

Abstract
Gene expression is regulated by promoters and enhancers marked by histone H3-lysine-27 acetylation (H3K27ac), which is established by the paralogous histone acetyltransferases (HATs), EP300 and CBP. These enzymes display overlapping regulatory roles in untransformed cells, but less characterized roles in cancer cells. We demonstrate that the majority of high-risk pediatric neuroblastoma (NB) depend on EP300, whereas CBP has a limited role. EP300 controls enhancer acetylation by interacting with TFAP2β, a transcription factor member of the lineage-defining transcriptional core regulatory circuitry (CRC) in NB. To disrupt EP300, we developed a proteolysis-targeted-chimaera (PROTAC) compound termed “JQAD1” that selectively targets EP300 for degradation. JQAD1 treatment causes loss of H3K27ac at CRC enhancers and rapid neuroblastoma apoptosis, with limited toxicity to untransformed cells where CBP may compensate. Further, JQAD1 activity is critically determined by cereblon (CRBN) expression across neuroblastoma cells.
INTRODUCTION
Gene transcription is controlled by networks of epigenetic regulators and master transcription factors (TFs) (reviewed in (1,2)). These proteins form complexes that modulate DNA accessibility and establish epigenetic marks to control the activity of specific gene enhancers and promoters (2,3). Gene enhancers are required to control the mRNA expression of the genes that establish cell fate (4). These identity-defining processes are dysregulated in disease states, including cancer, by the altered regulation of transcription, often through selection for mutations in epigenetic regulatory proteins (2,5–8).
The establishment of cell identity and fate requires high level of expression of key lineage-related master transcription factors (4,9). Master transcription factor loci are typically associated with enhancer elements marked by extensive stretches of acetylation on histone H3, lysine-27 (H3K27ac), termed “super-enhancer” or “stretch-enhancer” (SE) elements (10,11). H3K27ac is catalyzed by the activity of two paralogous enzymes, the E1A-binding protein (EP300, KAT3B) or the CREB-binding protein (CREBBP, CBP, KAT3A)(12,13). These two enzymes share a large degree of sequence homology, and both contain multiple homologous domains, including kinase-inducible domain interacting (KIX) domains, histone acetyltransferases (HAT) and bromodomains (14). Each protein can catalyze H3K27ac and acetylate lysine residues on many other proteins (15). Given the extensive homology between these related HAT enzymes, many review articles refer to them as one unit—“EP300/CBP”—and all available inhibitors of their HAT and bromodomains cross-react with both enzymes with nearly identical Kd values (13,16–19).
Various studies have suggested that EP300 and CBP play overlapping but distinct roles in the regulation of cell survival. Germline loss of EP300 or CBP results in murine embryonic lethality, with distinct phenotypes (20). Further, CBP is required for self-renewal, while EP300 is required for the differentiation of hematopoietic stem cells (21). Somatic mutations of either EP300 or CBP are found in a variety of malignancies (22), and in CBP-mutated tumor cells, loss of EP300 is synthetic lethal (23). Chromatin immunoprecipitation coupled to high-throughput sequencing (ChIP-Seq) studies have identified largely overlapping but distinct binding of EP300 and CBP genome-wide, indicating that these two proteins may function differently by regulating the enhancers of distinct genes (24,25). However, many studies interrogating EP300 and CBP have relied on genetic disruption or mRNA depletion of each gene, which does not permit a time-associated analysis, or alternatively have relied on the use of inhibitors with non-selective activity against both enzymes (13,16–19). The derivation of pharmacologic inhibitors targeting only one of these enzymes has thus been limited by the homology between these proteins (13,18). This has fundamentally limited our ability to rapidly dissect independent mechanisms regulated by each individual enzyme in cells.
One group of newly developed pharmacologic compounds has been used to induce targeted protein degradation mediated by E3 ligase receptor proteins and their small molecule binders. These include the E3 ligase receptor protein Cereblon (CRBN) and its binding molecules, the phthalimides (thalidomide, lenalidomide and pomalidomide, referred to as IMiDs) (26). Heterobifunctional molecules termed proteolysis-targeted chimaeras (PROTACs) induce ligand-dependent target protein degradation by recruitment of target proteins to the proteasome. PROTAC E3-binding heterobifunctional small molecules have been validated for a variety of targets including BRD4, FKBP12, ERRα, RIPK2, BRD9 and p38 MAP kinase, among others (reviewed in (27,28)). By catalyzing the formation of a ternary complex involving an E3 ligase receptor, protein of interest and small molecule, PROTACs may yield enhanced substrate specificity (29).
Here, we used functional and chemical genomics to show that EP300, but not CBP, is typically required for establishment of H3K27ac at essential gene enhancers in the high-risk pediatric cancer, neuroblastoma (NB). We identify that EP300 is an enhancer-regulating dependency in neuroblastoma, recruited to DNA by interactions with the AP2 transcription factor TFAP2β, a member of the lineage-defining core regulatory circuitry of NB. In contrast, CBP is dispensable for the malignant phenotype in most cases and does not interact with TFAP2β. Using a chemical biology strategy, we synthesized a novel PROTAC degrader, called JQAD1, which displays strong selectivity for EP300, and use this chimeric small molecule to demonstrate a time-dependent loss of EP300, enhancer acetylation and transcriptional output in NB cells both in vitro and in vivo. In contrast to catalytic inhibition, this degrader molecule rapidly drives neuroblastoma apoptotic cell death associated with MYCN downregulation. Further, it has limited toxicity to untransformed cells in vivo, while causing growth delay of neuroblastoma tumor xenografts. Finally, we demonstrate that enhanced dependency on EP300 or CBP is found across numerous cancer lineages and confirm that the mechanism of degrader function is highly dependent on expression of CRBN, providing a foundation that will enable the study of EP300-selective and other degraders in multiple distinct cellular contexts. Thus, this study identifies a critical role for EP300 in regulating the enhancer and transcriptional landscapes of high-risk neuroblastoma through physical interactions with the oncoprotein MYCN and TFAP2β, members of the core regulatory circuitry.
RESULTS
EP300 is required for high-risk neuroblastoma growth
Previously, we showed that high-risk neuroblastoma selectively requires a group of 147 genes for survival (9). One of these genes encodes the histone acetyltransferase enzyme EP300, but the gene encoding its paralog CBP is not required, which is surprising because EP300 is often redundant with CBP (13). Both EP300 and CBP acetylate the Lys-27 residue of histone H3 (H3K27ac), which is a mark associated with active gene transcription (9,13). We were intrigued that EP300 appeared to be uniquely required in neuroblastoma compared to CBP and therefore we sought to investigate the relative expression and dependency of these two genes across a panel of representative neuroblastoma cell lines. First, we examined the relative dependency of EP300 or CBP in 19 high-risk neuroblastoma cell lines using the DepMap exome-wide CRISPR-Cas9 deletion dataset (30). This demonstrated that most high-risk neuroblastoma cell lines require EP300 for cell growth (Fig. 1A). Interestingly, in four of the cell lines with a high level of dependency on EP300, we also observed dependency on CBP, indicating that each protein was essential (Fig. 1A). An additional four of the cell lines were not dependent on either EP300 or CBP, potentially indicating redundancy of these two acetyltransferases in these cell lines (Fig. 1A). To extend these findings, we performed CRISPR-Cas9-mediated knockout of EP300 and CBP in two MYCN-amplified NB cell lines, Kelly and BE2C (Fig. 1B, S1A). Although both EP300 and CBP are expressed in these two cell lines, only loss of EP300 caused a profound reduction of H3K27ac expression levels, while loss of CBP had a more minor effect, indicating that in these cell lines, most enhancers and promoters rely on EP300 to catalyze H3K27ac. Further, expression of MYCN, which is a well-known dependency in MYCN-amplified neuroblastoma, was almost completely dependent on EP300 and much less so on CBP (Fig. 1B, S1A). Accordingly, CRISPR-Cas9-mediated deletion of EP300 markedly reduced colony formation in each cell line, while CBP loss did not (Fig. 1C). The residual colonies formed by EP300 CRISPR knockout cells did not express GFP, which was co-expressed in the vector containing the guide RNA, indicating that they represent cells that were not infected with the vector containing EP300-targeted guide RNAs.
Figure 1. EP300 but not CBP is required for neuroblastoma cell growth.
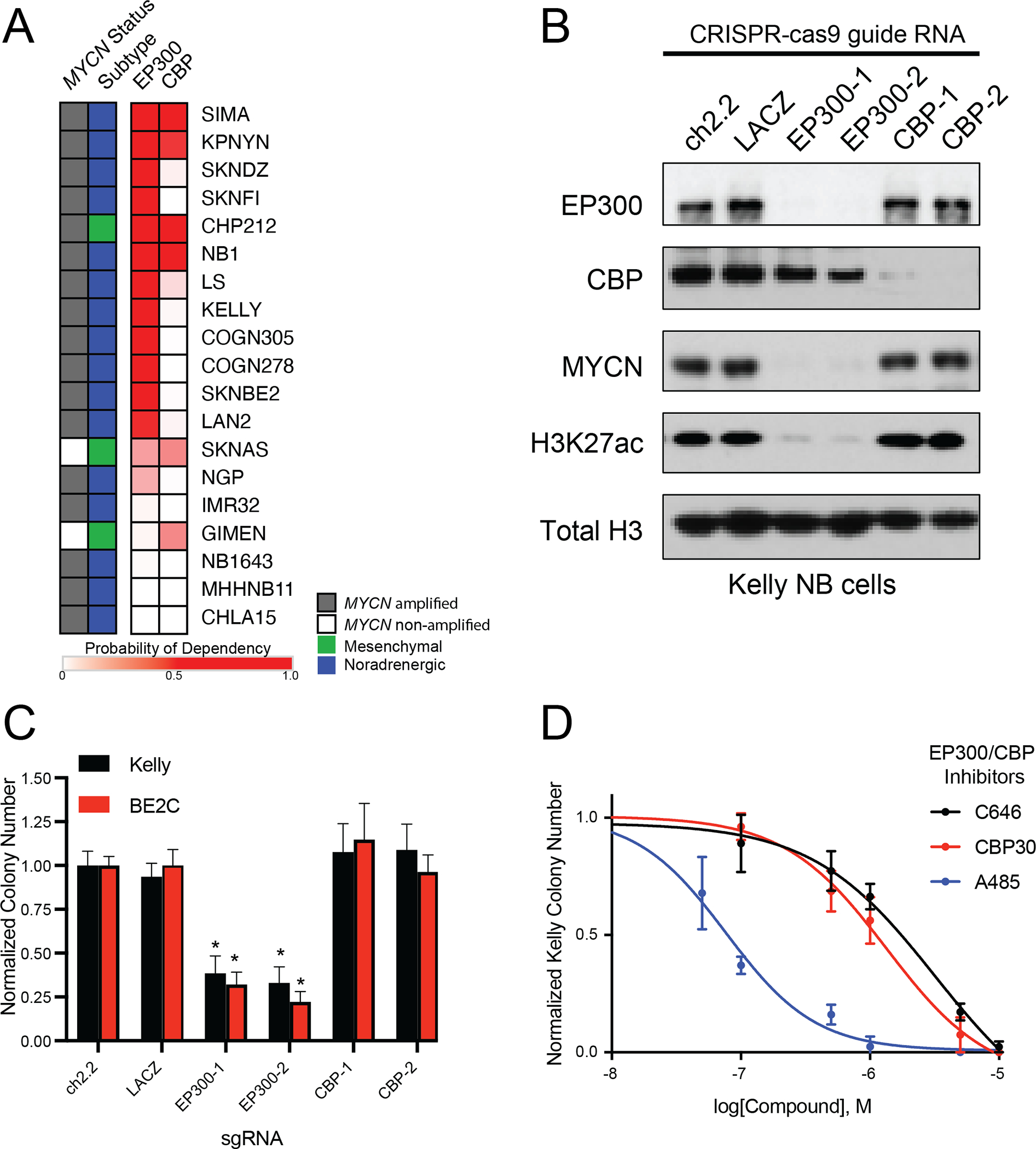
A. Heatmap of probability of dependency on neuroblastoma cell lines (n=19) in the DepMap 20Q2 data release demonstrates that most NB cell lines depend on EP300 (darker red color) compared with only few requiring CBP.
B. Kelly cells stably expressing Cas9 were infected with sgRNAs targeting EP300 (EP300–1,2), CBP (CBP-1,2) or controls (ch2.2, LACZ) for five days, prior to western blotting to the noted targets. Data is representative of three independent sgRNA infections and lysates.
C. Colony formation assays were performed following sgRNA infection as in C, in Kelly and BE2C cells. Cells were cultured for 10 days after infection. n=3 independent replicates per cell line, per treatment. * p<0.05. Bars represent S.E.M.
D. Kelly NB cells were treated in colony formation assays with a range of concentrations of the EP300/CBP combined inhibitors C646, CBP30, and A485. n=3 independent replicates per cell line, per treatment. Bars represent S.E.M. See also Fig. S1.
Analysis of EP300 and CBP mRNA expression in primary neuroblastoma tumors revealed a positive correlation (Fig. S1B). Further, analysis of publicly available sequencing data in primary neuroblastoma tumors demonstrated that while inactivating mutations in EP300 or CBP did occur in human high-risk neuroblastoma, they were extremely rare (31). By western blotting, EP300 and CBP levels were generally correlated across a panel of neuroblastoma cell lines (Fig. S1C). Analysis of cancer cell lines in the Cancer Cell Line Encyclopedia (CCLE) proteomics and mRNA expression datasets also showed correlated expression levels of EP300 and CBP at both the RNA and protein levels, including the cell lines from patients with neuroblastoma (in red), indicating that these findings pertain across multiple tumor lineages (Fig. S1D,E).
To test our genetic findings using small molecule probes, we next performed colony formation assays of NB cells using known EP300 and CBP inhibitors that are non-selective between these two proteins. This included two inhibitors targeting the EP300 and CBP HAT domain – A485 and C646 (18,19), and one targeting the bromodomain – CBP30 (16). In multiple NB cell lines, the most potent compound in reducing neuroblastoma colony formation was the HAT domain inhibitor A485 (Fig. 1D, S1F–H). Non-selective inhibition of both EP300 and CBP with A485 caused G1 cell cycle arrest within 24 hours (Fig. S1I–K), similar to the effects of knockout of EP300, but not CBP (Fig. S1L,M). After seven days, A485 treatment led to global loss of the H3K27ac modification, loss of MYCN expression, induction of cleaved caspase 3 and PARP1, and increased cells in the subG1 peak, all indicative of apoptotic cell death (Fig. S1N,O). Though both cell cycle progression and cell survival are impaired by inhibition of the HAT activity of both EP300 and CBP, our genetic studies indicate that this is due to a dependency on EP300, not CBP, for the growth and survival of most MYCN-amplified neuroblastoma cell lines.
EP300 facilitates adrenergic NB CRC-driven transcription through binding to TFAP2β
Next, we sought to determine the mechanism by which EP300, but not CBP, was required for growth of MYCN-amplified neuroblastoma cell lines. Previously we demonstrated that core regulatory circuitry (CRC) transcription factors are critically important in determining cell fate in neuroblastoma and are marked and regulated by H3K27ac-marked super-enhancers (9,32,33). This analysis uncovered that the master transcription factors of the adrenergic NB subtype include HAND2, ISL1, PHOX2B, GATA3, TBX2, and ASCL1. Thus, we sought to understand the mechanism by which EP300 collaborates with the NB CRC-driven gene expression program. We began by using the STRING database to perform an interaction analysis of all expressed nuclear neuroblastoma dependency proteins (34). This demonstrated that EP300 was found in a densely interacting network of proteins enriched for CRC transcription factors (Fig. 2A) (9,35). To understand the genome-wide binding patterns of EP300 and CBP, we performed chromatin immunoprecipitation coupled to massively parallel high-throughput sequencing (ChIP-seq) experiments using antibodies recognizing EP300 and CBP in two separate MYCN-amplified neuroblastoma cell lines, BE2C and Kelly (Fig. 2B, S2A). The majority of sites genome-wide bound by EP300 were also bound by CBP, based on genome-wide scatterplot analysis (Fig 2B, S2A). Intriguingly, a small number of sites were preferentially bound by either EP300 or CBP (Fig 2B, S2A). Next, to examine the importance of EP300 and CBP in regulating gene expression programs in neuroblastoma cells, we assayed the degree of co-localization of EP300 and CBP with the targets of the previously defined neuroblastoma CRC transcription factors and H3K27ac (Fig. 2C, S2B). Genome-wide heatmap analysis demonstrated that both EP300 and CBP displayed a similar pattern of binding as each of the CRC TFs (Fig. 2C, S2B). Previously, we identified that some of the most heavily bound sites marked by H3K27ac in neuroblastoma cells included the CRC transcription factor loci(9). Focused evaluation of EP300 and CBP binding at the loci encoding the CRC TFs demonstrated co-localization of EP300 with CRC transcription factors (Fig. 2D, Fig. S2C, red tracks). CBP, however, was minimally enriched at these loci (Fig. 2D, Fig. S2C, green tracks). These data indicate that EP300, but not CBP, is preferentially localized at sites that control the expression of adrenergic CRC genes in NB cells (9).
