

Abstract
Background –
Arrhythmogenic Cardiomyopathy (ACM) is characterized by progressive loss of cardiomyocytes with fibrofatty tissue replacement, systolic dysfunction and life-threatening arrhythmias. A substantial proportion of ACM is caused by mutations in genes of the desmosomal cell-cell adhesion complex, but the underlying mechanisms are not well understood. So far, treatment options are only symptomatic. Here, we investigate the relevance of defective desmosomal adhesion for ACM development and progression.
Methods –
We mutated the binding site of desmoglein-2 (DSG2), a crucial desmosomal adhesion molecule in cardiomyocytes. This DSG2-W2A mutation abrogates the tryptophan swap, a central interaction mechanism of DSG2 based on structural data. Impaired adhesive function of DSG2-W2A was confirmed by cell-cell dissociation assays and force spectroscopy measurements by Atomic Force Microscopy. The DSG2-W2A knock-in mouse model was analyzed by echocardiography, ECG, histological and bio-molecular techniques including RNA sequencing, transmission electron and super-resolution microscopy. The results were compared to ACM patient samples and their relevance was confirmed in vivo and in cardiac slice cultures by inhibitor studies applying the small molecule EMD527040 or an inhibitory integrin-αVβ6 antibody.
Results –
The DSG2-W2A mutation impaired binding on molecular level and compromised intercellular adhesive function. Mice bearing this mutation develop a severe cardiac phenotype recalling the characteristics of ACM, including cardiac fibrosis, impaired systolic function and arrhythmia. A comparison of the transcriptome of mutant mice with ACM patient data suggested deregulated integrin-αVβ6 and subsequent TGF-β signaling as driver of cardiac fibrosis. Accordingly, blocking integrin-αVβ6 led to reduced expression of pro-fibrotic markers and reduced fibrosis formation in mutant animals in vivo.
Conclusions –
Here, we show that disruption of desmosomal adhesion is sufficient to induce a phenotype which fulfils the clinical criteria to establish the diagnosis of ACM, confirming the dysfunctional adhesion hypothesis. Mechanistically, deregulation of integrin-αVβ6 and TGF-β signaling was identified as a central step towards fibrosis. A pilot in vivo drug test revealed this pathway as promising target to ameliorate fibrosis. This highlights the value of this model to discern mechanisms of cardiac fibrosis and to identify and test novel treatment options for ACM.
Keywords: Arrhythmogenic Cardiomyopathy, Intercellular Adhesion, Desmosome, Intercalated Disc, TGF-β signaling, Cardiac Fibrosis, Integrin
3. Introduction
Patients suffering from Arrhythmogenic Cardiomyopathy (ACM) present with impaired cardiac function and ventricular arrhythmia up to sudden cardiac death. This inherited disease with a prevalence of 1:1’000 – 1:5’000 becomes typically evident in young adults. The therapeutic options for ACM are limited to symptomatic treatments like beta-adrenergic receptor blockers, restriction of physical exercise, implantation of a cardiac defibrillator, or transplantation as ultima ratio.1 Structurally, ACM is characterized by progressive loss of cardiomyocytes, cardiac fibrosis with fibrofatty tissue replacement and ventricular dilatation. In the majority of cases, mutations in components of the desmosomal complex are causative for ACM.1,2
In cardiomyocytes, this cell-cell adhesion complex is composed of the desmosomal adhesion molecules desmoglein-2 (DSG2) and desmocollin-2, which are linked to desmin intermediate filaments via plakoglobin (PG), plakophilin-2 (PKP2) and desmoplakin (DSP).3 These components are an integral part of the intercalated disc (ICD) where they form a functional unit with molecules of adherens junctions, gap junctions, and others such as sodium channels.4 The ICD is essential to provide mechanical and electrical coupling to adjacent cardiomyocytes, which is a basis for coordinated contraction and function of the heart. In ACM, a variety of mutations in genes encoding for desmosomal molecules were described.2 Together with the finding of disrupted ICD ultrastructure,5 this formed the hypothesis of impaired cardiomyocyte cohesion as central step in ACM pathogenesis. However, data on the role of cell-cell adhesion are conflicting and the impact of disrupted mechanical coupling for disease induction and progression is not finally resolved.6–8
In this study, we address the role of disrupted desmosomal adhesion in ACM development by interfering with the binding mechanism of DSG2. The crystal structure of DSG2 suggests the extracellular trans-interaction of desmosomal adhesion molecules to be mediated by a so-called tryptophan swap.9 Here, a tryptophan residue at position 2 is inserting into a hydrophobic pocket of the first extracellular domain of the respective molecule from the opposing cell and vice versa. Importantly, mutations of this tryptophan (ClinVar data bank: VCV000199796, VCV000044282 VCV000420241)10 as well as of residues being part of the corresponding hydrophobic binding pocket (VCV000518623, VCV000925370, VCV001345680, VCV000044316)9,10 were reported in ACM patients.
To interfere profoundly with this mechanism, we inserted a mutation substituting the tryptophan-2 with an alanine (DSG2-W2A) and characterized its functional implication in vitro, in cultivated cells, and in a knock-in mouse model. Further, we applied this in vivo model to identify mechanisms underlying ACM aiming for new therapeutic approaches.
4. Methods
For detailed methods, please refer to the Supplemental Material.
Human heart samples
Heart samples of ACM patients and healthy controls were derived from forensic autopsy. This study was conducted according to the tenets of the Declaration of Helsinki and approved by the local ethic committees, licence number 494–16 (ethics committee LMU Munich, Germany) and 152/15 (ethics committee Goethe University, Frankfurt am Main, Germany). Samples were fixed and embedded in paraffin according to standard procedures.
Animal experiments
Mouse experiments were approved by the Cantonal Veterinary Office of Basel-Stadt (License number 2973_32878 and 3070_32419). Mice were housed under specific pathogen-free conditions with standard chow and bedding with 12 hours day/night cycle according to institutional guidelines. Animals of both sexes were applied without bias. For inhibitor treatments, mice from the same litter and sex were paired and randomly allocated to the groups.
Statistics and data compilation
Figures were compiled with Adobe Photoshop CC 2017 and Adobe Illustrator CC 2017 (both Adobe, San José, CA). Statistical computations were performed with Prism 8 (GraphPad Software, La Jolla, CA). For comparison of two or multiple groups, distribution of data was analysed by Shapiro-Wilk normality test and group variances were analysed by f-test or Brown-Forsythe test, respectively. According to the results of these tests, a parametric or non-parametric test with or without Welch’s correction for unequal variances was applied. For Kaplan-Meier survival analysis, significance was assessed by the Gehan-Breslow-Wilcoxon test. The statistical test used to compare the respective data sets is described in the corresponding figure legend. Statistical significance was assumed at P < 0.05. Unless otherwise stated, data are presented as dot blot, with each dot representing the mean of the respective technical replicates of one biological replicate. Each animal or independent seeding of cells were taken as biological replicate, or as indicated in the legend. The mean value of these dots is shown as bar diagram ± standard deviation.
Data availability
RNA-Seq data were deposited in the public database GEO accession number GSE181868. Additional data pertaining to the current article are available from the corresponding author upon request.
5. Results
W2A mutation abrogates DSG2 interactions and impairs cell-cell adhesion in vitro and in vivo.
To study the functional consequences of impaired DSG2 binding, we generated the W2A point mutation (DSG2-W2A) to abrogate the proposed interaction mechanism of the only desmoglein expressed in cardiomyocytes (Figure 1A). First, we investigated in a cell-free setup whether DSG2-W2A is indeed affecting the binding properties of DSG2 interaction by applying single molecule force spectroscopy. Here, molecules are coupled to the tip of an Atomic Force Microscopy (AFM) probe and the surface of a mica sheet via a PEG linker. By measuring the deflection of the probe during repeated approach to and retraction from the surface, binding events can be quantitatively assessed. From these data, properties such as binding frequency, binding force, and bond half-life time can be determined. Constructs were generated for expression of wildtype (WT) and mutant (W2A) DSG2 extracellular domains fused to a human Fc-fragment and tested for homo- (WT:WT, W2A:W2A) and heterotypic (WT:W2A) interaction properties (Figure 1B). These experiments showed a significant reduction of the frequency of W2A:W2A as well as WT:W2A interactions compared to WT:WT (Figure 1C). The frequency of the remaining W2A bindings was comparable to probing Fc alone, which indicates that these are mostly non-specific interactions. Homotypic WT interactions display a clear binding force peak, while the remaining W2A interaction forces were widely spread, again pointing to the non-specificity of these interactions (Figure 1D). The lifetime of DSG2 interactions under zero force (τ0) was determined by fitting the binding forces detected at different loading rates11, yielding τ0=1.088s with R=0.993. In contrast, no sufficient fit (R=0.509) was possible for W2A interactions (Figure 1E). Together, these data outline that the tryptophan swap is the major interaction mechanism of DSG2.
Figure 1. Desmoglein-2 adhesion is mediated via tryptophan swap at position 2.
