Urea cycle | 1.93 (1.34 to 2.80) | <0.001 | 1.58 (0.93 to 2.69) | 0.09 | 1.66 (0.94 to 2.91) | 0.08 |
| Bile-acid synthesis | 0.99 (0.82 to 1.20) | 0.92 | 1.09 (0.84 to 1.41) | 0.51 | 1.64 (0.97 to 2.75) | 0.06 |
Demographics included age, sex, and race. Full models were adjusted for demographics and statin use for arginine/proline metabolism and urea cycle pathways; for the bile-acid synthesis pathway, full models were adjusted for demographics; coronary artery disease; and use of calcium-based phosphate binder, RAAS blockade, and statin. CAC, coronary artery calcification; OR, odds ratio; RAAS, renin-angiotensin-aldosterone system.
Per 1 unit higher in the first principal component score of each pathway.
Correlations with Significant Metabolites and Pathways
We then examined the correlations of bile-acid synthesis and arginine/proline-metabolism pathways and their key metabolites with serum markers of mineral metabolism and inflammation, circulating inhibitors of cardiovascular calcification, and properties of calciprotein particles. Results are shown in a heat map (Figure 3) and in Supplemental Table 5. The PC1 score of bile-acid synthesis was positively correlated with FGF-23 and osteoprotegerin levels, and negatively correlated with T50. Chenodeoxycholic, deoxycholic, and glycolithocholic acids correlated negatively with C-reactive protein, but were not correlated with serum markers of mineral metabolism or circulating inhibitors of calcification. The PC1 score of arginine/proline metabolism correlated positively with levels of serum phosphorous, FGF-23, osteoprotegerin, and dp-ucMGP.
Figure 3. |. Correlations of bile-acid synthesis and arginine/proline-metabolism pathways and their key metabolites with serum markers of mineral metabolism (Ca, Phos, Mg, PTH, FGF23, klotho), circulating inhibitors of calcification (osteoprotegerin, dp-ucMGP, fetuin-A, CPP2, T50), and inflammation (CRP).
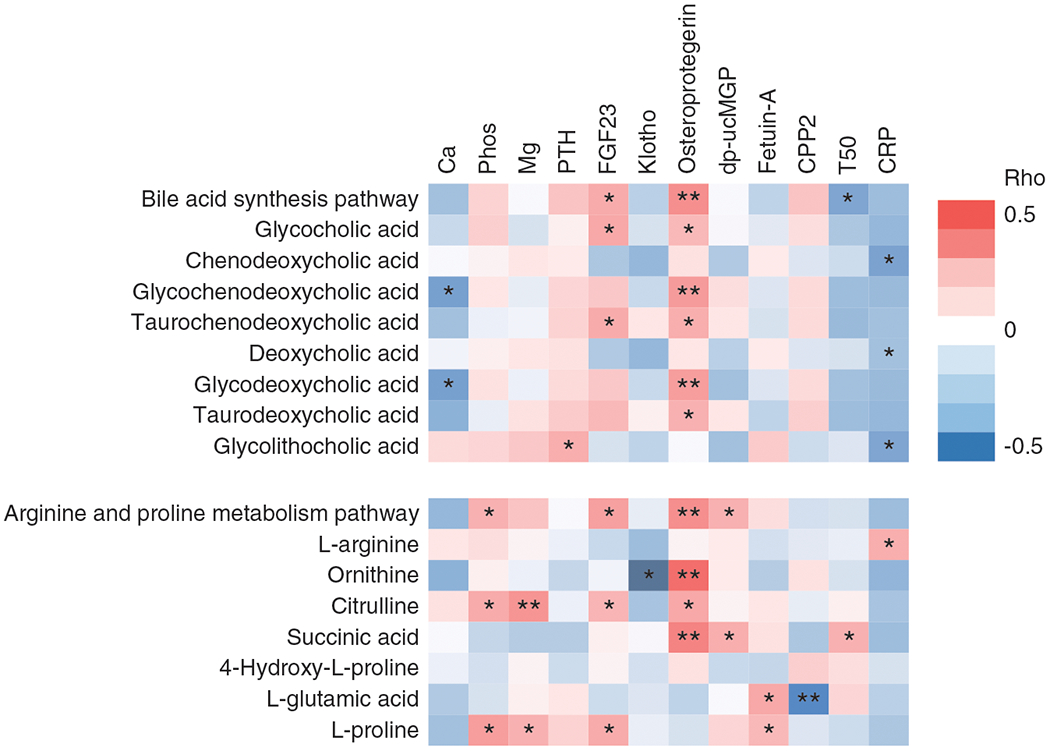
The first principal component scores were used to represent the pathways. Heat map shows Spearman rank correlations. Correlation strength is represented by color bar (red, positive correlation; white, no correlation; blue, negative correlation). *P<0.05; **P<0.005. Ca, calcium; CPP2, secondary calciprotein particle; CRP, C-reactive protein; dp-ucMGP, dephosphorylated and uncarboxylated matrix glutamate protein; FGF-23, fibroblast growth factor-23; klotho, soluble klotho; Mg, magnesium; phos, phosphorous; PTH, intact parathyroid hormone; T50, time of half maximal transformation from primary to secondary calciprotein particle.
Discussion