Figure 2. EP300 regulates the neuroblastoma core regulatory circuitry directed by TFAP2β.
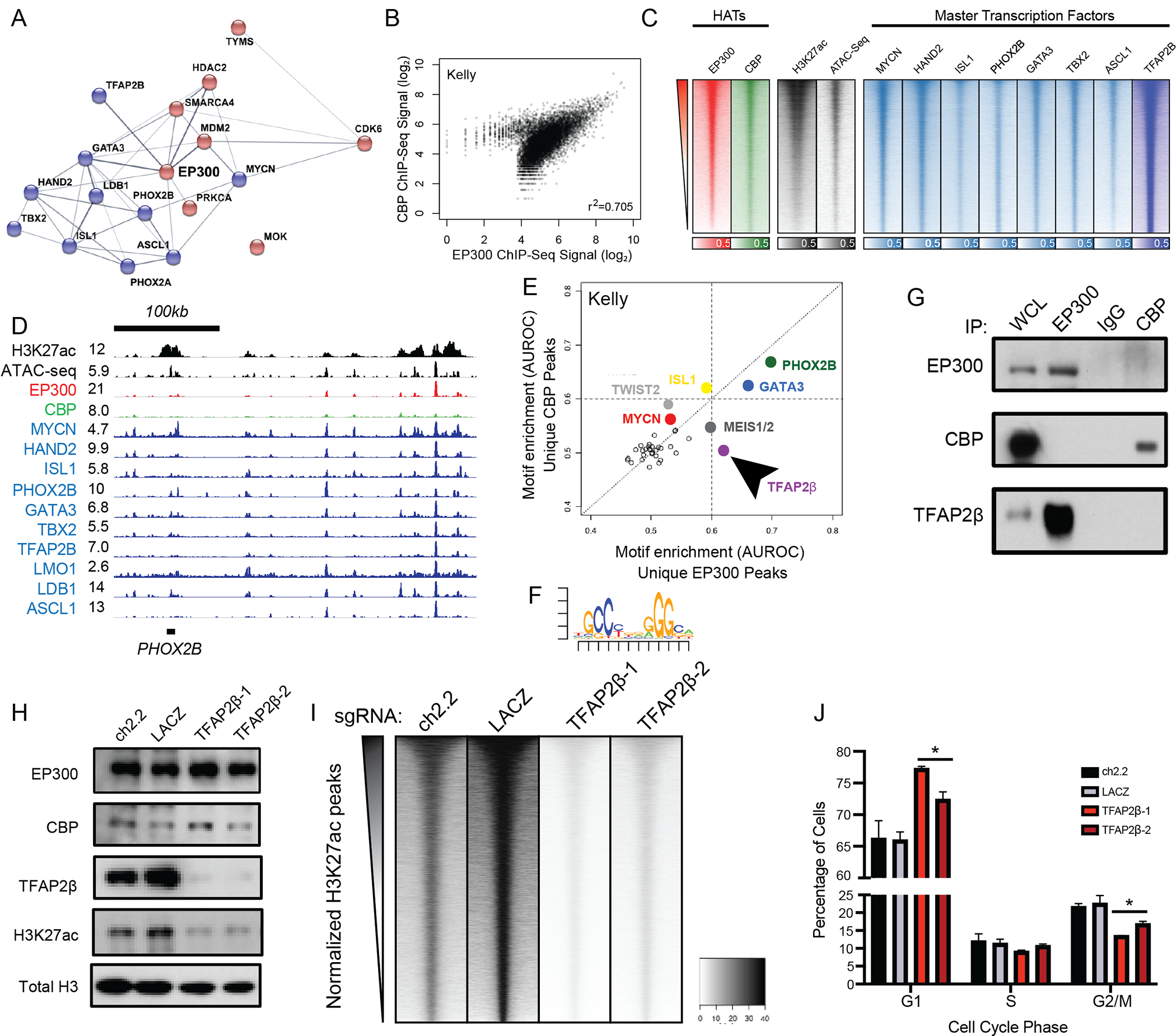
A. STRING database interaction plot of nuclear dependency genes in NB cells. Data is derived from (9). Displayed are core regulatory circuitry transcription factors in blue and proteins with available targeting compounds in red. Connecting lines indicate previously demonstrated protein-protein interactions.
B. Scatterplot of log2 transformed read counts of EP300 or CBP ChIP-seq in the collapsed union of separately identified high-confidence CBP and EP300 binding sites in Kelly cells. R indicates the Spearman correlation coefficient demonstrating a strong linear relationship in coverage. Similar analysis in BE2C cells as Fig. S2A.
C. Genome-wide heatmap analysis of chromatin composition at the collapsed union of separately identified high-confidence CRC transcription factor-binding sites in Kelly cells. Rows ordered by EP300 signal. Similar analysis in BE2C cells as Fig. S2B.
D. Representative ChIP-seq plots demonstrating binding of CRC factors (blue), CBP (green), EP300 (red) at the PHOX2B core regulatory circuitry transcription factor locus in Kelly NB cells. Also shown is the PHOX2B super-enhancer (H3K27ac) and open chromatin (ATAC-seq) (black). Data is representative of both Kelly and BE2C cells and all CRC loci.
E. Motif enrichment analysis of ChIP-seq to EP300 and CBP in Kelly NB cells. Data was restricted to the top 500 bound peaks by EP300 or CBP in Kelly NB cells. Colored dots indicate known enriched transcription factors. Arrow indicates specifically enriched motif, corresponding to TFAP2β.
F. Position-weight matrix from analysis in D demonstrates the top enriched specific sequence under EP300 peaks, compared with CBP peaks, corresponding to the consensus binding sequence for TFAP2β.
G. Co-immunoprecipitation followed by western blotting analysis of EP300 and CBP in Kelly NB cells. WCL = whole cell lysate. IgG = isotype-matched rabbit IgG antibody. Data is representative of three independent co-IP-western blots.
H. Kelly NB cells expressing Cas9 were infected with sgRNAs targeting TFAP2β (TFAP2β−1,2) or control loci (ch2.2, LACZ), followed by western blotting to the shown targets. Data is representative of three independent lysates and blots.
I. Genome-wide heatmap analysis of H3K27ac coverage in wild-type and TFAP2β-knockout Kelly cells using cell number- and E.coli-spike-in normalized CUT&RUN-sequencing. Rows represent 6kb regions centered on the center of the collapsed union of high-confidence peaks separately identified in each condition and are ordered by control (ch2.2) signal.
J. Propidium-iodide flow cytometry of Kelly NB cells expressing Cas9 and infected with sgRNAs targeting TFAP2β (TFAP2β−1,2) or control loci (ch2.2, LACZ). n=3 independent infections and flow analyses. * p<0.05. Bars represent S.E.M. See also Fig. S2.
Both EP300 and CBP lack sequence-specific DNA-binding activity and require association with a DNA-binding factor to achieve locus-specific binding (36). Thus, we sought to identify how EP300 is targeted to chromatin loci associated with enhancers of the CRC. To identify proteins involved in EP300 recruitment to DNA in NB cells, we performed a motif enrichment analysis of the top 500 peaks bound specifically by either EP300 or CBP in Kelly and BE2C cells. Consistent with prior evidence indicating that EP300 proteins form interactions with several TFs to nucleate higher-order enhanceosome structures (37), this analysis demonstrated enrichment for several transcription factor consensus binding motifs preferentially associated with EP300 and/or CBP binding (Fig. 2E,F S2D,E). Two motifs were selectively enriched under EP300-bound peaks in both cell lines, corresponding to consensus binding sequences for the transcription factors GATA3 and TFAP2β (Fig. 2E,F S2D,E). To validate that these transcription factors associate with H3K27ac-marked chromatin, we next performed co-immunoprecipitation of H3K27ac from nuclear extracts of Kelly and BE2C cells, followed by mass spectrometry of the isolated protein. As expected, in both Kelly and BE2C cells, we detected peptides corresponding to EP300 and CBP that co-immunoprecipitated with H3K27ac. This experiment also identified that four transcription factors, including GATA3 and TFAP2β, physically interact with H3K27ac-marked nucleosomes in both cell lines (Table S1, Fig. S2F). GATA3 is a known member of the CRC of high-risk NB cells (9,32). Previously, we and others identified TFAP2β as a possible CRC member, since it is a transcription factor and growth dependency in NB cells that is commonly regulated by a super-enhancer (9,33,35); however, we could not prove this because suitable antibodies for ChIP-seq were not available to determine sites of chromatin binding. Using new antibodies against TFAP2β, we performed ChIP-seq for TFAP2β binding, and identified that it binds to the super-enhancers and regulates other members of the CRC (Fig. 2C,D, S2B,C). Thus, TFAP2β represents a newly identified member of the adrenergic core regulatory circuitry in MYCN-amplified NB cells.
Because EP300 binding was enriched at sites containing GATA3 and TFAP2β motifs, and these proteins bound to H3K27ac-marked chromatin, we sought to determine whether EP300 physically associates with GATA3 and TFAP2β. Immunoprecipitation of EP300 and CBP in Kelly NB cells, followed by western blotting for TFAP2β and GATA3 demonstrated that EP300, but not CBP, physically interacts with both TFAP2β and GATA3 (Fig. 2G, S2G). Additionally, reciprocal co-immunoprecipitation of TFAP2β in Kelly cells demonstrated the presence of EP300 proteins, but not CBP (Fig. S2G). To determine whether these transcription factors are able to control localization of EP300, we performed CRISPR-Cas9-based knockout of TFAP2β or GATA3 in Kelly NB cells (Fig. 2H, S2H,I). As a control, we also performed knockout of HAND2, a CRC factor that did not display selective motif enrichment under EP300 peaks (Fig. S2J). Day 5 after infection with lentiviruses encoding sgRNAs to knockout TFAP2β, we observed loss of H3K27ac levels, without effects on expression levels of EP300 or CBP (Fig. 2H, S2H). In contrast, knockout of either GATA3 or HAND2 at the same timepoints had no or minor effects, respectively, on the levels of H3K27ac (Fig. S2I,J). Consistent with these findings, genome-wide analysis of TFAP2β binding demonstrated higher correlation with EP300 than CBP in both Kelly and BE2C cells (Spearman’s Rho for TFAP2β to EP300 0.628 (Kelly), 0.589 (BE2C); to CBP 0.481 (Kelly), 0.492 (BE2C)). To identify whether knockout of TFAP2β resulted in site-specific or genome-wide loss of H3K27ac, we performed knockout of TFAP2β in Kelly cells using two distinct sgRNAs and then performed CUT&RUN sequencing against H3K27ac with exogenous spike-in E.coli DNA controls. Loss of TFAP2β, but not control loci, resulted in genome-wide loss of H3K27ac (Fig 2I), without detectable differences between subsets of sites including promoters or non-promoter regions (Fig. S2K). Finally, as with knockout of EP300 but not with CBP, knockout of TFAP2β also resulted in G1 cell cycle arrest in Kelly and NGP NB cells (Fig. 2J, S2L). In sum, these data indicate that EP300 is targeted to DNA through a physical interaction with the CRC transcription factor TFAP2β in neuroblastoma.
EP300 is selectively degraded by a novel chemical PROTAC, JQAD1
Each currently available small molecule that inhibits the HAT activity of EP300 also inhibits the HAT activity of CBP with nearly an equivalent Kd (13,16,18,19). This includes A485, the most potent and specific HAT inhibitory compound toward EP300/CBP developed to date (18,38). One approach to selectively target EP300 in neuroblastoma may be to disrupt the interaction between TFAP2β and EP300; however, a strategy like this has typically been difficult to implement (reviewed in (28)). Recently, alternative approaches to develop selective compounds have been described by developing small molecule degraders, termed “PROTACs.” PROTACs are heterobifunctional small molecules that bind the target protein and mediate the formation of a ternary complex between the target protein and an E3 ligase receptor (reviewed in (27)). The ternary complex formed by the PROTAC bridges the target protein to an E3 ubiquitin ligase, which polyubiquitinates the target protein and directs it to the proteasome for degradation and recycling(27). To develop a potential selective compound to target EP300, we used A485 as a bait molecule, since A485 was the most potent small molecule inhibitor in neuroblastoma cells and has the lowest Kd value for EP300 and CBP of all small molecules targeting these proteins(18) (Fig. 3A). Computational structural modeling of the interaction between the HAT domain of EP300 and the iMiD binding region of CRBN indicated that an optimal linker length between A485 and the E3 ligase would be a distance of 8–12 atoms. We therefore designed and synthesized an optimized compound containing the two chiral centers found within the A485 molecule (38), and a 12-carbon linking chain, termed JQAD1 (Fig. 3A, Scheme S1). We synthesized both the (R,S) and (S,S) stereoisomers of this molecule in parallel, and treated three neuroblastoma cell lines that express high levels of CRBN (Fig. S1C) with these compounds. The (R,S) diastereomer had the lowest IC50 concentration in intact Kelly, NGP and SIMA NB cells, and this IC50 was lower than that of the parental molecule A485 (Fig. 3B, S3A–F). Therefore, the name JQAD1 in this article will refer to the more active (R,S) diastereomer of the PROTAC compound, unless the (S,S) stereoisomer is used as a negative control, as specified.
Figure 3. JQAD1 is a selective EP300 degrader.
