

Abstract
In the face of constant genomic insults, the DNA damage response (DDR) is initiated to preserve genome integrity; its disruption is a classic hallmark of cancer. Protein Phosphatase Mg2+/Mn2+–Dependent 1D (PPM1D) is a central negative regulator of the DDR that is mutated or amplified in many solid cancers. PPM1D overexpression is associated with increased proliferative and metastatic behavior in multiple solid tumor types and patients with PPM1D-mutated malignancies have poorer prognoses. Recent findings have sparked an interest in the role of PPM1D in hematologic malignancies. Acquired somatic mutations may provide hematopoietic stem cells with a competitive advantage, leading to a substantial proportion of mutant progeny in the peripheral blood, an age-associated phenomenon termed “clonal hematopoiesis” (CH). Recent large-scale genomic studies have identified PPM1D to be among the most frequently mutated genes found in individuals with CH. While PPM1D mutations are particularly enriched in patients with therapy-related myeloid neoplasms, their role in driving leukemic transformation remains uncertain. Here, we examine the mechanisms through which PPM1D overexpression or mutation may drive malignancy by suppression of DNA repair, cell cycle arrest and apoptosis. We also discuss the divergent roles of PPM1D in the oncogenesis of solid vs. hematologic cancers with a view to clinical implications and new therapeutic avenues.
Introduction
Our cells constantly acquire somatic mutations from endogenous and environmental sources and must rely on the DNA damage response (DDR) to preserve genomic integrity. The DDR is a complex network of cellular pathways that function to sense DNA damage, signal the presence of damage, and mediate DNA repair. These signaling networks bring about various cellular outcomes including cell cycle arrest, DNA repair, and apoptosis when the damage is too extensive for repair. The tumor suppressor, p53, plays a central role in activating the DDR and via regulation of multiple nodes of the DDR signaling cascade (1,2). Early studies identified genes transcriptionally regulated by p53, one of which was called WIP1 (wild-type p53-induced protein), later known as PPM1D (protein phosphatase Mg2+/Mn2+-dependent 1D) or PP2Cδ (3). Following DNA repair, p53 induces the expression of PPM1D which in turn acts as a negative regulator of the DDR to restore cellular homeostasis (4–6).
Over the last 20 years, mutations, and amplifications in PPM1D have been identified in several cancer-associated clinical contexts implicating it as a proto-oncogene (7–12). PPM1D-overexpressing solid cancers exhibit advanced tumor stage, increased metastatic potential, and poorer prognosis (8,13–16). In the blood, mutations in PPM1D are often found in individuals with clonal hematopoiesis (17), a pre-malignant expansion of mutant hematopoietic stem cells. PPM1D mutations in the blood are enriched in individuals who were treated with chemotherapy for solid tumors, suggesting mutation of PPM1D offers a selective advantage (12,18). However, whether these mutations promote hematologic malignancies is still unclear.
In this review, we will highlight the DDR pathways modulated by PPM1D and reflect on the degree to which the oncogenic properties of PPM1D mutations can be accounted for by its role in regulating p53. We will discuss the contexts and mechanisms in which PPM1D mutant cells gain dominance over wild type cells and the relevance of this so-called “clonal emergence” for cancer development. We will highlight remaining questions in the field about the conflicting clinical implications and divergent roles of PPM1D in solid and hematologic cancers.
PPM1D Amplifications and Mutations
The PPM1D gene consists of 6 exons on chromosome 17q23 in humans (19). The three domains of the PPM1D protein include the N-terminus, the phosphatase domain, and the C-terminus. The phosphatase domain of PPM1D is evolutionarily conserved with that of the other members in the protein phosphatase 2C (PP2C) family of Ser/Thr phosphatases (3). Genomic aberrations of PPM1D can present as amplifications of chromosome 17q as seen in ovarian and breast cancer (Figure 1A) (4,7,20–22). This results in increased expression of wildtype PPM1D that is correlated with the gene dosage and copy number variation which can range from 4 to 27 (23). It is also important to note that several other cancer-associated genes are also located on chromosome 17q including BRCA1, ERBB2, NF1, RAD51C, BRIP1 and BIRC5 (24). Therefore, overexpression of PPM1D in 17q amplifications may act cooperatively with the increased expression of other oncogenes to promote tumorigenesis.
Figure 1. PPM1D truncating mutations in solid cancers.
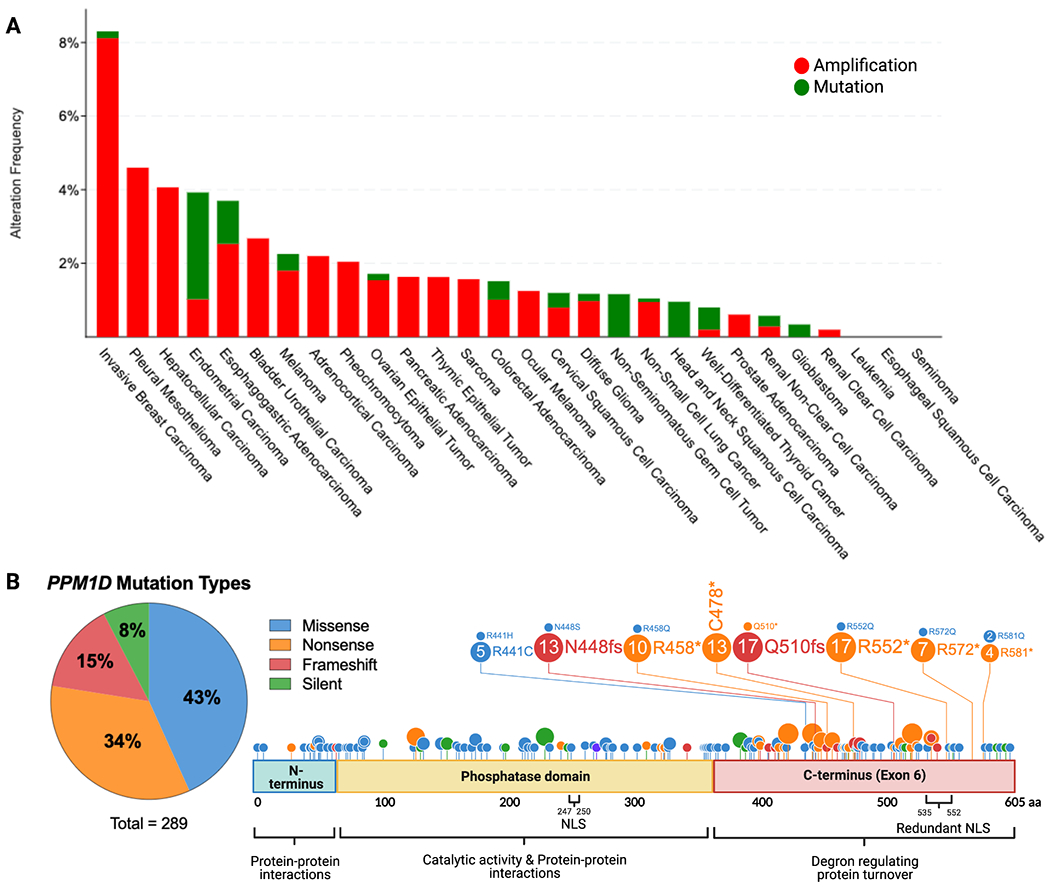
(a) Histogram showing the prevalence of PPM1D amplifications and c-terminal truncating mutations by cancer type, as indicated on the x-axis. The percentage of cases with PPM1D genomic alterations is indicated on the y-axis. The data was obtained from Pan-Cancer studies available in the cBioPortal database (10967 total samples), which was then filtered to show only cancer types with more than 50 cases. (b) Lollipop plot showing the location of the truncating mutations in the context of the domains of the PPM1D gene. A total of 289 PPM1D mutations were identified across 43 histology types from the COSMIC database. A pie chart of the mutation types is included with missense and nonsense mutations being the most common. The phosphatase domain and exon 6 of PPM1D are shown. Several mutation hotspots are noted.
With the advent of next generation sequencing, mutations in PPM1D were first reported in 2013 (25). Strikingly, almost all mutations are nonsense or frameshift mutations spanning across the terminal exon of the PPM1D gene with no clear hotspot (Figure 1B). Importantly, exon 6 mutations are all located downstream of the catalytic domain, and studies have demonstrated that truncation of the protein has minimal effect on the phosphatase activity of PPM1D. Instead, these mutations result in the loss of a C-terminal degradation motif leading to the stabilization and accumulation of the mutant protein (12,18,26). Truncated PPM1D protein can accumulate in the cell up to 16 times the level of full-length PPM1D even in the absence of stressors where wild-type PPM1D levels would be low (12). This finding suggests that PPM1D truncating mutations could mimic the effect of PPM1D amplifications, as both alterations increase levels of PPM1D protein in the cell. However, it remains an open question as to whether the truncation variant has neomorphic effects or different interaction partners than full-length PPM1D. Two common cancer cells lines, HCT116 (colon cancer) and U2OS (osteosarcoma), both harbor heterozygous PPM1D truncating mutations (25). We have curated a list of additional cell lines with PPM1D truncating mutations and amplifications (Table 1) using the Cell Model Passports database from the Sanger Institute (27). In the next section, we will discuss the overall consequences of increased PPM1D activity on the DNA damage response, cell cycle, and apoptosis and how dysregulation of these pathways can promote the formation of solid vs. hematologic malignancies.
Table 1.
List of cell lines with PPM1D truncating mutations, amplifications, and copy number gains curated from the Cell Model Passports database.
