

Abstract
Black and Hispanic cancer patients have higher incidence of cancer mortality. Many factors (e.g., socioeconomic differences, insufficient access to healthcare) contribute to racial disparity. Emerging research implicates biological disparity in cancer outcomes. Studies show distinct differences in the tumor immune microenvironment (TIME) in Black cancer patients. Studies also have linked altered mitochondrial metabolism to changes in immune cell activation in TIME. Recent publications revealed a novel immunomodulatory role for triphenylphosphonium-based mitochondrial-targeted drugs (MTDs). These are synthetically modified, naturally occurring molecules (e.g., honokiol, magnolol, metformin) or FDA-approved small molecule drugs (e.g., atovaquone, hydroxyurea). Modifications involve conjugating the parent molecule via an alkyl linker chain to a triphenylphosphonium moiety. These modified molecules (e.g., Mito-honokiol, Mito-magnolol, Mito-metformin, Mito-atovaquone, Mito-hydroxyurea) accumulate in tumor cell mitochondria more effectively than in normal cells and inhibit mitochondrial respiration, induce reactive oxygen species, activate AMPK and redox transcription factors, and inhibit cancer cell proliferation. Besides these intrinsic effects of MTDs in redox signaling and proliferation in tumors, MTDs induced extrinsic effects in the TIME of mouse xenografts. MTD treatment inhibited tumor suppressive immune cells, myeloid-derived suppressor cells, and regulatory T cells, and activated T cells and antitumor immune effects. One key biological disparity in Black cancer patients was related to altered mitochondrial oxidative metabolism; MTDs targeting vulnerabilities in tumor cells and the TIME may help us understand this biological disparity. Clinical trials should include an appropriate number of Black and Hispanic cancer patients, and should validate the intratumoral, antihypoxic effects of MTDs with imaging.
Keywords: mitochondrial drugs, OXPHOS inhibitors, tumor microenvironment, racial disparity
1.0. Introduction
Most cancer patients do not respond positively to immunotherapy. Strategic development of additional combinatorial drug regimens is necessary. Tumor cells evade the immune system through upregulation of programmed death receptor ligand 1 (PD-L1) expression that binds to programmed cell death protein 1 (PD-1) in T cells, resulting in immunosuppression. One way to prevent cancer cells from immune evasion is to decrease the expression of PD-L1 or hinder the binding of PD-L1 to PD-1.1,2 Therefore, a major challenge is to target specific metabolic pathways in cancer cells without adversely impairing immune cells while enhancing antitumor immunity. Tumor cell metabolism is primarily governed by the Warburg effect, or aerobic glycolysis. However, glucose metabolism is essential not only for cancer cells but also for T cells and macrophages. Thus, targeting cancer cells alone using 2-deoxy-d-glucose and other glycolytic inhibitors will also adversely affect immune cells. Targeting oxidative phosphorylation (OXPHOS), in particular mitochondrial complex I of the mitochondrial electron transport chain, is emerging as a potent and selective antiproliferative strategy in tumor cells.3–12 Hypoxic tumors with a reduced capacity for compensatory glycolysis are more susceptible to OXPHOS inhibitors.12 Modulators of glutamine metabolism in the Krebs cycle are being developed in cancer therapy.13
Mitochondrial OXPHOS inhibitors also target cancer-associated immune cells in the tumor immune microenvironment (TIME)10 and play an important role in cancer immune evasion.14 Inhibition of the OXPHOS function in cancer cells and the concomitant decrease in tumor hypoxia induced remodeling of the TIME and the antitumor response.8 Recent reports suggest that triphenylphosphonium (TPP+)-containing mitochondrial drugs inhibit tumor suppressive cells, such as the myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs), in the TIME.15,16 MDSCs suppress T cells that destroy tumor cells.17 Targeting MDSCs and Tregs is emerging as an antitumor therapeutic strategy.18 Current chemotherapeutics (e.g., gemcitabine and 5-fluorouracil) used to inhibit MDSCs cause bone marrow suppression; therefore, less toxic and more targeted agents are needed to suppress MDSCs and/or suppressive neutrophils and enhance the cytotoxic antitumor function of T cells.19–21 Investigating the immunomodulatory effects of mitochondria-targeted drugs (MTDs), especially TPP+-based drugs, is an area of intense research.16,22 Emerging research indicates that the TIME in Black cancer patients consists of more protumorigenic and immunosuppressive factors than in white cancer patients.23,24 Developing potent yet nontoxic MTDs may help overcome this biological disparity.
2.0. MTDs conjugated to TPP+, OXPHOS inhibition, and redox signaling
The most studied and widely used mitochondria-targeting vector is TPP+.3,25–28 TPP+ possesses a single positive charge that is delocalized over three phenyl groups, stabilizing resonance. In addition to the charge, the hydrophobicity of the lipophilic cation favors the interaction with the hydrophobic inner mitochondrial membrane. Driven by the membrane potential, the concentration of the TPP+ in the cytoplasm increases by about 5–10 fold, compared with that of the extracellular space. The resulting accumulation of TPP+ in the cytoplasm is about 100–500 times that in the extracellular space (Fig. 1). This provides a highly targeted and effective mitochondrial vector. The advantages of TPP+-based targeting molecules are the stability of TPP+ in the biological system, low chemical reactivity toward cellular components, combination of lipophilic and hydrophilic moieties, and ease of synthesizing large quantities of molecules for in vivo work.28
Figure 1. A hypothetical picture showing how MTDs could potentially change the immunosuppressive microenvironment to an antitumor microenvironment and cause a decrease in breast tumor metastasis.
