

Abstract
Background&Aims:
NLR family pyrin domain-containing 3 (NLRP3) inflammasome activation has been shown to result in liver fibrosis. Mechanisms and downstream signaling remain incompletely understood. Here, we studied the role of interleukin 18 (IL-18) in hepatic stellate cells (HSCs) and its impact on liver fibrosis.
Approach&Results:
We observed significantly increased serum levels of IL-18 (128.4pg/ml vs. 74.9pg/ml) and IL-18 binding protein (BP; 46.50ng/ml vs. 15.35ng/ml) in patients with liver cirrhosis compared to healthy controls. Single cell RNA sequencing data showed that an immunoregulatory subset of murine HSCs expresses IL-18 and the IL-18 receptor. Treatment of cultured primary murine HSC with rmIL-18 accelerated their transdifferentiation into myofibroblasts. In vivo, IL-18 receptor deficient mice had reduced liver fibrosis in a model of fibrosis induced by HSC-specific NLRP3 overactivation. Whole liver RNA sequencing analysis from a murine model of severe NASH-induced fibrosis by feeding choline-deficient, L-amino acid-defined high-fat diet showed that genes related to Il18 and its downstream signaling were significantly upregulated and Il18−/− mice receiving this diet for 10 weeks showed protection from fibrotic changes with decreased number of αSMA-positive cells and collagen deposition. HSC activation triggered by NLRP3 inflammasome activation was abrogated when IL-18 signaling was blocked by its naturally occurring antagonist IL-18BP. Accordingly, we observed that the severe inflammatory phenotype associated with myeloid cell-specific Nlrp3 gain-of-function was rescued by IL-18BP.
Conclusions:
Our study highlights the role of IL-18 in the development of liver fibrosis by its direct effect on HSC activation identifying IL-18 as a novel target to treat liver fibrosis.
Graphiacal Abstract
The cytokine interleukin 18 (IL-18) was initially described as interferon gamma (IFNγ)-inducing proinflammatory factor in a septic shock model, which stimulated T cell proliferation. (1) Like IL-1β, IL-18 belongs to the IL-1 superfamily and is synthesized as biologically inactive 24-kDa precursor protein (pro-IL-18) lacking a signal peptide required for secretion. An intracellular cysteine protease called caspase-1 that is also known as IL-1β–converting enzyme, cleaves pro-IL-18 into its mature and active form. (2) Activation of caspase-1 itself is mediated by multiprotein complexes called inflammasomes. The most studied member of the nucleotide-binding oligomerization domain-like receptor (NLR) family is the NLR family pyrin domain containing 3 (NLRP3) inflammasome, which detects a broad range of endogenous or exogenous danger signals including pathogens and sterile triggers. (3) IL-18 precursors were shown to be constitutively expressed in peripheral blood mononuclear cells, macrophages, monocytes, keratinocytes, intestinal epithelial cells, Kupffer cells as well as in spleen cells. The transcription of IL-18 was not induced by inflammatory stimuli in these cells demonstrating that IL-18 plays a role in homeostasis and is not regulated by inflammation like IL-1β. (4–9) IL-18 signaling through its receptor is mediated by a unique extracellular receptor complex that consists of the ligand-binding chain IL-18Rα and the nonbinding signaling-transducing chain IL-18Rβ. (10, 11)
Besides transcription and caspase-1 processing, IL-18 is also regulated by IL-18 binding protein (IL-18BP), which acts as an endogenous antagonist with extremely high affinity. (12) The 40 kDa glycoprotein lacks a transmembrane domain and impedes the binding of IL-18 to its receptor, blocking the initiation of signal transduction. In healthy humans about 2–3 ng/ml of IL-18BP are found in blood circulation showing a 20-fold molar excess in comparison to IL-18. (13)
Hepatic fibrosis is a progressive form of chronic liver injury characterized by an excessive deposition of extracellular matrix (ECM) like type I collagen, which has been shown to contribute to the development of cirrhosis and hepatocellular carcinoma. (14) Activation of hepatic stellate cells (HSCs) by other liver cells, predominantly liver macrophages, has been identified to have a pivotal role in fibrogenesis. Under physiological conditions, HSCs maintain in a quiescent state located in the space of Disse and participate in the homeostasis of ECM, regeneration, repair as well as control of retinoid storage and metabolism. Upon liver damage, quiescent HSCs transdifferentiate into an activated and proliferating state characterized by a myofibroblast-like appearance, the loss of retinoid droplets, the expression of alpha smooth muscle actin (αSMA), and the secretion of extracellular matrix (ECM) and cytokines. (15) However, HSCs can also contribute to the regression of liver fibrosis by undergoing apoptosis or reverting to an inactive HSC state with the capacity of rapid reactivation in the case of pro-fibrotic stimuli. (16, 17)
Recently, our research group demonstrated that NLRP3 activation and pyroptosis are important contributors to liver inflammation and fibrosis. (18–20) Specifically, we discovered that hyperactivation of NLRP3 in murine HSCs provoked transdifferentiation into an activated myofibroblast phenotype. Moreover, using a genetic gain-of-function mouse model we found that persistent Nlrp3 activation in HSCs resulted in increased expression of pro-fibrotic genes and proteins and liver fibrosis by 24 weeks of age. These results imply a direct role of NLRP3 inflammasome activation in HSCs and hepatic fibrogenesis. (21)
Several inflammatory diseases are associated with significantly increased IL-18 expression in serum such as type 2 diabetes mellitus (22), rheumatoid arthritis (23), Crohńs disease (24) and multiple sclerosis (25). Growing evidence suggests an important role of IL-18 in liver diseases. Elevated amounts of IL-18 in serum have been observed for patients with primary biliary cholangitis (26), biliary atresia (27) and chronic liver disease (CLD) of different etiologies (12) and are correlated with disease severity. Further studies demonstrated that IL-18 inhibition by exogenous IL-18BP, neutralizing antibodies or caspase-1 deficiency protected mice from endotoxin-induced liver injury or non-alcoholic fatty liver disease, whereas IL-1β blockade does not rescue the phenotype. (1, 28–30) Therefore, we initiated this study to elucidate the role of the proinflammatory cytokine IL-18 on HSC activation and the development of liver fibrosis.
Material and Methods
Patient cohort
Patients were recruited from the Department of Hepatology and Gastroenterology, Charité University Medicine Berlin. The study protocol was approved by the ethics committee of Charité, University Medicine Berlin, Germany (ethical approval number EA2/091/19) and was in accordance with the Declaration of Helsinki. Diagnosis of liver fibrosis/cirrhosis was based on liver imaging consistent with liver cirrhosis (e.g. nodular hepatic contour, changes in volume distribution indicating portal hypertension in the absence of portal vein thrombosis, secondary phenomena of portal hypertension such as splenomegaly, enlarged caudate lobe and left lobe lateral segment, regenerative nodules) together with clinical and laboratory signs of portal hypertension/cirrhosis (e.g. low platelets, albumin and prothrombin time, esophageal varices). For all patients with cirrhosis individual MELD score was calculated. Healthy individuals without evidence of liver disease were recruited as control group. Written informed consent was obtained from all patients.
Single cell RNA sequencing
Gene expression analysis of Il18 and Il18 receptor (Il18r) in HSCs was performed by analyzing recently published data with permission of the authors. (31) Raw data are available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE132662. In this previously published dataset, activated HSCs were isolated from wild type mice (WT; C57BL6/J) receiving carbon tetrachloride (CCl4) for 3 weeks. Quiescent HSCs were isolated from untreated animals and processed as described in the previous manuscript. Feature plots were generated by using the R package “Seurat” (v2.3.2) package for R (v3.5) (https://www.r-project.org/). (31, 32)
Mouse strains
Different mutant Nlrp3 gain-of-function mouse models of increasing severity were used including: Nlrp3D301NneoR mice (B6N.129-Nlrp3tm3Hhf/J) carrying the aspartic acid 301 to asparagine D301N), Nlrp3A350VneoR mice (B6N.129-Nlrp3tm1Hhf/J) carrying the alanine 350 to valine (A350V) substitution, and Nlrp3L351PneoR mice (B6N.129-Nlrp3tm2Hhf/J) carrying the leucine 351 to proline (L351P) substitution. (33) Nlrp3A350VneoR mice were crossed to mice expressing Cre recombinase under control of an endogenous Lyz2 promoter (CreL; B6.129P2-Lyz2tm1(cre)Ifo/J) so that mutant Nlrp3 expression is only restricted to myeloid lineage cells (Nlrp3A350V CreL). Furthermore, Nlrp3A350V CreL mice were bred to Il18−/− (B6.129P2-Il18tm1Aki/J) or Il1r−/− (B6.129S7-Il1r1tm1Imx/J) and Il18r−/− (B6.129P2-Il18r1tm1Aki/J) mice. Nlrp3L351PneoR mice were crossed with transgenic mice expressing lecithin retinol acyltransferase-driven Cre (Lrat) kindly provided by the laboratory of R.F. Schwabe (Columbia University, New York, US), in which expression of mutant Nlrp3L351P is restricted to HSCs (Nlrp3L351P/+CreLrat). Nlrp3L351P/+CreLrat mice were additionally bred to Il18r−/− mice to inhibit the IL-18 signaling cascade. The group size (n = 3–5) was based on a former publication and the 3R principle and governmental regulations. (21) The all-male mice were maintained on a standard rodent chow for 24 weeks. At this time point, mice were sacrificed and liver tissue and serum were harvested. Cre-negative littermates were used as control mice in all Cre-inducible models, WT mice with C57BL/6 background for all other experiments. Genetically modified mice were purchased from The Jackson Laboratory and have been bred to mice with C57BL/6 background over several generations. All experiments were approved by German local governmental authorities (Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen, LANUV NRW) and the Institutional Animal Care and Use Committee at the University of California, San Diego. Mice were housed in filter top cages on a 12-hours light/dark cycle with wood shaving bedding, ad libitum access to rodent chow and water unless otherwise noted.
