

Abstract
Background:
Prostatic cancers include a diverse microenvironment of tumor cells, cancer-associated fibroblasts, and immune components. This tumor microenvironment (TME) is a known driving force of tumor survival after treatment, but the standard-of-care tissue freezing or fixation in pathology practice limit the use of available approaches/tools to study the TME’s functionality in tumor resistance. Thus, there is a need for approaches that satisfy both clinical and laboratory endpoints for TME study. Here we present methods for clinical case identification, tissue processing, and analytical workflow that are compatible with standard histopathology while enabling molecular and functional interrogation of prostate TME components.
Methods:
We first performed a small retrospective review to identify cases where submission of alternate prostate tissue slices and a parallel live tissue processing protocol complement traditional histopathology and enable viable multi-compartment analysis of the TME. Then, we tested its compatibility with commonly employed methods to study the microenvironment including quantification of components both in-situ and after tissue dissociation. We also evaluated tissue digestion conditions and cell isolation techniques to aid various molecular and functional endpoints.
Results:
We identified Gleason Grade Group 3+ clinical cases where tumor volume was sufficient to allow slicing of unfixed tissue and distribution of alternating tissue slices to standard-of-care histopathology and viable multi-modal TME analyses. No single method was found that preserved cellular sub-types for all downstream readouts; instead, tissues were further divided so techniques could be catered to each endpoint. For instance, we show that incorporating the protease dispase into tissue dissociation improves viability for culture and functional analyses but hinders immune cell analysis by flow cytometry. We also found that FACS provides highly pure cell populations for qRT-PCR and RNA-seq while isolation using antibody-labeled paramagnetic particles facilitated functional co-culture experiments.
Conclusions:
The identification of candidate cases and use of these techniques enables translational research and the development of molecular and functional assays to facilitate prostate TME study without compromising standard-of-care histopathological diagnosis. This allows bridging clinical histopathology and further interrogation of the prostate TME and promises to advance our understanding of tumor biology and unveil new predictive and prognostic markers of prostate cancer progression.
Keywords: tumor microenvironment, prostate, tissue processing, functional analyses, molecular analyses
1.0. Introduction
In spite of advances in diagnosis and treatment, prostate adenocarcinoma remains the second leading cause of cancer-related deaths among men (1). Men who present with locally advanced disease are still at high risk for recurrence and mortality. Disease-free survival at 5 years for men with Gleason 8+ disease is only 47%, markedly lower than 93% for those diagnosed with Gleason 6 disease (2). This emphasizes the need for treatment advancements in high-risk prostatic disease. A major hurdle that impedes the success of these treatments is the lack of good prognostic and predictive metrics/markers to guide treatment. For example, the Gleason score is an accepted metric for determining prostate cancer prognosis, but its positive predictive value for estimating the risk of disease-related death can be as low as 17% (3). This highlights the need to identify better predictive and prognostic metrics/markers to guide clinical treatment.
The tumor microenvironment (TME) has been identified as a driving force of tumor survival after treatment but the standard tissue processing methods in pathology practice limit the use of available approaches/tools to study the tumor microenvironment functionally and molecularly (4). Prostatic cancer lesions are multifaceted, complex, and consist of many cell types such as tumor cells, cancer-associated fibroblasts (CAFs), smooth muscle cells, vasculature, and infiltrating immune types. Functional assays have shown that CAFs increase tumor progression through inducing cancer cell proliferation and the release of pro-survival signals (5–7). Flow cytometry studies have found that inflammatory immune cells can modulate prostate cancer progression through the generation of reactive oxygen species, tumor-promoting cytokines, and the suppression of anti-tumor immune response (8). However, clinical translation of these studies is hampered because the standard approach to handling tissues post-prostatectomy is immediate fixation or freezing of the specimen. Even then, typically only about 60% of the prostate sample is embedded for sectioning, while the remaining tissue is discarded in two weeks after the final pathology report is signed out in many clinical centers (9, 10). To improve translational research in prostate cancer, there is a need to develop tissue procuring and processing techniques that better utilize patient tissue.
The complexity and diversity within the TME pose another hurdle to studying each of its components using a variety of desired endpoints. Mechanical and enzymatic tissue digestion has been employed to isolate various individual cell components from whole tissue for further study; however, these protocols are plagued by low cell viability and cleavage of surface proteins typically used to identify cellular subtypes (11, 12). The many cell types in the TME have diverse properties and means of attachment to the extracellular matrix which makes it difficult to develop a system for investigating each microenvironmental component without harming another component. Consequently, the vast proportion of tissue processing, dissociation, and cell separation protocols have been designed for the isolation and study of only a single cell type from a large tissue sample (13, 14). While some approaches have shown promise in defining digestion conditions to interrogate multiple cell types of the microenvironment, there is an unmet need to better characterize the compatibility of these conditions for downstream molecular and functional readouts (15). For example, differential sedimentation is a simple, low-cost method for separating prostate epithelial and stromal populations but is greatly limited by the number of cellular subtypes it can separate (16). Flow activated cell sorting (FACS) expands the ability to simultaneously isolate multiple cell types and is commonly used to identify and separate specific cells of interest (17). However, FACS is costly and solely limited to phenotypic analysis that is hampered by high background and autofluorescence from debris in solid tumor processing, as well as low cell viability. Magnetic isolation using PMPs is quicker, cheaper, and preserves better cellular viability; however, it requires serial isolations to obtain multiple cellular subtypes and isolation is based on either positive or negative surface protein expression, rather than a defined threshold as with FACS. Overall, the poor characterization of digestion and cellular isolation techniques and their effects on various prostate cellular components and downstream endpoints has hampered the ability to robustly interrogate the molecular and functional study of the prostate tumor microenvironment.
Here we present an integrated “histology and TME friendly” process for clinical case identification, tissue procuring, processing and analytical workflow that is compatible with the standard clinical pathology practice and enables the investigation of the prostate TME and its distinct cellular components. This technique utilizes patient-specific 3D printed prostate molds to stabilize and evenly slice the prostate to procure fresh tissue. We then optimized tissue dissociation conditions for culture and cytometric needs, maintained viable microenvironment components, and isolated desired cellular subtypes for molecular and functional characterization. We found that not one protocol was able to satisfy each aspect of TME analysis, and that protocols should be tailored depending on the experiment and desired endpoints. Application of these techniques bridges clinical pathology with TME molecular and functional characterization and promises to advance our understanding of the role each cellular compartment plays in prostate cancer.
2.0. Materials and Methods
2.1. Prostate Mold Development and Tissue Slicing:
All prostate tissue procurement for these experiments was performed in compliance with the laws and institutional guidelines as approved by the Institutional Review Board committee from the University of Wisconsin-Madison. Prostate tissue was obtained from patients undergoing radical prostatectomy with informed consent through the Translational Science BioCore Biobank at the Carbone Cancer Center at the University of Wisconsin-Madison. Depending on the protocol, the unfixed gland was sectioned free hand (only the first two slices) or later a 3-dimensional prostate mold was used to slice the gland as previously described (18, 19). Briefly, patients underwent preoperative MRI scans to obtain T2W images, three planes (sagittal, axial, and coronal); then patient-specific prostate molds were modeled in Analyze (AnalyzeDirect, Inc.) and 3-dimensionally printed. Post-surgery, unfixed prostate tissue was then spatial oriented, inked, placed in 3-dimensional molds and manually sliced into 2.5mm sections using a tissue slicer blade (Thomas Scientific) along the slits of the mold; first, last and odd numbered slices were retained for standard histopathology and even numbered slices were released for research. Areas identified by radiographic imaging as tumor foci were then localized and targeted for further analysis.
2.2. Live Tissue Sectioning, Staining, Imaging, and Analysis:
Tissue sectioning was done as previously described (20). Briefly, a small portion (10%) of each tissue sample (acquired in accordance with the UW-Madison Institutional Review Board) was superglued to the cutting stage of a VF-300 Compresstome (Precisionary Instruments, Greenville, NC, USA). Then, tissue was covered in low melt agarose, immersed in sterile media, and sectioned to 100um thickness. Before imaging, slices were stained in screw-top vials with fluorescent dyes. Slices were stained as previously described with viability markers (Cellometer ViaStain AOPI Staining Solution) and fluorescent antibodies (Hoechst, CD45 – FITC, EpCAM (epithelial cell adhesion molecule) – PE, CD49a – Alexa Fluor™ 647) (20). Reagent specifics are listed in Supplementary Table S1. Viability and tissue cellular composition was quantified via Fiji automated image analysis. Area fraction (percent of total tissue area covered) of each cell type was used to estimate the percentage of live and dead cells, as well as the percentage of epithelial, stromal, and immune cell types.