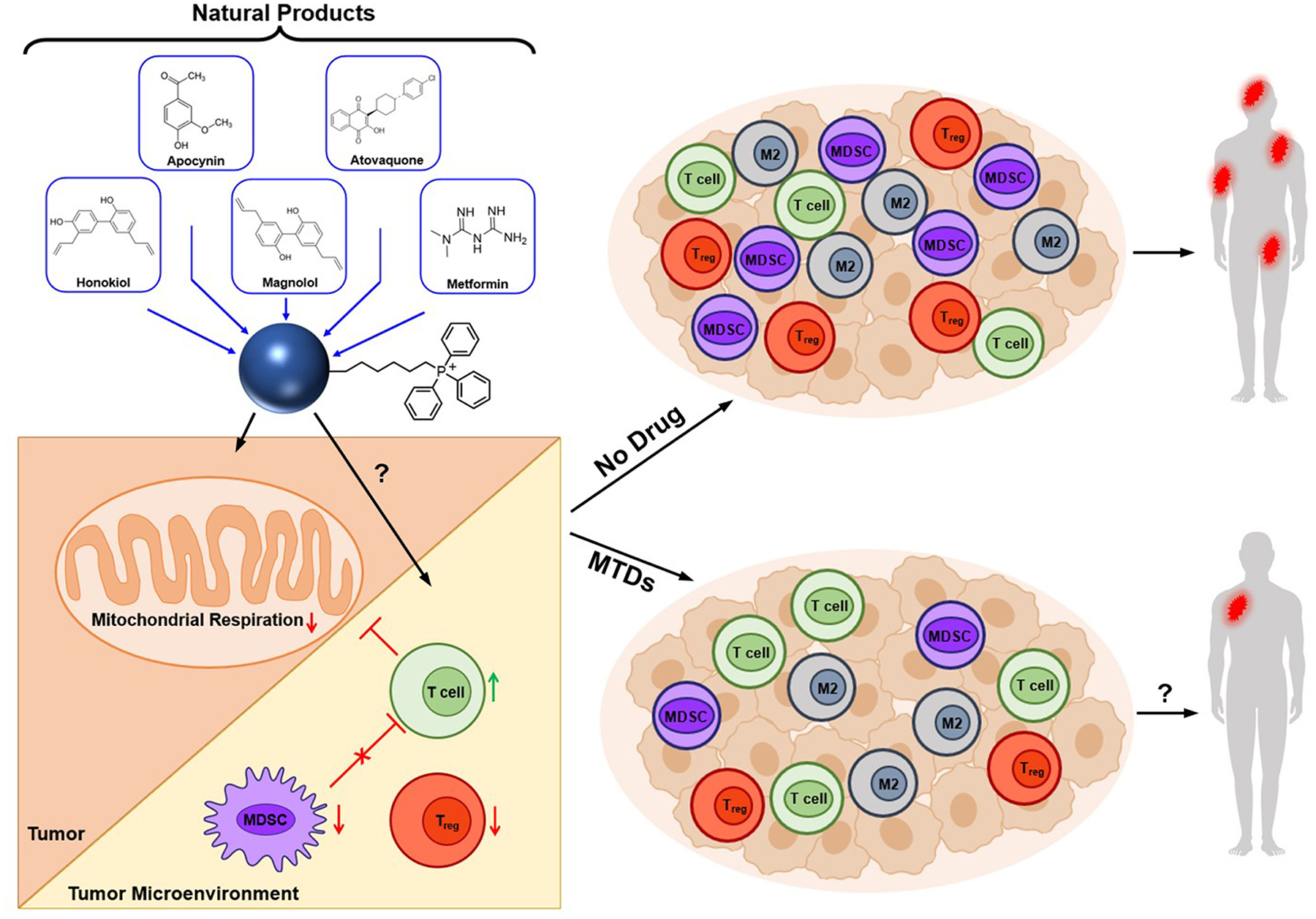
M2, tumor promoting; MDSCs, myeloid-derived suppressor cells; MTDs, mitochondria-targeted drugs; Treg, regulatory T cells. A portion of this figure was Reprinted from iScience, 24, Cheng G, Hardy M, Topchyan P, Zander R, Volberding P, Cui W, Kalyanaraman B., Mitochondria-targeted hydroxyurea inhibits OXPHOS and induces antiproliferative and immunomodulatory effects, Pages No. 102673, Copyright 2021, with permission from Elsevier; a portion of this figure was reprinted with permission from Zielonka J, Joseph J, Sikora A, Hardy M, Ouari O, Vasquez-Vivar J, Cheng G, Lopez M, Kalyanaraman B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem Rev. 2017 Aug 9;117(15):10043–10120. doi: 10.1021/acs.chemrev.7b00042. Copyright 2017 American Chemical Society; and a portion of the figure is licensed under CC BY, copyright ©2020 Kim G, Pastoriza JM, Condeelis JS, Sparano JA, Filippou PS, Karagiannis GS, Oktay MH. The Contribution of Race to Breast Tumor Microenvironment Composition and Disease Progression. Front Oncol. 2020 Jun 30;10:1022. doi: 10.3389/fonc.2020.01022.
TPP+-based MTDs conjugated to natural products or FDA-approved drugs are potent, tumor selective, and relatively nontoxic (with minimal off-target pharmacology) in cells and preclinical tumor xenografts.3–10,15,16 Several natural products (e.g., honokiol, magnolol, metformin) and FDA-approved drugs (e.g., atovaquone, hydroxyurea) conjugated to the TPP+ moiety (e.g., Mito-honokiol, Mito-magnolol, Mito-metformin, Mito-atovaquone, Mito-hydroxyurea) (Fig. 1) exhibit significantly more antiproliferative potency than other non-TPP+ mitochondrial inhibitors (e.g., metformin, phenformin, atovaquone, IACS-010759) in different cancer cells.
3.0. Mito-honokiol inhibits lung cancer metastasis to the brain
Previously, using low-temperature electron paramagnetic resonance, we determined the mitochondrial redox changes in pancreatic cancer cells treated with TPP+-containing mitochondria-targeted agents.29 Based on the electron paramagnetic resonance spectral changes of mitochondrial complex I iron-sulfur (FeS) clusters, [2Fe2S]+ and [4Fe-4S]+, we surmised that TPP+-containing MTDs (mitochondrial complex I inhibitors) bind closer to the NADH-dehydrogenase site in the mitochondrial complex I dictated by the NAD+/NADH couple.30 Mito-honokiol is synthesized by conjugating TPP+ through an alkyl side chain to honokiol, a component of magnolia tree bark extract and a widely used nutritional supplement. Mito-honokiol inhibits mitochondrial complex I-induced oxygen consumption, induces superoxide and hydrogen peroxide formation and activation of AMPK, inhibits signal transducer and activator of transcription 3 (STAT3) phosphorylation, and inhibits proliferation of cancer cells.9 Further, Mito-honokiol inhibits lung cancer progression and prevents metastasis of lung cancer cells to lymph nodes and to the brain.9 From a mechanistic standpoint, the antitumor and antimetastatic effects were shown to be mediated by the STAT3 pathway. Knockdown of STAT3 abrogated both the antiproliferative and antimetastatic effects of Mito-honokiol.9 Mito-honokiol inhibits STAT3 phosphorylation irrespective of the epidermal growth factor receptor mutation status in lung cancer cells.9 Reports indicate that a decrease in the core body temperature and death result from the excessive inhibition of OXPHOS.12 Mito-honokiol did not elicit these effects.9 Currently, treatment of lung cancer metastasis does not start until after diagnosis of brain cancer. Mito-honokiol treatment inhibits metastasis of primary cancer to brain. Emerging research is focused on metabolic reprogramming of metastatic lung cancer cells and their vulnerability to MTDs.7–10,31
4.0. Antiproliferative effects of Mito-magnolol in drug resistant melanoma cells
Drug resistance to kinase inhibitors is attributed to metabolic reprogramming from glycolysis to mitochondrial oxidative metabolism. Bioenergetic mapping results showed that tumor cells with enhanced mitochondrial OXPHOS were more sensitive to TPP+-based MTDs and other OXPHOS inhibitors.32 There are currently no effective drugs for treating melanoma, an aggressive form of skin cancer. B-Raf serine/threonine kinase, or BRAF, inhibitor antiglycolytic drugs induce a rapid onset of drug resistance. BRAF inhibitors cause metabolic reprogramming from a glycolytic phenotype to an OXPHOS phenotype that is attributed to resistance against antiglycolytic kinase inhibiting drugs (e.g., vemurafenib). The increased dependence on OXPHOS for energy makes OXPHOS a vulnerable target in drug-resistant melanoma cells. Increased mitochondrial biogenesis and upregulated OXPHOS genes are associated with enhanced mitochondrial respiration in drug-resistant melanoma cells. A mitochondria-targeted analog of magnolol (Mito-magnolol) was shown to potently inhibit melanoma cell proliferation and tumor growth in murine melanoma xenografts.22,33 Mito-magnolol is synthesized by conjugating a TPP+ moiety via an alkyl side chain to magnolol.33 Magnolol is present in abundance in magnolia extract, a traditional herbal medicine used effectively for centuries in East Asia to treat inflammatory diseases. Mito-magnolol belongs to a new class of mitochondria-targeted polyphenolic drugs.
