

Abstract
Disease recurrence in high-grade serous ovarian cancer may be due to cancer stem-like cells (CSCs) that are resistant to chemotherapy and capable of reestablishing heterogeneous tumors. The alternative NF-κB signaling pathway is implicated in this process, however the mechanism is unknown. Here we show that TNF-like weak inducer of apoptosis (TWEAK) and its receptor, Fn14, are strong inducers of alternative NF-κB signaling and are enriched in ovarian tumors following chemotherapy treatment. We further show that TWEAK enhances spheroid formation ability, asymmetric division capacity, and expression of SOX2 and EMT genes VIM and ZEB1 in ovarian cancer cells, phenotypes that are enhanced when TWEAK is combined with carboplatin. Moreover, TWEAK in combination with chemotherapy induces expression of the CSC marker CD117 in CD117− cells. Blocking the TWEAK-Fn14-RelB signaling cascade with a small molecule inhibitor of Fn14 prolongs survival following carboplatin chemotherapy in a mouse model of ovarian cancer. These data provide new insights into ovarian cancer CSC biology and highlight a signaling axis that should be explored for therapeutic development.
Keywords: Ovarian cancer, CSCs, TWEAK, Fn14, RelB
INTRODUCTION
Ovarian cancer is the most-deadly gynecological malignancy in the United States, resulting in over 15,000 deaths each year (1). Although most high-grade serous ovarian cancer (HGSOC) patients initially respond to platinum-based chemotherapy, over 80% of patients with advanced-stage disease relapse within 24 months and eventually develop chemoresistance (2). Thus, much research has focused on clarifying mechanisms of chemoresistance and the processes involved in tumor relapse. Over a decade of research has implicated tumor-initiating cells, here referred to as cancer stem-like cells (CSCs), in mediating these activities; efforts to characterize and target these cells are a promising avenue for preventing relapse and improving survival for women with ovarian cancer (3–9).
As a master regulator of several cellular processes including proliferation, apoptosis, and immune response, the NF-κB signaling network has a central role in cancer progression (10,11) and contributes to metastasis and poor outcomes in ovarian cancer (12–15). NF-κB follows classical (canonical) or alternative (non-canonical) signaling pathways leading to activation of either p65(RelA):p50 or RelB:52 NF-κB heterodimers, respectively. Whereas induction of NF-κB activity through the classical pathway is rapid, transient, and typically a consequence of inflammatory cytokines such as TNF-α, IL-1β, IL-6, and IL-8, alternative pathway signaling promotes more delayed and persistent NF-κB activity in response to cytokines such as LT-β, TWEAK, CD40, BAFF, and RANK (16,17). Although classical NF-κB signaling has been observed previously in ovarian cancers, involvement of NF-κB through the alternative pathway has been investigated only recently (17–20). For example, one recent study examining 196 ovarian cancer tissues found that p52 expression, independent of p65, is associated with significantly lower progression-free and overall survival (19).
Our previous work implicates alternative NF-κB in maintaining ovarian cancer CSCs phenotypes (18). Relative to 2-D monolayer cultured cells, 3-D cultured cells exhibit stem-like properties including enhanced tumor initiation and chemoresistance, elevated expression of SOX2, OCT4, and NANOG genes, and an enrichment of CD117, CD44, and CD133 surface markers and aldehyde-dehydrogenase (ALDH) activity (9,18,21). We found that these cells exhibit increased activation of both classical and alternative NF-κB pathways that promote either a proliferative or quiescent phenotype, respectively (18).
Given its ubiquitous activity and pleiotropic effects on a variety of normal cells, targeting NF-κB has remained clinically unfeasible. However, identifying and targeting specific inducers of this pathway is a promising approach. The upstream activators of alternative NF-κB signaling in ovarian cancer are unclear, but include a variety of factors from the inflammatory but immunosuppressive tumor microenvironment (TME) that characterizes this disease (14). To begin to elucidate these, we investigated the hypothesis that specific inducers of alternative NF-κB are enriched in the ovarian TME and contribute to disease progression and relapse through induction of NF-κB activity. We show that TNF-like weak inducer of apoptosis (TWEAK), a multifunctional cytokine important for tissue repair, is enriched following chemotherapy, induces activation of alternative NF-κB, enhances stem-like features, and contributes to relapse in an ovarian cancer mouse model. This study provides novel insight into NF-κB signaling and CSC biology and uncovers a potential new therapeutic target for inhibiting relapse in ovarian cancer.
MATERIALS AND METHODS
General Cell Culture Conditions
Cell line information presented in Supplemental Table 1. OVCAR8 and CAOV4 cells were obtained and authenticated from NCI-Frederick DCTD tumor/cell line repository. OV90 was obtained and authenticated by ATCC. De-identified ascites specimen VBCF004 was collected as pathological waste from post-treatment ovarian cancer patients and was determined to be outside the federal regulations for protection of human subjects [45 CFR 46 (OHSRP #12727 and #12797)] (18). Viable tumor cells and non-cell fractions from ascites were purified using Ficoll separation as previously reported (18). All adherent cultures were maintained in standard media (RPMI for OVCAR8, OV90, and CAOV4; DMEM:F12 for VBCF004), supplemented with 10% FBS and 1% 10,000 U/ml Pen/Strep. When using inducible CRISPR-Cas9 cell lines, tetracycline-free FBS was substituted for standard FBS. Cells are tested for mycoplasma annually. Cells were maintained in culture for a maximum of 15 passages.
Molecules
TWEAK (human and mouse), BAFF, CD40L, CRP, IL-1β, IL-6, IL-8, Leptin, TGFβ1, TGFβ3, and TNF-α cytokines were purchased from R & D Systems and reconstituted per manufacturer’s instructions (Table S1). Carboplatin for in vitro assays was reconstituted per manufacturer’s instructions (R & D Systems, Table S1). FN14 inhibitor L542-0366 was reconstituted in DMSO with sonication until dissolved per experiment (Millipore, Table S2).
NF-κB Luciferase Assay
The NF-κB Luciferase Cignal Lenti Reporter Assay was purchased from Qiagen, transduced per the manufacturer’s protocol at MOI of 50, selected with 2μg/ml puromycin, and expanded to generate stably expressing reporter cell lines. Cells were plated in 96-well, opaque white, clear-bottom plates at 1500-6000 cells/well depending on cell line with cytokines added the following day. The cells were allowed to incubate for 24 hours before analysis using the Promega ONE-Glo EX Luciferase Assay System per manufacturer’s protocol. Luminescence was read on a SpectraMax iD3 plate reader.
NF-κB Binding Assay
Cells were plated in 10 cm dishes and allowed to rest overnight before the addition of TWEAK for either 2 or 24 hours. NF-κB proteins were quantified in whole cell lysates using the TransAM® NF-κB Activation Assay from Active Motif per manufacturer’s instructions.
Western Blots
Cells were plated in culture dishes and allowed to rest overnight before treating. Whole cell lysates were collected using NP-40 cell lysis buffer with HALT protease and phosphatase inhibitor. Nuclear and cytosolic fractions were collected using Active Motif Nuclear Extract Kit per manufacturer’s instructions. Proteins were separated by SDS-PAGE using 4-12% Bis-Tris gels and MOPS running buffer and transferred to a PVDF membrane before blocking and incubation overnight at 4°C with primary antibodies (Table S2). Secondary antibodies were incubated for 1 hour at room temperature (Table S2). Bands were visualized on the Invitrogen iBright CL1000 Imaging System with analysis using the iBright Analysis Software.
Cell Viability Assay
Cells were seeded into 96-well, opaque white, clear-bottom plates and allowed to rest overnight before being treated with TWEAK and allowed to grow for 72 hours. Viability was assessed after 72 hours with CellTiter-Glo 2.0 reagent per manufacturer’s instructions. Luminescence was detected using a SpectraMax iD3 plate reader.
Proliferation
For Ki-67, cells were seeded into culture dishes and incubated overnight before treatment with TWEAK for 72 hours. Cells were then fixed in 1% formalin followed by Triton-X permeabilization. Cells were stained with primary antibody to Ki-67 followed by secondary antibody (Table S2) and quantified by flow cytometry.
For EdU, 500-1000 cells were seeded into black 96-well, clear-bottom plates overnight prior to treatment with TWEAK and EdU for 72 hours. EdU incorporation was assessed with the Click-iT Plus EdU Alexa Fluor 647 assay per manufacturer’s instructions. Hoechst 33342 and EdU incorporation were imaged on the ImageXpress Pico plate imager and analyzed with CellReporterXpress software (Table S2).
TUNEL Assay
2500-4000 cells were seeded into black 96-well, clear-bottom plates and incubated overnight prior to treatment with either vehicle, TWEAK, or carboplatin and allowed to incubate for 6 hours. EdUTP incorporation was assessed with the Click-iT Plus TUNEL Alexa Fluor 488 assay per manufacturer’s instructions. Hoechst 33342 and EdUTP incorporation were imaged on the Molecular Devices ImageXpress Pico plate imager and analyzed with CellReporterXpress software. Percent EdUTP incorporation was quantified blindly using automated imaging analysis with CellProfiler for counting and scoring (22).
Caspase Assay
Cells were plated into black 96-well, clear-bottom plates and incubated overnight prior to treatment with either vehicle, TWEAK, or carboplatin and incubated for 72 hours. Caspase-3/7 activity was assessed using the Invitrogen CellEvent Caspase-3/7 Green Detection Reagent per manufacturer’s instructions (Table S2). Hoechst 33342 and caspase-3/7 cleavage was imaged on the Molecular Devices ImageXpress Pico plate imager and analyzed with CellReporterXpress software.
RNA Extraction and Quantitative Reverse Transcription PCR (qRT-PCR)
Total RNA was isolated using the NucleoSpin RNA Plus kit per manufacturer’s instructions. cDNA synthesis was carried out using the High-Capacity cDNA Reverse Transcription kit per manufacturer’s protocol. Quantitation and normalization of gene expression was performed using Taqman Fast Advanced Master Mix and Taqman probes (Table S2). Experiments were run on a QuantStudio 3 instrument and analyzed with the QuantStudio Design and Analysis software using the delta-delta Ct method with GAPDH endogenous control.
Spheroid Formation Assay
Cells were plated at 100-500 cells/well in ultra-low attachment, flat bottom 96-well plates cultured in RPMI media supplemented with 5-10% FBS, 1% 10,000 U/ml Pen/Strep, and either TWEAK or vehicle. Spheroids were allowed to grow for 4–7 days, with TWEAK supplementation every 3-4 days, followed by staining with Hoechst 33342. Spheroids were imaged using the ImageXpress Pico and size 50–500 μm were counted using the CellReporterXpress software (Table S2). Spheroid efficiency is defined as (# of spheroids)/(# of cells per well).
Pulse Chase Assay
Cells were plated at 500-3000 cells per well in a 96-well, black well, clear-bottom plate and allowed to rest overnight. For the pulse, cells were grown for at least two divisions in 0.5-1μM EdU prior to the chase, in which cells were removed from EdU and allowed to divide in the presence of either TWEAK or vehicle for 60-72 hours. EdU incorporation was assessed with the Click-iT Plus EdU Alexa Fluor 647 assay and F-actin was also assessed using Phalloidin-Alexa Fluor 488, both used per manufacturer’s instructions. Hoechst 33342 and EdU incorporation or F-actin staining were imaged on the Molecular Devices ImageXpress Pico plate imager and analyzed with CellReporterXpress software (Table S2).
siRNA Transfections
Cells were plated the day before to achieve a confluency of around 60% for transfection. 30nM pools of 4 siRNAs targeting either FN14 or a non-targeting control were transfected using Lipofectamine RNAiMAX per manufacturer’s instructions (Table S2). Cells were incubated for 24 hours before being collected for downstream experiments. Cells were replated and cultured for an additional 48 hours for validation of Fn14 silencing.