Dietary-related fibrosis induction
8 weeks old WT (n = 7–8) or Il18−/− (n = 5–6) male mice were either fed with choline-deficient, L-amino acid-defined high-fat diet (CDAA-HFD; 60%; #A06071302; Research Diets, New Brunswick, NJ, USA) or with choline-supplemented, L-amino acid-defined control diet (CD) over a period of 10 weeks. Mice were randomly assigned to CDAA-HFD or CD groups so that all groups include mice from several litters.
IL-18BP treatment in mice
Recombinant human IL-18BP (rhIL-18BP; Tadekinig alfa) was provided by AB2 Bio LTD (Lausanne, Switzerland) due to the ability of human IL-18BPa to also neutralize murine IL-18. (34) In this study, Nlrp3D301NneoR+/− (B6N.129-Nlrp3tm3Hhf/J) bred to Lyz2tm1(cre)Ifo+/− mice (Nlrp3D301N CreL) to generate myeloid specific Nlrp3 gain-of-function mutants (male and female) were injected beginning at the age of 2 days subcutaneously with 6 μg/g body weight rhIL-18BP (n = 6) or PBS (n = 5) six times per week. After 14 days of treatment mice were euthanized and livers and sera were collected. Mice were randomly assigned to control and treatment groups from several litters.
HSC isolation and in vitro stimulation
For isolation of HSCs, 16–24 week old wild type (C57BL/6J) female or male mice were used. Briefly, mice were anesthetized by ketamine/xylazine injection and perfused in situ through the inferior vena cava with sequential Pronase E (0.4 mg/ml) and Collagenase D (0.8 mg/ml) solutions. Liver was removed and digested in vitro with Collagenase D (0.5 mg/ml), Pronase E (0.5 mg/ml) and DNase I (0.02 mg/ml). After 20 minutes, tissue was filtered through a 70 μm mesh. Cells were separated using a Nycodenz gradient centrifugation. The isolated HSCs were cultured in DMEM supplemented with 10% sera plus (PAN Biotech) and 100 U/ml penicillin/streptomycin. After cells attached overnight, HSCs were washed with PBS and stimulated with recombinant mouse (rm)IL-18 (100 ng/ml and 400 ng/ml; R&D, 9139-IL, Minneapolis, MN, US) in sera-free medium for 24 h. In an independent experiment, inflammasome activation in HSCs was induced by LPS (1 μg/ml) for 3 h and adenosine triphosphate (ATP; 5 mM) for 1 h. In addition, HSCs were co-incubated with 400 ng/ml rmIL-18BPd (R&D, 122-BP) to inhibit binding of IL-18 to its receptor. As all substances were either dissolved in PBS or water, culture medium without serum was used as vehicle. In all experiments, cells were incubated at 37°C and 5% CO2.
Statistical analysis
Statistical analysis was performed with GraphPad Prism 8.0 (GraphPad Software Inc., CA, US). Unpaired two-tailed Student’s t test was used for two-group comparison, whereas ANOVA followed by Tukeýs multiple comparison was used in order to analyze three or more groups. The significance level was set at α = 5% for all comparisons. Unless otherwise stated, data are presented as means ± standard deviation.
Other materials and methods are described in the Supplemental Materials and Methods.
Results
IL-18 signaling is a key regulator of liver fibrosis/cirrhosis in humans and mice
To investigate the potential role of IL-18 and IL-18BP as serum-based biomarkers in patients with NASH-induced liver cirrhosis we measured the serum levels of IL-18 and IL-18BP in 38 patients and 50 healthy volunteers. Patient and control characteristics are summarized in Supp. table 1. Interestingly, the level of circulating IL-18 was elevated in patients with liver cirrhosis (128.4 pg/ml; 76.48 – 209.8 pg/ml) in comparison to healthy controls (74.9 pg/ml; 47.73 – 103.1 pg/ml) (Fig. 1A). A significant increase was also observed for IL-18BP in serum of cirrhotic patients (46.50 ng/ml; 34.89 – 64.07 ng/ml) when compared to healthy individuals (15.36 ng/ml; 13.50 – 17.84 ng/ml) (Fig. 1B).
Fig. 1: IL-18 signaling impacts liver fibrosis/cirrhosis in humans and mice.
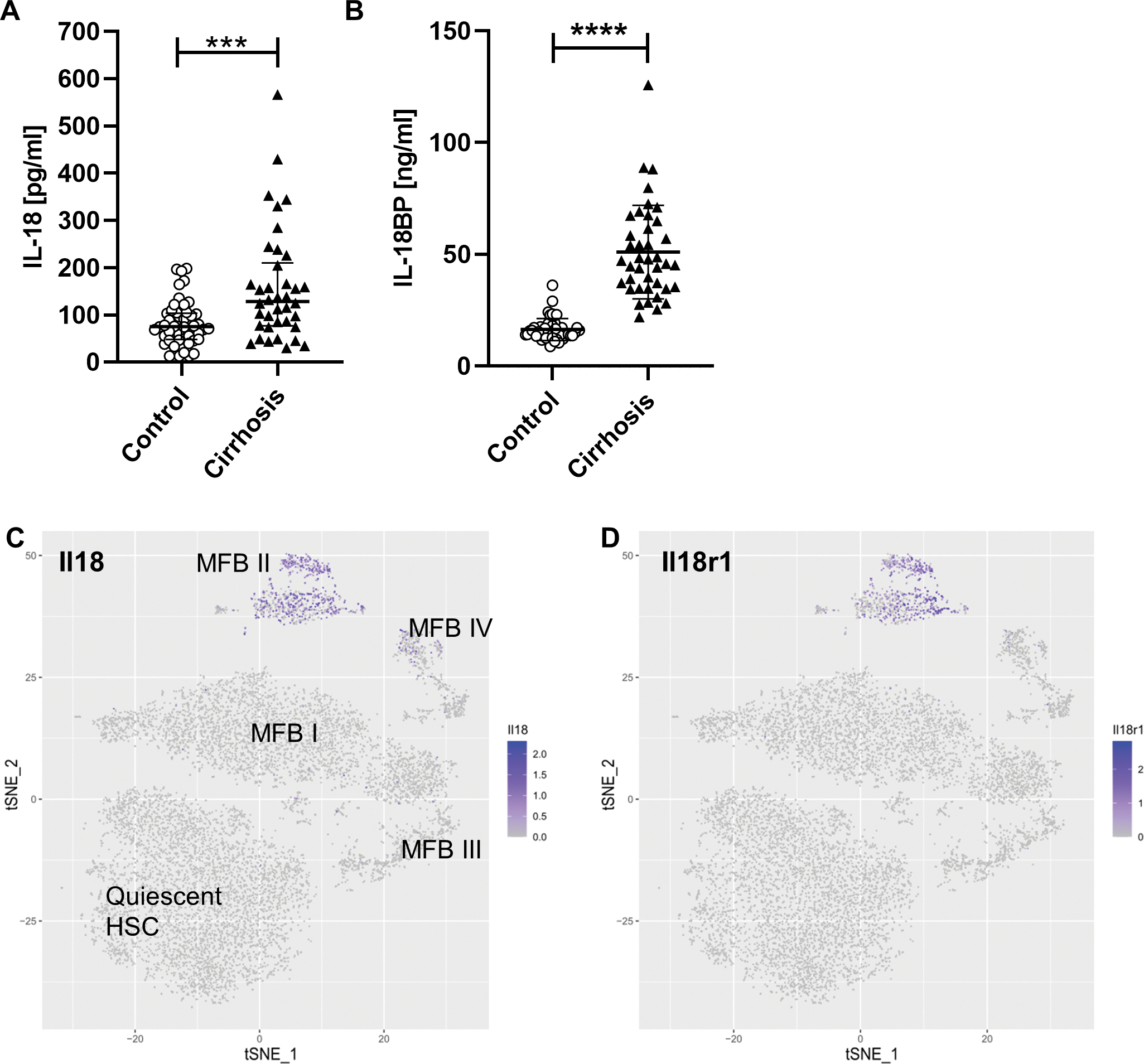
(A) Circulating IL-18 levels in patients with NASH-induced liver cirrhosis (LC; (128.4 pg/ml; 76.48 – 209.8 pg/ml) compared to healthy controls (74.9 pg/ml; 47.73 – 103.1 pg/ml). (B) Serum IL-18BP of LC patients (46.50 ng/ml; 34.89 – 64.07 ng/ml) in comparison to healthy individuals (15.36 ng/ml; 13.50 – 17.84 ng/ml). Data shown median; IQR. (Control, n = 50; LC, n = 38; ****P < 0.0001; Mann-Whitney U test).