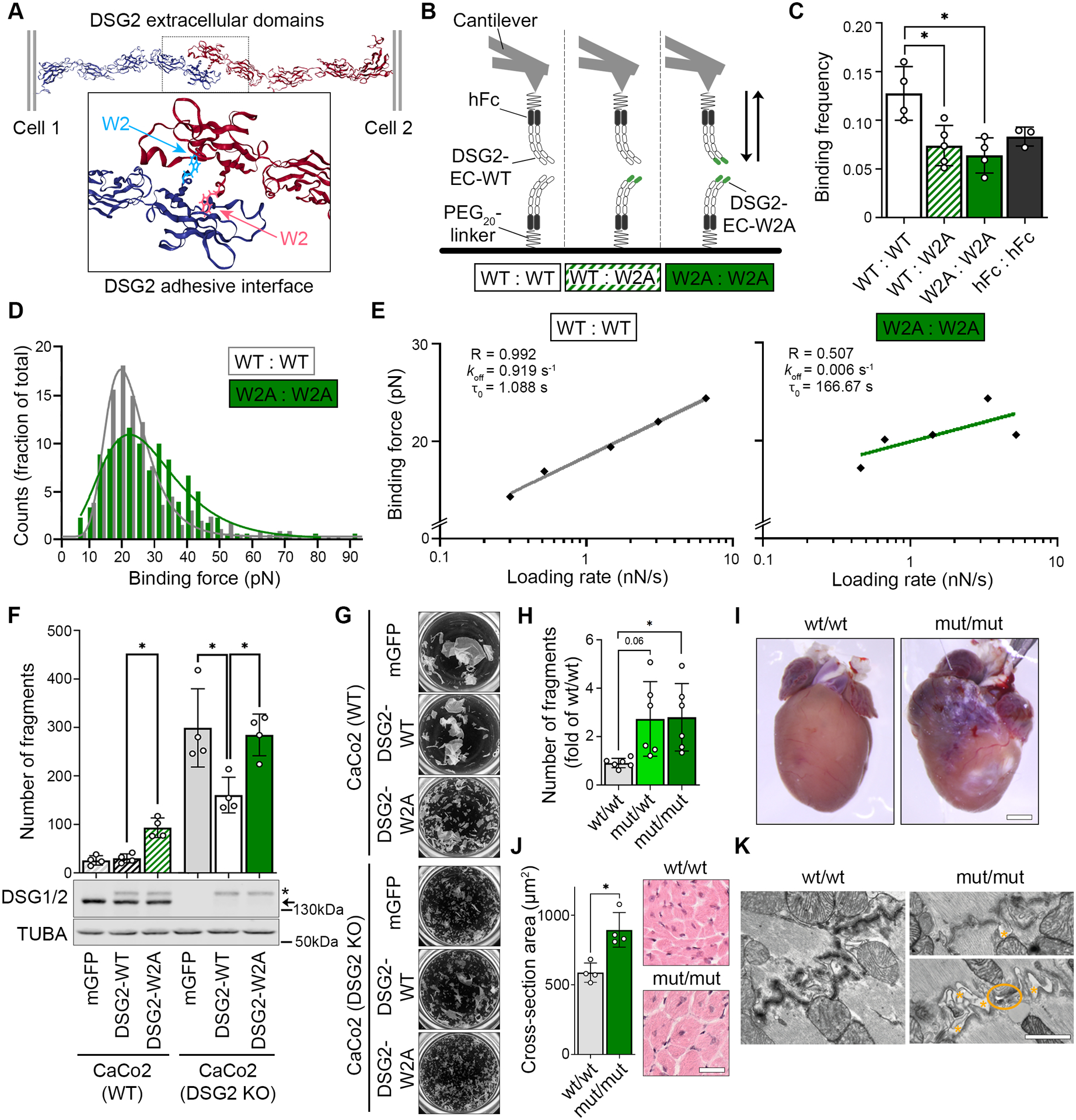
(A) Predicted interaction model of desmoglein-2 (DSG2) extracellular domains via exchange of tryptophan residue at position 2 into a hydrophobic pocket of the opposing molecule. Cartoon 3D presentation of PDB entry 5ERD9, tryptophan-2 is highlighted by ball and stick presentation. (B) Schematic of single molecule force spectroscopy experiments. Recombinant extracellular domains (EC) of DSG2-WT or DSG2-W2A protein were coupled to a mica surface and AFM cantilever via a human Fc-tag (hFc) and a PEG20-linker and probed as indicated. (C) Binding frequency of DSG2-W2A/DSG2-WT heterotypic and homotypic interactions at a pulling speed of 2 μm/s are shown. hFc served as control for unspecific binding. *P< 0.05, one-way ANOVA, Dunnett’s post hoc test. Each independent coating procedure with minimum 625 force curves is taken as biological replicate. (D) Histogram of binding forces distribution with peak fit at a pulling speed of 2 μm/s corresponding to data in C. (E) Determination of the bond half lifetime via Bell’s equation11 of mean loading rates and binding forces analysed from data of pulling speeds at 0.5, 1, 2, 5, and 7.5 μm/s. Average of values from four independent coating procedures with minimum 625 force curves each. R = R squared, koff = off rate constant, τ0 = bond half lifetime under zero force. (F) Dissociation assays to determine cell-cell adhesion were performed in CaCo2 cells (WT or DSG2 KO background) expressing DSG2-WT-mGFP or DSG2-W2A-mGFP constructs. mGFP empty vector served as control. *P< 0.05, one-way ANOVA, Sidak’s post hoc test. Corresponding Western blot analysis confirmed effective expression of DSG2 constructs (*) vs. the endogenous protein (arrow) in CaCo2 cells. α-tubulin (TUBA) served as loading control. (G) displays representative images of monolayer fragmentation from experiments in F. (H) Dissociation assays in immortalized keratinocytes isolated from neonatal murine skin of the respective genotype. *P< 0.05 or as indicated, Welch’s ANOVA, Dunnett’s post hoc test. (I) Macroscopic cardiac phenotype of DSG2-W2A mut/mut mice at the age of 15 weeks. (J) Cardiac hypertrophy was analysed as mean cross-sectional area of cardiomyocytes in haematoxylin/eosin stained sections. Scale bar: 30 μm. *P< 0.05, unpaired Student’s t-test. (K) Representative images of ICDs acquired by TEM, 3 mice per genotype. Orange asterisks mark intercellular widening, orange circle marks a ruptured junction. Scale bar: 1 μm.
To determine the effect of the W2A mutation on cellular level, DSG2-WT and DSG2-W2A constructs were stably expressed in the epithelial cell line CaCo2 either in a WT or DSG2-deficient background [CaCo2(WT) or CaCo2(DSG2 KO)]12. Cell-cell dissociation assays were performed, in which a confluent cell monolayer is detached and exposed to defined mechanical stress. Expression of DSG2-W2A in CaCo2(WT) cells significantly reduced intercellular adhesion as indicated by an increased number of fragments (Figure 1F–G). Importantly, while expressing DSG2-WT in the CaCo2(DSG2 KO) line significantly rescued the impaired intercellular adhesion in response to DSG2 loss, the expression of DSG2-W2A had no effect on fragment numbers.
As these data demonstrate the DSG2 tryptophan swap to be the central adhesive mechanism, we next generated a CRISPR/Cas9 based knock-in mouse model for DSG2-W2A to investigate the consequences of impaired DSG2 adhesion in vivo (Figure S1). In line with the studies in human cells, dissociation assays in keratinocyte cell lines generated from DSG2-W2A pups revealed reduced cell-cell adhesion in heterozygous (mut/wt) and homozygous (mut/mut) mutants (Figure 1H). Moreover, DSG2-W2A mice presented with a pronounced cardiac phenotype. Adult mut/mut mice demonstrated ventricular deformation, fibrotic and calcified areas and hypertrophic cardiomyocytes (Figure 1I, J). In line with reduced intercellular adhesion of cardiomyocytes, Transmission Electron Microscopy (TEM) revealed disturbed ICDs with widened intercellular space and occasionally completely ruptured junctions in mut/mut hearts (Figure 1K).
A cardiac phenotype was also detectable during embryogenesis. Here, mating of heterozygous mice revealed a reduction of mut/mut offspring to 2.8% compared to the expected Mendelian ratio of 25%. Embryo dissections showed loss of the mut/mut animals between E12 and E14 (Figure S2A). Macroscopically, mut/mut embryos at day E12.5 appeared pale with accumulation of blood in the cardiac area, while the heart was still beating (Figure S2B). Cells suggestive for blood precursors were detectable in the pericardial space (Figure S2C). This finding suggests a rupture of the cardiac wall leading to pericardial bleeding and loss of mut/mut animals.
Together, these in vitro and in vivo data outline an essential role of the DSG2 tryptophan swap for cell-cell adhesion and demonstrate severe pathologies associated with impaired DSG2 interaction.
DSG2-W2A mutant mice resemble the phenotype of ACM
Given (I) the cardiac affection in mutant mice, (II) the notion that a majority of ACM patients are reported with mutations in desmosomal genes2 and (III) that mutations specifically of W2 have already been associated with ACM10, we examined homozygous and heterozygous mice including both sexes with regard to histological and clinical ACM parameters in an age range of 12 to 80 weeks.
Histological analysis showed cardiac fibrosis, a major hallmark of ACM, in the myocardium of the right (RV) and left ventricle (LV) in mut/mut animals (Figure 2A–D), which did not profoundly increase over time. In contrast, heterozygous animals demonstrated a milder phenotype, with a fraction of animals (36%) starting to develop fibrosis in the RV around 6 months, while LV was unaffected. To better address differences over time, we pooled animals into groups with a mean age of 4 and 6 months for homozygous and 6 and 12 months for heterozygous mice and performed functional analyses by echocardiography and electrocardiogram (ECG) parallel to histological evaluation. Moreover, we separated 12-months old heterozygous animals according to RV fibrosis into a group without and “with fibrosis” (> 10% fibrotic area). Mut/mut animals showed impaired RV systolic function at both timepoints with reduced tricuspid annular plane systolic excursion (TAPSE), and fractional shortening (Figure 2E–G, Table S1). Interestingly, LV systolic impairment with reduction of the ejection fraction and corresponding parameters worsened over time and became significant after 6 months, although no increase in fibrosis was detectable (Figure 2D, H, Table S1). In mut/wt mice, RV fractional shortening was significantly reduced in 12-months animals with fibrosis. In ECG recordings of mut/mut mice for 30 min under anaesthesia, we noted deformation of the QRS complex with elongated QRS interval and reduced amplitude of the S peak with effects more pronounced in the older group, while heterozygous animals with RV fibrosis showed similar effects or trends (Figure 2I–K, Table S1). As the T wave was not clearly detectable in mutants, the J peak amplitude as indicator for early repolarisation was determined. This parameter was reduced in the mut/mut groups with same trend for fibrotic mut/wt hearts (Figure 2L). Parallel to these de- and repolarization abnormalities, we also noted the occurrence of ventricular arrhythmia, ranging from single premature ventricular contractions (PVCs) to multiple, multifocal PVCs and even non-sustained ventricular tachycardia in one mouse (Figure 2M–O). The fraction of homo- and heterozygous animals with PVCs and the frequency of these events correlated with age and fibrosis. Moreover, the notion that a significant fraction (16%) of mut/mut animals was found dead during the observation period strongly suggests the occurrence of malignant arrhythmia with sudden death (Figure 2P). Investigating sex related differences, no effect was observed on the extent of fibrosis or echocardiography parameters. However, in mut/wt mice PVCs were found in males only and male mut/mut mice were more prone to premature sudden death compared to females (Figure S3).
Figure 2. DSG2-W2A mutant mice develop characteristics of ACM.
