

Abstract
The ATP-binding cassette (ABC) transporters P-glycoprotein (P-gp) and ABCG2 are multidrug transporters that confer drug resistance to numerous anti-cancer therapeutics in cell culture. These findings initially created great excitement in the medical oncology community, as inhibitors of these transporters held the promise of overcoming clinical multidrug resistance in cancer patients. However, clinical trials of P-gp and ABCG2 inhibitors in combination with cancer chemotherapeutics have not been successful due, in part, to flawed clinical trial designs resulting from an incomplete molecular understanding of the multifactorial basis of multidrug resistance (MDR) in the cancers examined. The field was also stymied by the lack of high-resolution structural information for P-gp and ABCG2 for use in the rational structure-based drug design of inhibitors. Recent advances in structural biology have led to numerous structures of both ABCG2 and P-gp that elucidated more clearly the mechanism of transport and the polyspecific nature of their substrate and inhibitor binding sites. These data should prove useful helpful for developing even more potent and specific inhibitors of both transporters. As such, although possible pharmacokinetic interactions would need to be evaluated, these inhibitors may show greater effectiveness in overcoming ABC-dependent multidrug resistance in combination with chemotherapeutics in carefully selected subsets of cancers. Another perhaps even more compelling use of these inhibitors may be in reversibly inhibiting endogenously expressed P-gp and ABCG2, which serve a protective role at various blood-tissue barriers. Inhibition of these transporters at sanctuary sites such as the brain and gut could lead to increased penetration by chemotherapeutics used to treat brain cancers or other brain disorders and increased oral bioavailability of these agents, respectively.
Keywords: P-glycoprotein, ABCG2, ABC transporters, multidrug resistance, inhibitor
INTRODUCTION
Multidrug resistance (MDR) in cancer is a complex, multifactorial problem in which tumors are either intrinsically resistant to chemotherapeutic treatment or acquire resistance to drugs over the course of treatment. Cancer cells can develop MDR through various mechanisms, including drug sequestration in intracellular compartments, downregulation of cell death mechanisms, microenvironmental changes, and expression of ATP-binding cassette (ABC) transporters[1–9]. Unlike other types of resistance that alter the effects of only one drug, ABC transporter expression confers cellular resistance to a wide variety of chemically and structurally unrelated anti-cancer agents, a phenomenon known as MDR[3,6,10–13]. Most notably, the expression of ABCB1, commonly known as P-glycoprotein (P-gp), and ABCG2, also known as BCRP or the breast cancer resistance protein, in tumor cells has been correlated to poor patients prognosis in numerous studies. However, it was unclear whether these correlations were causal since expression levels in patient samples did not reach those observed in cultured cells[14–17]. Although overexpression of other ABC transporters, including MRP1/ABCC1, has also been implicated in cancer MDR[18], this review focuses on P-gp and ABCG2. Though found in different subfamilies of the ABC transporter superfamily, these two transporters have similar overlapping substrate and inhibitor specificities. This knowledge, coupled with recent structures of both ABCG2 and P-gp with inhibitors bound, offer the opportunity to explore the future of rational structure-based drug design of either specific or dual inhibitors for the reversal of MDR clinically or for use in delivering therapeutic agents to sanctuary sites in the body protected by the endogenous expression of these transporters.
Given this clinical correlation, it was hypothesized, in retrospect naïvely so, that inhibition of these transporters would result in the reversal of the MDR phenotype clinically in patients. As such, over the past three decades, three generations of agents have been developed that showed great promise as potent inhibitors in in vitro and some in vivo model systems[19–24]. However, no inhibitor to date has been shown to significantly reverse MDR in human clinical trials[17,24–29]. For a comprehensive list and discussion of these human clinical trials, please see references[17,23,26,29–32]. This lack of success and a greater understanding of the molecular complexity of MDR in tumors has led to substantial and, to a great extent, warranted skepticism in the field regarding whether this approach will ever be successful in reversing MDR in patients[17,24,28,29,33].
However, as is discussed more in-depth herein, these trials were flawed in many ways, including poor patient selection, a lack of understanding of the different subtypes of cancers within a patient population, the presence of other mechanisms of MDR, the effects of expression of other transporters in the cells and P-gp transporter polymorphisms[17,24,29]. Coupled with the lack of high-resolution structures of the transporter with substrates or inhibitors, which severely limited rational drug design efforts for P-gp and ABCG2, enthusiasm for this approach waned considerably. However, recent advances in structural biology, especially in cryo-EM, have resulted in high-resolution three-dimensional structures of P-gp and ABCG2[34], allowing for a complete dissection of their molecular mechanisms. As discussed in detail herein, the emergence of the high-resolution structures of these transporters in complex with inhibitors should finally allow for true rational structure-based drug design of more potent inhibitors with enhanced specificity. In turn, better inhibitors and future rigorously designed clinical trials specifically targeting patient populations with tumors expressing P-gp, may offer more promising results in some subsets of cancer patients[17,29]. Other methods such as controlling ABC transporter expression through microRNAs and small interfering RNAs (siRNAs) and monoclonal antibodies are also currently being researched as possible ways to inhibit P-gp and ABCG2[35].
P-gp and ABCG2 are endogenously expressed at the blood-brain barrier (BBB) and the intestinal epithelium, where they serve a protective role in limiting the entry of xenobiotics into the brain and bloodstream respectively[36–38]. However, during chemotherapeutic treatment, this protective role can be thought of as a form of drug resistance since the transporters limit the number of drugs that can reach target cancer cells. Therefore, even if inhibitors of P-gp and ABCG2 do not ultimately prove useful for MDR cancers directly, they may prove to be efficacious as tools to increase the bioavailability of chemotherapeutic agents to sites protected by endogenously expressed ABC transporters, such as the central nervous system and the gut.
ABC TRANSPORTERS AND CULTURED CELL MULTIDRUG RESISTANCE
The failure of chemotherapy in cancer is often due to either intrinsic or acquired resistance to many structurally and chemically unrelated compounds over the course of the treatment, a phenomenon called MDR[1–3]. One mechanism studied extensively is ABC transporters’ expression limiting the number of drugs that can enter cells by effluxing the agent from the cells in an ATP-dependent manner[3,6–9,25]. This phenotype was initially described in a subculture of HeLa cells grown in selective media containing actinomycin D[39]. Several Chinese hamster ovary (CHO) cell lines cultured with increasing concentrations of actinomycin D were also shown to be resistant to some other drugs, including daunomycin, vincristine, and vinblastine[40]. In further studies of the nature of this acquired resistance, Danø demonstrated active transport of actinomycin D in drug-resistant Erlich ascites tumor cells, leading to the hypothesis that the protein responsible for the MDR phenotype was a transporter[41]. This transporter was subsequently identified in CHO cells that were shown to be resistant to some unrelated drugs[42]. Due to observed glycosylation and its role in reducing the permeability of many unrelated drugs in resistant cell lines, the 170 kDa transporter was given the name permeability glycoprotein or, as it is more commonly referred, P-glycoprotein or P-gp[42]. P-gp was subsequently purified from plasma membrane-derived vesicles of colchicine-resistant CHO cells[43]. The mdr gene encoding P-gp was first cloned from colchicine-resistant CHO cells and soon after in human KB carcinoma cells[44–46]. Since the discovery of P-gp, an entire superfamily of transporters has been discovered and continues to be studied[47–52]. P-gp is a member of this family of ABC transporters and has been given the gene classification of ABC subfamily B member 1, ABCB1[47]. More recently, another member of the ABC superfamily that has been shown to confer multidrug resistance in cancer cells in culture is the BCRP or ABCG2. ABCG2 was discovered in adriamycin-resistant MCF-7 breast cancer cells and mitoxantrone-resistant S1-M1–80 colon carcinoma cells as well as in the placenta, where it is endogenously expressed[53–55]. Further study of the ABC transporter superfamily revealed 48 human membrane transporters with diverse functions[47,56].
CLINICAL TRIALS
The results of the cell culture model studies and studies correlating P-gp expression to poor clinical outcome fueled great excitement for the potential therapeutic use of P-gp inhibitors to reverse MDR clinically[17,29]. As a result, numerous clinical trials were performed to determine if adding a P-gp inhibitor to drug regimens would improve therapeutic efficacy. Unfortunately, many inhibitors of P-gp and ABCG2 that were successful in vitro did not prove to be efficacious in these human trials[3,17,19,27,29,57,58]. However, many of these clinical studies were flawed from the outset in significant ways[3,17,27,29,58]. Perhaps most importantly, in most trials, patients were not selected for inclusion in the study based on tumor expression of P-gp[17,58].
More recently, robust analyses of drug-resistant tumors using new genomic tools and diagnostic methods suggest that resistance caused by P-gp, ABCG2, and perhaps other ABC transporters may be limited to subsets of tumors and/or subsets of patient populations. Further developments in flow cytometry, positron emission tomography (PET), FISH analysis, RNA-seq, and next generation sequencing, will more readily allow for further in-depth evaluation of samples that may aid in selecting the subset of patients who would most likely respond to the use of a P-gp or ABCG2 inhibitor. In a genome-wide characterization of chemoresistant ovarian carcinoma patients, it was shown that only a small subset demonstrated the recurrent promoter fusion event associated with P-gp overexpression[59], suggesting, perhaps, that this subset of patients might benefit from this clinical approach rather than of the entire cohort. In a recent study where flow cytometry was used to determine P-gp expression in the bone marrow and peripheral blood samples from 346 patients diagnosed with AML, the patients were further stratified based on age, gender, and disease status[60]. Although overall, P-gp was expressed in only 32.1% of patients, a greater prevalence of P-gp expression was found in older patients and those with the refractory, recurrent and secondary disease as compared to de novo AML. While P-gp expression was only found in 60/261 (22.9%) of de novo AML patients, it was found in 23/24 (95.8%) of patients with the drug-refractory disease and 24/30 (80.0%) of those with relapsing AML[60]. Positron emission tomography (TEM) is another powerful tool used to detect and/or measure the expression of P-gp using radio-labeled substrates and inhibitors. PET was first used to measure P-gp function at the BBB non-invasively in 1998 in mdr1a(−/−) knockout mice with [11C]verapamil and cyclosporin A[61,62]. Since this initial work, PET has also been used to evaluate whether alterations of P-gp function at the BBB are implicated in the pharmacoresistance of some HIV and epilepsy patients to therapeutics or if P-gp function is either compromised or enhanced in several other central nervous system disorders[62]. In total, although the results of many clinical trials generally dampened enthusiasm tremendously for a P-gp inhibitor approach to treating MDR cancers, in the new era of personalized medicine and advanced diagnostic techniques as described above, it is worthwhile to repeat these trials with more stringent requirements for inclusion and the use of more specific and more potent inhibitors.
ENDOGENOUS EXPRESSION OF P-GP AND ABCG2: LIMITING ORAL BIOAVAILABILITY AND DRUG PENETRATION OF THE CNS
As described above, ABC transporters present in the plasma membrane of multidrug-resistant cancer cells may be responsible for significant alterations in the pharmacokinetics and efficacy of many chemotherapeutics[3,6,10–13]. Additionally, endogenous expression of P-gp and ABCG2 transporters at blood-tissue barriers is known to lead to reduced uptake of therapeutics across these barriers[11,36–38,63–66]. In our view, the limited bioavailability of therapeutics to these sanctuary sites constitutes an alternate form of MDR that, if overcome, could result in more efficacious treatments of not only cancer but other diseases.
At the BBB, the ABC transporters P-gp and, to a somewhat lesser extent, ABCG2 are thought to serve a protective function by limiting the accumulation of lipophilic agents in the brain by effluxing the compounds back into the bloodstream[36,67–69]. To probe if P-gp and ABCG2 limit the accumulation of chemotherapeutic agents in the brain, P-gp-null, and ABCG2-null mice have been studied[11,37,69–76]. Using a mouse knockout model, P-gp was found to be an important determinant in the brain penetration and pharmacological activity of many drugs[11,69]. For example, many of the chemotherapeutic agents used to treat glioblastomas are, in fact, P-gp or ABCG2 substrates, including Gleevec, topotecan, and paclitaxel[74,77,78]. Another study demonstrated in vivo and in vitro that co-administration of paclitaxel with the potent P-gp inhibitor valspodar (PSC833) increased the levels of fluorescently labeled paclitaxel in normal mouse brains[79]. Furthermore, it was demonstrated that co-administration of these two drugs reduced the growth of implanted human glioblastomas by 90% in nude mice, whereas treatment with paclitaxel or valspodar alone did not affect tumor size[79].