T-distributed stochastic neighbor embedding (t-SNE) plots mapping distribution of HSCs expressing (C) Il18 and (D) Il18r1. Activated HSCs from carbon tetrachloride (CCl4)-treated mice that induces liver fibrosis were differentiated into four sub-populations (myofibroblasts; MFB I to MFB IV) according to their different gene expression pattern. (31) t-SNE plots show that especially HSCs from the immunoregulatory MFB II cluster express Il18 and Il18r1.
To delineate the expression pattern of IL-18 in quiescent and active HSCs during liver fibrosis, we leveraged single cell RNA sequencing data from a recent publication by Krenkel et al. (31) In this study, activated HSCs were isolated from mice treated with CCl4 to induce liver fibrosis. Based on the 50 most significant principal components they could be divided into four sub-populations (myofibroblasts; MFB I to MFB IV). T-SNE analysis show that the expression of Il18 and Il18r1 were found to be highly expressed in a subset of activated HSCs (named MFB II) but not in quiescent HSC (Fig. 1C and D). Interestingly, this specific cluster of activated HSC was characterized as an immunoregulatory subset that expressed various collagens as well as inflammatory mediators suggesting a potential role in modulating the inflammatory environment in their surroundings. (31)
Stimulation with IL-18 activated HSCs whereas IL-18BP mitigated LPS-induced HSC activation
To characterize the direct effect of IL-18 on HSCs, we stimulated primary mouse HSC from WT livers with two different concentrations of recombinant mouse (rm)IL-18 (100 ng/ml and 400 ng/ml) for 24 h. Immunofluorescence staining demonstrated an increase in αSMA expression but not for collagen I in primary HSCs treated with rmIL-18. These findings were observed especially for the higher dosage (Fig. 2A, B), where some cells already show αSMA-positive stress fibers, which was not observed for the lower concentration. Correspondingly, IL-18 treatment induced the expression of pro-fibrotic genes including Ctgf, Col1a1 and Acta2 (Fig. 2C).
Fig. 2: Stimulation with recombinant IL-18 accelerates HSC activation in vitro whereas blockage of IL-18 signaling inhibits HSC activation.
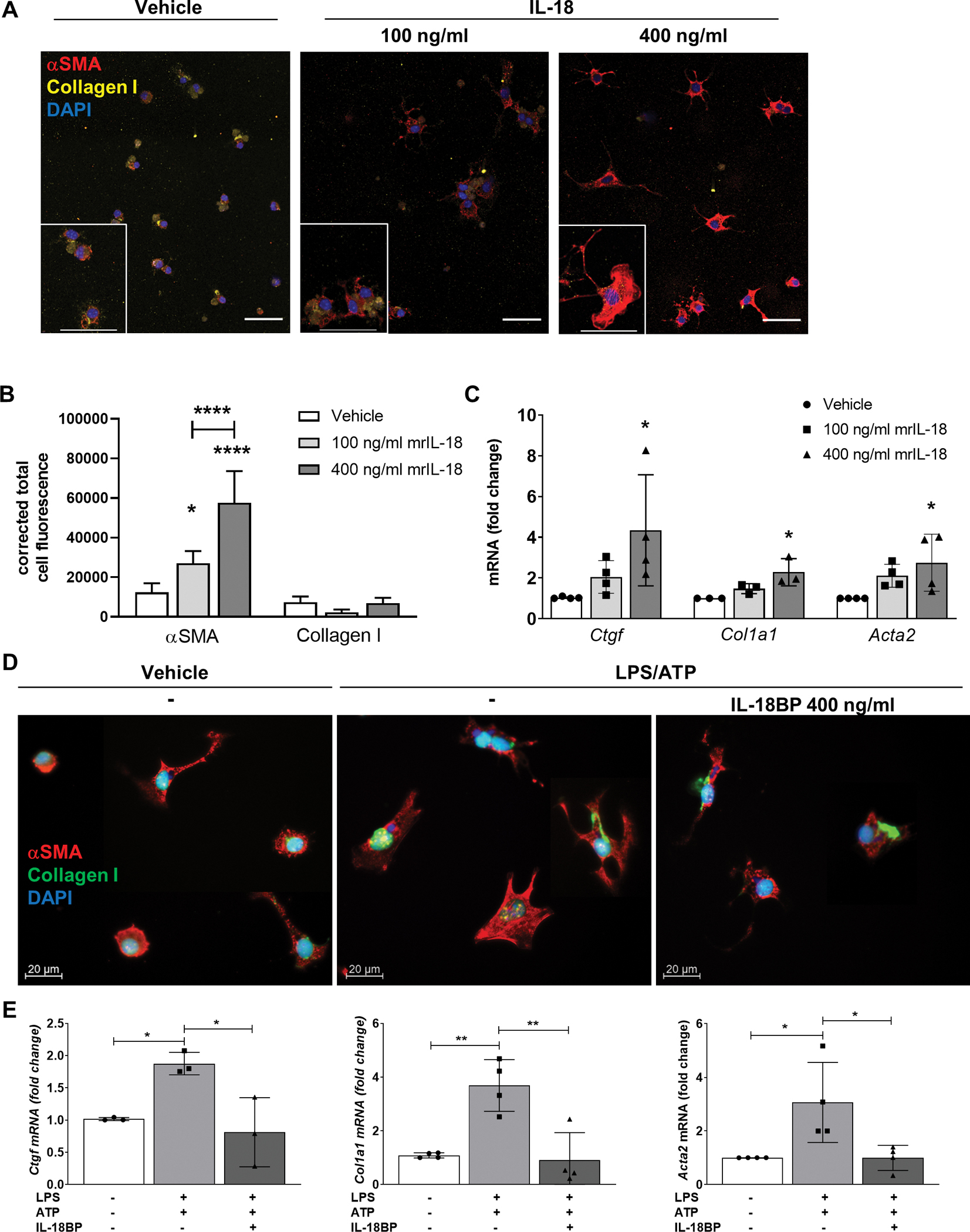
(A) Representative immunofluorescence images and (B) their quantification by the corrected total cell fluorescence showing αSMA and Collagen I of HSCs after treatment with either 100 ng/ml or 400 ng/ml rmIL-18 after 24 h. (C) mRNA levels of Ctgf, Col1a1 and Acta2 of primary murine HSCs after rmIL-18 stimulation. (D) immunofluorescence images and (E) mRNA levels of Ctgf, Col1a1 and Acta2 of primary HSCs treated with LPS/ATP and additional pre-treatment with rmIL-18BP. (3–4 independent replicates were included for all measured values; upper scale bars represent 50 μm *P < 0.05; **P < 0.01; ****P < 0.0001).
Next, we investigated the impact of inhibiting IL-18 signaling using its naturally occurring antagonist IL-18BP on primary HSCs from mouse livers. We found that HSC activation induced by LPS/ATP treatment was attenuated by IL-18BP, as evidenced by decreased expression of αSMA protein (Fig. 2D) and significant down-regulated levels of Ctgf, Col1a1 and Acta2 close to basal conditions (Fig. 2E). Taken together, these in vitro results suggest that IL-18 signaling plays a role in HSC activation and that IL-18BP inhibits self-amplifying activation, which might have an impact on fibrogenesis.
IL-18 signaling deficiency prevented liver fibrosis induced by persistent NLRP3 expression in HSCs
As described previously by our group, the enzyme LRAT is uniquely expressed in liver by HSCs and breeding of Lrat Cre expressing mice to Nlrp3L351P/+ (Nlrp3L351P/+CreLrat) generates a model of NLRP3 hyperactivation in HSCs that is associated with “spontaneous” liver fibrosis. (21) We used this model to examine IL-18 signaling deficiency in mice with early fibrosis by crossing these mice to IL-18 receptor knockout animals (Il18r−/−). In Il18r−/− mice, mRNA levels of αSma and Col1a1 were significantly reduced compared to Nlrp3L351P/+CreLrat Il18r+/+ littermates (Fig. 3A). Sirius Red liver staining displayed increased collagen deposition in Nlrp3L351P/+CreLrat Il18r+/+ compared to WT control, which was reduced to baseline conditions in Nlrp3L351P/+CreLrat Il18r−/− (Fig. 3B, D). Interestingly, mice with IL-18 signaling deficiency revealed a trend towards reduced macrophage infiltration represented by quantification of F4/80 positive cells in liver sections (Fig. 3B, E). Importantly, serum levels of ALT/AST and H&E staining of liver tissue indicated that liver injury was similar between the genotypes (Fig. 3B, C).
Fig. 3: IL-18 receptor deficiency significantly reduced expression of HSC activation marker in 24 weeks old mice with HSC-specific NLRP3 hyperactivation.