CRISPR Cell Line Production
Knockout clonal cell lines for RelB were derived per the manufacturer’s protocol for Horizon’s Edit-R Inducible Lentiviral Cas9 Nuclease with Edit-R Lentiviral sgRNA (Table S2). 10μg/ml blasticidin and 2μg/ml puromycin was used to select for cells with the integrated Cas9 nuclease and sgRNA guides followed by induction of genomic editing with 3μg/ml doxycycline for 7 days. Serially diluted clonal lines were expanded and screened for the loss of RelB by western blot.
Flow Cytometry and CD117 Sorting
Flow cytometry experiments were completed as previously described (9) (Table S2). For sorting experiments, CAOV4 cells were seeded for 6 days in Ultra Low-Attachment T-75cm2 flasks with stem cell media (DMEM/F12, 1% Knock-out serum replacement, 0.4% BSA, 0.1% Insulin-Transferrin-Selenium-X (ITS), 1% 10,000 U/ml Pen/Strep and supplemented every 2-3 days with 20ng/ml EGF and 10ng/ml FGF). After 6 days, cells were collected and prepared into single cell suspensions using CellStripper Dissociation Reagent and needle dissociation using 25-gauge needle. Cells were stained for CD117 or propidium iodide (PI), sorted for CD117− and/or CD117+ cells using BD FACSMelody™ Cell Sorter, and then plated for 24 hours prior to treatment with either vehicle, TWEAK, Carboplatin, or TWEAK and Carboplatin (Table S2). For assessing gene expression, CD117+ or CD117− cells were harvested after 72 hours of treatment with TWEAK and immediately processed for downstream qRT-PCR. For assessing viability, CD117+ cells were harvested after 120 hours and processed for the cell viability assay as described above. 120-hour treatments consisted of 72 hours carboplatin followed by 48 hours vehicle; 72 hours carboplatin and TWEAK followed by 48 hours vehicle; 48 hours TWEAK followed by 72 hours carboplatin; or 72 hours carboplatin followed by 48 hours TWEAK. For assessing marker expression of CD117 in sorted cells, either mock sorted or CD117− cells were cultured for 14 days with two doses of sequential treatments of vehicle or carboplatin and TWEAK, as previously reported (9). Briefly, cells received an initial treatment of carboplatin and TWEAK for 24 hours with subsequent two-fold dilution with media.
To assess CD117 growth characteristics and chemoresistance, OV90 cells were enriched for CD117+ and CD117− using the Miltenyi Biotec CD117 microbead kit and the quadroMACS separator with LS columns. Separated cell populations were either plated for cell viability assay or for spheroid formation assay as described above.
Animal Experiments
All animal studies were approved by the SDSU Animal Care and Use Committee (protocol approval numbers 18-04-006H and 21-05-003H). Power analysis indicate using 6-8 mice/grp to achieve a conservative effect size of 0.4. For subcutaneous xenografts, 50,000 - 500,000 OV90 cells in 1:1 Matrigel in PBS were subcutaneously injected into the left flank of 8-week-old female athymic Nu/Nu mice. Mice were weighed and tumors measured with blinded groups twice weekly. Once tumors reached 150mm3, mice were treated with either vehicle or carboplatin (50mg/kg) delivered via intraperitoneal (IP) injection once per week for three weeks. Mice were sacrificed in two groups: on average 5 days (group 1) or 12 days (group 2) after the third dose of carboplatin to evaluate progressive changes.
For IP xenografts, 4x106 CAOV4 cells in 500μl PBS were injected into 8 week-old female athymic Nu/Nu mice. Mice were weighed and monitored for clinical signs twice weekly. Tumor formation was assessed by palpation and visual inspection of the abdomen. 7 days post injection, mice were randomly assigned to either group 1: vehicle, group 2: carboplatin (50mg/kg) IP once per week for three weeks, group 3: carboplatin (as administered in group 2) combined with FN14 inhibitor (9mg/kg) in 5% DMSO/95% Corn oil delivered by IP every other day for three weeks, or group 4: carboplatin (as administered in group 2) followed by FN14 inhibitor (as administered in group 3) for 6 weeks or until humane endpoints. Tumor burden for mice was assessed after necropsy with collection of peritoneal wall, spleen, liver, omentum, spleen, mesentery, ovaries, and diaphragm. To avoid endpoint bias, mice were blinded to treatment groups, randomized to separate cages, weighed twice weekly, and assessed for survival endpoints four times a week by the same observer.
Enzyme-Linked Immunosorbent Assay (ELISA)
The concentration of cytokines IL-6 and TWEAK (Table S2) was assessed using DuoSet sandwich ELISA per manufacturer’s instruction using non-cell fractions from ascites samples and tumor tissue samples which were flash frozen immediately following collection. For the tissue, 1-3mg sections were dissected, weighed, and protein was dissociated on gentleMACS M tubes in RIPA lysis buffer. Samples were prepared and analyzed per manufacturer’s instructions.
Immunohistochemistry (IHC) and Immunofluorescence (IF)
For IHC, tumors were resected, fixed in 10% neutral buffered formalin, and stored in 70% ethanol before processing. Tumors were embedded in paraffin and sectioned at 5μm. Antigen retrieval was performed in citrate buffer and quenched with hydrogen peroxide. Slides were incubated with primary antibodies overnight at 4°C followed by an HRP-linked secondary for 1 hour at room temperature and processed using DAB (Table S2). Four randomly selected, blinded images per slide were acquired with an Olympus BX51 with CellSens Standard software. A digital quantification of DAB staining was performed using ImageJ with a FIJI deconvolution package as described previously (9).
For IF, tumors were fixed, embedded and antigen retrieval was done as above. Slides were blocked with 1% goat serum in PBS and then incubated with RelB-FITC primary antibody overnight (Table S2). Slides were mounted with Fluoroshield antifade medium with DAPI and four randomly selected, blinded images per slide were acquired within 1-3 days with an Olympus BX63 with CellSens Standard software. Exposure time and beam intensity were kept consistent. Nuclear and cytosolic digital quantification of IF staining was performed blinded using automated imaging analysis with CellProfiler (22) and modeled with image set BBC014v1 (23). Nuclear:Cytosolic ratio was calculated as described previously (24).
Public Database Analysis
Public databases were queried as clarified (Table S3).
Statistical Analysis
Statistics were generated using Prism 9.3.1 with data acquired from at least three independent biological replicates. Results are presented as mean +/− SEM. Significance was calculated using either student’s t-test for comparisons of two means or ANOVA for comparisons of three or more means with a post-hoc test to identify differences between groups as described in figure legends. Differences between means are considered statistically significant at the 95% level (p < 0.05). Statistics were completed as described here or as otherwise noted in the figure legends.
Data Availability
The data generated in this study are available upon request from the corresponding author.
RESULTS
To identify factors that sufficiently activate NF-κB in ovarian cancer we first exposed three different HGSOC ovarian cancer cell lines, including primary tumor-derived line OVCAR8, metastatic site-derived CAOV4, and ascites-derived OV90 (25,26) (Table S1), each stably expressing an NF-κB luciferase reporter to increasing concentrations of known NF-κB inducers. We found consistent elevated signaling in response to TNF-α and TWEAK across all three cell lines tested (Figure 1A). TNF-α-induced activation of classical NF-κB has been shown in several studies to contribute to ovarian cancer progression and metastasis (27–29), however few studies to date have investigated the role of TWEAK in this disease (30,31). To expand on these findings, we next performed an NF-κB DNA binding assay to determine the relative abundance of activated NF-κB transcription factors in nuclear lysates collected after 2 and 24 hours of TWEAK stimulation. All three cell lines tested show significantly enhanced activation of both classical (p50 and p65) and alternative (p52 and RelB) NF-κB proteins after 24 hour stimulation relative to vehicle; however, activation of RelB is highest across the three cell lines tested, relative to the other NF-κB subunits (Figure 1B). To lend further support to these findings we performed a western blot on nuclear and cytosolic lysates collected from ovarian cancer cell lines treated with TWEAK for 2 or 24 hours. TWEAK stimulation leads to strong nuclear localization of alternative NF-κB subunit factors p52 and RelB (Figure 1C). RelA nuclear localization was minimally increased with TWEAK stimulation. Similar results were found in a primary cell line (VBCF004) derived from the ascites of a HGSOC patient (Figure 1D). Together these data suggest TWEAK is a strong inducer of alternative NF-κB activation that can also induce some classical NF-κB activity in ovarian cancer cells.
Figure 1. TWEAK activates classical and alternative NF-κB pathways in ovarian cancer cell lines and patient-derived cells.
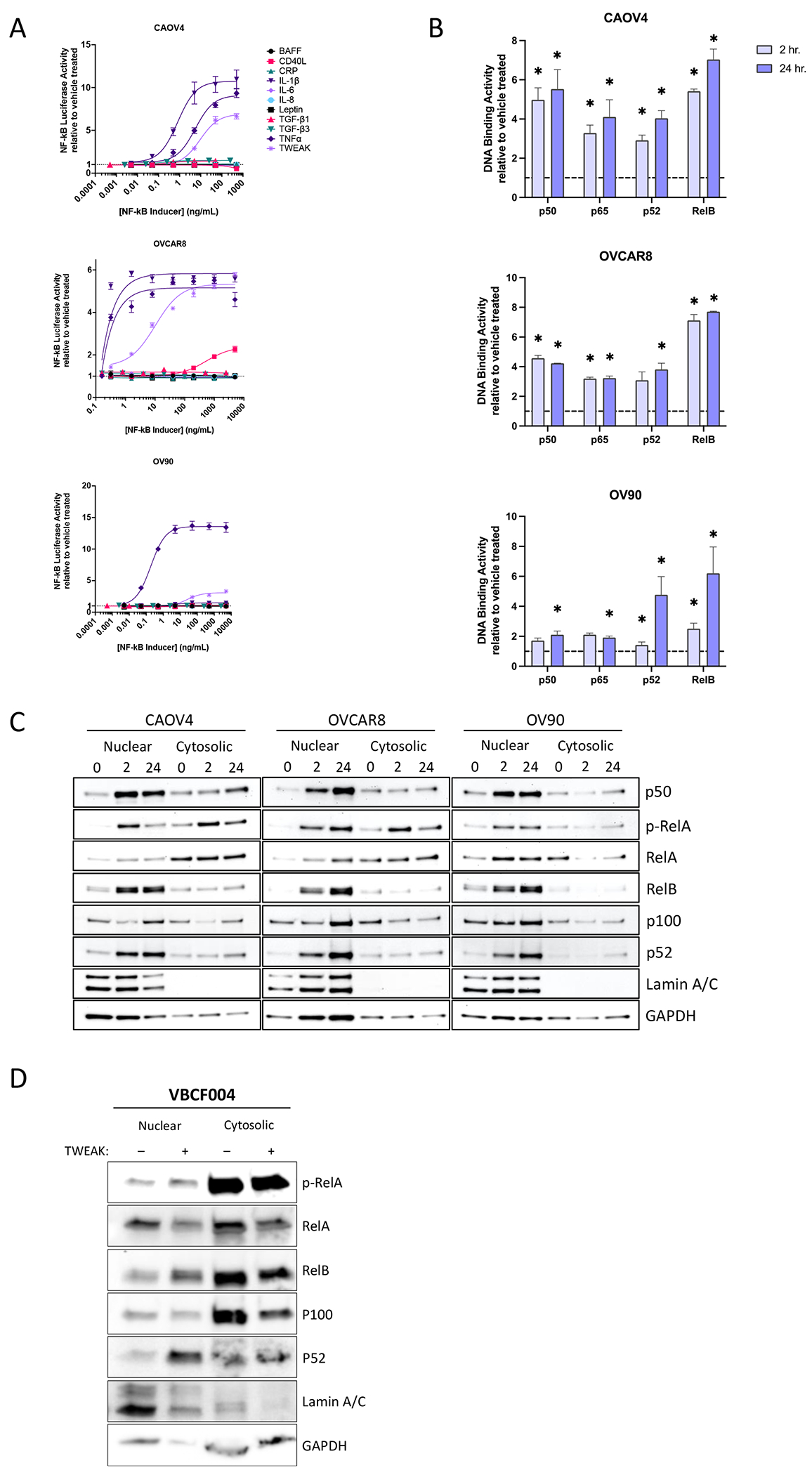
(A) Luciferase reporter assay showing NF-κB activity in response to 24 hour stimulation with different cytokines (n=3). (B) DNA-binding of NF-κB proteins with TWEAK stimulation (25ng/ml) for 2 or 24 hours relative to vehicle. One-way ANOVA with Dunnett’s post-hoc (n=3). (C) Nuclear and Cytosolic western blots of NF-κB proteins with TWEAK (25ng/ml, 0, 2, or 24 hours) in CAOV4, OVCAR8, and OV90. (D) Nuclear and Cytosolic western blots of NF-κB proteins with TWEAK (100ng/ml, 24 hours) stimulation in ascites sample VBCF004. Data represent mean and SEM. * p<0.05.