In ABCG2 knockout mice, increased brain accumulation of drugs has been observed when only ABCG2 was eliminated, especially for compounds that are not substrates for both transporters[69,78,80]. Interestingly, however, synergistic effects on drug brain accumulation are observed in double ABCG2/P-gp knockout mice for drugs that are substrates of both transporters, further enforcing their functional redundancy in vivo[78,81–83]. For example, whereas the brain concentration of the kinase inhibitor ceritinib was approximately 38-fold higher in P-gp-only knockout mice compared to wild-type, the concentration increased to 90-fold in brains of mice lacking both P-gp and ABCG2[82]. Another study demonstrated an approximately 20-fold increase of vemurafenib in the brains of P-gp/ABCG2 knockout mice compared to no increase in an ABCG2 knockout and a 1.7-fold increase in a P-gp knockout[81]. Thus, although P-gp appears to often be the dominant ABC transporter at the blood-brain barrier, these data suggest that dual P-gp/ABCG2 inhibitors may have even greater therapeutic promise for brain diseases[23,84–86].
P-gp and ABCG2 are also expressed endogenously on the apical side of the intestine and colon epithelial cells, where they are thought to protect from ingested xenobiotics[87–89]. Given this localization, these transporters have also been implicated in limiting the bioavailability of many oral therapeutics[38,90,91]. For example, studies in P-gp mdr1a(−/−) knockout mice showed an approximate 25% increase in the oral bioavailability of paclitaxel as compared to wild-type mice[92]. Furthermore, in wild-type mice, co-administration of elacridar (GF120918) with paclitaxel increased the plasma values for orally dosed paclitaxel in wild-type mice by 6.6-fold. Importantly, these results were also observed in the plasma of patients treated with paclitaxel combined with elacridar or cyclosporin A[93].
Similarly, the uptake of therapeutics, such as topotecan and quercetin, has been shown to be limited in ABCG2 homozygous knockout mice and rats[94,95]. In addition, the co-administration of gefitinib, an ABCG2 inhibitor, has also been shown to increase the oral uptake of sulfasalazine by 13-fold compared to sulfasalazine alone in a mouse model[96]. These data together suggest that inhibition of P-gp and ABCG2 in the gastrointestinal tract is a viable method for increasing the bioavailability of oral chemotherapeutic agents. However, as the pharmacokinetic and pharmacodynamic properties of the agents will be altered upon systemic inhibition of P-gp, whole-body toxicity and especially toxicity at sanctuary sites protected by endogenous P-gp, such as the brain, must be considered. Thus, the dose of the therapeutic agent would necessarily have to be optimized, and likely lowered to maximize drug exposure but minimize whole-body toxicity.
P-GP AND ABCG2 STRUCTURE AND MECHANISM
P-gp and ABCG2, members of the ABC transporter superfamily, are transmembrane proteins that use the energy of ATP to efflux substrates across a membrane[13,34,48,97,98]. Structurally, P-gp and ABCG2 consist of four common domains to the ABC transporter superfamily, although many other ABC transporters also contain additional auxiliary domains or only nucleotide-binding domains[47]. Both P-gp and ABCG2 transporter have two cytoplasmic nucleotide-binding domains (NBDs) that hydrolyze ATP and two transmembrane domains (TMDs) that recognize and bind the substrate molecules and form a pathway for their translocation across the membrane[34,97,99]. These domains can be encoded by a single gene that produces a full-length transporter, like P-gp, or by two genes that encode a half-transporter with one NBD and one TMD that homodimerize to produce a full-length transporter, like ABCG2[34,47,48]. In addition, some full-length ABC transporters are heterodimers of two distinct half-transporters (e.g., ABCG5/ABCG8). The NBDs are highly conserved across the superfamily and fall into the P-loop NTPase superfamily with a RecA-type binding core along with a unique ABC signature motif, LSGGQ[34]. The TMDs are far less conserved, allowing for a range of substrates to be recognized[47].
As informed first by a plethora of past biochemical and pharmacological experiments[13,100–109], and currently by the three-dimensional structures[110–121], the basic scheme of ATP-dependent substrate transport in P-gp involves large conformational changes during the efflux cycle [Figure 1]. The hydrophobic P-gp substrates diffuse into the membrane and interact with the transporter via the inner leaflet. Substrate binding signals P-gp to adopt a state in which substrate is bound at the apex of the TMDs, creating an occluded binding pocket[112]. Interestingly, this study also showed that while only one molecule of the substrate vincristine was able to bind, inhibitors such as elacridar, tariquidar, or zosuquidar were bound in pairs.
Figure 1.
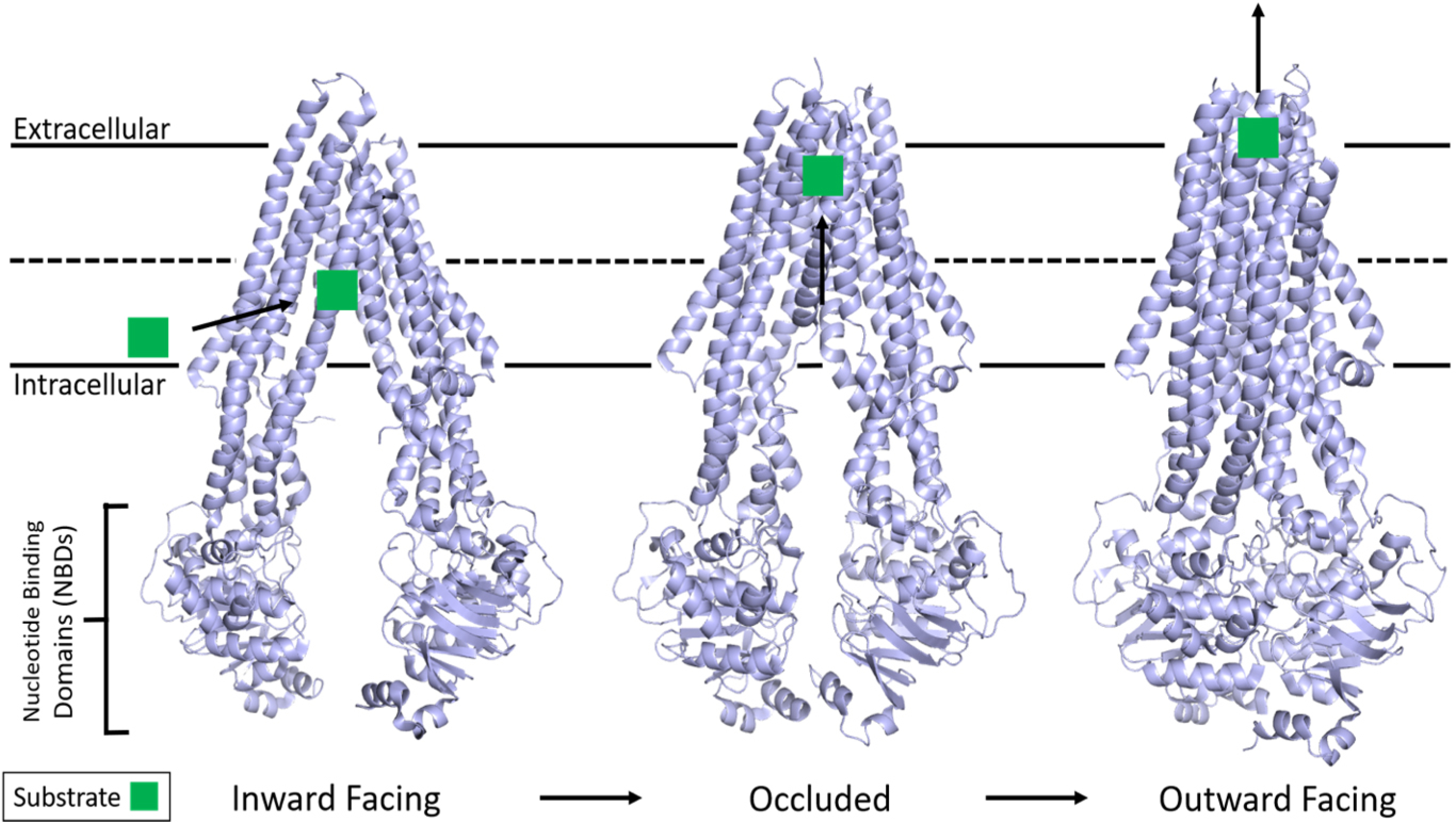
Structure of P-gp and the basic mechanism of ATP hydrolysis and transport. P-gp undergoes large conformational changes in its transport mechanism. The membrane is delineated by solid black lines. Substrate from the inner leaflet first interacts with an inward-facing conformation of P-gp while two ATP molecules bind the free NBDs. The structure then adopts an occluded state with substrate bound at the apex of the TMDs while TM helices 4 and 10 significantly kink inwards, occluding the binding pocket. The outward-facing conformation is correlated with asymmetric ATP hydrolysis and solvent-exposed substrate diffusion into extracellular space before ADP release and the mechanism resetting. Adapted from PDB: 5KPI (inward)[94], 7A6C (occluded)[96], and 6C0V (outward)[95]. NBDs: Nucleotide-binding domains; TMDs: transmembrane domains.
It is clear that the energy from ATP hydrolysis is required to complete a substrate transport cycle and that, although both NBDs are catalytically active, they are functionally asymmetric, with only one hydrolysis event occurring at a time[107–109,122]. For human P-gp, the three-dimensional structure demonstrated that ATP binding drives dimerization of the NBDs, causes the transition to the outward-facing conformation, and promotes subsequent substrate release[111]. However, recent double electron-electron resonance electron paramagnetic resonance (DEER EPR) experiments with P-gp demonstrated that substrate transport could not proceed until at least one ATP is hydrolyzed[123]. It has been further postulated that ATP hydrolysis and/or subsequent Pi or ADP release is required for resetting the transporter back to the inward-facing conformation to accept another substrate molecule[34,111]. Interestingly, the basal ATP hydrolysis observed in P-gp is hypothesized to be an intrinsic property that allows it to sample a multitude of conformations as a mechanism to recognize a diverse set of substrates[110].
Concurrently, the structures of human ABCG2 in various states have also been solved[124–127]. Unlike P-gp, ABCG2 does not show large conformational changes during the transport cycle, as the NBDs are in close proximity in the nucleotide-free state. In ABCG2 structures, ATP binding has been shown to cause the NBDs to dimerize more closely, which, in turn, collapses the inward-facing drug-binding cavity and opens the outward-facing cavity to the extracellular space[125]. Further, no occluded intermediate conformation in the presence of substrate was observed for ABCG2. As such, it is proposed that ATP binding alone might be sufficient to move the substrate to the external cavity and subsequent release. Further, it is proposed that ATP hydrolysis and/or subsequent Pi or ADP release would be required for resetting the transporter. At present, neither the structural nor functional reason for the proximity of the NBDs in ABCG2 compared to P-gp has been elucidated.
P-GP AND ABCG2 SUBSTRATE AND INHIBITOR BINDING SITES
Due to the promiscuous and adaptive nature of the drug binding sites of both P-gp and ABCG2, these transporters are able to recognize, bind, and transport a wide variety of substrates. For example, P-gp recognizes many unrelated neutral and cationic hydrophobic anti-cancer agents, including anthracyclines, camptothecins, epipodophyllotoxins, taxanes, tyrosine kinase inhibitors, and vinca alkaloids in cell culture models[13]. Substrate recognition by ABCG2 has overlap with P-gp, but with some notable differences, including transport of the anti-cancer drugs flavopiridol, irinotecan, methotrexate, and mitoxantrone[128,129]. In addition, numerous biochemical studies have demonstrated the existence of at least three spatially distinct substrate binding sites within the transmembrane regions of P-gp that function in substrate transport or regulation of substrate transport[130–141]. In total, these studies revealed a mostly hydrophobic region of residues from multiple transmembrane domains that are capable of binding not only a large variety of substrates but also are able to bind multiple substrates simultaneously.
Recent cryo-EM structures of human P-gp have provided additional information about the substrate as well as inhibitor binding sites. The structures of inhibitor bound P-gp are of particular interest in the development of future modulators of P-gp in relation to MDR, oral bioavailability, and the ability to access reservoirs beyond blood-tissue barriers. Using a human P-gp complexed with the Fab fragment of the inhibiting antibody MRK-16, cryo-EM structures of complexes with bound substrates and inhibitors (vincristine, paclitaxel, elacridar, tariquidar, and zosuquidar) were observed in an occluded state, which is characterized by the kinking of TMs 4 and 10 that also brings the nucleotide-binding domains closer together[112,113]. These structures revealed not only a large hydrophobic binding pocket within P-gp, consistent with evidence from many biochemical studies, but also a vestibule and access tunnel formed by transmembrane helices 7, 8, 9, and 12. Whereas substrates only occupy the central hydrophobic pocket, it was found that two inhibitor molecules can bind simultaneously to P-gp [Figure 2A][112,113]. While one inhibitor molecule binds to the central binding pocket of P-gp, the second binds allosterically in the adjacent vestibule area extending away from the binding pocket and perhaps further into a tunnel that accesses the cytoplasm. The presence of inhibitor molecules in the vestibule and/or access tunnel is thought to either sterically inhibit conformational changes involving TM9 to an outward-facing conformation or trap the P-gp molecule in high energy occluded conformation.