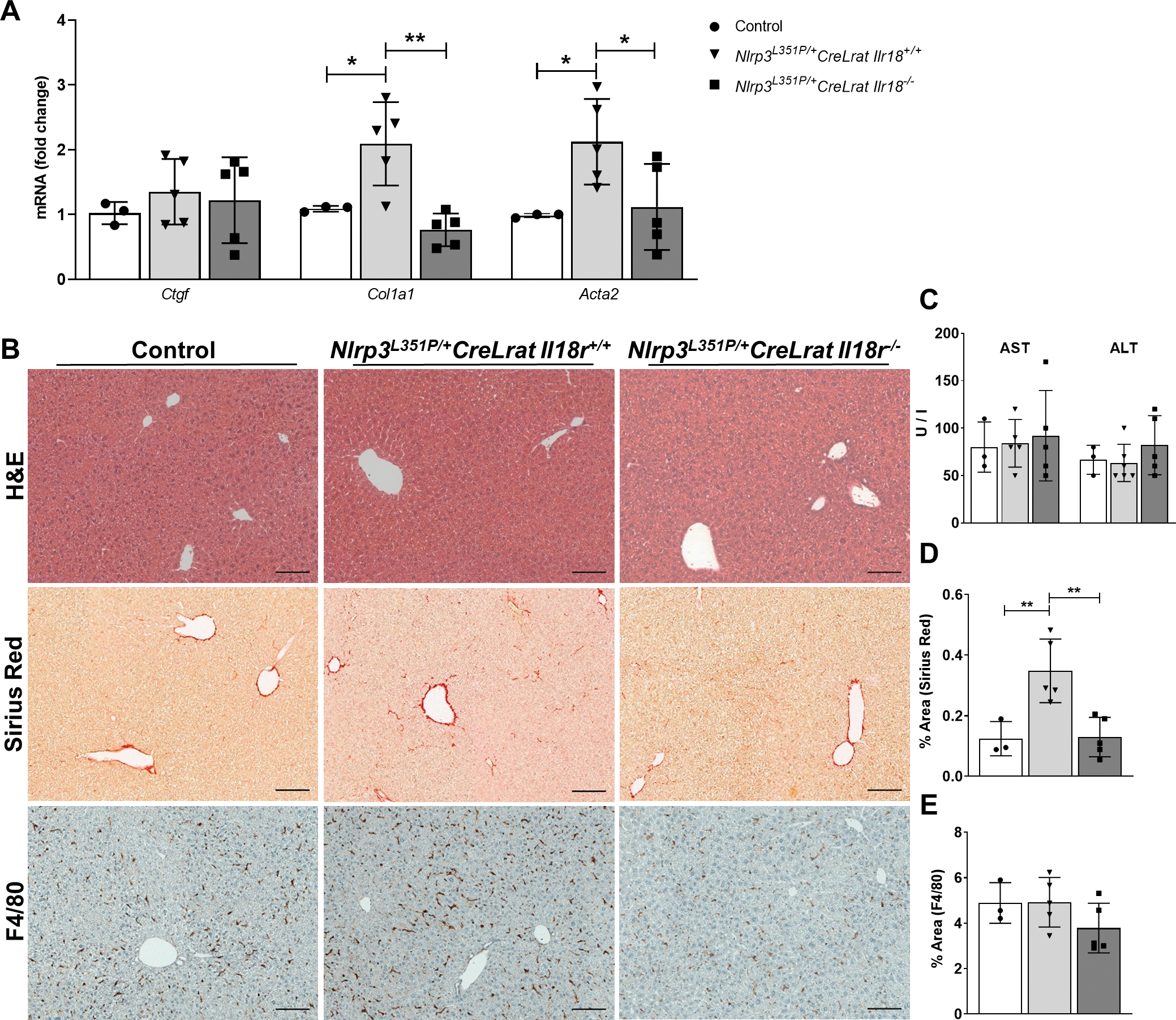
(A) mRNA levels of Ctgf, Col1a1 and αSma in control, Nlrp3L351P/+CreLrat Il18r+/+ and Nlrp3L351P/+CreLrat Il18r−/−mice. (B) Representative pictures of H&E, Sirius Red- and F4/80-stained liver sections. (C) Liver transaminases and quantification of (D) Sirius Red and (E) F4/80-positive cells in mice with Il-18R deficiency compared to Nlrp3L351P/+CreLrat Il18r+/+ mice. (n = 3–5 mice per group for all measured values; scale bars represent 100 μm; *P < 0.05; **P < 0.01).
IL-18 signaling and downstream regulatory network is activated in CDAA-HFD-induced fibrotic NASH
To assess the relevance of IL-18 signaling in a model of diet-induced fibrosis, we chose to use CDAA-HFD as this diet recapitulates many features of human NASH with fibrosis. (35) Mice were put on CDAA-HFD or control diet (CD) for 10 weeks and the gene expression profile of whole liver tissue was analyzed by RNAseq (Fig. 4A). IL-18 signaling was activated in mice fed a CDAA-HFD compared to mice fed a CD (Fig. 4B). Genes including Il18r1, Il18 receptor accessory protein (Il18rap) and Il18bp were upregulated in mice fed a CDAA-HFD, associated with increased expression of genes downstream of IL-18 signaling such as Cxcl10, Il16 and Fth1 (ferritin). In addition, inflammasome components that act upstream of IL-18 activation were also upregulated including Nlrp3 and Casp1. Fibrosis development is a major characteristic of NASH. Mice fed a CDAA-HFD showed a strong activation of fibrosis related genes including Col1a1, Col3a1, Vimentin, Timp1, Acta2, and Tgfb (Fig. 4C). These data highlights that many molecular players involved in IL-18 pathway are transcriptionally induced in fibrotic NASH.
Fig. 4: IL-18 signaling and downstream regulatory network is activated in CDAA-HFD-induced fibrotic NASH.
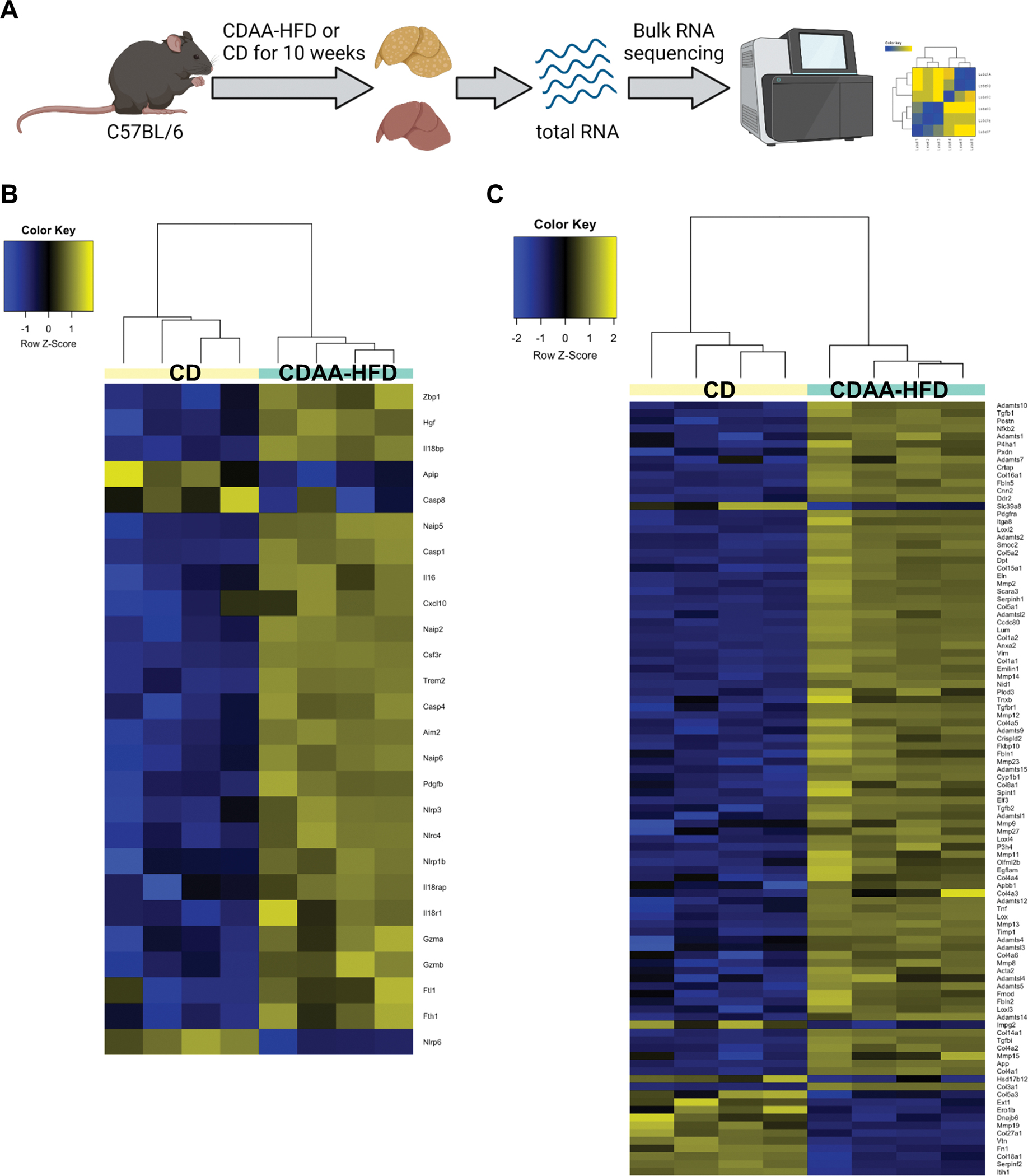
Bulk RNA sequencing analysis of liver tissue after 10 weeks of CDAA-HFD or CD. (A) An overview of experimental design. (B) Clustering of IL-18 signaling-related gene transcripts. (C) Clustering of fibrosis-related gene transcripts. (n = 4 mice per group).