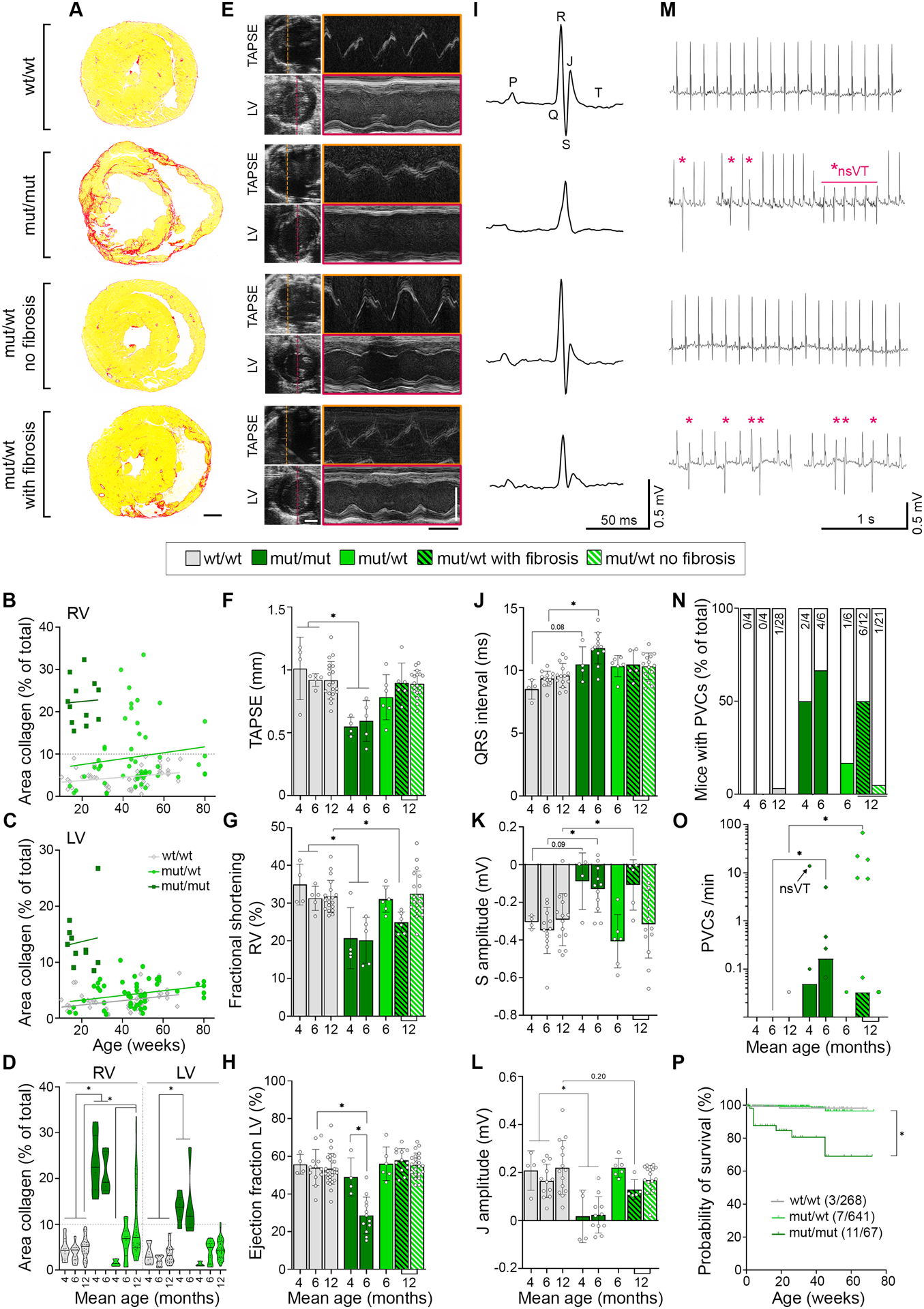
(A) Cardiac fibrosis detected by picrosirius red collagen staining with representative images of sections from 6-months old DSG2-W2A mut/mut and 12-months old wt/wt and mut/wt animals. Scale bar = 1 mm. (B - D) Corresponding analysis of the area of collagen in the right (RV) and left ventricle (LV). Continuous lines indicate the simple linear regression between age and area of collagen. Hearts with more than 10% of collagen in the RV (grey dotted line) were defined as “with fibrosis”. Each dot represents one animal. *P< 0.05, mixed effects analysis with LV and RV matched per animal, Sidak’s post hoc test. Lines indicate median and quartile values. (E) Representative echocardiography images for measurements of the tricuspid annular plane systolic excursion (TAPSE) and LV function in 6-months old mut/mut and 12-months old wt/wt and mut/wt animals. Left side: 2D images from apical four chamber view for TAPSE and parasternal short axis view for LV. M-mode tracings on the right were performed along the line indicated on the left. Scale bars: white 2 mm; black 100 ms. Corresponding analysis of (F) TAPSE, *P< 0.05, Kruskal-Wallis test with Dunn’s post hoc test, (G) fractional shortening of the RV, *P< 0.05, Kruskal-Wallis test with Dunn’s post hoc test, and (H) ejection fraction of the LV, *P< 0.05, one-way ANOVA, Sidak’s post hoc test. (I) Electrocardiogram (ECG) recoded in lead II with representative curves from 6-months old mut/mut and 12-months old wt/wt and mut/wt animals. Definition of respective peaks is indicated in the wt/wt curve. Scale bars: vertical 0.5 mV; horizontal 50 ms. Corresponding analysis of (J) QRS interval, *P< 0.05 or as indicated, Kruskal-Wallis test with Dunn’s post hoc test, (K) amplitude of the S peak, *P< 0.05 or as indicated, one-way ANOVA, Sidak’s post hoc test, and (L) amplitude of the J peak (early repolarization), *P< 0.05 or as indicated, Kruskal-Wallis test with Dunn’s post hoc test. (M) Ventricular arrythmia detected by ECG during 30 min of baseline measurements with example curves from 6-months old mut/mut and 12-months old wt/wt and mut/wt animals. Asterisks mark premature ventricular contractions (PVC), *nsVT indicates a non-sustained ventricular tachycardia detected in one mut/mut animal. Scale bars: vertical 0.5 mV; horizontal 1 s. Corresponding analysis of (N) percentage of mice presenting with PVCs. Values in bars indicate corresponding absolute number of mice with PVC (colored bars) compared to total number of mice evaluated (empty bar), and (O) PVC burden depicted as number of PVCs per minute, *P< 0.05, Kruskal-Wallis test with Dunn’s post hoc test. The black arrow indicates the animal presenting with non-sustained ventricular tachycardia. (P) Kaplan-Meier survival plot of DSG2-W2A mice from an analysis period of 3 years. Vertical lines indicate drop-outs due to unrelated elimination (end of experiment, breeding, injuries). Values indicate corresponding absolute number of mice with sudden death compared to total number of mice evaluated. *P< 0.05, Gehan-Breslow-Wilcoxon test. Box with color indications of respective groups in the middle apply to the entire figure.
In summary, mutant animals show histopathological features with echocardiography and ECG abnormalities similar to ACM patients. Within the limits of a murine model, both genotypes fulfil the Padua criteria13 to establish the diagnosis of ACM (Table S2). Here, DSG2-W2A mut/mut animals resemble the phenotype of biventricular ACM, while in mut/wt mice the phenotype is milder with variable penetrance and age-dependent occurrence only in the RV. According to the Padua criteria,13 the heterozygous genotype recalls the characteristics of a right-dominant ventricular ACM, as long as fibrosis was present. Together, these data demonstrate that loss of desmosomal adhesion is sufficient to induce an ACM phenotype and that the DSG2-W2A model may resemble two different variants of the disease.
Integrin-β6 expression is altered in ACM patient and DSG2-W2A mouse samples
Next, we applied this mouse model to investigate mechanisms leading to ACM. RNA sequencing was performed from ventricles of mut/mut and wt/wt hearts both before onset of fibrosis (age of 5 days), and when the biventricular fibrosis and ACM phenotype was established (after 9 weeks). First, we compared sequencing data from 9-weeks-old mice to the top differentially expressed genes derived from published transcriptomic data sets of ACM patient hearts (GEO data base, GSE107157/GSE10748014 and GSE2981915). Murine samples clustered according to their genotype and mut/mut hearts resembled the gene expression pattern of patients (Figure S4), further supporting the observation that mutant mice faithfully reproduce major aspects of the disease.
To identify common genes deregulated during ACM pathogenesis, we compared the differentially expressed genes from patient data with the results from mutant mice. Here, we included data from 5-days-old mice to identify changes already present before onset of secondary effects due to fibrosis. Interestingly, integrin-β6 (Itgb6) was the only transcript consistently deregulated in all data sets (Figure 3A). Its common down-regulation in ACM patients and mutant mice on RNA levels was further confirmed for heterozygous DSG2-W2A mutants compared to wildtype (Figure 3B). However, protein levels were unaltered in mutant heart samples (Figure 3C). By immunostaining, we noted that in mutant hearts ITGB6, which mainly localized to a compartment suggestive for the transverse tubules and costameres in wt/wt animals, was enriched at the ICD (Figure 3D, E). Together, these data demonstrate altered ITGB6 mRNA expression as common feature in ACM patients and DSG2-W2A hearts and an enrichment of the protein at the ICD in mutant murine samples.
Figure 3. Integrin-β6 is deregulated in DSG2-W2A mutants.
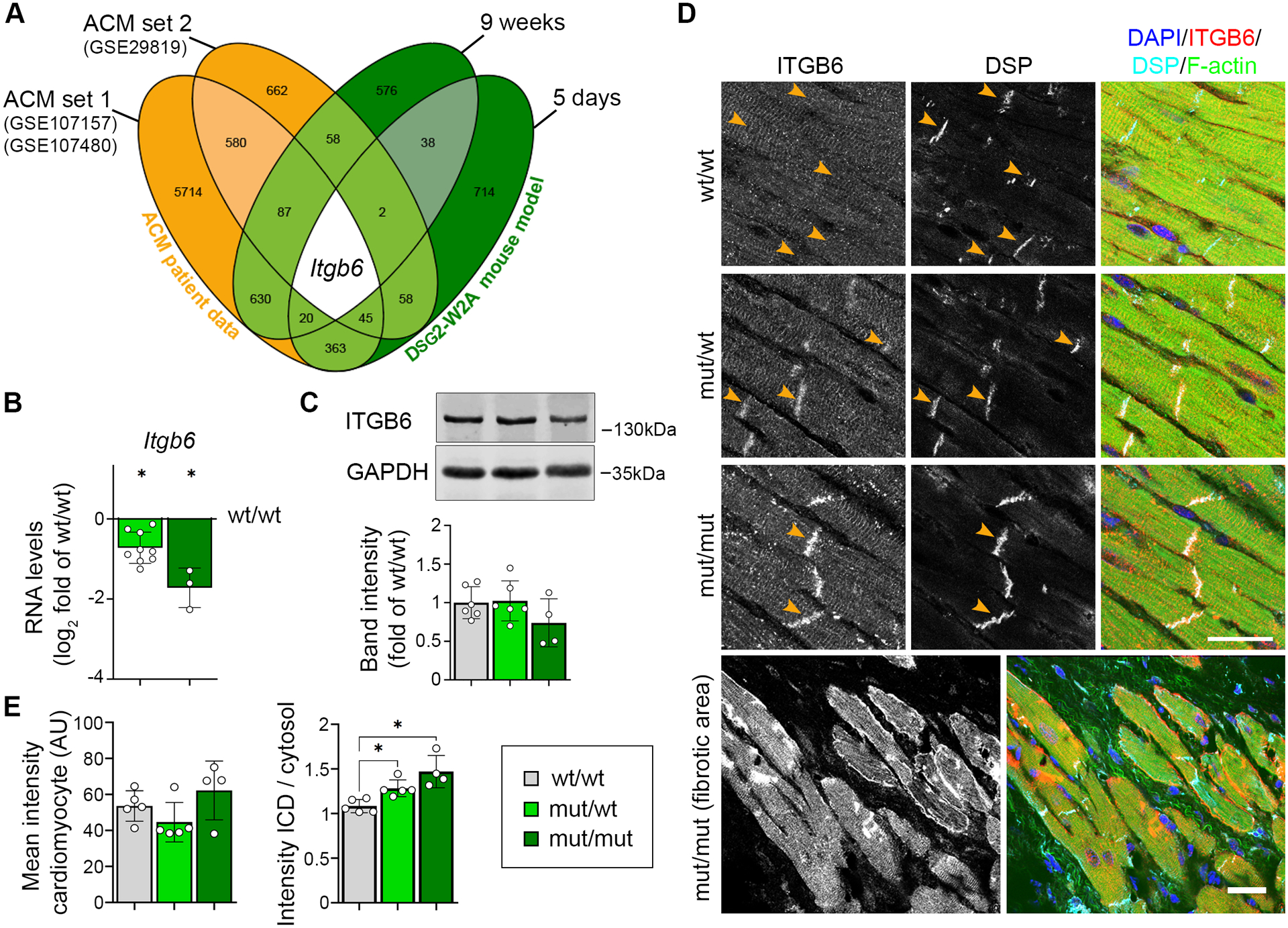
(A) Venn diagram of significantly altered genes from indicated ACM patient data sets (ACM vs. healthy control) and DSG2-W2A mice at the age of 5 days and 9 weeks (mut/mut vs. wt/wt) highlighting integrin-β6 (Itgb6) as only overlapping gene with same direction of expression in all data sets. Numbers indicate the amount of overlapping genes for the respective overlays. (B) RNA expression of Itgb6 analysed via qRT-PCR in adult DSG2-W2A mouse hearts. *P< 0.05, unpaired Student’s t-test vs. wt/wt. Gapdh served as reference gene. (C) Representative Western blot and respective analysis of band intensity of ITGB6 in DSG2-W2A hearts. GAPDH served as loading control. One-way ANOVA, Dunnett’s post hoc test. (D, E) Immunostaining of ITGB6 (red in overlay) in DSG2-W2A hearts with corresponding analysis of staining intensity in cardiomyocytes in total and ratio of staining intensity at the ICD area (orange arrow heads) vs. cardiomyocytes’ cytosolic area. Desmoplakin (DSP, cyan) marks ICDs, DAPI (blue) nuclei and F-actin (green) the sarcomere system. Lower row shows an overview image of a fibrotic area in mut/mut hearts. Scale bars: 25 μm. *P< 0.05, left graph: Kruskal-Wallis test with Dunn’s post hoc test; right graph: one-way ANOVA, Dunnett’s post hoc test. Box with color indications of respective groups apply to the entire figure.