Mito-magnolol potently inhibits mitochondrial complex I-induced mitochondrial respiration, blocks cell cycle progression, and inhibits proliferation of melanoma cells, primarily through downregulation of mTOR/AKT signaling and mitophagy.33
Mito-magnolol induced AMPK–threonine 172 phosphorylation, activating AMPK signaling, mitophagy, and energy-related proteins in melanoma cells. Mito-magnolol treatment was equally effective in inhibiting drug-resistant melanoma cells (with enhanced OXPHOS).33
Mito-magnolol inhibited tumor progression in an immune-competent mouse xenograft model.22 Also, Mito-magnolol remodeled the TIME in a mouse melanoma model. Mito-magnolol induced infiltration of T cells, decreased MDSCs, and decreased tumor-associated macrophages in melanoma tumors.22 The antitumor effect of Mito-magnolol is inhibited by immune depletion.22 The antitumor immunity effect of mitochondria-targeted polyphenolics is an exciting area of therapeutic drug targeting and TIME remodeling.
5.0. Antitumor immune effects of Mito-Atovaquone
Recent reports indicate that selective targeting and inhibiting of mitochondrial complex III mitigate and reverse immunosuppression by Tregs, promoting the function of effector T cells.34 Tregs suppress the antitumor immunity that greatly hampers immunotherapy. Inhibitors of mitochondrial complex III (e.g., antimycin A) and not complex I (e.g., rotenone) reversed the immunosuppressive function of Tregs.34 Although several relatively nontoxic mitochondrial complex I inhibitors exist (excluding rotenone that is toxic), the availability of mitochondrial complex III inhibitors is relatively scarce except for antimycin A and atovaquone. Mito10-atovaquone is synthesized by conjugating TPP+ via an alkyl side chain to atovaquone, an FDA-approved antimalarial drug.15 Mito10-atovaquone inhibits both mitochondrial complex III- and complex I-induced oxygen consumption.15 We showed, for the first time, that conjugating atovaquone to TPP+ and increasing the aliphatic linker side chain length generates Mito-atovaquone analogs (e.g., Mito4-atovaquone and Mito10-atovaquone) that are potent inhibitors of mitochondrial complex I- and complex III-induced oxygen consumption in cancer cells.15 Mito4-atovaquone and Mito10-atovaquone effectively inhibit Treg differentiation and survival while stimulating effector T cell response. These compounds represent a new class of antitumor and immunoregulatory drugs. The TIME is a potentially vulnerable target in cancer therapy. MTDs (e.g., Mito-honokiol, Mito-magnolol, Mito-metformin, Mito-atovaquone, Mito-hydroxyurea) inhibit immunosuppressive cells (e.g., MDSCs and Tregs) and increase the infiltration of cytolytic T cells in the TIME as well as Mito-magnolol and Tregs.15,16,33
6.0. OXPHOS inhibitors, MDSCs, and metastatic cancer
The metabolic reprogramming (enhanced OXPHOS) that occurs in metastatic cancer cells likely plays a major role in metastatic cancer cell survival and progression.31,35 Reports indicate that an OXPHOS inhibitor, IACS-010759, inhibits melanoma brain metastasis.10 The mitochondrial complex I inhibitor also inhibits MDSCs in the metastatic TIME. TPP+-conjugated OXPHOS inhibitors of mitochondrial complex I and complex III—Mito-magnolol, Mito-atovaquone, and Mito-hydroxyurea—are potentially suitable antimetastatic drugs.15,33 It was reported that brain metastases from patients with melanoma displayed a considerable degree of immunosuppression and increased expression of genes related to OXPHOS. IACS-010759, a reported mitochondrial complex I inhibitor, blocked metastasis formation in mouse models.10 Mito-atovaquone and Mito-lonidamine are potent OXPHOS inhibitors and inhibit lung cancer metastasis to the brain in mouse models.15,36
7.0. Oxidative metabolism, a barrier to immunotherapy
Hypoxia (a lack of oxygen) is a key hallmark of tumors and the TIME. Hypoxia is associated with decreased metabolic function of T cells in the TIME and inhibits antitumor immunity. Studies showed that tumors with enhanced oxidative metabolism (due to metabolic reprogramming) responded poorly to immunotherapy (PD-1 blockade). Tumors with decreased mitochondrial respiration and oxidative metabolism responded more positively to immunotherapy.37–39 This was attributed to enhanced T cell exhaustion in the TIME of tumors with enhanced oxidative metabolism (increased hypoxia in the TIME) in contrast to tumors with less mitochondrial oxidative metabolism (decreased hypoxia in the TIME). These findings suggest that it may be possible to manipulate tumor hypoxia and remodel the TIME using MTDs.