2.3. Tissue Digestion and Cell Staining:
Remaining tissue not used for tissue slice analysis (90%) was used for tissue digestion and cell isolation. Tissue digestion enzymes (Worthington Biochemical, Lakewood, NJ USA) and conditions are described in the table seen in Figure 3A. All digestion buffers were made in 1mL Hepatocyte Wash Buffer (ThermoFisher Scientific) and digested in 5mL round bottom tubes (Corning). The digestion reaction was neutralized by adding equal volume (1mL) of 10% fetal bovine serum diluted in hepatocyte wash buffer (VWR, Radnor, PA USA). Samples were then strained through a 100 uM tube top filter (Corning) and washed with 500 uL 1X PBS. Cell suspensions were centrifuged at 1000 RPM (200xG) for 3 minutes and re-suspended in 300 uL MACS Buffer for cell isolation. This re-suspended sample is referred to as the “raw digestate”. 20 uL of the raw digestate was removed for viability and cellular composition analysis. Cells were stained with viability markers and fluorescent antibodies as described above and reagent specifics can be found in Supplementary Table S1. The remaining 280uL of raw digestate was used for cell isolation.
2.4. Cell Isolation:
All magnetic cell isolations were performed on an Extractman (Gilson, Middleton, WI USA) using the commercial paramagnetic particle (PMP) isolation kits FlowComp Dynabead Flexi Kit (ThermoFisher) or SeraMag SpeedBeads Blocked Streptavidin (GE Healthcare, Madison, WI USA). Biotinylation of EpCAM (R&D Systems, Minneapolis, MN USA) and integrin alpha 1/CD49a (R&D Systems) antibodies was performed using the standard DSB-x Biotin Protein Labeling Kit (Molecular Probes, Eugene, OR USA) protocol.
To prepare commercial beads for isolation, PMPs were bound to DSB-x labeled antibody (EpCAM or CD49a). For each isolation, 10 ul of beads were washed 3 times with 25 uL of 0.1% Tween20 diluted in PBS. Beads were then re-suspended in 25 uL of 0.1% Tween20. 1.5 uL of DSB-x labeled antibody was added to paramagnetic bead suspension and shaken (1000 rpm) at room temperature for 30 minutes. Labeled beads were washed 3 times with 25 uL of 0.1% BSA diluted in PBS. Finally, beads were re-suspended in 25 uL of 0.1% BSA. Reagent specifics for Cell Isolation are listed in Supplementary Table S2.
Cell type specific isolations were performed on the raw digestate in serial isolation format, where multiple cell types of interest are removed sequentially one after another. For isolations, 25 uL of prepared EpCAM-labeled beads were added to an aliquot of the raw digestate cell suspension and tumbled at 4°C for 20 minutes. Gilson Extractman was then used to remove bead bound cells from the cell suspension and into the desired buffer: MACS buffer for antibody staining and imaging, lysis buffer for mRNA extraction, or FlowComp release buffer for culture or downstream flow cytometry analysis. These processes were then repeated using CD49a-coated beads for the capture of stromal cells.
The remaining cell suspension after sequential isolation of epithelium and stromal cells is referred to as the “remaining input.” This negatively selected suspension was used to analyze the immune cell population from the sample. The remaining input was centrifuged at 1100 rpm (200xg) for 3 minutes and resuspended into desired buffer: MACS buffer for antibody staining and flow cytometry or lysis buffer for mRNA extraction.
FACS live cell sorting -
Single cell preparations were stained with Ghost Dye™ Violet 510 fixable Live/Dead stain, CD45, EpCAM, Cd49a and Fc-blocker antibodies. Reagent specifics can be found in Supplementary Table S3. Samples were acquired on a 5-laser BD FACS Aria SORP sorter. Debris and dead cells were excluded, and target cell populations were sorted from within the live/singlet (for CD45+) or live/singlet/CD45- (for epithelial and stromal cells) gate. To achieve optimal precision of deflection, yield and purity, sort precision was set at 4-way purity mode (Yield Mask 0, Purity Mask set at maximum 32, Phase Mask 0). The cells were sorted into RLT Plus lysis buffer, frozen immediately and stored at −80C until RNA isolation.
2.5. Cell Suspension and Isolation Analysis:
Fluorescent Microscopy Analysis -
The automated image-processing program imageJ, was used to analyze fluorescent microscopy images and to identify cell populations. First, a rolling ball background subtraction was conducted. Cell nuclei, stained by Hoechst (ThermoFisher Scientific), were identified and an ellipse with radius 8um was drawn around the nuclei to create a ROI. Fluorescent intensities of antibodies (CD45-FITC, EpCAM-PE, and CD49a-Alexa Fluor™ 647) at identified ROIs were then quantified and used to identify different cell types.
Flow Cytometry Analysis -
Multi-parameter flow cytometry was utilized to analyze surface expression of prostate-resident and prostate-infiltrating cellular subsets. Single cell preparations were stained with Ghost Dye™ Violet 510 fixable Live/Dead stain (Tonbo Biosciences) and CD45, CD11b, EpCAM, Cd49a, CD4 and CD8 antibodies. Samples were acquired on a 5-laser BD LSR Fortessa instrument and data was analyzed by the FlowJo software v9.9 (BD Bioscience). Gating strategy included exclusion of dead cells, debris and aggregates,Fluorescent Minus One and internal negative controls. Reagent specifics can be found in Supplementary Table S4.
2.6. qRT-PCR and RNA Sequencing:
mRNA was extracted using the standard Dynabead mRNA Direct Purification Kit (ThermoFisher) protocol. Reverse transcription was performed using the High-Capacity cDNA Reverse Transcription Kit (ThermoFisher) standard protocol. The cycle threshold value was used to evaluate relative gene expression of epithelial markers EpCAM [Hs00901885_m1] and cytokeratin 8 [Hs01595539_g1], immune marker CD45 [Hs04189704_m1], and stromal fibroblast markers integrin 1alpha (CD49a) [Hs00235006_m1], alpha-smooth muscle actin [Hs00426835_g1], and vimentin [Hs00958111_m1]. Housekeeping genes GAPDH [hs99999905_m1] and RPLP0 (hs00420895_gH) were used for normalization.
cDNA synthesis, library preparation, and mRNA sequencing were performed by the UW-Madison Biotechnology Center. cDNA synthesis was performed using the standard SmartSeq v4 Ultra-Input RNA kit protocol (Takara Bio USA, Mountain View, CA). An 18-cycle full length ds cDNA amplification was performed via LD-PCR. Amplified cDNA was purified using standard AMPure XP beads protocol (Beckman Coulter, Brea, CA). Nextera XT DNA library preparation (Illumina, San Diego, CA) was performed, and samples were submitted for Agilent QC and sequencing. Volcano plots of RNA sequencing plots were made in RStudio using Bonferroni adjusted p-values.
2.7. Cell Culture:
Isolated cells in FlowComp release buffer were centrifuged and resuspended in media. Fibroblasts were cultured in Fibroblast Medium (ScienCell, Carlsbad, CA USA, Cat. 2301) and epithelial cells were grown in Prostate Epithelial Cell Medium (ScienCell, Cat. 4411). Cells were plated at 25,000 cells per well in a standard 96 well TC treated plate (Corning) and cultured at 37C with 5% CO2. After 24 hours, media was removed and replaced. Media changes were then performed every 3 days.
2.8. Dose-Response Experiments:
Patient-derived epithelial cells were plated at 10,000 cells per well in a 96-well plate (Corning); 5,000 fibroblasts were added in a transwell insert for co-culture conditions. Cells were cultured at 37°C with 5% CO2 for 24 hours. Then, a range of docetaxel doses (Sigma Aldrich, Darmstadt, Germany) were added to the media conditions and cells were cultured at 37 C⁰ with 5% CO2 for 72 hours. Following 72-hour culture period, epithelial cell viability was measured on a PHERAstar FSX plate reader (BMG Labtech, Ortenberg, Germany) using RealTime-Glo Luminescent Viability Assay (Promega, Madison, WI USA).
3.0. Results
3.1. Histopathology identifies prostatectomy tissues for translational research -
Traditional histopathology uses Gleason grading to identify and give prognostic information for prostate cancer cases. In many clinical centers, this is typically achieved via fixation of the entire prostate gland following prostatectomy for at least a few hours to overnight followed by submission for histological analysis where roughly 60% of the specimen is processed for clinical endpoints, and the remaining tissue is discarded in two weeks after the final pathology report is signed out. We performed a retrospective analysis of 88 prostate adenocarcinoma cases to identify cases where hospital pathology disposes of excess prostate tissue that could otherwise be used for research. We found that in 11% of Gleason 3+3 cases (n = 26), pathologists needed excess tissue for further analysis (Figure 1E). However, in 97% of Gleason 3+4 cases (n = 34), pathologists did not need excess process tissue for clinical analysis. Similarly, in all Gleason Grade Group 3+ (Gleason score 4+3 and 8+) cases (n =26), pathologists did not use excess prostate tissue slices and instead disposed of the tissue (Figure 1E).
Figure 1: Live tissue slicing enables standard of care histopathology as well as live cell functional and molecular readouts in a wet lab.