In patients with ESKD on HD, CAC is a prevalent and independent predictor of cardiovascular morbidity and mortality (2–6,19). In this nested, case-control study, we compared metabolites and pathway scores in 99 incident patients on HD who were nondiabetic with a CAC score >100 and a CAC score of zero. Compared with those with a CAC score of zero, patients with a CAC score >100 have more than three times the risk of coronary events (13,20). We identified three metabolites in bile-acid synthesis (chenodeoxycholic, deoxycholic, and glycolithocholic acids) and one metabolic pathway (arginine/proline metabolism) that were associated with CAC.
We found that higher levels of chenodeoxycholic, deoxycholic, and glycolithocholic acids were associated with CAC after adjusting for demographics. Similarly, in a study of patients with predialysis CKD, higher serum deoxycholic acid levels were associated with greater CAC after adjusting for demographics, comorbidities, and kidney function (21). However, after we further adjusted for comorbidities and medication use, the association between bile acids and CAC status was no longer significant. The lack of association in the fully adjusted logistic models could be due to small sample size. In the sensitivity analyses using Tobit regression, chenodeoxycholic acid remained significant after adjusting for demographics, CAD, and medication use. Compared with logistic regression, Tobit regression is more likely to identify predictors for cardiovascular calcification (17).
Elevated bile-acid levels may reflect disruption of intestinal barrier function or alteration in the intestinal microbiome, making them potential candidates as biomarkers for CAC in patients on HD. Bile acids are important for intestinal nutrient absorption and biliary secretion of lipid and toxic metabolites (Supplemental Figure 7A) (22). Chenodeoxycholic acid is a primary bile acid, because it is directly synthesized from cholesterol in liver (23). After synthesis, chenodeoxycholic acid is secreted into the intestinal tract, where it is converted by intestinal bacteria to lithocholic acid, a secondary bile acid (24). Conjugation of lithocholic acid with glycine forms glycolithocholic acid, increasing solubility and decreasing toxicity (22,25). Cholic acid, another primary bile acid, is also converted in the intestinal tract to deoxycholic acid, a secondary bile acid. After bile acids are secreted into the intestinal tract, they are reabsorbed in the ileum, or by portal circulation, back to the liver. This enterohepatic circulation of bile acids is generally highly efficient (22). Patients on HD may have a disrupted intestinal barrier, which results in translocation of bile acids, endotoxins, and bacterial metabolites in the systemic circulation (26–28). These translocated metabolites and toxins may then contribute to chronic inflammation and, ultimately, to cardiovascular disease (29–32).