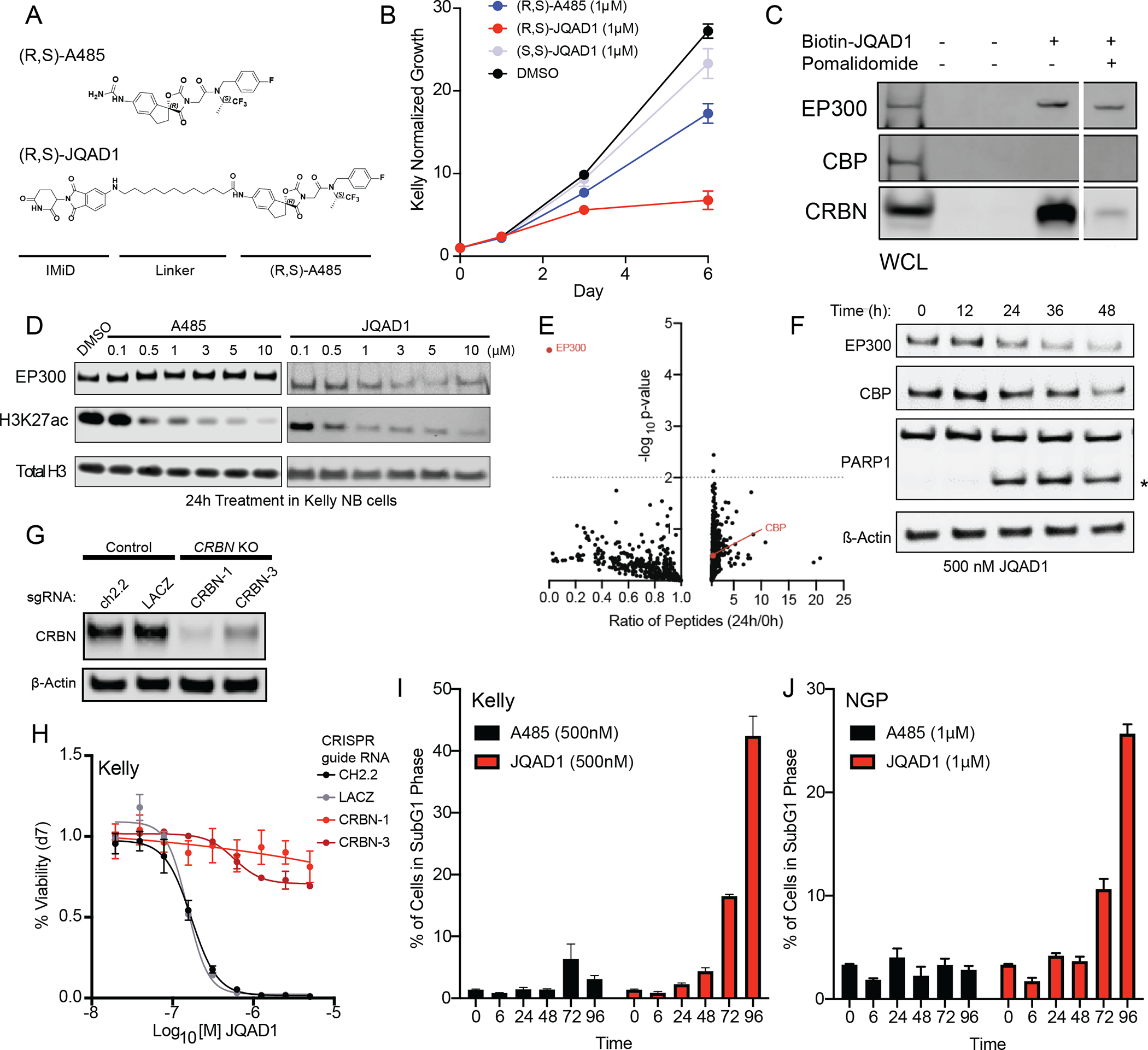
A. Chemical structure of (R,S)-A485 and (R,S)-JQAD1.
B. Kelly cells were treated with 1μM (R,S)-A485, (R,S)-JQAD1, (S,S)-JQAD1 or DMSO for 6 days and growth measured by CellTiter-Glo assay. n=3 independent experiments and measurements at each timepoint. Bars represent S.E.M.
C. Kelly cell lysates were treated with combinations of Biotin-JQAD1 or pomalidomide, prior to streptavidin-bead purification and western blotting of protein isolates demonstrating enriched interaction of JQAD1 with CRBN and EP300 proteins. WCL=whole cell lysate. Data are representative of three independent experiments and blots.
D. Kelly NB cells were treated with DMSO, A485 or JQAD1 at the noted concentrations (in μM), for 24h prior to lysis for western blotting. Data are representative of three independent biological repeats.
E. SILAC labelled-Kelly NB cells were treated with JQAD1 at 500 nM or DMSO vehicle for 24h, prior to nuclear extraction and analysis by mass spectrometry. Ratio of detected peptides at 0h vs. 24h is demonstrated. Data represents the sum ratio of heavy:light labelled protein detected in triplicate at 24h, compared to 0h. Dotted line indicates a p-value of 0.01. Red labelled points indicate EP300 and CBP. n=3 independent treatments, lysates and mass spectrometry reactions.
F. Kelly NB cells were treated with JQAD1 at 500 nM for the noted timepoints prior to lysis for western blotting. Data is representative of three independent experiments and blots. * indicates cleaved PARP1 species.
G. Kelly cells stably expressing Cas9 were infected with sgRNAs targeting CRBN (CRBN-1,3) or control loci (ch2.2, LACZ) and pools of knockout cells established. Western blotting was performed with antibodies against CRBN. Actin is shown as a loading control. Data is representative of three independent western blots.
H. Kelly-Cas9 control or CRBN knockout cells were treated with a range of doses of JQAD1 for seven days, prior to assay by CellTiter-Glo. n=3 independent replicates per dose and timepoint.
I,J. PI-flow cytometry of sub-G1 events in Kelly (I) and NGP (J) cells treated with JQAD1 or A485 for the noted timepoints (in hours). Data is a summary of n>3 independent flow experiments. Compound treatment was performed at 500nM (Kelly) and 1μM (NGP). Similar results were obtained in SIMA cells treated with compounds at 1μM. Bars represent S.E.M. See also Fig. S3.
Next, we sought to determine the interactions of JQAD1 with EP300 and CBP using synthesized biotinylated JQAD1 (Biotin-JQAD1, Scheme S2) incubated in Kelly cell lysates, followed by streptavidin-based bead purification. Western blotting of biotin-JQAD1-purified lysates demonstrated the presence of EP300 and CRBN, but surprisingly not CBP proteins (Fig. 3C). This surprising finding indicated that JQAD1 may be selective for EP300, relative to CBP. To further characterize the interaction of JQAD1 with EP300 and CBP, we co-treated Kelly cell lysates with JQAD1 and excess pomalidomide, to compete for binding to CRBN, which resulted in a partial loss of the interaction between JQAD1, CRBN and EP300 (Fig. 3C). These data indicated that these three proteins form a ternary complex. We were excited by the apparent specificity of this PROTAC for binding to EP300, because EP300, but not CBP, is the dominant mediator of H3K27ac in high-risk neuroblastoma. PROTACs may acquire preferential specificity for one of two possible target HATs due to restricted three-dimensional interactions (27). In our case, the preferential targeting of EP300 by this PROTAC is an advantage, because neuroblastoma cells are often exclusively dependent on EP300, while normal cells of different lineages may require CBP, and thus would be spared from toxicity by the specificity of this PROTAC.
Because JQAD1 interacts preferentially with both EP300 and CRBN, we next examined whether JQAD1 preferentially induces degradation of EP300 compared to CBP in MYCN-amplified neuroblastoma cells. Treatment of Kelly cells for 24h with JQAD1 demonstrated a dose-dependent decrease in EP300 expression, along with a parallel loss of the H3K27ac modification (Fig. 3D). Similar treatment of Kelly cells with A485 caused a loss of H3K27ac, due to catalytic inhibition of EP300 enzymatic activity (Fig. 3D). Treatment of Kelly cells with (R,S)-JQAD1 and the control (S,S)-JQAD1 for 24h revealed that (S,S)-JQAD1 had limited effects on H3K27ac or EP300 expression levels, while (R,S)-JQAD1 suppressed both H3K27ac and EP300 expression levels (Fig. S3G). Consistent with the specificity of JQAD1 for EP300, we noted that neither compound had significant effects on CBP expression levels at this timepoint (Fig. S3G). To comprehensively characterize the specificity of JQAD1 for EP300 at this timepoint, we performed an analysis of the effects of JQAD1 by stable isotope labeling of amino acids in cell culture (SILAC) (Fig. 3E). Kelly cells were cultured with SILAC media containing heavy or light-labelled arginine and lysine. Heavy labelled cells were treated with 500 nM JQAD1, and light-labelled cells treated with DMSO for 24h, prior to nuclear extraction and protein lysis. As a control, we performed nuclear extraction and lysis on untreated heavy and light-labelled Kelly cells. Protein abundance was then analyzed by mass spectrometry, to determine global changes in the nuclear proteome. Following 24h of treatment with JQAD1, EP300 protein was significantly decreased (p=3.3 ×10−5), while CBP and other proteins within the nuclear proteome remained detectable at similar levels as controls (Fig. 3E). Next, we treated three NB cell lines, Kelly, NGP and SIMA that have high protein expression levels of CRBN (Fig. S1C) with JQAD1 and then measured the effects on specific proteins by western blotting. In all three cell lines, JQAD1 induced selective loss of EP300 expression coincident with cleavage of PARP1, signaling the onset of apoptosis (Fig. 3F, S3H). At this time point in all three cell lines, CBP could still be detected. With extended treatment, we also noted loss of CBP expression, though markers of apoptosis (cleaved PARP1) could be detected prior to the loss of CBP protein expression (Fig. 3F, S3H).
JQAD1 contains an IMiD moiety that interacts with the E3 ligase receptor CRBN (Fig. 3A,C). To further demonstrate this interaction, we used the AlphaLISA platform (39) to perform AlphaLISA fluorescent assays using bead-bound biotinylated pomalidomide and His-tagged CRBN. Multiple iMiD-containing compounds, including JQAD1 and free pomalidomide, efficiently interacted with CRBN by the AlphaLISA assay, while the parental compound A485 did not (Fig. S3I). We next sought to establish if CRBN proteins are required for JQAD1-mediated EP300 degradation and cellular effects. To do so, we used CRISPR-Cas9 gene editing to produce Kelly cells with stable disruption of the CRBN gene. Western blotting of lysates prepared from control or CRBN-edited Kelly cells demonstrated loss of CRBN expression in CRBN edited cells, with retained expression in control-edited cells (Fig. 3G). Control-edited Kelly cells were potently killed by JQAD1, however CRBN-knockout cells were resistant to the effects of JQAD1, indicating that CRBN expression was required for JQAD1 growth suppressive activity (Fig. 3H). In contrast, A485 equivalently inhibited the growth of both CRBN-knockout cells and controls (Fig. S3J), indicating that loss of CRBN has no effect on the enzymatic function of EP300. Western blotting of lysates prepared from control and CRBN-knockout Kelly cells treated with JQAD1 or DMSO demonstrated that JQAD1 suppressed EP300 expression, the H3K27ac modification and induced apoptosis, marked by PARP1 cleavage in control cells, but not in CRBN-edited cells (Fig. S3K). Thus, CRBN is required for JQAD1-mediated EP300 degradation and induction of apoptosis. Since treatment of CRBN-edited cells with JQAD1 had no effect on H3K27ac, we hypothesize that the structure of JQAD1 prevents its A485 moiety from competitively inhibiting EP300 HAT activity, and therefore it acts as a CRBN-dependent protein degrader, without significant catalytic inhibitory activity (Fig. S3K). To further probe the pathway involved in JQAD1-mediated EP300 degradation, we next performed western blotting on Kelly cells co-treated with JQAD1 and other compounds predicted to disrupt JQAD1 function. Degradation of EP300 was blocked by co-treatment with excess A485, IMiD (pomalidomide), and E3 ubiquitin ligases neddylation (MLN4924) and more minimally by inhibition of the proteasome (bortezomib) (Fig. S3L). These data indicate that JQAD1 functions by binding to EP300, which leads to CRBN-dependent proteasomal degradation of EP300 and cell death.
JQAD1 causes apoptosis concurrent with MYCN downregulation
Next, we sought to evaluate the mechanism by which JQAD1 reduced cell growth. We treated Kelly and NGP cells with JQAD1, A485 or vehicle control and performed propidium-iodide DNA flow cytometry. Cells treated with A485 to inhibit EP300/CBP catalytic activity underwent G1 cell cycle arrest (Fig. S3M). Strikingly, (R,S)-JQAD1 treatment resulted in early time-dependent induction of a subG1 peak, suggestive of apoptotic cell death (Fig. 3I,J, S3N). To more deeply characterize the differences between HAT inhibition and EP300-selective degradation on neuroblastoma apoptosis, we treated Kelly and NGP cells for 12–36h with equal concentrations of either A485 or JQAD1, prior to extracting protein to analyze effects on apoptosis. Treatment of both cell lines with JQAD1 resulted in cellular apoptosis, marked by cleavage of caspase-3 and PARP1. In contrast, A485 treatment had little effect on these parameters at these timepoints (Fig. 4A, S4A). To identify the mechanism underlying this difference in response between degradation of EP300 and catalytic inhibition of both EP300 and CBP, we next treated Kelly NB cells with equivalent concentrations of DMSO, JQAD1 or A485 for 24h and performed RNA-seq analysis with exogenous spike-in RNA normalization. RNA-seq results for JQAD1 and A485 treated samples were then compared by gene set enrichment (GSEA) analysis. Consistent with our DNA flow cytometry studies, analysis of GSEA results with the molecular signatures database (MSigDB) hallmark gene sets demonstrated enrichment of the “apoptosis” gene set in JQAD1-treated cells, compared with A485-treated cells (Fig. 4B). Furthermore, JQAD1-treated cells exhibited upregulation of the proapoptotic BH3-only effectors BIM, BID and PUMA together with the proapoptotic mediator BAX and its inhibitors BCL2 and MCL1, while transcript levels for each of these mRNAs was unaffected in A485-treated cells at this timepoint (Fig. 4C). The induction of apoptotic cell death at an early time point with JQAD1, compared to A485 treatment, suggests that there may be a HAT-independent activity contributing to the apoptosis observed with EP300 degradation.
Figure 4. EP300 degradation rapidly disrupts MYCN expression and causes apoptosis.
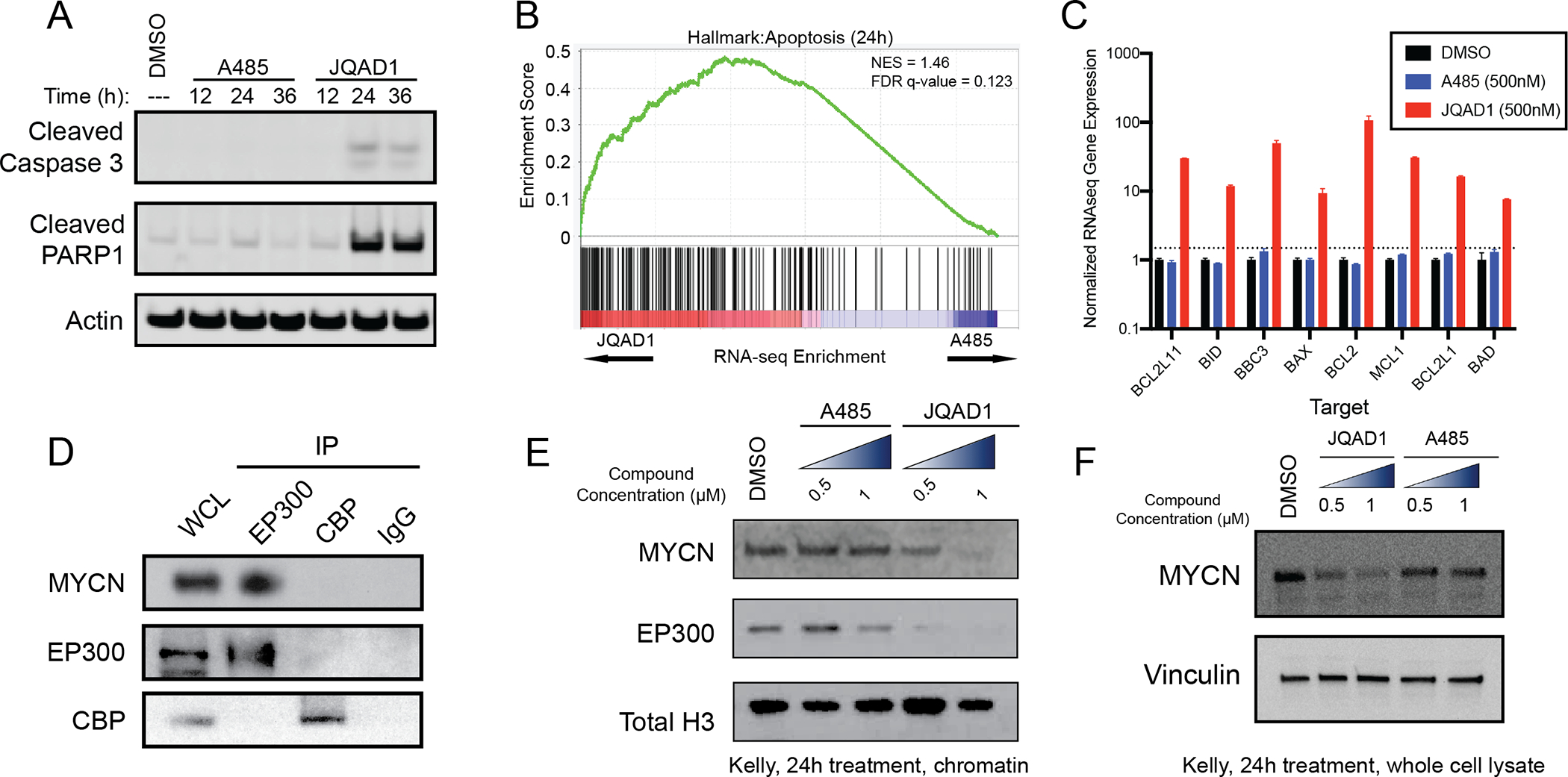
A. Kelly NB cells were treated with 1μM JQAD1, A485 or DMSO control for 12, 24, or 36h, prior to lysis and western blotting for the markers of apoptosis: cleaved caspase-3 and cleaved PARP1. Actin is demonstrated as a loading control. Data is representative of three independent treatments and analyses in Kelly and NGP cells.