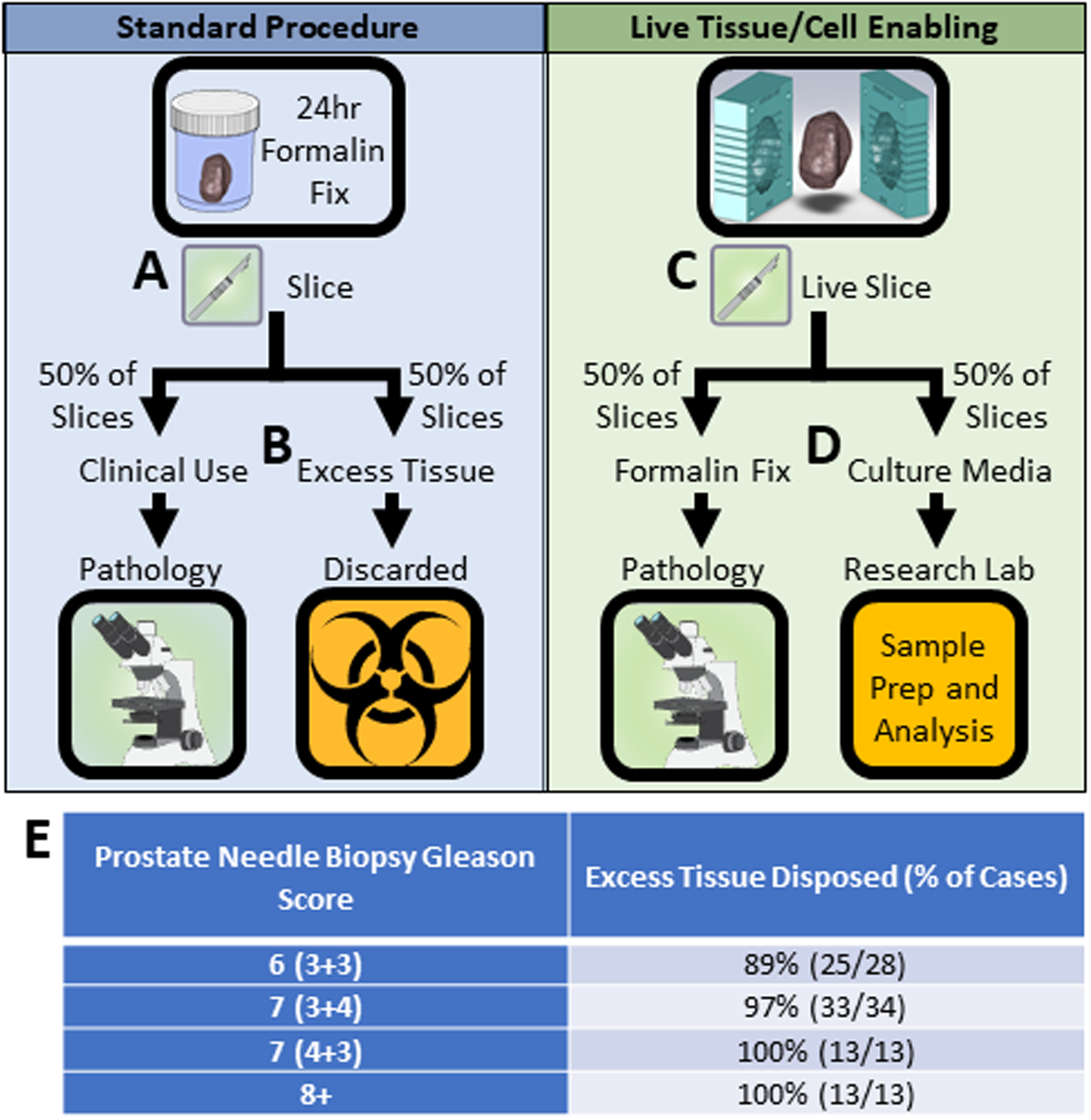
(A) In standard hospital procedure, prostate tissue is fixed in formalin for 24 hours then cut into 4mm slices. (B) Tissue needed for standard of care histology (~50% of tissue slices) is sent to hospital pathology, excess tissue is discarded. (C) In live tissue/cell enabled protocols, prostate tissue is placed in a slicing mold and cut into 4mm slices before formalin fixation. (D) Tissue needed for standard of care histology (~50% of tissue slices) is formalin fixed and sent to hospital pathology, excess tissue is placed in live culture media and remains viable for sample processing and analysis at the research wet lab. (see Figure 2). (E) Prostatectomy cases (n=88) at University of Wisconsin – Hospital and Clinics in which excess prostate tissue was disposed of after clinical analyses were complete.
We next sought to develop a method that could meet the needs for both standard clinical practice and translational wet lab research. Fresh, unfixed prostate tissue was immediately brought to Surgical Pathology after surgical resection. The apex and base margins of the prostate were manually sliced and the remaining tissue was placed in patient-specific 3-dimensional molds designed from an axial-oblique T2 MRI sequence of the prostate (18, 19) to help stabilize and maintain even slicing. The prostate was sliced in the 3D-mold as described in section 2.1. and alternate slices were designated for standard of care histopathology or fresh tissue digestion and downstream cell isolation, culture, and transcriptomics. This workflow is depicted in Figure 2 and discussed below.
Figure 2: Overview of tissue processing workflow.
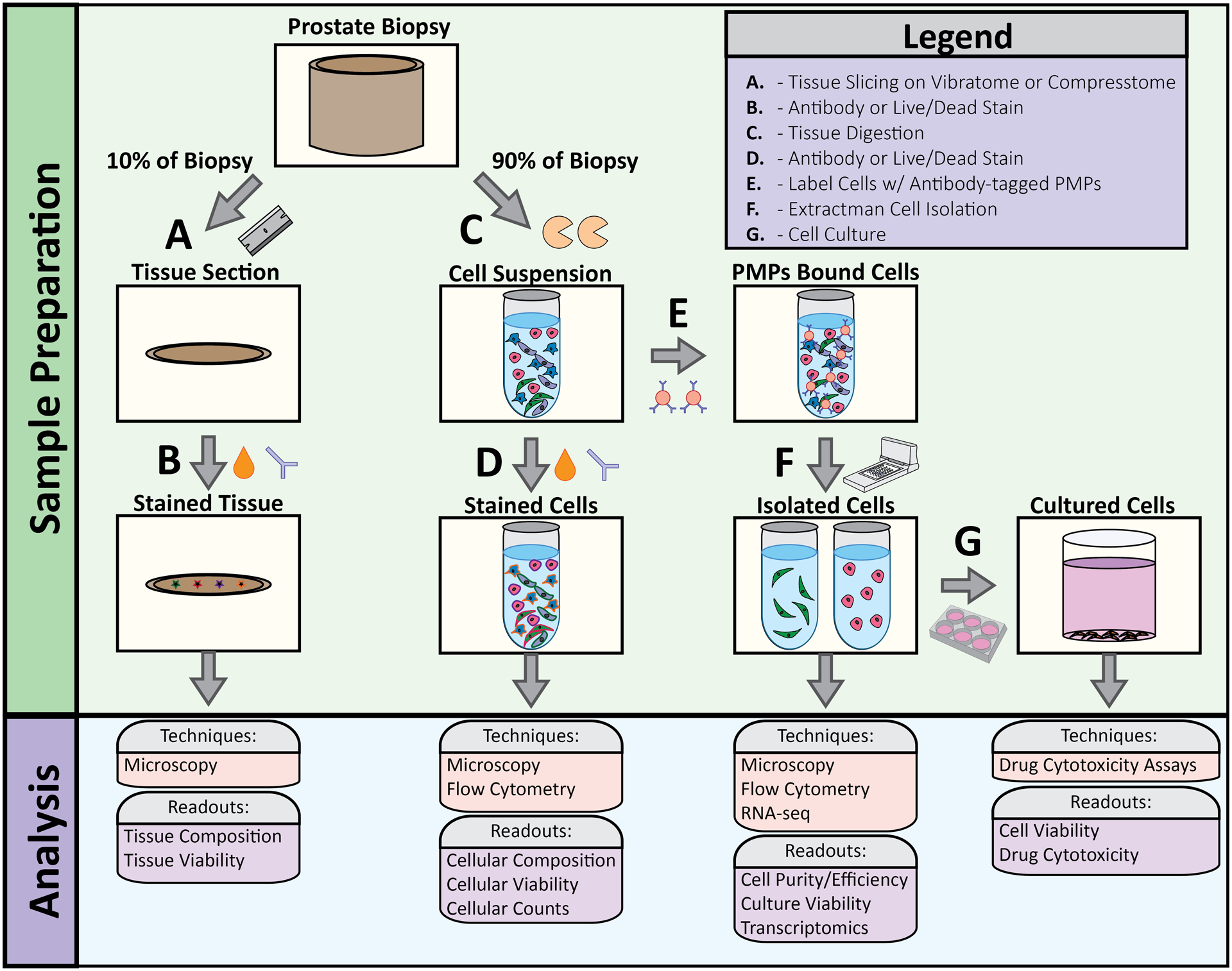
(A) 10% of the tissue biopsy was removed with a hand razor then sliced to 75um sections using a VF-300 Compresstome. (B) 75um tissue sections were stained with viability markers and cell-specific antibodies to evaluate tissue viability and cellular composition. (C) 90% of the tissue biopsy was used for enzymatic digestion to obtain single cell population. (D) Cell suspensions were stained with viability markers and cell-specific antibodies to evaluate cell viability and cell type. (E) Antibody-bound PMPs were added to cell suspension to bind specific cell types. (F) Extractman was used to isolate PMP-bound cell cells for flow analysis, RNA-seq, (G) cell culture, and cytotoxicity assays.