Animal studies have shown that bile acids may have direct effects on cardiovascular calcification via the farnesoid X receptor, a bile acid nuclear receptor (33,34). In our study, bile-acid synthesis was one of the top three pathways identified by the pathway-enrichment analyses, but its pathway impact was low compared with that of arginine/proline metabolism and urea cycle (0.01 versus 0.21 and 0.23), and the association between the bile-acid synthesis pathway and CAC status was not significant. We also did not observe any significant correlation of bile acids with serum markers of mineral metabolism or circulating inhibitors of calcification. Our findings do not support a causal relationship between bile-acid synthesis and the development of CAC.
We found that arginine/proline metabolism was associated with CAC in both fully adjusted logistic and Tobit regression models. The urea-cycle pathway has three common metabolites with arginine/proline metabolism (i.e., l-arginine, ornithine, and citrulline), and was associated with CAC only in Tobit regression. Supplemental Figure 7B illustrates the intracellular arginine/proline metabolism, one of the central pathways for the biosynthesis of arginine and proline from glutamine (35–37). Compared with controls, cases had higher levels of l-arginine, ornithine, citrulline, and succinic acid, and lower levels of 4-hydroxy-l-proline. None of the amino acids were associated with CAC after adjusting for demographics, but their relative betweenness centrality resulted in a high pathway impact. Assuming intracellular metabolism of these amino acids mirrors their extracellular metabolism and serum levels, their directionality seems to suggest there was an increased synthesis of l-arginine and decreased metabolism of proline in participants with CAC.
Patients on HD with CAC might have increased synthesis of l-arginine and decreased metabolism of proline to counteract the calcifying milieu. There are few studies that examined the effect of arginine on cardiovascular calcification. In an adenine-induced model of renal failure and arterial calcification, dietary l-arginine supplementation suppressed arterial calcification in seven out of ten rats (38). The exact mechanism of how arginine may attenuate arterial calcification in unclear. In vitro, l-arginine attenuated precipitation of calcium and phosphate (38). Arginine is a precursor for nitric oxide (Supplemental Figure 7B), and its effect on arterial calcification may be mediated by nitric oxide (39). To the best of our knowledge, no animal study has examined the effect of l-proline on arterial calcification. In cell culture, l-proline inhibited the apoptosis of human vascular smooth muscle cells induced by calcium and phosphate (38). Patients with CKD can develop both intimal calcification, which is an indicator of atherosclerosis, and medial calcification, which is characterized by diffuse calcium and phosphate deposition. Unfortunately, CT scans cannot differentiate between intimal and medial calcification. Altered arginine/proline metabolism may reflect atherosclerosis rather than mineralization of calcium and phosphate. In rabbits fed with a high-cholesterol diet, arginine supplementation limited the development of atherosclerosis (40). In our exploratory analyses, we found that arginine/proline metabolism was positively correlated with serum phosphorous, FGF-23, osteoprotegerin, and dp-ucMGP. Arginine/proline metabolism may serve as a scaffold on which a variety of regulatory mechanisms for arterial calcification are integrated. Although controversial, arginine supplementation has been used in a wide variety of conditions, such as hypertension and erectile dysfunction (41). Because the arginine/proline metabolic pathway could be a potential therapeutic target, the role of arginine in arterial calcification merits further investigation.
Our study has several limitations. Because metabolites and CAC were both measured at baseline, the temporal relationship between serum metabolites and CAC could not be studied, thus limiting the inference of potential causal relationships. We were not able to address all potential or unmeasured confounders. Diabetes and hypertension are major potential confounders because both are associated with arterial calcification and metabolic dysregulation (12,42,43). All participants in our study were incident to HD, nondiabetic, and diagnosed with hypertension. Liver and residual renal function may influence the association of serum metabolites with CAC. Unfortunately, liver-function tests were not available in PACE, and only approximately a third of the study population had available data on residual renal function. Due to the limitation in sample size, we did not include residual renal function in our analyses. Last, the study cohort is not representative of the ESKD population in the United States and may need validation in a larger and more diverse ESKD population.