| Cell Line | Cancer Type | Nucleotide Change | Amino Acid Change | Classification | TP53 Status |
|---|---|---|---|---|---|
| SUP-T1 | TALL | c.1528_1529insA | p.N512fs*16 | Frameshift | 1 |
| MN-60 | BALL | p.N512fs*2 | c.1529delA | Frameshift | 0 |
| HCC1569 | BRCA | c.1344delT | p.L450fs*1 | Frameshift | 1 |
| PA-1 | OV | c.1370delC | p.A457fs*8 | Frameshift | 0 |
| PA-1 | OV | p.A457fs*8 | c.1370delC | Frameshift | 0 |
| U2OS | OS | c.1372C>T | p.R458* | Nonsense | 0 |
| BB58-HNC | HNC | p.N477fs*6 | c.1427delA | Frameshift | 0 |
| CCK-81 | CRC | p.N512fs*2 | c.1529delA | Frameshift | 1 |
| CL-34 | CRC | p.R572* | c.1714C>T | Nonsense | 1 |
| CW-2 | CRC | p.K336fs*3 | c.1003delA | Frameshift | 0 |
| HCM-SANG-520-C18 | CRC | p.N512fs*2 | c.1529delA | Frameshift | 1 |
| HCT-116 | CRC | c.1344delT | p.L450fs*1 | Frameshift | 0 |
| SNU-175 | CRC | p.N512fs*16 | c.1528_1529insA | Frameshift | 0 |
| HEC-108 | EC | p.L546fs*1 | c.1632delC | Frameshift | 1 |
| HEC-6 | EC | p.N512fs*16 | c.1528_1529insA | Frameshift | 1 |
| NCI-H3122 | NSCLC | p.N512fs*16 | c.1528_1529insA | Frameshift | 1 |
| Cell Line | Cancer Type | Copy Number | Cell Line | Cancer Type | Copy Number |
| ZR-75-30 | BRCA | 40 | NCI-H650 | NSCLC | 5 |
| MCF7 | BRCA | 30 | LN-229 | GBM | 5 |
| HCC2218 | BRCA | 23 | NCI-H2081 | SCLC | 5 |
| BT-474 | BRCA | 23 | KATOIII | GC | 5 |
| MDA-MB-361 | BRCA | 9 | CFPAC-1 | PC | 5 |
| SK-MEL-5 | MEL | 7 | NCI-H64 | SCLC | 5 |
| HCC1428 | BRCA | 7 | NCI-H740 | SCLC | 5 |
| UACC-893 | BRCA | 7 | SK-MEL-3 | MEL | 5 |
| NCI-H508 | CRC | 7 | DAN-G | PC | 5 |
| CAKI-1 | RCC | 6 | SK-MES-1 | SqCLC | 5 |
| NCI-H28 | MS | 6 | SW780 | BC | 5 |
| U-2-OS | OS | 6 | SK-N-DZ | NB | 5 |
| NCI-H1993 | NSCLC | 6 | NCI-H2009 | NSCLC | 4 |
| PANC-02-03 | PaC | 6 | MDA-MB-453 | BRCA | 4 |
| SK-MEL-1 | MEL | 6 | MHH-ES-1 | EW | 3 |
| PC-3 | PrC | 5 | NCI-H2405 | NSCLC | 3 |
| MDA-MB-330 | BRCA | 5 | MOLT-4 | TALL | 3 |
TALL = T-cell acute lymphoblastic leukemia
BALL = B-cell acute myeloid leukemia
BRCA = breast cancer
OV = ovarian carcinoma
OS = osteosarcoma
MEL = melanoma
NSCLC = non-small cell lung carcinoma
CRC = colorectal
RCC = renal cell carcinoma
GBM = glioblastoma
PaC = pancreatic cancer
EC = endometrial carcinoma
GC = gastric cancer
HNSC = head & neck squamous cell carcinoma
PrC = prostate cancer
MS = mesothelioma
BC = bladder cancer
NB = neuroblastoma
SCLC = small cell lung cancer
PPM1D is a negative regulator of the DNA damage response
The DDR is integral to maintaining genome integrity by coordinating the arrest of normal cellular functions and cell cycling to recruit downstream effectors that repair damaged DNA. As a homeostatic regulator of the DDR, PPM1D is activated in response to exogenous (i.e., radiation, chemicals, or chemotherapy) and endogenous (i.e., reactive oxygen species or DNA replication errors) stimuli (28). When activated, PPM1D attenuates the stress response through dephosphorylation of p53 (5), DNA damage sensors (ATM, ATR) (29), cell cycle checkpoint proteins (CHK1, CHK2, p21) (30,31), apoptotic proteins (BAX, DAXX) (32), among others. Through this coordinated network of events, the DDR is inactivated, and the cell resumes normal cell cycling and homeostasis. Defects in the DDR lead to genomic instability and allow for the accumulation of driver aberrations that promote neoplastic growth. It is important to understand the role of PPM1D in modulating DNA repair to contextualize how its overexpression can lead to a blunted DNA damage response (33–37) to promote malignant transformation. There are three DNA damage repair pathways that PPM1D is known to regulate: double-stranded break (DSB) repair, nucleotide excision repair (NER), and base excision repair (BER). An in-depth graphical summary of dephosphorylation sites is provided in Figure 2.
Figure 2. The role of PPM1D in DNA repair.
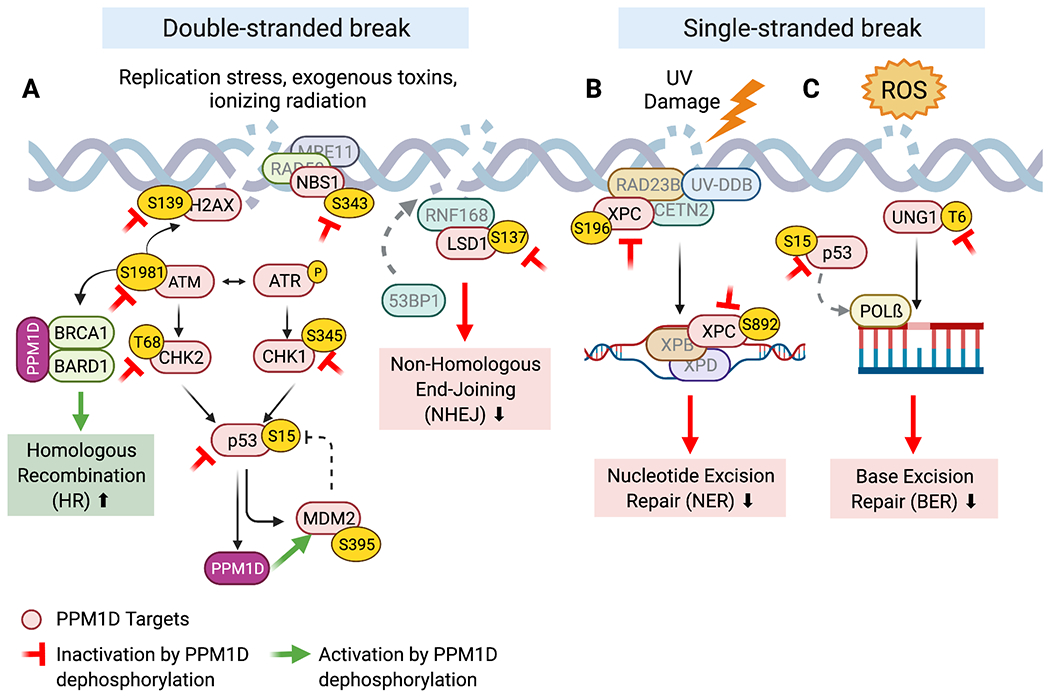
The red proteins are known PPM1D targets. The red inhibitory symbol denotes the inactivation of that protein upon PPM1D dephosphorylation. The green arrow represents activation of the protein upon dephosphorylation. A) PPM1D inhibits key players involved in double-stranded break repair resulting in decreased NHEJ and increased HR. B) PPM1D inhibits several BER and NER repair proteins leading to decreased single-stranded break repair.
Double-stranded Break (DSB) Repair
DSBs result from exposure to ionizing radiation, chemicals (i.e., bleomycin and specific chemotherapeutic agents) and endogenous replication stress. DSB repair begins when ataxia telangiectasia mutated (ATM) undergoes autophosphorylation and orchestrates DSB repair by recruiting downstream effectors. Importantly, p-ATM phosphorylates H2AX at Ser139, which then becomes referred to as γ-H2AX. γ-H2AX is a highly specific and sensitive molecular marker for the initiation of the DNA damage response, as it serves as a docking site for the recruitment of DNA repair proteins to DSB sites (33). As part of a negative feedback loop, PPM1D suppresses the activation of the ATM-dependent signaling cascade through dephosphorylation (29). PPM1D also directly dephosphorylates γ-H2AX, which further inhibits the recruitment of DNA repair factors after damage is successfully repaired. Notably, premature dephosphorylation of γ-H2AX by PPM1D can lead to failure in the recruitment of DNA repair proteins and delayed DNA repair (37).
Independent of γ-H2AX, the MRE11-RAD50-NBS1 (MRN) complex also plays a crucial role as an initial sensor of and responder to DNA damage by modulating the activity of ATM at DSBs (38). In addition to its role in the DDR, the MRN complex also mediates cell cycle checkpoints and telomere maintenance. Yamaguchi et al. have shown that NBS1 is dephosphorylated by PPM1D at Ser343 in vitro (39). While it remains unclear how NBS1-S343 dephosphorylation may affect the DDR-specific role of the MRN complex, S343-mutant variants of NBS have defective CHK2 activation and inappropriate cell cycle progression following genotoxic stress (40). Therefore, constitutive downregulation of NBS1 at S343 by PPM1D overexpression may promote tumorigenesis due to increased cell cycling and mutagenesis.
There is some evidence that PPM1D regulates certain aspects of non-homologous end joining (NHEJ) and homologous recombination (HR), the two major pathways that mediate DSB repair. In the context of NHEJ, PPM1D has been shown to dephosphorylate LSD1 resulting in impaired recruitment of 53BP1 to DSBs after bleomycin exposure (35). On the contrary, PPM1D was recently shown to promote HR by forming a stable complex with BRCA1-BARD1, which is critical for the timely recruitment of these substrates to DSBs (41). In summary, PPM1D appears to inhibit NHEJ while promoting HR. However, given the complexity of events in DSB repair, the net effect of PPM1D mutations on HR and NHEJ activity remains to be explored.
Nucleotide Excision Repair (NER)
NER is the key pathway involved in the repair of bulky, helix-distorting DNA damage including UV-induced genomic lesions. Following UV damage, p53 is phosphorylated by ATR and induces expression of XPC, p48XPE, and GADD45 to facilitate NER (42). Since p53 enhances NER, suppression of p53 activity by PPM1D should inhibit expression of these NER effectors. However, overexpression of PPM1D has been shown to inhibit NER activity in both p53-proficient and p53-deficient cell lines. Inhibition of NER activity was not observed with overexpression of a phosphatase-dead form of PPM1D, suggesting that the catalytic activity of PPM1D plays a direct role in regulating NER. Furthermore, mouse embryonic fibroblasts (MEFs) derived from Ppm1d-deficient mice exhibited the opposite phenotype, with faster genome-wide resolution of damage after UV exposure (34). PPM1D was found to regulate NER via dephosphorylation of XPC and XPA. This activity appeared to be specific to PPM1D, as members of other classes of phosphatases failed to dephosphorylate XPA at the same sites (34).
Base excision repair (BER)
BER is responsible for repairing non helix-distorting base lesions that typically result from deamination, oxidation, or alkylation of nuclear and mitochondrial DNA. BER is initiated by DNA glycosylases, which recognize, and excise mismatched or damaged bases. P53 has been shown to promote BER through the regulation of several BER glycosylases as well as DNA polymerase ß, the main polymerase involved in short-patch BER (2). Ppm1d−/− MEFs were shown to have 6-fold higher BER activity compared to WT MEFs, independent of p53 status. These results suggest that PPM1D suppresses BER activity. Indeed, it was later discovered that PPM1D dephosphorylates UNG2, a key uracil-DNA glycosylase that excises uracil bases from DNA, to shut down BER and return the cell to homeostasis (36). However, BER can be initiated by other DNA glycosylases, each with a substrate specificity for a particular type of damaged DNA base. Therefore, while PPM1D may decrease BER in response to UV-mediated damage, it remains unknown whether it decreases the repair of other types of damaged bases.
Consequences of Mutant PPM1D in the DDR
Overall, PPM1D inhibits several DNA repair pathways involved in double-stranded break repair, NER, and BER. In contrast, PPM1D seems to promote homologous recombination (HR) a distinct DNA repair mode. This finding is further bolstered by studies demonstrating that inhibition of PPM1D sensitizes cancer cells to PARP1 inhibition, a key player in mediating DNA repair and HR (41,43). One caveat of the DNA repair studies described above is that many were performed by comparing WT cells to PPM1D knockout models. While one could infer that PPM1D overexpression models behave in an opposite manner, whether this is the case remains an open question. Similarly, as mentioned previously, while PPM1D truncation variants found in cancer have preserved phosphatase activity, it remains unclear whether it interacts with the same targets as wild type PPM1D. Furthermore, the genomic consequences of the combined suppression of DSB repair, NER, and BER activity that is presumed to occur with excess PPM1D is still unknown. One possibility is excess PPM1D simply leads to delayed DNA repair that occurs with normal fidelity. Another possibility is that fidelity of DNA repair is compromised, leading to accumulation of mutations in the genome. This may increase the risk of a “second hit” mutation activating an oncogene or inhibiting a tumor suppressor. If such is the case, we may expect PPM1D mutant cells to harbor a unique mutation signature representing a combination of distinct signatures reflective of the corresponding defective DNA repair pathways. Addressing these critical unanswered questions will shed light on the mechanism and potency of PPM1D mutations and amplifications in driving cancer.