B. Kelly cells were treated with 500nM JQAD1, A485 or DMSO control for 24h prior to ERCC-controlled spike in RNAseq. Gene set enrichment analysis of RNAseq results was performed with the MSigDB Hallmarks dataset. n=3 biological replicates and independent RNA extractions per treatment.
C. Normalized RNAseq gene expression of pro- and anti-apopotic mRNA transcripts from Kelly cells treated as in B. Log10 transcript expression is shown, normalized against DMSO and ERCC controls. n=3 biological replicates and independent RNA extractions per treatment. Bars represent S.E.M.
D. Nuclear lysates from Kelly cells were immunoprecipitated with anti-EP300, anti-CBP or IgG control antibodies. WCL=whole cell lysate. Data is representative of >3 independent co-immunoprecipitation/western blots.
E,F. Kelly cells were treated with DMSO control, A485 (0.5, 1 μM) or JQAD1 (0.5, 1 μM), followed by extraction of chromatin (E) or whole cell lysates (F) and western blotting. Total H3 is shown as a loading control. Data is representative of 3 independent biological replicates. See also Fig. S4.
One mechanism by which NB cells repress apoptosis is through high level expression and transcriptional activity of the MYCN oncoprotein, sometimes referred to as “oncogene addiction” (reviewed in (40,41)). EP300 and CBP regulate the MYCN family member c-MYC protein through protein-protein interactions (42–44), so we hypothesized that a similar physical interaction between MYCN and EP300 might exist, resulting in MYCN localization at chromatin. To examine this hypothesis, we performed co-immunoprecipitation assays with antibodies targeting either endogenous EP300 or CBP in Kelly NB cells. Immunoprecipitation of protein from Kelly nuclear lysates with anti-EP300 antibodies followed by western blotting demonstrated pronounced association with MYCN protein. In contrast, immunoprecipitation of CBP, like IgG controls, did not reveal detectable MYCN protein (Fig. 4D). Thus, in Kelly NB cells, EP300, but not CBP, physically interacts with the MYCN oncoprotein.
To evaluate the functional significance of this interaction, we next treated Kelly NB cells with DMSO or two concentrations of either A485 or JQAD1 for 24h and isolated proteins associated with chromatin (Fig 4E) or whole-cell lysate (Fig 4F). Kelly cells treated with A485 demonstrated stable levels of MYCN proteins in chromatin extracts and whole-cell lysates up to 1.0 μM (Fig. 4E,F). In contrast, cells treated with JQAD1 to degrade EP300 showed loss of both EP300 and MYCN proteins from the chromatin-associated protein fraction and whole-cell lysate (Fig. 4E,F). To examine whether MYCN is the primary mediator of EP300 function, we over-expressed in Kelly cells either MYCN cDNA or EGFP control cDNA tagged with a nuclear localization sequence using lentiviral infection. Western blotting demonstrated that MYCN-overexpressing cells had higher detectable levels of MYCN, and these two infected pools expressed equivalent amounts of EP300 and CBP (Fig. S4B). We then examined the response of these cells to JQAD1 in CellTiter-Glo growth assays and found that slightly higher dosages of JQAD1 are required to cause reduced growth in neuroblastoma cells overexpressing MYCN, after treatment with JQAD1. The IC50 of the MYCN overexpressing cells was shifted slightly to 121 nM from 95 nM in EGFP overexpressing cells (Fig. S4C). MYCN-overexpressing Kelly cells were somewhat less responsive to JQAD1 treatment by CellTiter-Glo analysis, indicating that MYCN re-expression is insufficient to correct for the loss of EP300. These data suggest that one effect of EP300 degradation is rapid loss of MYCN expression, whereas inhibition of EP300 HAT activity requires much more prolonged treatment to induce the same levels of MYCN repression (Fig. S1N).
JQAD1 causes loss of H3K27ac at chromatin
Since JQAD1 selectively degrades EP300 with minimal effects on CBP until 48 hours in NB cell lines, we next used this compound to identify the effects of EP300 loss on genome-wide H3K27ac. We performed ChIP-seq with antibodies recognizing H3K27ac in Kelly NB cells, over a time course from 0–24 hours after exposure to (R,S)-JQAD1. These samples were externally normalized using spiked-in Drosophila melanogaster chromatin. Comparison of H3K27ac-marked sites to untreated samples demonstrated approximately 2-fold general suppression genome-wide by 24h of treatment, at a time when EP300 was degraded and CBP was retained (Fig. 5A). While comparison of H3K27ac signal at earlier timepoints (6h) to 0h controls demonstrated no consistent changes in acetylation, by 24h of treatment, there was general loss of H3K27ac signal genome-wide, which was most pronounced at densely acetylated super-enhancers, including those regulating the core regulatory circuitry. (Fig. 5B–D, S4D). These data indicate that super-enhancer loci in Kelly cells are regulated predominantly by EP300 and not CBP, because at this timepoint, EP300 is degraded without effects on the levels of CBP protein expression (Fig. 5D, S4D).
Figure 5. JQAD1 causes genome-wide loss of histone H3K27-acetylation, enriched at super-enhancers.
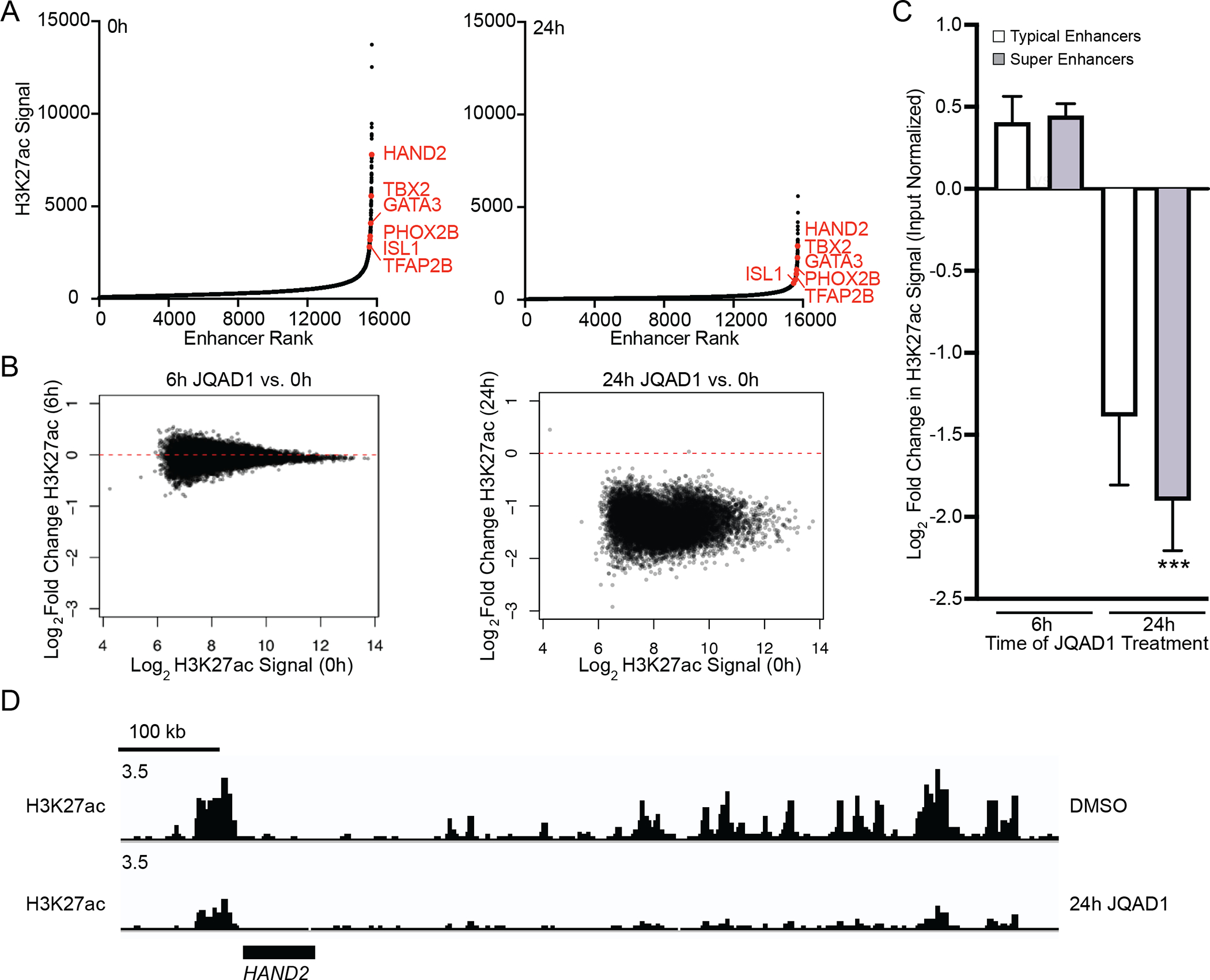
A. Enhancers were ranked by H3K27ac signal at 0h (left) and 24h (right) after treatment of Kelly cells with 500nM JQAD1. Data are representative of two independent treatments and ChIP-seq experiments.
B. Log2 fold change in enhancer H3K27ac signal resolved by H3K27ac ChIP-seq in Kelly NB cells at 0 vs. 6h (left) and 0 vs. 24h (right).
C. Log2 fold change in enhancer H3K27ac signal stratified by super-enhancers and typical enhancers at 6h and 24h after treatment of Kelly cells with 500 nM JQAD1. *** indicates p<0.0001 by students t-test, comparing super-enhancer and typical enhancer-regulated genes at 24h. Bars represent S.E.M.
D. Representative gene tracks of Kelly cells treated with JQAD1 at 500 nM for 0 and 24h at the HAND2 core regulatory circuitry factor locus. Data is representative of the adrenergic CRC factor loci (HAND2, ISL1, PHOX2B, GATA3, TBX2, ASCL1 and TFAP2β) and two independent treatments and ChIP-seq experiments. See also Fig. S4.
JQAD1 is effective with limited toxicity in vivo
Some CRBN-based PROTAC agents cause target protein degradation in vivo (reviewed in (27)). Thus, we next sought to identify whether JQAD1 would degrade EP300 in vivo in human neuroblastoma xenografts. First, we performed pharmacokinetic analysis after a single intraperitoneal (I.P.) dose of JQAD1 at 10 mg/kg, to identify the half-life and maximum serum concentration of the compound. We found that after 10 mg/kg intraperitoneal dosage, JQAD1 has a half-life of 13.3 (+/−3.37 SD) h in murine serum, with a Cmax of 7 μM (Fig. S5A), which is well above the IC50 of human neuroblastoma cells in vitro (Fig. S3A–C). Then we sought to identify the maximum tolerated dose (MTD) in murine models. To do so, we performed daily I.P. injection of JQAD1 at increasing doses in CD1 mice. Daily treatment with JQAD1 was well tolerated with no signs of animal weight loss (Fig. S5B). At doses of JQAD1 higher than 40 mg/kg, we experienced problems with the compound forming precipitates in the peritoneal cavity. Thus, 40 mg/kg of JQAD1 was determined to be the maximal dosage we could administer I.P., without evident toxicity in the mouse.
Next, we established subcutaneous xenografts of Kelly cells in the flanks of NSG mice, and treated mice with either vehicle control or JQAD1 at 40 mg/kg I.P. once daily (Fig. 6A). JQAD1 treatment suppressed xenograft tumor growth by day 3 of treatment (p<0.0001 for suppressed growth rates in JQAD1-treated tumors by mixed-effects analysis with post-hoc Tukey’s multiple comparisons test), and prolongation of survival (log-rank test p=0.0003 for JQAD1-treated tumors compared with vehicle control) (Fig. 6A,B). We also monitored for effects of JQAD1 treatment on animal weight. Body weight was maintained over 15 days of treatment, prior to when control animals began to display rapidly enlarging tumors (Fig. 6C). Separately, NSG mice were xenografted with Kelly cells and treated with vehicle control or JQAD1 daily at 40 mg/kg for 10 days. Animals were then sacrificed, and the tumors were extracted. Tumor material was fixed for IHC and processed for ERCC-controlled RNA-seq analysis (Fig. 6D,E). Tumors recovered from animals treated with JQAD1 displayed a loss of EP300, but retained CBP immunostaining, compared with vehicle control tumors (Fig. 6D). Consistent with our in vitro studies, RNA expression profiles of tumor cells from mice treated with JQAD1, compared with vehicle control demonstrated preferential downregulation of genes regulated by super-enhancers compared with those regulated by typical enhancers (Fig. 6E, p<0.0001).
Figure 6. JQAD1 causes tumor growth suppression and loss of EP300 in vivo.
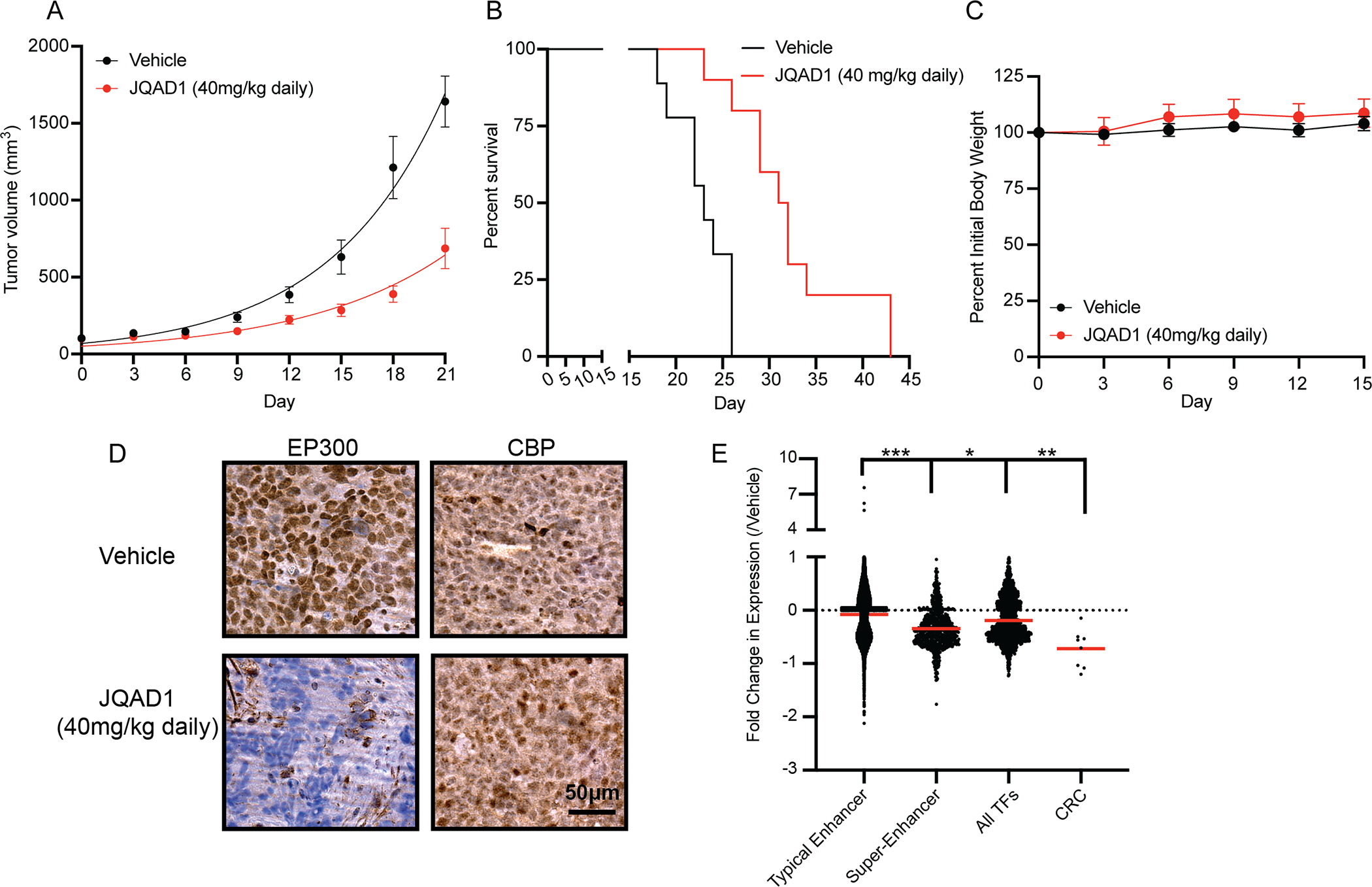
A. Kelly NB cell xenografts were established in NSG mice, and mice treated with vehicle control (n=9), or JQAD1 at 40 mg/kg I.P. daily (n=10). Tumor growth curve kinetics were also analyzed by two-way ANOVA with mixed-effects analysis, demonstrating that JQAD1 suppresses tumor growth (p<0.0001 for vehicle vs. JQAD1 treatment groups).