3.2. Viability and antibody stains characterize in-situ cell populations -
Punch biopsies of fresh prostate slices were sliced to 75um thickness on a VF-300 Compresstome and in-situ antibody and chemical stains were used to analyze tissue slice viability and cellular composition prior to digestion (Figure 3A, B). Calculations of area fraction covered by each dye provided estimates of percent tissue viability and percent tissue composition of each labeled cell type (Figure 3C, D). Figures 3C and D display the variability in whole tissue viability and cellular composition between five patient tissues. Tissue viability ranged from 36 to 80%, with an average of 59% live cells and standard deviation of 15.6% (Figure 3C). Percentage of epithelial cell composition within in-situ differed as much as 43% between patient tissues with a standard deviation of 14.6%; stromal cell percentage varied by as much as 44% between patients with a standard deviation of 15.7% (Figure 3D).
Figure 3: Viable prostate tissue slice staining benchmarks tissue viability and cellular composition.
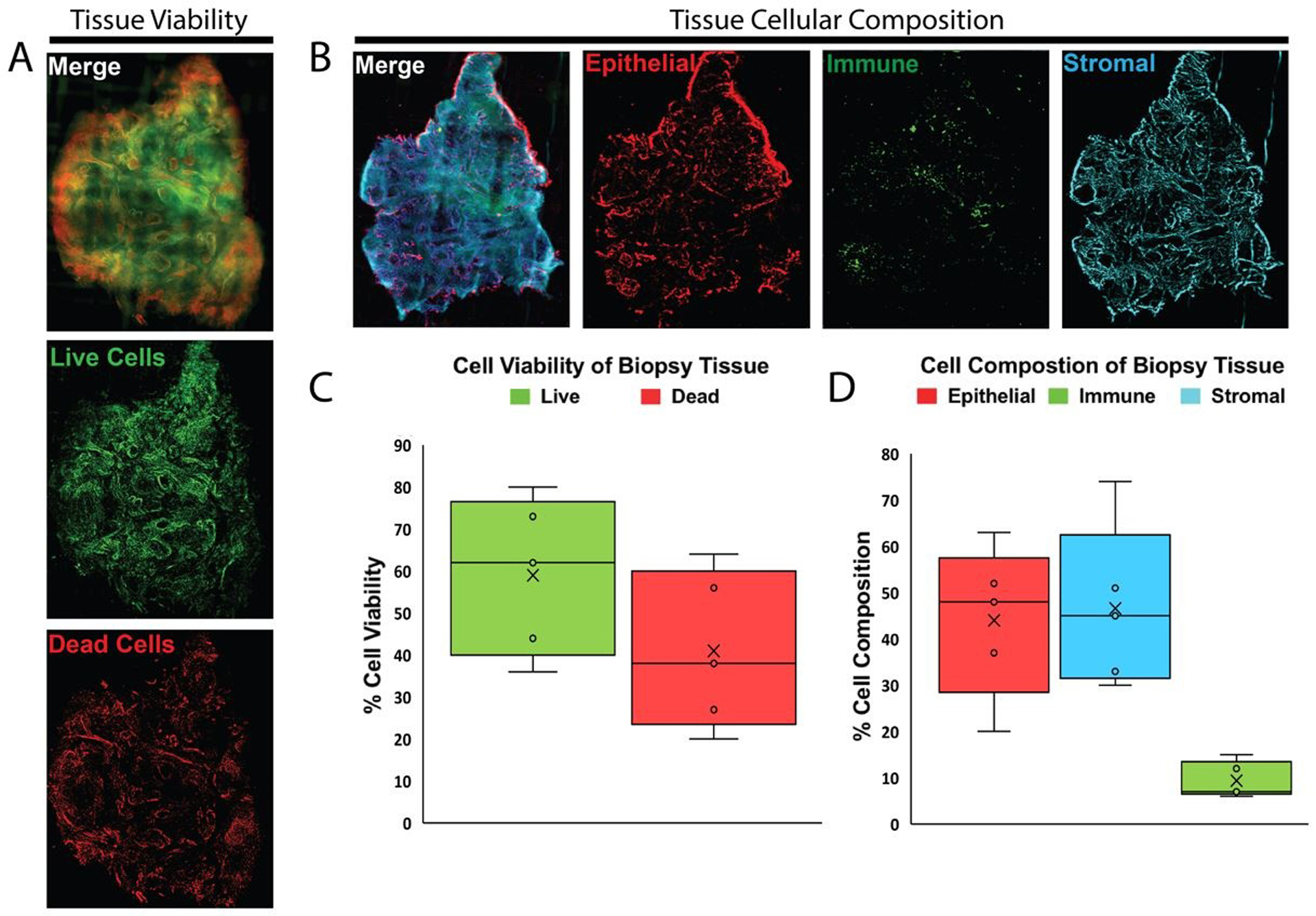
(A) Fluorescent images of compresstome sectioned tissue slices from PB027 prostatectomy biopsy punches. Slices are stained with ViaStain AOPI Staining Solution labeling live cells in green (acridine orange) and dead cells in red (propidium iodide). (B) Fluorescent images of compresstome sectioned tissue slices from PB027 prostatectomy biopsy punches. Slices are stained with fluorescent-tagged antibodies for CD45-FITC (immune cell marker), EpCAM-PE (epithelial cell marker), CD49a-Alexa-647 (fibroblast marker), and nuclei labeled with Hoechst 33342-blue. (C) Box and whisker plots show the quantified in-situ tissue viability from patient tissues (n=5). Whiskers represent maximum and minimum values; other patient values are labeled with open circles “o”. Mean value is labeled with the “X” and boxes represent interquartile range and median. (D) Box and whisker plots show the quantified in-situ tissue cellular composition from patient tissues (n=5). Whiskers represent maximum and minimum values; other patient values are labeled with open circles “o”. Mean value is labeled with the “X” and boxes represent interquartile range and median.
3.3. Tailoring tissue digestion conditions to allow multi-modal TME analyses -
Hemocytometer cell counts, fluorescent microscopy, flow cytometry, and qRT-PCR were used to evaluate tissue digestion effects on cell yield and viability, cellular composition, and gene expression. To optimize digestion conditions and achieve single cell suspensions, we tested combinations of mechanical disruption and enzymes that degrade extracellular matrix proteins. When digesting prostate core biopsies, a 10-minute digestion protocol using heavy mechanical mincing with a scalpel (pieces <100um) and collagenase type IV reduced cellular yield by 24% (p-value <0.05), when compared to a 4-hour digestion protocol using light mincing (1mm3 pieces) and collagenase type I (Figure 4A). However, 4-hour digestion led to far greater changes in gene expression when compared to the shorter 10-minute digestion. qRT-PCR of cells after 4-hour tissue digestion, compared to non-digested tissue, showed >4-fold increases in androgen receptor pathway transcripts (AR4/5, KLK3, FOLH1, and TMPRSS2) and >700-fold increases in immune cell activation transcripts (IL-6, IL-1B, and IL-8), whereas 10-minute digestion elicited far smaller fold changes in gene expression (Figure 4C).
Figure 4: Varying tissue digestion enzymes impacts cell yield, viability, and cellular populations.