We next investigated the significance of TWEAK in tumor samples from ovarian cancer patients and mouse models of ovarian cancer. We found that soluble TWEAK in ascites derived from HGSOC patients with recurrent, chemoresistant disease had an average concentration of 4241pg/ml (288-14798pg/ml) and, while lower, is analogous to the average IL-6 concentration of 7677pg/ml (2527-12427pg/ml) (Figure 2A). IL-6 is a cytokine shown to be important and present at high levels in ovarian cancer progression and relapse (32,33). These data suggest TWEAK is an appreciable component of the ovarian TME.
Figure 2. Following chemotherapy, TWEAK and its receptor, Fn14, are significantly elevated in patient samples and mouse tumors.
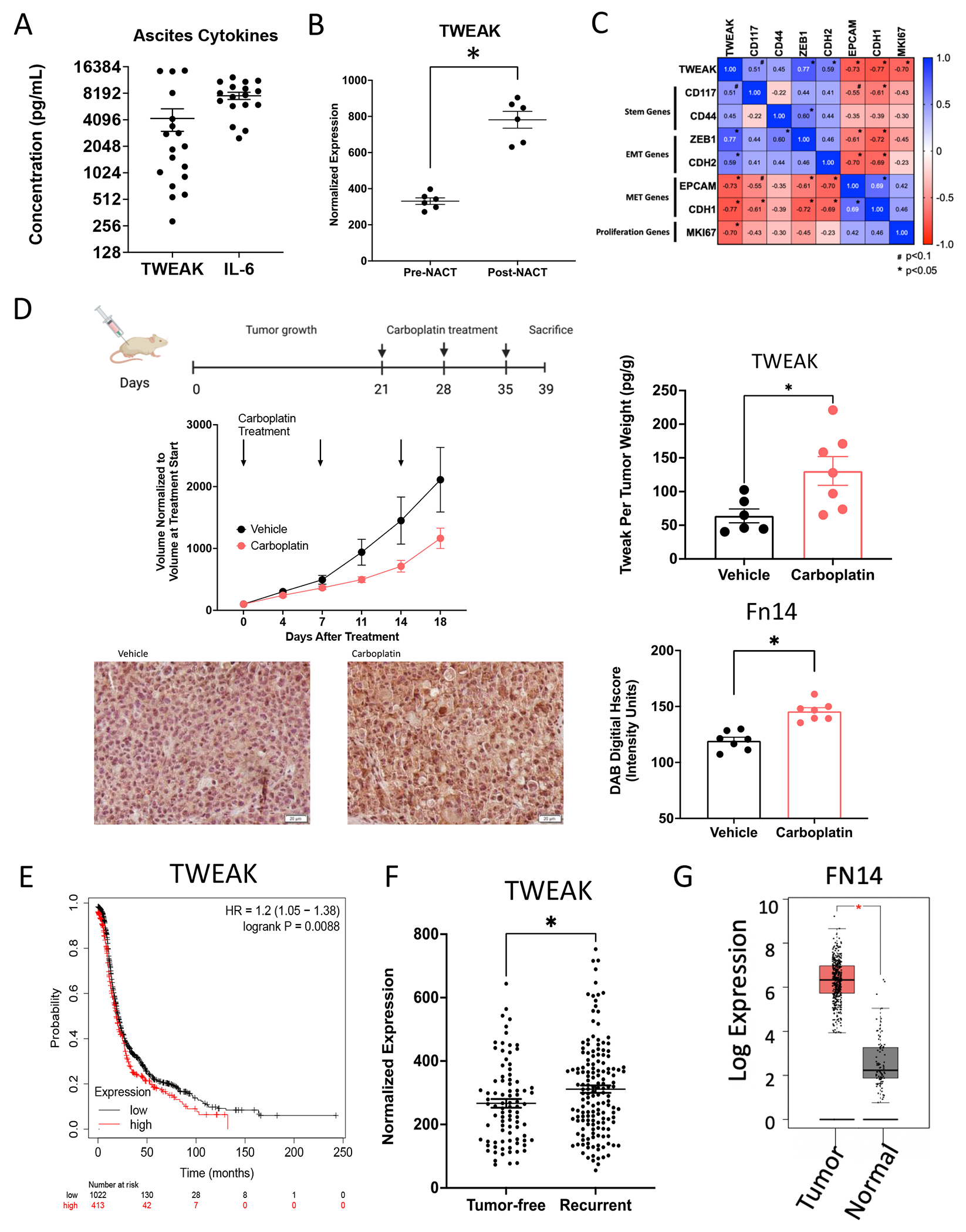
(A) TWEAK and IL-6 concentration in noncellular ascites fraction collected from recurrent HGSOC patients (n=18). (B) TWEAK gene expression in ovarian cancer tumors collected pre- and post- neoadjuvant chemotherapy (NACT). Paired t-test (n=6). (C) Pearson’s correlation of select genes to TWEAK expression for all collected samples (pre- and post-NACT) (n=6). (D) Subcutaneous tumors were generated in nude mice using OV90 cells. Tumors were allowed to grow for 21 days followed by 3 cycles of carboplatin (50mg/kg) or vehicle and resected 5 days after the final carboplatin treatment. Experimental design created with BioRender.com (top). Tumor volume was measured twice weekly and graphed normalized to the volume at treatment start (center, left). TWEAK concentration per tumor weight measured using ELISA (center, right). Unpaired t-test (n=6,7). Fixed tumor fractions were histologically stained for Fn14. Representative images (bottom, left) and Fn14 digital Hscore quantification using ImageJ (bottom, right). Unpaired t-test (n=7,7). (E) Kaplan-Meier plots from KMPlotter comparing high expression (red) and low expression (black) of TWEAK and associated progression free survival (PFS) of ovarian cancer patients (n=1435). (F) TCGA analysis of TWEAK in tumors from HGSOC patients that recurred (tumor <5 years after follow-up) or remained tumor-free. Unpaired t-test (n=585). (G) FN14 gene expression by Gepia2 of normal ovarian surface epithelium (black) and ovarian tumor (red). Unpaired t-test (n=88 normal, n=426 tumor). Data represent mean and SEM. * p<0.05.
To establish TWEAK expression in clinical samples, we assessed TWEAK transcripts in tumor samples derived from HGSOC patients undergoing neoadjuvant chemotherapy (NACT) (33). Post-NACT tumor samples had significantly higher levels of TWEAK mRNA relative to patient matched pre-NACT tumor samples (Figure 2B). These findings were confirmed using another recently published dataset (Supplemental Figure 1A, method in Supplemental Table 3) (34). Further analysis of these samples shows that TWEAK mRNA levels positively correlate with CSC marker CD117, and the epithelial-to-mesenchymal transition (EMT) markers, ZEB1 and CDH2 (N-cadherin). Conversely, TWEAK mRNA negatively correlates with epithelial marker genes EPCAM and CDH1 (E-cadherin), and the proliferation marker MKI67 (Ki-67) (Figure 2C).
Given its established role in tissue repair and its elevated expression post-NACT, we reasoned that TWEAK levels might be higher in cytotoxic chemotherapy-treated tumors relative to vehicle-treated tumors. Indeed, subcutaneous xenograft tumors resected from mice receiving three cycles of carboplatin chemotherapy exhibited higher levels of soluble TWEAK relative to mice receiving vehicle (Figure 2D). Moreover, fixed tumors resected from chemotherapy-treated mice showed significantly higher expression of the TWEAK receptor, Fn14, relative to tumors from vehicle-treated mice (Figure 2D). Altogether these findings support the notion that TWEAK is present in the ovarian TME, that it correlates with the expression of stem and EMT genes, and that it is elevated after chemotherapy treatment.
Analysis of ovarian cancer datasets, including TCGA, revealed a significant decrease in progression-free and overall survival of HGSOC patients whose tumors have high expression of TWEAK transcripts (Figure 2E, Supplemental Figure 1B–C). Moreover, tumors from TCGA patients that had a recurrence had significantly higher levels of TWEAK transcripts relative to patients that remained tumor-free (Figure 2F). Given the specificity of TWEAK for its receptor, Fn14 (35), we confirmed the elevated expression of FN14 transcripts in ovarian cancer tissues relative to normal ovarian tissue (Figure 2G). Datasets, including TCGA, further show that FN14 expression is associated with a decrease in overall survival (Supplemental Figure 1D). We confirmed that TWEAK and FN14 were not comparatively high in normal ovarian tissues nor in our cell lines (Supplemental Figure 1E–F). We then confirmed that Fn14 was present on ovarian cancer cells by flow cytometry (Supplemental Figure 1G), and that TWEAK gene expression is not changed in ovarian cancer cells after carboplatin treatment (Supplemental Figure 1H), suggesting TWEAK is produced from other cells in the tumor microenvironment.
We next conducted several experiments to examine the effects of TWEAK stimulation on tumor cells. Exogenous TWEAK stimulation for 72 hours had minimal effects on viability (Supplemental Figure 2A) or proliferation as measured by Ki-67 staining (Supplemental Figure 2B) or EdU incorporation (Supplemental Figure 2C). Only the CAOV4 cell line exhibited a small but significant decrease in viability and proliferation when stimulated with TWEAK. Likewise, there was no significant effect on apoptosis when cells were treated with TWEAK relative to vehicle-treated cells as demonstrated by TUNEL assay (Supplemental Figure 2D) and caspase activity assay (Supplemental Figure 2E). By contrast carboplatin significantly induced apoptosis in these cells. Given these findings and its ability to activate alternative NF-κB signaling, its correlation with stem cell marker gene expression, and its enrichment with chemotherapy, we hypothesized that TWEAK may contribute to phenotypes associated with CSCs.
We have previously shown that spheroid formation was at least partially dependent on NF-κB transcription factors (18), so we tested whether TWEAK stimulation enhances the ability of ovarian cancer cells to form spheroids and asymmetrically divide. Because these processes largely depend on activation of EMT genes and stem cell transcription factors that support long term self-renewal and asymmetric division, we first examined transcript levels of relevant genes after TWEAK stimulation (Figure 3A–B). TWEAK induced a significant increase in expression of NF-κB inducing kinase (NIK), a critical kinase in the alternative NF-κB signaling pathway, in all cells. Expression of the EMT gene Vimentin (VIM) also significantly increased in all cell lines tested, while ZEB1 increased in CAOV4 and OVCAR8. The epithelial marker gene CDH1 decreased in OVCAR8 and VBCF004. There was no significant difference in OCT4 or NANOG expression with TWEAK stimulation in any of the cell lines, however SOX2 was upregulated in CAOV4 and OVCAR8.
Figure 3. TWEAK stimulation enhances the development of stem-like features in ovarian cancer cell lines and patient-derived cells.