IL-18 knockout mice are protected from CDAA-HFD-induced fibrosis
Based on the RNAseq data we next determined the role of ablation of IL-18 in NASH associated liver fibrosis. CDAA-HFD-fed animals showed prominent macro-vesicular steatosis with incipient nonalcoholic steatohepatitis in both WT and Il18−/− mice (Suppl. fig. 1A). In addition, CDAA-HFD mice had increased liver weight and lower body weight (Suppl. fig. 1B) compared to those mice fed with CD. Liver sections from CDAA-HFD-fed Il18−/− mice displayed less intrahepatic collagen accumulation and bridging fibrosis (Fig. 5A, B) compared to WT mice on CDAA-HFD, which was supported by a trend toward reduced amount of hydroxyproline in the Il18−/− liver tissue (Fig. 5C). In line with the fibrosis data, the positive area of the HSC activation marker αSMA (Fig. 5D, E) was significantly reduced in Il18−/− liver tissue. The gene expression levels of Ctgf, Col3a, Timp1 and Vimentin increased in response to CDAA-HFD in both groups compared to CD, but to a reduced extent in Il18−/− mice (Fig. 5F). To analyze the impact of ductular reaction, which is a common feature in CDAA-HFD fed mice (36), the amount of CK19-positive cells (apart from mature bile ducts, reactive cholangiocytes and hepatic progenitor cells) were determined. Both, WT and Il18−/− mice show ductular reaction, but without significant difference (Suppl. fig. 1D, E).
Fig. 5: IL-18 deficiency reduced choline-deficient, L-amino acid-defined high fat diet (CDAA-HFD)-induced fibrosis.
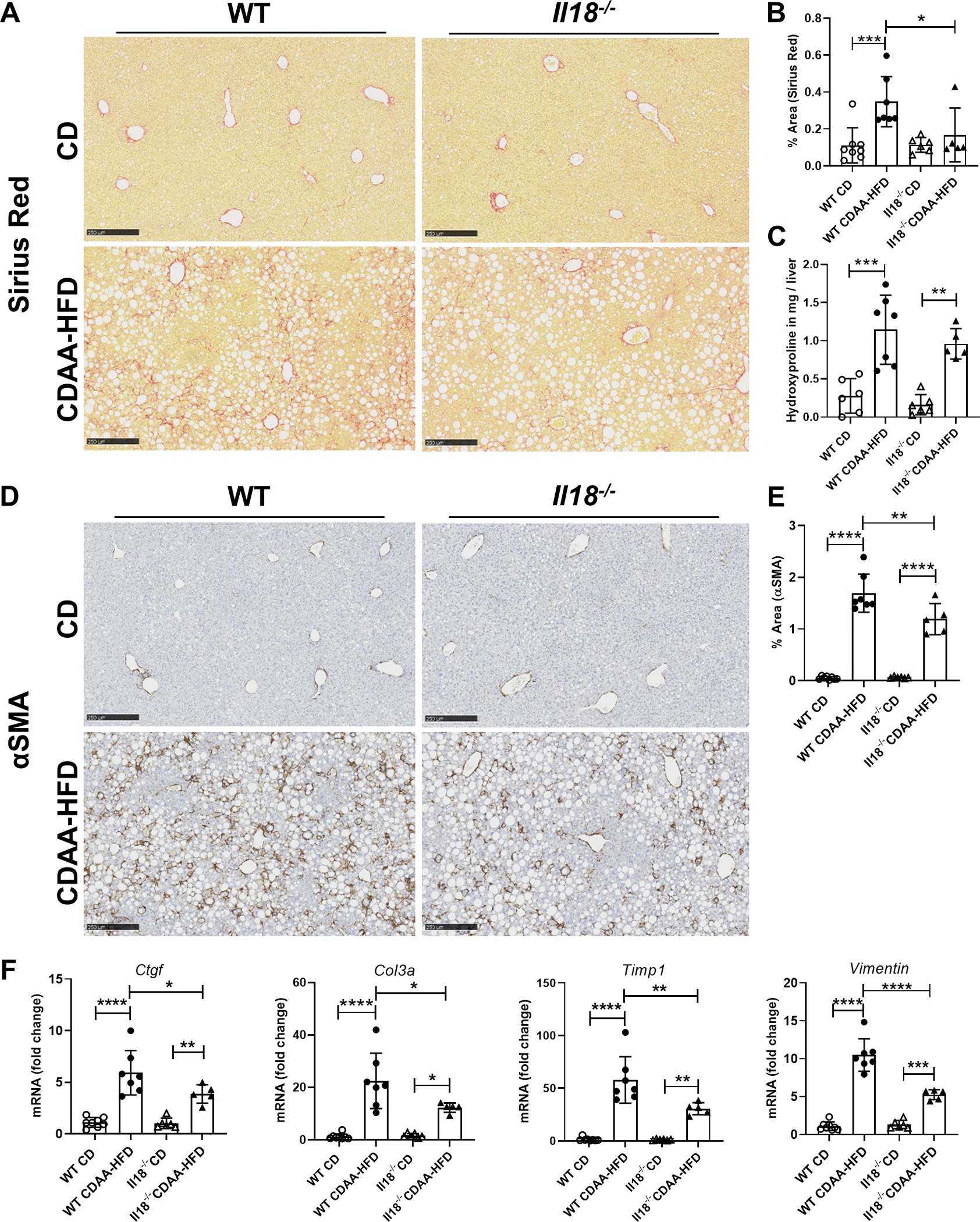
(A, B) Collagen deposition assessed by Sirius Red staining and (C) hydroxyproline, (D, E) αSMA-positive cells and (F) mRNA expression levels of the pro-fibrotic genes Ctgf, Col3a, Timp1 and Vimentin and in WT and Il18−/− mice fed with CDAA-HFD compared to mice on control diet (CD). (n = 5–8 mice per group for all measured values; scale bars represent 250 μm; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
IL-18BP treatment alleviated liver inflammation and fibrosis in myeloid cell-specific Nlrp3 mutant mice
In a previous study we demonstrated that disruption of IL-1β signaling partially rescued the hepatic and systemic inflammatory phenotype concomitant with growth impairment and shortened lifespan of myeloid cell-specific Nlrp3 mutant mice (Suppl. fig. 2). (18) Interestingly, IL-18 ablation combined with IL-1R deficiency by generating a double knockout (Nlrp3A350V CreL Il1r−/−Il18−/−) displayed significantly mitigated liver fibrosis (Suppl. fig. 2).
Based on the critical role of endogenous IL-18 for HSC activation and fibrosis, we hypothesized that IL-18 antagonism, e.g. by its natural inhibitor IL-18BP, could be a strategy to attenuate liver fibrosis. To investigate therapeutic effects of rhIL-18BP, we injected Nlrp3D301N CreL mice with IL-18BP or PBS (vehicle) subcutaneously for 14 days. The characteristic growth retardation and the urticarial-like rash of Nlrp3D301N CreL mice were ameliorated by rhIL-18BP treatment (Fig. 6A). In the Nlrp3D301N CreL mice treated with vehicle, liver histology demonstrated severe inflammation with numerous inflammatory foci and infiltrating cells. However, the mice treated with rhIL-18BP had a substantial attenuation of the liver inflammatory response (Fig. 6B). In particular, infiltrating neutrophils were prevalent in liver of non-treated mice visualized by MPO staining, but not in rhIL-18BP-treated ones (Suppl. fig. 3A, B). Consistently, liver mRNA levels of immune cell markers and chemoattractants (Tnfa, Cxcl1, Ly6C, Ly6G, Mpo) as well as matrix metalloproteinase 8 (Mmp8, neutrophil collagenase) were significantly down-regulated for Nlrp3D301N CreL mice injected with IL-18BP, which are abundantly expressed by macrophages, granulocytes and/or neutrophils (Suppl. fig. 3C). Furthermore, we observed a reduction of collagen deposition in treated mice compared to vehicle visualized by Sirius Red staining (Fig. 6C) as well as significantly decreased gene expression level of the extracellular matrix regulating enzyme Timp1 (Fig. 6D). In contrast, Mmp10 and Mmp13 were increased in treated mice compared to vehicle group (Fig. 6D). Interestingly, immunofluorescence staining for αSMA and PDGFR show that IL-18BP-treated mice show less activated, αSMA-expressing HSCs, suggesting that IL-18 is a mediator of HSC activation (Fig.6E, F).