DSG2-W2A mutant hearts present with structurally impaired ICDs
This result together with the TEM data (Figure 1K) suggest structural changes of the ICD in mutant hearts, which we addressed further. Using DSP as marker, we reconstructed the outlines of ICDs in wt/wt and mut/mut hearts from 3D stacks captured with Structured Illumination Microscopy (SIM). This analysis yielded an increase in ICD volume, in particular due to an enhanced width between two adjacent cells (Figure 4A). Interestingly, the majority of desmosomal molecules as well as N-cadherin were regularly distributed at mutant ICDs (Figure S5A, B). This was further corroborated by the RNA-sequencing data from both, 5-days- and 9-weeks-old animals, which showed no consistent changes of desmosomal, adherens, gap, or tight junction molecules (Figure S5C–E). The only adhesion molecule reduced globally was DSG2 (Figure S5A, B). Even though, DSG2 was still present at the ICD, detailed analysis by SIM revealed reduced number and volume of DSG2 signals (Figure 4B). Moreover, Fluorescence Recovery After Photobleaching (FRAP) experiments in neonatal murine cardiomyocytes transduced with DSG2-WT-mGFP or DSG2-W2A-mGFP showed a higher mobility of the mutant protein at cardiomyocyte junctions (Figure 4C), indicating reduced membrane stability of DSG2-W2A. To further investigate the possibility that the stability and integrity of ICD molecules is compromised, we performed Triton-X-100 separation assays for proteins with unaltered ICD localization in mutant hearts. Cytosolic and unanchored membrane proteins are found in the Triton-X-100 soluble fraction, while cytoskeleton-bound molecules are mainly detectable in the non-soluble fraction. Both, desmosomal molecules (PG, PKP2) as well as N-cadherin showed increased levels in the soluble pool, indicating reduced cytoskeletal anchorage and impaired ICD integrity in mutants (Figure 4D). Moreover, the gap junction molecule connexin-43, which is required for electrical coupling of cardiomyocytes was delocalized to the lateral membrane (Figure 4E), which was described as feature of disrupted ICDs.8
Figure 4. Disrupted ICD structure in DSG2-W2A mutant mice.
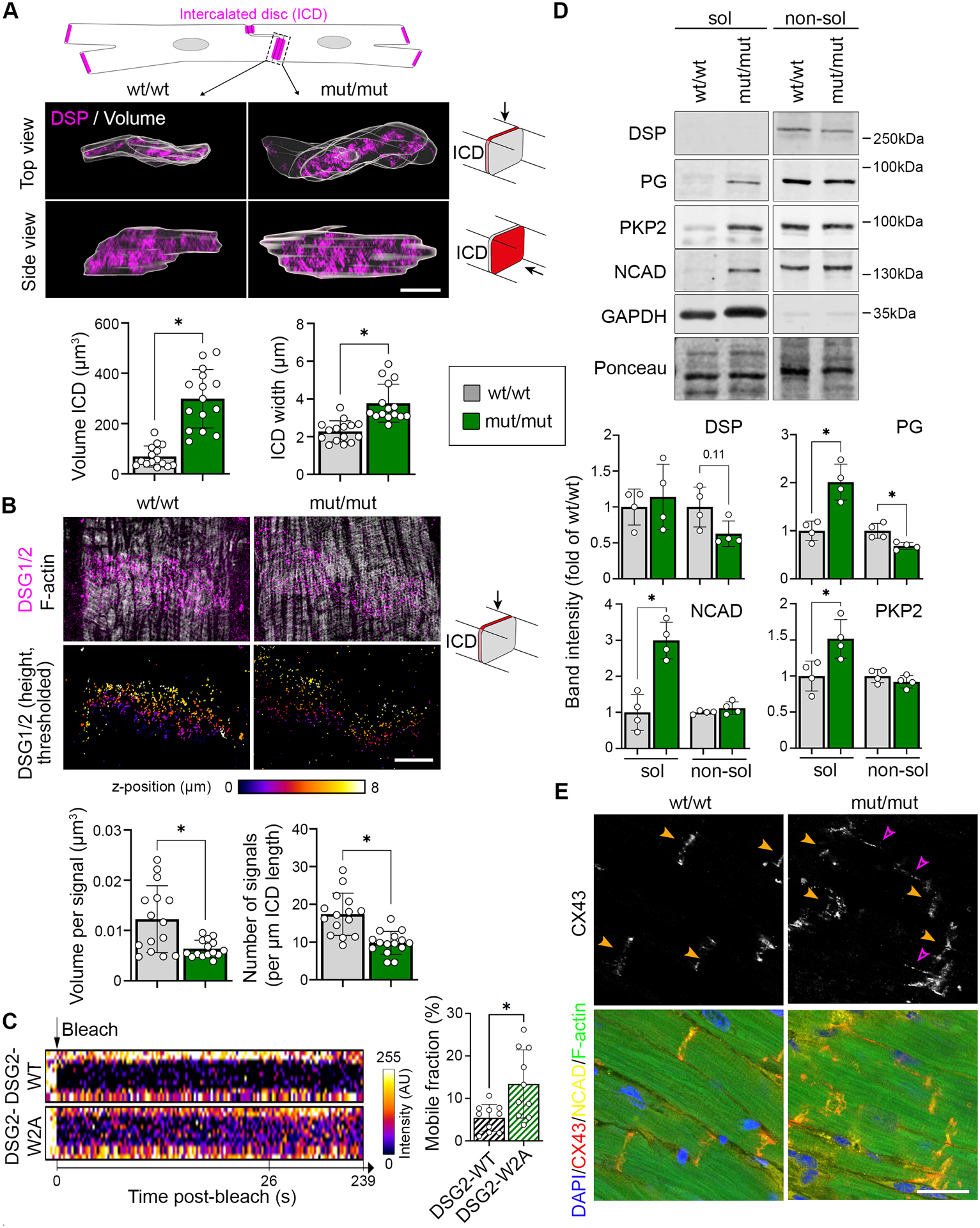
(A) Representative z-stack reconstruction of segmented ICDs in top and side view acquired with SIM. Overlay of analysed ICD volume is shown in grey. ICDs are marked by DSP (magenta). Scheme on top presents segmented area and pictograms on the right display the respective angle of view. Corresponding analysis of ICD volume and width of ICD between adjacent cardiomyocytes below. Scale bar: 5 μm. *P< 0.05, Mann-Whitney test. Each dot represents the value of one ICD from in total 4 mice per genotype. (B) Representative images of DSG2 (magenta) and filamentary actin (f-actin, white) z-stacks acquired by SIM and presented as maximum intensity projection. Lower row shows color-coded height projection of DSG2 signals in z-stack after signal thresholding as performed for analysis. Related analysis of DSG2 signal volume and number per ICD length is shown below. Pictograms on the right display the respective angle of view. Scale bar: 5 μm. *P< 0.05, unpaired Student’s t-test, with Welch’s correction. Each dot represents the value of one ICD from in total 3 mice per genotype. (C) FRAP analysis of DSG2-WT and DSG2-W2A-mGFP fusion proteins at the cell-cell junction of neonatal cardiomyocytes with representative intensity kymographs of bleached areas on the left. Time point 0 = bleach as indicated by the black arrow. Analysis of the mobile fraction of the indicated mGFP-fusion proteins is shown on the right. *P< 0.05, unpaired Student’s t-test, with Welch’s correction. Each dot represents the mean value of one heart from in total 3 isolations. (D) Representative triton-X-100 assay immunoblot with separation of a soluble (sol), non-cytoskeletal bound protein fraction from a non-soluble (non-sol), cytoskeletal-anchored fraction and corresponding analysis shown below. Plakoglobin (PG), plakophilin-2 (PKP2) and N-cadherin (NCAD) were analysed. Intensity of proteins was normalized to the total amount of protein detected by ponceau staining. GAPDH and desmoplakin (DSP) served as separation control. *P< 0.05 or as indicated, unpaired Student’s t-test (PG, NCAD, PKP2) or Mann-Whitney test (DSP). Each dot represents the result from one mouse. (E) Immunostaining of connexin-43 (CX43) (red in overlay) in DSG2-W2A hearts. N-cadherin (NCAD, yellow) marks ICDs, DAPI (blue) nuclei and F-actin (green) the sarcomere system. Orange arrows mark ICD, pink arrows highlight lateralization of CX43 staining. Scale bars: 25 μm. Images representative for 5 mice per genotype. Box with color indications of respective groups apply to the entire figure.
These data together with the TEM results demonstrate structurally severely altered ICDs in response to the DSG2-W2A mutation and suggest a molecular basis leading to the functional alterations detectable by echocardiography and ECG.
Integrin-αV/β6 is activated in DSG2-W2A mutant hearts
In contrast to DSG2, we detected ITGB6 to be increased at the ICD (Figure 3D, E), which was further confirmed by analysis of SIM images (Figure 5A). ITGB6 needs to heterodimerize with integrin-αV (ITGAV) in order to be activated and bind to the extracellular matrix.16–18 Thus, we analyzed the expression of the counterpart ITGAV, which was significantly upregulated on protein level and increased at the ICD and costameres of mutant hearts (Figure 5B, C). In contrast, the localization and intensity of integrin-β1, as classical representative of the integrin group, was not altered (Figure S6). Importantly, staining with an antibody specifically recognizing the heterodimer of ITGAV/B6 revealed increased amount of dimer formation at the ICD and costameres in mutant mice (Figure 5D). Moreover, increased ITGAV/B6 staining intensity was also detectable in an ACM patient sample with DSP mutation (Figure 5E). In order to stabilize active integrins, recruitment of the cytoskeletal adapters talin and vinculin is required.19 Accordingly, the expression of talin-2 was increased in DSG2-W2A mut/mut myocardium, with vinculin being enriched at the ICD (Figure 5F, G). This effect is similar to what was shown before in response to increased intracellular traction forces on junctions.20 Thus, these data suggest that in response to a dysfunctional, non-adhesive ICD with impaired cytoskeletal anchorage, ITGAV/B6 dimers are recruited and activated at the compromised ICD as well as at the costamere region.