Figure 2.
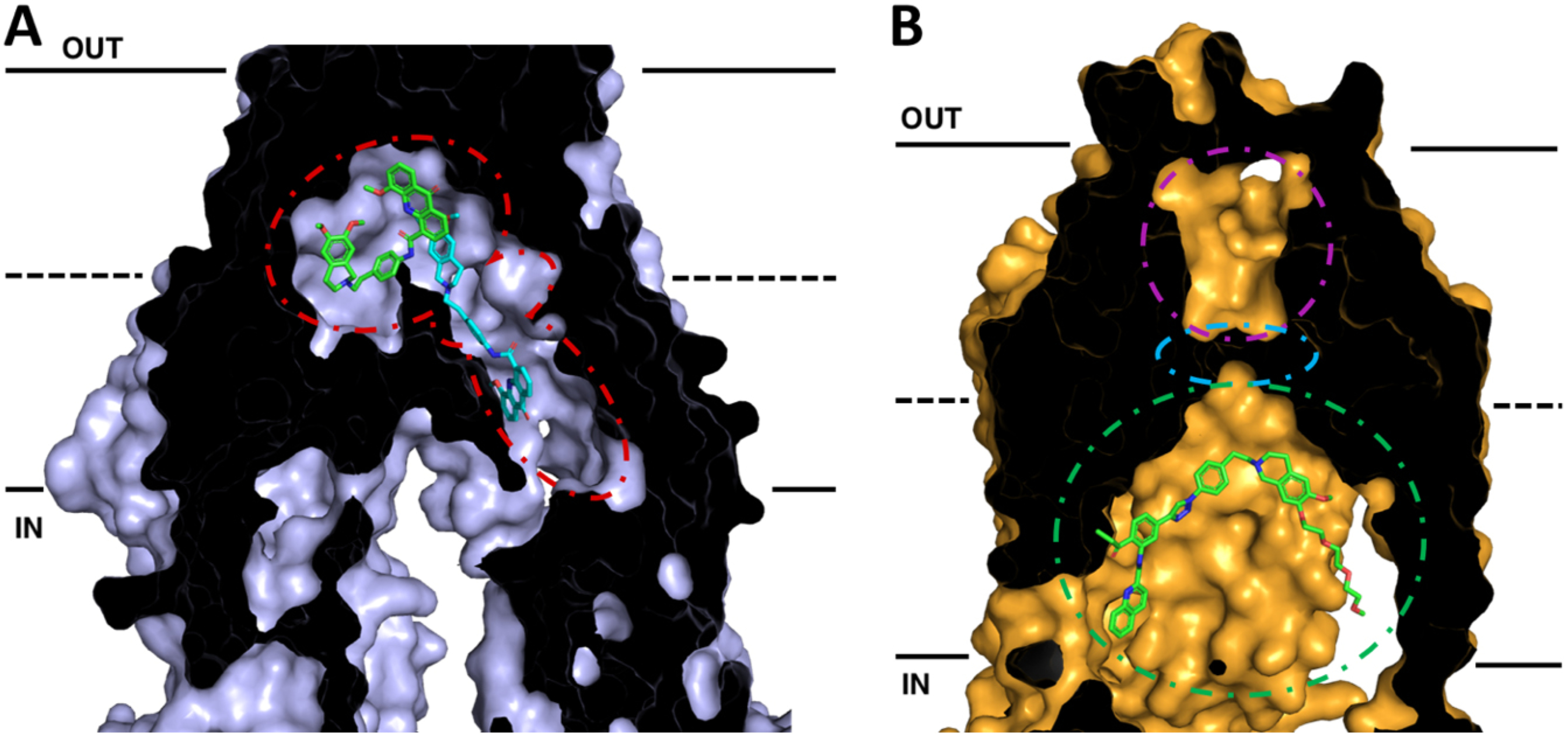
Cross-sections of cryo-EM structures of P-gp and ABCG2 with bound inhibitors in inward-facing conformations. The membrane is delineated by solid black lines. The extracellular space (OUT) is at the top of the image, and the intracellular space (IN) is at the bottom. (A) Cryo-EM structure of P-gp bound with two molecules of the inhibitor elacridar. One molecule (green) is bound to the drug-binding pocket while the other (cyan) passes through a smaller binding region, the vestibule, before extending into an access tunnel. (B) Cryo-EM structure of ABCG2 with bound the tariquidar analog MB136. MB136 is bound to the top of cavity 1 (green) under the leucine gate (cyan) and is believed to stop conformational changes that would allow the substrate to pass into cavity 2 (magenta) and be effluxed. Adapted from PDB: 7A6C (P-gp)[96] and 6FEQ (ABCG2)[112]
With this in mind, inhibitors and/or prodrugs could be designed to exploit this feature of P-gp by occupying the binding pocket, vestibule, and/or access tunnel simultaneously or stabilizing high energy occluded state through stabilization of TMHs 5, 7, 8, 9 and 12 at residues exposed to the access tunnel. Alternatively, inhibitors and or substrates could be joined by a linker moiety so that upon binding of P-gp, they would interact simultaneously with the binding pocket and extend into the access tunnel to stabilize a non-transporting conformation of P-gp. Several dimers of P-gp substrates and other molecules linked by variable-length tethers act as potent inhibitors and may be functioning to block P-gp function in such a manner[84,86,142–154]. The structures of P-gp with multiple inhibitors bound in the binding pocket and the vestibule provide interesting insights for strategies into inhibitor design. For instance, it may be possible to append additional aromatic moieties onto the binding pocket inhibitor that extend into the vestibule and access channel in such a way as to mimic portions of the inhibitor in that channel. In this way, increased contact with the transporters and increased binding affinity could be achieved.
Advances in cryo-EM have also allowed for precise structural determination of the homodimer of human ABCG2[124–127]. The ABCG2 structures revealed two cavities for substrate binding. A central hydrophobic slit-like cavity (cavity 1) faces the cytoplasmic side of the membrane and binds the substrate. Cavity 2 is smaller with less hydrophobic character with access to the extracellular space, thus promoting substrate release following transport. These two cavities are separated by a “leucine plug” that undergoes conformational changes during the transport cycle. Further conformational changes shift transmembrane helices 5b, 5c, and 6a, exposing cavity 2 to the extracellular space.
Recent structures of inhibitor bound ABCG2 could lead to future rationally designed inhibitors to exploit critical features of the inhibited conformation of ABCG2. Recent studies with the Ko143-derived ABCG2 inhibitor MZ29 demonstrated inhibition of ABCG2 occurs via binding of two molecules in cavity 1 and subsequent stabilization of the inward-facing conformation, thus not allowing the substrate to move across the membrane[124]. Additionally, structures with the tariquidar analog MB136 bound only one molecule of the inhibitor in cavity 1, and stabilized the transporter in the inward-facing conformation [Figure 2B]. Corroborative biochemical data also demonstrated that stoichiometric ratios of 1:1 for ABCG2:MB136 and 1:2 for MZ29 were needed for complete inhibition of ATPase activity. These inhibitors showed additional interactions and contacts with the transporter itself, including additional interactions in TM 1b, 2, and 5a that are not observed with molecules that are substrates for the transporter. It appears that the shape, size, and ability to fill the cavity to make these additional contacts are some of the critical features that discriminate inhibitor molecules from substrates, allowing them to form a tightly bound wedge to immobilize the transporter[124,125]. More recently, the anti-cancer drug imatinib was also found to lock the inward open structure of ABCG2[127]. In this study, one molecule of imatinib bound directly under the leucine plug in cavity 1 and was sandwiched between the interface of the ABCG2 monomers, similarly acting as a wedge to stabilize the inhibited, inward-facing conformation. These structural insights into how substrates and inhibitors interact with ABCG2 should be invaluable in designing future inhibitors, giving chemists an excellent platform for rational structure-based drug design of new inhibitors with higher binding affinities relative to substrates exploting these additional contacts in the TM domains.
INHIBITOR DESIGN AND EVALUATION
After the discovery of P-gp, some substrates that could act as inhibitors were identified, including amiodarone, cyclosporine A, quinidine, and verapamil, and implemented in chemotherapy regimes in clinical trials[3,17,19,23,24,27,29,57,58,155]. The promising results observed in in vitro cell culture models were not replicated in patients, as this first generation of inhibitors, though pharmacologically active, was not potent and potentially toxic [Table 1][3,17,23,155,156]. Second generation inhibitors that lacked pharmacological activity, such as valspodar and dexverapamil, were more potent than the first generation but resulted in no better outcomes due to off-target effects[23,24,155–157]. Third generation inhibitors including biricodar, elacridar, dofequidar, tariquidar, and zosuquidar were designed to have higher specificity for P-gp and lower toxicity in the hopes of better outcomes with co-administration with chemotherapeutics[23,155,156]. Tariquidar was shown to reverse drug resistance to doxorubicin caused by P-gp upregulation in a mouse model of breast cancer[158]. However, in a human clinical trial in non-small cell lung cancer patients, co-administration of tariquidar with chemotherapy led to increased toxicity and a halt to the study[19,29].
Table 1.
Representative inhibitors of P-gp and ABCG2
| First generation | Cyclosporin A*, Verapamil*, Nifedipine*,†, Quinidine*, Quinine*, Tamoxifen*, Reserpine*,†, Prazosin*,†, Propafenone*,†, Erythromycin*, Itraconazole*,†, Ritonavir*,†, Benzquinamide*[23] |
| Second generation | Dexverapamil*, MM36*, Valspodar*,†, BIBW22BS*, Toremifene*, Quinine Homodimer (Q2)*, S9788*,†[23] |
| Third generation | Elacridar*,†, Tariquidar*,†, Zosuquidar*, Laniquidar*, Ontogen*, DP7*[23] |
| Fourth generation | Natural product derivatives: Flavonoids (Curcumin*[163], Apigenin*[164]), Polyphenols (Quercetin*[165], Resveratrol*,†[166]), Antimalarials (Chloroquine*,†[167]), Alkaloids (Galegine*[168], Nitensidine A*[169]), Fungal (Ko143†[170], Tryprostatin A†[171]), Chinese herbal medicines (Artemisinin*[172], Tetrandrine*[173]), Marine (Kendarimide* A, Sipholenol A*)[174] Repurposing FDA-approved drugs[175]: Imatinib*,†, Vardenafil*, Azithromycin*,†, Chlorpromazine*,† Peptidomimetics[23] Dual activity ligands[23] |
P glycoprotein inhibitor
ABCG2 inhibitor.
Additionally, and perhaps even more importantly, this trial was also seemingly flawed in that there is inconclusive evidence that non-small cell lung carcinomas express P-gp or that P-gp expression correlates with outcome clinically[29,159]. Further, once thought to be specific only for P-gp, tariquidar has also been shown to inhibit ABCG2, complicating interpretation of the results, perhaps partially accounting for the increased toxicity[160,161]. Thus, there remains room for improvement for even the best of the third generation inhibitors such as tariquidar. To that end, the structures of both ABCG2 and P-gp solved in the presence of tariquidar or a tariquidar analog[112,124] offer a great opportunity to determine how differences in binding of the same molecule to the two different transporters may be exploited to generate even more specific inhibitors with higher affinity, and less cross-reactivity. Fourth generation inhibitor design is focused on natural product derivatives, peptidomimetics, dual activity ligands, and the potential of repurposing current FDA-approved drugs (see below for further discussion)[23,162].
Due to the clinical importance of this area, many small-molecule inhibitors and modulators have continued to be developed for P-gp and ABCG2[23,24,176–180]. These designs focus on different aspects of blocking transport through either direct interaction with the drug binding site(s) or interaction with allosteric sites. To this end, different classes of inhibitors are being investigated including, but not limited to, dimeric substrates[84,86,142–150], aromatic or heterocyclic dimers[181,182], bifendates[85], condensed ring derivatives[183–185], 1,4-dihydropyridines and 1,4-dihydroquinolines[186–188], dimeric flavonoids[151–154], peptides[189,190], steroids[191–193], and tetrahydroisoquinolines[194–196]. For example, homodimers of the P-gp substrate quinine were potent inhibitors of P-gp and were found to reverse the drug resistance phenotype for taxol in a breast cancer cell line overexpressing P-gp[149]. In addition, homodimers of the P-gp substrate quetiapine demonstrated inhibition of P-gp in human brain capillary endothelial cells, in rat brain capillaries, and at the BBB in an in situ rat brain model[144]. Flavonoid dimers linked by polyethylene glycol units have also been shown to be inhibitors of P-gp[151–153,197]. Dual inhibitors of P-gp and ABCG2 were also developed using this strategy; homodimers of paliperidone with dual inhibition in a blood-brain barrier cellular model have been reported.