Fig. 6: Treatment of Nlrp3D301N CreL mice with IL-18BP rescued highly inflammatory phenotype.
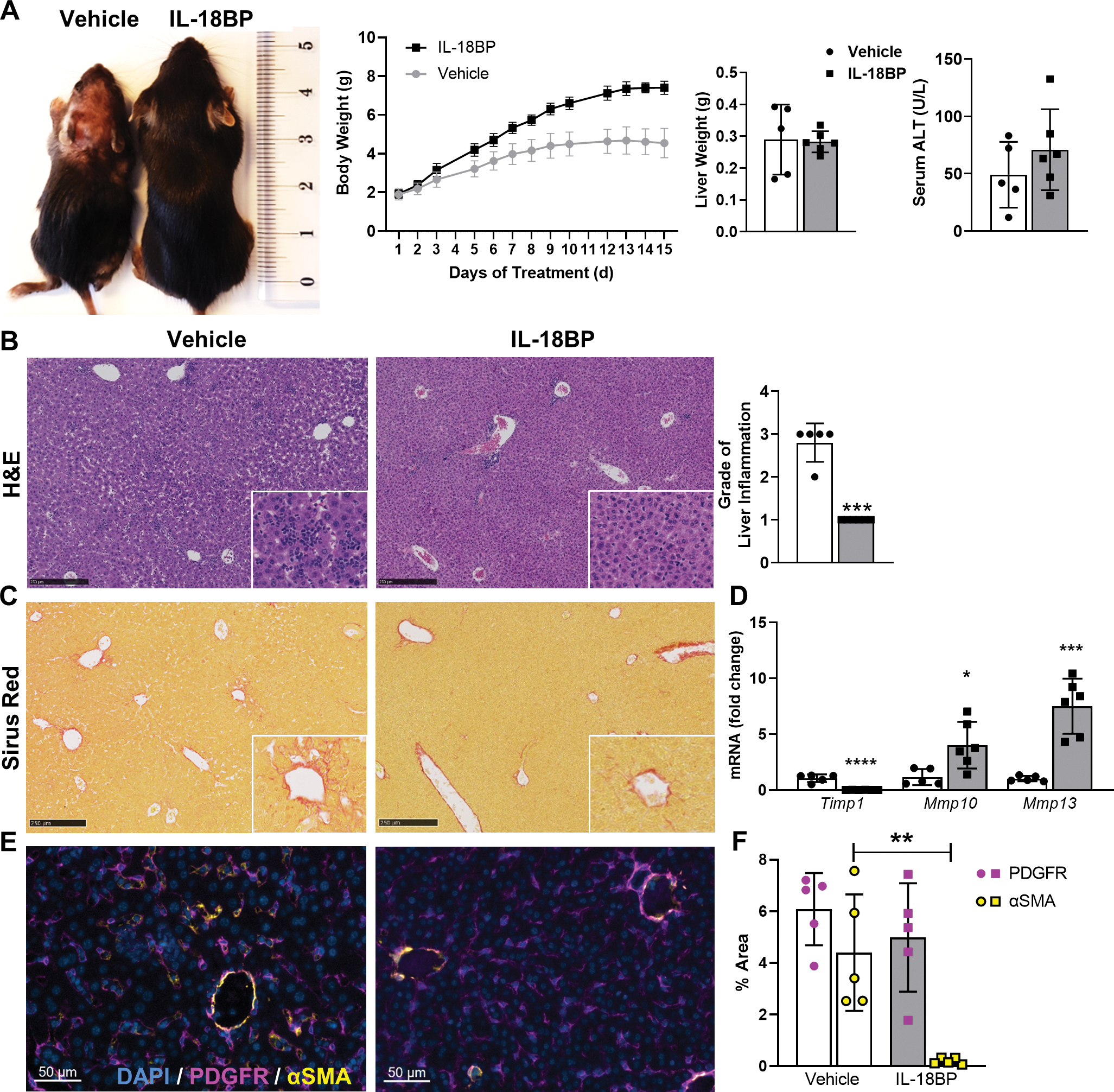
Nlrp3D301N CreL mice were treated with 6 μg/g rhIL-18BP (Tadekinig Alfa) or PBS from day 2 to 14. (A) Growth impairment, rash and reduced body weight, consistent with the phenotype of Nlrp3D301N CreL mice, were clearly normalized by IL-18BP treatment, whereas serum ALT did not significantly changed. (B) H&E- stained liver sections and quantification showing less inflammation in IL-18BP-treated mice. (C) Sirius Red-staining. (D) mRNA expression of extracellular matrix regulating enzymes (Timp1, Mmp10, Mmp13) were measured from total liver lysate. (E) Immunofluorescence staining and (F) quantification of PDGFR and αSMA showing more activated HSCs in the non-treated mice. (n = 5–6 mice per group for all measured values; scale bars represent 250 μm; *P < 0.05; **P < 0.01; ***P < 0.001).
Discussion
In the present study, we found a significant elevation of circulating IL-18 and IL-18BP in patients with liver cirrhosis in comparison to healthy controls. Subsequently, we provided strong evidence that IL-18 signaling and the corresponding downstream signaling cascade play a pivotal role in experimental NASH-induced liver fibrogenesis, which was confirmed by Il18−/− mice that showed protection from the fibrotic changes in a NASH model. Finally, we identified IL18BP as a novel therapeutic approach that has potential for the treatment of liver fibrosis.
IL-18 is a pleiotropic cytokine with proinflammatory properties, whose biological activity is controlled by IL-18BP. It has been reported that IL-18 is involved in the activation and differentiation of T helper cells in concert with IL-12 or IL-15 as well as in activation of natural killer cells (38). In line with our results, Ludwiczek and colleagues indicated a parallel elevation of IL-18 and IL-18BP in patients’ serum with different stages of liver cirrhosis induced by different etiologies. Interestingly, patients with end stage liver disease (Child-Pugh stage C) presented a decreased level of IL-18BP and therefore significantly elevated amounts of unbound IL-18, suggesting a causative role of IL-18 in the development and progression of liver diseases (12). Of importance, in cirrhotic patients who undergo liver transplantation the serum level of IL-18 normalized indicating a key role of the liver in IL-18 production (39). Furthermore, IL-18 has been associated previously with progression of cardiac (40), pulmonary (41) and renal fibrosis (42, 43). Activation of HSCs is well studied and essential event in fibrogenesis by their transdifferentiation into a proliferative and migratory myofibroblast-like cell type. Current data indicate that stimulation with IL-18 in vitro directly induced HSC activation by upregulation of Ctgf, Col1a1 and Acta2 as well as protein expression of αSMA in a concentration-dependent manner (Fig. 2). These observations were supported in vivo by using an HSC-specific NLRP3 overactivation fibrosis model, which is independent of inflammation and liver injury (37). We demonstrated that collagen deposition (Sirius Red) and key fibrosis-associated genes (αSma, Col1a1) are decreased in the absence of IL-18 signaling in Nlrp3L351P/+CreLrat mice (Fig. 3).
In line with our results that IL-18 induces transition of quiescent HSCs to αSMA-expressing myofibroblasts, inhibition of IL-18 signaling showed a protective effect on renal as well as pulmonary fibrosis due to inhibition of cell transdifferentiation into myofibroblasts (41, 43). This leads to the assumption that IL-18 might drive cell differentiation. It has been reported that hepatic IL-18 expression and its release mainly originated from Kupffer cells and macrophages (1, 44). Inflammasome activation has previously been implicated in the development of fibrosis (18, 20, 45). To this end, fibrogenesis in different experimental mouse models was suppressed by inflammasome-related (Nlrp3, caspase-1, Il1β) knockout mice in which the production and gene expression of collagen I and αSMA was diminished (20, 46, 47). Correspondingly, persistent global expression of NLRP3 resulted in severe liver inflammation and advanced stages of liver fibrosis by increased HSC activation and remarkable collagen deposition. Blocking IL-1β signaling by an IL-1 receptor antagonist (anakinra) decreased liver inflammation, but did not change HSC activation level or the degree of liver fibrosis (18). Further studies have observed bivalent results regarding the role of IL-1β in liver fibrosis depending on the mouse model. In ATP-binding cassette transporter b4 knockout animals, fibrosis was associated with increased gene expression of IL-1β. In vitro experiments with primary HSCs revealed decreased proliferation when treated with anakinra, but anakinra failed to improve liver injury or fibrosis in vivo (48). In another study, IL-1 receptor antagonist knockout mice exhibited more fibrosis in the bile duct ligation model, but less fibrosis in the CCl4 model. In contrast, when WT mice were treated with anakinra, fibrosis was improved after bile duct ligation, but aggravated after CCl4 injections (49).