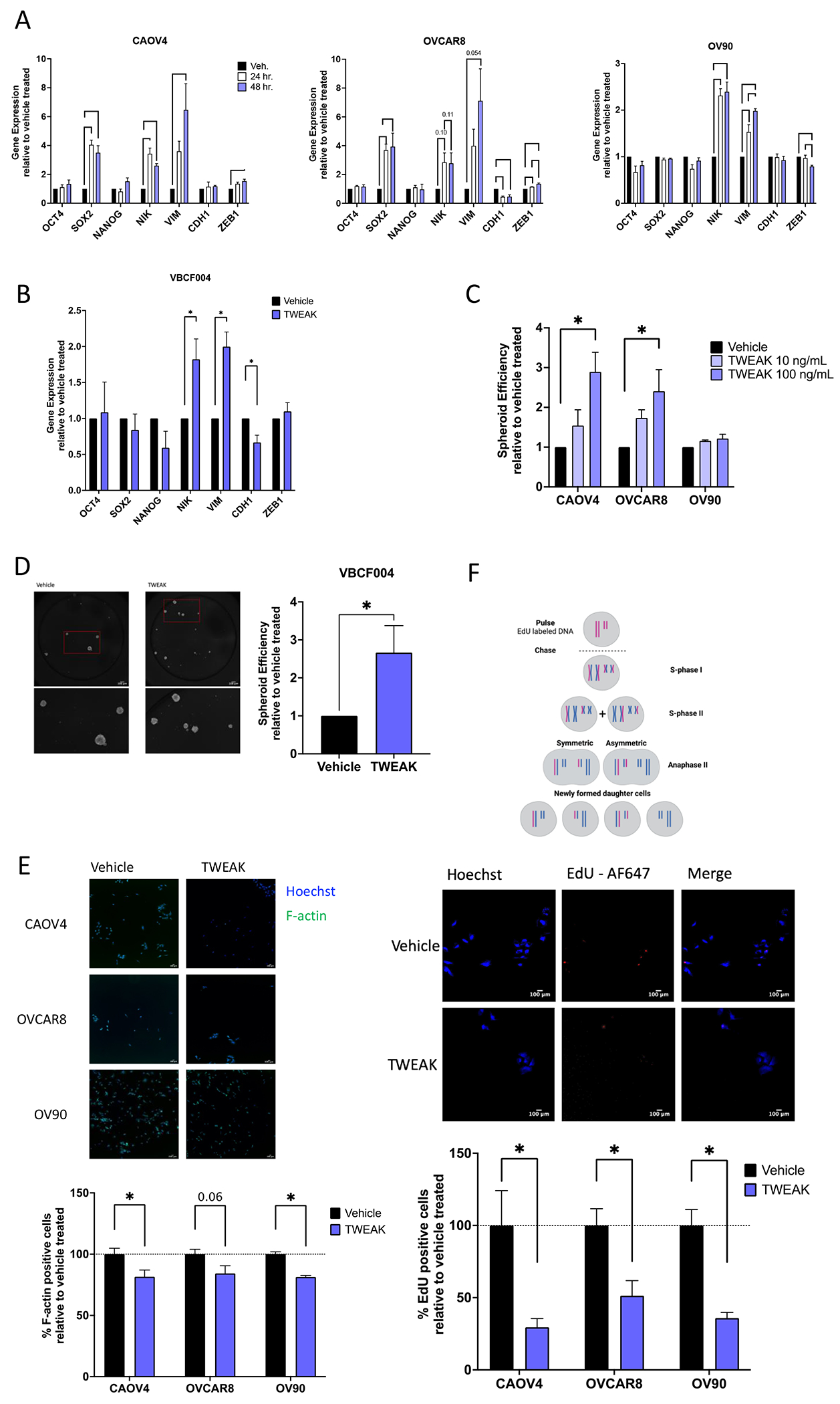
(A) qRT-PCR for select stemness and EMT genes in HGSOC cells with vehicle or TWEAK treatment (100ng/ml) for 24 or 48 hours. One-way ANOVA with Tukey’s post-hoc test (n=3). (B) qRT-PCR on ascites-derived cells VBCF004 for select stemness and EMT genes with TWEAK treatment (100ng/ml) for 48 hours. Unpaired t-test (n=3). (C) Spheroid efficiency of HGSOC cell lines with TWEAK treatment (10 or 100ng/ml) relative to vehicle for 6 days. One-way ANOVA, Dunnett’s post-hoc test (n=4). (D) Spheroid efficiency of ascites derived cells VBCF004 with TWEAK treatment (100ng/ml) relative to vehicle for 4 days. Representative images (left) and quantification with Hoechst (right). Unpaired t-test (n=3). (E) F-actin staining of HGSOC cells with TWEAK (100ng/ml) treatment for 72 hours. Representative images (top) and quantification per well relative to vehicle treated (bottom). Unpaired t-test (n=6). (F) EdU incorporation pulse-chase assay of HGSOC cells with TWEAK (100ng/ml) treatment for 72 hours. Experimental Design created with BioRender.com (top), representative images (center) and quantification per well relative to vehicle treated (bottom). Unpaired t-test (n=6). Data represent mean and SEM. * p<0.05.
We then examined spheroid formation and asymmetric division. Relative to vehicle, TWEAK significantly enhanced spheroid formation efficiency in CAOV4 and OVCAR8 cells, but not in OV90 cells (Figure 3C). TWEAK treatment also promoted spheroid formation in the primary cells (Figure 3D). There was a significant decrease in F-actin staining, a factor that increases during cell differentiation (36,37) (Figure 3E) and a corresponding decrease in symmetric division after TWEAK stimulation relative to vehicle (Figure 3F). These data indicate that TWEAK can induce expression of genes associated with stemness and/or EMT and can promote spheroid formation and asymmetric division in a variety of ovarian cancer cells.
To better characterize the effects of TWEAK-Fn14 signaling on CSC populations we examined ovarian cancer cells expressing known markers of ovarian CSCs (CD117, CD133, and ALDH) with TWEAK stimulation in the presence or absence of chemotherapy. Relative to vehicle, there was no significant difference in the percentage of cells expressing traditional CSC markers when stimulated with TWEAK, with the exception of OV90 which showed an increase of cells with ALDH activity (Figure 4A). As expected, with carboplatin treatment there was a significant enrichment of CD117+ and/or CD133+ cells in the CAOV4 and OVCAR8 cell lines. In agreement with our previous findings, we observe that CD133+ALDH+ double-positive cells are enriched in OV90 cells after chemotherapy treatment (9). We have shown in other studies that CAOV4 and OVCAR8 do not express sufficient CD133+ALDH+ cells (9,25). Interestingly, carboplatin combined with TWEAK led to the largest enrichment of CD117 and CD133 expressing cells in two of the three lines tested. Notably, the combination treatment led to an almost complete elimination of CD117− cells, as CD117+ cells made up 65-85% of the surviving population in CAOV4 and OVCAR8 cells (Figure 4A). We examined gene expression changes in cells undergoing these treatments and found that while TWEAK stimulation alone enhances expression of SOX2, NIK, and VIM genes, combining it with chemotherapy leads to the largest increase in expression of these genes (Figure 4B). This treatment combination similarly led to the greatest increase in spheroid development (Figure 4C), in agreement with other studies (38–41), presumably due to enrichment of CSCs. Interestingly, OV90 cells do not respond to TWEAK as strongly as the other cell lines and this is likely due the high endogenous levels of SOX2 previously established in this line (9) and the relatively low level of Fn14 expression we discover in this study. Taken together these data suggest that TWEAK enhances either the development of new CSCs during chemotherapy, or it improves the ability of carboplatin to eliminate non-CSCs. Either scenario would lead to an enrichment of CSCs after chemotherapy.
Figure 4. TWEAK in combination with carboplatin enriches for CSCs and eliminates non-CSCs.
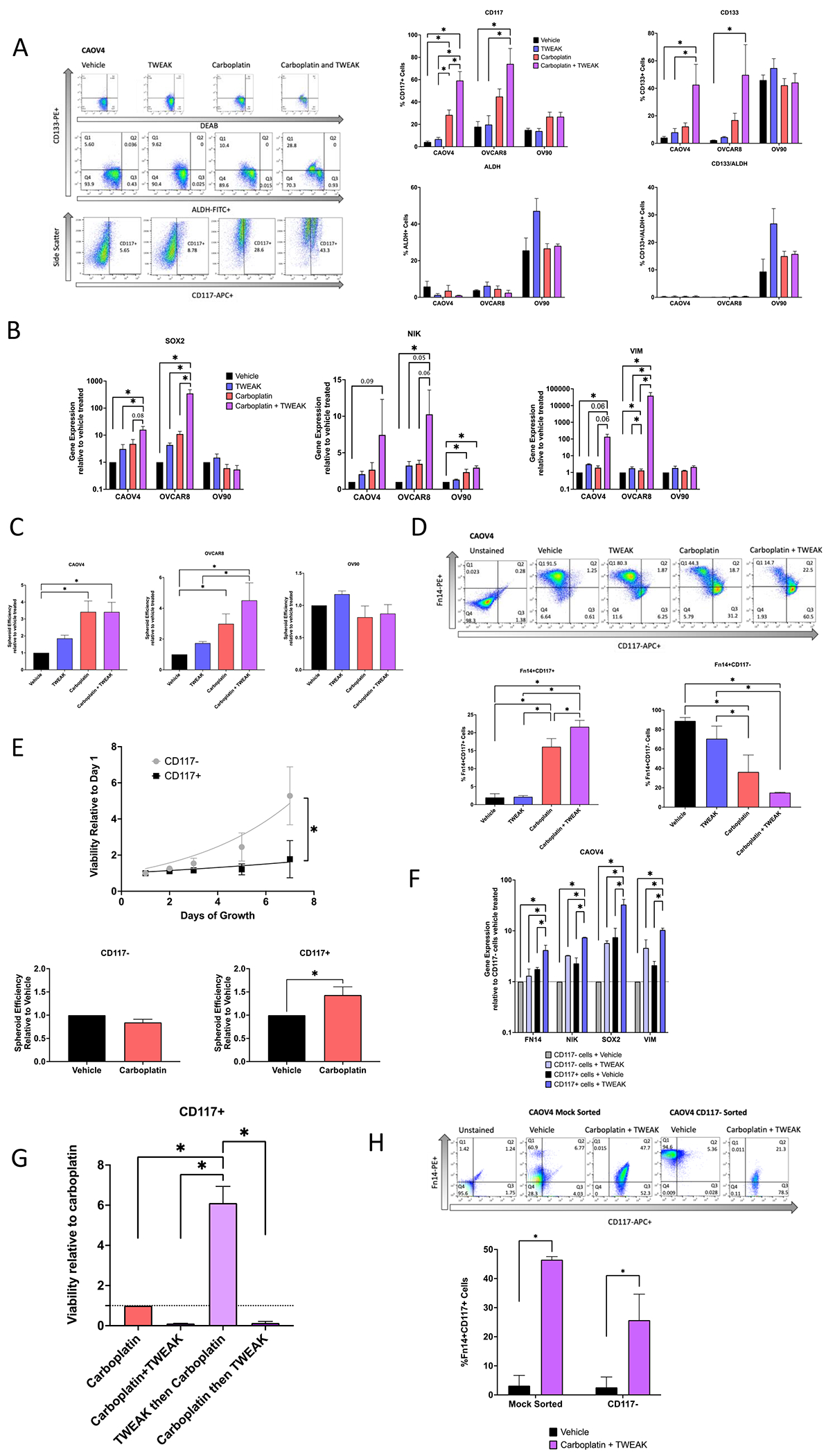
(A) Stem cell marker flow cytometry of HGSOC cells treated with vehicle, TWEAK (100ng/ml), carboplatin (125μM), or TWEAK and carboplatin for 72 hours. Representative gating for CAOV4 (left), percent positive cells representing mean plus SD (right). One-way ANOVA, Tukey’s post-hoc test (n=3). (B) qRT-PCR for select stemness genes in HGSOC cells with vehicle, TWEAK (100ng/ml), carboplatin (125μM), or TWEAK and carboplatin for 72 hours represented relative to vehicle. One-way ANOVA, Tukey’s post-hoc test (n=3). (C) Spheroid efficiency of HGSOC cells treated with vehicle, TWEAK (100ng/ml), carboplatin (125μM), or TWEAK and carboplatin for 4 days represented relative to vehicle. One-way ANOVA, Tukey’s post-hoc test (n=4). (D) Flow cytometry analysis of CD117 expression on Fn14+ cells treated with vehicle, TWEAK (100ng/ml), carboplatin (125μM), or TWEAK and carboplatin for 72 hours. Representative gating (left) and percent positive cells representing mean plus SD (right). One-way ANOVA, Tukey’s post-hoc test (n=3). (E) Viability of sorted OV90 CD117+ or CD117− cells grown for 8 days normal media (top). Two-way repeated measures ANOVA, Sidak’s post-hoc test (n=4). Spheroid efficiency of OV90 CD117+ or CD117− cells treated with carboplatin (90μM) for 4 days represented relative to vehicle (bottom). Unpaired t-test (n=4). (F) qRT-PCR of select genes for sorted CD117+ or CD117− cells treated with vehicle or TWEAK (100ng/ml) for 72 hours. One-way ANOVA, Tukey’s post-hoc test (n=4). (G) Viability of CD117+ sorted CAOV4 cells treated with either Carboplatin (125μM), Carboplatin and TWEAK (100ng/ml), TWEAK then Carboplatin, and Carboplatin then TWEAK for 120 hours. One-way ANOVA, Tukey’s post-hoc test (n=3). (H) Flow cytometry analysis of CD117 and Fn14 expression CAOV4 cells mock sorted or CD117− sorted cells treated with vehicle or TWEAK (100ng/ml) and carboplatin (30μM) for two cycles of 7 days, where initial treatment is for 24 hours with subsequent two-fold dilution with media. Representative gating (top) and percent Fn14+CD117+ cells representing mean plus SD. Two-way ANOVA, Sidak’s post-hoc test (n=4 mock, 5 CD117− sorted). Data represent mean and SEM. * p<0.05.