Interestingly, these homodimers, as well as dimers of abacavir, were designed to also revert to the monomeric drug within the reducing environment of the cell, thus acting as both ABC transporter inhibitors and prodrugs[84,146]. Recently, homodimers of triazole-bridged flavonoids were also shown to potently inhibit ABCG2 in a highly selective manner over P-gp[154]. These data suggest that exploiting the polyvalent nature of the drug binding sites of P-gp and ABCG2 may be an effective strategy for developing potent inhibitors of these transporters. Interestingly, as described above, structural studies demonstrated that several inhibitors were shown to bind in pairs to ABCG2 and P-gp[112,113,124,125], suggesting that these dimeric inhibitors may have similar binding mechanisms. Thus, new inhibitor-bound structures of P-gp and ABCG2 should provide a more comprehensive understanding of inhibition, leading to better structure-based rational drug design for additional P-gp and ABCG2 inhibitors.
Another interesting prospect for inhibitor development is repurposing already FDA-approved drugs known as modulators P-gp and ABCG2 activity to improve the disposition of cancer chemotherapeutic drugs or, perhaps, agents used to treat other disorders[175]. These agents are also of particular interest as they can potentially provide both modulations of ABC transporters as well as their original FDA-approved function. As these FDA-approved drugs have already been rigorously tested clinically, have well-understood toxicities, and established mechanisms of action, co-administration with a chemotherapeutic drug may offer clinical benefits, barring unforeseen drug-drug interactions and changes in the pharmacokinetic and pharmacodynamic properties of either agent. In search of the Drugbank database of FDA-approved drugs, Lai et al.[175] determined a set of 67 agents that have activity against P-gp but do not have anti-cancer activity. Interestingly, this list includes the anti-emetic/antipsychotic chlorpromazine[198], the antibiotic macrolides erythromycin, azithromycin, and clarithromycin[199], and the phosphodiesterases inhibitors sildenafil and vardenafil[200].
Repurposing anti-cancer agents that are also P-gp and ABCG2 modulators have recently emerged as an area of more intense investigation, especially for tyrosine kinase inhibitors[201]. These agents, including elacridar, imatinib, sorafenib, dasatinib, gefitinib, nilotinib, erlotinib, and afatinib, are used in the treatment of a variety of cancers, including chronic myelogenous leukemia, gastrointestinal stromal tumors, renal cell carcinoma, and non-small cell lung cancer[80,202–207]. As these are also known modulators of P-gp and/or ABCG2, they could be used as chemosensitizers combined with other cytotoxic agents to increase the treatment efficacy. Care must be taken, however, as the inclusion of these agents as P-gp or ABCG2 inhibitors can also affect the pharmacodynamics and pharmacokinetics of any co-administered chemotherapeutic drug. For example, the addition of nilotinib in a mouse xenograft model of BCR-ABL chronic myelogenous leukemia treated with doxorubicin resulted in an increase in the cardiotoxicity of doxorubicin[207], likely due to the inhibition of P-gp. Other anti-cancer agents being explored for repurposing as P-gp and ABCG2 inhibitors include PARP inihibitors[208,209], CDK4/6 inhibitors[210,211], and taxanes[212,213].
OTHER INHIBITORY APPROACHES
siRNAs downregulate protein expression by binding mRNA and forming RNA-induced silencing complexes recognized and digested by dicer RNases. Such siRNAs have been developed specifically for MDR1 or ABCG2 to reduce transporter expression and thus the MDR phenotype in tumor cells in culture[214–216]. In one study, siRNAs downregulated P-gp mRNA expression in drug-resistant breast carcinoma and osteosarcoma cell lines[216]. Further, transfection of the siRNA appeared to sensitize osteosarcoma cells to the anti-proliferative effects of doxorubicin, decreasing the IC50 10-fold. In another study, transfection of drug-resistant NIH3T3-MDR fibroblasts with P-gp-targeted altritol-modified siRNAs resulted in a reduction of P-gp expression, a parallel reduction in P-gp mRNA levels, an increased accumulation of the P-gp substrate rhodamine 123, and increased sensitivity to doxorubicin[214]. Similarly, an ABCG2-targeted siRNA transfected into drug-resistant human liver HepG2 cells showed reduced levels of AGCG2 mRNA and increased sensitivity to doxorubicin, a known substrate of ABCG2[215].
miRNAs are short noncoding RNAs, approximately 20–25 nucleotides, that modulate gene expression by binding the 3’ untranslated region (UTR) of target mRNA and have been explored as modulators of MDR in cancer cells. Expression of the miRNA miR-296 was shown to affect survival and MDR in esophageal squamous cell carcinoma[217]. In this study, cells treated with antagomirs demonstrated increased survival due to decreased tumorigenesis. Further, it was shown that expression of P-gp decreased, as measured by immunoblotting and RT-PCR, and cells were more sensitive to treatment with therapeutics known to be P-gp substrates[217]. In another study, over-expression of the miRNA miR-298 was shown to increase doxorubicin accumulation at the nucleus of resistant breast cancer cells through downregulation of P-gp expression via binding at the 3’ untranslated region of the MDR1 gene[218,219]. Downregulation of miR-298 in sensitive breast cancer cells showed increased expression of P-gp and conferred resistance to doxorubicin[218]. Transfection of miR-298 into human brain microvascular endothelial cells downregulated surface expression of P-gp and increased accumulation of antiepileptic drugs known to be substrates of P-gp[219]. miR-298 binding of the 3’ UTR of MDR1 was confirmed by cotransformation of P-gp non-expressing HEK293T cells with plasmids containing either the 3’ UTR of MDR1, miR-298, or controls and quantified through luciferase assay[219]. These studies demonstrate the potential utility of miRNAs in downregulating P-gp expression, either combat MDR, increase the bioavailability of P-gp substrates or allow the entry of therapeutic agents into sanctuary sites protected by P-gp, such as the brain.
Antisense oligonucleotides, which are short DNA or RNA sequences designed to bind target mRNA, provide another strategy to inhibit the expression of P-gp and ABCG2 at the translational level. Antisense oligonucleotides were shown to reduce the amount of MDR1/mRNA, and P-gp expression in human chronic myelogenous leukemia cells (K562) expressing P-gp[220] and were also shown to increase doxorubicin accumulation in human glioblastoma and endothelial cell lines expressing P-gp[221].
Gene manipulation using the CRISPR-Cas9 gene-editing technology is a newer approach that also affords an opportunity for treating drug-resistant cancers caused by ABC transporter expression or overexpression. For example, the CRISPR-Cas9 system could be used for downregulating or inactivating the genes that encode the ABC transporters implicated in multidrug resistance, such as MRP1, P-gp, and ABCG2, before treatment with chemotherapy[222]. A proof-of-concept experiment was performed successfully in a doxorubicin-resistant human epithelial ovarian cell line A2780/ADR, in which chemosensitivity to doxorubicin was restored following CRISPR-Cas9 knockdown of the human MDR1 gene that encodes P-gp[223]. CRISPR-Cas9 was also used in an MDCK canine kidney cell line to knock out endogenous P-gp expression. This new cell line could then be used to study exogenously expressed human or other P-gp’s, without the confounding effects of endogenous P-gp expression[224]. Using CRISPR-Cas9 to knock down ABC drug transporter expression may also be useful at the BBB where endogenous ABC transporters are of concern in limiting drug bioavailability to the brain. However, as permanent changes made by the CRISPR-Cas9 system to the expression of ABC transporters at the BBB may not be advantageous or even desirable, RNA editing techniques may offer more tunable systems for modulating the expression of these transporters in the future.
Monoclonal antibodies also have the potential to reverse MDR by binding to P-gp. MRK16, for example, was developed in the 1980s as a possible way to reverse drug resistance[225–228]. MRK16, which recognizes external epitopes of P-gp, was shown to work in concert with cyclosporin A treatment, operating synergistically to increase the accumulation of vincristine and doxorubicin in human leukemia K562 cells[229]. In another study, MRK-16 treatment was shown to partially restore sensitivity to vincristine-resistant tumor cells both in vitro and in a mouse xenograft model in vivo[230]. Furthermore, the monoclonal antibody UIC2 potently reverses P-gp-mediated drug resistance in cells in culture and recognizes extracellular epitopes of P-gp[231,232]. Interestingly, UIC2 was later determined also to be conformationally sensitive, binding to multiple conformations of the transporter[231,233–235]. Most recently, the structure of a chimeric human-mouse P-gp in complex with zosuquidar and UIC2 revealed that the antibody recognizes a conformational epitope of ABCB1 that involves residues from TM1 and TM2 and the extracellular loops EL1, EL3, and EL4[114], which is in agreement with previous biochemical data[232,234,235]. The structure also gives insight into how the antibody acts as a transport inhibitor. By clamping the external loops together, UIC2 prevents P-gp from moving to the outward-open conformation required to release substrate to the extracellular space[114]. Unfortunately, although antibodies have great potential for selective and tight binding to ABC transporters, this field has not seen significant advancement since these early reports.
CONCLUSIONS
An extensive effort has focused on multidrug resistance in cancer over the past 60 years, leading to the discovery of the ABC transporter superfamily. While the role of ABC transporters, including P-gp and ABCG2, has been studied extensively in vitro, there is still much to learn about the clinical importance of ABC transporter expression in cancer. While the early discovery of P-gp and ABCG2 inhibitors led to high expectations for clinical use, an FDA approved agent targeting P-gp or ABCG2 for combating multidrug resistance has been elusive, confounded by both other mechanisms of drug resistance and poorly designed clinical trials. Major advancements in structural biology in the last decade have led to valuable insights into the structures of human P-gp and ABCG2, and more recently, inhibitor-bound structures. These understandings will hopefully lead to more potent and specific inhibitors that, combined with more selective and rigorous clinical trials, lead to a deeper appreciation of the role of these transporters in the MDR phenotype in patients. Further, these inhibitors may have a positive impact on drug delivery efforts to sanctuary sites protected by endogenous expression of these transporters such as the brain and offer more effective oral bioavailability at the gut for many classes of chemotherapeutics.
Footnotes
Conflicts of interest
All authors declared that there are no conflicts of interest.