Figure 5. Increased activation of ITGB6/AV at ICDs of DSG2-W2A mutant mice.
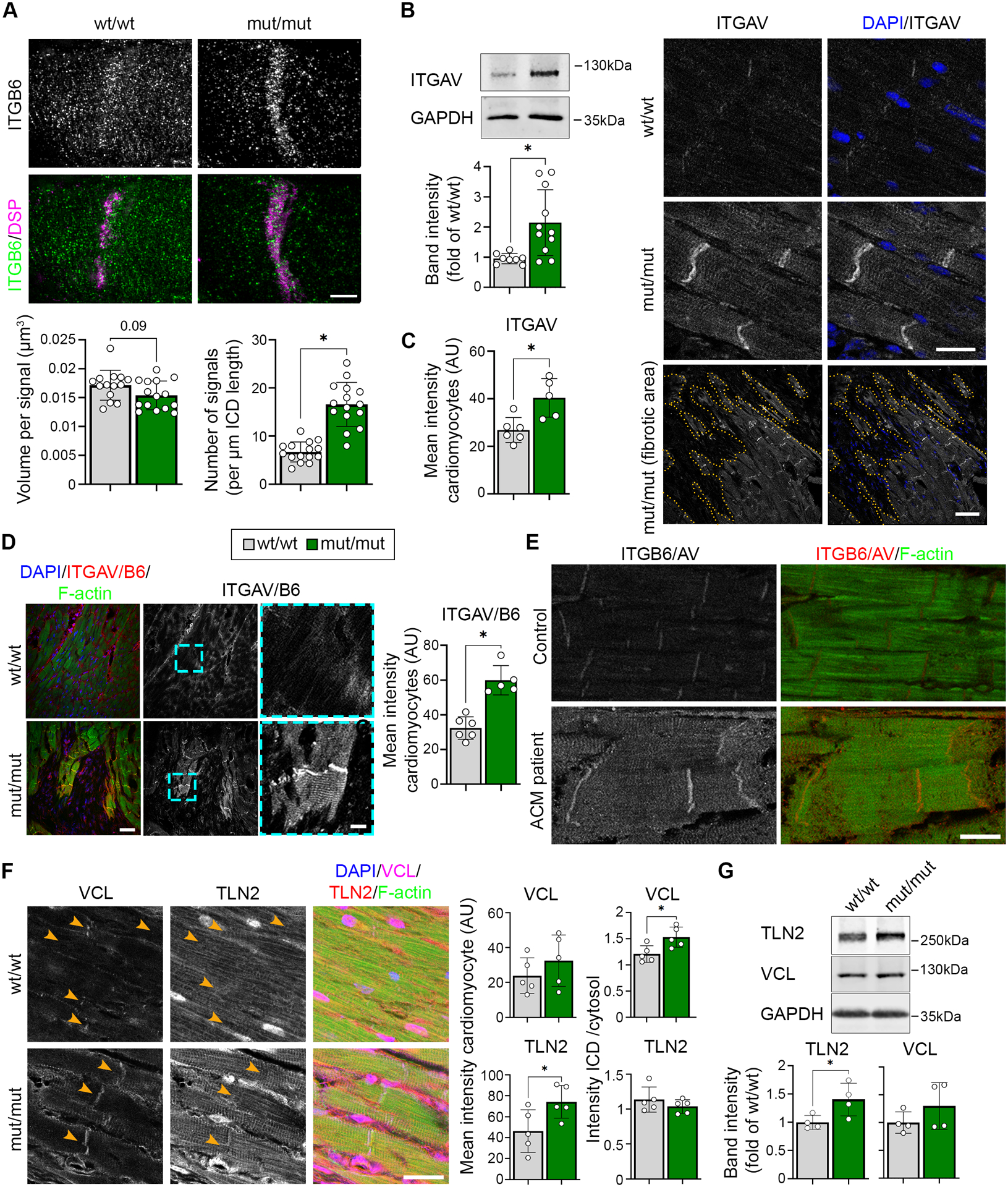
(A) Representative ICD reconstruction from z-stacks of ITGB6 (green) and DSP (magenta) immunostaining acquired by SIM and presented as maximum intensity projection. Related analysis of ITGB6 signal volume and number per ICD length is shown below. Scale bar: 5 μm. *P< 0.05 or as indicated, unpaired Student’s t-test with Welch’s correction (left graph) and Mann-Whitney test (right graph). Each dot represents the value of one ICD from in total 4 mice per genotype. (B) Representative western blot and analysis of band intensity of Integrin-αV (ITGAV) in DSG2-W2A hearts. GAPDH served as loading control. *P< 0.05, unpaired Student’s t-test with Welch’s correction. (C) Immunostaining of ITGAV in DSG2-W2A hearts on the right with respective analysis on the left. DAPI (blue) marks nuclei. Lower row shows an overview image of a fibrotic area in mut/mut hearts. Dotted orange line marks the edge of fibrotic area. Scale bars: upper rows: 20 μm, lower row: 50 μm. *P< 0.05, unpaired Student’s t-test. (D) Immunostaining of ITGAV/B6 heterodimer in DSG2-W2A mutant hearts with respective analysis of staining intensity on the right. Cyan rectangle marks zoomed area on the right. Scale bars: overview 50 μm; insert 10 μm. *P< 0.05, unpaired Student’s t-test. (E) Representative immunostaining images of ITGAV/B6 heterodimer staining (red) in an ACM patient (DSP-E952X, heterozygous) and healthy control sample. F-actin (green) stains the sarcomere system. For the ACM patient, 4 different tissue samples were analysed and compared to 2 tissue samples from 2 healthy controls. Scale bar: 20 μm. (F) Immunostaining of vinculin (VCL, magenta) and talin-2 (TLN2, red) in DSG2-W2A hearts on the right with respective analysis of the mean staining intensity in cardiomyocytes and ratio of the staining intensity at the ICD vs. cytosolic area on the left. DAPI (blue) marks nuclei. F-actin (green) stains the sarcomere system. Scale bar: 25 μm, *P< 0.05, unpaired Student’s t-test. (G) Representative western blot and analysis of band intensity of VCL and TLN2 in DSG2-W2A hearts. GAPDH served as loading control. *P< 0.05, unpaired Student’s t-test for TLN2, Mann-Whitney test for VCL. Box with color indications of respective groups apply to the entire figure.
TGF-β signaling is upregulated in DSG2-W2A mutant hearts
ITGAV/B6 dimers have the ability to activate the pro-fibrotic cytokine TGF-β by binding and removal of the latency associate peptide (LAP).17 Gene set enrichment analyses revealed an upregulation of genes associated with TGF-β signaling in both 9-weeks-old mutants as well as ACM patients (Figure 6A). Moreover, direct targets of receptor-regulated SMADs involved in TGF-β signaling21 were upregulated in DSG2-W2A mutant hearts (Figure 6B). Accordingly, increased amounts of nuclear SMAD2/3 phosphorylated at the activation sites S465/S467 or S423/S425, respectively, were detected in these hearts (Figure 6C). Interestingly, increased levels of pSMAD2/3 were not only found in fibroblasts but also in cardiomyocytes mainly adjacent to fibrotic areas. These data indicate upregulation of the pro-fibrotic TGF-β pathway and its downstream targets, which include extracellular matrix proteins such as collagens, laminin or fibronectin.
Figure 6. Elevated TGF-β signaling in DSG2-W2A hearts as result of ITGAV/B6 activity.
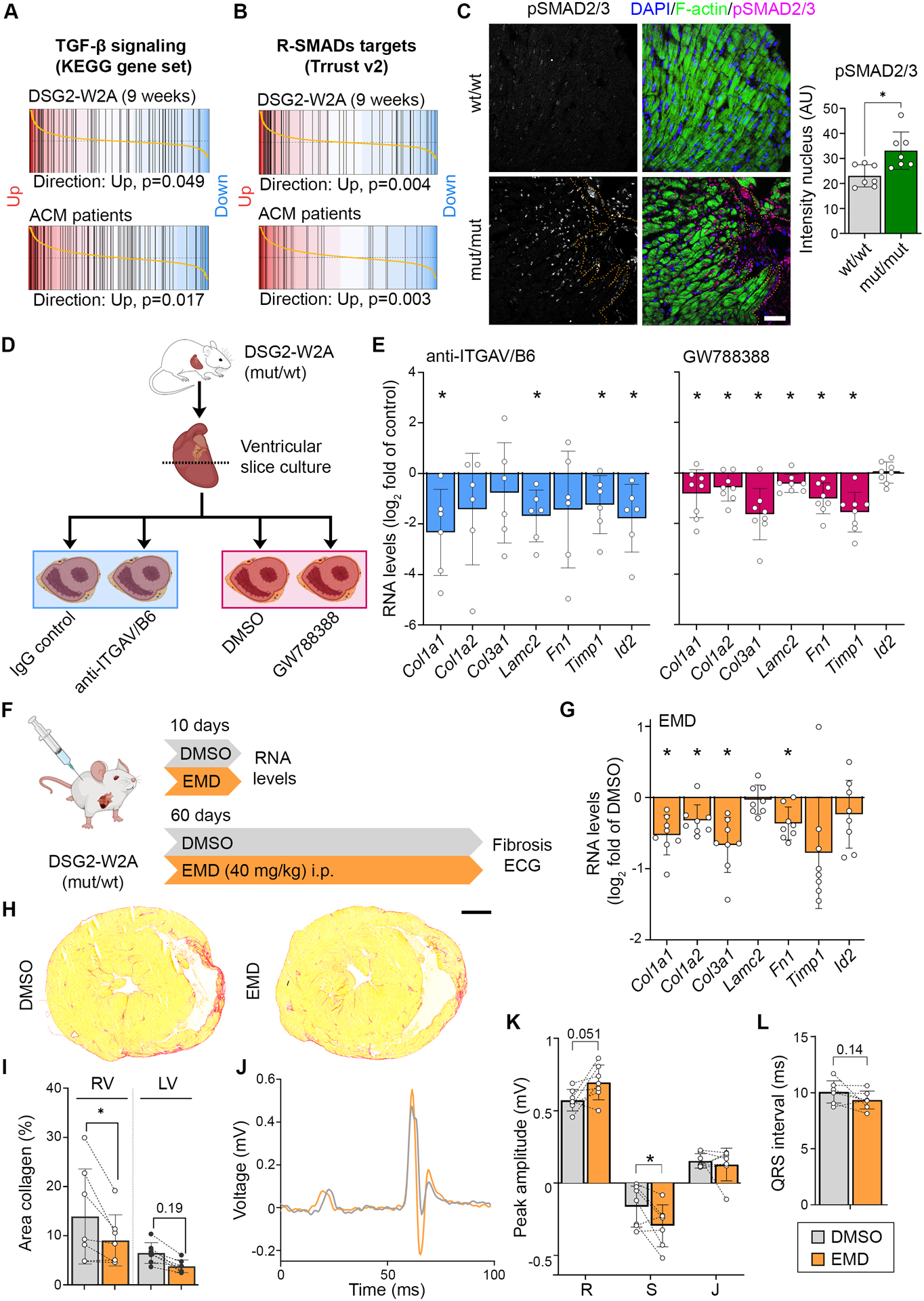
Barcode plots of gene set enrichment analysis of (A) the KEGG_TGF_BETA_SIGNALING_PATHWAY data set (systematic name: M2642),41 or (B) genes directly regulated by receptor-regulated SMADs (R-SMADs, includes SMAD1/2/3/5/9) as published in the TRRUST data base21 in 9-weeks-old DSG2-W2A (mut/mut vs wt/wt) or ACM patient data set 1 (ACM vs. healthy control, GEO: GSE107157/GSE107480). Indicated p-values are calculate by function cameraPR of the R package limma. (C) Immunostainings of phosphorylated SMAD2/3 (magenta, S465/S467 or S423/S425, respectively) in sections of DSG2-W2A hearts and related analysis of nuclear staining intensity. Nuclei are stained with DAPI (blue), cardiomyocytes are marked with f-actin (green). Dotted orange line marks edge of fibrotic area. Scale bar: 50 μm. *P< 0.05, unpaired Student’s t-test. (D) Schematic of experimental set-up for ITGAV/B6 blocking experiments in cardiac slice culture with related results in E. Icons are derived from BioRender. (E) qRT-PCR analysis of expression of genes downstream of TGF-β signaling in cardiac slices cultures treated with inhibiting anti-ITGAV/B6 (1:15) or 10 μmol/l GW788388, an inhibitor of TGF-β receptor I, for 24 hours. *P< 0.05, paired Student’s t-test vs. indicated control condition. (F) Schematic of experimental set-up for in vivo ITGAV/B6 blocking experiments by injection of 40 mg/kg EMD527040 (EMD) i.p. daily. DMSO was applied as vehicle control. Icons are derived from BioRender. (G) qRT-PCR expression analysis of genes downstream of TGF-β signaling in hearts of mice treated with EMD or respective amount of DMSO for 10 days. *P< 0.05, unpaired Student’s t-test vs. DMSO. (H, I) Cardiac fibrosis detected by picrosirius red collagen staining with representative images and corresponding analysis of the area of collagen in the right (RV) and left ventricle (LV). Lines indicate littermates. Each dot represents one animal. *P< 0.05 or as indicated, grouped two-way RM ANOVA with LV/RV and experimental pairs matched, Sidak’s post hoc test. Scale bar: 1 mm. (J – L) ECG recoded in lead II with representative curves shown in J. Corresponding analysis of R, S, and J peak amplitude and QRS interval, *P< 0.05 or as indicated, Paired student’s t-test. Lines indicate littermates. Each dot represents one animal.