Relationship between PPM1D and TP53
The tumor suppressor p53 has long been known to be the “guardian of the genome” and transcriptionally regulates hundreds of downstream effectors to promote cell cycle arrest (CDKN1A and GADD45A), DNA damage repair (XPC, DDB2, etc), and cell fate pathways including apoptosis (PUMA, BAX) and senescence. Mutations in p53 lead to the dysregulation of these critical cellular pathways and allow the neoplastic transformation of cells into cancer (1). In normal cells, p53 becomes activated and turns on these genome-protective pathways. A major role of PPM1D is to attenuate this activation. P53 in fact transcriptionally activates the expression of PPM1D, which then dephosphorylates p53 at Ser15, which is a critical post-translational modification required to stimulate transactivation of p53-responsive promoters (44); thus, PPM1D dephosphorylation of p53 directly de-escalates the DDR. PPM1D also indirectly inhibits p53 by dephosphorylating upstream activating kinases such as ATM, ATR, CHEK1, and CHEK2. In addition, PPM1D dephosphorylates MDM2, an E3 ubiquitin ligase, which in turn tags p53 for proteasomal degradation (6). Together, all these actions serve to turn down the DDR broadly, and p53 specifically.
Given the suppressive role of PPM1D on p53 activity, PPM1D amplifications and mutations are thought to mimic partial loss of TP53. Nevertheless, p53 undergoes more than 300 different PTMs including ubiquitination, acetylation, and phosphorylation, which instigate programs independent of PPM1D (45). Therefore, while PPM1D directly inhibits p53-Ser15-dependent roles, other p53-initiated programs that are dependent on ubiquitination or acetylation may not be affected. Thus, to understand the impact of PPM1D alterations, it is important to determine the phenotypic, as well as mechanistic relationship to TP53 alterations. A recent study found that germline overexpression of human PPM1D in mice could induce tumors that were phenotypically similar to those developed in mouse models with TP53 mutations (24). Here, they exposed PPM1D mice to sub-lethal whole-body irradiation. Interestingly, they observed that the tumor spectrum was more comparable to that of TP53 loss-of-function mouse models rather than TP53 knockout mice. These findings support the hypothesis that PPM1D overexpression leads to only partial impairment of p53.
As PPM1D and TP53 mutations both act through similar signaling pathways, we would expect functional redundancy to having both a knockdown of p53 and an upregulation of PPM1D within the same cell. Indeed, early studies showed that PPM1D genetic alterations and TP53 mutations appear to be mutually exclusive in solid cancers. For example, PPM1D amplifications were almost exclusively found in TP53 wild-type tumors in one breast cancer study (21), and PPM1D truncating mutations and TP53 inactivating mutations were mutually exclusive in brainstem gliomas (8,46). These reports suggest that, in certain contexts, mutations in both genes confer minimal additional advantage over mutations in either gene alone. Similarly, a recent analysis of 10,225 patients from The Cancer Genome Atlas database revealed that PPM1D is amplified significantly more often in TP53 wild-type than TP53-mutant tumors (47). Yet, other studies have observed PPM1D amplifications and mutations to co-occur with TP53 mutations in some tumor samples (20,48). This conflict suggests that PPM1D may confer additional advantages to cancer cells through p53-independent mechanisms, including through mTOR signaling pathways, DNA repair pathways (NER and BER), and NF-kB signaling pathway, among others (34,36,49).
In the blood, true co-mutations are more difficult to identify, as bulk sample sequencing does not distinguish between mutations in separate sub-clones versus within the same cell. PPM1D mutations have been reported to be co-mutated with TP53 more frequently than expected by chance alone in therapy-related myelodysplastic syndrome (t-MDS) (11), and mutations in both genes are enriched after exposure to chemotherapy (12). Yet, single cell genome sequencing studies recently revealed that PPM1D and TP53 mutations were typically present in separate clones in the blood (50). This finding is consistent with the hypothesis of functional redundancy. If PPM1D acts through similar pathways as p53, one would expect PPM1D and TP53 mutant cells to have similar mutational profiles and prognosis. However, in t-MDS, the presence of TP53 mutations was strongly associated with complex chromosomal abnormalities, whereas the presence of PPM1D mutations without concurrent TP53 mutations was associated with lower frequencies of complex karyotypes at frequencies comparable to TP53 and PPM1D wildtype cases. It is possible that partial suppression of p53 activity by PPM1D results in less genome instability than complete loss-of-function TP53 mutations. Additionally, complex karyotype is only one form of genetic alteration, and PPM1D mutants could have additional alterations or mutational signatures distinct from TP53 mutants. Comparing the mutation burden and signatures between PPM1D and TP53 mutants through whole genome sequencing studies would shed light on the degree of functional overlap between the two genes.
Lastly, TP53 mutations are associated with a poorer overall prognosis than PPM1D mutations in t-MDS and are also much more prevalent in de novo leukemias (11). These findings suggest that loss of TP53 is a more potent oncogenic driver than excess PPM1D, at least in the blood. As both mutations are enriched following exposure to chemotherapy and are highly prevalent in therapy-related acute myeloid leukemia (t-AML) and t-MDS, clonal expansion of PPM1D mutants may be preferable. In fact, an expanded PPM1D clone with high fitness may help suppress the rise of more potent oncogenic clones in the bone marrow and blood. Perhaps, in this way, PPM1D mutants can be viewed as a “friend” compared to TP53 mutant clones which have higher potential for malignant transformation. It would be interesting to experimentally compete TP53 and PPM1D mutant clones head-to-head in the blood under varying conditions of stress. If PPM1D mutants do have a competitive advantage in certain stress conditions, we could infer that these cells function through p53-independent mechanisms. In contrast, if PPM1D acts predominantly through TP53, one would expect PPM1D mutants to have similar, if not lower, competitiveness compared to TP53 mutants. Overall, these studies may help to illuminate why PPM1D mutations are more enriched in t-AML, but not in other types of de novo hematologic and solid malignancies compared to TP53 mutations.
PPM1D in Solid Cancers
PPM1D amplifications were first found in human cancers in the early 2000s, shortly after the gene was discovered. Initial studies used microarray-based comparative genomic hybridization to identify amplification of chromosome region 17q23 harboring the PPM1D locus in 11-16% of primary breast cancer samples (4,7,21). Importantly, PPM1D amplifications were found to be associated with poorer survival and more aggressive disease in breast cancer patients (4,21). These early findings established PPM1D as a potential oncogene in cancer research and were soon followed by numerous studies that identified either PPM1D genomic amplifications or increased PPM1D gene expression in a wide variety of other solid tumors including neuroblastoma (1,36) medulloblastoma (51,52), esophageal squamous cell carcinoma (53), and more (summarized in Table 1). Several recurring features and characteristics are notable across PPM1D-overexpressing cancer types. For example, PPM1D overexpression is associated with significantly decreased overall, recurrence-free, or 5-year survival (4,54–57), lymph node metastases as well as distant metastases, and advanced tumor stage across several different solid cancer types (14–16,23,52,55,58–60). Broadly, these findings suggest that PPM1D overexpression can serve as a valuable prognostic factor for risk stratification of solid cancer patients.
Like PPM1D amplifications, PPM1D truncating mutations have also been identified across solid cancers (8,25,48,61). PPM1D mutations have been established as oncogenic drivers in de novo diffuse midline glioma formation and is required for in vivo gliomagenesis (62). Sequencing tumor tissue from larger patient cohorts will illuminate whether, like PPM1D amplifications, patients with tumors harboring truncating PPM1D mutations have worse survival outcomes and metastatic potential than those without. In other solid cancer types, PPM1D mutations seems to play more of a supporting oncogenic role. In a colorectal cancer study, mice with truncated Ppm1d (Ppm1dT/+) were crossed with mice harboring an inactivating mutation in the tumor suppressor Apc (Apcmin). Double mutant mice had significantly increased colonic polyps, accelerated tumor formation, and greater tumor penetrance compared to Apcmin mice alone. However, no intestinal polyps were found in Ppm1dT/+ mice, suggesting that Ppm1d mutations alone are not potent enough to drive tumor initiation. Furthermore, organoids derived from Ppm1dT/+/Apcmin mice were resistant to 5-fluorouracil (5-FU) treatment and sensitivity was restored after pharmacological inhibition of PPM1D (63). Overall, PPM1D-mutant cancer cells appear to have a similar chemoresistance phenotype as PPM1D-overexpressing cancer cells, and PPM1D inhibitors can potentially work synergistically with traditional chemotherapeutic agents in both instances.
PPM1D Mutations in the Blood – a top hit in clonal hematopoiesis
In 2014, multiple landmark studies described the phenomenon of clonal hematopoiesis (CH), where large clones derived from single hematopoietic stem cells (HSCs) were found to comprise a significant proportion of peripheral blood in individuals with no history of hematologic diseases. These expanded cell populations often harbor somatic mutations in one of several recurrently mutated genes. In some studies, PPM1D was found to be one of the most mutated genes in individuals with CH (17,64). CH has since been found to be associated with an increased risk of hematologic malignancies (17), cardiovascular disease (65), and increased all-cause mortality (66). Around the same time as the discovery of CH, a series of large-cohort studies reported an enrichment of PPM1D truncating mutations in the peripheral blood of solid cancer patients compared to control patients without cancer. Since PPM1D mutations were previously associated with various solid tumors, and the blood samples were thought to represent “germline” variants, the studies speculated that PPM1D germline mutations could be a risk factor or biomarker for development of these solid cancers (9,25). However, additional studies that included analysis of matched blood and lung tumor samples revealed a discordance; PPM1D mutations were detected in the blood but not in the tumor (10). These findings suggested that PPM1D mutations had a hematopoietic origin and reflected somatic, not germline, mutations of PPM1D followed by clonal expansion of mutant hematopoietic cells.
Indeed, the observed frequency of PPM1D mutations in the blood of these solid cancer patients (0.2-1.5%) was similar to that reported in the general population (0.12%) (17). Furthermore, a breast cancer cohort study noted that the presence of PPM1D truncating variants in the blood was positively correlated with age (67), which is consistent with age-related CH. However, the enrichment of PPM1D mutations in the blood in association with patients having solid tumors suggested that other variables besides age may contribute. Indeed, it was noted that all ovarian cancer patients reported to exhibit PPM1D mutations had previously received chemotherapy treatment (68). Subsequent large-scale sequencing studies in patients who had been treated for solid tumors (MSK-IMPACT) validated the significant association between somatic PPM1D mutations in the blood and prior chemotherapy exposure (69). Together, these studies pointed to the concept of therapy-related clonal hematopoiesis and the view that PPM1D mutations can occur in aging-related CH, but that they are highly enriched after exposure to chemotherapy due to clear positive selection for mutant clones in the blood.
PPM1D-related CH not only has implications for leukemia predisposition but has also been shown to be associated with worse outcomes after autologous stem cell transplantation (66), promote heart failure (70), and alter immune cell function (71) in murine models. Recently, Ppm1d overexpression in murine immune cells was found to alter the degree of immunosuppression within the tumor microenvironment to increase tumor progression. This finding would suggest that PPM1D clonal hematopoiesis could impact disease progression and outcomes for patients that have PPM1D-wild-type solid tumors. Pharmacologic inhibition of PPM1D could reprogram Ppm1d-mutant neutrophils towards a higher antitumor potential by promoting tumor infiltration (71). These findings expand on the clinical relevance of PPM1D CH and highlight the potential therapeutic benefits of PPM1D inhibitors.