DECLARATIONS
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
REFERENCES
- 1.Lehnert M Clinical mutidrug resistance in cancer: a multifactorial problem. Eur J Cancer 1996;32A:912–20. [DOI] [PubMed] [Google Scholar]
- 2.Saraswathy M, Gong S. Different strategies to overcome multidrug resistance in cancer. Biotechnol Adv 2013;31:1397–407. [DOI] [PubMed] [Google Scholar]
- 3.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nature Rev Cancer 2002;2:48–58. [DOI] [PubMed] [Google Scholar]
- 4.Tarasov VV, Chubarev VN, Ashraf GM, et al. How cancer cells resist chemotherapy: design and development of drugs targeting protein-protein interactions. Curr Top Med Chem 2019;19:394–412. [DOI] [PubMed] [Google Scholar]
- 5.Shankaraiah N, Nekkanti S, Ommi O, P S LS. Diverse targeted approaches to battle multidrug resistance in cancer. Curr Med Chem 2019;26:7059–80. [DOI] [PubMed] [Google Scholar]
- 6.Bukowski K, Kciuk M, Kontek R. Mechanisms of multidrug resistance in cancer chemotherapy. Int J Mol Sci 2020;21:3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Higgins CF. The multidrug resistance P-glycoprotein. Curr Opin Cell Biol 1993;5:684–7. [DOI] [PubMed] [Google Scholar]
- 8.Breier A, Gibalova L, Seres M, Barancik M, Sulova Z. New insight into p-glycoprotein as a drug target. Anticancer Agents Med Chem 2013;13:159–70. [PubMed] [Google Scholar]
- 9.Scheper RJ, Broxterman HJ, Scheffer GL, Meijer CJLM, Pinedo HM. Drug-transporter proteins in clinical multidrug resistance. Clin Chim Acta 1992;206:25–32. [DOI] [PubMed] [Google Scholar]
- 10.Hrycyna CA. Molecular genetic analysis and biochemical characterization of mammalian P-glycoproteins involved in multidrug resistance. Semin Cell Dev Biol 2001;12:247–56. [DOI] [PubMed] [Google Scholar]
- 11.Borst P, Schinkel AH. P-glycoprotein ABCB1: a major player in drug handling by mammals. J Clin Invest 2013;123:4131–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amawi H, Sim HM, Tiwari AK, Ambudkar SV, Shukla S. ABC transporter-mediated multidrug-resistant cancer. Adv Exp Med Biol 2019;1141:549–80. [DOI] [PubMed] [Google Scholar]
- 13.Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol 1999;39:361–98. [DOI] [PubMed] [Google Scholar]
- 14.Khalifa MA, Abdoh AA, Mannel RS, Walker JL, Angros LH, Min KW. P-glycoprotein as a prognostic indicator in pre- and postchemotherapy ovarian adenocarcinoma. Int J Gynecol Pathol 1997;16:69–75. [DOI] [PubMed] [Google Scholar]
- 15.Soini Y, Virkajarvi N, Raunio H, Paakko P. Expression of P-glycoprotein in hepatocellular carcinoma: a potential marker of prognosis. J Clin Pathol 1996;49:470–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu D, Knaust E, Pisa P, et al. Levels of mdr1 and mrp mRNA in leukaemic cell populations from patients with acute myelocytic leukaemia are heterogenous and inversely correlated to cellular daunorubicin accumulation. Br J Haematol 1996;92:847–54. [DOI] [PubMed] [Google Scholar]
- 17.Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, Gottesman MM. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat Rev Cancer 2018;18:452–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hipfner DR, Deeley RG, Cole SP. Structural, mechanistic and clinical aspects of MRP1. Biochim Biophys Acta 1999;1461:359–76. [DOI] [PubMed] [Google Scholar]
- 19.Binkhathlan Z, Lavasanifar A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: current status and future perspectives. Curr Cancer Drug Targets 2013;13:326–46. [DOI] [PubMed] [Google Scholar]
- 20.Darby RA, Callaghan R, McMahon RM. P-glycoprotein inhibition: the past, the present and the future. Curr Drug Metab 2011;12:722–31. [DOI] [PubMed] [Google Scholar]
- 21.Dong J, Qin Z, Zhang WD, et al. Medicinal chemistry strategies to discover P-glycoprotein inhibitors: an update. Drug Resist Updat 2020;49:100681. [DOI] [PubMed] [Google Scholar]
- 22.Krishna R, Mayer LD. Modulation of P-glycoprotein (PGP) mediated multidrug resistance (MDR) using chemosensitizers: recent advances in the design of selective MDR modulators. Curr Med Chem Anti-Canc Agents 2001;1:163–74. [DOI] [PubMed] [Google Scholar]
- 23.Palmeira A, Sousa E, Vasconcelos MH, Pinto MM. Three decades of P-gp inhibitors: skimming through several generations and scaffolds. Curr Med Chem 2012;19:1946–2025. [DOI] [PubMed] [Google Scholar]
- 24.Jaramillo AC, Saig FA, Cloos J, Jansen G, Peters GJ. How to overcome ATP-binding cassette drug efflux transporter-mediated drug resistance? Cancer Drug Resistance 2018;1:6–29. [Google Scholar]
- 25.Sharom FJ. ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics 2008;9:105–27. [DOI] [PubMed] [Google Scholar]
- 26.Amiri-Kordestani L, Basseville A, Kurdziel K, Fojo AT, Bates SE. Targeting MDR in breast and lung cancer: discriminating its potential importance from the failure of drug resistance reversal studies. Drug Resist Updat 2012;15:50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leonard GD, Fojo T, Bates SE. The role of ABC transporters in clinical practice. Oncologist 2003;8:411–24. [DOI] [PubMed] [Google Scholar]
- 28.Borst P Looking back at multidrug resistance (MDR) research and ten mistakes to be avoided when writing about ABC transporters in MDR. FEBS Lett 2020;594:4001–11. [DOI] [PubMed] [Google Scholar]
- 29.Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov 2006;5:219–34. [DOI] [PubMed] [Google Scholar]
- 30.Bradshaw DM, Arceci RJ. Clinical relevance of transmembrane drug efflux as a mechanism of multidrug resistance. J Clin Oncol 1998;16:3674–90. [DOI] [PubMed] [Google Scholar]
- 31.Ferry DR, Traunecker H, Kerr DJ. Clinical trials of P-glycoprotein reversal in solid tumours. Eur J Cancer 1996;32A:1070–81. [DOI] [PubMed] [Google Scholar]
- 32.Fisher GA, Sikic BI. Clinical studies with modulators of multidrug resistance. Hematol Oncol Clin North Am 1995;9:363–82. [PubMed] [Google Scholar]
- 33.Libby E, Hromas R. Dismounting the MDR horse. Blood 2010;116:4037–8. [DOI] [PubMed] [Google Scholar]
- 34.Thomas C, Tampe R. Structural and mechanistic principles of ABC transporters. Annu Rev Biochem 2020;89:605–36. [DOI] [PubMed] [Google Scholar]
- 35.Niazi M, Zakeri-Milani P, Najafi Hajivar S, et al. Nano-based strategies to overcome p-glycoprotein-mediated drug resistance. Expert Opin Drug Metab Toxicol 2016;12:1021–33. [DOI] [PubMed] [Google Scholar]
- 36.Schinkel AH. P-Glycoprotein, a gatekeeper in the blood-brain barrier. Adv Drug Deliv Rev 1999;36:179–94. [DOI] [PubMed] [Google Scholar]
- 37.Schinkel AH, Wagenaar E, Mol CA, van Deemter L. P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J Clin Invest 1996;97:2517–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dietrich CG, Geier A, Oude Elferink RP. ABC of oral bioavailability: transporters as gatekeepers in the gut. Gut 2003;52:1788–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldstein MN, Slotnick IJ, Journey LJ. In vitro studies with HeLa cell line sensitive and resistant to actinomycin D. Ann N Y Acad Sci 1960;89:474–83. [DOI] [PubMed] [Google Scholar]
- 40.Biedler JL, Riehm H. Cellular resistance to actinomycin D in Chinese hamster cells in vitro: cross-resistance, radioautographic, and cytogenetic studies. Cancer Res 1970;30:1174–84. [PubMed] [Google Scholar]
- 41.Danø K Active outward transport of daunomycin in resistant Ehrlich ascites tumor cells. Biochem Biophys Acta 1973;323:466–83. [DOI] [PubMed] [Google Scholar]
- 42.Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta 1976;455:152–62. [DOI] [PubMed] [Google Scholar]
- 43.Riordan JR, Ling V. Purification of P-glycoprotein from plasma membrane vesicles of Chinese hamster ovary cell mutants with reduced colchicine permeability. J Biol Chem 1979;254:12701–5. [PubMed] [Google Scholar]
- 44.Gros P, Croop J, Roninson IB, Varshavsky A, Housman DE. Isolation and characterization of DNA sequences amplified in multidrug-resistant hamster cells. Proc Natl Acad Sci U S A 1986;83:337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roninson IB, Chin JE, Choi K, et al. Isolation of human mdr DNA sequences amplified in multidrug-resistant KB carcinoma cells. Proc Natl Acad Sci U S A 1986;83:4538–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ueda K, Cornwell MM, Gottesman MM, et al. The mdr1 gene, responsible for multidrug-resistance, codes for P-glycoprotein. Biochem Biophys Res Comm 1986;141:956–62. [DOI] [PubMed] [Google Scholar]
- 47.Dean M, Hamon Y, Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. J Lipid Res 2001;42:1007–17. [PubMed] [Google Scholar]
- 48.Higgins CF. ABC transporters: from microorganisms to man. Annu Rev Cell Biol 1992;8:67–113. [DOI] [PubMed] [Google Scholar]
- 49.Ouellette M, Legare D, Papadopoulou B. Microbial multidrug-resistance ABC transporters. Trends Microbiol 1994;2:407–11. [DOI] [PubMed] [Google Scholar]
- 50.Theodoulou FL. Plant ABC transporters. Biochim Biophys Acta 2000;1465:79–103. [DOI] [PubMed] [Google Scholar]
- 51.Allikmets R, Gerrard B, Hutchinson A, Dean M. Characterization of the human ABC superfamily: isolation and mapping of 21 new genes using the expressed sequence tags database. Hum Mol Genet 1996;5:1649–55. [DOI] [PubMed] [Google Scholar]
- 52.Balzi E, Goffeau A. Yeast multidrug resistance: the PDR network. J Bioenerg Biomembr 1995;27:71–6. [DOI] [PubMed] [Google Scholar]
- 53.Allikmets R, Schriml LM, Hutchinson A, Romano-Spica V, Dean M. A human placenta-specific ATP-binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res 1998;58:5337–9. [PubMed] [Google Scholar]
- 54.Doyle LA, Yang W, Abruzzo LV, et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci U S A 1998;95:15665–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miyake K, Mickley L, Litman T, et al. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: demonstration of homology to ABC transport genes. Cancer Res 1999;59:8–13. [PubMed] [Google Scholar]
- 56.Dean M, Annilo T. Evolution of the ATP-binding cassette (ABC) transporter superfamily in vertebrates. Annu Rev Genomics Hum Genet 2005;6:123–42. [DOI] [PubMed] [Google Scholar]
- 57.Thomas H, Coley HM. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control 2003;10:159–65. [DOI] [PubMed] [Google Scholar]
- 58.Robey RW, Massey PR, Amiri-Kordestani L, Bates SE. ABC transporters: unvalidated therapeutic targets in cancer and the CNS. Anticancer Agents Med Chem 2010;10:625–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patch AM, Christie EL, Etemadmoghadam D, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015;521:489–94. [DOI] [PubMed] [Google Scholar]
- 60.da Silveira Júnior LS, Soares VL, Jardim da Silva AS, et al. P-glycoprotein and multidrug resistance-associated protein-1 expression in acute myeloid leukemia: biological and prognosis implications. Int J Lab Hematol 2020;42:594–603. [DOI] [PubMed] [Google Scholar]
- 61.Hendrikse NH, Schinkel AH, de Vries EG, et al. Complete in vivo reversal of P-glycoprotein pump function in the blood-brain barrier visualized with positron emission tomography. Br J Pharmacol 1998;124:1413–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Syvanen S, Eriksson J. Advances in PET imaging of P-glycoprotein function at the blood-brain barrier. ACS Chem Neurosci 2013;4:225–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Begley DJ. ABC transporters and the blood-brain barrier. Curr Pharm Des 2004;10:1295–312. [DOI] [PubMed] [Google Scholar]
- 64.Cardon-Cardo C, O’Brien JP, Casals D, et al. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc Natl Acad Sci U S A 1989;86:695–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Takano M, Yumoto R, Murakami T. Expression and function of efflux drug transporters in the intestine. Pharmacol Ther 2006;109:137–61. [DOI] [PubMed] [Google Scholar]
- 66.Gottesman MM, Hrycyna CA, Schoenlein PV, Germann UA, Pastan I. Genetic analysis of the multidrug transporter. Annu Rev Genet 1995;29:607–49. [DOI] [PubMed] [Google Scholar]
- 67.AL. Blood-brain barrier P-glycoprotein function in neurodegenerative disease. Curr Pharm Des 2011;17:2771–7. [DOI] [PubMed] [Google Scholar]
- 68.Loscher W, Potschka H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx 2005;2:86–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vlaming ML, Lagas JS, Schinkel AH. Physiological and pharmacological roles of ABCG2 (BCRP): recent findings in Abcg2 knockout mice. Adv Drug Deliv Rev 2009;61:14–25. [DOI] [PubMed] [Google Scholar]