Fibrosis in DSG2-W2A mutant animals is reduced by inhibition of ITGAV/B6-dependent release of TGF-β
Based on the known pro-fibrotic role of TGF-β in cardiomyopathies2 and the established function of ITGAV/B6 dimers to activate TGF-β17, we investigated whether inhibition of ITGAV/B6 was efficient to reduce fibrosis in DSG2-W2A mice. Initially, cardiac slice cultures were generated from the ventricles of heterozygous mice at the age of 40–50 weeks and treated for 24 hours with anti-ITGAV/B6, which was shown to neutralize the function of the dimer.22 In comparison, slices were treated with the TGF-β receptor I inhibitor GW788388 to directly block TGF-β signaling (Figure 6D). Significant downregulation in the expression of the pro-fibrotic molecules collagen-I type-α1 (Col1a1), laminin subunit-γ2 (Lamc2), metalloproteinase inhibitor-1 (Timp1), and ID-2 (Id2) was induced in the mutant mice in response to inhibition of ITGAV/B6 (Figure 6E). Other extracellular matrix proteins such as fibronectin (Fn1) or collagens (Col3a1, Col1a2) showed the same tendency. Importantly, all of these fibrotic markers were significantly upregulated in adult mutant hearts as detected by RNA-Sequencing (Figure 6A, B). Further, direct inhibition of TGF-β led to a similar downregulation of these markers as the blocking antibody.
To investigate the in vivo relevance of this pathway, we applied the small molecule EMD527040 (EMD), which is an established inhibitor of ITGAV/B6-dependent TGF-β release, to the DSG2-W2A mouse model.23 For these experiments, we chose wt/mut mice as they reflect the heterozygosity most commonly found in ACM patients and develop the phenotype during adulthood, when compound application is feasible. Littermates between the age of 28–38 weeks were sex-matched and i.p.-injected daily for 10 days with EMD or DMSO control (Figure 6F). In this short-time approach, myocardial samples of EMD-treated animals showed a similar reduction of pro-fibrotic markers as reported for slice cultures (Figure 6G). Having established an effective dose, we finally performed in vivo studies for a duration of 60 days during the time period when RV fibrosis establishes. At the end of the treatment period, ECG and histological analysis were performed. EMD-treated mice demonstrated reduced RV fibrosis (Figure 6H, I). Moreover, EMD animals showed a mitigation of ECG abnormalities, with significantly higher S amplitude and trends towards an increased R amplitude and shorter QRS interval compared to the vehicle-treated control.
In conclusion, these experiments link ITGAV/B6 dimerization in response to abrogated DSG2 binding and ICD dysfunction to TGF-β-dependent fibrosis generation. Moreover, the pilot inhibitor studies suggest ITGAV/B6 as promising treatment target to ameliorate the ACM phenotype.
6. Discussion
In this study, we generated a knock-in mouse model with defective binding function of the adhesion molecule DSG2, which demonstrated that impaired desmosomal adhesion is sufficient to induce a phenotype mimicking the characteristics of ACM. Our data suggest a cascade of defective desmosomal adhesion, disrupted ICD structure and subsequent activation of ITGAV/B6 with pro-fibrotic TGF-β signaling as important underlying mechanism leading to this phenotype (Figure 7). Moreover, our pilot study indicates a beneficial effect of ITGAV/B6 inhibition by EMD treatment with regard to fibrosis and several ECG parameters, suggesting that this pathway can be successfully targeted by drug treatment.
Figure 7. Schematic conclusion of data.
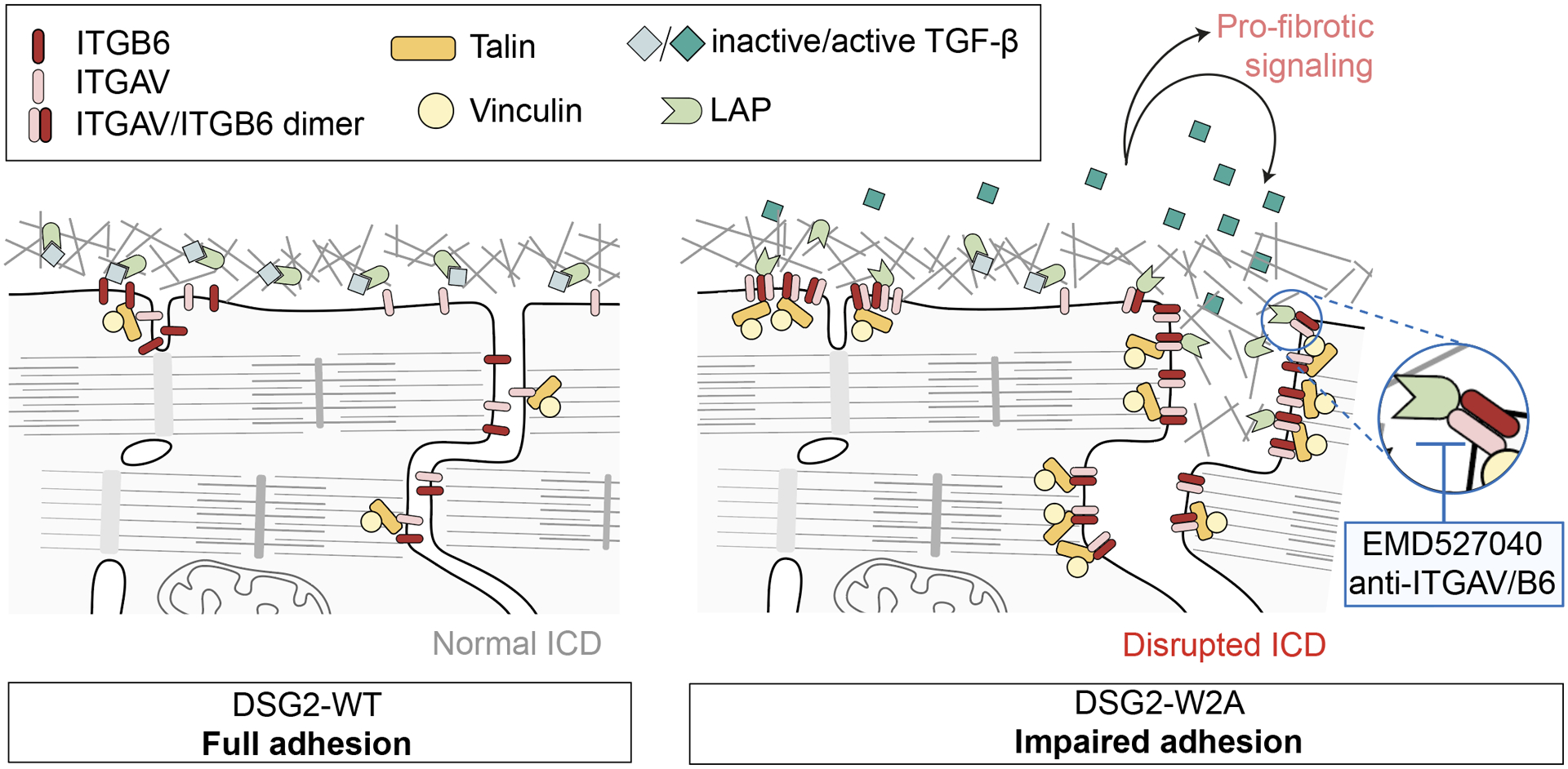
DSG2-W2A mutation with loss of desmosomal adhesion leads to impaired ICD structure with deregulation of ITGB6 and enhanced heterodimerization with ITGAV. The dimer efficiently binds to the extracellular matrix and activates TGF-β by removal of the latency-associated peptide (LAP). Active TGF-β can then induce pro-fibrotic downstream signaling via SMAD molecules. In our experiments, this cascade was blocked by different approaches to inhibit ITGAV/B6.
Defective desmosomal adhesion by DSG2-W2A mutation is inducing an ACM phenotype
Due to mutations mainly affecting desmosomal genes and evidence of disrupted ICDs, the hypothesis of dysfunctional desmosomes with loss of cell-cell adhesion as central pathological step was soon adopted in the field.2 However, experimental data on this topic are contradictory.6–8 We show that W2 of DSG2 is central for binding and intercellular adhesion in vitro and in vivo. Importantly, disrupted desmosomal adhesion is sufficient to induce an ACM phenotype fulfilling the Padua criteria13, which are used to establish the diagnosis in patients, including ventricular fibrosis, de- and repolarization abnormalities, arrhythmia, and impaired ventricular function. In line with this, three mutations were described in ACM patients directly affecting the DSG2 binding mechanism by exchange of the tryptophan with a serine, leucine, or arginine (ClinVar data bank VCV000199796, VCV000044282, VCV000420241)10. This indicates a disruption of the tryptophan swap independent from the substituting amino acid and supports defective desmosomal adhesion as important factor in ACM.2,24 Moreover, different published DSG2 mutant mouse models show a phenotype resembling the features of DSG2-W2A mutants. These include (I) a DSG2 mutant in which parts of the two outermost DSG2 extracellular domains were deleted (but leaving W2A intact),25 (II) two mouse lines with loss of DSG2,25,26 and (III) mice overexpressing a patient mutation (DSG2-N271S)27, which also showed that ICD structural aberrations precede functional abnormalities and fibrosis generation. These data fuel the hypothesis that disruption and rearrangement of the ICD in response to impaired adhesion between cardiomyocytes is a crucial initial step, at least in case of mutations in desmosomal molecules. Other mechanisms described in patients or translational models, such as aberrant WNT or Hippo/YAP signaling, or immune cell infiltrations2,28 may be secondary responses and in part even represent adaptive attempts to rescue the functional consequences of such severe structural aberrations.