PPM1D Mutations in the Blood – a bystander in AML?
From the perspective of hematologic malignancies, PPM1D mutations appear in specific subsets of disease. Notably, PPM1D mutations are significantly more common in t-AML/t-MDS compared to primary AML and MDS (11,12). In-depth analysis revealed that PPM1D mutations are significantly associated with prior exposure to specific genotoxic agents, including platinum therapy, topoisomerase I and II inhibitors, and radiation therapy (69,72). Comparable to its frequency in CH, PPM1D is the eighth most mutated gene in myeloproliferative neoplasms (MPNs). One study found PPM1D mutations in 19% of MPN patients, of whom 10 carried the PPM1D mutation only after treatment with hydroxyurea. Generally, PPM1D mutations were found to be acquired later in disease based on mutational tree mapping (73).
Regarding the prognostic impact of these mutations, we found no significant difference in overall survival between PPM1D-mutated and non-PPM1D mutated cases of t-AML/t-MDS, respectively (12). Similarly, while PPM1D mutations were associated with a significant hazard ratio of death of 1.64 (adjusted p=0.002), further stratification revealed that PPM1D mutations were not associated with adverse prognoses in patients without co-existing TP53 mutations (11). Even in de novo AML and MDS, PPM1D mutations do not appear to be associated with worse overall outcomes. Consistent with earlier studies, Al Hinai et. al identified PPM1D truncating mutations in 0.6% of newly diagnosed AML, with sizable clones (VAF~45%) in 3 of the cases. As this cohort was followed into clinical remission after chemotherapy treatment, the frequency of PPM1D mutations increased to 4% cases. Yet, the presence PPM1D mutations in clinical remission do not appear to predict AML relapse (74). Another study of MDS patients with deletion of chromosome 5q identified PPM1D truncating mutations in 5.6% cases and TP53 monoallelic mutations in 15% cases, but observed that the rate of disease progression and lenalidomide resistance was independent of mutation status in either gene. However, lenalidomide resistance was associated with the acquisition and expansion of novel PPM1D and TP53 mutations (75). The findings from both studies suggest that treatment may induce the acquisition of new mutations and confer a selective fitness advantage to hematopoietic clones harboring PPM1D mutations. However, the contribution of these mutant clones to disease progression is unclear. Several cases of PPM1D truncating mutations have been identified in pediatric therapy-related myeloid neoplasms, although it remains unknown whether these PPM1D mutations were present pre-treatment (76). These observations lead to two models on the origin and evolution of these mutations: 1) cytotoxic therapies directly induce the mutations, which then clonally expand; or 2) the mutations were initially present at low variant allele frequencies, which may be undetectable depending on depth of sequencing, but preferentially survive and repopulate the hematopoietic compartment after exposure to cytotoxic therapy. Recent studies utilizing whole-genome sequencing of single-cell derived hematopoietic colonies have suggested that PPM1D CH-associated mutations can occur early in life, and even in utero (77). Additionally, the detection of the same somatic mutations with deep sequencing before and after cytotoxic exposure in multiple cases appears to support the latter model (78–80), but the models are not necessary mutually exclusive.
The Role of PPM1D in Oncogenesis
PPM1D has emerged as an oncogenic candidate due to its inhibitory effects on multiple tumor suppressors and DDR regulators. Several studies have experimentally demonstrated that Ppm1d overexpression accelerates transformation of mouse embryonic fibroblasts (MEFs) in cooperation with other classic oncogenes such as Hras, Neu1, and Myc, compared to either Ppm1d overexpression or activated oncogenes alone (7,81,82). Conversely, deletion of Ppm1d yielded the opposite phenotype, with suppression of oncogene-driven transformation of MEFs. Loss of Ppm1d impairs carcinogenesis in two mammary tumor models (82) and Ppm1d knock-out mice have lower lifetime incidences of cancer (9–11,13,51). Therefore, PPM1D overexpression and PPM1D deletion represent opposite ends of the oncogenic spectrum in cancer: overexpression confers a pro-oncogenic effect, whereas deletion confers an anti-oncogenic effect, supporting a significant contributing role of PPM1D in oncogenesis.
The oncogenic effects of PPM1D do not necessarily require genetic modification via gene duplication or activating mutations. Numerous solid cancer cell lines and patient samples have been shown to upregulate PPM1D at the mRNA level without copy number gains (14,15,52,83,84). This is also true in several leukemia cell lines and primary human AML samples (13). However, PPM1D expression in AML seemed to vary according to cytogenetic and molecular status, owing to the heterogeneity and complexity of leukemia development (85). Nevertheless, these findings indicate that upregulation of PPM1D supports survival and disease progression in both solid and hematologic malignancies. While mechanisms of PPM1D-mediate oncogenesis based on cancer type has been reviewed recently by others (86), in this section, we will explore the overarching mechanisms in which PPM1D overexpression promotes tumorigenesis. These mechanisms converge on decreased cell cycle arrest, resistance to apoptosis, and increased metastatic potential.
Decreased cell cycle arrest
One of the key mechanisms by which PPM1D-overexpressing cells can gain proliferative capacity is through the loss of cycle cell arrest. Upon sensing DNA damage, the DDR is activated to stop normal cellular functions to allow the resolution of damaged DNA prior to DNA replication. This process is critical to maintaining genome integrity by preventing the propagation of harmful genetic lesions. PPM1D-overexpressing cells dysregulate cell cycle checkpoints by persistent dephosphorylation and inactivation of several cell cycle regulators including ATM (29), CHK1 (5), CHK2 (30), and p53 (5) (Figure 3a).
Figure 3. PPM1D-mediated suppression of cell cycle arrest and apoptosis.
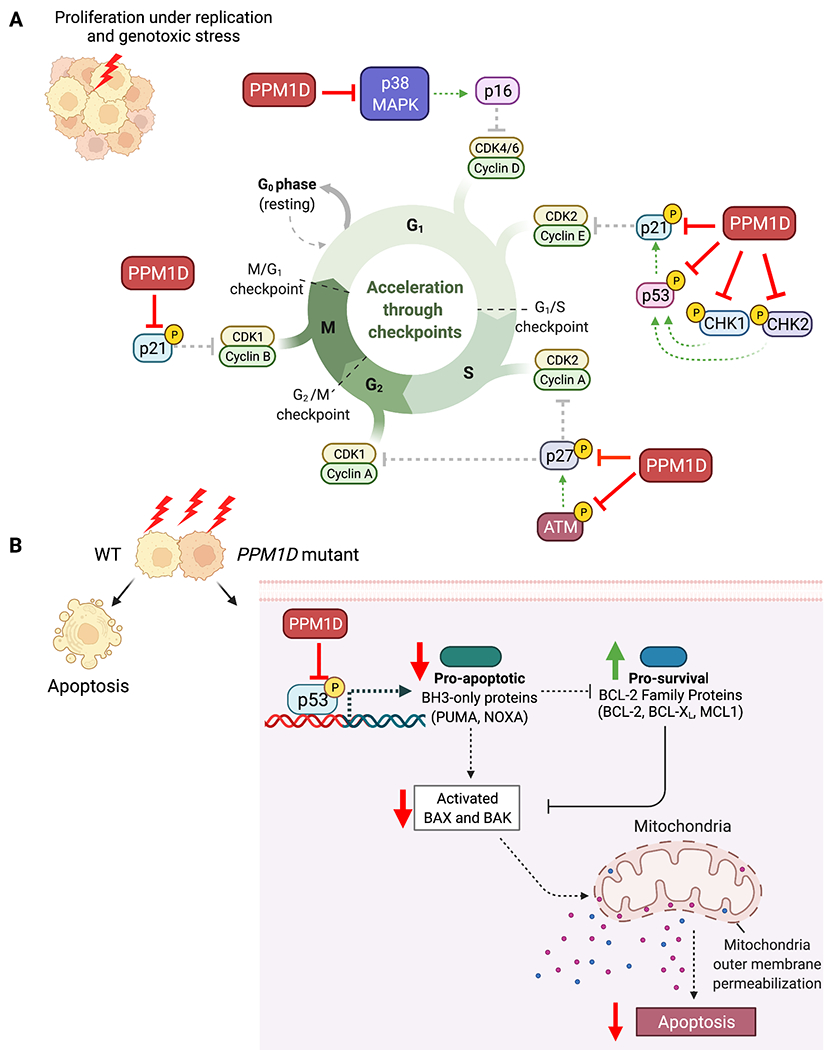
A) PPM1D inhibits key cell cycle regulators including p53, CHK, CHK2, p21, p27, and p38 leading to loss of cell cycle arrest. B) PPM1D inhibits p53 leading to loss of pro-apoptotic factors resulting in suppression of apoptosis.
One of the downstream targets of ATM is the cyclin-dependent kinase (CDK) inhibitor, p27Kip1, which binds to CDK2 and inhibits G1 progression into S phase. PPM1D not only impairs this cell cycle checkpoint through ATM dephosphorylation, but it also dephosphorylates p27Kip1 at S140 (87). Additionally, CHK1 and CHK2 both phosphorylate p53, leading to upregulation of another CDK inhibitor, p21Cip1 (88). Truncated PPM1D was found to suppress the expression of Cdkn1a (the gene encoding p21) after ionizing radiation exposure in the mouse colon (63) due to the inhibition of p53. PPM1D also dephosphorylates p38 mitogen-activated protein kinase (MAPK) which leads to decreased expression of the CDK inhibitor, p16, in human breast cancer (82,89) and non-small cell lung cancer patients (90).
Several studies in various PPM1D-mutated solid cancer cell lines also exhibited impaired cell cycle arrest in vitro (14,15,58,84). U2OS (osteosarcoma) and HCT116 (colorectal) cell lines harboring endogenous PPM1D mutations have impaired G1 cell cycle arrest after IR (25). Similarly, PPM1D-mutated retinal pigmented epithelial (RPE) cell lines and primary mouse neuronal stem cells (mNSCs) also failed to arrest at the G1 and G2 checkpoint after IR and continued to proliferate under genotoxic stress (62,63). Phosphoproteomic studies done in PPM1D-mutant mNSCs and patient-derived PPM1D mutant glioma cell lines showed that proteins related to cell cycling and DNA damage were differentially dephosphorylated in the PPM1D-mutated samples (62). In hematologic malignancies, PPM1D mutant leukemia cells also exhibit increased cell cycle progression to G2/M and proliferative advantages following exposure to cytarabine (18). Normal HSCs must persist throughout one’s lifetime in order to reconstitute the hematopoietic system. Therefore, cell cycle regulation is critical to maintaining normal HSC function over time. It is currently unknown how PPM1D mutations may affect HSC cell cycling, activation, and quiescence. However, given the dormant nature of HSCs, this could potentially give us clues as to why PPM1D mutations are not as prevalent in hematologic malignancies.
Several studies have shown that inhibition of PPM1D by RNA interference leads to reduced cell proliferation and colony formation ability in thyroid, colorectal, and lung cancer cell lines (84,90–92). This loss of proliferation was accompanied by increased G0/G1 cell cycle arrest and accumulation of cells in the sub-G1 phase. In addition, there was a significant downregulation of cyclin B1 in both lung and colorectal cell lines, suggesting another possible mechanism by which PPM1D mutant cells escape cell cycle arrest (91,92). In papillary thyroid cancer cell lines, siRNA knockdown of PPM1D decreased proliferation with a concurrent increase in phospho-p38 MAPK and p53. Interestingly, chemical inhibition of p38 restored the proliferative and colony-forming abilities of PPM1D knockdown cells, indicating that suppression of the p38 MAPK pathway is a mechanism by which PPM1D overexpression promotes proliferation. Overall, PPM1D overexpression can affect multiple pathways that result in abnormal cellular proliferation under external stress that can lead to the accumulation and propagation of genetic mutations.