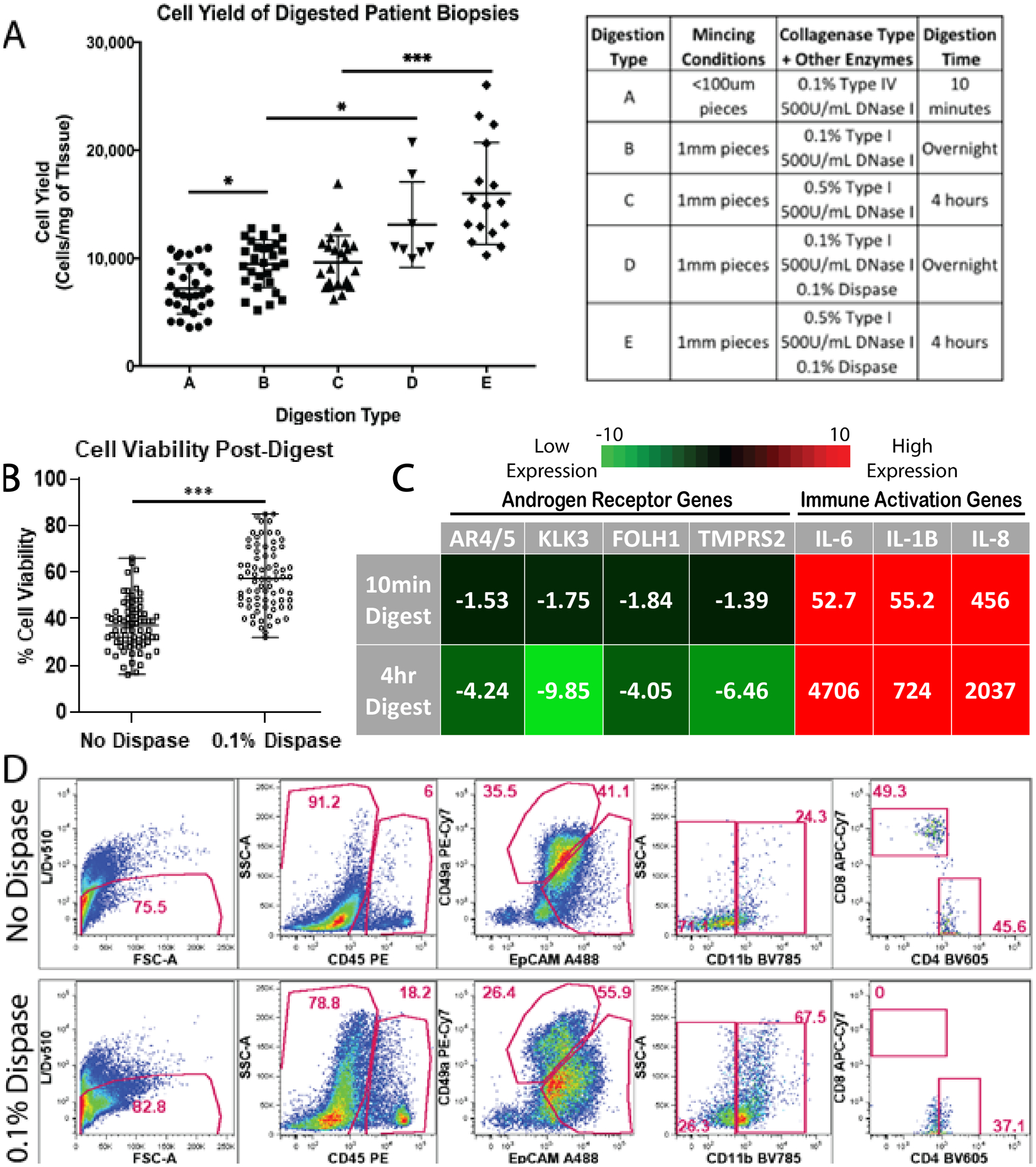
(A) Scatter plot displays mean cellular yields, with standard deviation, from individual prostate biopsies digested using numerous tissue digestion conditions. Digestion conditions A, B, C, D, and E are listed in the corresponding table (* indicates p-value <0.05, *** indicates p-value <0.001). (B) Scatter plot displays mean cell viability, with standard deviation, from individual prostate biopsies with and without 0.1% dispase added to digestion buffers (*** indicates p-value <0.001). (C) q-PCR measures androgen receptor transcripts (AR4/5, KLK3, FOLH1, TMPRSS2) and immune cell activation transcripts (IL-6, IL-1B, IL-8) from cells for both 10-minute digestion protocols and 4-hour digestion protocols. Fold change values are normalized to undigested tissue and averaged, n=3. (D) Gated flow cytometry dot plots display cell populations present in digested prostate samples from patient PB054 biopsies, with and without 0.1% dispase added (top row and bottom row respectively). The first column represents the live/single cell content of the isolates when is then projected to show CD45 expression in the second column. The third column represents EpCAM and CD49a expression within the CD45- fraction. The fourth and fifth columns show CD11b expression within the CD45+ subset and CD4/CD8 expression within the CD45+/CD11b- gate, respectively. Data shows frequency within the parent gates.
To improve cell viability, we tested the effect of supplementing our digestion media with dispase, a neutral protease which cleaves fibronectin and is reported to improve cell isolation from various tissues (12, 21, 22). Adding 0.1% dispase increased cellular yield by 38% (p-value <0.05) in overnight digestion conditions and 66% (p-value <0.001) in 4-hour digestion conditions (Figure 4A). Supplementing digestion buffers with 0.1% dispase improved cell viability by over 20% on average (Figure 4B). When evaluating specific cell populations, the addition of dispase also showed a 2.75-fold increase in myeloid compartment (CD11b) retrieval, as well as an improvement in the distinction of epithelial (EpCAM) and stromal (CD49a) cells into more definitive populations (Figure 4D). However, in contrast to the positive effects seen by dispase addition to epithelial, fibroblast, and myeloid compartments, negative effects were seen on CD8+ lymphoid populations. Figure 4D shows the complete loss of CD8+ staining when 0.1% dispase is supplemented into digestion conditions, as well as a shift in CD4 staining.
3.4. Tailoring cell isolation to desired molecular and functional endpoints -
To isolate specific cellular subsets for culture and downstream analyses, we tested both fluorescent-activated cell sorting (FACS) and paramagnetic particles sorting (PMPs). FACS is a well-established method for sorting desired cell types using fluorochrome-labeled antibodies that bind cell surface receptors with high specificity, purity and accuracy. Using known surface markers of epithelial cells (EpCAM), prostatic stroma (CD49a) and immune cells (CD45 panmarker), we isolated desired cell types and compared the lysates from the sorted subsets to the total lysates of unsorted bulk cellular preparations. Debris, dead cells were excluded. CD45+ cells were then sorted from the live, singlet gate, while epithelial and stromal cells were sorted from the live/singlet/CD45- gate. Although qRT-PCR did not show a significant increase epithelial transcripts (EpCAM and cytokeratin 8) in the cell lysates in the EpCAM+-sorted samples after sorting, depletion of stromal transcripts CD49a/ITGA1 (2-fold) and vimentin (19-fold), as well as a 45-fold decrease immune cell transcript (CD45) indicate epithelial enrichment of EpCAM+ cells by FACS (Figure 5A). Similarly, cell sorting of CD49a+ stromal fibroblasts resulted in enriched stromal transcripts including integrin alpha 1, vimentin, and alpha-smooth muscle actin (8-fold, 3-fold, and 5.5-fold respectively), and led to >300-fold reductions in epithelial and immune transcripts. The CD45+ immune population showed a 9-fold enrichment of CD45 transcript, >900-fold reductions in epithelial transcripts, as well as a 23-fold reduction in alpha-smooth muscle actin (Figure 5A).
Figure 5: Antibody-labeled PMPs capture cell types of interest from digested cell suspensions.