To further investigate this, we assessed changes in the percentage of cells expressing both the Fn14 receptor (Fn14+) and CD117 (CD117+) under these same treatment conditions. While there is a significant increase in the percentage of Fn14+CD117+ cells with TWEAK in combination with carboplatin relative to all other treatments, there is a significant decrease in the percentage of Fn14+CD117− cells under this treatment regimen (Figure 4D). These findings suggest that the effects of TWEAK on non-CSCs (CD117−) are different from those on CSCs (CD117+). As further validation of CD117 as an ovarian cancer CSC marker, we performed column separation of CD117+ and CD117− cells and assessed growth rates and spheroid chemosensitivity. CD117+ cells grew significantly more slowly when compared to CD117− cells, and CD117+ spheroids were chemoresistant to carboplatin treatment, while CD117− spheroids were somewhat more chemosensitive (Figure 4E). To explore the possibility that TWEAK could act differently on non-CSCs and CSCs, we performed FACS on CD117+ and CD117− cells and subsequently treated with TWEAK to evaluate changes in gene expression relative to vehicle. TWEAK treatment of CD117− cells enhances expression of FN14, NIK, SOX2, and VIM genes relative to vehicle-treated CD117− cells (Figure 4F). The levels of gene expression in CD117− cells treated with TWEAK are comparable to those endogenously expressed in vehicle-treated CD117+ cells. However, stimulation of CD117+ cells with TWEAK led to the highest expression of these genes, relative to vehicle-treated CD117+ cells.
To evaluate TWEAK-induced chemoresistance of CD117+ cells associated with these gene changes, we compared different delivery times of TWEAK treatment in a chemosensitivity assay (Figure 4G). We found that CD117+ cells treated with TWEAK before introduction of carboplatin had a 6-fold survival advantage, when compared to CD117+ carboplatin-treated cells, CD117+ cells treated with carboplatin and TWEAK at the same time, and CD117+ cells treated with TWEAK after carboplatin treatment. This suggests that TWEAK-induced gene changes can confer chemoresistance on CD117+ cells. To evaluate whether TWEAK and chemotherapy can promote the expression of CD117 on CD117− cells, we treated either mock sorted cells or CD117− sorted cells with two sequential treatments of serum concentration of carboplatin and TWEAK over 14 days, as we have done previously (9). Mock sorted cells recapitulated our findings in Figure 4D showing a significant increase in the percentage of Fn14+CD117+ cells when treated with TWEAK in combination with carboplatin (Figure 4H). CD117− sorted cells treated with TWEAK in combination with carboplatin significantly upregulated CD117 relative to vehicle treated cells. These experiments strongly support the idea that TWEAK acts on CD117+ cells to enhance chemoresistance and, when combined with chemotherapy, enhances the development of CD117+ cells from CD117− cells, both which would facilitate survival of CSCs during chemotherapy.
We next evaluated the specificity of TWEAK-Fn14 dependent differences in gene expression and spheroid formation. Expression of the Fn14 receptor is dramatically increased in CSC-enriching 3-D cultures, relative to 2-D monolayer cultures in CAOV4 and OVCAR8 cells (Figure 5A). We verified that this enriched expression was not due to fibroblast growth factor, a component of 3-D culture media (Figure 5B). Knockdown of the receptor using siRNA or FN14 inhibitor (L542-0366) eliminated the TWEAK induced NF-κB activation (Figure 5C–E), spheroid formation (Figure 5F), and SOX2 and NIK expression (Figure 5G), implicating TWEAK-Fn14 as a vital upstream contributor to the CSC phenotype observed in ovarian cancer cells. We also confirmed that the upregulation of SOX2 and NIK that was most enhanced with combination of carboplatin and TWEAK, was lost when the treatment included an FN14 inhibitor (Figure 5H).
Figure 5. TWEAK receptor Fn14 is required for TWEAK mediated stem-like features.
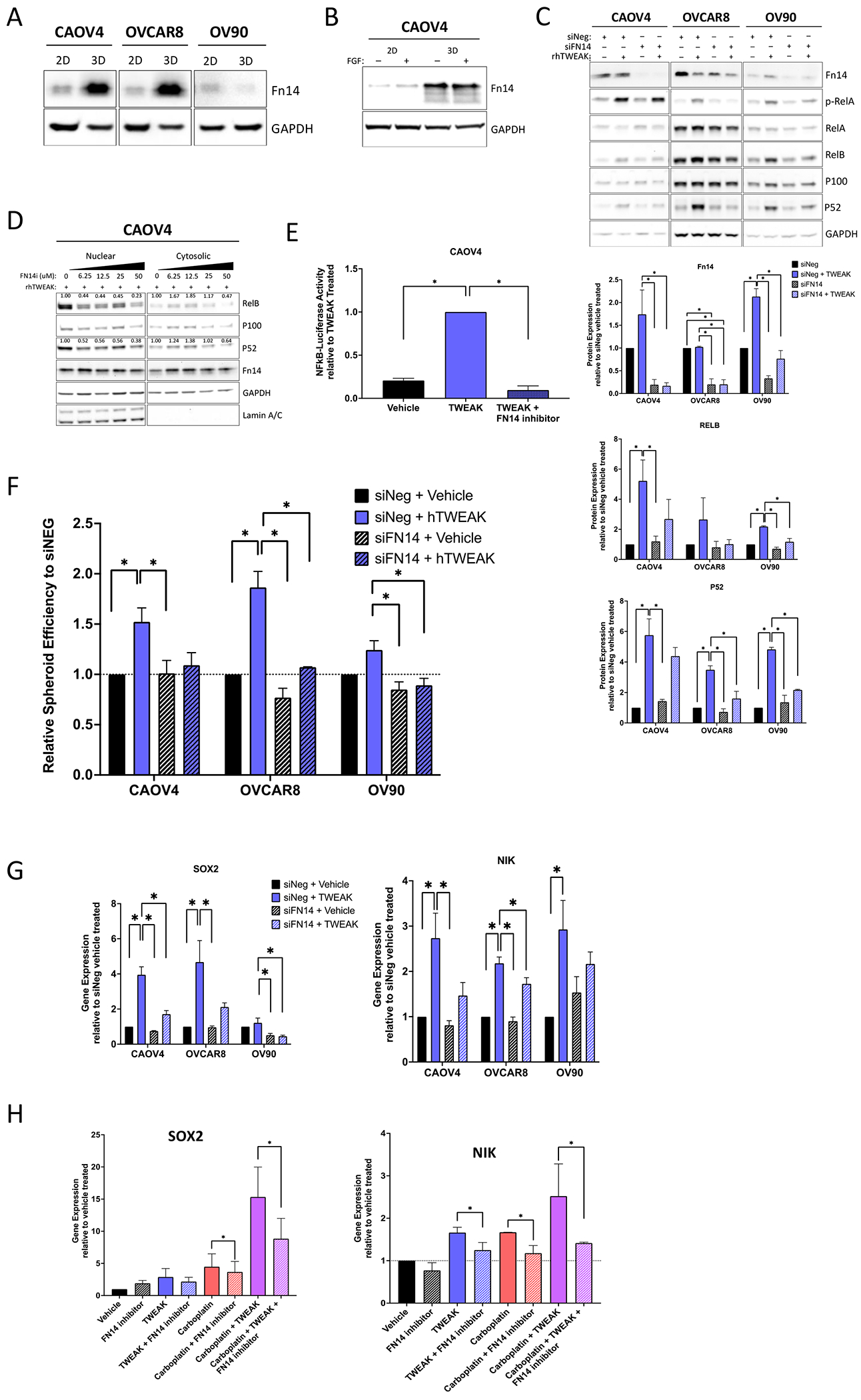
(A) Western blot for Fn14 from cells grown in standard adherent conditions or in spheroid conditions. (B) Western blot for Fn14 from cells grown in standard monolayer conditions or in spheroid suspension conditions with or without FGF. (C) Western blot of Fn14 and NF-κB proteins in siNeg or siFN14 HGSOC cells treated with vehicle or TWEAK (100ng/ml) for 72 hours. Representative image (top) and quantification of band intensities (bottom). One-way ANOVA, Tukey’s post-hoc test (n=3). (D) Nuclear and cytosolic western blots for indicated proteins in CAOV4 cells treated with TWEAK (100ng/ml), or TWEAK plus FN14 inhibitor (L542-0366, 6.25-50μM). Quantification values normalized to Lamin A/C for nuclear fraction and GAPDH for cytosolic fraction. (E) Luciferase reporter assay showing NF-κB activity is lost in response to TWEAK (100ng/ml) plus FN14i (L542-0366, 50μM) in CAOV4 cells (n=4). (F) Spheroid efficiency of siNeg or siFN14 HGSOC cells treated with vehicle or TWEAK (100ng/ml) for 4 days, represented relative to vehicle. One-way ANOVA, Tukey’s post-hoc test (n=4). (G) qRT-PCR for SOX2 or NIK in siNeg or siFN14 HGSOC cells treated with TWEAK (100ng/ml) for 48 hours, relative to vehicle. One-way ANOVA, Tukey’s post-hoc test (n=3). (H) qRT-PCR for SOX2 or NIK in CAOV4 cells treated with FN14 inhibitor (L542-0366, 50μM) for 72 hours in combination with vehicle, TWEAK (100ng/ml), carboplatin (125μM), or TWEAK and carboplatin conditions. Unpaired t-test for condition versus condition plus FN14 inhibitor (n=3). Data represent mean and SEM. * p<0.05.
We have previously shown that RelB expression is enhanced in ovarian cancer cells after chemotherapy or when grown in 3-D conditions (18). In this study, we discovered that Fn14 expression is also enhanced with chemotherapy and 3-D culture conditions (Figure 5A and 6A). Moreover, TWEAK treatment leads to higher expression of RelB relative to carboplatin (Figure 6A). To assess the delayed effect of TWEAK on downstream targets like CSC features and RelB in post-chemotherapy tumors, we resected xenograft tumors 12 days after three cycles of carboplatin were completed in a relapsing subcutaneous mouse model, in contrast to 5 days post-chemotherapy presented in Figure 2G. Soluble TWEAK remained high in chemotherapy-treated mice relative to mice receiving vehicle (Figure 6B). As expected, there was a significant enrichment for CD117+ expressing cells (Figure 6C), as well as RelB nuclear localization following chemotherapy (Figure 6D). To determine if the TWEAK-induced CSC features are dependent on RelB, we used CRISPR-Cas9 editing technology to knockout the RelB transcription factor in CAOV4 cells (Figure 6E). Loss of RelB prevented the upregulation of SOX2, VIM, and ZEB1 genes induced by TWEAK (Figure 6F). Moreover, the downregulation of CDH1 induced by TWEAK was partially rescued with knockout of RelB. Loss of RelB also diminished TWEAK-induced spheroid forming ability (Figure 6G) and asymmetric division (Figure 6H). The percentage of Fn14+CD117+ cells surviving carboplatin alone and carboplatin combined with TWEAK was significantly hampered with knockout of RelB (Figure 6I). Similarly, loss of RelB led to a significant enrichment in the percentage of Fn14+CD117− cells, indicating the diverse effects of TWEAK on CD117+ and CD117− cells are at least partially dependent on RelB. Thus, TWEAK may be a promising upstream target for inhibiting NF-κB dependent phenotypes, such as chemoresistance in CSCs.