Emerging research suggests that targeted therapy to remodel the TIME and enhance T cell function would increase antitumor effect and improve the efficacy of immunotherapeutic effects of cancer.40–42 Remodeling processes enhancing the oxygen tension of the TIME was proposed as a viable therapy.43 Higher metabolic rates in tumors result in tumor hypoxia, especially in solid tumors with disorganized vasculature.44,45 One approach to decrease tumor hypoxia (i.e., to enhance oxygen concentration in tumors) is to decrease tumor oxygen consumption.11 Decreased hypoxia in tumors is a pharmacodynamic response of MTDs that can be quantitated by positron emission tomography imaging.46,47 Immunotherapeutic efficacy was potentiated by a metformin-induced decrease in tumor hypoxia.48 Previously, we showed that inhibition of OXPHOS by Mito-metformin enhances radiation-induced pancreatic cancer cell killing, which is attributable to increased oxygen tension or decreased hypoxia in pancreatic cancer cells.4
8.0. MTDs and checkpoint inhibitors
Tumor hypoxia facilitates the recruitment of immunosuppressive cells (MDSCs, Tregs, tumor associated macrophages) to the TIME.49 In addition, immunosuppressive metabolites and cytokines are released by both tumor cells and immune cells under hypoxia. Immune checkpoint molecules (e.g., PD-L1, cytotoxic T-lymphocyte-associated protein 4) are also upregulated. All of these adaptations to tumor hypoxia blunt an effective immune response. Restoring the oxygen supply to tumors was shown to reactivate the antitumor response because of decreased immunosuppressive cells and increased effector cytotoxic T cells in the TIME.50,51 In a paradoxical study, oxygen was shown to impair the anticancer activity of T cells in mice, and inhibiting the oxygen-sensing capability of immune cells prevented lung metastasis.52 Recent studies show that radiotherapy combined with inhibition of OXPHOS is an effective strategy to overcome the barrier to PD-1 immunotherapy.53,54 The combination of IACS-010759, a mitochondrial complex I inhibitor, with radiotherapy proved to be a promising strategy to treat PD-1-resistant lung cancer. Several clinical trials are underway investigating the antihypoxic effects of FDA-approved drugs for other diseases (e.g., metformin, atovaquone, papaverine) in combination with radio and immunotherapies of cancer. Metformin, a weak inhibitor of mitochondrial complex I, was more effective in treating Black cancer patients.55,56 Metformin was shown to decrease PD-L1 expression through activating the Hippo signaling pathway in colorectal cancer cells.57 A clinical trial investigating the use of metformin to reduce disparities in breast cancer is ongoing.58 Mito-metformin containing longer aliphatic side chains that are significantly more potent than metformin in inhibiting pancreatic cancer cell proliferation and growth may be a viable candidate drug for future clinical trials designed to decrease racial disparities in breast cancer.
9.0. OXPHOS inhibition, AMPK activation, and enhanced immunosuppressive TIME
AMPK, a master regulator of cellular energy homeostasis, is typically activated by increased intracellular AMP.59,60 We previously showed that OXPHOS inhibitors stimulate a signaling pathway for antiproliferative effects, linking mitochondrial complex I inhibition to AMPK activation4,6,9,33,36 and leading to inhibition of STAT3 ser727 phosphorylation.61 AMPK activation inhibits the functions of MDSCs.62–65 The MTD, phenformin, inhibits MDSCs and enhances the antitumor activity.66 Cumulative evidence suggests that STAT3 activation leads to immunosuppression, and inhibiting STAT3 signaling is an effective strategy to improve antitumor immunity.67,68 Mitochondria-targeted polyphenolics (e.g., Mito-honokiol, Mito-magnolol) and Mito-metformin activate AMPK phosphorylation in multiple cancer cells and inhibit immunosuppressive cells in the TIME.
10.0. Mitochondrial biomarkers and personalized therapy
Developing novel therapeutic strategies targeting mitochondria might decrease or prevent racial health disparities.69,70 Mitochondrial determinants of cancer health and the mitochondrial basis of cancer disparities are unknown.70 However, recent reports suggest that mitochondrial biomarkers could predict tumor progression and outcome.71,72 Atovaquone decreased tumor hypoxia or increased tumor oxygenation and inhibited hypoxia-regulated gene expression in lung cancer patients.73 In hypoxia PET-CT (positron emission tomography–computed tomography), a key pharmacodynamics endpoint was reduction in hypoxia-regulated genes, which were downregulated in atovaquone treatment of non-small-cell lung cancer patients.73 OXPHOS targeting is an effective way to inhibit hypoxic cancer cells. Hypoxic monitoring may, therefore, serve as an effective biomarker in therapeutic selection and treatment.
11.0. Mitochondrial dysfunction and racial disparities
Increasing evidence supports the existence of racial and ethnic disparities in the breast cancer immune microenvironment.23 Higher levels of pro-tumorigenic factors (e.g., macrophages, Tregs, exhausted T cells) were identified in the TIME of Black breast cancer patients as compared with white counterparts.23,24,74 Upregulation of genes associated with OXPHOS was identified in tumor samples obtained from Black cancer patients.75 Tumors from Black cancer patients have more mitochondria, ERR-1 (estrogen-related receptor 1), and peroxisome PGC-1α (proliferator-activated receptor gamma coactivator 1-alpha).76 Clinical trial data show that Black cancer patients respond better to mitochondrial inhibitors (e.g., metformin) than white cancer patients.55 Developing the next generation of mitochondrial inhibitors was perceived to be a promising therapeutic strategy to mitigate or prevent enhanced mortality in Black cancer patients.75 A newly developed mitochondria-targeted atovaquone (i.e., Mito10-ATO) inhibits MDSCs and Tregs.15 It is conceivable that newly developed, mitochondria-targeted modified natural products and FDA-approved drugs (e.g., Mito-honokiol, Mito-magnolol, Mito-metformin, Mito-atovaquone, and Mito-hydroxyurea) and their analogs potentially could be useful in understanding the biological racial disparity in cancer mortality. Additional research centered on understanding the role of mutations in epidermal growth factor receptor, epidermal growth factor receptor tyrosine kinase inhibitors, OXPHOS pathway,77 and racial disparity in cancer patients is needed.
A recent report highlights that changes in mitochondria (enrichment of OXPHOS in tumors from Black patients) could be a biomarker and provides a rationale for the repurposing of mitochondrial inhibitors to treat cancers in Black patients.76 Hypoxic gene expression signatures using RNA sequencing may be used as a biomarker for patient selection and treatment with MTDs.73 A recent report suggests a biomarker-based approach to patient selection to overcome or mitigate racial disparities in clinical cancer trials.78
12.0. Unanswered questions
How mitochondrial inhibition of tumor tissues decreases immunosuppression in the TIME is not known. Whether this effect is tumor intrinsic, tumor extrinsic, or both remains to be determined. Dynamic variations in the mitochondrial membrane potentials of immune cells (i.e., T cells, Tregs, natural killer cells, macrophages) that determine the toxicity of MTDs are not known. The effect of MTDs on PD-L1 and other checkpoint protein expression in cancer cells needs to be examined in detail.