Our study has several strengths. First, we studied the key metabolites and pathways in detail in regard to their relationships with serum markers of mineral metabolism, inflammation, and circulating inhibitors of calcification. Our findings provide an important framework for future studies that investigate the roles of these novel metabolites and arginine/proline metabolism pathway on CAC. Second, all samples were collected after 8 hours of fasting, thus minimizing the effects of diet on metabolites (44). Third, by focusing on incident patients on HD who had similar characteristics, including the status of diabetes and hypertension, we minimized potential residual confounding.
In this nested, case-control study of incident patients on HD who were nondiabetic, we used a relatively unbiased approach of metabolomics profiling and identified three novel metabolites (chenodeoxycholic, deoxycholic, and glycolithocholic acids) and one pathway (arginine/proline metabolism) that were associated with CAC. Chenodeoxycholic, deoxycholic, and glycolithocholic acids may be potential serum biomarkers for CAC, and arginine and proline metabolism may emerge as a new pathway in the pathogenesis of CAC and could be a potential treatment target. Our findings provide new insight into pathophysiology of CAC in patients on HD and lay an important framework for future studies that investigate the roles of bile acids and arginine/proline metabolism on CAC.
Supplementary Material
Supplemental Methods.
Supplemental Table 1. Comparing baseline characteristics of study population with unselected participants.
Supplemental Table 2. Summary of metabolites that differed between cases and controls.
Supplemental Table 3. Tobit regression of log-transformed CAC score.
Supplemental Table 4. Metabolized analyzed in arginine/proline metabolism, urea cycle and bile acid synthesis.
Supplemental Table 5. Correlations of bile acid synthesis and arginine/proline metabolism pathways and their key metabolites with serum markers of mineral metabolism (Ca, Phos, Mg, PTH, FGF23, klotho), circulating inhibitors of calcification (osteoprotegerin, dp-ucMGP, fetuin-A, CPP2, T50) and inflammation (CRP).
Supplemental Figure 1. (A) Distribution of coefficients of variation (CVs) for all metabolites measured (m=452). (B) Pathways and number of metabolites (m=247) analyzed.
Supplemental Figure 2. Volcano plot identified metabolites that differed between cases and controls.
Supplemental Figure 3. Synchronized 3-dimensional plot of partial least squares-discriminant analyses. Cases are in green and controls are in red.
Supplemental Figure 4. Top 15 serum metabolites listed by their variable importance in projection (VIP) scores using partial least squares discriminant analyses.
Supplemental Figure 5. Correlation among 3 bile acids.
Supplemental Figure 6. Scree plots of arginine/proline metabolism (m=15), urea cycle (m=10) and bile acid synthesis (m=12).
Supplemental Figure 7. Bile acid synthesis and intracellular arginine and proline metabolism, simplified version.
Supplemental References.
Acknowledgments
We thank participants, nephrologists, and staff of the DaVita and MedStar dialysis units in the Baltimore region who contributed to the PACE study.
Each author contributed important intellectual content during manuscript drafting or revision and accepts accountability for the overall work by ensuring that questions pertaining to the accuracy or integrity of any portion of the work are appropriately investigated and resolved.
Funding
The research described was supported by the National Center for Advancing Translational Science grant UL1 TR002556. The PACE study was funded by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grants R01 DK072367 and DK090070 and the National Kidney Foundation Serving Maryland and Delaware. W. Chen is supported by the American Society of Nephrology Carl W. Gottschalk Research Grant and NIDDK grant K23 DK114476. D. Bushinsky is supported by NIDDK grant R01 DK075462. R. S. Parekh is supported by the Canada Research Chair in Chronic Kidney Disease Epidemiology. D. Riascos-Bernal is supported by the American Heart Association career development award 19CDA34660217. N. Sibinga is supported by National Heart, Lung, and Blood Institute grants R01 HL133861 and R01 HL149921, American Heart Association grant 19TPA34890070, and the Irma T. Hirschl-Monique Weill-Caulier Charitable Trust. The Stable Isotope and Metabolomics Core Facility of the Diabetes Research and Training Center of the Albert Einstein College of Medicine is supported by National Cancer Institute grant P60DK020541.