DSG2-W2A mice as model to study ACM
Our results suggest the DSG2-W2A model as a valuable tool to study ACM mechanisms. Within the limitations of a murine model, it reproduces a majority of features found in ACM patients (Table S2). It is interesting to note that, while in homozygous animals both ventricles are affected from early on in life, heterozygous animals develop a milder phenotype with fibrosis occurring only in the RV. So far it is unclear if this milder phenotype would extend to the LV over time, or whether a gene dose effect underlies the structural differences. LV and RV remodel differently in response to loading and injury29 and it is possible that the RV is less able to compensate a partial reduction of cardiomyocyte cohesion in contrast to the LV. Interestingly, a fraction of mut/wt animals did not develop the phenotype at all within the observation period of up to 80 weeks. Thus, the model might help to understand why patients with the same mutation (i.e. in the same family) develop the disease with variable penetrance.1 Interestingly, similar to patients,1 male mutant mice appear to suffer more from arrhythmia and sudden death than females.
We can interpret from the combined histology, echocardiography, and ECG data that the changes in heart function are secondary to fibrosis generation. This is indicated by the notion that functional parameters (ejection fraction, QRS interval) worsened over time in homozygous mutants, while no increase was detectable for fibrosis. Moreover, functional changes in mut/wt animals were only observed when fibrosis was present. This is in line with a patient cohort with DSP mutations, in which LV fibrosis preceded systolic dysfunction,30 while a different study in DSG2-N271S-expressing ex vivo-paced hearts demonstrated conduction velocity impairments before onset of fibrosis.27 More longitudinal studies using different mutational backgrounds and sensitive detection methods, e.g. cardiac MRI and ECG under exercise, are necessary to define the role of fibrosis as cause or consequence of functional changes.
As all murine models, the DSG2-W2A line has limitations in its ability to fully recapitulate the human disease phenotype. Mutant mice do not show pronounced replacement with adipose tissue, which is a common characteristic in patient heart samples, but in general is not fully modelled in mice.24 Moreover, at least in ECGs under anesthesia, not all mutant animals develop arrhythmia, even if fibrosis is present. However, in contrast to most other ACM models, this knock-in model has the advantage of a single amino acid exchange under the endogenous promotor, thus reducing the possibility of secondary unwanted effects due to complete protein absence or overexpression. As in patients, the mutation is present in all cell types and not limited to cardiomyocytes.2,24
DSG2-W2A leads to dysfunctional ICDs and ITGAV/B6 rearrangement
DSG2-W2A led to a severely altered ICD structure consistent with impaired cytoskeletal attachment and ruptured junctions. While these changes provide explanations for compromised intercellular adhesion, we also noted alterations in cell-matrix protein distribution. RNA-Sequencing before and after onset of the disease phenotype and a comparison with ACM patient datasets identified ITGB6 as commonly deregulated in ACM patients and DSG2-W2A mice. While we noted reduced mRNA levels in mutant hearts, the overall protein content was unaltered. This suggests pronounced posttranslational regulation of ITGB6, which was already demonstrated in skeletal muscle.31 In mutant hearts, ITGB6 was increased at the ICD together with elevated levels of ITGAV and increased heterodimerization of both molecules. By mechanical force exerted through ITGAV/B6, TGF-β is detached from LAP and can induce signaling via TGF-β receptors.17,32 In line with higher intracellular forces, talin-2 and vinculin were increased at ICDs, as binding of these molecules activates and stabilizes integrins. A mutual regulation of both adhesive compartments is well established. As example, upon loss of N-cadherin, integrins are activated and induce fibronectin deposition.33 A similar mechanism is conceivable in DSG2-W2A hearts, either by loss of DSG2 or due to reduced N-cadherin anchorage. So far, only limited data are available on the role of integrins in ACM. A recent study showed downregulation of integrin-β1D leading to ventricular arrhythmia.34 Further, knock-down of PKP2 in HL-1 cardiomyocytes was described to deregulate focal adhesions including integrin-α1.35
Activation of TGF-β signaling by ITGAV/B6 as potential therapeutic target in ACM
The ITGAV/B6 heterodimer is described as one of the major activators of latent TGF-β1 and TGF-β3.17 Although TGF-β signaling is known as general driver of cardiac fibrosis36 and more specifically was implicated in ACM,2 to our knowledge no data are available on the role of ITGAV/B6 and their regulation of TGF-β signaling in cardiac fibrosis. Uncovering this pathway is of high interest, as it offers the possibility to target TGF-β with reduced risk of severe side effects occurring in response to direct inhibition.37 Accordingly, we demonstrate a reduction of pro-fibrotic gene expression under ACM conditions in response to ITGAV/B6 inhibition and our pilot study data suggest that a small molecule blocking ITGAV/B6-dependent TGF-β release diminished the generation of fibrosis. Similar approaches using this small molecule or neutralizing antibodies were shown to be protective in murine models of lung, liver and biliary fibrosis.23,38,39 However, based on the heterogeneity of the phenotype in heterozygous animals, more detailed studies with larger sample sizes are required to further substantiate this finding.
Currently it is not fully clear whether fibrosis generation is a contributor to the disease or rather a protective mechanism to ensure integrity of the heart under conditions of compromised adhesion. In case of the latter, pharmacological inhibition of fibrosis generation might be detrimental. Nevertheless, fibrosis is a driver of arrhythmia generation,40 which is also supported by our results that ECG abnormalities were reduced by inhibition of fibrosis. Careful studies using different therapeutic approaches to reduce fibrosis in appropriate model organisms need to address this aspect in detail.
In conclusion, we established a new mouse model phenocopying many aspects of ACM and uncovered a novel pathway of fibrosis induction. Furthermore, we identified an approach to target this mechanism with future implication as potential therapeutic option in patients.
Supplementary Material
Clinical Perspective.
What is new?
The tryptophan residue at position 2 of the desmosomal adhesion molecule desmoglein-2 (DSG2) is central for its binding function and cell-cell adhesion in vitro and in vivo.
Mice with abrogated DSG2 binding function (DSG2-W2A) develop a cardiac phenotype recalling Arrhythmogenic Cardiomyopathy with fibrosis, impaired systolic function, ECG abnormalities and ventricular arrhythmia.
Increased integrin-αVβ6-dependent TGF-β signaling was identified as driver of fibrosis.
What are the Clinical implications?
We provide a new mouse model reproducing major features of Arrhythmogenic Cardiomyopathy which can be applied to study disease mechanisms and test therapeutic approaches.
The model suggests that loss of mechanical cardiomyocyte coupling is a central and early event causing Arrhythmogenic Cardiomyopathy.
Our pilot in vivo study highlights the applicability and potential to target integrin-αVβ6-dependent TGF-β release as new therapeutic approach to diminish the development of fibrosis in Arrhythmogenic Cardiomyopathy.
Acknowledgement
We are grateful to Alain Brühlhart and the team from the Animal Facility (University of Basel, Switzerland), Dr. Cinzia Tiberi, Center for Cellular Imaging and NanoAnalytics (C-CINA, University of Basel), Dr. Alexia Loynton-Ferrand, Imaging Core Facility (IMCF, University of Basel), Dr. Diego Calabrese, Histology Core Facility, Dr. Mike Abanto, Dr. Beat Erne, Pascal Lorentz and Ewelina Bartoszek, all Microscopy Core Facility (all DBM, University of Basel), and Dr. Christian Beisel and Philippe Demougin, Genomics Facility Basel (D-BSSE, ETH Zürich, and University of Basel) for excellent support. We thank Nicolas Schlegel (Department of Surgery, University Hospital Würzburg, Germany) for providing CaCo2 cells, and Nikola Golenhofen (Institute of Anatomy, University of Ulm, Germany) for providing the Fc-His-pEGFP-N3 plasmid. We thank Anja Fuchs for excellent technical assistance. Calculations were performed at sciCORE (http://scicore.unibas.ch/) scientific computing center at University of Basel.
Sources of Funding
This project was supported by the German Research Council (SP1300-3/1, to VS), the Swiss National Science foundation (#197764 to VS), the Swiss heart foundation (FF21098 to CS, VS), the Research Fund Junior Researchers, University of Basel (3BM1079 to CS), and the Theiler-Haag foundation (VS). FS is supported by National Institutes of Health (HL142251) and Department of Defense (W81XWH1810380) grants.
Non-standard Abbreviations and Acronyms
- ACM
Arrhythmogenic Cardiomyopathy
- AFM
Atomic Force Microscopy
- CaCo2(DSG2 KO)
CaCo2 desmoglein-2 knock-out cells
- CaCo2(WT)
CaCo2 wildtype cells
- DSG2
desmoglein-2
- DSP
desmoplakin
- ECG
Electrocardiogram
- EMD
EMD527040
- FRAP
Fluorescence Recovery After Photobleaching
- ICD
intercalated disc
- ITGAV
integrin-αV
- ITGB6
integrin-β6
- LAP
latency associate peptide
- LV
left ventricle
- mut
DSG2-W2A mutant
- PG
plakoglobin
- PKP2
plakophilin-2
- PVC
premature ventricular contraction
- RV
right ventricle
- SIM
Structured Illumination Microscopy
- TAPSE
tricuspid annular plane systolic excursion
- TEM
Transmission Electron Microscopy
- wt
wildtype
Footnotes
Disclosures
FS is a co-founder and equity shareholder of Papillon Therapeutics Inc. and is a consultant and equity shareholder of LEXEO Therapeutics Inc. All other authors have nothing to disclose.