Figure 6. RelB mediates TWEAK-Fn14 dependent enrichment of CSCs and elimination of non-CSCs.
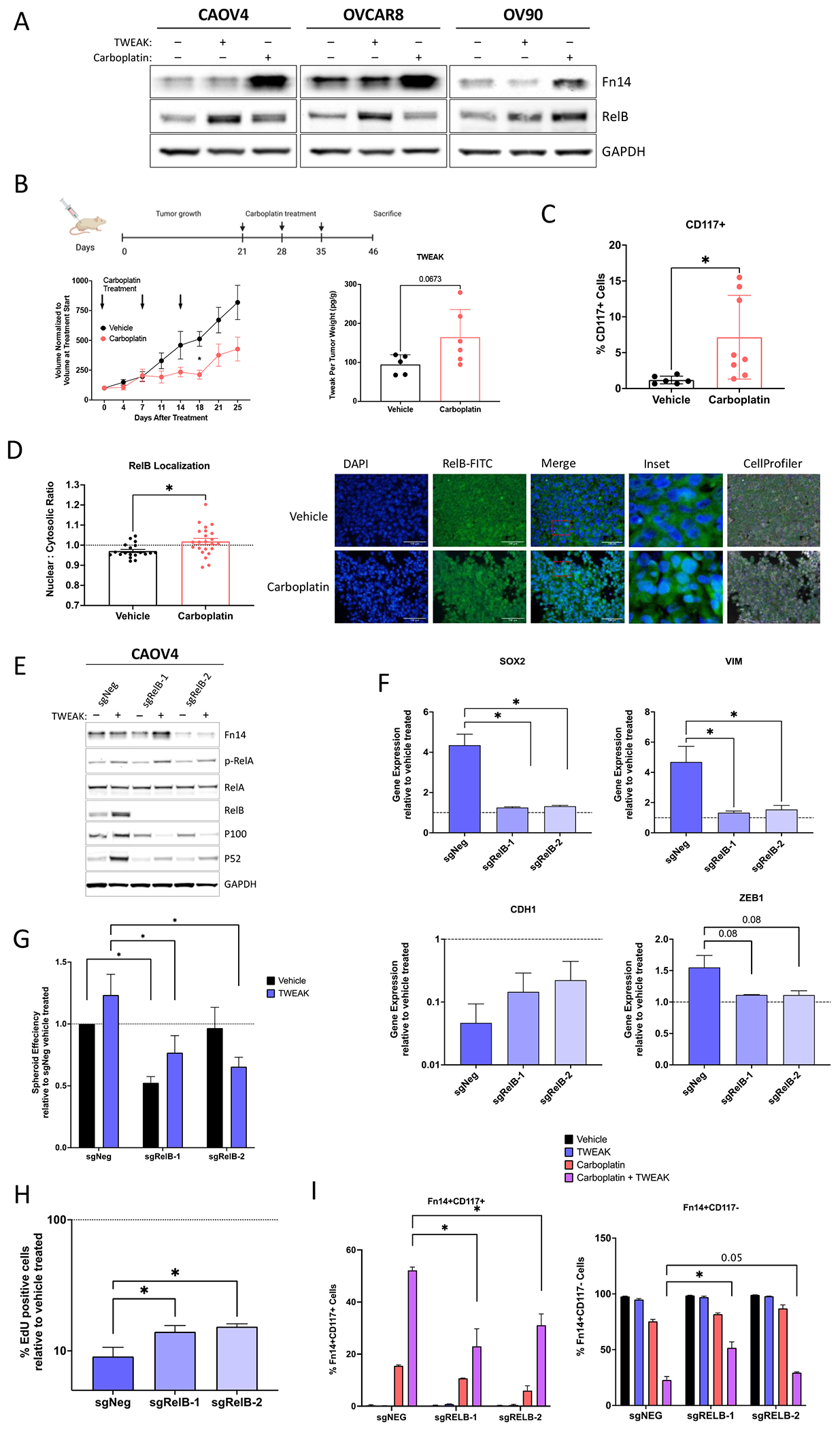
(A) Western blot for Fn14 and RelB from cells treated with vehicle, TWEAK, or Carboplatin for 72 hours. (B-C) Subcutaneous tumors were generated in nude mice using OV90 cells. Tumors were allowed to grow for 21 days followed by 3 cycles of carboplatin (50mg/kg) or vehicle and resected 12 days after the final carboplatin treatment. (B) Experimental design created with BioRender.com (left). Tumor volume was measured twice weekly and graphed normalized to the volume at treatment start (center). Two-way ANOVA, Tukey’s post-hoc test (n=6). TWEAK concentration per tumor weight measured using ELISA (right). Unpaired t-test (n=6). (C) Flow cytometry analysis of CD117 expression on excised tumors following dissociation into single-cell suspension. Unpaired t-test (n=8). (D) Fixed tumor fractions were histologically stained for nuclei (DAPI) and RelB. Representative images (left) and nuclear relative to cytosolic RelB was quantified per field using Cellprofiler (right). Unpaired t-test (n=3). (E) Western blot of NF-κB proteins in CAOV4-RelB knockout (KO) lines with TWEAK treatment (100ng/ml) for 48 hours. (F) qRT-PCR for select genes in CAOV4-RelB KO lines treated with TWEAK (100ng/ml) for 48 hours, relative to vehicle. One-way ANOVA, Tukey’s post-hoc test (n=3). (G) Spheroid efficiency of CAOV4-RelB KO lines treated with TWEAK (100ng/ml) for 7 days, relative to vehicle. Two-way ANOVA, Tukey’s post-hoc test (n=6). (H) EdU incorporation pulse-chase assay of CAOV4-RelB KO lines treated with TWEAK (100ng/ml) for 60 hours, relative to vehicle. One-way ANOVA, Tukey’s post-hoc test (n=6). (I) Flow cytometry of CD117 expression on Fn14+ CAOV4-RelB KO lines treated with TWEAK (100ng/ml), carboplatin (125μM), or TWEAK and carboplatin for 72 hours, represented relative to vehicle as mean plus SD. One-way ANOVA, Tukey’s post-hoc test (n=3). Data represent mean and SEM. * p<0.05.
We next tested whether inhibiting TWEAK activity in vivo could prolong survival in an IP xenograft mouse model of ovarian cancer. Given that TWEAK is likely secreted by stromal cells (35,42), we first verified that mouse TWEAK is structurally homologous to human TWEAK (Supplemental Figure 3A–B), can sufficiently activate NF-κB (Supplemental Figure 3C–D), and induce spheroid formation (Supplemental Figure 3E) in human ovarian cancer cells, can induce expression of SOX2 and NIK as observed with human TWEAK (Supplemental Figure 3F), and that these phenotypes are dependent on the human Fn14 receptor (Supplemental Figure 3D–F).
Since TWEAK levels appear to be highest with chemotherapy administration, we assessed whether inhibiting TWEAK-Fn14 signaling in combination with carboplatin chemotherapy or as a maintenance therapy after completion of 3 cycles of carboplatin would prolong overall survival in an IP model (Figure 7A). Administration of the FN14 inhibitor, L542-0366, in combination with carboplatin significantly prolonged overall survival (Figure 7B). The longest overall survival was observed when the L542-0366 was given as a maintenance therapy following three cycles of carboplatin chemotherapy (Figure 7B). Consistent with our subcutaneous xenograft tumor model, TWEAK was enriched in chemotherapy-treated IP tumors relative to vehicle (Figure 7C). Tumor weights at necropsy were significantly lower for mice treated with L542-0366 as a maintenance, relative to carboplatin alone (Figure 7D–E). Furthermore, FN14 inhibition leads to lower tumor burden at the ovary (Figure 7F). IHC analysis of resected tumors showed that the overall levels of RelB at necropsy are not significantly different among any of the treatment groups (Figure 7G), however blinded automated imaging analysis with CellProfiler as previously described (22–24), demonstrates reduced nuclear localization of RelB in L524-0366-treated tumors, relative to vehicle or chemotherapy alone (Figure 7H). These data indicate that inhibiting TWEAK-Fn14 signaling following standard 3-cycle chemotherapy provides a survival benefit in mice. Our in vitro data suggest this may be due to the ability of TWEAK to promote expression of stemness genes and corresponding phenotypes in CSCs via alternative pathway NF-κB activity during chemotherapy. Thus, blocking the TWEAK-Fn14-NF-κB signaling cascade should be further explored as a potential clinical approach to prolong remission in ovarian cancer patients.
Figure 7. A small molecule inhibitor of Fn14, as a maintenance therapy following chemotherapy, inhibits RelB activation and significantly slows ovarian cancer relapse.