Footnotes
Disclosures
D.A. Bushinsky is a consultant for Amgen, Relypsa/Vifor/Fresenius, Sanofi Genzyme, and Tricida, and has an equity interest in Amgen and Tricida. All remaining authors have nothing to disclose.
Supplemental Material
This article contains the following supplemental material online at http://kidney360.asnjournals.org/lookup/suppl/doi:10.34067/KID.K3602020000442/DCSupplemental.
References
- 1.United States Renal Data System, 2019. edition. Available at https://www.usrds.org/annual-data-report/. Accessed December 16, 2020
- 2.Chen W, Fitzpatrick J, Monroy-Trujillo JM, Sozio SM, Jaar BG, Estrella MM, Wu TT, Melamed ML, Parekh RS, Bushinsky DA: Diabetes mellitus modifies the associations of serum magnesium concentration With arterial calcification and stiffness in incident hemodialysis patients. Kidney Int Rep 4: 806–813, 2019. 10.1016/j.ekir.2019.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raggi P, Boulay A, Chasan-Taber S, Amin N, Dillon M, Burke SK, Chertow GM: Cardiac calcification in adult hemodialysis patients. A link between end-stage renal disease and cardiovascular disease? J Am Coll Cardiol 39: 695–701, 2002. 10.1016/S0735-1097(01)01781-8 [DOI] [PubMed] [Google Scholar]
- 4.Chen J, Budoff MJ, Reilly MP, Yang W, Rosas SE, Rahman M, Zhang X, Roy JA, Lustigova E, Nessel L, Ford V, Raj D, Porter AC, Soliman EZ, Wright JT Jr, Wolf M, He J; CRIC Investigators: Coronary artery calcification and risk of cardiovascular disease and death among patients with chronic kidney disease. JAMA Cardiol 2: 635–643, 2017. 10.1001/jamacardio.2017.0363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peters SA, Bakker M, den Ruijter HM, Bots ML: Added value of CAC in risk stratification for cardiovascular events: A systematic review. Eur J Clin Invest 42: 110–116, 2012. 10.1111/j.1365-2362.2011.02555.x [DOI] [PubMed] [Google Scholar]
- 6.Ohtake T, Ishioka K, Honda K, Oka M, Maesato K, Mano T, Ikee R, Moriya H, Hidaka S, Kobayashi S: Impact of coronary artery calcification in hemodialysis patients: Risk factors and associations with prognosis. Hemodial Int 14: 218–225, 2010. 10.1111/j.1542-4758.2009.00423.x [DOI] [PubMed] [Google Scholar]
- 7.Kalim S, Rhee EP: An overview of renal metabolomics. Kidney Int 91: 61–69, 2017. 10.1016/j.kint.2016.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dubin RF, Rhee EP: Proteomics and metabolomics in kidney disease, including insights into etiology, treatment, and prevention. Clin J Am Soc Nephrol 15: 404–411, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalim S, Clish CB, Wenger J, Elmariah S, Yeh RW, Deferio JJ, Pierce K, Deik A, Gerszten RE, Thadhani R, Rhee EP: A plasma long-chain acylcarnitine predicts cardiovascular mortality in incident dialysis patients. J Am Heart Assoc 2: e000542, 2013. 10.1161/JAHA.113.000542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu JR, Grams ME, Coresh J, Hwang S, Kovesdy CP, Guallar E, Rhee EP, Shafi T: Serum metabolites and cardiac death in patients on hemodialysis. Clin J Am Soc Nephrol 14: 747–749, 2019. 10.2215/CJN.12691018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parekh RS, Meoni LA, Jaar BG, Sozio SM, Shafi T, Tomaselli GF, Lima JA, Tereshchenko LG, Estrella MM, Kao WH: Rationale and design for the Predictors of Arrhythmic and Cardiovascular Risk in End Stage Renal Disease (PACE) study. BMC Nephrol 16: 63, 2015. 