- 70.Schinkel AH, Mayer U, Wagenaar E, et al. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc Natl Acad Sci U S A 1997;94:4028–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schinkel AH, Mol CA, Wagenaar E, van Deemter L, Smit JJ, Borst P. Multidrug resistance and the role of P-glycoprotein knockout mice. Eur J Cancer 1995;31A:1295–8. [DOI] [PubMed] [Google Scholar]
- 72.Schinkel AH, Smit JJ, van Tellingen O, et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell 1994;77:491–502. [DOI] [PubMed] [Google Scholar]
- 73.Schinkel AH, Wagenaar E, van Deemter L, Mol CA, Borst P. Absence of the mdr1a P-Glycoprotein in mice affects tissue distribution and pharmacokinetics of dexamethasone, digoxin, and cyclosporin A. J Clin Invest 1995;96:1698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Breedveld P, Pluim D, Cipriani G, et al. The effect of Bcrp1 (Abcg2) on the in vivo pharmacokinetics and brain penetration of imatinib mesylate (Gleevec): implications for the use of breast cancer resistance protein and P-glycoprotein inhibitors to enable the brain penetration of imatinib in patients. Cancer Res 2005;65:2577–82. [DOI] [PubMed] [Google Scholar]
- 75.Jonker JW, Buitelaar M, Wagenaar E, et al. The breast cancer resistance protein protects against a major chlorophyll-derived dietary phototoxin and protoporphyria. Proc Natl Acad Sci U S A 2002;99:15649–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou S, Morris JJ, Barnes Y, Lan L, Schuetz JD, Sorrentino BP. Bcrp1 gene expression is required for normal numbers of side population stem cells in mice, and confers relative protection to mitoxantrone in hematopoietic cells in vivo. Proc Natl Acad Sci U S A 2002;99:12339–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Deliv Rev 2003;55:3–29. [DOI] [PubMed] [Google Scholar]
- 78.Oostendorp RL, Buckle T, Beijnen JH, van Tellingen O, Schellens JH. The effect of P-gp (Mdr1a/1b), BCRP (Bcrp1) and P-gp/BCRP inhibitors on the in vivo absorption, distribution, metabolism and excretion of imatinib. Invest New Drugs 2009;27:31–40. [DOI] [PubMed] [Google Scholar]
- 79.Fellner S, Bauer B, Miller DS, et al. Transport of paclitaxel (Taxol) across the blood-brain barrier in vitro and in vivo. J Clin Invest 2002;110:1309–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lagas JS, van Waterschoot RA, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Breast cancer resistance protein and P-glycoprotein limit sorafenib brain accumulation. Mol Cancer Ther 2010;9:319–26. [DOI] [PubMed] [Google Scholar]
- 81.Durmus S, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Oral availability and brain penetration of the B-RAFV600E inhibitor vemurafenib can be enhanced by the P-GLYCOprotein (ABCB1) and breast cancer resistance protein (ABCG2) inhibitor elacridar. Mol Pharm 2012;9:3236–45. [DOI] [PubMed] [Google Scholar]
- 82.Kort A, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Brain accumulation of the EML4-ALK inhibitor ceritinib is restricted by P-glycoprotein (P-GP/ABCB1) and breast cancer resistance protein (BCRP/ABCG2). Pharmacol Res 2015;102:200–7. [DOI] [PubMed] [Google Scholar]
- 83.Tang SC, Lagas JS, Lankheet NA, et al. Brain accumulation of sunitinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by oral elacridar and sunitinib coadministration. Int J Cancer 2012;130:223–33. [DOI] [PubMed] [Google Scholar]
- 84.Bohn K, Lange A, Chmielewski J, Hrycyna CA. Dual modulation of human P-glycoprotein and ABCG2 with prodrug dimers of the atypical antipsychotic agent paliperidone in a model of the blood-brain barrier. Mol Pharm 2017;14:1107–19. [DOI] [PubMed] [Google Scholar]
- 85.Gu X, Ren Z, Peng H, Peng S, Zhang Y. Bifendate-chalcone hybrids: a new class of potential dual inhibitors of P-glycoprotein and breast cancer resistance protein. Biochem Biophys Res Commun 2014;455:318–22. [DOI] [PubMed] [Google Scholar]
- 86.Namanja-Magliano HA, Bohn K, Agrawal N, Willoughby ME, Hrycyna CA, Chmielewski J. Dual inhibitors of the human blood-brain barrier drug efflux transporters P-glycoprotein and ABCG2 based on the antiviral azidothymidine. Bioorg Med Chem 2017;25:5128–32. [DOI] [PubMed] [Google Scholar]
- 87.Berggren S, Gall C, Wollnitz N, et al. Gene and protein expression of P-glycoprotein, MRP1, MRP2, and CYP3A4 in the small and large human intestine. Mol Pharm 2007;4:252–7. [DOI] [PubMed] [Google Scholar]
- 88.Maliepaard M, Scheffer GL, Faneyte IF, et al. Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res 2001;61:3458–64. [PubMed] [Google Scholar]
- 89.Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC. Cellular localization of the multidrug resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci U S A 1987;84:7735–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lepper ER, Nooter K, Verweij J, Acharya MR, Figg WD, Sparreboom A. Mechanisms of resistance to anticancer drugs: the role of the polymorphic ABC transporters ABCB1 and ABCG2. Pharmacogenomics 2005;6:115–38. [DOI] [PubMed] [Google Scholar]
- 91.Sparreboom A, Danesi R, Ando Y, Chan J, Figg WD. Pharmacogenomics of ABC transporters and its role in cancer chemotherapy. Drug Resist Updat 2003;6:71–84. [DOI] [PubMed] [Google Scholar]
- 92.Malingré MM, Beijnen JH, Rosing H, et al. Co-administration of GF120918 significantly increases the systemic exposure to oral paclitaxel in cancer patients. Br J Cancer 2001;84:42–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Meerum Terwogt JM, Malingré MM, Beijnen JH, et al. Coadministration of oral cyclosporin A enables oral therapy with paclitaxel. Clin Cancer Res 1999;5:3379–84. [PubMed] [Google Scholar]
- 94.Jonker JW, Smit JW, Brinkhuis RF, et al. Role of breast cancer resistance protein in the bioavailability and fetal penetration of topotecan. J Natl Cancer Inst 2000;92:1651–6. [DOI] [PubMed] [Google Scholar]
- 95.Sesink AL, Arts IC, de Boer VC, Breedveld P, Schellens JH et al. Breast cancer resistance protein (Bcrp1/Abcg2) limits net intestinal uptake of quercetin in rats by facilitating apical efflux of glucuronides. Mol Pharmacol 2005;67:1999–2006. [DOI] [PubMed] [Google Scholar]
- 96.Zaher H, Khan AA, Palandra J, Brayman TG, Yu L, Ware JA. Breast cancer resistance protein (Bcrp/abcg2) is a major determinant of sulfasalazine absorption and elimination in the mouse. Mol Pharm 2006;3:55–61. [DOI] [PubMed] [Google Scholar]
- 97.Gottesman MM, Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem 1993;62:385–427. [DOI] [PubMed] [Google Scholar]
- 98.Borst P, Elferink RO. Mammalian ABC transporters in health and disease. Annu Rev Biochem 2002;71:537–92. [DOI] [PubMed] [Google Scholar]
- 99.Rees DC, Johnson E, Lewinson O. ABC transporters: the power to change. Nat Rev Mol Cell Biol 2009;10:218–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hrycyna CA, Ramachandra M, Germann UA, Cheng PW, Pastan I, Gottesman MM. Both ATP sites of human P-glycoprotein are essential but not symmetric. Biochemistry 1999;38:13887–99. [DOI] [PubMed] [Google Scholar]
- 101.Julien M, Gros P. Nucleotide-induced conformational changes in P-glycoprotein and in nucleotide binding site mutants monitored by trypsin sensitivity. Biochemistry 2000;39:4559–68. [DOI] [PubMed] [Google Scholar]
- 102.Loo TW, Bartlett MC, Clarke DM. Drug binding in human P-glycoprotein causes conformational changes in both nucleotide-binding domains. J Biol Chem 2003;278:1575–8. [DOI] [PubMed] [Google Scholar]
- 103.Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM. P-glycoprotein: from genomics to mechanism. Oncogene 2003;22:7468–85. [DOI] [PubMed] [Google Scholar]
- 104.Sauna ZE, Ambudkar SV. Characterization of the catalytic cycle of ATP hydrolysis by human P-glycoprotein. The two ATP hydrolysis events in a single catalytic cycle are kinetically similar but affect different functional outcomes. J Biol Chem 2001;276:11653–61. [DOI] [PubMed] [Google Scholar]
- 105.Ramachandra M, Ambudkar SV, Chen D, et al. Human P-glycoprotein exhibits reduced affinity for substrates during a catalytic transition state. Biochemistry 1998;37:5010–9. [DOI] [PubMed] [Google Scholar]
- 106.Sauna ZE, Ambudkar SV. Evidence for a requirement for ATP hydrolysis at two distinct steps during a single turnover of the catalytic cycle of human P-glycoprotein. Proc Natl Acad Sci U S A 2000;97:2515–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Senior AE, al-Shawi MK, Urbatsch IL. The catalytic cycle of P-glycoprotein. FEBS Lett 1995;377:285–9. [DOI] [PubMed] [Google Scholar]
- 108.Urbatsch IL, Sankaran B, Bhagat S, Senior AE. Both P-glycoprotein nucleotide-binding sites are catalytically active. J Biol Chem 1995;270:26956–61. [DOI] [PubMed] [Google Scholar]
- 109.Urbatsch IL, Sankaran B, Weber J, Senior AE. P-glycoprotein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J Biol Chem 1995;270:19383–90. [DOI] [PubMed] [Google Scholar]
- 110.Esser L, Zhou F, Pluchino KM, et al. Structures of the multidrug transporter P-glycoprotein reveal asymmetric ATP binding and the mechanism of polyspecificity. J Biol Chem 2017;292:446–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kim Y, Chen J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018;359:915–9. [DOI] [PubMed] [Google Scholar]
- 112.Nosol K, Romane K, Irobalieva RN, et al. Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. Proc Natl Acad Sci U S A 2020;117:26245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Alam A, Kowal J, Broude E, Roninson I, Locher KP. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019;363:753–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Alam A, Küng R, Kowal J, et al. Structure of a zosuquidar and UIC2-bound human-mouse chimeric ABCB1. Proc Natl Acad Sci U S A 2018;115:E1973–E82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Thonghin N, Collins RF, Barbieri A, Shafi T, Siebert A, Ford RC. Novel features in the structure of P-glycoprotein (ABCB1) in the post-hydrolytic state as determined at 7.9 Å resolution. BMC Struct Biol 2018;18:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nicklisch SC, Rees SD, McGrath AP, et al. Global marine pollutants inhibit P-glycoprotein: Environmental levels, inhibitory effects, and cocrystal structure. Sci Adv 2016;2:e1600001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Szewczyk P, Tao H, McGrath AP, et al. Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Crystallogr D Biol Crystallogr 2015;71:732–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ward AB, Szewczyk P, Grimard V, et al. Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc Natl Acad Sci U S A 2013;110:13386–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Le CA, Harvey DS, Aller SG. Structural definition of polyspecific compensatory ligand recognition by P-glycoprotein. IUCrJ 2020;7:663–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li J, Jaimes KF, Aller SG. Refined structures of mouse P-glycoprotein. Protein Sci 2014;23:34–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jin MS, Oldham ML, Zhang Q, Chen J. Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature 2012;490:566–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hrycyna CA, Ramachandra M, Ambudkar SV, et al. Mechanism of action of human P-glycoprotein ATPase activity. Photochemical cleavage during a catalytic transition state using orthovanadate reveals cross-talk between the two ATP sites. J Biol Chem 1998;273:16631–4. [DOI] [PubMed] [Google Scholar]
- 123.Dastvan R, Mishra S, Peskova YB, Nakamoto RK, McHaourab HS. Mechanism of allosteric modulation of P-glycoprotein by transport substrates and inhibitors. Science 2019;364:689–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jackson SM, Manolaridis I, Kowal J, et al. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat Struct Mol Biol 2018;25:333–40. [DOI] [PubMed] [Google Scholar]
- 125.Manolaridis I, Jackson SM, Taylor NMI, Kowal J, Stahlberg H, Locher KP. Cryo-EM structures of a human ABCG2 mutant trapped in ATP-bound and substrate-bound states. Nature 2018;563:426–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Taylor NMI, Manolaridis I, Jackson SM, Kowal J, Stahlberg H, Locher KP. Structure of the human multidrug transporter ABCG2. Nature 2017;546:504–9. [DOI] [PubMed] [Google Scholar]