However, the role of IL-18 was not investigated in these prior studies. Our findings indicate the pro-fibrotic capability of IL-18 in liver disease. Sequencing data demonstrated that the gene regulatory network both up- and downstream of IL-18 including compartments of inflammasomes and IL-18-related ligands are significantly increased in our murine fibrotic-NASH model. Further experiments elucidated that IL-18 deficiency was protective against the fibrotic changes when fed with CDAA-HFD. Especially the area of collagen- and αSMA-positive area as well as pro-fibrotic genes were significantly reduced in the Il18−/− mice compared to WT, which highlights again the impact of IL-18 signaling on HSC activation and fibrogenesis. Hohenester et al. showed that liver injury in non-alcoholic fatty liver disease, which increases the risk for fibrosis, was dependent on IL-18, but not IL-1β signaling (30). Il18r−/− mice were protected from hepatocellular injury assessed by reduced ALT levels in an American lifestyle-induced obesity syndrome-diet mouse model but it was not further supported by histological stainings or other methods (30). In contrast, in our study serum ALT levels showed no difference when comparing WT and Il18−/− on CDAA-HFD which might be caused by the strong inflammatory phenotype related to this diet.
Using mice that have persistent hyperactivation of NLRP3 in myeloid cells (Nlrp3A350V CreL), a double knockout of Il-1r and Il18 prevented liver fibrosis whereas the Il1r single knockout had no protection. Brydges et al. demonstrated that 9-day old Nlrp3A350V mice on an Il18r−/− background feature almost no neutrophilic inflammation compared to Nlrp3A350V mice on an Il1r−/− background, which might drive the development of fibrosis in these mice when aging. (50) These findings suggest that IL-18 signaling may have a more prominent role in fibrogenesis than IL-1β. In our experiments, administration of IL-18BP not only alleviated inflammation and liver fibrosis in the highly inflammatory Nlrp3D301N CreL mouse model, (Fig. 6B, C) but also significantly reduced neutrophil infiltration assessed by MPO staining (Suppl. fig. 3). Interestingly, treatment with IL-18BP also resulted in remarkably lower number of αSMA-positive HSCs in the liver (Fig. 6E), which points out the correlation of IL-18BP on HSC activation. Accordingly, in vitro experiments with IL-18BP-treated primary HSCs inhibited their activation suggesting that IL-18BP binds IL-18 released in response to NLRP3 inflammasome activation by LPS/ATP treatment in order to interrupt their self-amplifying activation (Fig. 2). Consistent with our results, other studies have shown that IL-18 neutralization resulted in reduced neutrophil accumulation in experimental hepatic ischemia/reperfusion injury and lethal endotoxemia (29, 51). IL-18BP prevents mature IL-18 from binding to the IL-18 receptor by virtue of its exceptionally high binding affinity, which is greater than IL-18Rα. (10, 11) Recently, rhIL-18BP (Tadekinig Alfa) was tested in clinical trial phase II for the autoinflammatory disorder Adult-onset Still’s disease (52). In summary, our results highlight the pivotal role of IL-18 signaling in liver fibrogenesis via activation of HSCs in vitro and in vivo in three different mouse models. Its blockage by use of IL-18BP as well as potential other approaches such a neutralizing antibodies may provide a novel therapeutic strategy for the treatment of liver fibrosis.
Supplementary Material
Acknowledgements:
The authors wish to thank AB2 Bio LTD (Lausanne, Switzerland) for providing recombinant human rhIL-18BP (Tadekinig alfa) and discussions related to its use and Dr. Cem Gabay for advice on the experimental conditions.
Financial Support:
This work was funded by NIH grants R01 DK113592 to HMH and AEF, U01 AA024206 to AEF, German Research Foundation (WR173/3–1 TR285/10–2 and SFB/TRR57) to AW and CT, German Cancer Aid (Deutsche Krebshilfe 70113000) to AW and CT, German Research Foundation (DFG-Grant KA 5089/1–1) to BK. In addition, the project described was partially supported by the NIH grant UL1TR001442 of CTSA. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Abbreviations
- αSma
alpha smooth muscle actin
- ALT
alanine aminotransferase
- ATP
adenosine triphosphate
- CCl4
carbon tetrachloride
- Col1a1
collagen type I alpha 1
- Ctgf
connective tissue growth factor
- DAMPs
damage-associated molecular pattern molecule
- DAPI
4′,6-diamidino-2-phenylindole
- ECM
extracellular matrix
- ELISA
enzyme-linked immunosorbent assay
- F4/80
murine macrophage marker
- FACS
fluorescence activated cell sorting
- Gapdh
glyceraldehyde 3-phosphate dehydrogenase
- H&E
hematoxylin-eosin
- HSCs
hepatic stellate cells
- IL
interleukin
- IL-18BP
interleukin 18 binding protein
- LPS
lipopolysaccharide
- Lrat
lecithin retinol acyltransferase
- MFI
mean fluorescence intensity
- Mmp
matrix metalloproteinase
- MPO
myeloperoxidase
- NLRs
nucleotide binding oligomerization domain leucine rich repeat containing receptors
- NLRP3
NLR family pyrin domain-containing 3
- PAMPs
pathogen-associated molecular patterns
- Timp1
tissue inhibitor of matrix metalloproteinase 1
- TNF
tumor necrosis factor
- Tgfβ
transforming growth factor beta
- WT
wild type
References
- 1.Okamura H, Tsutsi H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature. 1995;378(6552):88–91. [DOI] [PubMed] [Google Scholar]
- 2.Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature. 1997;386(6625):619–623. [DOI] [PubMed] [Google Scholar]
- 3.Guo H, Callaway JB, Ting JPY. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nature Medicine. 2015;21(7):677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Puren AJ, Fantuzzi G, Dinarello CA. Gene expression, synthesis, and secretion of interleukin 18 and interleukin 1beta are differentially regulated in human blood mononuclear cells and mouse spleen cells. Proc Natl Acad Sci U S A. 1999;96(5):2256–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mehta VB, Hart J, Wewers MD. ATP-stimulated release of interleukin (IL)-1beta and IL-18 requires priming by lipopolysaccharide and is independent of caspase-1 cleavage. J Biol Chem. 2001;276(6):3820–3826. [DOI] [PubMed] [Google Scholar]
- 6.Gulinelli S, Salaro E, Vuerich M, Bozzato D, Pizzirani C, Bolognesi G, et al. IL-18 associates to microvesicles shed from human macrophages by a LPS/TLR-4 independent mechanism in response to P2X receptor stimulation. European Journal of Immunology. 2012;42(12):3334–3345. [DOI] [PubMed] [Google Scholar]
- 7.Naik SM, Cannon G, Burbach GJ, Singh SR, Swerlick RA, Ansel JC, et al. Human Keratinocytes Constitutively Express Interleukin-18 and Secrete Biologically Active Interleukin-18 After Treatment with Pro-Inflammatory Mediators and Dinitrochlorobenzene. Journal of Investigative Dermatology. 1999;113(5):766–772. [DOI] [PubMed] [Google Scholar]
- 8.Pizarro TT, Michie MH, Bentz M, Woraratanadharm J, Smith MF, Foley E, et al. IL-18, a Novel Immunoregulatory Cytokine, Is Up-Regulated in Crohn’s Disease: Expression and Localization in Intestinal Mucosal Cells. The Journal of Immunology. 1999;162(11):6829–6835. [PubMed] [Google Scholar]
- 9.Matsui K, Yoshimoto T, Tsutsui H, Hyodo Y, Hayashi N, Hiroishi K, et al. Propionibacterium acnes treatment diminishes CD4+ NK1.1+ T cells but induces type I T cells in the liver by induction of IL-12 and IL-18 production from Kupffer cells. J Immunol. 1997;159(1):97–106. [PubMed] [Google Scholar]
- 10.Born TL, Thomassen E, Bird TA, Sims JE. Cloning of a novel receptor subunit, AcPL, required for interleukin-18 signaling. J Biol Chem. 1998;273(45):29445–29450. [DOI] [PubMed] [Google Scholar]
- 11.Tsutsumi N, Kimura T, Arita K, Ariyoshi M, Ohnishi H, Yamamoto T, et al. The structural basis for receptor recognition of human interleukin-18. Nature Communications. 2014;5(1):5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ludwiczek O, Kaser A, Novick D, Dinarello CA, Rubinstein M, Vogel W, et al. Plasma Levels of Interleukin-18 and Interleukin-18 Binding Protein Are Elevated in Patients with Chronic Liver Disease. Journal of Clinical Immunology. 2002;22(6):331–337. [DOI] [PubMed] [Google Scholar]
- 13.Novick D, Schwartsburd B, Pinkus R, Suissa D, Belzer I, Sthoeger Z, et al. A novel IL-18BP ELISA shows elevated serum IL-18BP in sepsis and extensive decrease of free IL-18. Cytokine. 2001;14(6):334–342. [DOI] [PubMed] [Google Scholar]
- 14.Yin C, Evason KJ, Asahina K, Stainier DY. Hepatic stellate cells in liver development, regeneration, and cancer. J Clin Invest. 2013;123(5):1902–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14(7):397–411. [DOI] [PubMed] [Google Scholar]