How MTD-mediated inhibition of complex I-induced mitochondrial respiration affects antigen presentation on major histocompatibility class 1 molecules, including tumor peptides, needs to be investigated in detail. Studies suggest a compelling role for mitochondria in antigen processing and presentation79,80 as well as in cancer immune evasion.81 Important considerations include strategies to counteract the inactivation of the major histocompatibility class 1 pathway, and how MTD-induced inhibition of respiration affects cytokine (e.g., interferon gamma) response in antigen presenting cells.82,83
How epigenetic modifications (DNA methylations, histone modifications, mRNA expression modulation) that affect changes in the gene expression (not caused by changes in the DNA sequences) are influenced by MTD-mediated repression of mitochondrial respiration in cancer cells is not known. Epigenetic modifications have been used as predictive biomarkers in cancer.84 The importance of epigenetic events in racial disparity is increasingly recognized.85–88 One of the primary characteristics of cancer cells is altered metabolism (i.e., the Warburg effect). Inhibitors of glycolytic metabolism in tumors affect epigenetic modifications.89 Understanding the interplay between alterations in DNA methylations, histone modifications, chromatin remodeling in light of altered tumor metabolism, and metabolic reprogramming is critical to understanding the implications of mitochondrial dysfunction and racial disparity in cancer treatment.90
Mitochondrial reactive oxygen species (ROS) and their significance in MTD-dependent antiproliferative and antitumor effects are not known. N-acetylcysteine, a membrane-permeable cysteine precursor, is used as an ROS scavenger and a potent antioxidant. Its cytoprotective effect and/or inhibition of oxidation of fluorescent dye were related to its ability to scavenge ROS (superoxide or hydrogen peroxide) and modulate redox signaling effects in cancer cells treated with TPP+-containing agents.91 However, neither superoxide nor hydrogen peroxide react at an appreciable rate with N-acetylcysteine. This calls into question the ROS scavenging as an antioxidant mechanism of N-acetylcysteine. N-acetylcysteine can enhance intracellular glutathione and glutathione-dependent hydroperoxide-removing antioxidant enzyme machinery. Thus, the proposed antioxidant mechanism of N-acetylcysteine is not related to direct scavenging of ROS.
Although most of the previous preclinical work with OXPHOS inhibitors was performed in immunodeficient mice, the involvement of both tumor mitochondria and the TIME in antitumor mechanism of MTDs was demonstrated in immune competent mice.22 Mitochondrial transfer from the stromal cells in the TIME to tumor cells was shown to occur in several cancers, including acute myeloid lukemia.92,93 However, the mechanism of transfer needs to be determined. Although an ROS mechanism has been proposed, the identity of the species responsible for the transfer has not been determined. Rigorous characterization of diagnostic marker products of fluorescent dyes, as previously described,30 need to be determined.
13.0. Conclusions and future perspectives
The TIME in Black cancer patients consists of more pro-tumorigenic factors and mitochondrial dysfunction than in white cancer patients. Thus, developing a highly potent, less toxic, and tumor/TIME selective next generation of mitochondrial OXPHOS inhibitors is timely and critical in overcoming racial and ethnic disparities in cancer treatment. Poor accrual of Black and Hispanic cancer patients in clinical trials has hindered our understanding of the biological basis of racial disparity. Clinical trials should include Black and Hispanic cancer patients and combinatorial treatments (potent OXPHOS inhibitors alleviating hypoxia and radiation, immunotherapy). Patient selection should be based on imaging studies that validate hypoxic modification of drugs in Black and Hispanic cancer patients.
It is conceivable that newly developed, mitochondria-targeted modified natural products and FDA-approved drugs (e.g., Mito-honokiol, Mito-magnolol, Mito-metformin, Mito-atovaquone, Mito-hydroxyurea), as well as those developed in other labs,94,95 potentially could be useful in understanding the biological racial disparity in cancer mortality.
Recently, mono-alkyl lipophilic cations (also referred to as cationic surfactants) consisting of a dimethyl sulfonium cation and a long alkyl side chain were reported to inhibit mitochondrial respiration of fungi and exert a strong antifungal mechanism.13,96 To establish the generality of the OXPHOS inhibition mechanism, additional cationic molecules should be tested.
Do all complex I inhibitors have the potential to become anticancer agents? The lack of effect of complex I inhibitors on normal cells must be studied in all cases.
MTDs combined with standard-of-care chemotherapy, radiation therapy, and immunotherapy are likely to have potentiating effects. Ongoing research in preclinical models suggests that MTDs augment the efficacy of PD-L1 inhibitors. These combinational modalities may counteract the immunosuppressive TIME and enhance immunotherapy using the checkpoint inhibitors.97
Emerging research reports nanotube-mediated transfer of mitochondria from T cells to cancer cells as an immune evasion mechanism,98 further substantiating the urgent need to develop more potent MTDs. Future research should also focus on the effect of MTDs on nanotube or nanotube assembly machinery formation. Previous research has demonstrated that dual targeting of mitochondria with MTDs and antiglycolytics (e.g., 2-deoxy-d-glucose) significantly inhibits generation of adenosine triphosphate in breast cancer cells.27 This combinatorial therapeutic approach may hinder nanotube-mediated mitochondrial trafficking between immune cells and cancer cells.
Acknowledgements
Thank you to Lydia Washechek for preparing and proofreading the manuscript.
Thanks to my collaborators and coworkers for their contributions in advancing the preclinical scope of mitochondria-targeted drugs in cancer therapy, cancer prevention, and immune-oncology.
This work was supported by the National Cancer Institute of the National Institutes of Health under Award Numbers R01CA208648 and R01CA232433. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Nonstandard Abbreviation List
- ERR-1
estrogen-related receptor 1
- FeS
iron-sulfur
- M1
tumor inhibitory
- M2
tumor promoting
- MDSC
myeloid-derived suppressor cell
- Mito
mitochondria
- MTD
mitochondria-targeted drug
- OXPHOS
oxidative phosphorylation
- PD-1
programmed cell death protein 1
- PD-L1
programmed death receptor ligand 1
- PET-CT
positron emission tomography–computed tomography
- PGC-1α
proliferator-activated receptor gamma coactivator 1-alpha
- ROS
reactive oxygen species
- STAT3
signal transducer and activator of transcription 3
- TIL
tumor-associated lymphocytes
- TIME
tumor immune microenvironment
- TPP+
Triphenylphosphonium cation
- Tregs
regulatory T cells
Footnotes
Conflicts of Interest
B.K. is an inventor of US Patent No. 10,836,782 (issued November 17, 2020)/European Patent No. 3307254 (issued August 5, 2020), “Mito-honokiol compounds and methods of synthesis and use thereof”; US Patent No. 11,083,739 (issued August 10, 2021), “Mito-magnolol compounds and methods of synthesis and use thereof”; and US Patent No. 9,956,233 (issued May 1, 2018), “Neuroprotection by mitochondria-targeted metformin.”