References
- 1.Corrado D, Link MS, Calkins H. Arrhythmogenic Right Ventricular Cardiomyopathy. N Engl J Med. 2017;376:1489–1490. doi: 10.1056/NEJMc1701400 [DOI] [PubMed] [Google Scholar]
- 2.Austin KM, Trembley MA, Chandler SF, Sanders SP, Saffitz JE, Abrams DJ, Pu WT. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat Rev Cardiol. 2019;16:519–537. doi: 10.1038/s41569-019-0200-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Delva E, Tucker DK, Kowalczyk AP. The desmosome. Cold Spring Harb Perspect Biol. 2009;1:a002543. doi: 10.1101/cshperspect.a002543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delmar M, McKenna WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res. 2010;107:700–714. doi: 10.1161/CIRCRESAHA.110.223412 [DOI] [PubMed] [Google Scholar]
- 5.Basso C, Czarnowska E, Della Barbera M, Bauce B, Beffagna G, Wlodarska EK, Pilichou K, Ramondo A, Lorenzon A, Wozniek O, et al. Ultrastructural evidence of intercalated disc remodelling in arrhythmogenic right ventricular cardiomyopathy: an electron microscopy investigation on endomyocardial biopsies. Eur Heart J. 2006;27:1847–1854. doi: 10.1093/eurheartj/ehl095 [DOI] [PubMed] [Google Scholar]
- 6.Hariharan V, Asimaki A, Michaelson JE, Plovie E, MacRae CA, Saffitz JE, Huang H. Arrhythmogenic right ventricular cardiomyopathy mutations alter shear response without changes in cell-cell adhesion. Cardiovasc Res. 2014;104:280–289. doi: 10.1093/cvr/cvu212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang H, Asimaki A, Lo D, McKenna W, Saffitz J. Disparate effects of different mutations in plakoglobin on cell mechanical behavior. Cell Motil Cytoskeleton. 2008;65:964–978. doi: 10.1002/cm.20319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schlipp A, Schinner C, Spindler V, Vielmuth F, Gehmlich K, Syrris P, McKenna WJ, Dendorfer A, Hartlieb E, Waschke J. Desmoglein-2 interaction is crucial for cardiomyocyte cohesion and function. Cardiovasc Res. 2014;104:245–257. doi: 10.1093/cvr/cvu206 [DOI] [PubMed] [Google Scholar]
- 9.Harrison OJ, Brasch J, Lasso G, Katsamba PS, Ahlsen G, Honig B, Shapiro L. Structural basis of adhesive binding by desmocollins and desmogleins. Proc Natl Acad Sci U S A. 2016;113:7160–7165. doi: 10.1073/pnas.1606272113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.NM_001943.5(DSG2). In: ClinVar database (version 06/01/2022). [Google Scholar]
- 11.Bell GI. Models for the specific adhesion of cells to cells. Science. 1978;200:618–627. doi: 10.1126/science.347575 [DOI] [PubMed] [Google Scholar]
- 12.Meir M, Burkard N, Ungewiss H, Diefenbacher M, Flemming S, Kannapin F, Germer CT, Schweinlin M, Metzger M, Waschke J, et al. Neurotrophic factor GDNF regulates intestinal barrier function in inflammatory bowel disease. J Clin Invest. 2019;129:2824–2840. doi: 10.1172/JCI120261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corrado D, Perazzolo Marra M, Zorzi A, Beffagna G, Cipriani A, Lazzari M, Migliore F, Pilichou K, Rampazzo A, Rigato I, et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int J Cardiol. 2020;319:106–114. doi: 10.1016/j.ijcard.2020.06.005 [DOI] [PubMed] [Google Scholar]
- 14.Song JS, Wang RS, Leopold JA, Loscalzo J. Network determinants of cardiovascular calcification and repositioned drug treatments. FASEB J. 2020;34:11087–11100. doi: 10.1096/fj.202001062R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaertner A, Schwientek P, Ellinghaus P, Summer H, Golz S, Kassner A, Schulz U, Gummert J, Milting H. Myocardial transcriptome analysis of human arrhythmogenic right ventricular cardiomyopathy. Physiol Genomics. 2012;44:99–109. doi: 10.1152/physiolgenomics.00094.2011 [DOI] [PubMed] [Google Scholar]
- 16.Margadant C, Sonnenberg A. Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010;11:97–105. doi: 10.1038/embor.2009.276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Munger JS, Sheppard D. Cross talk among TGF-beta signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb Perspect Biol. 2011;3:a005017. doi: 10.1101/cshperspect.a005017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koivisto L, Bi J, Hakkinen L, Larjava H. Integrin alphavbeta6: Structure, function and role in health and disease. Int J Biochem Cell Biol. 2018;99:186–196. doi: 10.1016/j.biocel.2018.04.013 [DOI] [PubMed] [Google Scholar]
- 19.Horton ER, Humphries JD, James J, Jones MC, Askari JA, Humphries MJ. The integrin adhesome network at a glance. J Cell Sci. 2016;129:4159–4163. doi: 10.1242/jcs.192054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merkel CD, Li Y, Raza Q, Stolz DB, Kwiatkowski AV. Vinculin anchors contractile actin to the cardiomyocyte adherens junction. Mol Biol Cell. 2019;30:2639–2650. doi: 10.1091/mbc.E19-04-0216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han H, Cho JW, Lee S, Yun A, Kim H, Bae D, Yang S, Kim CY, Lee M, Kim E, et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018;46:D380–D386. doi: 10.1093/nar/gkx1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weinreb PH, Simon KJ, Rayhorn P, Yang WJ, Leone DR, Dolinski BM, Pearse BR, Yokota Y, Kawakatsu H, Atakilit A, et al. Function-blocking integrin alphavbeta6 monoclonal antibodies: distinct ligand-mimetic and nonligand-mimetic classes. J Biol Chem. 2004;279:17875–17887. doi: 10.1074/jbc.M312103200 [DOI] [PubMed] [Google Scholar]
- 23.Patsenker E, Popov Y, Stickel F, Jonczyk A, Goodman SL, Schuppan D. Inhibition of integrin alphavbeta6 on cholangiocytes blocks transforming growth factor-beta activation and retards biliary fibrosis progression. Gastroenterology. 2008;135:660–670. doi: 10.1053/j.gastro.2008.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerull B, Brodehl A. Genetic Animal Models for Arrhythmogenic Cardiomyopathy. Front Physiol. 2020;11:624. doi: 10.3389/fphys.2020.00624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kant S, Holthofer B, Magin TM, Krusche CA, Leube RE. Desmoglein 2-Dependent Arrhythmogenic Cardiomyopathy Is Caused by a Loss of Adhesive Function. Circ Cardiovasc Genet. 2015;8:553–563. doi: 10.1161/CIRCGENETICS.114.000974 [DOI] [PubMed] [Google Scholar]
- 26.Chelko SP, Asimaki A, Andersen P, Bedja D, Amat-Alarcon N, DeMazumder D, Jasti R, MacRae CA, Leber R, Kleber AG, et al. Central role for GSK3beta in the pathogenesis of arrhythmogenic cardiomyopathy. JCI Insight. 2016;1. doi: 10.1172/jci.insight.85923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rizzo S, Lodder EM, Verkerk AO, Wolswinkel R, Beekman L, Pilichou K, Basso C, Remme CA, Thiene G, Bezzina CR. Intercalated disc abnormalities, reduced Na(+) current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc Res. 2012;95:409–418. doi: 10.1093/cvr/cvs219 [DOI] [PubMed] [Google Scholar]
- 28.Meraviglia V, Alcalde M, Campuzano O, Bellin M. Inflammation in the Pathogenesis of Arrhythmogenic Cardiomyopathy: Secondary Event or Active Driver? Front Cardiovasc Med. 2021;8:784715. doi: 10.3389/fcvm.2021.784715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friedberg MK, Redington AN. Right versus left ventricular failure: differences, similarities, and interactions. Circulation. 2014;129:1033–1044. doi: 10.1161/CIRCULATIONAHA.113.001375 [DOI] [PubMed] [Google Scholar]
- 30.Smith ED, Lakdawala NK, Papoutsidakis N, Aubert G, Mazzanti A, McCanta AC, Agarwal PP, Arscott P, Dellefave-Castillo LM, Vorovich EE, et al. Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct From Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation. 2020;141:1872–1884. doi: 10.1161/CIRCULATIONAHA.119.044934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ducceschi M, Clifton LG, Stimpson SA, Billin AN. Post-transcriptional regulation of ITGB6 protein levels in damaged skeletal muscle. J Mol Histol. 2014;45:329–336. doi: 10.1007/s10735-014-9567-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong X, Zhao B, Iacob RE, Zhu J, Koksal AC, Lu C, Engen JR, Springer TA. Force interacts with macromolecular structure in activation of TGF-beta. Nature. 2017;542:55–59. doi: 10.1038/nature21035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Julich D, Cobb G, Melo AM, McMillen P, Lawton AK, Mochrie SG, Rhoades E, Holley SA. Cross-Scale Integrin Regulation Organizes ECM and Tissue Topology. Dev Cell. 2015;34:33–44. doi: 10.1016/j.devcel.2015.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Li C, Shi L, Chen X, Cui C, Huang J, Chen B, Hall DD, Pan Z, Lu M, et al. Integrin beta1D Deficiency-Mediated RyR2 Dysfunction Contributes to Catecholamine-Sensitive Ventricular Tachycardia in Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation. 2020;141:1477–1493. doi: 10.1161/CIRCULATIONAHA.119.043504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Puzzi L, Borin D, Gurha P, Lombardi R, Martinelli V, Weiss M, Andolfi L, Lazzarino M, Mestroni L, Marian AJ, et al. Knock Down of Plakophillin 2 Dysregulates Adhesion Pathway through Upregulation of miR200b and Alters the Mechanical Properties in Cardiac Cells. Cells. 2019;8. doi: 10.3390/cells8121639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frangogiannis NG. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Aspects Med. 2019;65:70–99. doi: 10.1016/j.mam.2018.07.001 [DOI] [PubMed] [Google Scholar]
- 37.Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A. 1993;90:770–774. doi: 10.1073/pnas.90.2.770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Puthawala K, Hadjiangelis N, Jacoby SC, Bayongan E, Zhao Z, Yang Z, Devitt ML, Horan GS, Weinreb PH, Lukashev ME, et al. Inhibition of integrin alpha(v)beta6, an activator of latent transforming growth factor-beta, prevents radiation-induced lung fibrosis. Am J Respir Crit Care Med. 2008;177:82–90. doi: 10.1164/rccm.200706-806OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.John AE, Graves RH, Pun KT, Vitulli G, Forty EJ, Mercer PF, Morrell JL, Barrett JW, Rogers RF, Hafeji M, et al. Translational pharmacology of an inhaled small molecule alphavbeta6 integrin inhibitor for idiopathic pulmonary fibrosis. Nat Commun. 2020;11:4659. doi: 10.1038/s41467-020-18397-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maione AS, Pilato CA, Casella M, Gasperetti A, Stadiotti I, Pompilio G, Sommariva E. Fibrosis in Arrhythmogenic Cardiomyopathy: The Phantom Thread in the Fibro-Adipose Tissue. Front Physiol. 2020;11:279. doi: 10.3389/fphys.2020.00279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Concordet JP, Haeussler M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018;46:W242–W245. doi: 10.1093/nar/gky354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ebner A, Wildling L, Kamruzzahan AS, Rankl C, Wruss J, Hahn CD, Holzl M, Zhu R, Kienberger F, Blaas D, et al. A new, simple method for linking of antibodies to atomic force microscopy tips. Bioconjug Chem. 2007;18:1176–1184. doi: 10.1021/bc070030s [DOI] [PubMed] [Google Scholar]
- 44.Hutter JL, Bechhoefer J. Calibration of atomic‐force microscope tips. Rev Sci Instrum. 1993;64:1868–1873. doi: 10.1063/1.1143970 [DOI] [Google Scholar]
- 45.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- 47.Chopin M, Preston SP, Lun ATL, Tellier J, Smyth GK, Pellegrini M, Belz GT, Corcoran LM, Visvader JE, Wu L, et al. RUNX2 Mediates Plasmacytoid Dendritic Cell Egress from the Bone Marrow and Controls Viral Immunity. Cell Rep. 2016;15:866–878. doi: 10.1016/j.celrep.2016.03.066 [DOI] [PubMed] [Google Scholar]
- 48.Ehler E, Moore-Morris T, Lange S. Isolation and culture of neonatal mouse cardiomyocytes. J Vis Exp. 2013. doi: 10.3791/50154 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-Seq data were deposited in the public database GEO accession number GSE181868. Additional data pertaining to the current article are available from the corresponding author upon request.