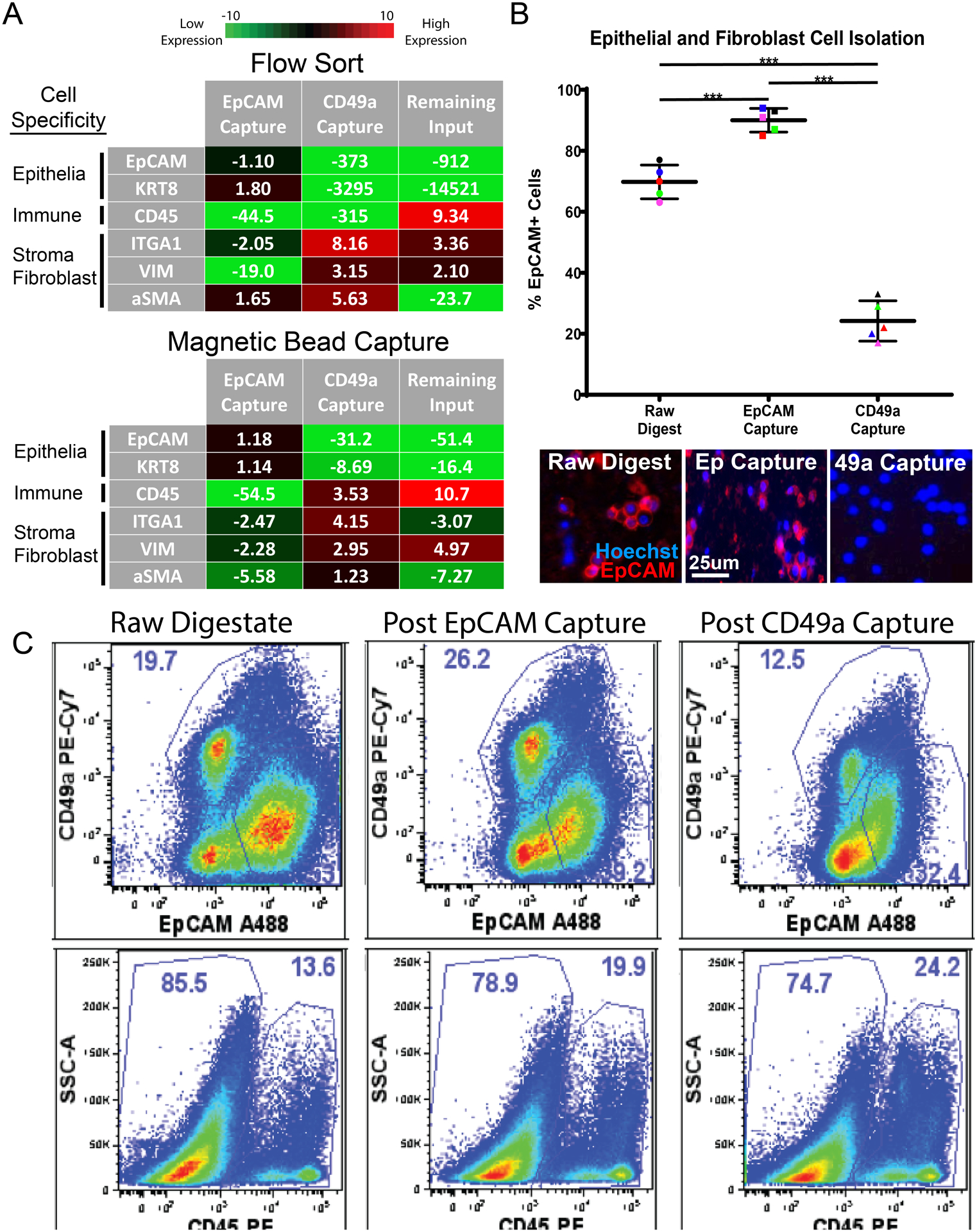
(A) q-PCR measures epithelial transcripts (EpCAM, KRT8), immune cell transcripts (CD45), and fibroblast transcripts (ITGA1, VIM, aSMA) from cells isolated via FACS and magnetic bead capture. EpCAM Capture represents cells isolated using EpCAM antibody, CD49a Capture represents cells isolated using CD49a antibody, and Remaining Input represents cells left over after EpCAM and CD49a capture. Fold change values are normalized to raw cell suspensions before cell isolation and averaged, n=3. (B) Scatter plot showing the percentage of EpCAM-positive cells throughout magnetic bead cell isolation experiments, quantified via fluorescent microscopy. Representative fluorescent images of digested cell suspensions are shown below. “Raw digestate” represents digested cell suspensions before magnetic bead capture, while “EpCAM Capture” represents cells isolated using EpCAM-labeled PMPs and “CD49a Capture” represents cells isolated using CD49a-labeled PMPs. Cells are stained with EpCAM-PE (epithelial cell marker) and cell nuclei are labeled with hoechst 33342-blue. (*** indicates p-value <0.01). (D) Gated flow cytometry plots display cell populations present during cell isolation experiments. “Raw digestate” columns represent digested cell suspensions before magnetic bead capture, “Post EpCAM Capture” columns represent cell populations remaining after cell capture using EpCAM-labeled PMPs, and “Post CD49a Capture” columns represent cell populations remaining after the second isolation step (cell capture using CD49a-labeled PMPs). The first row represents EpCAM and CD49a expression within the single cell/live/CD45- fraction, the second row shows CD45 expression within the total single/live cells. Data represents frequencies within the parent gate.
Next, we tested cell isolation using paramagnetic particles (PMPs) for transcriptional enrichment and culture. Using commercial PMPs (FlowComp and SeraMag) conjugated to an EpCAM or CD49a antibody, it was possible to serially remove epithelial cells and stromal fibroblasts, respectively. Similar to FACS, large increases in qRT-PCR epithelial transcripts (EpCAM and cytokeratin 8) were not seen in EpCAM-captured cell lysates. However, decreases in stromal markers CD49a/ITGA1, alpha-smooth muscle actin, and vimentin (2.5-fold, 5.6-fold, and 2.3-fold respectively) as well as 55-fold reduction in CD45 immune cell transcript indicate epithelial enrichment by EpCAM capture (Figure 5A). PMP isolation of CD49a+ cells resulted in 4-fold and 3-fold enrichment of fibroblast transcripts ITGA1 and Vimentin, respectively, as well as reductions in epithelial cell transcripts EpCAM and Cytokeratin 8 (31-fold and 8.5-fold, respectively). The negatively selected immune population showed a 11-fold increase in CD45 immune cell transcript, as well as depletion of epithelial cell transcripts, and stromal fibroblast transcripts (ITGA1 and alpha-smooth muscle actin).
To further characterize PMP cell isolation, we performed fluorescent microscopy to monitor the presence of epithelial cells (EpCAM+) throughout the cell capture process. PMP capture using EpCAM-labeled SeraMag beads resulted in a 20% epithelial enrichment of the raw cell digestate population, and an overall cell purity of 90% epithelial cells (Figure 5B). Cell isolation using CD49a-labeled PMPs resulted in a cell population containing as few as 18% epithelial cells (EpCAM+), a 3.2-fold reduction from the raw cell digestate. Finally, we also evaluated PMP cell isolation using flow cytometry to qualitatively assess the cell populations removed from the raw cell digestate. The cells remaining in the raw cell digestate after EpCAM-PMP capture showed a substantial reduction in the amount of EpCAM+ cells, leaving higher percentages CD49a+ stroma and CD45+ immune cells (Figure 5C). Similarly, CD49a-PMPs removed a considerable population of CD49a+ stromal fibroblasts and further enriched the CD45+ immune cell population.
Cell populations isolated using PMPs remained viable for culture and functional studies as well as RNA sequencing. Isolated populations of epithelial cells and fibroblasts were cultured individually and maintained in selective medias. Epithelial cells survived and propagated successfully in standard 2D culture format as well as spheroids (Figure 6A). To demonstrate primary cells were sufficient for use in functional assays, isolated prostate epithelial cells were exposed to the chemotherapeutic docetaxel when grown as a monoculture or in co-culture with patient-matched primary fibroblasts. After 72 hours a RealTime Glo assay was performed, which revealed a dose-dependent relationship between docetaxel concentration and primary cell viability in both the monoculture and co-culture. Interestingly, the IC50 was increased 2-fold in the co-culture (p-value <0.05) (Figure 6A). RNA sequencing on each cellular component was conducted to provide transcriptomic data. RNA sequencing of EpCAM-captured cells (Figure 6C-i) show an upregulation of epithelial markers (EpCAM, KRT8, and AR pathway genes) and a downregulation of stromal markers (ACTA2, IGTA1, and VIM). In contrast, stromal markers were upregulated and epithelial markers were downregulated in CD49a-captured cells (Figure 6C-ii).
Figure 6: Isolated cell populations of interest remain viable for cell culture and Next Gen RNA-sequencing.
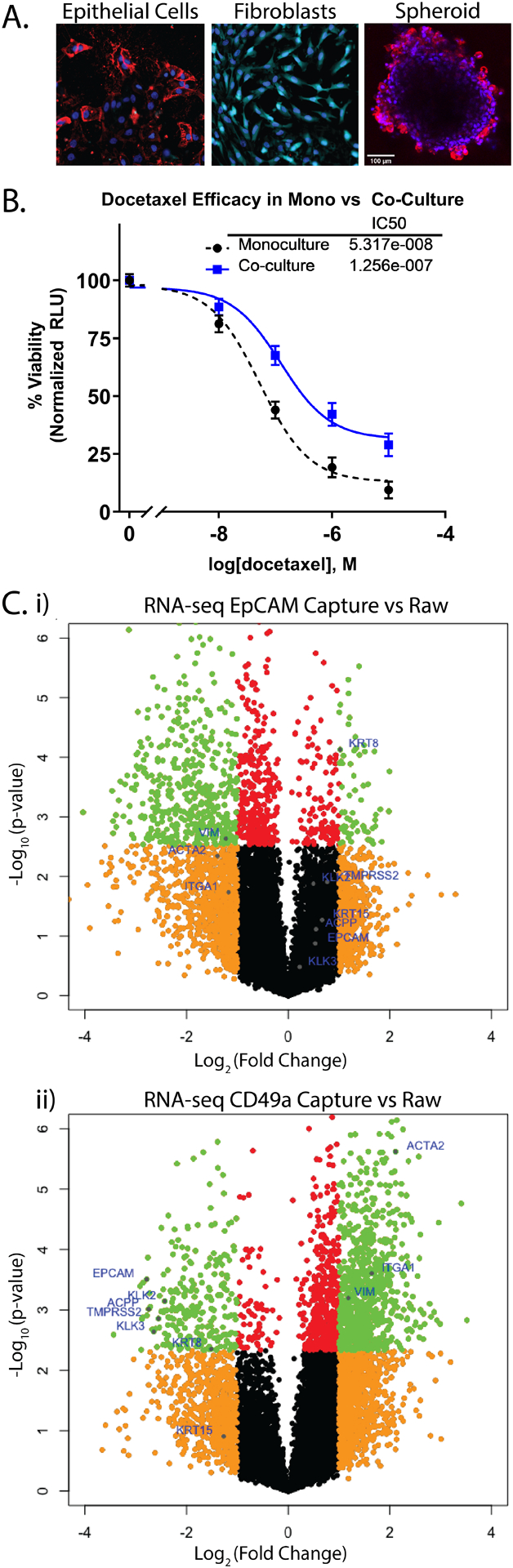
(A) Fluorescent images of isolated cells at day 12 of cell culture. Cells are stained with fluorescent-tagged antibodies for EpCAM-PE (epithelial cell marker), CD49a-Alexa-647 (fibroblast marker), and nuclei labeled with hoechst 33342-blue. (B) Dose-response plot shows cell viability, measured by RealTime Glo luminescence, of primary epithelial cells cultured in monoculture and in co-culture with patient-matched primary fibroblasts after 72-hours exposure to docetaxel chemotherapeutic. (C) Volcano plots from cell isolation experiments show average RNA-seq transcript fold changes across 4 patients. “EpCAM-Capture vs Raw” volcano plot shows transcripts up-/down-regulated in isolated epithelial cells compared to the raw digestate (i), while “CD49a-Capture vs Raw” shows transcripts up-/down-regulated in isolated stromal cells (ii) with known epithelial and stromal markers labeled with blue dots (orange dots represent transcripts up- or down-regulated >2-fold with p-value >0.005, green dots represent transcripts up- or down-regulated >2-fold with p-value <0.005, red dots represent transcripts up- or down-regulated with p-value <0.005 but by fold change <2, and black dots represent transcripts up- or down-regulated by fold change <2 and p-value >0.005).