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References
- 1.Cha JH, Chan LC, Li CW, Hsu JL, Hung MC. Mechanisms Controlling PD-L1 Expression in Cancer. Mol Cell. 2019;76(3):359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Han Y, Liu D, Li L. PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res. 2020;10(3):727–742. [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng G, Zielonka J, McAllister DM, et al. Mitochondria-targeted vitamin E analogs inhibit breast cancer cell energy metabolism and promote cell death. BMC Cancer. 2013;13:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng G, Zielonka J, Ouari O, et al. Mitochondria-Targeted Analogues of Metformin Exhibit Enhanced Antiproliferative and Radiosensitizing Effects in Pancreatic Cancer Cells. Cancer Res. 2016;76(13):3904–3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng G, Zielonka J, Hardy M, et al. Synergistic inhibition of tumor cell proliferation by metformin and mito-metformin in the presence of iron chelators. Oncotarget. 2019;10(37):3518–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boyle KA, Van Wickle J, Hill RB, Marchese A, Kalyanaraman B, Dwinell MB. Mitochondria-targeted drugs stimulate mitophagy and abrogate colon cancer cell proliferation. J Biol Chem. 2018;293(38):14891–14904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015;11(1):9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu Y, Xue D, Bankhead A 3rd, Neamati N. Why All the Fuss about Oxidative Phosphorylation (OXPHOS)? J Med Chem. 2020;63(23):14276–14307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pan J, Lee Y, Cheng G, et al. Mitochondria-Targeted Honokiol Confers a Striking Inhibitory Effect on Lung Cancer via Inhibiting Complex I Activity. iScience. 2018;3:192–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischer GM, Jalali A, Kircher DA, et al. Molecular Profiling Reveals Unique Immune and Metabolic Features of Melanoma Brain Metastases. Cancer Discov. 2019;9(5):628–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin Cancer Res. 2018;24(11):2482–2490. [DOI] [PubMed] [Google Scholar]
- 12.Molina JR, Sun Y, Protopopova M, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med. 2018;24(7):1036–1046. [DOI] [PubMed] [Google Scholar]
- 13.Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16(10):619–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klein K, He K, Younes AI, et al. Role of Mitochondria in Cancer Immune Evasion and Potential Therapeutic Approaches. Front Immunol. 2020;11:573326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng G, Hardy M, Topchyan P, et al. Potent inhibition of tumour cell proliferation and immunoregulatory function by mitochondria-targeted atovaquone Sci Rep. 2020;10(1):17872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng G, Hardy M, Topchyan P, et al. Mitochondria-targeted hydroxyurea inhibits OXPHOS and induces antiproliferative and immunomodulatory effects. iScience. 2021;24(6):102673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagaraj S, Youn JI, Gabrilovich DI. Reciprocal relationship between myeloid-derived suppressor cells and T cells. J Immunol 2013;191(1):17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawano Y, Moschetta M, Manier S, et al. Targeting the bone marrow microenvironment in multiple myeloma. Immunol Rev. 2015;263(1):160–172. [DOI] [PubMed] [Google Scholar]
- 19.Sawant A, Schafer CC, Jin TH, et al. Enhancement of antitumor immunity in lung cancer by targeting myeloid-derived suppressor cell pathways. Cancer Res. 2013;73(22):6609–6620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Law AMK, Valdes-Mora F, Gallego-Ortega D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells. 2020;9(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hossain F, Al-Khami AA, Wyczechowska D, et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol Res. 2015;3(11):1236–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.AbuEid M, McAllister DM, McOlash L, et al. Synchronous effects of targeted mitochondrial complex I inhibitors on tumor and immune cells abrogate melanoma progression. iScience. 2021;24(6):102653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim G, Pastoriza JM, Condeelis JS, et al. The Contribution of Race to Breast Tumor Microenvironment Composition and Disease Progression. Front Oncol. 2020;10:1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deshmukh SK, Srivastava SK, Tyagi N, et al. Emerging evidence for the role of differential tumor microenvironment in breast cancer racial disparity: a closer look at the surroundings. Carcinogenesis. 2017;38(8):757–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656. [DOI] [PubMed] [Google Scholar]
- 26.Dhanasekaran A, Kotamraju S, Karunakaran C, et al. Mitochondria superoxide dismutase mimetic inhibits peroxide-induced oxidative damage and apoptosis: Role of mitochondrial superoxide. Free Radical Biol Med. 2005;39(5):567–583. [DOI] [PubMed] [Google Scholar]
- 27.Cheng G, Zielonka J, Dranka BP, et al. Mitochondria-targeted drugs synergize with 2-deoxyglucose to trigger breast cancer cell death. Cancer Res. 2012;72(10):2634–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zielonka J, Joseph J, Sikora A, et al. Mitochondria-targeted triphenylphosphonium-based compounds: Syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem Rev. 2017;117(15):10043–10120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalyanaraman B, Cheng G, Zielonka J, Bennett B. Low-Temperature EPR Spectroscopy as a Probe-Free Technique for Monitoring Oxidants Formed in Tumor Cells and Tissues: Implications in Drug Resistance and OXPHOS-Targeted Therapies. Cell Biochem Biophys. 2019;77(1):89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng G, Zielonka M, Dranka B, et al. Detection of mitochondria-generated reactive oxygen species in cells using multiple probes and methods: Potentials, pitfalls, and the future. J Biol Chem. 2018;293(26):10363–10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. 2020;368(6487). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng G, Zielonka J, McAllister D, Tsai S, Dwinell MB, Kalyanaraman B. Profiling and targeting of cellular bioenergetics: Inhibition of pancreatic cancer cell proliferation. Br J Cancer. 2014;111(1):85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng G, Hardy M, Zielonka J, et al. Mitochondria-Targeted Magnolol Inhibits OXPHOS, Proliferation, and Tumor Growth via Modulation of Energetics and Autophagy in Melanoma Cells. Cancer Research and Treatment Communications. 2020;25:100210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weinberg SE, Singer BD, Steinert EM, et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature. 