10.1186/s12882-015-0050-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katz R, Budoff MJ, O’Brien KD, Wong ND, Nasir K: The metabolic syndrome and diabetes mellitus as predictors of thoracic aortic calcification as detected by non-contrast computed tomography in the Multi-Ethnic Study of Atherosclerosis. Diabet Med 33: 912–919, 2016. 10.1111/dme.12958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greenland F, Bonow RO, Brundage BH, Budoff MJ, Eisenberg MJ, Grundy SM, Lauer MS, Post WS, Raggi P, Redberg Rf, Rodgers GP, Shaw LJ, Taylor AJ, Weintraub WS; American College of Cardiology Foundation Clinical Expert Consensus Task Force (ACCF/AHA Writing Committee to Update the 2000 Expert Consensus Document on Electron Beam Computed Tomography); Society of Atherosclerosis Imaging and PreventionSociety of Cardiovascular Computed Tomography: ACCF/AHA 2007 clinical expert consensus document on coronary artery calcium scoring by computed tomography in global cardiovascular risk assessment and in evaluation of patients with chest pain: A report of the American College of Cardiology Foundation Clinical Expert Consensus Task Force (ACCF/AHA writing committee to update the 2000 expert consensus document on electron beam computed tomography) developed in collaboration with the Society of Atherosclerosis Imaging and Prevention and the Society of Cardiovascular Computed Tomography. J Am Coll Cardiol 49: 378–402, 2007. 10.1016/j.jacc.2006.10.001 [DOI] [PubMed] [Google Scholar]
- 14.Haberl R, Becker A, Leber A, Knez A, Becker C, Lang C, Brüning R, Reiser M, Steinbeck G: Correlation of coronary calcification and angiographically documented stenoses in patients with suspected coronary artery disease: Results of 1,764 patients. J Am Coll Cardiol 37: 451–457, 2001. 10.1016/S0735-1097(00)01119-0 [DOI] [PubMed] [Google Scholar]
- 15.Yuan M, Breitkopf SB, Yang X, Asara JM: A positive/negative ionswitching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc 7: 872–881, 2012. 10.1038/nprot.2012.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gromski PS, Muhamadali H, Ellis DI, Xu Y, Correa E, Turner ML, Goodacre R: A tutorial review: Metabolomics and partial least squares-discriminant analysis--a marriage of convenience or a shotgun wedding. Anal Chim Acta 879: 10–23, 2015. 10.1016/j.aca.2015.02.012 [DOI] [PubMed] [Google Scholar]
- 17.Reilly MP, Wolfe ML, Localio AR, Rader DJ: Coronary artery calcification and cardiovascular risk factors: Impact of the analytic approach. Atherosclerosis 173: 69–78, 2004. 10.1016/j.atherosclerosis.2003.10.010 [DOI] [PubMed] [Google Scholar]
- 18.Chong J, Wishart DS, Xia J: Using MetaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr Protoc Bioinformatics 68: e86, 2019. 10.1002/cpbi.86 [DOI] [PubMed] [Google Scholar]
- 19.Goodman WG, Goldin J, Kuizon BD, Yoon C, Gales B, Sider D, Wang Y, Chung J, Emerick A, Greaser L, Elashoff RM, Salusky IB: Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N Engl J Med 342: 1478–1483, 2000. 10.1056/NEJM200005183422003 [DOI] [PubMed] [Google Scholar]
- 20.Pletcher MJ, Tice JA, Pignone M, Browner WS: Using the coronary artery calcium score to predict coronary heart disease events: A systematic review and meta-analysis. Arch Intern Med 164: 1285–1292, 2004. 10.1001/archinte.164.12.1285 [DOI] [PubMed] [Google Scholar]
- 21.Jovanovich A, Isakova T, Block G, Stubbs J, Smits G, Chonchol M, Miyazaki M: Deoxycholic acid, a metabolite of circulating bile acids, and coronary artery vascular calcification in CKD. Am J Kidney Dis 71: 27–34, 2018. 10.1053/j.ajkd.2017.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiang JY: Bile acid metabolism and signaling. Compr Physiol 3: 1191–1212, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Small Molecule Pathway Database: Bile acid biosynthesis pathway description. Available at: http://smpdb.ca/view/SMP0000035. Accessed January 28, 2020