Resistance to apoptosis
Resistance to apoptosis is another key feature of PPM1D overexpressing cells. P53 induces apoptosis by transcriptional activation of the pro-apoptotic BH3-only family members PUMA and NOXA. These proteins control cell death by inhibiting the pro-survival BCL2 family proteins, resulting in the de-repression of the cell death effectors, BAX and BAK. Activation of BAX and BAK lead to mitochondrial outer membrane permeabilization leading to caspase activation and subsequent apoptosis (Figure 3B) (93). Activation of apoptosis during the DNA damage response allows the elimination of cells with unrepaired DNA lesions. Therefore, PPM1D overexpression prevents p53-mediated induction of these pro-apoptotic factors to allow cells to escape apoptosis. In acute myeloid leukemia, pancreatic, ovarian, and papillary thyroid cancer cell lines, PPM1D was also found to suppress apoptosis through dephosphorylation of p38 MAPK, which cross-talks with the p53 pathway (13,84,94,95). Furthermore, PPM1D knockdown increased apoptosis, which was reversed by inhibition of p38 phosphorylation (94). This finding once again demonstrates how PPM1D-overexpressing cells act through the p38 MAPK pathway to escape both apoptosis and cell cycle arrest.
The resistance of PPM1D-overexpressing cells to apoptosis also extends to stress conditions, such as exposure to chemotherapy and IR. In medulloblastoma cells, PPM1D overexpression inhibited p53-mediated apoptosis and cell cycle arrest following etoposide treatment (52). We and others have demonstrated that PPM1D mutant HSCs undergo less apoptosis compared to wildtype cells after chemotherapeutic insults and ionizing radiation (12,96). Strikingly, this difference in apoptosis is compounded with multiple rounds of chemotherapy, such that a small proportion of PPM1D mutant HSCs can significantly outcompete wildtype cells following multiple treatments. In Ppm1d-truncating mutant mouse models, mutant thymocytes were found to not only have impaired DDR and cell cycle arrest, but also decreased apoptosis in response to IR. The net effects of these impairments allowed the propagation of cells with improperly repaired lesions and promoted the formation of IR-induced lymphoma (96).
Conversely, PPM1D inhibition has been shown to promote chemosensitivity in colon cancer cells through increased apoptosis following exposure to oxaliplatin, 5-FU, and adriamycin (97). In MCF-7 breast cancer cells, downregulation of PPM1D was also able to sensitize cells to doxorubicin-induced apoptosis through p53 activation of Bax (98). Whether PPM1D amplifications or mutations are enriched in the subset of patients who are refractory to cancer treatment or have disease recurrence remains an open question of great clinical interest. Addressing this gap may highlight the need to develop clinically effective PPM1D inhibitors to resensitize PPM1D-overexpressing tumors to chemotherapy.
Increased metastatic potential
Lastly, PPM1D overexpressing solid cancers have an increased tendency to metastasize to lymph nodes and distant sites and there are several proposed mechanisms by which PPM1D promotes this invasive behavior and migration. Buss et. al observed that PPM1D expression is significantly increased in metastatic medulloblastoma, and that cells with high PPM1D expression have increased levels of CXCR4, a cell surface chemokine receptor that is associated with metastatic behavior. Stimulation of medulloblastoma cells with the CXCR4-ligand, SDF, activated PI-3K signaling and promoted growth and invasion in a p53-dependent manner. In contrast, knocking-out PPM1D decreased cell surface accumulation of CXCR4 and inhibited migration and invasion (55).
In pancreatic cancer, PPM1D was shown to promote cell migration and invasion through the Wnt/B-catenin pathway via downregulation of the tumor suppressor ASPP2 (94). Studies in other solid cancer cell lines have also shown that PPM1D expression is positively correlated with the expression of matrix metallopeptidase 2 and 9 (MMP-2 and MMP-9), enzymes that degrade extracellular matrix, plakophilin-2 (PKP-2), a positive regulator of Endothelial Growth Factor Receptor signaling, and vascular endothelial growth factor C (VEGF-C), a potent angiogenic factor that mediates metastasis to lymph nodes. Consistent with these findings, knockdown of PPM1D reduced MMP-9, PKP2 and VEGF-C expression and inhibited invasion and migration. On the contrary, overexpression of MMP-9, PKP2, or VEGF-C in a PPM1D knockdown background restored the invasive and migratory phenotype. These studies point to MMP-9, PKP2, and VEGF-C as likely downstream targets of PPM1D activity that are yet to be explored (57–59,99).
Although the hematopoietic system does not fall under the same physical restraints as solid cancers, the effects of PPM1D-mutant cells on the bone marrow microenvironment has yet to be elucidated. Studies have shown that Ppm1d-mutant macrophages exhibit a more proinflammatory profile (70). On the other hand, PPM1D has been shown to negatively regulate NFkB and TGFß signaling (100,101). It would be of interest to study how Ppm1d-mutant progenitors in the bone marrow might modulate the activation and differentiation of neighboring HSCs to either promote or suppress growth of other malignant clones.
PPM1D in Solid vs. Hematologic Malignancies
The role of PPM1D activating mutations as a supporting oncogene in solid cancer is bolstered by compelling clinical evidence demonstrating that increased PPM1D mRNA expression is not only present in a significant subset of tumors but is also associated with more aggressive disease and worse survival outcomes. In contrast to solid malignancies, PPM1D mutations appear to play a distinct, more passive role in the hematologic realm. In cases where the frequency of PPM1D mutations is lower than the leukemia blast percentage, PPM1D likely plays the role of a passive bystander that is positively selected for following exposure to therapy and is clonally distinct from the driver clone (Figure 4). Another possibility is that PPM1D mutant clones could act as active bystanders, where they indirectly promote disease progression through alterations of the microenvironment or cell competition dynamics in the bone marrow, although this remains an open question. Future work is needed to clarify the precise role of PPM1D in hematologic malignancies, particularly the discrepancy between its prevalence in clonal expansion and in de novo malignancy.
Figure 4. Potential roles of PPM1D mutations in the blood.
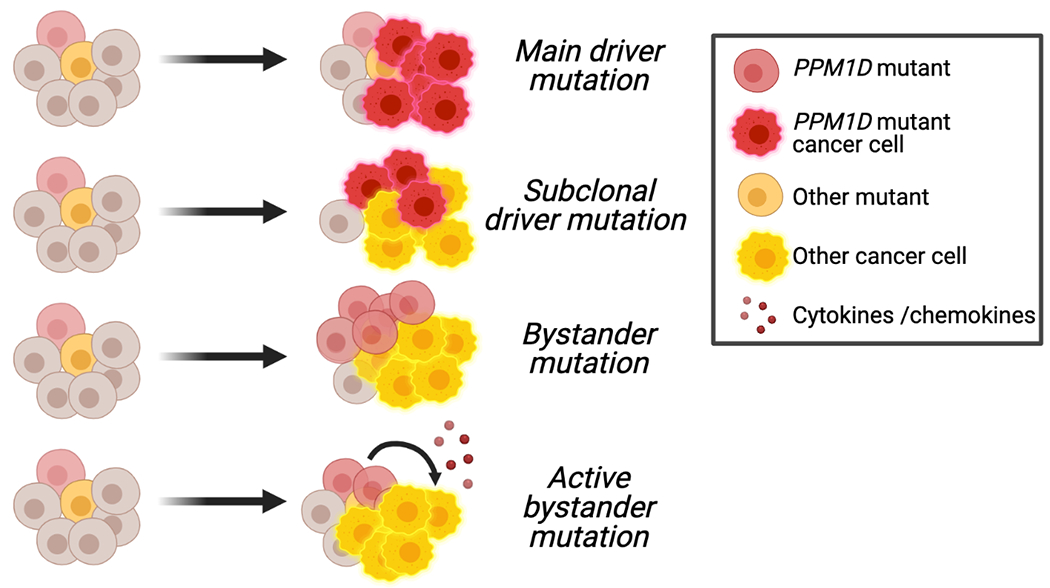
Schematic showing the potential roles of PPM1D mutations as driver, subclonal, bystander, or active bystander mutations in leukemogenesis. The precise role is unknown.
Given that PPM1D affects many DDR and stress response pathways, it is possible that PPM1D mutants have increased suppression of these pathways leading to genomic instability and accumulation of DNA damage over time. Whether PPM1D mutants have an increased overall mutation burden at baseline and following exposure to genotoxic stresses is under active investigation. One plausible hypothesis is that PPM1D mutations potentiate the acquisition of additional mutations, which then work cooperatively with PPM1D to accelerate the initiation or progression of cancer. Indeed, PPM1D had been found to broadly co-mutate with multiple other genes in solid tumors, t-AML and t-MDS (12). Systematic study of the effect of different cooperating mutations on the rate of transformation in different cancer contexts will enable us to better understand the potency of PPM1D amplification and mutations as an oncogenic driver in hematologic malignancies.
The frequent appearance of PPM1D mutations and amplifications in cancer also suggests an underlying competitive advantage that may precede the manifestation of disease. Broadly, there are two possibilities for this fitness advantage: an intrinsic advantage independent of stressors and/or an advantage that is dependent on the presence of external stressors. We have discussed the intrinsic cellular advantages in the previous sections in which PPM1D overexpression can drive suppression of cell cycle arrest and apoptosis. In contrast to solid tumors, where PPM1D overexpression promotes fitness in cooperation with other oncogenes, external stressors appear to play a more major role in the expansion of PPM1D mutant clones in the hematologic setting (Figure 5). Selection of PPM1D mutant clones under cytotoxic therapies in the hematopoietic setting raises an interesting question of whether PPM1D mutant cells are similarly selected for in solid tumors following exposure to chemotherapy. While solid tumors (i.e., ovarian cancer) refractory to cisplatin treatment may be enriched for PPM1D mutations, this has not yet been explored. In support of this possibility, pre-malignant PPM1D clonal expansion has recently been observed in non-hematopoietic tissues, including the esophageal lining. One study identified clones harboring PPM1D somatic mutations in 13% of normal esophageal epithelial samples (102). In several elderly individuals, the mutated clones were found to have expanded and replaced the majority of normal epithelium. Of note, the authors observed that heavy alcohol consumption and tobacco use, both of which are known mutagenic agents, substantially accelerate clonal expansion in the esophagus. Interestingly, the aldehyde metabolites from alcohol have been shown to cause DNA DSBs and chromosomal rearrangements, reminiscent of genotoxic therapy (103). Therefore, environmental exposures may also play a significant role in the expansion of premalignant PPM1D-mutant clones in normal, non-hematopoietic tissues. Altogether, these findings suggest that various environmental exposures associated with cellular or genotoxic stress can affect PPM1D clonal dynamics and transformation into malignancy.
Figure 5. Environmental conditions that promote the selection of PPM1D mutants in the blood and esophageal lining.