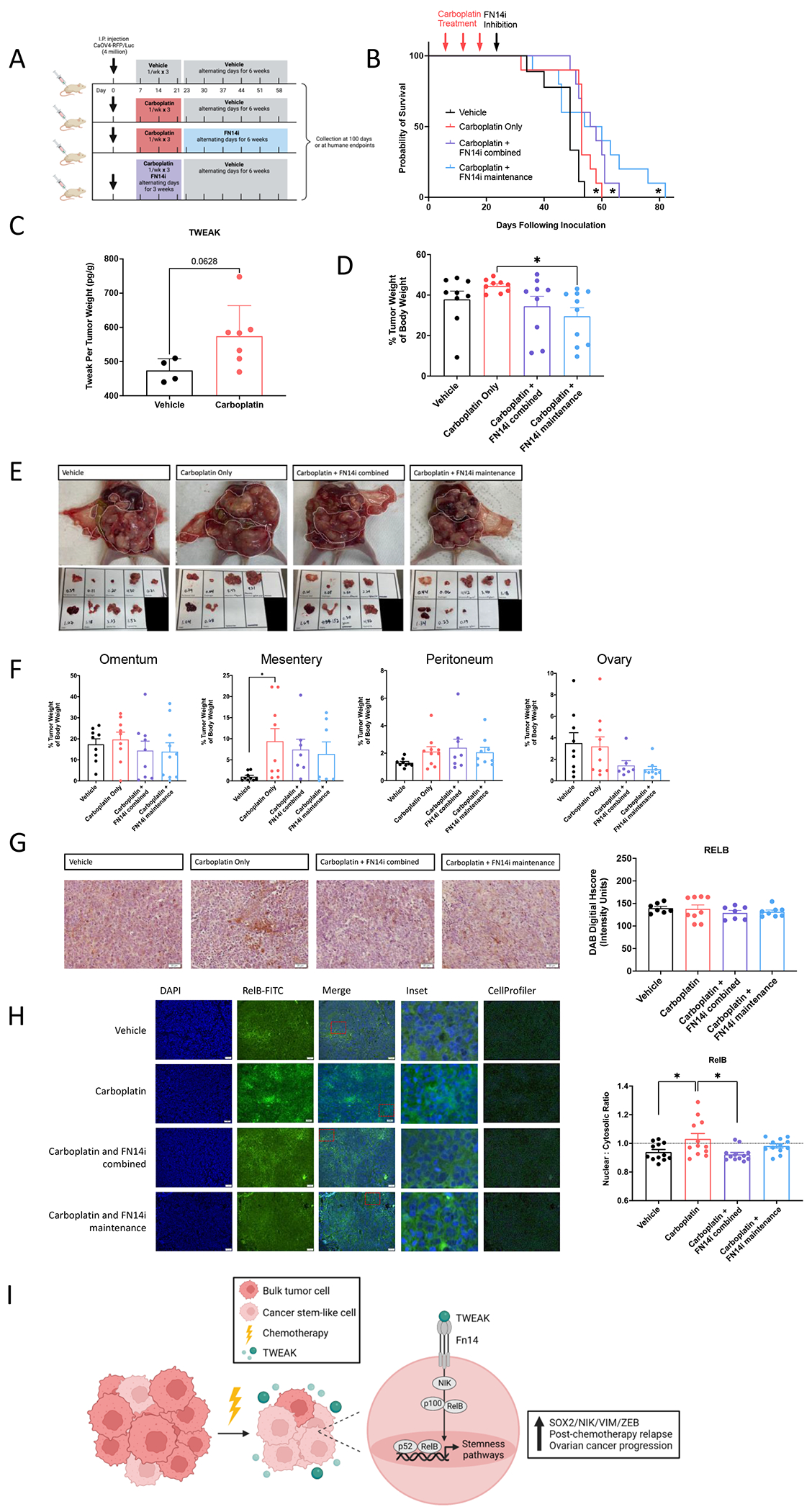
(A) Experimental design created with BioRender.com. (B) Kaplan-Meier survival of intraperitoneal xenograft tumors with CAOV4 cells treated with either vehicle, carboplatin (50mg/kg), carboplatin and FN14i (L542-0366, 9mg/kg) combined, or carboplatin followed by FN14i maintenance. Log-rank test (n=10). (C) TWEAK concentration per tumor weight. Unpaired t-test (n=4 vehicle, n=7 treatment). (D) Tumors were resected and weighed relative to body weight. One-way ANOVA, Tukey’s post-hoc test. (E) Representative images of tumors from different treatment groups at necropsy. (F) Tumors were resected and weighed by organ, represented relative to body weight. One-way ANOVA, Tukey’s post-hoc test (n=10). (G) Tumors were resected, fixed, and histologically stained RelB-DAB. Representative images (left) and quantification using ImageJ (right). One-way ANOVA, Tukey’s post-hoc test (n=10). Data represent mean and SEM. * p<0.05. (H) Fixed tumor fractions were histologically stained for nuclei (DAPI) and RelB. Representative images (left) and nuclear relative to cytosolic RelB was quantified per field using Cellprofiler (right). One-way ANOVA, Tukey’s post-hoc test (n=3). Data represent mean and SEM. * p<0.05. (I) Proposed mechanism created with BioRender.com to illustrate TWEAK-Fn14-RelB signaling supports post-chemotherapy ovarian cancer progression.
DISCUSSION
In addition to tumor cells, the ovarian TME is characterized by a variety of non-tumor cells including tumor-associated macrophages, cancer-associated adipocytes, and cancer-associated fibroblasts, all of which secrete cytokines that can activate NF-κB and/or stemness signaling pathways in cancer cells (14,43). Emerging studies in ovarian cancer implicate the TME in inducing stemness through secretion of cytokines that alter signaling pathways and/or epigenetic patterns (32,44). NF-κB, a master regulator of diverse cellular processes, is persistently activated in ovarian cancer and is associated with a poor prognosis (12,19,45). We previously identified a novel role for the alternative NF-κB transcription factor subunit, RelB, in maintaining ovarian CSCs with low proliferative potential, tumor initiation capacity, and chemotherapy resistance (18). Given its robust activation by various stimuli, in this study we sought to identify consistent inducers of alternative NF-κB that may serve as promising targets for eliminating CSCs and preventing recurrence in ovarian cancer.
We discovered that TWEAK is a strong inducer of the alternative NF-κB pathway in HGSOC cells and is enriched in tumors following chemotherapy. A previous study showed that TWEAK-Fn14 signaling contributes to ovarian cancer metastasis via NF-κB-mediated upregulation of vascular endothelial growth factor (VEGF) (30). This study focused on the role of classical NF-κB as a downstream effector of TWEAK-Fn14 signaling and did not investigate alternative NF-κB mechanisms. Although TWEAK can activate both classical and alternative NF-κB pathways, activation of the alternative pathway is more long-term and may be restricted to specific cell types (35,46–48). Our findings suggest TWEAK is a strong inducer of alternative NF-κB in ovarian cancer cells and promotes a stem-like phenotype. Interestingly, activation of either classical or alternative NF-κB by TWEAK can provoke contrasting phenotypes. For example, TWEAK-mediated activation of classical NF-κB leads to myoblast proliferation and suppression of myogenesis in muscle regeneration, whereas TWEAK-dependent activation of alternative NF-κB leads to myogenesis and suppression of myoblast proliferation (46). These findings are intriguing given our previous work in ovarian cancer showing that classical NF-κB signaling supports a proliferative phenotype, whereas alternative NF-κB supports a quiescent phenotype (18). Although the current study focuses on alternative NF-κB pathway-dependent processes promoted by TWEAK, future studies to unravel features facilitated by classical NF-κB would be worthwhile and may uncover a novel mechanism for maintaining heterogeneity in ovarian cancer. While activation of NF-κB transcription factors are the most common effectors of TWEAK-Fn14 signaling, other pathways including ERK, JNK, and PI3K/AKT have been shown to be induced by TWEAK in specific cell types, which together may impact ovarian cancer biology (35,49,50).
TWEAK is a cytokine of the TNF superfamily and its activity is mediated by binding, with high specificity, to its receptor, a type I transmembrane protein known as Fn14 (49–51). TWEAK and Fn14 are normally expressed at low levels in healthy tissues but can be upregulated during tissue injury, for example in response to IFNγ (52–54). Transient TWEAK-Fn14 signaling is critical for efficient wound repair, however constitutive expression of Fn14 is implicated in several diseases including cardiovascular disease, rheumatoid arthritis, and cancer (52,53,55). Our results show an upregulation of Fn14 on ovarian tumor cells following chemotherapy in vitro and in vivo. TWEAK is primarily secreted by macrophages and stromal cells, (56–60) and given the tissue remodeling activities that underscore tumor formation and responses to cytotoxic treatments, it is conceivable that TWEAK is elevated in tumors and may be increased further following chemotherapy.
Our findings indicate that TWEAK-Fn14 signaling has diverse effects on subpopulations of ovarian cancer cells. Given that CD117 expression is a marker of chemoresistant CSCs (9,61), we examined the effects of TWEAK on CD117+ and CD117− cells. TWEAK-Fn14 signaling in CD117− bulk tumor cells appears to sensitize them to carboplatin chemotherapy as the percentage of CD117− cells decreases when TWEAK is combined with carboplatin. These findings are in agreement with previous studies showing that Fn14 expression sensitizes ovarian cancer cells to chemotherapy, although this study did not examine CSCs nor the role of TWEAK in this process (31). Conversely, we found that the percentage of CD117+ cells increases with TWEAK and carboplatin treatment. Moreover, TWEAK stimulation of CD117+ cells induces the highest expression of stemness genes, thus enhancing their CSC characteristics including their ability to survive in the presence of chemotherapy, and likely their ability to initiate relapse. Interestingly, TWEAK stimulation during carboplatin treatment of CD117− cells induced CD117 expression, suggesting a possible reprogramming of non-CSCs into CSCs in the presence of TWEAK. Our results align with several studies that implicate TWEAK in inhibiting differentiation, inducing EMT, and maintaining stemness in a variety of tissue types (52,62–65).
Given the enrichment of TWEAK with chemotherapy and its ability to enhance CSC features, we propose that the TWEAK-Fn14-RelB signaling axis contributes to CSC development thereby enhancing relapse potential in ovarian cancer (Figure 7I). We show that a maintenance therapy inhibiting TWEAK signaling following carboplatin administration significantly prolonged survival in a mouse model of ovarian cancer. Our in vitro studies suggest this may be at least partially due to the inhibition of alternative NF-κB signaling, although we cannot rule out the involvement of other TWEAK signaling pathways. Future studies investigating the regulation of TWEAK in the ovarian TME may enable the development of new therapeutic approaches for preventing tumor recurrence in ovarian cancer.
Supplementary Material
Implications:
This study identifies a unique mechanism for the induction of ovarian cancer stem cells that may serve as a novel therapeutic target for preventing relapse.
ACKNOWLEDGEMENTS
This work was funded by the National Institute on Minority Health and Health Disparities at the NIH under award number U54MD012397. This work was also supported by the National Cancer Institute at the NIH under award numbers R00 CA20472703 and R01CA260281, and by the SDSU/UCSD Cancer Partnership funded by the National Cancer Institute at the NIH under award numbers U54CA132384 (SDSU) & U54CA132379 (UC San Diego). RH was supported by the Arne N. Wick Predoctoral Fellowship from the California Metabolic Research Foundation.
We thank Dr. Christina M. Annunziata and the National Cancer Institute for kind gift of clinical ascites samples, Benjamin Canter of the SDSU FACS core facility for his excellent assistance with Flow cytometry experiments, Larkin Slater of the SDSU vivarium for her assistance with mouse studies, and Natalie Gude for her assistance with histology studies.