4.0. Discussion
Mounting studies have implicated the tumor microenvironment in prostate cancer progression and treatment resistance (23, 24). However, there exists few methods to obtain and process viable prostate tissue for molecular and functional interrogation of major microenvironment components, that are compatible with standard clinical pathology. The advancement of techniques to tease apart the prostate tumor microenvironment ex-vivo that also satisfy standard clinical readouts promises to improve our knowledge of tumor microenvironment interactions and biology.
We developed a tissue processing workflow that made available substantial amounts of viable prostate tissue for molecular and functional analysis without impeding standard of care. Standard clinical practice uses formalin-fixed-paraffin-embedded (FFPE) prostate tissue to evaluate tissue morphology to form a histological diagnosis. However, FFPE limits functional and molecular analysis of tumor microenvironment that have shown promise in identifying new prognostic and predictive biomarkers (25–29). When evaluating clinical cases, we found the vast majority, in fact 100% of Gleason Grade Group (GGG) 3+ cases, discarded excess tissue not needed for pathologic diagnosis due to the high volume of tumor in these patients. Those discarded prostate tissue slices are a valuable research resource that could be used without disrupting standard of care. Patients with low grade or low volume on biopsy are likely not appropriate for these methods as there is a small chance that the excess tissue sent for research may actually need to be retrieved at a later time for diagnostic purposes, which could potentially hamper patient care and treatment decisions. Thus, it is our strategy to carefully select cases based on biopsy results showing high tumor grade and volume; in our institutional experience, excess tissue was never needed in GGG 3+ cases making them ideal candidates live prostate slicing methods depicted here. Moreover, these patients diagnosed with advanced disease are more likely to develop resistant disease and encounter poor outcomes, elucidating the need to identify new prognostic markers of progression and therapeutic resistance in these tissues (3). Furthermore, the use of patient-specific 3D prostate molds in this live-slicing method allows for precise lesion targeting. Although not presented in this manuscript, in a clinical trial of 30 patients with multi-focal prostate cancer, we were able to use these 3D molds to isolate at least 4 lesions per patient. This allowed interrogation of specific biology across individual tumors which will continue to facilitate the study of multi-focal prostatic disease and support precision medicine efforts.
We evaluated tissue viability and cellular composition in prostate tissue slices in-situ to determine the initial cellular composition and viability of the tissue samples. Using these previously described tissue slicing techniques (20), we were able to robustly identify live and dead cells as well as major TME components; epithelial cells, stroma, and immune cells in-situ. These results displayed significant variation in tissue viability and cellular composition from patient sample to patient sample indicating that single downstream measurements of post-digestion viability and cellular composition can be skewed by upstream quality of the source tissue. This whole-mount tissue slicing method can also be used to identify functional heterogeneity and pinpoint focal areas of interest for further study as previously described (20). Yet, these in-situ tissue-level analyses are often omitted in traditional tissue processing protocols and TME studies. Overall, whole-mount tissue slicing and staining provides an in-situ measure of tissue viability and cellular composition that can be used to benchmark tissue digestion conditions and identifies specific microenvironment components and functionality for further study.
Multiple variations of enzymes, concentrations, combinations, and commercial kits are available for tissue digestions, each with their own pros and cons (15). Multi-compartment and multi-modal analysis of the tumor microenvironment presents unique challenges due to the multi-parametric multiple endpoints from a variety of different microenvironmental cell types. With this in mind, we evaluated digestion recipes to identify a condition that was compatible with downstream molecular analyses, cell isolation, and culture. We found that cellular yield was increased with less mechanical mincing (1mm3 tissue pieces vs 100um3 tissue pieces), the addition of 0.1% dispase enzyme, and longer tissue digestion time (4 hours vs 10 minutes). However, a 4-hour digestion time significantly impacted gene expression in the androgen receptor pathway and immune activation pathway. We also found that supplementing with 0.1% dispase significantly improved cell viability and retrieval of certain cell populations such as EpCAM+ epithelial cells, CD49a+ fibroblasts, and CD11b+ leukocytes. Yet, dispase also showed negative effects such as the complete loss of CD8 binding on T-cells. These findings are consistent with previous studies (12) and suggest that evaluating cell yield and viability alone are not sufficient when pinpointing ideal digestion conditions, but rather a combined comparison of cell viability, cellular marker characterization, and gene expression. These data also advocate that digestion conditions not be standardized but tailored to downstream experiments. For instance, shorter digestion times are ideal for gene expression and molecular studies. Supplementing 0.1% dispase increases viability for downstream cell culture and functional analysis, while excluding dispase improves immune cell phenotypic study via flow cytometry. The analytical tools seen here provide a simple platform to optimize tissue digestion conditions for desired readouts and multi-compartment analysis of solid prostate tumor tissues.
Fluorescent-activated cell sorting (FACS) and paramagnetic particles sorting (PMPs) strategies are used for separating cell subpopulations from tissue digests. The majority of existing cell isolation protocols target only one cell type from a given cell suspension, neglecting the study of remaining populations of interest (13, 14). Antibody-bound PMPs that target cell surface markers have been extensively explored as a means of separating cellular subtypes from the prostate microenvironment (15, 30). When comparing these two methods, we found that both resulted in highly enriched populations of epithelial cells, fibroblasts, and immune cells, but FACS-based live cell sorting achieved increased purity of cell isolates. FACS resulted in >300-fold reductions in epithelial and immune transcripts when isolating stromal fibroblasts and >900-fold reductions in epithelial transcript in immune selected populations, while the PMP-based approach showed only 8 to 50-fold enrichment during fibroblast and immune cell isolation. However, FACS requires trained users, may be limited by timely instrument accessibility, and faces known difficulties with sample loss and cell viability that hamper downstream culture and functional analyses (30, 31). Cells isolated via the PMP-based approach showed high purity populations, up to 90% purity measured by fluorescent microscopy, that were able to robustly propagate in 2D and spheroid conditions.
Interestingly, neither FACS or PMP-based methods sufficiently enriched epithelial transcripts from the whole tissue. This is likely because epithelial transcripts make up a large majority of RNA in the prostate. With this in mind and given AR-driven transcripts were decreased even during a 10-minute digestion, it may be better to use RNA from the whole tissue to represent the epithelial fraction. These data suggest that cell isolation methods should be evaluated and catered to desired readouts. For example, FACS can be used to achieve high purity cell samples and satisfy molecular readouts, but PMP-based methods can be used as a more gentle isolation technique to conserve cellular viability, mitigate environmental stress (ie. avoid exposure to sub-optimal pressure conditions in the sort chamber), facilitate cell culture and functional experiments.
To advance our understanding of the role individual components of the TME play in prostate cancer progression, an ideal goal would fulfill clinical pathology while allowing parallel study of TME molecular and functional characterization. This is best achieved via building workflows satisfying standard clinical endpoints while maintaining viable cells that can be used for both molecular analyses and ex-vivo isolation to recombine into co-culture environments that reconstitute a translationally more accurate reflection of the primary TME. Here, we evaluated if isolated epithelial and fibroblast cells could satisfy each of those needs and found that cells remained viable for RNA-sequencing and functional cytotoxicity experiments. RNA-sequencing provided a rich transcriptomic analysis of the stromal and epithelial cell populations, as well as it allowed for assessment of cell isolation purity. Single-cell RNA-sequencing (SS RNAseq) is an emerging area of study in the TME and while not performed in this study, we show that we can obtain live cell suspensions that can undergo live cell sorting while maintaining good RNA quality, so these methods should also be easily translatable to SS RNAseq. Epithelial cells and fibroblasts isolated by the PMP-based approach were also suitable for co-culture experiments to evaluate chemotherapeutic efficacy, where the presence of fibroblasts was found to have a significant impact on cancer cell sensitivity to docetaxel. These results highlight both the need for further studies evaluating TME-mediated effects on cancer cell therapeutic resistance, as well as the functionality of the proposed methods in facilitating molecular and functional readouts. Together, these orthogonal analyses can provide a rich characterization of the tumor microenvironment and allow further exploration of new biology in each cell type.