- 16.Kisseleva T, Cong M, Paik Y, Scholten D, Jiang C, Benner C, et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci U S A. 2012;109(24):9448–9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Troeger JS, Mederacke I, Gwak GY, Dapito DH, Mu X, Hsu CC, et al. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology. 2012;143(4):1073–1083.e1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology (Baltimore, Md). 2014;59(3):898–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wree A, McGeough MD, Inzaugarat ME, Eguchi A, Schuster S, Johnson CD, et al. NLRP3 inflammasome driven liver injury and fibrosis: Roles of IL-17 and TNF in mice. Hepatology. 2018;67(2):736–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wree A, McGeough MD, Peña CA, Schlattjan M, Li H, Inzaugarat ME, et al. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J Mol Med (Berl). 2014;92(10):1069–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inzaugarat ME, Johnson CD, Holtmann TM, McGeough MD, Trautwein C, Papouchado BG, et al. NLR Family Pyrin Domain-Containing 3 Inflammasome Activation in Hepatic Stellate Cells Induces Liver Fibrosis in Mice. Hepatology. 2019;69(2):845–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moriwaki Y, Yamamoto T, Shibutani Y, Aoki E, Tsutsumi Z, Takahashi S, et al. Elevated levels of interleukin-18 and tumor necrosis factor-alpha in serum of patients with type 2 diabetes mellitus: relationship with diabetic nephropathy. Metabolism. 2003;52(5):605–608. [DOI] [PubMed] [Google Scholar]
- 23.Yamamura M, Kawashima M, Taniai M, Yamauchi H, Tanimoto T, Kurimoto M, et al. Interferon-gamma-inducing activity of interleukin-18 in the joint with rheumatoid arthritis. Arthritis Rheum. 2001;44(2):275–285. [DOI] [PubMed] [Google Scholar]
- 24.Ludwiczek O, Kaser A, Novick D, Dinarello CA, Rubinstein M, Tilg H. Elevated systemic levels of free interleukin-18 (IL-18) in patients with Crohn’s disease. Eur Cytokine Netw. 2005;16(1):27–33. [PubMed] [Google Scholar]
- 25.Losy J, Niezgoda A. IL-18 in patients with multiple sclerosis. Acta Neurol Scand. 2001;104(3):171–173. [DOI] [PubMed] [Google Scholar]
- 26.Yamano T, Higashi T, Nouso K, Nakatsukasa H, Kariyama K, Yumoto E, et al. Serum interferon-gamma-inducing factor/IL-18 levels in primary biliary cirrhosis. Clin Exp Immunol. 2000;122(2):227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Urushihara N, Iwagaki H, Yagi T, Kohka H, Kobashi K, Morimoto Y, et al. Elevation of serum interleukin-18 levels and activation of kupffer cells in biliary atresia. Journal of Pediatric Surgery. 2000;35(3):446–449. [DOI] [PubMed] [Google Scholar]
- 28.Faggioni R, Cattley RC, Guo J, Flores S, Brown H, Qi M, et al. IL-18-Binding Protein Protects Against Lipopolysaccharide- Induced Lethality and Prevents the Development of Fas/Fas Ligand-Mediated Models of Liver Disease in Mice. The Journal of Immunology. 2001;167(10):5913–5920. [DOI] [PubMed] [Google Scholar]
- 29.Netea MG, Fantuzzi G, Kullberg BJ, Stuyt RJL, Pulido EJ, McIntyre RC, et al. Neutralization of IL-18 Reduces Neutrophil Tissue Accumulation and Protects Mice Against Lethal Escherichia coli and Salmonella typhimurium Endotoxemia. The Journal of Immunology. 2000;164(5):2644–2649. [DOI] [PubMed] [Google Scholar]
- 30.Hohenester S, Kanitz V, Schiergens T, Einer C, Nagel J, Wimmer R, et al. IL-18 but Not IL-1 Signaling Is Pivotal for the Initiation of Liver Injury in Murine Non-Alcoholic Fatty Liver Disease. Int J Mol Sci. 2020;21(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krenkel O, Hundertmark J, Ritz TP, Weiskirchen R, Tacke F. Single Cell RNA Sequencing Identifies Subsets of Hepatic Stellate Cells and Myofibroblasts in Liver Fibrosis. Cells. 2019;8(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nature Biotechnology. 2015;33(5):495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brydges SD, Mueller JL, McGeough MD, Pena CA, Misaghi A, Gandhi C, et al. Inflammasome-mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity. 2009;30(6):875–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim SH, Eisenstein M, Reznikov L, Fantuzzi G, Novick D, Rubinstein M, et al. Structural requirements of six naturally occurring isoforms of the IL-18 binding protein to inhibit IL-18. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(3):1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsumoto M, Hada N, Sakamaki Y, Uno A, Shiga T, Tanaka C, et al. An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. International journal of experimental pathology. 2013;94(2):93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei G, An P, Vaid KA, Nasser I, Huang P, Tan L, et al. Comparison of murine steatohepatitis models identifies a dietary intervention with robust fibrosis, ductular reaction, and rapid progression to cirrhosis and cancer. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2020;318(1):G174–G188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Inzaugarat ME, Johnson CD, Holtmann TM, McGeough MD, Trautwein C, Papouchado BG, et al. NLR Family Pyrin Domain-Containing 3 Inflammasome Activation in Hepatic Stellate Cells Induces Liver Fibrosis in Mice. Hepatology (Baltimore, Md). 2019;69(2):845–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaplanski G Interleukin-18: Biological properties and role in disease pathogenesis. Immunol Rev. 2018;281(1):138–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yasudo H, Ando T, Maehara A, Ando T, Izawa K, Tanabe A, et al. A possible association between a novel NLRP1 mutation and an autoinflammatory disease involving liver cirrhosis. Hepatology. 2021. [DOI] [PubMed] [Google Scholar]
- 40.Xiao H, Li H, Wang JJ, Zhang JS, Shen J, An XB, et al. IL-18 cleavage triggers cardiac inflammation and fibrosis upon β-adrenergic insult. Eur Heart J. 2018;39(1):60–69. [DOI] [PubMed] [Google Scholar]
- 41.Zhang LM, Zhang Y, Fei C, Zhang J, Wang L, Yi ZW, et al. Neutralization of IL-18 by IL-18 binding protein ameliorates bleomycin-induced pulmonary fibrosis via inhibition of epithelial-mesenchymal transition. Biochem Biophys Res Commun. 2019;508(2):660–666. [DOI] [PubMed] [Google Scholar]
- 42.Liang H, Xu F, Zhang T, Huang J, Guan Q, Wang H, et al. Inhibition of IL-18 reduces renal fibrosis after ischemia-reperfusion. Biomed Pharmacother. 2018;106:879–889. [DOI] [PubMed] [Google Scholar]
- 43.Bani-Hani AH, Leslie JA, Asanuma H, Dinarello CA, Campbell MT, Meldrum DR, et al. IL-18 neutralization ameliorates obstruction-induced epithelial–mesenchymal transition and renal fibrosis. Kidney International. 2009;76(5):500–511. [DOI] [PubMed] [Google Scholar]
- 44.Seki E, Tsutsui H, Nakano H, Tsuji NM, Hoshino K, Adachi O, et al. Lipopolysaccharide-Induced IL-18 Secretion from Murine Kupffer Cells Independently of Myeloid Differentiation Factor 88 That Is Critically Involved in Induction of Production of IL-12 and IL-1β. The Journal of Immunology. 2001;166(4):2651–2657. [DOI] [PubMed] [Google Scholar]
- 45.Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol. 2017;66(5):1037–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dixon LJ, Berk M, Thapaliya S, Papouchado BG, Feldstein AE. Caspase-1-mediated regulation of fibrogenesis in diet-induced steatohepatitis. Laboratory investigation; a journal of technical methods and pathology. 2012;92(5):713–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gieling RG, Wallace K, Han Y-P. Interleukin-1 participates in the progression from liver injury to fibrosis. American journal of physiology Gastrointestinal and liver physiology. 2009;296(6):G1324–G1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reiter FP, Wimmer R, Wottke L, Artmann R, Nagel JM, Carranza MO, et al. Role of interleukin-1 and its antagonism of hepatic stellate cell proliferation and liver fibrosis in the Abcb4(−/−) mouse model. World journal of hepatology. 2016;8(8):401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meier RPH, Meyer J, Montanari E, Lacotte S, Balaphas A, Muller YD, et al. Interleukin-1 Receptor Antagonist Modulates Liver Inflammation and Fibrosis in Mice in a Model-Dependent Manner. International journal of molecular sciences. 2019;20(6):1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brydges SD, Broderick L, McGeough MD, Pena CA, Mueller JL, Hoffman HM. Divergence of IL-1, IL-18, and cell death in NLRP3 inflammasomopathies. J Clin Invest. 2013;123(11):4695–4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takeuchi D, Yoshidome H, Kato A, Ito H, Kimura F, Shimizu H, et al. Interleukin 18 causes hepatic ischemia/reperfusion injury by suppressing anti-inflammatory cytokine expression in mice. Hepatology. 2004;39(3):699–710. [DOI] [PubMed] [Google Scholar]
- 52.Gabay C, Fautrel B, Rech J, Spertini F, Feist E, Kötter I, et al. Open-label, multicentre, dose-escalating phase II clinical trial on the safety and efficacy of tadekinig alfa (IL-18BP) in adult-onset Still’s disease. Ann Rheum Dis. 2018;77(6):840–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.