- 127.Orlando BJ, Liao M. ABCG2 transports anticancer drugs via a closed-to-open switch. Nat Commun 2020;11:2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Litman T, Druley TE, Stein WD, Bates SE. From MDR to MXR: new understanding of multidrug resistance systems, their properties and clinical significance. Cell Mol Life Sci 2001;58:931–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Robey RW, To KK, Polgar O, et al. ABCG2: a perspective. Adv Drug Deliv Rev 2009;61:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chufan EE, Sim HM, Ambudkar SV. Molecular basis of the polyspecificity of P-glycoprotein (ABCB1): recent biochemical and structural studies. Adv Cancer Res 2015;125:71–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dey S, Ramachandra M, Pastan I, Gottesman MM, Ambudkar SV. Evidence for two nonidentical drug-interaction sites in the human P-glycoprotein. Proc Natl Acad Sci U S A 1997;94:10594–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Loo TW, Clarke DM. Determining the structure and mechanism of the human multidrug resistance P-glycoprotein using cysteine-scanning mutagenesis and thiol-modification techniques. Biochim Biophys Acta 1999;1461:315–25. [DOI] [PubMed] [Google Scholar]
- 133.Loo TW, Clarke DM. The transmembrane domains of the human multidrug resistance P-glycoprotein are sufficient to mediate drug binding and trafficking to the cell surface. J Biol Chem 1999;274:24759–65. [DOI] [PubMed] [Google Scholar]
- 134.Loo TW, Clarke DM. Defining the drug-binding site in the human multidrug resistance P-glycoprotein using a methanethiosulfonate analog of verapamil, MTS-verapamil. J Biol Chem 2001;276:14972–9. [DOI] [PubMed] [Google Scholar]
- 135.Loo TW, Clarke DM. Location of the rhodamine-binding site in the human multidrug resistance P-glycoprotein. J Biol Chem 2002;277:44332–8. [DOI] [PubMed] [Google Scholar]
- 136.Martin C, Berridge G, Higgins CF, Mistry P, Charlton P, Callaghan R. Communication between multiple drug binding sites on P-glycoprotein. Mol Pharmacol 2000;58:624–32. [DOI] [PubMed] [Google Scholar]
- 137.Mittra R, Pavy M, Subramanian N, et al. Location of contact residues in pharmacologically distinct drug binding sites on P-glycoprotein. Biochem Pharmacol 2017;123:19–28. [DOI] [PubMed] [Google Scholar]
- 138.Shapiro AB, Corder AB, Ling V. P-glycoprotein-mediated Hoechst 33342 transport out of the lipid bilayer. Eur J Biochem 1997;250:115–21. [DOI] [PubMed] [Google Scholar]
- 139.Shapiro AB, Fox K, Lam P, Ling V. Stimulation of P-glycoprotein-mediated drug transport by prazosin and progesterone. Evidence for a third drug-binding site. Eur J Biochem 1999;259:841–50. [DOI] [PubMed] [Google Scholar]
- 140.Bruggemann EP, Currier SJ, Gottesman MM, Pastan I. Characterization of the azidopine and vinblastine binding site of P-glycoprotein. J Biol Chem 1992;267:21020–6. [PubMed] [Google Scholar]
- 141.Bruggemann EP, Germann UA, Gottesman MM, Pastan I. Two different regions of P-glycoprotein are photoaffinity labeled by azidopine. J Biol Chem 1989;264:15483–8. [PubMed] [Google Scholar]
- 142.Sauna ZE, Andrus MB, Turner TM, Ambudkar SV. Biochemical basis of polyvalency as a strategy for enhancing the efficacy of P-glycoprotein (ABCB1) modulators: stipiamide homodimers separated with defined-length spacers reverse drug efflux with greater efficacy. Biochemistry 2004;43:2262–71. [DOI] [PubMed] [Google Scholar]
- 143.Chmielewski J, Hrycyna C. Tools for eradicating HIV in the brain: prodrug dimeric inhibitors of P-gp. Ther Deliv 2012;3:689–92. [DOI] [PubMed] [Google Scholar]
- 144.Emmert D, Campos CR, Ward D, et al. Reversible dimers of the atypical antipsychotic quetiapine inhibit p-glycoprotein-mediated efflux in vitro with increased binding affinity and in situ at the blood-brain barrier. ACS Chem Neurosci 2014;5:305–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kuriakose J, Hrycyna CA, Chmielewski J. Click chemistry-derived bivalent quinine inhibitors of P-glycoprotein-mediated cellular efflux. Bioorg Med Chem Lett 2012;22:4410–2. [DOI] [PubMed] [Google Scholar]
- 146.Namanja HA, Emmert D, Davis DA, et al. Toward eradicating HIV reservoirs in the brain: inhibiting P-glycoprotein at the blood-brain barrier with prodrug abacavir dimers. J Am Chem Soc 2012;134:2976–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Namanja HA, Emmert D, Hrycyna CA, Chmielewski J. Homodimers of the antiviral abacavir as modulators of P-glycoprotein transport in cell culture: Probing Tether Length. Medchemcomm 2013;4: 10.1039/C3MD00196B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Namanja HA, Emmert D, Pires MM, Hrycyna CA, Chmielewski J. Inhibition of human P-glycoprotein transport and substrate binding using a galantamine dimer. Biochem Biophys Res Commun 2009;388:672–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pires MM, Emmert D, Hrycyna CA, Chmielewski J. Inhibition of P-glycoprotein-mediated paclitaxel resistance by reversibly linked quinine homodimers. Mol Pharmacol 2009;75:92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Pires MM, Hrycyna CA, Chmielewski J. Bivalent probes of the human multidrug transporter P-glycoprotein. Biochemistry 2006;45:11695–702. [DOI] [PubMed] [Google Scholar]
- 151.Chan KF, Zhao Y, Burkett BA, Wong IL, Chow LM, Chan TH. Flavonoid dimers as bivalent modulators for P-glycoprotein-based multidrug resistance: synthetic apigenin homodimers linked with defined-length poly(ethylene glycol) spacers increase drug retention and enhance chemosensitivity in resistant cancer cells. J Med Chem 2006;49:6742–59. [DOI] [PubMed] [Google Scholar]
- 152.Chan KF, Zhao Y, Chow TW, et al. Flavonoid dimers as bivalent modulators for p-glycoprotein-based multidrug resistance: structure-activity relationships. ChemMedChem 2009;4:594–614. [DOI] [PubMed] [Google Scholar]
- 153.Cui J, Liu X, Chow LMC. Flavonoids as P-gp inhibitors: a systematic review of SARs. Curr Med Chem 2019;26:4799–831. [DOI] [PubMed] [Google Scholar]
- 154.Zhu X, Wong ILK, Chan KF, et al. Triazole bridged flavonoid dimers as potent, nontoxic, and highly selective breast cancer resistance protein (BCRP/ABCG2) inhibitors. J Med Chem 2019;62:8578–608. [DOI] [PubMed] [Google Scholar]
- 155.Amin ML. P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights 2013;7:27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Tamaki A, Ierano C, Szakacs G, Robey RW, Bates SE. The controversial role of ABC transporters in clinical oncology. Essays Biochem 2011;50:209–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Leonard GD, Polgar O, Bates SE. ABC transporters and inhibitors: new targets, new agents. Curr Opin Investig Drugs 2002;3:1652–9. [PubMed] [Google Scholar]
- 158.Pajic M, Iyer JK, Kersbergen A, et al. Moderate increase in Mdr1a/1b expression causes in vivo resistance to doxorubicin in a mouse model for hereditary breast cancer. Cancer Res 2009;69:6396–404. [DOI] [PubMed] [Google Scholar]
- 159.Stewart DJ. Tumor and host factors that may limit efficacy of chemotherapy in non-small cell and small cell lung cancer. Crit Rev Oncol Hematol 2010;75:173–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kannan P, Telu S, Shukla S, et al. The “specific” P-glycoprotein inhibitor Tariquidar is also a substrate and an inhibitor for breast cancer resistance protein (BCRP/ABCG2). ACS Chem Neurosci 2011;2:82–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kühnle M, Egger M, Müller C, et al. Potent and selective inhibitors of breast cancer resistance protein (ABCG2) derived from the p-glycoprotein (ABCB1) modulator tariquidar. J Med Chem 2009;52:1190–7. [DOI] [PubMed] [Google Scholar]
- 162.Karthikeyan S, Hoti SL. Development of fourth generation ABC inhibitors from natural products: a novel approach to overcome cancer multidrug resistance. Anticancer Agents Med Chem 2015;15:605–15. [DOI] [PubMed] [Google Scholar]
- 163.Lopes-Rodrigues V, Sousa E, Vasconcelos MH. Curcumin as a modulator of P-glycoprotein in cancer: challenges and perspectives. Pharmaceuticals (Basel) 2016;9:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kumar KK, Priyanka L, Gnananath K, Babu PR, Sujatha S. Pharmacokinetic drug interactions between apigenin, rutin and paclitaxel mediated by P-glycoprotein in rats. Eur J Drug Metab Pharmacokinet 2015;40:267–76. [DOI] [PubMed] [Google Scholar]
- 165.Kim MK, Kim Y, Choo H, Chong Y. Quercetin-glutamic acid conjugate with a non-hydrolysable linker; a novel scaffold for multidrug resistance reversal agents through inhibition of P-glycoprotein. Bioorg Med Chem 2017;25:1219–26. [DOI] [PubMed] [Google Scholar]
- 166.El-Readi MZ, Eid S, Abdelghany AA, Al-Amoudi HS, Efferth T, Wink M. Resveratrol mediated cancer cell apoptosis, and modulation of multidrug resistance proteins and metabolic enzymes. Phytomedicine 2019;55:269–81. [DOI] [PubMed] [Google Scholar]
- 167.Sun JH, Ye C, Bai EH, et al. Co-delivery nanoparticles of doxorubicin and chloroquine for improving the anti-cancer effect in vitro. Nanotechnology 2019;30:085101. [DOI] [PubMed] [Google Scholar]
- 168.Xue C, Wang C, Liu Q, et al. Targeting P-glycoprotein expression and cancer cell energy metabolism: combination of metformin and 2-deoxyglucose reverses the multidrug resistance of K562/Dox cells to doxorubicin. Tumour Biol 2016;37:8587–97. [DOI] [PubMed] [Google Scholar]
- 169.Tajima Y, Nakagawa H, Tamura A, et al. Nitensidine A, a guanidine alkaloid from Pterogyne nitens, is a novel substrate for human ABC transporter ABCB1. Phytomedicine 2014;21:323–32. [DOI] [PubMed] [Google Scholar]
- 170.Zhai Q, Xu Y, Li C, et al. Inhibition of breast cancer resistance protein (BCRP) by Ko143 can affect pharmacokinetics of enrofloxacin in exopalaemon carinicauda. Journal of Ocean University of China volume 2020;19:1124. [Google Scholar]
- 171.Woehlecke H, Osada H, Herrmann A, Lage H. Reversal of breast cancer resistance protein-mediated drug resistance by tryprostatin A. Int J Cancer 2003;107:721–8. [DOI] [PubMed] [Google Scholar]
- 172.Wang Y, Li Y, Shang D, Efferth T. Interactions between artemisinin derivatives and P-glycoprotein. Phytomedicine 2019;60:152998. [DOI] [PubMed] [Google Scholar]
- 173.Xu W, Wang X, Chen S, et al. Tetrandrine enhances glucocorticoid receptor translocation possibly via inhibition of P-glycoprotein in daunorubicin-resistant human T lymphoblastoid leukemia cells. Eur J Pharmacol 2020;881:173232. [DOI] [PubMed] [Google Scholar]
- 174.Lopez D, Martinez-Luis S. Marine natural products with P-glycoprotein inhibitor properties. Mar Drugs 2014;12:525–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lai JI, Tseng YJ, Chen MH, Huang CF, Chang PM. Clinical perspective of FDA approved drugs with P-glycoprotein inhibition activities for potential cancer therapeutics. Front Oncol 2020;10:561936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mollazadeh S, Sahebkar A, Hadizadeh F, Behravan J, Arabzadeh S. Structural and functional aspects of P-glycoprotein and its inhibitors. Life Sci 2018;214:118–23. [DOI] [PubMed] [Google Scholar]
- 177.Juvale K, Wiese M. Design of inhibitors of BCRP/ABCG2. Future Med Chem 2015;7:1521–7. [DOI] [PubMed] [Google Scholar]
- 178.Wiese M BCRP/ABCG2 inhibitors: a patent review (2009-present). Expert Opin Ther Pat 2015;25:1229–37. [DOI] [PubMed] [Google Scholar]
- 179.Leopoldo M, Nardulli P, Contino M, Leonetti F, Luurtsema G, Colabufo NA. An updated patent review on P-glycoprotein inhibitors (2011–2018). Expert Opin Ther Pat 2019;29:455–61. [DOI] [PubMed] [Google Scholar]
- 180.Toyoda Y, Takada T, Suzuki H. Inhibitors of human ABCG2: from technical background to recent updates with clinical implications. Front Pharmacol 2019;10:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Laiolo J, Tomašič T, Vera DMA, et al. Analogues of the Lignan Pinoresinol as Novel Lead Compounds for P-glycoprotein (P-gp) Inhibitors. ACS Med Chem Lett 2018;9:1186–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Sachs J, Kadioglu O, Weber A, et al. Selective inhibition of P-gp transporter by goniothalamin derivatives sensitizes resistant cancer cells to chemotherapy. J Nat Med 2019;73:226–35. [DOI] [PubMed] [Google Scholar]
- 183.Chae SW, Lee J, Park JH, Kwon Y, Na Y, Lee HJ. Intestinal P-glycoprotein inhibitors, benzoxanthone analogues. J Pharm Pharmacol 2018;70:234–41. [DOI] [PubMed] [Google Scholar]
- 184.Chen CY, Liu NY, Lin HC, Lee CY, Hung CC, Chang CS. Synthesis and bioevaluation of novel benzodipyranone derivatives as P-glycoprotein inhibitors for multidrug resistance reversal agents. Eur J Med Chem 2016;118:219–29. [DOI] [PubMed] [Google Scholar]