5.0. Conclusions
The tumor microenvironment is a known regulator of prostate cancer pathogenesis(4–8, 32–34). However, current clinical and wet lab methods and logistics have hindered the study of the microenvironment and its role in cancer progression. In order to investigate the genomic and functional input of each cellular subset, new methods to tease apart ex vivo patient tissue for orthogonal analyses are needed. Here, we present an approach to optimize tissue digestion conditions for interrogation of various cell types and endpoints from excess clinical specimen without disturbing standard clinical diagnostic and prognostic methods such as histopathology. We also present methods to isolate enriched populations of distinct microenvironment components and have optimized protocols for desired downstream applications, as well as assessed tools to verify isolation efficacy. We showed that isolated epithelial and fibroblast populations remained viable for RNA sequencing as well as culture and functional interrogation. Importantly, we demonstrate that no single method can adequately satisfy a full TME analysis; instead, methods need to be tailored to specific TME components and readout. Alternatively, if there is sufficient tissue, the sample can be aliquoted and processed separately in order to satisfy multiple TME components and endpoints for a more complete TME analysis. Although future work is required to optimize protocols to study other rare cell populations in the prostate tumor microenvironment such as cancer stem cells and vascular endothelium, the lessons learned in this study can hopefully guide their development. These methods provide a basis to build translationally relevant platforms to facilitate controlled functional assays and molecular studies of the prostate tumor microenvironment while correlating with standard clinical readouts to further our understanding of prostate cancer biology.
Supplementary Material
Acknowledgements -
cDNA synthesis, library preparation, and mRNA sequencing were performed by the UW-Madison Biotechnology Center. by the National Institute of Health for the UWCCC Flow Cytometry Lab and the Translational Science Biocore at UWCCC.
Funding Statement -
This research was supported by NIH awards T32 CA157322 and K99ES028744 to Brian Johnson, as well as a Prostate Cancer Foundation Challenge Award to David Beebe and Joshua Lang. This work was also supported by the shared resources grant titled ‘University of Wisconsin Carbone Cancer Center Support Grant P30 CA014520’ and Grant#: 1S100OD018202-01, Project Title: Special BD LSR Fortessa by the National Institute of Health for the UWCCC Flow Cytometry Lab and the Translational Science Biocore at UWCCC. The U.S. Army Medical Research Acquisition Activity, 820 Chandler Street, Fort Detrick MD 21702-5014 is the awarding and administering acquisition office. This work was supported by the Assistant Secretary of Defense for Health Affairs through the Prostate Cancer Research Program, Award Nos. W81XWH-16-1-0511, W81XWH-16-1-0514, and W81XWH-16-1-0515. Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the Department of Defense.
Footnotes
Conflict of Interest Statement – David Beebe holds equity in Bellbrook Labs, LLC, Tasso, Inc., Stacks to the Future, LLC, Salus Discovery, LLC, Lynx Biosciences, Inc. Flambeau Diagnostics LLC, Onexio Biosystems LLC. Brian Johnson holds equity in Onexio Biosystems LLC. Joshua Lang holds equity in Salus Discovery, LLC. Wei Huang holds equity in PathomIQ, LLC.
Ethics and Patient Consent Statement - All tissue procurement for these experiments was performed in compliance with the laws and institutional guidelines as approved by the Institutional Review Board committee from the University of Wisconsin-Madison. Prostate tissue was obtained from patients undergoing radical prostatectomy with informed consent through the Translational Science BioCore Biobank at the Carbone Cancer Center at the University of Wisconsin-Madison.
Data Availability Statement -
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Siegel RL, Miller KD, Jemal A, Cancer statistics, 2016. CA Cancer J Clin 66, 7–30 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Egevad L, Granfors T, Karlberg L, Bergh A, Stattin P, Prognostic value of the Gleason score in prostate cancer. BJU Int 89, 538–542 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Andrén O et al. , How well does the Gleason score predict prostate cancer death? A 20-year followup of a population based cohort in Sweden. J Urol 175, 1337–1340 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Junttila MR, de Sauvage FJ, Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 501, 346–354 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Barron DA, Rowley DR, The reactive stroma microenvironment and prostate cancer progression. Endocr Relat Cancer 19, R187–204 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalluri R, The biology and function of fibroblasts in cancer. Nat Rev Cancer 16, 582–598 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Augsten M, Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front Oncol 4, 62 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dai J et al. , Immune mediators in the tumor microenvironment of prostate cancer. Chin J Cancer 36, 29 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Samaratunga H, Montironi R, True L, et al. International Society of Urological Pathology (ISUP) Consensus Conference on Handling and Staging of Radical Prostatectomy Specimens. Working group 1: specimen handling. Mod Pathol. 24, 6–15 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Sehdev AE, Pan CC, Epstein JI. Comparative analysis of sampling methods for grossing radical prostatectomy specimens performed for nonpalpable (stage T1c) prostatic adenocarcinoma. Hum Pathol. 32, 494–499 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Autengruber A, Gereke M, Hansen G, Hennig C, Bruder D, Impact of enzymatic tissue disintegration on the level of surface molecule expression and immune cell function. Eur J Microbiol Immunol (Bp) 2, 112–120 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ford AL, Foulcher E, Goodsall AL, Sedgwick JD, Tissue digestion with dispase substantially reduces lymphocyte and macrophage cell-surface antigen expression. J Immunol Methods 194, 71–75 (1996). [DOI] [PubMed] [Google Scholar]
- 13.Höfner T et al. , Defined conditions for the isolation and expansion of basal prostate progenitor cells of mouse and human origin. Stem Cell Reports 4, 503–518 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang Y et al. , Isolation of Fibroblast-Activation Protein-Specific Cancer-Associated Fibroblasts. Biomed Res Int 2017, 4825108 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strand DW, Aaron L, Henry G, Franco OE, Hayward SW, Isolation and analysis of discreet human prostate cellular populations. Differentiation 91, 139–151 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lasfargues EY, Coutinho WG, Redfield ES, Isolation of two human tumor epithelial cell lines from solid breast carcinomas. J Natl Cancer Inst 61, 967–978 (1978). [PubMed] [Google Scholar]
- 17.Yu YR et al. , A Protocol for the Comprehensive Flow Cytometric Analysis of Immune Cells in Normal and Inflamed Murine Non-Lymphoid Tissues. PLoS One 11, e0150606 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shah V et al. , A method for correlating in vivo prostate magnetic resonance imaging and histopathology using individualized magnetic resonance-based molds. Rev Sci Instrum 80, 104301 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rutkowski DR, Wells SA, Johnson B, et al. Mri-based cancer lesion analysis with 3d printed patient specific prostate cutting guides. Am J Clin Exp Urol. 7, 215–227 (2019). [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson BP et al. , Vital ex vivo tissue labeling and pathology-guided micropunching to characterize cellular heterogeneity in the tissue microenvironment. Biotechniques 64, 13–19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seach N, Wong K, Hammett M, Boyd RL, Chidgey AP, Purified enzymes improve isolation and characterization of the adult thymic epithelium. J Immunol Methods 385, 23–34 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Wang H, Van Blitterswijk CA, Bertrand-De Haas M, Schuurman AH, Lamme EN, Improved enzymatic isolation of fibroblasts for the creation of autologous skin substitutes. In Vitro Cell Dev Biol Anim 40, 268–277 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Correia AL, Bissell MJ, The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist Updat 15, 39–49 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cunha GR, Hayward SW, Wang YZ, Ricke WA, Role of the stromal microenvironment in carcinogenesis of the prostate. Int J Cancer 107, 1–10 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Van Allen EM et al. , Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsu J et al. , Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J Clin Invest 128, 4654–4668 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fong L et al. , Potentiating endogenous antitumor immunity to prostate cancer through combination immunotherapy with CTLA4 blockade and GM-CSF. Cancer Res 69, 609–615 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Kwek SS et al. , Diversity of antigen-specific responses induced in vivo with CTLA-4 blockade in prostate cancer patients. J Immunol 189, 3759–3766 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drake RR, Angel PM, Wu J, Pachynski RK, Ippolito JE, How else can we approach prostate cancer biomarker discovery? Expert Rev Mol Diagn, 1–3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Allinen M et al. , Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell 6, 17–32 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Guckenberger DJ et al. , Magnetic System for Automated Manipulation of Paramagnetic Particles. Anal Chem 88, 9902–9907 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quail DF, Joyce JA, Microenvironmental regulation of tumor progression and metastasis. Nat Med 19, 1423–1437 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun Y et al. , Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat Med 18, 1359–1368 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corn PG, The tumor microenvironment in prostate cancer: elucidating molecular pathways for therapy development. Cancer Manag Res 4, 183–193 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.