B. Kaplan-Meier survival analysis of mice in A. JQAD1 prolongs survival, log-rank test p=0.0003 for JQAD1-treated mice compared with vehicle.
C. Normalized body weights of animals from A,B.
D. Immunohistochemistry to EP300 and CBP in Kelly cell xenografts treated with either vehicle control or JQAD1 (40 mg/kg I.P. daily) for 14 days. Data are representative of 3 independent animals per treatment. Bar = 50μm.
E. ERCC-spike in RNA-seq was performed on tumor cells recovered from animals treated in D. Results are shown as the fold change in expression of animals treated with 40 mg/kg JQAD1 daily (n=3) compared with vehicle control (n=4) at day 14. RNA-seq groups of genes are stratified by their regulation by typical or super-enhancers, and gene identity of transcription factor or CRC gene. *** p<0.0001 between typical enhancer and super-enhancer groups and between typical enhancer and CRC gene expression, * p=0.0223 between super-enhancer groups and CRC gene expression, ** p=0.0013 between all TFs and CRC gene expression. See also Fig. S5.
Human CRBN differs from mouse at a key residue, CRBNVal388 compared to CrbnIle391 in the mouse, which is important for binding, ubiquitinating and degrading key substrates (45,46). To more rigorously assess the potential activity and toxicities of JQAD1 on murine tissues, we administered JQAD1 at 40 mg/kg I.P. daily for 21 days to Balb/c CrbnILE391VAL-humanized knock--in mice(46). JQAD1 at this dosage was well tolerated, with no effects on grooming, behavior, weight, peripheral blood counts, liver function tests or creatinine measurements performed after 14 days of treatment (Table S2, Fig. S5C). After 14 days of treatment, three mice per each treatment group were sacrificed and skin, brain, heart, lung, liver, spleen, kidney, pancreas, small intestine, colon, adrenal gland and bladder were extracted and processed for pathologic analysis. Tissues were evaluated by an independently blinded pathologist, by hematoxylin and eosin staining for evidence of toxicity. This revealed no gross changes in tissue architecture or immune infiltrate, consistent with a lack of toxicity. To establish whether JQAD1 was selective in degrading EP300 and not CBP in vivo, we then performed immunohistochemistry against EP300 and CBP on liver tissues from Balb/c CrbnILE391VAL mice treated with vehicle control or JQAD1. This analysis demonstrated that JQAD1-treated animals had reduced EP300, but not CBP protein expression levels, in liver cell nuclei compared with vehicle treated controls (Fig. S5D). Consistent with the hypothesis that CBP could partially compensate for loss of EP300, JQAD1-treated animals displayed no histologic or biochemical evidence of toxicity in the liver.
Previously, we identified that MYCN and each of the master transcription factor members of the adrenergic CRC are dependencies in neuroblastoma(9). Since EP300 dominantly catalyzes the H3K27ac mark, we hypothesized that EP300 might preferentially be responsible for the high levels of expression of CRC master transcription factors. Further, since JQAD1 preferentially degrades EP300, the HAT that primarily catalyzes H3K27ac seen at super-enhancers and those of the CRC in particular, we reasoned that treatment with JQAD1 might have major effects on the expression levels of genes in the CRC. Therefore, we compared the effects of JQAD1 given daily for 14 days on the expression levels of several different classes of mRNAs, including those regulated by typical enhancers, super-enhancers, and all TFs as well as TFs that encoded known CRC members (Fig. 6E, S5E). This revealed that the CRC genes, along with MYCN, were among the most downregulated genes in tumors treated with JQAD1, compared with genes in the other categories. Gene set enrichment analysis of the MSigDB Cancer Hallmarks dataset revealed 5/50 cancer hallmarks downregulated in JQAD1 treated tumor samples with an FDR<0.25 and NOM p-value<0.05 (Fig. S5F). Consistent with our finding that MYCN was among the most downregulated genes in tumors treated with JQAD1 in vivo, these hallmark gene sets included an enrichment for MYC target genes (Fig. S5G,H). These results demonstrate that JQAD1 treatment in vivo strongly down-regulates the expression of the very important subset of genes that encode transcription factors comprising the CRC, including MYCN, which are neuroblastoma growth dependencies (9,35).
JQAD1 has broad CRBN-dependent anti-neoplastic activity across cancer cell lines
Epigenetic and enhancer-mediated control of gene expression is required for normal cellular and tissue developmental processes and is dysregulated in different cancer subtypes (reviewed in (2,28)). We therefore hypothesized that in addition to neuroblastoma, there may be a preferential reliance on EP300 or CBP in other cancer types as well. Thus, we examined the relative dependence of all available cell lines on EP300 or CBP using the DepMap genome-scale CRISPR-Cas9 loss-of-function screening dataset (30). Comparison of the probability of dependency on EP300 and CBP across a total of 757 human cancer cell lines, representing 36 distinct tumor lineages, demonstrated a higher probability of dependency on EP300 than CBP, across many cancer cell lines (p<0.0001, Fig. 7A). We also stratified all cell lines in DepMap by tumor lineage and examined the probability of dependency on EP300 and CBP in each lineage. By this analysis, many tumor lineages displayed an enhanced probability of dependency on EP300, compared with CBP (Fig. 7B). Few tumor cell lineages, notably thyroid, pancreatic and cervical carcinomas displayed enhanced dependency on CBP, compared with EP300 (Fig. 7B).
Figure 7. Cancer cells display increased dependency on EP300, compared to CBP.
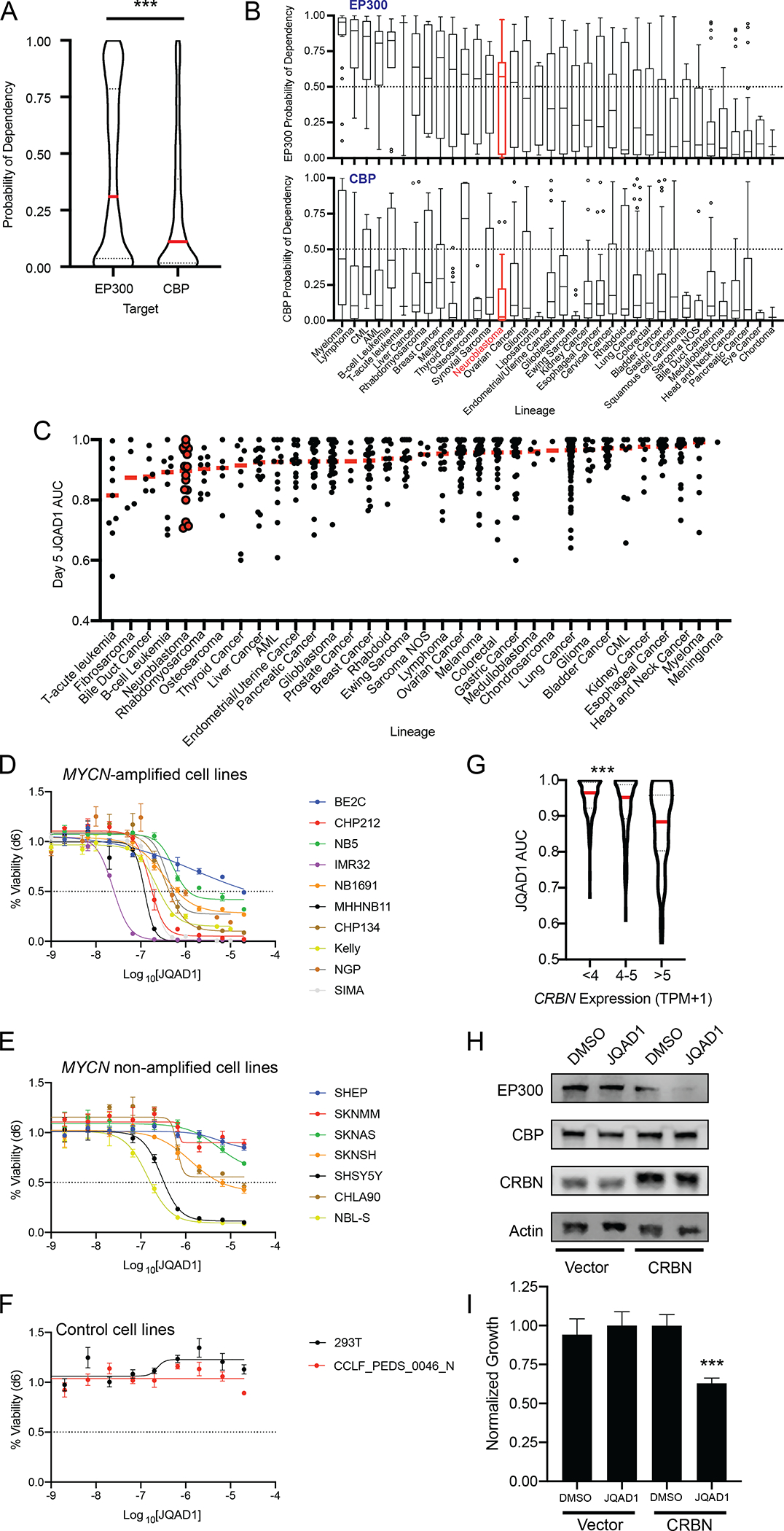
A. Probability of dependency of all cell lines in DepMap (n=757, 20Q2 release), on EP300 and CBP were compared, demonstrating dependency on EP300 in 308/757 (40.7%) and CBP in 140/757 (18.5%) of all cell lines, determined by probability of dependency >0.5. *** p<0.0001 by two-tailed Student’s t-test.
B. Individual lineages of cell lines from A were identified, and average probability of dependency on EP300 and CBP were plotted. Red indicates neuroblastoma, black indicates other tumor lineages. Bar in box indicates median, whiskers indicate 10th-90th centiles. Dots indicate outliers.
C. Barcoded cancer cell lines (n=557) were treated with a concentration range of (R,S)-JQAD1 of 1.2nM to 20μM for 5 days prior to sequencing of barcodes. Cell lines were individually classified by lineages, and area under the curve (AUC) of the dose-response relationship was plotted. Red bars = median, individual black dots = individual cell lines, Red dots = neuroblastoma cell lines. AUC was calculated from triplicate measurements at each dose at time = 120h.
D,E,F. NB and control cell lines were grown for 6 days in the presence of JQAD1 in a dose range from 4.3 nM to 20 μM, prior to CellTiter-Glo analysis. Dose-response curves are based on three independent replicates per cell line at each dose. Bars represent S.E.M. Analysis was performed on MYCN-amplified (D), non-amplified (E) and control (F) cell lines, including 293T cells and primary human fibroblasts (CCLF_PEDS_0046_N). Cell lines are all of the adrenergic subtype except for NB5 and SKNMM (unknown), and CHP212, SHEP and SKNAS (mesenchymal). Adrenergic or mesenchymal cell state annotations are derived from(33,69).
G. JQAD1 AUC values from C were plotted against CRBN expression from the Cancer Cell Line Encyclopedia (CCLE). *** p<0.001 by ANOVA for >5 TPM compared against <4 and 4–5 TPM groups with post-hoc Bonferroni correction.
H. BE2C cells stably expressing control (zsGreen) or CRBN (CRBN) were established and pools of cells were treated with DMSO or 10 μM JQAD1 for 24h. Cell lysates were subjected to western blotting for EP300, CBP and CRBN. Actin is shown as a loading control. Data is representative of three independent treatments and analyses.
I. BE2C cells stably expressing control (zsGreen) or CRBN (CRBN) were treated with DMSO or 10 μM JQAD1 for 6 days prior to CellTiter-Glo analysis for cell growth. Data was normalized against BE2C-zsGreen, DMSO treated cells. *** p = 0.008 by student’s T-test comparing BE2C-CRBN DMSO and JQAD1 treated cells. n=3 biological replicates. See Fig. S6.
Because the probability of dependency on EP300 was higher for many tumor lineages than that of CBP, we next sought to determine whether JQAD1 would display antineoplastic effects across multiple tumor lineages. To do so, we analyzed the response to JQAD1 in a pooled, barcoded 5-day cell viability PRISM screen of 557 cancer cell lines (Fig. 7C) (47). These results demonstrated that JQAD1 treatment had antineoplastic activity across multiple tumor lineages, many of which displayed enhanced dependency on EP300, compared with CBP. Within most tumor lineages, there were cell lines that displayed growth inhibition with JQAD1 treatment (area under the curve, AUC<0.85) (Fig. 7C). Validation of these results in a wide array of neuroblastoma cell lines by CellTiter-Glo analysis demonstrated that many were sensitive to JQAD1, including MYCN-amplified and MYCN non-amplified cell lines (Fig. 7D,E). Further, two control cell lines, 293T and primary human fibroblasts, were unaffected by JQAD1 to a maximum dose of 20μM, suggesting that the low in vivo toxicity we identified may reflect lower toxicity in non-tumorigenic cells (Fig 7F). Further study will be required to determine whether the specificity of JQAD1 will translate to lower toxicity and a broader therapeutic index for cancers selectively dependent on EP300 compared to CBP.
Since cell lines from multiple lineages displayed growth suppression with JQAD1 treatment, we next sought to identify whether predictors of JQAD1 activity could be determined. To do so, we performed an analysis of RNA expression profiles of all cell lines treated with JQAD1. Consistent with its mechanism of action, we noted that higher expression levels of CRBN were correlated with higher JQAD1-mediated antineoplastic activity, as reflected by a lower AUC measurement of JQAD1 dose-response (Fig. 7G). This indicated that JQAD1 activity is at least partially determined by CRBN expression levels, which is consistent with the requirement by JQAD1 for CRBN to target EP300 for degradation. To further investigate this requirement, we hypothesized that increasing the expression levels of CRBN in JQAD1-resistant neuroblastoma cells may result in restoration of sensitivity. Thus, we examined BE2C neuroblastoma cells, which have lower CRBN protein expression and are less sensitive to JQAD1 than many other neuroblastoma cell lines (Fig. S1C, 7D). We established BE2C cells with stable overexpression of CRBN (BE2C-CRBN) or, as a control, zsGreen (BE2C-zsGreen), and treated these cells with JQAD1 or vehicle control (Fig. 7H). EP300 was degraded within 24h of treatment with JQAD1 in BE2C-CRBN cells, while expression of EP300 in control cells was unaffected (Fig. 7H). Further, the growth of BE2C-CRBN cells was suppressed by JQAD1 treatment, while untreated BE2C-CRBN cells grew at similar rates as BE2C-zsGreen cells treated with DMSO or JQAD1 (Fig. 7I). Thus, CRBN overexpression in JQAD1-insensitive BE2C cells is sufficient to restore sensitivity to JQAD1. Finally, to understand whether CRBN expression was variable in primary neuroblastoma tumors, we examined the inter-tumoral expression of CRBN in two large neuroblastoma cohorts using the R2 database. This demonstrated that CRBN has similar mean expression values with overlapping 95% confidence intervals across patient groups divided according to prognostic variables such as patient age (Fig. S6A,F), tumor stage (S6B,G) or MYCN-amplification status (S6C,H). Further, there was no apparent correlation between expression levels of CRBN and levels of either MYCN (S6D,I) or c-MYC (S6E,J). Then, to assess for potential intra-tumoral heterogeneity of CRBN expression in patient tumor samples, we re-analyzed a recently published, publicly available dataset of single-cell RNAseq from 16 primary neuroblastoma tumors. Here, CRBN expression was found diffusely throughout the entire malignant cell population, without clear subsets of tumor cells with differential expression (Fig. S6K). Thus, there does not appear to be significant heterogeneity in CRBN expression across or within primary neuroblastoma tumors. These data underscore that two important considerations for using degraders across distinct cell models are 1) individual cell line dependency on the PROTAC target, and 2) expression levels of key components of the PROTAC machinery, such as CRBN. In summary, these data indicate that cancer cells beyond neuroblastoma display enhanced dependency on EP300, compared to CBP, and that JQAD1 represents a potential method to capitalize on this enhanced dependency, especially in individual tumors with elevated CRBN expression levels.