- 185.D Dinić J, Podolski-Renić A, Jovanović M, et al. Novel heat shock protein 90 inhibitors suppress P-glycoprotein activity and overcome multidrug resistance in cancer cells. Int J Mol Sci 2019;20:4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hemmer M, Krawczyk S, Simon I, Hilgeroth A. Discovery of substituted 1,4-dihydroquinolines as novel promising class of P-glycoprotein inhibitors: first structure-activity relationships and bioanalytical studies. Bioorg Med Chem Lett 2015;25:3005–8. [DOI] [PubMed] [Google Scholar]
- 187.Mollazadeh S, Sahebkar A, Kalalinia F, Behravan J, Hadizadeh F. Synthesis, in silico and in vitro studies of new 1,4-dihydropiridine derivatives for antitumor and P-glycoprotein inhibitory activity. Bioorg Chem 2019;91:103156. [DOI] [PubMed] [Google Scholar]
- 188.Ranjbar S, Khonkarn R, Moreno A, et al. 5-Oxo-hexahydroquinoline derivatives as modulators of P-gp, MRP1 and BCRP transporters to overcome multidrug resistance in cancer cells. Toxicol Appl Pharmacol 2019;362:136–49. [DOI] [PubMed] [Google Scholar]
- 189.Liu CP, Xie CY, Zhao JX, et al. Dysoxylactam A: a macrocyclolipopeptide reverses P-glycoprotein-mediated multidrug resistance in cancer cells. J Am Chem Soc 2019;141:6812–6. [DOI] [PubMed] [Google Scholar]
- 190.Singh S, Prasad NR, Chufan EE, et al. Design and synthesis of human ABCB1 (P-glycoprotein) inhibitors by peptide coupling of diverse chemical scaffolds on carboxyl and amino termini of (S)-valine-derived thiazole amino acid. J Med Chem 2014;57:4058–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.de Ravel MR, Alameh G, Melikian M, et al. Synthesis of new steroidal inhibitors of P-glycoprotein-mediated multidrug resistance and biological evaluation on K562/R7 erythroleukemia cells. J Med Chem 2015;58:1832–45. [DOI] [PubMed] [Google Scholar]
- 192.Rocheblave L, de Ravel MR, Monniot E, et al. Deoxycholic acid derivatives as inhibitors of P-glycoprotein-mediated multidrug efflux. Steroids 2016;116:5–12. [DOI] [PubMed] [Google Scholar]
- 193.Zeino M, Paulsen MS, Zehl M, Urban E, Kopp B, Efferth T. Identification of new P-glycoprotein inhibitors derived from cardiotonic steroids. Biochem Pharmacol 2015;93:11–24. [DOI] [PubMed] [Google Scholar]
- 194.Bonandi E, Christodoulou MS, Fumagalli G, Perdicchia D, Rastelli G, Passarella D. The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discov Today 2017;22:1572–81. [DOI] [PubMed] [Google Scholar]
- 195.Jekerle V, Klinkhammer W, Scollard DA, et al. In vitro and in vivo evaluation of WK-X-34, a novel inhibitor of P-glycoprotein and BCRP, using radio imaging techniques. Int J Cancer 2006;119:414–22. [DOI] [PubMed] [Google Scholar]
- 196.Mistry P, Stewart AJ, Dangerfield W, et al. In vitro and in vivo reversal of P-glycoprotein-mediated multidrug resistance by a novel potent modulator, XR9576. Cancer Res 2001;61:749–58. [PubMed] [Google Scholar]
- 197.Chan KF, Wong IL, Kan JW, Yan CS, Chow LM, Chan TH. Amine linked flavonoid dimers as modulators for P-glycoprotein-based multidrug resistance: structure-activity relationship and mechanism of modulation. J Med Chem 2012;55:1999–2014. [DOI] [PubMed] [Google Scholar]
- 198.Wang JS, Zhu HJ, Markowitz JS, Donovan JL, DeVane CL. Evaluation of antipsychotic drugs as inhibitors of multidrug resistance transporter P-glycoprotein. Psychopharmacology (Berl) 2006;187:415–23. [DOI] [PubMed] [Google Scholar]
- 199.Munic V, Kelneric Z, Mikac L, Erakovic Haber V. Differences in assessment of macrolide interaction with human MDR1 (ABCB1, P-gp) using rhodamine-123 efflux, ATPase activity and cellular accumulation assays. Eur J Pharm Sci 2010;41:86–95. [DOI] [PubMed] [Google Scholar]
- 200.Domvri K, Zarogoulidis K, Zogas N, et al. Potential synergistic effect of phosphodiesterase inhibitors with chemotherapy in lung cancer. J Cancer 2017;8:3648–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Krchniakova M, Skoda J, Neradil J, Chlapek P, Veselska R. Repurposing tyrosine kinase inhibitors to overcome multidrug resistance in cancer: a focus on transporters and lysosomal sequestration. Int J Mol Sci 2020;21:3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Agarwal S, Sane R, Gallardo JL, Ohlfest JR, Elmquist WF. Distribution of gefitinib to the brain is limited by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. J Pharmacol Exp Ther 2010;334:147–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Chen Y, Agarwal S, Shaik NM, Chen C, Yang Z, Elmquist WF. P-glycoprotein and breast cancer resistance protein influence brain distribution of dasatinib. J Pharmacol Exp Ther 2009;330:956–63. [DOI] [PubMed] [Google Scholar]
- 204.Shukla S, Sauna ZE, Ambudkar SV. Evidence for the interaction of imatinib at the transport-substrate site(s) of the multidrug-resistance-linked ABC drug transporters ABCB1 (P-glycoprotein) and ABCG2. Leukemia 2008;22:445–7. [DOI] [PubMed] [Google Scholar]
- 205.Traxl A, Wanek T, Mairinger S, et al. Breast cancer resistance protein and P-glycoprotein influence in vivo disposition of 11C-Erlotinib. J Nucl Med 2015;56:1930–6. [DOI] [PubMed] [Google Scholar]
- 206.Hoppe S, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Breast cancer resistance protein (BCRP/ABCG2) and P-glycoprotein (P-gp/ABCB1) transport afatinib and restrict its oral availability and brain accumulation. Pharmacol Res 2017;120:43–50. [DOI] [PubMed] [Google Scholar]
- 207.Zhou ZY, Wan LL, Yang QJ, et al. Nilotinib reverses ABCB1/P-glycoprotein-mediated multidrug resistance but increases cardiotoxicity of doxorubicin in a MDR xenograft model. Toxicol Lett 2016;259:124–32. [DOI] [PubMed] [Google Scholar]
- 208.Durmus S, Sparidans RW, van Esch A, Wagenaar E, Beijnen JH, Schinkel AH. Breast cancer resistance protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1) restrict oral availability and brain accumulation of the PARP inhibitor rucaparib (AG-014699). Pharm Res 2015;32:37–46. [DOI] [PubMed] [Google Scholar]
- 209.Vaidyanathan A, Sawers L, Gannon AL, et al. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br J Cancer 2016;115:431–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.de Gooijer MC, Zhang P, Thota N, et al. P-glycoprotein and breast cancer resistance protein restrict the brain penetration of the CDK4/6 inhibitor palbociclib. Invest New Drugs 2015;33:1012–9. [DOI] [PubMed] [Google Scholar]
- 211.Wu T, Chen Z, To KKW, et al. Effect of abemaciclib (LY2835219) on enhancement of chemotherapeutic agents in ABCB1 and ABCG2 overexpressing cells in vitro and in vivo. Biochem Pharmacol 2017;124:29–42. [DOI] [PubMed] [Google Scholar]
- 212.Gallo JM, Li S, Guo P, Reed K, Ma J. The effect of P-glycoprotein on paclitaxel brain and brain tumor distribution in mice. Cancer Res 2003;63:5114–7. [PubMed] [Google Scholar]
- 213.Tang SC, Kort A, Cheung KL, et al. P-glycoprotein, CYP3A, and plasma carboxylesterase determine brain disposition and oral availability of the novel taxane cabazitaxel (Jevtana) in mice. Mol Pharm 2015;12:3714–23. [DOI] [PubMed] [Google Scholar]
- 214.Fisher M, Abramov M, Van Aerschot A, et al. Inhibition of MDR1 expression with altritol-modified siRNAs. Nucleic Acids Res 2007;35:1064–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Li H, Zhou S, Li T, et al. Suppression of BCRP expression and restoration of sensitivity to chemotherapy in multidrug-resistant HCC cell line HEPG2/ADM by RNA interference. Hepatogastroenterology 2012;59:2238–42. [DOI] [PubMed] [Google Scholar]
- 216.Perez J, Bardin C, Rigal C, Anthony B, Rousseau R, Dutour A. Anti-MDR1 siRNA restores chemosensitivity in chemoresistant breast carcinoma and osteosarcoma cell lines. Anticancer Res 2011;31:2813–20. [PubMed] [Google Scholar]
- 217.Hong L, Han Y, Zhang H, et al. The prognostic and chemotherapeutic value of miR-296 in esophageal squamous cell carcinoma. Ann Surg 2010;251:1056–63. [DOI] [PubMed] [Google Scholar]
- 218.Bao L, Hazari S, Mehra S, Kaushal D, Moroz K, Dash S. Increased expression of P-glycoprotein and doxorubicin chemoresistance of metastatic breast cancer is regulated by miR-298. Am J Pathol 2012;180:2490–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Xie Y, Shao Y, Deng X, Wang M, Chen Y. MicroRNA-298 reverses multidrug resistance to antiepileptic drugs by suppressing MDR1/P-gp expression in vitro. Front Neurosci 2018;12:602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Nadali F, Pourfathollah AA, Alimoghaddam K, et al. Multidrug resistance inhibition by antisense oligonucleotide against MDR1/mRNA in P-glycoprotein expressing leukemic cells. Hematology 2007;12:393–401. [DOI] [PubMed] [Google Scholar]
- 221.Rittierodt M, Tschernig T, Harada K. Modulation of multidrug-resistance-associated P-glycoprotein in human U-87 MG and HUV-ECC cells with antisense oligodeoxynucleotides to MDR1 mRNA. Pathobiology 2004;71:123–8. [DOI] [PubMed] [Google Scholar]
- 222.White MK, Khalili K. CRISPR/Cas9 and cancer targets: future possibilities and present challenges. Oncotarget 2016;7:12305–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Norouzi-Barough L, Sarookhani M, Salehi R, Sharifi M, Moghbelinejad S. CRISPR/Cas9, a new approach to successful knockdown of ABCB1/P-glycoprotein and reversal of chemosensitivity in human epithelial ovarian cancer cell line. Iran J Basic Med Sci 2018;21:181–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Simoff I, Karlgren M, Backlund M, et al. Complete Knockout of Endogenous Mdr1 (Abcb1) in MDCK Cells by CRISPR-Cas9. J Pharm Sci 2016;105:1017–21. [DOI] [PubMed] [Google Scholar]
- 225.Broxterman HJ, Kuiper CM, Schuurhuis GJ, Tsuruo T, Pinedo HM, Lankelma J. Increase of daunorubicin and vincristine accumulation in multidrug resistant human ovarian carcinoma cells by a monoclonal antibody reacting with P-glycoprotein. Biochem Pharmacol 1988;37:2389–93. [DOI] [PubMed] [Google Scholar]
- 226.Pearson JW, Fogler WE, Volker K, et al. In vivo administration of MRK-16 monoclonal antibody reverses drug resistance in a human colon cancer xenograft expressin the MDR1 cDNA. J Natl Cancer Inst 1991;83:1386–91. [DOI] [PubMed] [Google Scholar]
- 227.Tsuruo T, Hamada H, Sato S, Heike Y. Inhibition of multidrug-resistant human tumor growth in athymic mice by anti-P-glycoprotein monoclonal antibodies. Jpn J Cancer Res 1989;80:627–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Hamada H, Tsuruo T. Functional role for the 170- to 180-kDa glycoprotein specific to drug-resistant tumor cells as revealed by monoclonal antibodies. Proc Natl Acad Sci U S A 1986;83:7785–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Naito M, Tsuge H, Kuroko C, et al. Enhancement of cellular accumulation of cyclosporine by Anti-P-Glycoprotein monoclonal antibody MRK-16 and synergistic modulation of multidrug resistance. J Nat Cancer Inst 1993;85:311–6. [DOI] [PubMed] [Google Scholar]
- 230.Pearson JW, Fogler WE, Volker K, et al. Reversal of drug resistance in a human colon cancer xenograft expressing MDR1 complementary DNA by in vivo administration of MRK-16 monoclonal antibody. J Natl Cancer Inst 1991;83:1386–91. [DOI] [PubMed] [Google Scholar]
- 231.Mechetner EB, Roninson IB. Efficient inhibition of P-glycoprotein-mediated multidrug resistance with a monoclonal antibody. Proc Natl Acad Sci U S A 1992;89:5824–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Goda K, Fenyvesi F, Bacsó Z, et al. Complete inhibition of P-glycoprotein by simultaneous treatment with a distinct class of modulators and the UIC2 monoclonal antibody. J Pharmacol Exp Ther 2007;320:81–8. [DOI] [PubMed] [Google Scholar]
- 233.Druley TE, Stein WD, Roninson IB. Analysis of MDR1 P-glycoprotein conformational changes in permeabilized cells using differential immunoreactivity. Biochemistry 2001;40:4312–22. [DOI] [PubMed] [Google Scholar]
- 234.Mechetner EB, Schott B, Morse BS, et al. P-glycoprotein function involves conformational transitions detectable by differential immunoreactivity. Proc Natl Acad Sci U S A 1997;94:12908–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Ritchie TK, Kwon H, Atkins WM. Conformational analysis of human ATP-binding cassette transporter ABCB1 in lipid nanodiscs and inhibition by the antibodies MRK16 and UIC2. J Biol Chem 2011;286:39489–96. [DOI] [PMC free article] [PubMed] [Google Scholar]