- 24.Engelking LR: Bile acids. In: Textbook of Veterinary Physiological Chemistry, 3rd Ed., Oxford, United Kingdom, Academic Press, 2015, pp 397–405 10.1016/B978-0-12-391909-0.50062-1 [DOI] [Google Scholar]
- 25.National Center for Biotechnology Information: PubChem compound summary for CID 115245, glycolithocholic acid. Available at: https://pubchem.ncbi.nlm.nih.gov/compound/Glycolithocholic-acid. Accessed January 21, 2020
- 26.Vaziri ND, Dure-Smith B, Miller R, Mirahmadi MK: Pathology of gastrointestinal tract in chronic hemodialysis patients: An autopsy study of 78 cases. Am J Gastroenterol 80: 608–611, 1985 [PubMed] [Google Scholar]
- 27.Ramezani A, Raj DS: The gut microbiome, kidney disease, and targeted interventions. J Am Soc Nephrol 25: 657–670, 2014. 10.1681/ASN.2013080905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li R, Zeng L, Xie S, Chen J, Yu Y, Zhong L: Targeted metabolomics study of serum bile acid profile in patients with end-stage renal disease undergoing hemodialysis. PeerJ 7: e7145, 2019. 10.7717/peerj.7145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang WH, Kitai T, Hazen SL: Gut microbiota in cardiovascular health and disease. Circ Res 120: 1183–1196, 2017. 10.1161/CIRCRESAHA.117.309715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jovanovich A, Isakova T, Stubbs J: Microbiome and cardiovascular disease in CKD. Clin J Am Soc Nephrol 13: 1598–1604, 2018. 10.2215/CJN.12691117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X, Li Y, Yang P, Liu X, Lu L, Chen Y, Zhong X, Li Z, Liu H, Ou C, Yan J, Chen M: Trimethylamine-N-oxide promotes vascular calcification through activation of NLRP3 (nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3) inflammasome and NF-κB (nuclear factor κB) signals. Arterioscler Thromb Vasc Biol 40: 751–765, 2020. 10.1161/ATVBAHA.119.313414 [DOI] [PubMed] [Google Scholar]
- 32.Miyake JH, Wang SL, Davis RA: Bile acid induction of cytokine expression by macrophages correlates with repression of hepatic cholesterol 7alpha-hydroxylase. J Biol Chem 275: 21805–21808, 2000. 10.1074/jbc.C000275200 [DOI] [PubMed] [Google Scholar]
- 33.Miyazaki-Anzai S, Levi M, Kratzer A, Ting TC, Lewis LB, Miyazaki M: Farnesoid X receptor activation prevents the development of vascular calcification in ApoE−/− mice with chronic kidney disease. Circ Res 106: 1807–1817, 2010. 10.1161/CIRCRESAHA.109.212969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miyazaki-Anzai S, Masuda M, Levi M, Keenan AL, Miyazaki M: Dual activation of the bile acid nuclear receptor FXR and G-protein-coupled receptor TGR5 protects mice against atherosclerosis. PLoS One 9: e108270, 2014. 10.1371/journal.pone.0108270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Small Molecule Pathway Database: Arginine and proline metabolism pathway description. Available at: http://smpdb.ca/view/SMP0000020. Accessed January 17, 2020
- 36.Wu G, Bazer FW, Davis TA, Kim SW, Li P, Marc Rhoads J, Carey Satterfield M, Smith SB, Spencer TE, Yin Y: Arginine metabolism and nutrition in growth, health and disease. Amino Acids 37: 153–168, 2009. 10.1007/s00726-008-0210-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morris SM Jr: Arginine metabolism: Boundaries of our knowledge. J Nutr 137[Suppl 2]: 1602S–1609S, 2007. 10.1093/jn/137.6.1602S [DOI] [PubMed] [Google Scholar]