DISCUSSION
EP300 and CBP are paralogous, multi-domain protein acetyltransferases with broad cellular functions mediated by protein-protein interactions and catalytic acetyltransferase activities (13). These proteins are independently mutated or translocated in a variety of human cancers, and studies have identified distinct but overlapping activities of these proteins in untransformed cell types, including embryonic and hematopoietic stem cells and more differentiated fibroblasts and T-cells (20,21,48–50). EP300 and CBP display largely overlapping but partially distinct binding patterns across the genome, indicating that these proteins exhibit only partial functional redundancy in transcriptional regulation (24,25). Due to their high degree of homology, especially in the HAT and bromodomains, it has been difficult to study their function independently and design small molecule inhibitors that are selective for either. To this end, studies have demonstrated that EP300 exhibits synthetic lethality in cell lines in which CBP is mutationally inactivated (23). However, both enzymes are expressed in most cell lines and primary tissues, making it difficult to distinguish between their individual functions.
Here, we demonstrate that most childhood neuroblastomas display selective dependency on EP300 and not on CBP. By studying this dependency on EP300, we identified that the transcription factor TFAP2β is a key member of the core regulatory circuitry that co-binds genome-wide with the known CRC factors. Core regulatory circuitries are lineage-defining autoregulatory transcription factor networks that establish the transcriptional landscapes of different types of cells (9,35,51–53). EP300 and CBP do not bind DNA in a sequence-specific manner, and so depend on transcription factors to localize them to their target loci. Importantly, the transcription factor TFAP2β specifically binds EP300, but not CBP, and is responsible for recruiting EP300 to its targets in neuroblastoma cells. Loss of TFAP2β, but not the CRC transcription factors HAND2 or GATA3, results in loss of the H3K27ac mark on CRC associated super-enhancers catalyzed by EP300 in neuroblastoma cells, thereby identifying TFAP2β as a primary mediator of EP300 localization to critical super-enhancers. This mechanism results in direct EP300-regulation of lineage-specifying and oncogenic loci in neuroblastoma by CRC-dependent recruitment. This function cannot be accomplished by CBP, because it does not physically interact with TFAP2β. In addition to transcription factors, core regulatory circuitries also include enhancer RNAs and linker proteins such as LDB1 and LMO1 that are integral components of this regulatory complex (35,51,54). With evidence that coactivator proteins are found at genomic loci bound by CRC transcription factors(55), and that loss of EP300 results in enhanced loss of CRC factor expression, compared with other transcription factors in vivo, we posit that coactivator enzymes such as EP300 are critical for the high levels of expression that define genes of the CRC extended regulatory network. Moreover, lineage- and tumor-specific CRC factors, such as TFAP2β in neuroblastoma, play a role in the CRC complex by recruiting EP300 to establish the malignant cell state.
There is a striking enrichment for dependency on EP300, compared to CBP, in various cancer subtypes, consistent with the hypothesis that these two paralogous genes may play context-dependent, distinct roles in regulating cancer cell survival. As a result, selective targeting of EP300 in different types of cancer cell lines that are dependent of EP300 may be effective for eliciting anti-tumor activity, with reduced toxicity because CBP is still active in normal cells and may be able in most normal cells to compensate for the loss of EP300. This attractive hypothesis has been hard to test, because of significant homology between these two proteins, which has prevented pharmacologic strategies to preferentially target one of these enzymes, while sparing the other.
Here, we generated a novel PROTAC, termed JQAD1, which relies on the non-selective EP300 and CBP-binding compound A485 as a binding molecule but is selective in its ability to degrade EP300 compared to CBP. This selectivity stands in marked contrast to the more promiscuous acetyltransferase inhibitory activity of A485 against both EP300 and CBP. PROTAC agents synthesized from bait molecules with binding to several closely related proteins can display substrate specificity, such as with BRD4 and p38 degraders (reviewed in (27)). Recently, a degrader molecule has been reported that degrades both EP300 and CBP indiscriminately, using a bait molecule that targets the bromodomain of these proteins(56). We therefore hypothesize that the mechanism of JQAD1 selectivity is likely to be related to three-dimensional interactions between the targeted protein and the E3 ligase complex, mediated by the different binding component of the PROTAC. Due to the size of full-length EP300 and CBP proteins, full-length crystal structures have not been resolved, and as a result, this hypothesis remains unproven. However, we note that Biotin-JQAD1 forms a ternary complex with EP300 and CRBN, which does not contain CBP. Thus, in contrast to A485, which has equivalent binding activity to EP300 and CBP, JQAD1 binds more avidly to EP300 than CBP in the presence of CRBN. Our data indicates that JQAD1 strongly and preferentially degrades EP300. With continuous prolonged treatment in vitro, however, we observed loss of CBP. This is not observed in vivo, where more complex mechanisms of compound processing, bioavailability and excretion are at play. The effects of JQAD1 on CBP in vitro may be related to concurrent apoptotic processes such as activation of nuclear proteases, or CBP may serve as a secondary target after depletion of EP300. Further exploration of the mechanism of kinetic selectivity of PROTACs, and indeed, co-crystallization of full-length CRBN with JQAD1 bridging to EP300 will be required to thoroughly resolve the basis for preferential degradation of EP300 by JQAD1.
JQAD1 has several intriguing properties: i) It demonstrates preference for EP300 relative to CBP in multiple neuroblastoma cell lines; ii) It has higher potency than the parental inhibitor in some cell lines; iii) It induces EP300 degradation in a time-dependent manner as early as 16 h, which contrasts with other methods of selective disruption of EP300, such as knockdown or knockouts; and iv) It is useful for degradation of EP300 with limited effects on CBP and limited toxicities in vivo. EP300 is degraded by JQAD1 in vivo in normal murine tissues that express humanized CRBN; however, CBP immunostaining is only minimally affected in these tissues. Further, these tissues display normal architecture. These findings support the hypothesis that CBP may compensate for the loss of EP300 in some normal tissues. Accordingly, we were not able to identify toxicities in mice treated twice daily with 40 mg/kg JQAD1 I.P. for 14 days after profiling blood counts, liver and kidney function tests, weight and grooming. Thus, we hypothesize that CBP-mediated activities partially compensate for loss of EP300 in untransformed cells.
Fundamentally, degradation of full-length EP300 causes rapid induction of apoptosis in neuroblastoma cells, compared with catalytic inhibition, where induction of apoptosis is a delayed event. In neuroblastoma, EP300 physically interacts with, and controls the expression of the dominant tumor oncoprotein MYCN. Degradation of EP300 results in rapid loss of MYCN expression and indeed, loss of MYCN-driven transcriptional activity. This is associated with apoptosis, though re-expression of MYCN is insufficient to fully blunt the response to EP300 degradation. We hypothesize that rapid degradation of EP300 may cause disruption of protein complexes that are required to sustain the malignant phenotype. Future study will aim to evaluate the precise mechanisms resulting in apoptosis driven by acute degradation or prolonged inhibition of EP300.
Thus, here we identify distinct roles for EP300 and CBP in the regulation of cell growth in high-risk pediatric neuroblastoma. These findings are similarly identified in a variety of other tumor types, indicating that enhanced dependency on EP300 is a common finding in human cancers. EP300, but not CBP, is required for regulation of H3K27ac and the gene expression landscape of a subset of high-risk neuroblastoma. This function is promoted by interactions between EP300 and the novel CRC transcription factor TFAP2β that mediates EP300 binding to enhancers and promoters associated with the CRC. In doing so, TFAP2β and EP300 collaborate with the CRC to determine gene expression patterns in the adrenergic subtype of high-risk neuroblastoma. We have capitalized on these findings by generating a novel PROTAC compound, JQAD1, which preferentially depletes full-length EP300 in vitro and in vivo, with limited toxicity to normal tissues. Importantly, EP300 degradation results in loss of the dominant neuroblastoma oncoprotein MYCN, suppression of CRC-based transcription and apoptosis. This tool compound will be further optimized to improve solubility and its ability to induce neuroblastoma cell apoptosis in vivo. These data provide key insights into enhancer control in high-risk neuroblastoma and highlight a novel paradigm for chemical epigenetic control of gene enhancers and mRNA expression in high-risk neuroblastoma, with implications for other types of human cancers.
METHODS
Material and Data Availability
Requests for resources and reagents should be directed to Dr. Jun Qi (jun_qi@dfci.harvard.edu).
RNA-seq, CUT&RUN and ChIP-seq data have been deposited in the Gene Expression Omnibus (GEO) database under SuperSeries accession number GSE159617. Code used in this study is described in the experimental details and is available upon request.
Cell Lines
Cell lines were obtained from the American Type Culture Collection (BE2C, CHP212, IMR32, SKNSH, SHSY5Y, SKNAS, 293T), European Collection of Authenticated Cell Cultures (NB69, CHP134) and DSMZ (Kelly, NGP, SIMA, MHHNB11, NBL-S, SHEP). Cell lines were gifted for this study by Dr. Michael Dyer (St. Jude Children’s Research Hospital - NB5, NB1691, SKNMM and CHLA90), the Cancer Cell Line Factory (Broad Institute - CCLF_PEDS_0046_N primary human fibroblasts) and Dr. Karen Adelman (Harvard Medical School – S2). RRIDs included CVCL_0529, CVCL_1125, CVCL_0346, CVCL_0531, CVCL_0019, CVCL_1700, CVCL_0063, CVCL_1448, CVCL_1124, CVCL_2092, CVCL_2141, CVCL_1695, CVCL_1412, CVCL_2136, CVCL_0524, CVCL_8822, CVCL_5628, CVCL_6610, CVCL_Z232. Cell lines used for the exome-scale CRISPR–Cas9 screen and PRISM analyses have been previously described(30,47). All cell lines were short tandem repeat (STR) tested for identity prior to use. Human cell lines were cultured in RPMI media containing 10% heat-inactivated fetal bovine serum and 1% penicillin-streptomycin. CCLF_PEDS_0046_N primary human fibroblasts were grown in DMEM media containing 10% heat-inactivated fetal bovine serum and 1% penicillin-streptomycin and used between passages 8–12. S2 cells were cultured in Schneider’s media containing 10% heat-inactivated fetal bovine serum and 1% penicillin-streptomycin. All cell lines were routinely validated to be free of Mycoplasma species and used within 10 passages after thawing.
Chemicals
C646 and CBP30 were obtained from Tocris Biosciences. Bortezomib, MLN4924 and thalidomide were obtained from Sigma Aldrich, and pomalidomide and lenalidomide were obtained from Target Molecule Corp. All other chemicals were synthesized and characterized in Qi Lab. Compound JQAD1 and Biotin-JQAD1 were designed and synthesized based on the scheme listed in the supporting information. Compound structure and purity was confirmed by NMR and LCMS.
Animals
8-week-old female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (Jackson Laboratories, RRID:IMSR_JAX:025216) were used for tumor xenograft studies. For maximally tolerated dose testing, C57BL/6-Crbntm1.1Ble/J (Jackson Laboratories, RRID:IMSR_JAX:032487) and Crl:CD1(ICR) mice (Charles River Laboratories, RRID:IMSR_CRL:022) were used.
Lentiviral Infection
Stable and inducible Cas9-expressing cell lines were generated using lentiviral particles produced in 293T cells. Briefly, lentiCas9, pCW-Cas9-Blast, pLKO.5-EGFP, pLC-zsGreen and pLC-CRBN plasmids were obtained from Addgene (RRIDs: Addgene_52962, Addgene_83481, Addgene_57822, Addgene_124301, Addgene_124303). Lentiviruses encoding MYCN or EGFP with in-frame nuclear localization sequences were synthesized by Vectorbuilder Inc. Plasmids were transfected using lipofectamine 2000 (Invitrogen) along with pMD2.G (Addgene_12259) and psPAX2 (Addgene_12260) into 293T cells to generate viral particles by standard methodologies. sgRNAs targeting individual genes were subcloned by standard methodologies within pLKO.5-EFGP. sgRNA sequences are found in the supplementary methods. Kelly, SIMA, BE2C and NGP cells were infected with lentiCas9 or pCW-Cas9 followed by blasticidin selection. Stable or doxycycline-inducible expression of Cas9 was established by western blotting of protein lysates using Cas9 antibody (RRID:AB_2750916). Following infection of pLKO.5-EGFP-sgRNA lentivirus, cells were cultured for greater than 5 days prior to evaluation as noted in the figure legend. BE2C cells were infected with pLC-zsgreen or pLC-CRBN lentiviruses and selected using 500ug/mL hygromycin (Invitrogen). Kelly cells were infected with pLV-EGFP or pLV-MYCN lentiviruses and selected with 5 μg/mL blasticidin (Invitrogen).
Cell Growth Assays
Cells were infected with lentiviruses encoding sgRNAs or treated with compounds as described. Colony assays were performed by replating cells at 500 cells per well in 6-well dishes and grown in regular growth media for 10 d before 100% methanol fixation, 0.05% crystal violet staining, and subsequent quantitation. CellTiter-Glo assay was performed as per the manufacturer’s instructions (Promega). Briefly, 1000 cells/well were plated into 96-well plates and treated with a range of compound concentrations. Cell viability was measured at the noted timepoints, based on luminescence by the CellTiter-Glo assay (Promega) and read on an Envision 2104 (PerkinElmer, USA), according to the manufacturer’s protocol.
Cell Cycle Analysis
Cells were treated as noted, and then resuspended in hypotonic citrate-propidium iodide (PI) solution for 30 minutes at 37°C, nuclei stabilized in 5M NaCl and analyzed on a FACSAria II (BD Biosciences) as in (57). Analysis was performed using FlowJo v10.7 (BD Biosciences).
Western Blotting, Immunoprecipitation and Proteomic Analyses
Cells growing in culture were lysed for whole cell lysates as previously described(9,35). Nuclear lysates were prepared using the NE-PER nuclear lysate kit (Thermo-Fisher Scientific) according to the manufacturer’s protocol. Chromatin lysates were prepared with the total histone extraction kit (Epigentek). Briefly, equivalent amounts of protein were resolved by western blotting using 4–12% Bis-Tris NuPAGE gels (Thermo-Fisher Scientific) prior to transfer and immunoblotting using primary antibodies to: MYCN, total H3, CBP, Cas9, cleaved-PARP1, cleaved Caspase-3, β-actin TFAP2β, CRBN, H3K27ac, EP300, GATA3, Vinculin and HAND2 (Santa Cruz Biotechnology) RRIDs: RRID:AB_2800038, RRID:AB_2756816, RRID:AB_2616020, RRID:AB_2750916, RRID:AB_2160739, RRID:AB_2341188, RRID:AB_330288, RRID:AB_2058198, RRID:AB_2799810, RRID:AB_2118291, RRID:AB_297224, RRID:AB_11212253, RRID:AB_309711. Secondary antibodies were HRP-conjugated anti-rabbit or anti-mouse (Santa Cruz Biotechnology), incubated prior to exposure to enhanced chemiluminescence reagents (GE Amersham). For immunoprecipitation, equal amounts of protein were diluted in buffer C as described(58), and incubated with antibodies covalently conjugated to dynabeads M-270 beads (Thermo-Fisher Scientific), overnight, according to the manufacturer’s directions. Antibodies used included: H3K27ac, EP300, CBP, TFAP2β, MYCN, rabbit IgG. RRIDs include: RRID:AB_2118291, RRID:AB_297224, RRID:AB_2616020, RRID:AB_2058198, RRID:AB_2800038, RRID:AB_737197. Immunoprecipitated protein was isolated as per the manufacturer’s directions and subjected to western blotting or mass spectrometry.