2019;565(7740):495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gouirand V, Guillaumond F, Vasseur S. Influence of the Tumor Microenvironment on Cancer Cells Metabolic Reprogramming. Front Oncol. 2018;8:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng G, Zhang Q, Pan J, et al. Targeting lonidamine to mitochondria mitigates lung tumorigenesis and brain metastasis. Nat Commun. 2019;10(1):2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Najjar YG, Menk AV, Sander C, et al. Tumor cell oxidative metabolism as a barrier to PD-1 blockade immunotherapy in melanoma. JCI Insight. 2019;4(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bader JE, Voss K, Rathmell JC. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol Cell. 2020;78(6):1019–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sugiura A, Rathmell JC. Metabolic Barriers to T Cell Function in Tumors. J Immunol. 2018;200(2):400–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang B, Zhao Q, Zhang Y, et al. Targeting hypoxia in the tumor microenvironment: a potential strategy to improve cancer immunotherapy. J Exp Clin Cancer Res. 2021;40(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Musetti S, Huang L. Nanoparticle-Mediated Remodeling of the Tumor Microenvironment to Enhance Immunotherapy. ACS Nano. 2018;12(12):11740–11755. [DOI] [PubMed] [Google Scholar]
- 42.Zheng Z, Zhang J, Jiang J, et al. Remodeling tumor immune microenvironment (TIME) for glioma therapy using multi-targeting liposomal codelivery. J Immunother Cancer. 2020;8(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen Q, Liu G, Liu S, et al. Remodeling the Tumor Microenvironment with Emerging Nanotherapeutics. Trends Pharmacol Sci. 2018;39(1):59–74. [DOI] [PubMed] [Google Scholar]
- 44.Thomlinson RH, Gray LH. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br J Cancer. 1955;9(4):539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res. 1998;58(7):1408–1416. [PubMed] [Google Scholar]
- 46.Gammon ST, Pisaneschi F, Bandi ML, et al. Mechanism-Specific Pharmacodynamics of a Novel Complex-I Inhibitor Quantified by Imaging Reversal of Consumptive Hypoxia with [(18)F]FAZA PET In Vivo. Cells. 2019;8(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vashisht Gopal YN, Gammon S, Prasad R, et al. A Novel Mitochondrial Inhibitor Blocks MAPK Pathway and Overcomes MAPK Inhibitor Resistance in Melanoma. Clin Cancer Res. 2019;25(21):6429–6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scharping NE, Menk AV, Whetstone RD, Zeng X, Delgoffe GM. Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer Immunol Res. 2017;5(1):9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Y, Patel SP, Roszik J, Qin Y. Hypoxia-Driven Immunosuppressive Metabolites in the Tumor Microenvironment: New Approaches for Combinational Immunotherapy. Front Immunol. 2018;9:1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Colombani T, Eggermont LJ, Hatfield SM, et al. Oxygen-Generating Cryogels Restore T Cell Mediated Cytotoxicity in Hypoxic Tumors. Adv Funct Mater. 2021;31(37):2102234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hatfield SM, Sitkovsky MV. Antihypoxic oxygenation agents with respiratory hyperoxia to improve cancer immunotherapy. J Clin Invest. 2020;130(11):5629–5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clever D, Roychoudhuri R, Constantinides MG, et al. Oxygen Sensing by T Cells Establishes an Immunologically Tolerant Metastatic Niche. Cell. 2016;166(5):1117–1131.e1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen D, Barsoumian HB, Fischer G, et al. Combination treatment with radiotherapy and a novel oxidative phosphorylation inhibitor overcomes PD-1 resistance and enhances antitumor immunity. J Immunother Cancer. 2020;8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boreel DF, Span PN, Heskamp S, Adema GJ, Bussink J. Targeting Oxidative Phosphorylation to Increase the Efficacy of Radio- and Immune-Combination Therapy. Clin Cancer Res. 2021;27(11):2970–2978. [DOI] [PubMed] [Google Scholar]
- 55.Williams LK, Padhukasahasram B, Ahmedani BK, et al. Differing effects of metformin on glycemic control by race-ethnicity. J Clin Endocrinol Metab. 2014;99(9):3160–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Feng T, Sun X, Howard LE, et al. Metformin use and risk of prostate cancer: results from the REDUCE study. Cancer Prev Res (Phila). 2015;8(11):1055–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang JJ, Zhang QS, Li ZQ, Zhou JW, Du J. Metformin attenuates PD-L1 expression through activating Hippo signaling pathway in colorectal cancer cells. Am J Transl Res. 2019;11(11):6965–6976. [PMC free article] [PubMed] [Google Scholar]
- 58.Woman’s Hospital, Our Lady of the Lake Regional Medical Center, American Cancer Society Inc., Pfizer. Metformin Use to Reduce Disparities in Newly Diagnosed Breast Cancer (METBC). 2021; Identifier NCT04741204. Available at: https://clinicaltrials.gov/ct2/show/NCT04741204. [Google Scholar]
- 59.Hardie DG, Ashford ML. AMPK: regulating energy balance at the cellular and whole body levels. Physiology (Bethesda). 2014;29(2):99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carling D AMPK signalling in health and disease. Curr Opin Cell Biol. 2017;45:31–37. [DOI] [PubMed] [Google Scholar]
- 61.Harada Y, Ishii I, Hatake K, Kasahara T. Pyrvinium pamoate inhibits proliferation of myeloma/erythroleukemia cells by suppressing mitochondrial respiratory complex I and STAT3. Cancer Lett. 2012;319(1):83–88. [DOI] [PubMed] [Google Scholar]
- 62.Salminen A, Kaarniranta K, Kauppinen A. Phytochemicals inhibit the immunosuppressive functions of myeloid-derived suppressor cells (MDSC): Impact on cancer and age-related chronic inflammatory disorders. Int Immunopharmacol. 2018;61:231–240. [DOI] [PubMed] [Google Scholar]
- 63.Ostrand-Rosenberg S Myeloid-Derived Suppressor Cells: Facilitators of Cancer and Obesity-Induced Cancer. Annual Review of Cancer Biology. 2021;5(1):17–38. [Google Scholar]
- 64.Zeng D, Long H, Zhu B. Antitumor effects of targeting myeloid-derived suppressive cells. Translational Cancer Research. 2020;9(9):5787–5797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Trillo-Tinoco J, Sierra RA, Mohamed E, et al. AMPK Alpha-1 Intrinsically Regulates the Function and Differentiation of Tumor Myeloid-Derived Suppressor Cells. Cancer Res. 2019;79(19):5034–5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim SH, Li M, Trousil S, et al. Phenformin Inhibits Myeloid-Derived Suppressor Cells and Enhances the Anti-Tumor Activity of PD-1 Blockade in Melanoma. J Invest Dermatol. 2017;137(8):1740–1748. [DOI] [PubMed] [Google Scholar]