Footnotes
Conflict of Interest Statement:
The authors declare no potential conflict of interest.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70:7–30 [DOI] [PubMed] [Google Scholar]
- 2.Matulonis UA, Sood AK, Fallowfield L, Howitt BE, Sehouli J, Karlan BY. Ovarian cancer. Nat Rev Dis Primers 2016;2:16061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burgos-Ojeda D, Rueda BR, Buckanovich RJ. Ovarian cancer stem cell markers: prognostic and therapeutic implications. Cancer Lett 2012;322:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM, et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res 2008;68:4311–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foster R, Buckanovich RJ, Rueda BR. Ovarian cancer stem cells: working towards the root of stemness. Cancer Lett 2013;338:147–57 [DOI] [PubMed] [Google Scholar]
- 6.Terraneo N, Jacob F, Dubrovska A, Grünberg J. Novel Therapeutic Strategies for Ovarian Cancer Stem Cells. Front Oncol 2020;10:319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harrington BS, Ozaki MK, Caminear MW, Hernandez LF, Jordan E, Kalinowski NJ, et al. Drugs Targeting Tumor-Initiating Cells Prolong Survival in a Post-Surgery, Post-Chemotherapy Ovarian Cancer Relapse Model. Cancers (Basel) 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hatina J, Boesch M, Sopper S, Kripnerova M, Wolf D, Reimer D, et al. Ovarian Cancer Stem Cell Heterogeneity. Adv Exp Med Biol 2019;1139:201–21 [DOI] [PubMed] [Google Scholar]
- 9.Robinson M, Gilbert SF, Waters JA, Lujano-Olazaba O, Lara J, Alexander LJ, et al. Characterization of SOX2, OCT4 and NANOG in Ovarian Cancer Tumor-Initiating Cells. Cancers (Basel) 2021;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bollrath J, Greten FR. IKK/NF-kappaB and STAT3 pathways: central signalling hubs in inflammation-mediated tumour promotion and metastasis. EMBO Rep 2009;10:1314–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karin M NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol 2009;1:a000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Annunziata CM, Stavnes HT, Kleinberg L, Berner A, Hernandez LF, Birrer MJ, et al. Nuclear factor kappaB transcription factors are coexpressed and convey a poor outcome in ovarian cancer. Cancer 2010;116:3276–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007;12:115–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harrington BS, Annunziata CM. NF-κB Signaling in Ovarian Cancer. Cancers (Basel) 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu S, Kim M, Hernandez L, Grajales V, Noonan A, Anver M, et al. IKK-ε coordinates invasion and metastasis of ovarian cancer. Cancer Res 2012;72:5494–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun SC. Non-canonical NF-κB signaling pathway. Cell Res 2011;21:71–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tegowski M, Baldwin A. Noncanonical NF-κB in Cancer. Biomedicines 2018;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.House CD, Jordan E, Hernandez L, Ozaki M, James JM, Kim M, et al. NFκB Promotes Ovarian Tumorigenesis via Classical Pathways That Support Proliferative Cancer Cells and Alternative Pathways That Support ALDH. Cancer Res 2017;77:6927–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hufnagel DH, Wilson AJ, Saxon J, Blackwell TS, Watkins J, Khabele D, et al. Expression of p52, a non-canonical NF-kappaB transcription factor, is associated with poor ovarian cancer prognosis. Biomark Res 2020;8:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uno M, Saitoh Y, Mochida K, Tsuruyama E, Kiyono T, Imoto I, et al. NF-κB inducing kinase, a central signaling component of the non-canonical pathway of NF-κB, contributes to ovarian cancer progression. PLoS One 2014;9:e88347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.House CD, Hernandez L, Annunziata CM. In vitro enrichment of ovarian cancer tumor-initiating cells. J Vis Exp 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stirling DR, Swain-Bowden MJ, Lucas AM, Carpenter AE, Cimini BA, Goodman A. CellProfiler 4: improvements in speed, utility and usability. BMC Bioinformatics 2021;22:433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ljosa V, Sokolnicki KL, Carpenter AE. Annotated high-throughput microscopy image sets for validation. Nat Methods 2012;9:637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J, Sinnett-Smith J, Stevens JV, Young SH, Rozengurt E. Biphasic Regulation of Yes-associated Protein (YAP) Cellular Localization, Phosphorylation, and Activity by G Protein-coupled Receptor Agonists in Intestinal Epithelial Cells: A NOVEL ROLE FOR PROTEIN KINASE D (PKD). J Biol Chem 2016;291:17988–8005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hernandez L, Kim MK, Lyle LT, Bunch KP, House CD, Ning F, et al. Characterization of ovarian cancer cell lines as in vivo models for preclinical studies. Gynecol Oncol 2016;142:332–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bairoch A The Cellosaurus, a Cell-Line Knowledge Resource. J Biomol Tech 2018;29:25–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hagemann T, Wilson J, Kulbe H, Li NF, Leinster DA, Charles K, et al. Macrophages induce invasiveness of epithelial cancer cells via NF-kappa B and JNK. J Immunol 2005;175:1197–205 [DOI] [PubMed] [Google Scholar]
- 28.Hong L, Wang S, Li W, Wu D, Chen W. Tumor-associated macrophages promote the metastasis of ovarian carcinoma cells by enhancing CXCL16/CXCR6 expression. Pathol Res Pract 2018;214:1345–51 [DOI] [PubMed] [Google Scholar]
- 29.Chen R, Alvero AB, Silasi DA, Kelly MG, Fest S, Visintin I, et al. Regulation of IKKbeta by miR-199a affects NF-kappaB activity in ovarian cancer cells. Oncogene 2008;27:4712–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dai L, Gu L, Ding C, Qiu L, Di W. TWEAK promotes ovarian cancer cell metastasis via NF-kappaB pathway activation and VEGF expression. Cancer Lett 2009;283:159–67 [DOI] [PubMed] [Google Scholar]
- 31.Wu AY, Gu LY, Cang W, Cheng MX, Wang WJ, Di W, et al. Fn14 overcomes cisplatin resistance of high-grade serous ovarian cancer by promoting Mdm2-mediated p53-R248Q ubiquitination and degradation. J Exp Clin Cancer Res 2019;38:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y, Zong X, Mitra S, Mitra AK, Matei D, Nephew KP. IL-6 mediates platinum-induced enrichment of ovarian cancer stem cells. JCI Insight 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jordan KR, Sikora MJ, Slansky JE, Minic A, Richer JK, Moroney MR, et al. The Capacity of the Ovarian Cancer Tumor Microenvironment to Integrate Inflammation Signaling Conveys a Shorter Disease-free Interval. Clin Cancer Res 2020;26:6362–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Javellana M, Eckert MA, Heide J, Zawieracz K, Weigert M, Ashley S, et al. Neoadjuvant Chemotherapy Induces Genomic and Transcriptomic Changes in Ovarian Cancer. Cancer Res 2022;82:169–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Winkles JA. The TWEAK-Fn14 cytokine-receptor axis: discovery, biology and therapeutic targeting. Nat Rev Drug Discov 2008;7:411–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sliogeryte K, Thorpe SD, Lee DA, Botto L, Knight MM. Stem cell differentiation increases membrane-actin adhesion regulating cell blebability, migration and mechanics. Sci Rep 2014;4:7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fan YL, Zhao HC, Li B, Zhao ZL, Feng XQ. Mechanical Roles of F-Actin in the Differentiation of Stem Cells: A Review. ACS Biomater Sci Eng 2019;5:3788–801 [DOI] [PubMed] [Google Scholar]
- 38.Patra B, Lateef MA, Brodeur MN, Fleury H, Carmona E, Péant B, et al. Carboplatin sensitivity in epithelial ovarian cancer cell lines: The impact of model systems. PLoS One 2020;15:e0244549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bilbao M, Katz C, Kass SL, Smith D, Hunter K, Warshal D, et al. Epigenetic Therapy Augments Classic Chemotherapy in Suppressing the Growth of 3D High-Grade Serous Ovarian Cancer Spheroids over an Extended Period of Time. Biomolecules 2021;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, Zhao G, Condello S, Huang H, Cardenas H, Tanner EJ, et al. Frizzled-7 Identifies Platinum-Tolerant Ovarian Cancer Cells Susceptible to Ferroptosis. Cancer Res 2021;81:384–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bensen RC, Gunay G, Finneran MC, Jhingan I, Acar H, Burgett AWG. Small Molecule Targeting of Oxysterol-Binding Protein (OSBP)-Related Protein 4 and OSBP Inhibits Ovarian Cancer Cell Proliferation in Monolayer and Spheroid Cell Models. ACS Pharmacol Transl Sci 2021;4:744–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng E, Armstrong CL, Galisteo R, Winkles JA. TWEAK/Fn14 Axis-Targeted Therapeutics: Moving Basic Science Discoveries to the Clinic. Front Immunol 2013;4:473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pogge von Strandmann E, Reinartz S, Wager U, Müller R. Tumor-Host Cell Interactions in Ovarian Cancer: Pathways to Therapy Failure. Trends Cancer 2017;3:137–48 [DOI] [PubMed] [Google Scholar]
- 44.Qian J, LeSavage BL, Hubka KM, Ma C, Natarajan S, Eggold JT, et al. Cancer-associated mesothelial cells promote ovarian cancer chemoresistance through paracrine osteopontin signaling. J Clin Invest 2021;131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hernandez L, Hsu SC, Davidson B, Birrer MJ, Kohn EC, Annunziata CM. Activation of NF-kappaB signaling by inhibitor of NF-kappaB kinase beta increases aggressiveness of ovarian cancer. Cancer Res 2010;70:4005–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Enwere EK, Lacasse EC, Adam NJ, Korneluk RG. Role of the TWEAK-Fn14-cIAP1-NF-κB Signaling Axis in the Regulation of Myogenesis and Muscle Homeostasis. Front Immunol 2014;5:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dogra C, Changotra H, Mohan S, Kumar A. Tumor necrosis factor-like weak inducer of apoptosis inhibits skeletal myogenesis through sustained activation of nuclear factor-kappaB and degradation of MyoD protein. J Biol Chem 2006;281:10327–36 [DOI] [PubMed] [Google Scholar]
- 48.Saitoh T, Nakayama M, Nakano H, Yagita H, Yamamoto N, Yamaoka S. TWEAK induces NF-kappaB2 p100 processing and long lasting NF-kappaB activation. J Biol Chem 2003;278:36005–12 [DOI] [PubMed] [Google Scholar]
- 49.Sanz AB, Sanchez-Niño MD, Ortiz A. TWEAK, a multifunctional cytokine in kidney injury. Kidney Int 2011;80:708–18 [DOI] [PubMed] [Google Scholar]
- 50.Winkles JA, Tran NL, Brown SA, Stains N, Cunliffe HE, Berens ME. Role of TWEAK and Fn14 in tumor biology. Front Biosci 2007;12:2761–71 [DOI] [PubMed] [Google Scholar]
- 51.Chicheportiche Y, Bourdon PR, Xu H, Hsu YM, Scott H, Hession C, et al. TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J Biol Chem 1997;272:32401–10 [DOI] [PubMed] [Google Scholar]
- 52.Burkly LC, Michaelson JS, Hahm K, Jakubowski A, Zheng TS. TWEAKing tissue remodeling by a multifunctional cytokine: role of TWEAK/Fn14 pathway in health and disease. Cytokine 2007;40:1–16 [DOI] [PubMed] [Google Scholar]
- 53.Perez JG, Tran NL, Rosenblum MG, Schneider CS, Connolly NP, Kim AJ, et al. The TWEAK receptor Fn14 is a potential cell surface portal for targeted delivery of glioblastoma therapeutics. Oncogene 2016;35:2145–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakayama M, Kayagaki N, Yamaguchi N, Okumura K, Yagita H. Involvement of TWEAK in interferon gamma-stimulated monocyte cytotoxicity. J Exp Med 2000;192:1373–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Burkly LC, Michaelson JS, Zheng TS. TWEAK/Fn14 pathway: an immunological switch for shaping tissue responses. Immunol Rev 2011;244:99–114 [DOI] [PubMed] [Google Scholar]
- 56.Méndez-Barbero N, Gutierrez-Muñoz C, Madrigal-Matute J, Mínguez P, Egido J, Michel JB, et al. A major role of TWEAK/Fn14 axis as a therapeutic target for post-angioplasty restenosis. EBioMedicine 2019;46:274–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bird TG, Lu WY, Boulter L, Gordon-Keylock S, Ridgway RA, Williams MJ, et al. Bone marrow injection stimulates hepatic ductular reactions in the absence of injury via macrophage-mediated TWEAK signaling. Proc Natl Acad Sci U S A 2013;110:6542–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maecker H, Varfolomeev E, Kischkel F, Lawrence D, LeBlanc H, Lee W, et al. TWEAK attenuates the transition from innate to adaptive immunity. Cell 2005;123:931–44 [DOI] [PubMed] [Google Scholar]
- 59.Maymó-Masip E, Fernández-Veledo S, Garcia España A, Vázquez-Carballo A, Tinahones FJ, García-Fuentes E, et al. The rise of soluble TWEAK levels in severely obese subjects after bariatric surgery may affect adipocyte-cytokine production induced by TNFα. J Clin Endocrinol Metab 2013;98:E1323–33 [DOI] [PubMed] [Google Scholar]
- 60.Vendrell J, Chacón MR. TWEAK: A New Player in Obesity and Diabetes. Front Immunol 2013;4:488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Luo L, Zeng J, Liang B, Zhao Z, Sun L, Cao D, et al. Ovarian cancer cells with the CD117 phenotype are highly tumorigenic and are related to chemotherapy outcome. Exp Mol Pathol 2011;91:596–602 [DOI] [PubMed] [Google Scholar]
- 62.Fukuda T, Fukuda R, Koinuma D, Moustakas A, Miyazono K, Heldin CH. BMP2-induction of FN14 promotes protumorigenic signaling in gynecologic cancer cells. Cell Signal 2021;87:110146. [DOI] [PubMed] [Google Scholar]
- 63.Whitsett TG, Mathews IT, Cardone MH, Lena RJ, Pierceall WE, Bittner M, et al. Mcl-1 mediates TWEAK/Fn14-induced non-small cell lung cancer survival and therapeutic response. Mol Cancer Res 2014;12:550–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Itoigawa Y, Harada N, Harada S, Katsura Y, Makino F, Ito J, et al. TWEAK enhances TGF-β-induced epithelial-mesenchymal transition in human bronchial epithelial cells. Respir Res 2015;16:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu JY, Jiang L, He T, Liu JJ, Fan JY, Xu XH, et al. NETO2 promotes invasion and metastasis of gastric cancer cells via activation of PI3K/Akt/NF-κB/Snail axis and predicts outcome of the patients. Cell Death Dis 2019;10:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available upon request from the corresponding author.