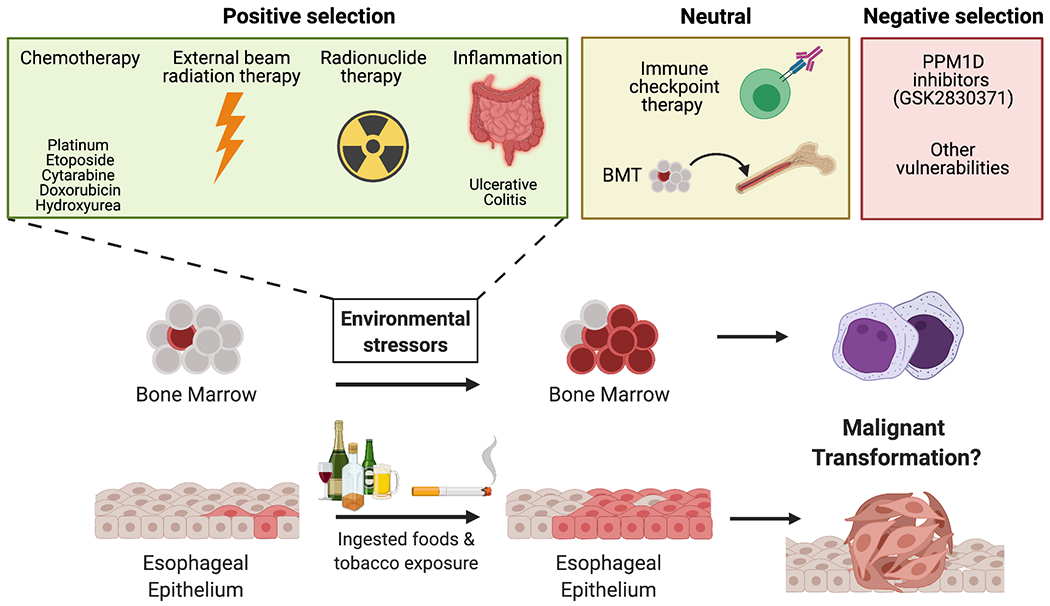
Schematic representation of how premalignant clonal expansion of PPM1D mutants in the blood and esophagus is shaped by multiple environmental stressors. These stressors can have a positive, neutral, negative effect on PPM1D mutant clonal dynamics.
Concluding Remarks
Through the suppression of DNA repair, cell cycle, and apoptosis, PPM1D overexpression can drive uncontrolled cellular growth to promote malignancy. Further studies on PPM1D will lead to a more complete understanding of the mechanisms through which PPM1D promotes oncogenesis, particularly in the context of its divergent role in solid and liquid malignancies. It remains an open question as to what the exact significance of PPM1D genomic aberrations is in normal tissues and pre-malignant states. Could PPM1D act as a “friend” by competing with and suppressing the growth of other, more “oncogenic” clones such as TP53 mutants? At what point does it turn from being a “friend” into a “foe”? Studies have demonstrated that PPM1D is more tumorigenic when it cooperates with other oncogenes (63). Inclusion of PPM1D in sequencing panels for normal and malignant tumor and blood samples will increase our knowledge of cooperating mutations and expand our understanding of the clinical implications of PPM1D overexpression.
PPM1D is an attractive therapeutic target given its prevalence in many cancers and its oncogenic potential. Yet, we are still lacking a clinically effective small molecular inhibitor. However, our understanding of PPM1D in the DNA damage response lends new strategies for cancer therapies. Several studies have demonstrated that inhibition of PPM1D in vitro can modulate the sensitivity of cancer cells to other DDR-targeted therapies including PARP inhibition (41) and MDM2 antagonists (85,104). Further investigations could yield additional druggable targets that either sensitize or confer synthetic lethality to cells bearing PPM1D mutations, an avenue that remains to be explored and would contribute greatly towards PPM1D-specific therapeutic development.
Table 2.
Summary of PPM1D genetic amplification and mutations in solid cancers.
| Cancer Type | PPM1D Status | % | Prognosis (if available) and Characteristics | Ref |
|---|---|---|---|---|
| Ovarian | Amplification | 10% | Silencing of PPM1D in vitro led to reduced cell survival | (22) |
| Breast | Amplification | 16% | Attenuation of apoptosis in vitro. Cooperated with RAS to transform primary MEFs. | (7) |
| Amplification | 11% | Associated with poor prognosis. | (4) | |
| Amplification | 6% | More prevalent in HER2+ breast cancers (19%) No association between PPM1D gene amplification or overexpression with disease-free, metastasis-free, or overall survival. | (20) | |
| Neuroblastoma | Amplification | 28% (9/32) | High expression of PPM1D correlated with significantly worse survival outcomes | (54) |
| Medulloblastoma | Amplification and Overexpression | 64% | Increased PPM1D expression associated with metastasis and decreased survival. Associated with CXCR4 and GRK5 upregulation. | (51,55,83) |
| Pancreatic Adenocarcinoma | Amplification | 51% (86/169) | 43% had metastatic disease at follow-up and harbored at least one mutation in MDM2, MDM4, or WIP1. | (23) |
| Overexpression | 55% | PPM1D expression positively correlated with tumor grade; promotes cell migration and invasion in vitro & tumor growth in vivo. | (94) | |
| Colorectal | Overexpression | 68% (252/368) | PPM1D expression significantly increased in tumors with nodal and distant metastasis and advanced TNM stages. | (14) |
| Papillary Thyroid | Overexpression | 63% (56/89) | PPM1D expression positively correlated with tumor size and lymph node metastasis. | (84) |
| Prostate Cancer | Overexpression | 56.4% (132/234) | PPM1D expression positively correlated with Gleason score, T-stage, lymph node status, and shorter biochemical recurrence-free survival, and decreased overall survival. | (15) |
| Salivary Carcinoma | Overexpression | 100% (82/82) | Correlated with malignant disease and poor prognosis. | (57) |
| Non-Small Cell Lung Cancer | Overexpression | 69% (52/75) | Positively correlated with clinical stage, lymph node metastasis, and pathological differentiation. | (16) |
| Nasopharyngeal Carcinoma | Overexpression | Unclear | Positively correlated with advanced clinical stage, lymph node metastasis, response to ionizing radiation; poor 5-year survival. | (58) |
| Renal Cell Carcinoma | Overexpression | 68% (53/78) | Positively correlated with T stages, lymph node metastasis, clinical stages and tumor differentiation, with poor overall survival. | (60) |
| Esophageal Squamous Cell Carcinoma | Overexpression | 69% (70/101) | Poor prognosis, lymph node metastasis, inferior 5-year survival. | (53) |
| Osteosarcoma | Overexpression | 51% (23/45) | Higher levels of PPM1D detected in patients with distant metastasis and unfavorable prognosis. | (59) |
| Glioma | Truncating Mutation | 23% (3/13) 18% |
PPM1D mutations mutually exclusive with TP53 mutations but always found in conjunction with NF1 mutations and frequently with H3F3A. | (8,46) |
Acknowledgements
Research focusing on PPM1D in M. Goodell’s laboratory is funded by the National Cancer Institute (CA237291) and supports L. Zhang and J. Hsu.
Footnotes
Authors’Disclosures: The authors declare no potential conflicts of interest.
References
- 1.Mantovani F, Collavin L, Del Sal G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ 2019;26(2):199–212 doi 10.1038/s41418-018-0246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams AB, Schumacher B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb Perspect Med 2016;6(5) doi 10.1101/cshperspect.a026070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fiscella M, Zhang H, Fan S, Sakaguchi K, Shen S, Mercer WE, et al. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc Natl Acad Sci U S A 1997;94(12):6048–53 doi 10.1073/pnas.94.12.6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bulavin DV, Demidov ON, Saito S, Kauraniemi P, Phillips C, Amundson SA, et al. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat Genet 2002;31(2):210–5 doi 10.1038/ng894. [DOI] [PubMed] [Google Scholar]
- 5.Lu X, Nannenga B, Donehower LA. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev 2005;19(10):1162–74 doi 10.1101/gad.1291305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu X, Ma O, Nguyen TA, Jones SN, Oren M, Donehower LA. The Wip1 Phosphatase acts as a gatekeeper in the p53-Mdm2 autoregulatory loop. Cancer Cell 2007;12(4):342–54 doi 10.1016/j.ccr.2007.08.033. [DOI] [PubMed] [Google Scholar]
- 7.Li J, Yang Y, Peng Y, Austin RJ, van Eyndhoven WG, Nguyen KC, et al. Oncogenic properties of PPM1D located within a breast cancer amplification epicenter at 17q23. Nat Genet 2002;31(2):133–4 doi 10.1038/ng888. [DOI] [PubMed] [Google Scholar]
- 8.Zhang L, Chen LH, Wan H, Yang R, Wang Z, Feng J, et al. Exome sequencing identifies somatic gain-of-function PPM1D mutations in brainstem gliomas. Nat Genet 2014;46(7):726–30 doi 10.1038/ng.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruark E, Snape K, Humburg P, Loveday C, Bajrami I, Brough R, et al. Mosaic PPM1D mutations are associated with predisposition to breast and ovarian cancer. Nature 2013;493(7432):406–10 doi 10.1038/nature11725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wen J, Lee J, Malhotra A, Nahta R, Arnold AR, Buss MC, et al. WIP1 modulates responsiveness to Sonic Hedgehog signaling in neuronal precursor cells and medulloblastoma. Oncogene 2016;35(42):5552–64 doi 10.1038/onc.2016.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindsley RC, Saber W, Mar BG, Redd R, Wang T, Haagenson MD, et al. Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. N Engl J Med 2017;376(6):536–47 doi 10.1056/NEJMoa1611604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsu JI, Dayaram T, Tovy A, De Braekeleer E, Jeong M, Wang F, et al. PPM1D Mutations Drive Clonal Hematopoiesis in Response to Cytotoxic Chemotherapy. Cell Stem Cell 2018;23(5):700–13 e6 doi 10.1016/j.stem.2018.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li B, Hu J, He D, Chen Q, Liu S, Zhu X, et al. PPM1D Knockdown Suppresses Cell Proliferation, Promotes Cell Apoptosis, and Activates p38 MAPK/p53 Signaling Pathway in Acute Myeloid Leukemia. Technol Cancer Res Treat 2020;19:1533033820942312 doi 10.1177/1533033820942312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peng TS, He YH, Nie T, Hu XD, Lu HY, Yi J, et al. PPM1D is a prognostic marker and therapeutic target in colorectal cancer. Exp Ther Med 2014;8(2):430–4 doi 10.3892/etm.2014.1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiao L, Shen D, Liu G, Jia J, Geng J, Wang H, et al. PPM1D as a novel biomarker for prostate cancer after radical prostatectomy. Anticancer Res 2014;34(6):2919–25. [PubMed] [Google Scholar]
- 16.Fu Z, Sun G, Gu T. Proto-oncogene Wip1, a member of a new family of proliferative genes in NSCLC and its clinical significance. Tumour Biol 2014;35(4):2975–81 doi 10.1007/s13277-013-1382-y. [DOI] [PubMed] [Google Scholar]
- 17.Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371(26):2477–87 doi 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kahn JD, Miller PG, Silver AJ, Sellar RS, Bhatt S, Gibson C, et al. PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood 2018;132(11):1095–105 doi 10.1182/blood-2018-05-850339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi J, Appella E, Donehower LA. The structure and expression of the murine wildtype p53-induced phosphatase 1 (Wip1) gene. Genomics 2000;64(3):298–306 doi 10.1006/geno.2000.6134. [DOI] [PubMed] [Google Scholar]
- 20.Lambros MB, Natrajan R, Geyer FC, Lopez-Garcia MA, Dedes KJ, Savage K, et al. PPM1D gene amplification and overexpression in breast cancer: a qRT-PCR and chromogenic in situ hybridization study. Mod Pathol 2010;23(10):1334–45 doi 10.1038/modpathol.2010.121. [DOI] [PubMed] [Google Scholar]
- 21.Rauta J, Alarmo EL, Kauraniemi P, Karhu R, Kuukasjarvi T, Kallioniemi A. The serine-threonine protein phosphatase PPM1D is frequently activated through amplification in aggressive primary breast tumours. Breast Cancer Res Treat 2006;95(3):257–63 doi 10.1007/s10549-005-9017-7. [DOI] [PubMed] [Google Scholar]