SILAC and H3K27ac co-IP Mass Spectrometry
For analysis of JQAD1 effects on the nuclear proteome, Kelly cells were labelled with both heavy 13C6 15N2 L-lysine and 13C6 15N4 L-arginine (“heavy” labelled cells) or normal L-lysine and L-arginine (“light” labelled cells). Heavy-labelled cells were treated with 1 μM JQAD1, and light-labelled cells were treated with equivalent concentrations of DMSO for 24h, prior to preparation of nuclear lysates using the NE-PER nuclear lysis kit (Thermo Fisher Scientific). Detailed SILAC methods are found in the Supplementary methods. Combined heavy and light nuclear lysate was pooled and subjected to trichloroacetic acid precipitation by standard methodologies, followed by SDS-PAGE. Gel pieces were processed using an LTQ Orbitrap Velos Pro ion-trap mass spectrometer (Thermo Fisher Scientific). Peptide sequences and protein identity were determined using Sequest (Thermo Fisher Scientific)(59). Protein abundance was determined by student’s t-test, comparing 0h abundance to 24h abundance.
For co-immunoprecipitation/mass spectrometry analysis of H3K27ac, BE2C and Kelly cells were treated to collect nuclear lysates as above. Equal amounts of nuclear protein was immunoprecipitated using Dynabeads M270 magnetic beads covalently bound with H3K27ac antibody (Abcam) or normal rabbit IgG (Santa Cruz Biotechnology) for >16h at 4°C, prior to washing and elution of immunoprecipitated protein as per the manufacturer’s instructions (Invitrogen). Eluted protein was trichloroacetic acid precipitated, trypsin-digested and subjected to mass spectrometry. Two independent co-immunoprecipitation/mass spectrometry experiments were performed in each of BE2C and Kelly cells. High confidence proteins (n=35, Table S1) were defined as the subset found in both Kelly and BE2C bound to H3K27ac and not to IgG. Gene identities/function were identified by Gene Ontology and PANTHER analyses(60,61).
In vivo Studies
We adhered to animal protocols approved by the Dana–Farber Cancer Institute Animal Care and Use Committee. Animals were maintained according to institutional guidelines, and animal experiments approved by local IACUC. For toxicity studies, four female CD1 mice (Charles River Laboratories) were injected intraperitoneally (i.p.) with single doses of 10 mg/kg (R,S)-JQAD1 solubilized in 10% hydroxypropyl β-cyclodextrin (Sigma-Aldrich) in sterile water. Blood concentration of (R,S)-JQAD1 was measured by serial measurements of serum at timepoints to 24h, by LC-MS/MS. Pharmacokinetics were performed at ChemPartner in Shanghai, China, using an (LC)/tandem mass spectrometry (MS-MS) method and pharmacokinetics parameters calculated with WinNonlin V 6.2 statistics software (Pharsight Corporation) using a noncompartmental model. For maximally tolerated dose (MTD) testing, six female CD1 mice were treated with daily I.P. doses of (R,S)-JQAD1 at 10, 20 or 40 mg/kg. Animals were monitored for animal weight, grooming and behavior daily without noted effects. For MTD testing in humanized CRBN knockin (Balb/c CRBNILE391VAL) (Jackson Laboratories), 6 mice per treatment group were treated with either vehicle control or (R,S)-JQAD1 at 40mg/kg/day by i.p. injection. Animal weights, behavior and grooming were monitored daily, for a total of 21 days. At day 14, three mice per treatment group were sacrificed and tissues fixed for immunohistochemistry. At this time, blood samples were obtained by retro-orbital puncture and blood analyzed at the Small Animal Imaging Facility at Beth Israel Deaconess Medical Center (Boston, MA), on a Hemavet 9500FS (Drew Scientific) for blood counts, creatinine, AST, ALT, ALP, GGTP and BUN measurements.
For tumor studies, eight-week-old female NSG mice (Jackson Laboratories) were subcutaneously implanted with 2.5 × 106 Kelly cells in 50% matrigel/PBS. Mice were assigned to two groups: vehicle (n=9), or JQAD1 (40 mg/kg/day) (n=10) by I.P. injection. Treatment with small-molecule inhibitors was initiated once tumors engrafted and reached 100–150 mm3. Mice were treated for 21d and then followed for survival. Tumors were measured by calipers and mice were weighed every three days. Animals were euthanized according to institutional guidelines when tumors reached 2,000 mm in length or width or if animals became moribund. Tumor sizes were compared at each timepoint by two-way ANOVA with post-hoc Tukey tests. Tumor growth curve kinetics were analyzed by both logistic regression and mixed-effects two-way ANOVA with post-hoc Tukey tests. Separately, eight animals were xenografted, and treated with vehicle (n=4) or JQAD1 (n=4) at 40 mg/kg i.p. daily for 14 days. Animals were sacrificed at day 14, with tumor being extracted, and divided for immunohistochemical or RNAseq analysis.
Immunohistochemistry
Immunohistochemistry was performed on the Leica Bond III automated staining platform. EP300 antibody (RRID:AB_2800077) was run at 1:50 dilution using the Leica Biosystems Refine Detection Kit with citrate antigen retrieval. KAT3A/CBP antibody (RRID: AB_303342) was run at 1:200 using the Leica Biosystems Refine Detection Kit with EDTA antigen retrieval.
RNA-seq and Analyses
Total RNA was extracted from control, A485 or JQAD1 treated Kelly cells using Trizol (Ambion). Prior to extraction, exogenous spike-in of synthetic ERCC RNA controls were added based on cell number (Ambion). DNAse I treated samples were were subjected to library construction with poly-adenylation preparation and sequencing using the Illumina NextSeq 500 (paired end, 75bp reads).
RNA-seq reads were aligned to a reference index (hg19 revision of the human reference genome), quantified comparing to ERCC spike-in probes, and converted to transcripts per million (TPM). ERCC-normalized expression of each gene after 24h of either A485 or JQAD1 treatment was compared against its expression in DMSO treated samples to create two-fold changes. These data were then analyzed by gene set enrichment analysis (GSEA)(RRID:SCR_003199) using the gene ontology hallmarks (H) collection in MSigDB(61,62).
For in vivo analyses, purified RNA samples were subjected to library construction with a low input RNA protocol, followed by poly-adenylation preparation and sequencing using the Illumina NextSeq 500 (paired end, 75bp reads). TPM were calculated as above. Genes were annotated as either controlled by super-enhancers (n=671) or typical enhancers (n=27116) using H3K27ac data derived from(9,35,63). For “transcription factor,” annotations, the list of 1639 high-confidence human transcription factors was obtained from(1). Data was compared by ANOVA with multiple hypothesis testing using the original method of Benjamini and Hochberg, comparing ERCC-controlled RNAseq expression in JQAD1-treated samples against vehicle-treated controls. GSEA was performed as above.
Biotin-JQAD1 Pulldown Assays
Biotin-JQAD1 (Supplementary Methods) was added to 500 μg of whole Kelly cell lysate prepared in IP lysis buffer (Pierce Biotechnology), and incubated for 16h at 4°C with end-over-end mixing. High-capacity streptavidin agarose resin (Pierce Biotechnology) was washed three times with cold PBS, prior to addition of cell lysate. Lysate was incubated at room temperature for 10 minutes prior to centrifugation, washing, and eluting in NuPAGE LDS sample buffer with reducing agent (Thermo-Fisher Scientific). Samples were processed by SDS-PAGE as blotted as above.
AlphaLISA Assay
Assays were performed with minimal modifications from the manufacturer’s protocol (PerkinElmer, USA). Briefly, CRBN-DDB1 (50 nM), Ni-coated Acceptor Beads (20 μg/ml), and biotinylated-pomalidomide (15 nM) were incubated with 100 nL of compound in 384-well plates (AlphaPlate-384, PerkinElmer, USA) using a Janus Workstation (PerkinElmer, USA). Streptavidin-coated donor beads (20 μg/ml) were added, incubated room temperature for 1 hour, and read on an Envision 2104 (PerkinElmer, USA), per the manufacturer’s protocol.
Genome-wide Occupancy Analysis
ChIP-seq was performed as previously described(9). Antibodies for ChIP-seq were: EP300, CBP, TFAP2β, ASCL1 and H3K27ac (RRIDs: AB_297224, AB_2616020, AB_2058198, AB_10918561, AB_2118291). Illumina sequencing, library construction and ChIP-seq analysis methods were previously described(51,58). Remaining ChIP-seq and ATAC-sequencing data was extracted from previously published datasets (GSE120074, GSE94822, GSE65664) available through the GEO portal (https://www.ncbi.nlm.nih.gov/gds). For experiments involving analysis of quantitative changes in H3K27ac, pellets of neuroblastoma cells were spiked in with similarly fixed and processed S2 cells at 1:10 ratio, prior to sonication.
ChIP-seq Enriched Regions and Heatmap Analysis
Regions enriched in ChIP-seq signal were identified using MACS 1.4.2 (RRID:SCR_013291) with corresponding control and parameters – keep-dup=auto and –p 1e-9. Scatterplots in Fig. 2B and S2A were created from the collapsed union of EP300 and CBP bound sites, while regions displayed in Fig. 2C and S2B were created from the collapsed union of master transcription factor (HAND2, ISL1, PHOX2B, GATA3, TBX2, ASCL1, TFAP2β) peaks from the respective cell line. ChIP-seq and ATAC-Seq signal was quantified for heatmap display in 4kb windows centered on the middle of each collapsed peak using bamToGFF (https://github.com/bradnerComputation) with parameters -m 50 -r -f 1. Putative PCR duplicates were removed using samtools (RRID:SCR_002105). Rows were ordered by EP300 signal in the whole displayed window (Fig. 2C, S2B).
ChIP-RX Alignment and Processing
ChIP-RX reads from Kelly cells treated with DMSO or 500nM JQAD1 were aligned the dm6 revision of the D. melanogaster reference genome with -k 1 –chunkmbs 256 --best to identify spiked-in DNA. Counts of fly reads were determined by counting unique read names in the aligned read file. Remaining non-fly reads were aligned to the hg19 revision of the human reference genome with parameters -k 2 -m 2 –chunkmbs 256 –best -l 75. Visualization files were constructed using macs 1.4 with parameters -w -S –space=50 –nomodel –shiftsize=200 to generate wiggle files, which were subsequently normalized by the millions of fly-mapped reads in the corresponding sample.
Super-Enhancer and Typical Enhancer Identification and Assignment
Super-enhancers in Kelly xenografts were identified using ROSE(RRID:SCR_017390) and the single-end BAMs generated as described(58). The collapsed union of regions called using both MACS parameter sets that do not contact the MYCN-proximal region were used as input for ROSE (https://bitbucket.org/young_computation/rose) as described in Mansour et al(58) with modifications. H3K27ac peaks were stitched computationally if they were within 12500bp of each other, though peaks fully contained within +/− 2000bp from a RefSeq promoter were excluded from stitching. Stitched enhancers (typical enhancers and super-enhancers) were assigned to the single active gene whose transcription start site is nearest the center of the stitched enhancer.
CUT&RUN Sequencing
CUT&RUN sequencing was performed by standard methodology and using reagents from Epicypher Inc. H3K27ac (RRID: AB_2893037, Millipore) and IgG (Epicypher) CUT&RUN validated antibodies were used. Briefly, 500,000 live cells per sample were permeabilized and processed by CUT&RUN by standard protocols. Samples were internally controlled by spiking in exogenous E.coli DNA relative to cell number, per the manufacturer’s protocol. Libraries were prepared using the Kapa Hyper Plus kit (Kapa Biosciences) and sequenced on a NovaSeq 6000 with 10 million paired-end 75bp reads per sample. Reads were aligned to the NC_008253.1 version of the E. coli reference genome using bowtie v1.2.2 (RRID:SCR_005476) in single-end mode with parameter -k 1 –best. Reads not aligned to E. coli were retained and aligned to the hg19 revision of the human reference genome using bowtie v1.2.2 in single-end mode with parameters -k 2 -m 2 –best. Mapped human reads were combined and used for H3K27ac peak-calling using MACS v1.4.1 with corresponding IgG control and -p 1e-9. Values were normalized by dividing each calculated value by the millions of E. coli-mapped reads.
Motif Enrichment Analysis
We compared the CBP and EP300 ChIP-seq peaks in Kelly and BE2C cells in order to remove the peaks bound by both factors. For each cell line, regions uniquely enriched in EP300 or CBP were determined using bedtools intersect -v -f 0.5 -r, which filters regions sharing 50% or more between factors. In each subset, we performed a motif enrichment analysis as previously(64). We quantified TF motif enrichment by using a well-established area under the receiver operator curve (AUROC)-based metric that assesses the presence of a TF motif among the 500 highest confidence peaks (foreground set) as compared to a background set of sequences(65). For the AUROC quantification, we used tools described for the analysis of TF ChIP-seq data from http://thebrain.bwh.harvard.edu/glossary-GENRE/download.html(64), which included the use of the R-function “matchPWM” (R-package “Biostrings”(RRID:SCR_016949)) to score each PWM against each sequence and the evaluation of an adjusted p-value to ensure statistical significance. In both cell lines, TFAP2β (PWM M5912_1 from CISBP databank Version 1.02) was the only PWM that showed a relevant differential enrichment, namely an enrichment above 0.6 AUROC (p-value <0.001) in EP300-unique peaks, and no enrichment (AUROC ~0.5, p-value>0.1) in CBP unique peaks.
PRISM Screening
PRISM barcoded pooled screening was performed using JQAD1 in 578 barcoded cell lines as described(47,66). Some cell lines included in the screen were genetically engineered to express exogenous genes, and these cell lines were removed, yielding 557 cell lines. Cells in pools of 20–25 were thawed and plated into 384-well plates (1250 cells/well for adherent cell pools, 2000 cells/well for suspension or mixed suspension/adherent cell pools) containing compound (top concentration: 10 μM, 8-point, threefold dilutions). All conditions were tested in triplicate. Cells were lysed after 5 days of treatment and mRNA-based Luminex detection of barcode abundance from lysates was carried out as described previously(47). Luminex median fluorescence intensity (MFI) data was input to a standard R pipeline (https://github.com/broadinstitute/prism_data_processing) to generate viability estimates relative to vehicle treatment for each cell line and treatment condition, and to fit dose-response curves from viability data.
STRING Database Analysis
Neuroblastoma-specific genetic dependencies (n=146) were identified previously(9), intersected with the Gene Ontology term “Cellular Component – nucleus” and input into the String database(RRID:SCR_005223)(34) to generate interaction plots. Network edges reflect evidence of interactions. Color indicates commercially available compounds targeting the protein (red = yes), or core regulatory circuitry member (blue).
DepMap Dependency Analysis
Analysis of dependency data was retrieved from the DepMap portal (www.depmap.org) using the 20Q2 dataset. Dependency data were extracted as probability of dependency for all cell lines (n=757), for EP300 and CBP. Details of individual cell lines are available at www.depmap.org and further analysis described in the Supplementary Methods.
Public Expression Analysis
Analyses of publicly available expression datasets was performed using either the R2 database (https://hgserver1.amc.nl/cgi-bin/r2/main.cgi#) or the DepMap portal (www.depmap.org). Cancer cell line encyclopedia analyses of RNA expression and proteomic expression was performed using the 20Q1 data release(67,68).
Quantification and Statistical Analyses
Data from ChIP-seq, CUT&RUN and CRISPR–Cas9 screens were analyzed as described. Animal experiments were analyzed by mixed-effects modeling and two-sided analysis of variance (ANOVA) for tumor volume and weight means, and by the log- rank test for survival. Other data were analyzed with one- or two-sided ANOVA with post hoc Tukey tests, two-sided t-tests, or one- or two-sided Fisher exact tests as appropriate for multiple or pair wise comparisons. Statistical significance was defined as a p < 0.05 unless otherwise stated. Data were analyzed with GraphPad Prism 7.01, and all error bars represent standard deviation unless otherwise noted.