- 67.Bu LL, Yu GT, Deng WW, et al. Targeting STAT3 signaling reduces immunosuppressive myeloid cells in head and neck squamous cell carcinoma. Oncoimmunology. 2016;5(5):e1130206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Trovato R, Fiore A, Sartori S, et al. Immunosuppression by monocytic myeloid-derived suppressor cells in patients with pancreatic ductal carcinoma is orchestrated by STAT3. J Immunother Cancer. 2019;7(1):255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shukla P, Singh KK. Uncovering Mitochondrial Determinants of Racial Disparities in Ovarian Cancer. Trends Cancer. 2021;7(2):93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Choudhury AR, Singh KK. Mitochondrial determinants of cancer health disparities. Semin Cancer Biol. 2017;47:125–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sotgia F, Lisanti MP. Mitochondrial biomarkers predict tumor progression and poor overall survival in gastric cancers: Companion diagnostics for personalized medicine. Oncotarget. 2017;8(40):67117–67128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Giedt RJ, Fumene Feruglio P, Pathania D, et al. Computational imaging reveals mitochondrial morphology as a biomarker of cancer phenotype and drug response. Sci Rep. 2016;6:32985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Skwarski M, McGowan DR, Belcher E, et al. Mitochondrial Inhibitor Atovaquone Increases Tumor Oxygenation and Inhibits Hypoxic Gene Expression in Patients with Non-Small Cell Lung Cancer. Clin Cancer Res. 2021;27(9):2459–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yao S, Cheng TD, Elkhanany A, et al. Breast Tumor Microenvironment in Black Women: A Distinct Signature of CD8+ T-Cell Exhaustion. J Natl Cancer Inst. 2021;113(8):1036–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Beebe-Dimmer JL, Cooney KA. Mitochondrial alterations may underlie race-specific differences in cancer risk and outcome. J Clin Invest. 2019;129(6):2187–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Piyarathna DWB, Balasubramanian A, Arnold JM, et al. ERR1 and PGC1α associated mitochondrial alterations correlate with pan-cancer disparity in African Americans. J Clin Invest. 2019;129(6):2351–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang CY, Hsu LH, Chen CY, et al. Inhibition of Alternative Cancer Cell Metabolism of EGFR Mutated Non-Small Cell Lung Cancer Serves as a Potential Therapeutic Strategy. Cancers (Basel). 2020;12(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Adashek JJ, Szeto CW, Reddy SK, Spiess PE. Real-world data validation for differential expression of immunoregulatory molecules and targetable cancer genes may provide therapeutic insights into agnostic-driven trial designs. Journal of Clinical Oncology. 2020;38(5_suppl):10–10. [Google Scholar]
- 79.Bonifaz L, Cervantes-Silva M, Ontiveros-Dotor E, López-Villegas E, Sánchez-García F. A Role For Mitochondria In Antigen Processing And Presentation. Immunology. 2014;144(3):461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Matheoud D, Sugiura A, Bellemare-Pelletier A, et al. Parkinson’s Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell. 2016;166(2):314–327. [DOI] [PubMed] [Google Scholar]
- 81.Dhatchinamoorthy K, Colbert JD, Rock KL. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front Immunol. 2021;12:636568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kiritsy MC, McCann K, Mott D, et al. Mitochondrial respiration contributes to the interferon gamma response in antigen-presenting cells. Elife. 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ankley LM, Kiritsy M, Trombley J, Huizinga G, Olive AJ. Defining the mechanisms of GSK3 and Med16 in the regulation of MHCII. The Journal of Immunology. 2021;206(1 Supplement):93.03–93.03. [Google Scholar]
- 84.Okugawa Y, Grady WM, Goel A. Epigenetic Alterations in Colorectal Cancer: Emerging Biomarkers. Gastroenterology. 2015;149(5):1204–1225.e1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ahmad A, Azim S, Zubair H, et al. Epigenetic basis of cancer health disparities: Looking beyond genetic differences. Biochim Biophys Acta Rev Cancer. 2017;1868(1):16–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Devall M, Sun X, Yuan F, et al. Racial Disparities in Epigenetic Aging of the Right vs Left Colon. J Natl Cancer Inst. 2020;113(12):1779–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rai R, Yadav SS, Pan H, et al. Epigenetic analysis identifies factors driving racial disparity in prostate cancer. Cancer Rep (Hoboken). 2019;2(2):e1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mohammed SI, Springfield S, Das R. Role of epigenetics in cancer health disparities. Methods Mol Biol. 2012;863:395–410. [DOI] [PubMed] [Google Scholar]
- 89.Miranda-Gonçalves V, Lameirinhas A, Henrique R, Jerónimo C. Metabolism and Epigenetic Interplay in Cancer: Regulation and Putative Therapeutic Targets. Front Genet. 2018;9:427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Morrison AJ. Cancer cell metabolism connects epigenetic modifications to transcriptional regulation. FEBS J. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kloepping KC, Kraus AS, Hedlund DK, et al. Triphenylphosphonium derivatives disrupt metabolism and inhibit melanoma growth in vivo when delivered via a thermosensitive hydrogel. PLoS One. 2020;15(12):e0244540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mistry JJ, Marlein CR, Moore JA, et al. ROS-mediated PI3K activation drives mitochondrial transfer from stromal cells to hematopoietic stem cells in response to infection. Proc Natl Acad Sci U S A. 2019;116(49):24610–24619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Marlein CR, Zaitseva L, Piddock RE, et al. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood. 2017;130(14):1649–1660. [DOI] [PubMed] [Google Scholar]
- 94.Rohlenova K, Sachaphibulkij K, Stursa J, et al. Selective Disruption of Respiratory Supercomplexes as a New Strategy to Suppress Her2(high) Breast Cancer. Antioxid Redox Signal. 2017;26(2):84–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Reddy CA, Somepalli V, Golakoti T, et al. Mitochondrial-targeted curcuminoids: a strategy to enhance bioavailability and anticancer efficacy of curcumin. PLoS One. 2014;9(3):e89351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Steinberg G, Schuster M, Gurr SJ, et al. A lipophilic cation protects crops against fungal pathogens by multiple modes of action. Nat Commun. 2020;11(1):1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tang T, Huang X, Zhang G, Hong Z, Bai X, Liang T. Advantages of targeting the tumor immune microenvironment over blocking immune checkpoint in cancer immunotherapy. Signal Transduct Target Ther. 2021;6(1):72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Saha T, Dash C, Jayabalan R, et al. Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nat Nanotechnol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.