- 22.Tan DS, Lambros MB, Rayter S, Natrajan R, Vatcheva R, Gao Q, et al. PPM1D is a potential therapeutic target in ovarian clear cell carcinomas. Clin Cancer Res 2009;15(7):2269–80 doi 10.1158/1078-0432.CCR-08-2403. [DOI] [PubMed] [Google Scholar]
- 23.Hu W, Feng Z, Modica I, Klimstra DS, Song L, Allen PJ, et al. Gene Amplifications in Well-Differentiated Pancreatic Neuroendocrine Tumors Inactivate the p53 Pathway. Genes Cancer 2010;1(4):360–8 doi 10.1177/1947601910371979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Milosevic J, Treis D, Fransson S, Gallo-Oller G, Sveinbjornsson B, Eissler N, et al. PPM1D Is a Therapeutic Target in Childhood Neural Tumors. Cancers (Basel) 2021;13(23) doi 10.3390/cancers13236042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kleiblova P, Shaltiel IA, Benada J, Sevcik J, Pechackova S, Pohlreich P, et al. Gain-of-function mutations of PPM1D/Wip1 impair the p53-dependent G1 checkpoint. J Cell Biol 2013;201(4):511–21 doi 10.1083/jcb.201210031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tokheim C, Wang X, Timms RT, Zhang B, Mena EL, Wang B, et al. Systematic characterization of mutations altering protein degradation in human cancers. Mol Cell 2021;81(6):1292–308 e11 doi 10.1016/j.molcel.2021.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van der Meer D, Barthorpe S, Yang W, Lightfoot H, Hall C, Gilbert J, et al. Cell Model Passports-a hub for clinical, genetic and functional datasets of preclinical cancer models. Nucleic Acids Res 2019;47(D1):D923–D9 doi 10.1093/nar/gky872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lowe J, Cha H, Lee MO, Mazur SJ, Appella E, Fornace AJ Jr., Regulation of the Wip1 phosphatase and its effects on the stress response. Front Biosci (Landmark Ed) 2012;17(4):1480–98 doi 10.2741/3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shreeram S, Demidov ON, Hee WK, Yamaguchi H, Onishi N, Kek C, et al. Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol Cell 2006;23(5):757–64 doi 10.1016/j.molcel.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 30.Fujimoto H, Onishi N, Kato N, Takekawa M, Xu XZ, Kosugi A, et al. Regulation of the antioncogenic Chk2 kinase by the oncogenic Wip1 phosphatase. Cell Death Differ 2006;13(7):1170–80 doi 10.1038/sj.cdd.4401801. [DOI] [PubMed] [Google Scholar]
- 31.Cao R, Zhang J, Zhang M, Chen X. PPM1D regulates p21 expression via dephoshporylation at serine 123. Cell Cycle 2015;14(4):641–7 doi 10.4161/15384101.2014.994922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brazina J, Svadlenka J, Macurek L, Andera L, Hodny Z, Bartek J, et al. DNA damage-induced regulatory interplay between DAXX, p53, ATM kinase and Wip1 phosphatase. Cell Cycle 2015;14(3):375–87 doi 10.4161/15384101.2014.988019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moon SH, Nguyen TA, Darlington Y, Lu X, Donehower LA. Dephosphorylation of gamma-H2AX by WIP1: an important homeostatic regulatory event in DNA repair and cell cycle control. Cell Cycle 2010;9(11):2092–6 doi 10.4161/cc.9.11.11810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen TA, Slattery SD, Moon SH, Darlington YF, Lu X, Donehower LA. The oncogenic phosphatase WIP1 negatively regulates nucleotide excision repair. DNA Repair (Amst) 2010;9(7):813–23 doi 10.1016/j.dnarep.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng B, Wang J, Hu Y, Zhao H, Hou W, Zhao H, et al. Modulation of LSD1 phosphorylation by CK2/WIP1 regulates RNF168-dependent 53BP1 recruitment in response to DNA damage. Nucleic Acids Res 2015;43(12):5936–47 doi 10.1093/nar/gkv528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu X, Bocangel D, Nannenga B, Yamaguchi H, Appella E, Donehower LA. The p53-induced oncogenic phosphatase PPM1D interacts with uracil DNA glycosylase and suppresses base excision repair. Mol Cell 2004;15(4):621–34 doi 10.1016/j.molcel.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 37.Cha H, Lowe JM, Li H, Lee JS, Belova GI, Bulavin DV, et al. Wip1 directly dephosphorylates gamma-H2AX and attenuates the DNA damage response. Cancer Res 2010;70(10):4112–22 doi 10.1158/0008-5472.CAN-09-4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yuan J, Chen J. MRE11-RAD50-NBS1 complex dictates DNA repair independent of H2AX. J Biol Chem 2010;285(2):1097–104 doi 10.1074/jbc.M109.078436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamaguchi H, Durell SR, Chatterjee DK, Anderson CW, Appella E. The Wip1 phosphatase PPM1D dephosphorylates SQ/TQ motifs in checkpoint substrates phosphorylated by PI3K-like kinases. Biochemistry 2007;46(44):12594–603 doi 10.1021/bi701096s. [DOI] [PubMed] [Google Scholar]
- 40.Buscemi G, Savio C, Zannini L, Micciche F, Masnada D, Nakanishi M, et al. Chk2 activation dependence on Nbs1 after DNA damage. Mol Cell Biol 2001;21(15):5214–22 doi 10.1128/MCB.21.15.5214-5222.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burdova K, Storchova R, Palek M, Macurek L. WIP1 Promotes Homologous Recombination and Modulates Sensitivity to PARP Inhibitors. Cells 2019;8(10) doi 10.3390/cells8101258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith ML, Seo YR. p53 regulation of DNA excision repair pathways. Mutagenesis 2002;17(2):149–56 doi 10.1093/mutage/17.2.149. [DOI] [PubMed] [Google Scholar]
- 43.Wang Z, Xu C, Diplas BH, Moure CJ, Chen CJ, Chen LH, et al. Targeting Mutant PPM1D Sensitizes Diffuse Intrinsic Pontine Glioma Cells to the PARP Inhibitor Olaparib. Mol Cancer Res 2020;18(7):968–80 doi 10.1158/1541-7786.MCR-19-0507. [DOI] [PubMed] [Google Scholar]
- 44.Loughery J, Cox M, Smith LM, Meek DW. Critical role for p53-serine 15 phosphorylation in stimulating transactivation at p53-responsive promoters. Nucleic Acids Res 2014;42(12):7666–80 doi 10.1093/nar/gku501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Y, Tavana O, Gu W. p53 modifications: exquisite decorations of the powerful guardian. J Mol Cell Biol 2019;11(7):564–77 doi 10.1093/jmcb/mjz060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alvi MA, Ida CM, Paolini MA, Kerezoudis P, Meyer J, Barr Fritcher EG, et al. Spinal cord high-grade infiltrating gliomas in adults: clinico-pathological and molecular evaluation. Mod Pathol 2019;32(9):1236–43 doi 10.1038/s41379-019-0271-3. [DOI] [PubMed] [Google Scholar]
- 47.Donehower LA, Soussi T, Korkut A, Liu Y, Schultz A, Cardenas M, et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep 2019;28(5):1370–84 e5 doi 10.1016/j.celrep.2019.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dudgeon C, Shreeram S, Tanoue K, Mazur SJ, Sayadi A, Robinson RC, et al. Genetic variants and mutations of PPM1D control the response to DNA damage. Cell Cycle 2013;12(16):2656–64 doi 10.4161/cc.25694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lowe JM, Cha H, Yang Q, Fornace AJ Jr., Nuclear factor-kappaB (NF-kappaB) is a novel positive transcriptional regulator of the oncogenic Wip1 phosphatase. J Biol Chem 2010;285(8):5249–57 doi 10.1074/jbc.M109.034579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morita K, Wang F, Jahn K, Hu T, Tanaka T, Sasaki Y, et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat Commun 2020;11(1):5327 doi 10.1038/s41467-020-19119-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ehrbrecht A, Muller U, Wolter M, Hoischen A, Koch A, Radlwimmer B, et al. Comprehensive genomic analysis of desmoplastic medulloblastomas: identification of novel amplified genes and separate evaluation of the different histological components. J Pathol 2006;208(4):554–63 doi 10.1002/path.1925. [DOI] [PubMed] [Google Scholar]
- 52.Castellino RC, De Bortoli M, Lu X, Moon SH, Nguyen TA, Shepard MA, et al. Medulloblastomas overexpress the p53-inactivating oncogene WIP1/PPM1D. J Neurooncol 2008;86(3):245–56 doi 10.1007/s11060-007-9470-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li K, Liu Y, Xu S, Wang J. PPM1D Functions as Oncogene and is Associated with Poor Prognosis in Esophageal Squamous Cell Carcinoma. Pathol Oncol Res 2018. doi 10.1007/s12253-018-0518-1. [DOI] [PubMed] [Google Scholar]
- 54.Saito-Ohara F, Imoto I, Inoue J, Hosoi H, Nakagawara A, Sugimoto T, et al. PPM1D is a potential target for 17q gain in neuroblastoma. Cancer Res 2003;63(8):1876–83. [PubMed] [Google Scholar]
- 55.Buss MC, Remke M, Lee J, Gandhi K, Schniederjan MJ, Kool M, et al. The WIP1 oncogene promotes progression and invasion of aggressive medulloblastoma variants. Oncogene 2015;34(9):1126–40 doi 10.1038/onc.2014.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qiu CW, Liu ZY, Hou K, Liu SY, Hu YX, Zhang L, et al. Wip1 knockout inhibits neurogenesis by affecting the Wnt/beta-catenin signaling pathway in focal cerebral ischemia in mice. Exp Neurol 2018;309:44–53 doi 10.1016/j.expneurol.2018.07.011. [DOI] [PubMed] [Google Scholar]
- 57.Tang YL, Liu X, Gao SY, Feng H, Jiang YP, Wang SS, et al. WIP1 stimulates migration and invasion of salivary adenoid cystic carcinoma by inducing MMP-9 and VEGF-C. Oncotarget 2015;6(11):9031–44 doi 10.18632/oncotarget.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y, Sun H, He G, Liu A, Wang F, Wang L. WIP1 regulates the proliferation and invasion of nasopharyngeal carcinoma in vitro. Tumour Biol 2014;35(8):7651–7 doi 10.1007/s13277-014-2034-6. [DOI] [PubMed] [Google Scholar]
- 59.He XL, Xiao Q, Zhou ZP, Hui CY. PPM1D accelerates proliferation and metastasis of osteosarcoma by activating PKP2. Eur Rev Med Pharmacol Sci 2021;25(1):78–85 doi 10.26355/eurrev_202101_24351. [DOI] [PubMed] [Google Scholar]
- 60.Sun GG, Wang YD, Liu Q, Hu WN. Expression of Wip1 in kidney carcinoma and its correlation with tumor metastasis and clinical significance. Pathol Oncol Res 2015;21(1):219–24 doi 10.1007/s12253-014-9811-9. [DOI] [PubMed] [Google Scholar]
- 61.Saliba J, Belsky N, Patel A, Thomas K, Carroll WL, Pierro J. From Favorable Histology to Relapse: The Clonal Evolution of a Wilms Tumor. Pediatr Dev Pathol 2020;23(2):167–71 doi 10.1177/1093526619875919. [DOI] [PubMed] [Google Scholar]