Supplementary Material
SIGNIFICANCE.
EP300, but not CBP, controls oncogenic core regulatory circuitry-driven transcription in high-risk neuroblastoma by binding TFAP2β. We developed JQAD1, a CRBN-dependent PROTAC degrader with preferential activity against EP300 and demonstrated its activity in neuroblastoma. JQAD1 has limited toxicity to untransformed cells and is effective in vivo in a CRBN-dependent manner.
ACKNOWLEDGEMENTS
We thank Dr. Richard A. Young (Whitehead Institute, MIT) and Dr. Stuart Orkin (DFCI) for critical discussion. We thank Dr. Ross Tomaino of the Taplin Mass Spectrometry facility at Harvard Medical School for assistance with mass spectrometry, and Drs Jennifer Roth, Ginevra Botta and, Andrew Boghossian of the PRISM screening group at the Broad Institute for assistance with analysis of PRISM data. We thank the Animal Resources Facility and Lurie Family Imaging Center at Dana-Farber Cancer Institute for assistance with animal studies. This work was supported by National Institutes of Health (NIH) grants R35-CA210064 (A.T.L.), R35-CA210030 (K.S.), R01-NS088355 (K.S.), P01 CA217959 (K.S.), P01-CA066996-19 (J.Q.), K08-CA245251 (A.D.D), R21-HG009268 (M.L.B.), T32-CA136432 (N.V.D), R25-CA23944 (M.M.) and P30-CA021765 (St. Jude Comprehensive Cancer Center). This work was supported by the Joey O’Neill Fund. A.D.D. was supported by the Damon Runyon Cancer Research Foundation (DRSG-24-18) and is the recipient of funds from the Rally Foundation for Childhood Cancer Research, and the CureSearch for Children’s Cancer Foundation. A.D.D. M.W.Z. and J.Q. are supported by funding from the Alex’s Lemonade Stand Foundation for Childhood Cancer. M.W.Z. was supported by grants from the Charles A. King Trust, and Claudia Adams Barr Program. N.V.D. was supported by the Julia’s Legacy of Hope St. Baldrick’s Foundation Fellowship. B.J.A., J.E and A.D.D. are supported by the American Lebanese Syrian Associated Charities (ALSAC).
DECLARATION OF INTERESTS
J.Q. is the scientific co-founder of and consultant for Epiphanes, member of the Scientific Advisory Board of Talus, and receives grant funding from Novartis. K.S. receives grant funding from Novartis and consults for and has stock options in Auron Therapeutics and has served as an advisor for Kronos Bio and AstraZeneca. A.T.L. is a shareholder in Jengu Therapeutics and is a consultant/advisory board member for Jengu Therapeutics and Omega Therapeutics. B.J.A. is a shareholder of Syros Pharmaceuticals. N.V.D is a current employee and shareholder of Genentech, Inc., a member of the Roche Group. No other potential conflicts of interest are declared.
REFERENCES
- 1.Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, et al. The Human Transcription Factors. Cell 2018;175(2):598–9 doi 10.1016/j.cell.2018.09.045. [DOI] [PubMed] [Google Scholar]
- 2.Bradner JE, Hnisz D, Young RA. Transcriptional Addiction in Cancer. Cell 2017;168(4):629–43 doi 10.1016/j.cell.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hnisz D, Schuijers J, Lin CY, Weintraub AS, Abraham BJ, Lee TI, et al. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol Cell 2015;58(2):362–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013;153(2):307–19 doi 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin CY, Lovén J, Rahl PB, Paranal RM, Burge CB, Bradner JE, et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012;151(1):56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012;151(1):68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature 2018;555(7696):321–7 doi 10.1038/nature25480. [DOI] [PubMed] [Google Scholar]
- 8.Ma X, Liu Y, Liu Y, Alexandrov LB, Edmonson MN, Gawad C, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018;555(7696):371–6 doi 10.1038/nature25795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Durbin AD, Zimmerman MW, Dharia NV, Abraham BJ, Iniguez AB, Weichert-Leahey N, et al. Selective gene dependencies in MYCN-amplified neuroblastoma include the core transcriptional regulatory circuitry. Nat Genet 2018;50(9):1240–6 doi 10.1038/s41588-018-0191-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parker SC, Stitzel ML, Taylor DL, Orozco JM, Erdos MR, Akiyama JA, et al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc Natl Acad Sci U S A 2013;110(44):17921–6 doi 10.1073/pnas.1317023110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell 2013;155(4):934–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 1996;87(5):953–9. [DOI] [PubMed] [Google Scholar]
- 13.Dancy BM, Cole PA. Protein lysine acetylation by p300/CBP. Chem Rev 2015;115(6):2419–52 doi 10.1021/cr500452k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arany Z, Sellers WR, Livingston DM, Eckner R. E1A-associated p300 and CREB-associated CBP belong to a conserved family of coactivators. Cell 1994;77(6):799–800. [DOI] [PubMed] [Google Scholar]
- 15.Weinert BT, Narita T, Satpathy S, Srinivasan B, Hansen BK, Scholz C, et al. Time-Resolved Analysis Reveals Rapid Dynamics and Broad Scope of the CBP/p300 Acetylome. Cell 2018;174(1):231–44 e12 doi 10.1016/j.cell.2018.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hammitzsch A, Tallant C, Fedorov O, O’Mahony A, Brennan PE, Hay DA, et al. CBP30, a selective CBP/p300 bromodomain inhibitor, suppresses human Th17 responses. Proceedings of the National Academy of Sciences 2015;112(34):10768–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zucconi BE, Luef B, Xu W, Henry RA, Nodelman IM, Bowman GD, et al. Modulation of p300/CBP Acetylation of Nucleosomes by Bromodomain Ligand I-CBP112. Biochemistry 2016;55(27):3727–34 doi 10.1021/acs.biochem.6b00480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lasko LM, Jakob CG, Edalji RP, Qiu W, Montgomery D, Digiammarino EL, et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017;550(7674):128–32 doi 10.1038/nature24028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan G, Eller MS, Elm C, Larocca CA, Ryu B, Panova IP, et al. Selective inhibition of p300 HAT blocks cell cycle progression, induces cellular senescence, and inhibits the DNA damage response in melanoma cells. J Invest Dermatol 2013;133(10):2444–52 doi 10.1038/jid.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yao TP, Oh SP, Fuchs M, Zhou ND, Ch’ng LE, Newsome D, et al. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell 1998;93(3):361–72. [DOI] [PubMed] [Google Scholar]
- 21.Rebel VI, Kung AL, Tanner EA, Yang H, Bronson RT, Livingston DM. Distinct roles for CREB-binding protein and p300 in hematopoietic stem cell self-renewal. Proc Natl Acad Sci U S A 2002;99(23):14789–94 doi 10.1073/pnas.232568499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483(7391):603–7 doi 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ogiwara H, Sasaki M, Mitachi T, Oike T, Higuchi S, Tominaga Y, et al. Targeting p300 Addiction in CBP-Deficient Cancers Causes Synthetic Lethality by Apoptotic Cell Death due to Abrogation of MYC Expression. Cancer Discov 2016;6(4):430–45 doi 10.1158/2159-8290.CD-15-0754. [DOI] [PubMed] [Google Scholar]
- 24.Martire S, Nguyen J, Sundaresan A, Banaszynski LA. Differential contribution of p300 and CBP to regulatory element acetylation in mESCs. BMC Mol Cell Biol 2020;21(1):55 doi 10.1186/s12860-020-00296-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramos YF, Hestand MS, Verlaan M, Krabbendam E, Ariyurek Y, van Galen M, et al. Genome-wide assessment of differential roles for p300 and CBP in transcription regulation. Nucleic Acids Res 2010;38(16):5396–408 doi 10.1093/nar/gkq184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015;348(6241):1376–81 doi 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burslem GM, Crews CM. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020;181(1):102–14 doi 10.1016/j.cell.2019.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wimalasena VK, Wang T, Sigua LH, Durbin AD, Qi J. Using Chemical Epigenetics to Target Cancer. Mol Cell 2020;78(6):1086–95 doi 10.1016/j.molcel.2020.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith BE, Wang SL, Jaime-Figueroa S, Harbin A, Wang J, Hamman BD, et al. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat Commun 2019;10(1):131 doi 10.1038/s41467-018-08027-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet 2017;49(12):1779–84 doi 10.1038/ng.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou X, Edmonson MN, Wilkinson MR, Patel A, Wu G, Liu Y, et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat Genet 2016;48(1):4–6 doi 10.1038/ng.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boeva V, Louis-Brennetot C, Peltier A, Durand S, Pierre-Eugene C, Raynal V, et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat Genet 2017;49(9):1408–13 doi 10.1038/ng.3921. [DOI] [PubMed] [Google Scholar]
- 33.van Groningen T, Koster J, Valentijn LJ, Zwijnenburg DA, Akogul N, Hasselt NE, et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat Genet 2017;49(8):1261–6 doi 10.1038/ng.3899. [DOI] [PubMed] [Google Scholar]
- 34.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015;43(Database issue):D447–52 doi 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang L, Tan TK, Durbin AD, Zimmerman MW, Abraham BJ, Tan SH, et al. ASCL1 is activated by LMO1 and MYCN as a core regulatory circuitry member in neuroblastoma. Nature Communications 2019;10(1):5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song CZ, Keller K, Chen Y, Murata K, Stamatoyannopoulos G. Transcription coactivator CBP has direct DNA binding activity and stimulates transcription factor DNA binding through small domains. Biochem Biophys Res Commun 2002;296(1):118–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He J, Ye J, Cai Y, Riquelme C, Liu JO, Liu X, et al. Structure of p300 bound to MEF2 on DNA reveals a mechanism of enhanceosome assembly. Nucleic Acids Res 2011;39(10):4464–74 doi 10.1093/nar/gkr030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Michaelides MR, Kluge A, Patane M, Van Drie JH, Wang C, Hansen TM, et al. Discovery of Spiro Oxazolidinediones as Selective, Orally Bioavailable Inhibitors of p300/CBP Histone Acetyltransferases. ACS Med Chem Lett 2018;9(1):28–33 doi 10.1021/acsmedchemlett.7b00395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yasgar A, Jadhav A, Simeonov A, Coussens NP. AlphaScreen-Based Assays: Ultra-High-Throughput Screening for Small-Molecule Inhibitors of Challenging Enzymes and Protein-Protein Interactions. Methods Mol Biol 2016;1439:77–98 doi 10.1007/978-1-4939-3673-1_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang M, Weiss WA. Neuroblastoma and MYCN. Cold Spring Harb Perspect Med 2013;3(10):a014415 doi 10.1101/cshperspect.a014415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gabay M, Li Y, Felsher DW. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med 2014;4(6) doi 10.1101/cshperspect.a014241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Faiola F, Liu X, Lo S, Pan S, Zhang K, Lymar E, et al. Dual regulation of c-Myc by p300 via acetylation-dependent control of Myc protein turnover and coactivation of Myc-induced transcription. Mol Cell Biol 2005;25(23):10220–34 doi 10.1128/MCB.25.23.10220-10234.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vervoorts J, Luscher-Firzlaff JM, Rottmann S, Lilischkis R, Walsemann G, Dohmann K, et al. Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep 2003;4(5):484–90 doi 10.1038/sj.embor.embor821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang K, Faiola F, Martinez E. Six lysine residues on c-Myc are direct substrates for acetylation by p300. Biochem Biophys Res Commun 2005;336(1):274–80 doi 10.1016/j.bbrc.2005.08.075. [DOI] [PubMed] [Google Scholar]
- 45.Donovan KA, An J, Nowak RP, Yuan JC, Fink EC, Berry BC, et al. Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome. Elife 2018;7 doi 10.7554/eLife.38430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fink EC, McConkey M, Adams DN, Haldar SD, Kennedy JA, Guirguis AA, et al. Crbn (I391V) is sufficient to confer in vivo sensitivity to thalidomide and its derivatives in mice. Blood 2018;132(14):1535–44 doi 10.1182/blood-2018-05-852798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Corsello SM, Nagari RT, Spangler RD, Rossen J, Kocak M, Bryan JG, et al. Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat Cancer 2020;1(2):235–48 doi 10.1038/s43018-019-0018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kasper LH, Fukuyama T, Biesen MA, Boussouar F, Tong C, de Pauw A, et al. Conditional knockout mice reveal distinct functions for the global transcriptional coactivators CBP and p300 in T-cell development. Mol Cell Biol 2006;26(3):789–809 doi 10.1128/MCB.26.3.789-809.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu Y, Wang L, Predina J, Han R, Beier UH, Wang LC, et al. Inhibition of p300 impairs Foxp3(+) T regulatory cell function and promotes antitumor immunity. Nat Med 2013;19(9):1173–7 doi 10.1038/nm.3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sen P, Lan Y, Li CY, Sidoli S, Donahue G, Dou Z, et al. Histone Acetyltransferase p300 Induces De Novo Super-Enhancers to Drive Cellular Senescence. Mol Cell 2019;73(4):684–98 e8 doi 10.1016/j.molcel.2019.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sanda T, Lawton LN, Barrasa MI, Fan ZP, Kohlhammer H, Gutierrez A, et al. Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell 2012;22(2):209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005;122(6):947–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saint-André V, Federation AJ, Lin CY, Abraham BJ, Reddy J, Lee TI, et al. Models of human core transcriptional regulatory circuitries. Genome Res 2016;26(3):385–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suzuki HI, Young RA, Sharp PA. Super-Enhancer-Mediated RNA Processing Revealed by Integrative MicroRNA Network Analysis. Cell 2017;168(6):1000–14 e15 doi 10.1016/j.cell.2017.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 2018;361(6400) doi 10.1126/science.aar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vannam R, Sayilgan J, Ojeda S, Karakyriakou B, Hu E, Kreuzer J, et al. Targeted degradation of the enhancer lysine acetyltransferases CBP and p300. Cell Chem Biol 2021;28(4):503–14 e12 doi 10.1016/j.chembiol.2020.12.004. [DOI] [PubMed] [Google Scholar]
- 57.Tate EH, Wilder ME, Cram LS, Wharton W. A method for staining 3T3 cell nuclei with propidium iodide in hypotonic solution. Cytometry 1983;4(3):211–5 doi 10.1002/cyto.990040304. [DOI] [PubMed] [Google Scholar]
- 58.Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 2014;346(6215):1373–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 1994;5(11):976–89 doi 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 60.Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc 2013;8(8):1551–66 doi 10.1038/nprot.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gene Ontology C Gene Ontology Consortium: going forward. Nucleic Acids Res 2015;43(Database issue):D1049–56 doi 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oldridge DA, Wood AC, Weichert-Leahey N, Crimmins I, Sussman R, Winter C, et al. Genetic predisposition to neuroblastoma mediated by a LMO1 super-enhancer polymorphism. Nature 2015;528(7582):418–21 doi 10.1038/nature15540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mariani L, Weinand K, Vedenko A, Barrera LA, Bulyk ML. Identification of Human Lineage-Specific Transcriptional Coregulators Enabled by a Glossary of Binding Modules and Tunable Genomic Backgrounds. Cell Syst 2017;5(3):187–201 e7 doi 10.1016/j.cels.2017.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gordan R, Hartemink AJ, Bulyk ML. Distinguishing direct versus indirect transcription factor-DNA interactions. Genome Res 2009;19(11):2090–100 doi 10.1101/gr.094144.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Corsello SM, Bittker JA, Liu Z, Gould J, McCarren P, Hirschman JE, et al. The Drug Repurposing Hub: a next-generation drug library and information resource. Nat Med 2017;23(4):405–8 doi 10.1038/nm.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ghandi M, Huang FW, Jane-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019;569(7757):503–8 doi 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nusinow DP, Szpyt J, Ghandi M, Rose CM, McDonald ER 3rd, Kalocsay M, et al. Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell 2020;180(2):387–402 e16 doi 10.1016/j.cell.2019.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gartlgruber M, Sharma AK, Quintero A, Dreidax D, Jansky S, Park Y, et al. Super enhancers define regulatory subtypes and cell identity in neuroblastoma. Nat Cancer 2020;2:114–28 doi 10.1038/s43018-020-00145-w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.