- 38.Shimomura A, Matsui I, Hamano T, Ishimoto T, Katou Y, Takehana K, Inoue K, Kusunoki Y, Mori D, Nakano C, Obi Y, Fujii N, Takabatake Y, Nakano T, Tsubakihara Y, Isaka Y, Rakugi H: Dietary L-lysine prevents arterial calcification in adenine-induced uremic rats. J Am Soc Nephrol 25: 1954–1965, 2014. 10.1681/ASN.2013090967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanno Y, Into T, Lowenstein CJ, Matsushita K: Nitric oxide regulates vascular calcification by interfering with TGF-β signalling. Cardiovasc Res 77: 221–230, 2008. 10.1093/cvr/cvm049 [DOI] [PubMed] [Google Scholar]
- 40.Jeremy RW, McCarron H, Sullivan D: Effects of dietary L-arginine on atherosclerosis and endothelium-dependent vasodilatation in the hypercholesterolemic rabbit. Response according to treatment duration, anatomic site, and sex. Circulation 94: 498–506, 1996. 10.1161/01.CIR.943.498 [DOI] [PubMed] [Google Scholar]
- 41.Rosenthal MD, Carrott PW, Patel J, Kiraly L, Martindale RG: Parenteral or enteral arginine supplementation safety and efficacy. J Nutr 146: 2594S–2600S, 2016. 10.3945/jn.115.228544 [DOI] [PubMed] [Google Scholar]
- 42.Smith ER, Ford ML, Tomlinson LA, Bodenham E, McMahon LP, Farese S, Rajkumar C, Holt SG, Pasch A: Serum calcification propensity predicts all-cause mortality in predialysis CKD. J Am Soc Nephrol 25: 339–348, 2014. 10.1681/ASN.2013060635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Newgard CB: Metabolomics and metabolic diseases: Where do we stand? Cell Metab 25: 43–56, 2017. 10.1016/j.cmet.2016.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rebholz CM, Lichtenstein AH, Zheng Z, Appel LJ, Coresh J: Serum untargeted metabolomic profile of the Dietary Approaches to Stop Hypertension (DASH) dietary pattern. Am J Clin Nutr 108: 243–255, 2018. 10.1093/ajcn/nqy099 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Methods.
Supplemental Table 1. Comparing baseline characteristics of study population with unselected participants.
Supplemental Table 2. Summary of metabolites that differed between cases and controls.
Supplemental Table 3. Tobit regression of log-transformed CAC score.
Supplemental Table 4. Metabolized analyzed in arginine/proline metabolism, urea cycle and bile acid synthesis.
Supplemental Table 5. Correlations of bile acid synthesis and arginine/proline metabolism pathways and their key metabolites with serum markers of mineral metabolism (Ca, Phos, Mg, PTH, FGF23, klotho), circulating inhibitors of calcification (osteoprotegerin, dp-ucMGP, fetuin-A, CPP2, T50) and inflammation (CRP).
Supplemental Figure 1. (A) Distribution of coefficients of variation (CVs) for all metabolites measured (m=452). (B) Pathways and number of metabolites (m=247) analyzed.
Supplemental Figure 2. Volcano plot identified metabolites that differed between cases and controls.
Supplemental Figure 3. Synchronized 3-dimensional plot of partial least squares-discriminant analyses. Cases are in green and controls are in red.
Supplemental Figure 4. Top 15 serum metabolites listed by their variable importance in projection (VIP) scores using partial least squares discriminant analyses.
Supplemental Figure 5. Correlation among 3 bile acids.
Supplemental Figure 6. Scree plots of arginine/proline metabolism (m=15), urea cycle (m=10) and bile acid synthesis (m=12).
Supplemental Figure 7. Bile acid synthesis and intracellular arginine and proline metabolism, simplified version.
Supplemental References.