- 62.Khadka P, Reitman ZJ, Lu S, Buchan G, Gionet G, Dubois F, et al. PPM1D mutations are oncogenic drivers of de novo diffuse midline glioma formation. Nat Commun 2022;13(1):604 doi 10.1038/s41467-022-28198-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Burocziova M, Burdova K, Martinikova AS, Kasparek P, Kleiblova P, Danielsen SA, et al. Truncated PPM1D impairs stem cell response to genotoxic stress and promotes growth of APC-deficient tumors in the mouse colon. Cell Death Dis 2019;10(11):818 doi 10.1038/s41419-019-2057-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zink F, Stacey SN, Norddahl GL, Frigge ML, Magnusson OT, Jonsdottir I, et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017;130(6):742–52 doi 10.1182/blood-2017-02-769869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med 2017;377(2):111–21 doi 10.1056/NEJMoa1701719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gibson CJ, Lindsley RC, Tchekmedyian V, Mar BG, Shi J, Jaiswal S, et al. Clonal Hematopoiesis Associated With Adverse Outcomes After Autologous Stem-Cell Transplantation for Lymphoma. J Clin Oncol 2017;35(14):1598–605 doi 10.1200/JCO.2016.71.6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Machiela MJ, Myers TA, Lyons CJ, Koster R, Figg WD Jr., , Colli LM, et al. Detectible mosaic truncating PPM1D mutations, age and breast cancer risk. J Hum Genet 2019;64(6):545–50 doi 10.1038/s10038-019-0589-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pharoah PDP, Song H, Dicks E, Intermaggio MP, Harrington P, Baynes C, et al. PPM1D Mosaic Truncating Variants in Ovarian Cancer Cases May Be Treatment-Related Somatic Mutations. J Natl Cancer Inst 2016;108(3) doi 10.1093/jnci/djv347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Coombs CC, Zehir A, Devlin SM, Kishtagari A, Syed A, Jonsson P, et al. Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell 2017;21(3):374–82 e4 doi 10.1016/j.stem.2017.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yura Y, Miura-Yura E, Katanasaka Y, Min KD, Chavkin NW, Polizio AH, et al. The Cancer Therapy-Related Clonal Hematopoiesis Driver Gene Ppm1d Promotes Inflammation and Non-Ischemic Heart Failure in Mice. Circ Res 2021. doi 10.1161/CIRCRESAHA.121.319314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Uyanik B, Goloudina AR, Akbarali A, Grigorash BB, Petukhov AV, Singhal S, et al. Inhibition of the DNA damage response phosphatase PPM1D reprograms neutrophils to enhance anti-tumor immune responses. Nat Commun 2021;12(1):3622 doi 10.1038/s41467-021-23330-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bolton KL, Ptashkin RN, Gao T, Braunstein L, Devlin SM, Kelly D, et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat Genet 2020;52(11):1219–26 doi 10.1038/s41588-020-00710-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Grinfeld J, Nangalia J, Baxter EJ, Wedge DC, Angelopoulos N, Cantrill R, et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N Engl J Med 2018;379(15):1416–30 doi 10.1056/NEJMoa1716614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Al Hinai ASA, Grob T, Rijken M, Kavelaars FG, Zeilemaker A, Erpelinck-Verschueren CAJ, et al. PPM1D mutations appear in complete remission after exposure to chemotherapy without predicting emerging AML relapse. Leukemia 2021. doi 10.1038/s41375-021-01155-y. [DOI] [PubMed] [Google Scholar]
- 75.Panagiota V, Meggendorfer M, Kubasch AS, Gabdoulline R, Kronke J, Mies A, et al. Impact of PPM1D Mutations in Patients with Myelodysplastic Syndrome and Deletion of Chromosome 5q. Am J Hematol 2021. doi 10.1002/ajh.26162. [DOI] [PubMed] [Google Scholar]
- 76.Schwartz JR, Ma J, Kamens J, Westover T, Walsh MP, Brady SW, et al. The acquisition of molecular drivers in pediatric therapy-related myeloid neoplasms. Nat Commun 2021;12(1):985 doi 10.1038/s41467-021-21255-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Williams N, Lee J, Mitchell E, Moore L, Baxter EJ, Hewinson J, et al. Life histories of myeloproliferative neoplasms inferred from phylogenies. Nature 2022;602(7895):162–8 doi 10.1038/s41586-021-04312-6. [DOI] [PubMed] [Google Scholar]
- 78.Wong TN, Miller CA, Jotte MRM, Bagegni N, Baty JD, Schmidt AP, et al. Cellular stressors contribute to the expansion of hematopoietic clones of varying leukemic potential. Nat Commun 2018;9(1):455 doi 10.1038/s41467-018-02858-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Takahashi K, Wang F, Kantarjian H, Doss D, Khanna K, Thompson E, et al. Preleukaemic clonal haemopoiesis and risk of therapy-related myeloid neoplasms: a case-control study. Lancet Oncol 2017;18(1):100–11 doi 10.1016/S1470-2045(16)30626-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gillis NK, Ball M, Zhang Q, Ma Z, Zhao Y, Yoder SJ, et al. Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: a proof-of-concept, case-control study. Lancet Oncol 2017;18(1):112–21 doi 10.1016/S1470-2045(16)30627-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fons NR, Sundaram RK, Breuer GA, Peng S, McLean RL, Kalathil AN, et al. PPM1D mutations silence NAPRT gene expression and confer NAMPT inhibitor sensitivity in glioma. Nat Commun 2019;10(1):3790 doi 10.1038/s41467-019-11732-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bulavin DV, Phillips C, Nannenga B, Timofeev O, Donehower LA, Anderson CW, et al. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat Genet 2004;36(4):343–50 doi 10.1038/ng1317. [DOI] [PubMed] [Google Scholar]
- 83.Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 2012;488(7409):49–56 doi 10.1038/nature11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lu ZW, Wen D, Wei WJ, Han LT, Xiang J, Wang YL, et al. Silencing of PPM1D inhibits cell proliferation and invasion through the p38 MAPK and p53 signaling pathway in papillary thyroid carcinoma. Oncol Rep 2020;43(3):783–94 doi 10.3892/or.2020.7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fontana MC, Nanni J, Ghelli Luserna di Rora A, Petracci E, Padella A, Ghetti M, et al. Pharmacological Inhibition of WIP1 Sensitizes Acute Myeloid Leukemia Cells to the MDM2 Inhibitor Nutlin-3a. Biomedicines 2021;9(4) doi 10.3390/biomedicines9040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Deng W, Li J, Dorrah K, Jimenez-Tapia D, Arriaga B, Hao Q, et al. The role of PPM1D in cancer and advances in studies of its inhibitors. Biomed Pharmacother 2020;125:109956 doi 10.1016/j.biopha.2020.109956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Choi BK, Fujiwara K, Dayaram T, Darlington Y, Dickerson J, Goodell MA, et al. WIP1 dephosphorylation of p27(Kip1) Serine 140 destabilizes p27(Kip1) and reverses anti-proliferative effects of ATM phosphorylation. Cell Cycle 2020;19(4):479–91 doi 10.1080/15384101.2020.1717025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, et al. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 2000;287(5459):1824–7 doi 10.1126/science.287.5459.1824. [DOI] [PubMed] [Google Scholar]
- 89.Yu E, Ahn YS, Jang SJ, Kim MJ, Yoon HS, Gong G, et al. Overexpression of the wip1 gene abrogates the p38 MAPK/p53/Wip1 pathway and silences p16 expression in human breast cancers. Breast Cancer Res Treat 2007;101(3):269–78 doi 10.1007/s10549-006-9304-y. [DOI] [PubMed] [Google Scholar]
- 90.Yang S, Dong S, Qu X, Zhong X, Zhang Q. Clinical significance of Wip1 overexpression and its association with the p38MAPK/p53/p16 pathway in NSCLC. Mol Med Rep 2017;15(2):719–23 doi 10.3892/mmr.2016.6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang C, Chen Y, Wang M, Chen X, Li Y, Song E, et al. PPM1D silencing by RNA interference inhibits the proliferation of lung cancer cells. World J Surg Oncol 2014;12:258 doi 10.1186/1477-7819-12-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yin H, Yan Z, Liang Y, Liu B, Su Q. Knockdown of protein phosphatase magnesium-dependent 1 (PPM1D) through lentivirus-mediated RNA silencing inhibits colorectal carcinoma cell proliferation. Technol Cancer Res Treat 2013;12(6):537–43 doi 10.7785/tcrt.2012.500349. [DOI] [PubMed] [Google Scholar]
- 93.Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene 2003;22(56):9030–40 doi 10.1038/sj.onc.1207116. [DOI] [PubMed] [Google Scholar]
- 94.Wu B, Guo BM, Kang J, Deng XZ, Fan YB, Zhang XP, et al. PPM1D exerts its oncogenic properties in human pancreatic cancer through multiple mechanisms. Apoptosis 2016;21(3):365–78 doi 10.1007/s10495-015-1211-4. [DOI] [PubMed] [Google Scholar]
- 95.Feng Y, Liu F, Du Z, Zhao D, Cheng J, Guo W. Wip1 regulates SKOV3 cell apoptosis through the p38 MAPK signaling pathway. Mol Med Rep 2017;15(6):3651–7 doi 10.3892/mmr.2017.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Martinikova AS, Burocziova M, Stoyanov M, Macurek L. Truncated PPM1D Prevents Apoptosis in the Murine Thymus and Promotes Ionizing Radiation-Induced Lymphoma. Cells 2020;9(9) doi 10.3390/cells9092068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xia ZS, Wu D, Zhong W, Lu XJ, Yu T, Chen QK. Wip1 gene silencing enhances the chemosensitivity of human colon cancer cells. Oncol Lett 2017;14(2):1875–83 doi 10.3892/ol.2017.6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kong W, Jiang X, Mercer WE. Downregulation of Wip-1 phosphatase expression in MCF-7 breast cancer cells enhances doxorubicin-induced apoptosis through p53-mediated transcriptional activation of Bax. Cancer Biol Ther 2009;8(6):555–63 doi 10.4161/cbt.8.6.7742. [DOI] [PubMed] [Google Scholar]
- 99.Liu S, Jiang B, Li H, He Z, Lv P, Peng C, et al. Wip1 is associated with tumorigenity and metastasis through MMP-2 in human intrahepatic cholangiocarcinoma. Oncotarget 2017;8(34):56672–83 doi 10.18632/oncotarget.18074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Park DS, Yoon GH, Kim EY, Lee T, Kim K, Lee PC, et al. Wip1 regulates Smad4 phosphorylation and inhibits TGF-beta signaling. EMBO Rep 2020;21(5):e48693 doi 10.15252/embr.201948693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chew J, Biswas S, Shreeram S, Humaidi M, Wong ET, Dhillion MK, et al. WIP1 phosphatase is a negative regulator of NF-kappaB signalling. Nat Cell Biol 2009;11(5):659–66 doi 10.1038/ncb1873. [DOI] [PubMed] [Google Scholar]
- 102.Yokoyama A, Kakiuchi N, Yoshizato T, Nannya Y, Suzuki H, Takeuchi Y, et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 2019;565(7739):312–7 doi 10.1038/s41586-018-0811-x. [DOI] [PubMed] [Google Scholar]
- 103.Garaycoechea JI, Crossan GP, Langevin F, Mulderrig L, Louzada S, Yang F, et al. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 2018;553(7687):171–7 doi 10.1038/nature25154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pechackova S, Burdova K, Benada J, Kleiblova P, Jenikova G, Macurek L. Inhibition of WIP1 phosphatase sensitizes breast cancer cells to genotoxic stress and to MDM2 antagonist nutlin-3. Oncotarget 2016;7(12):14458–75 doi 10.18632/oncotarget.7363. [DOI] [PMC free article] [PubMed] [Google Scholar]