

Abstract
Spontaneous preterm birth (sPTB) is a leading cause of maternal and neonatal morbidity and mortality, yet its prevention and early risk stratification are limited. Previous investigations have suggested that vaginal microbes and metabolites may be implicated in sPTB. Here, we performed untargeted metabolomics on 232 second trimester vaginal samples, 80 from pregnancies ending preterm. We find multiple associations between vaginal metabolites and subsequent preterm birth, and propose that several of these metabolites, including diethanolamine and ethyl-glucoside, are exogenous. We observe associations between the metabolome and microbiome profiles previously obtained using 16S rRNA amplicon sequencing, including correlations between bacteria considered sub-optimal, such as Gardnerella vaginalis, and metabolites enriched in term pregnancies, such as tyramine. These associations were investigated using metabolic models. We use machine learning models to predict sPTB risk from metabolite levels, weeks to months before birth, with good accuracy (auROC=0.78). These models, which we validate using two external cohorts, are more accurate than microbiome-based and maternal covariates-based models (auROC=0.55–0.59). Our results demonstrate the potential of vaginal metabolites as early biomarkers of sPTB and highlight exogenous exposures as potential risk factors for prematurity.
Introduction
Preterm birth (PTB), childbirth before 37 weeks of gestation, is the leading cause of neonatal death, and may lead to a variety of lifelong morbidities1,2. PTB also reflects a significant racial disparity, manifesting in a substantially higher PTB rate in Black women3. This disparity is driven by various factors, such as the persistent stress of systemic and environmental racism and a lack of access to maternal care4. Spontaneous preterm birth (sPTB), preterm birth not medically induced, accounts for two thirds of all PTBs1. Despite extensive efforts, methods for early prediction, prevention or treatment of PTB are lacking1,5,6, and its prevalence remains high1.
The human microbiome is a strong biomarker of many complex diseases7–11. The vaginal microbiome, specifically, has been repeatedly associated with sPTB and other adverse pregnancy outcomes12–17. However, a clear consensus on the relationship between the vaginal microbiome and sPTB has yet to emerge18, and our knowledge of specific mechanisms underlying potential host-microbiome interactions in sPTB is lacking.
Metabolites produced or modified by the microbiome have emerged as a prominent factor with potential local and systemic effects on the host19–22. Their study has been facilitated by metabolomics, which enables the measurement of thousands of small molecules present in an ecosystem, and paired microbiome-metabolome studies have yielded potential mechanistic insights into host-microbiome interactions in various pathologies23,24. A few studies of the vaginal metabolome described associations with the microbiome, inflammation, and PTB25,26. However, studies of demographic groups at high risk for sPTB, with measurements of a broad set of metabolites, and which generate robust prediction models for sPTB, are still needed to advance our understanding of the role of the vaginal ecosystem in prematurity and other pregnancy outcomes.
Here, we measured the second trimester vaginal metabolome of 232 pregnant women, for whom the microbiota was previously characterized using 16S rRNA gene amplicon sequencing14. We show that the vaginal metabolome partially corresponds to community state types (CSTs), reveal associations between metabolites measured in the middle of pregnancy and subsequent sPTB, and propose that some of these metabolites are of an exogenous source. Finally, we devise machine learning algorithms that use the vaginal metabolome to predict subsequent preterm birth an average of 3 months before delivery, which we validate on two external cohorts. Our results demonstrate a promising approach for studying potential causes of prematurity as well as for early risk stratification, and highlight the need to study environmental exposures as a risk factor for sPTB.
Results
Vaginal microbiota and metabolome from a pregnancy cohort
We used mass spectrometry to profile 232 vaginal samples collected between 20–24 weeks of gestation from women with singleton pregnancies, for which the microbiota was previously characterized from the same time point14 (Table S1; Methods). All women with subsequent sPTB and available samples (N=80), as well as similar term-birth controls (TB; N=152) were included (Table 1). As expected, PTB history was associated with sPTB (Fisher’s exact p=3x10−4).
Table 1: Cohort characteristics.
sPTB, spontaneous preterm birth; TB, term birth; BMI, body mass index; GA, gestational age; p – two-sided Fisher’s Exact or Mann-Whitney U test. Bold – p<0.05.
| sPTB | TB | Difference (p value) | |
|---|---|---|---|
| N | 80 | 152 | |
| Race [N (%)] | 0.417 | ||
| Black | 57 (71.25%) | 116 (76.3%) | 0.568 |
| White | 21 (26.25%) | 30 (19.7%) | 0.331 |
| Other | 2 (2.5%) | 6 (4%) | 0.666 |
| Nulliparous [N (%)] | 29.0 (36.2%) | 55.0 (36.2%) | 0.894 |
| PTB history [N (%)] | 34 (42.5%) | 28 (19.2%) | 0.0003 |
| GA at delivery [median weeks [range]] | 34 [21–36] | 39 [38–39] | <1e-10 |
| BMI [kg/m2 mean±SD] | 30.1±7.8 | 30.6±7.2 | 0.65 |
| Age [years mean±SD] | 29±6 | 28±6 | 0.28 |
We quantified 635 identified metabolites as well as 110 unnamed spectral features (Methods). Metabolites belonged to diverse biochemical classes, including amino acids, lipids, nucleotides, carbohydrates and xenobiotics. Most metabolites (549) were measured in over 50% of the cohort, and 108 metabolites were present in all samples (Extended Data Fig. 1; see Supplementary Note 1 and Extended Data Fig. 2 for discussion of batch processing of the samples.). We have previously shown that similar measurements are in excellent agreement with measurements by an independent certified medical laboratory27.
The vaginal metabolome partially preserves CST structure
The vaginal microbiome clusters to well-defined community state types (CSTs)28. We demonstrated the same for this cohort14 (PERMANOVA p<0.001; Fig. 1a), and investigated whether the vaginal metabolome recapitulates this structure. The metabolome is separated by CSTs (p<0.001; Fig. 1b), and is generally associated with the microbiome (Mantel p<0.001), as previously described29. However, specific CSTs are not as well separated. While the metabolomes of women with CST-I (Lactobacillus crispatus) and CST-IV (diverse anaerobes) microbiomes are well separated from the rest of the cohort (PERMANOVA p<0.001 for both), neither the metabolomes of women with CSTs IV-A and IV-B, nor with CST-II (Lactobacillus gasseri) and CST-III (Lactobacillus iners), were well separated from one another (p=0.158 and p=0.155, respectively). Overall, these results demonstrate a strong but imperfect correspondence between the vaginal microbiome and metabolome.
Figure 1 |. Vaginal metabolome clusters are associated with preterm birth.
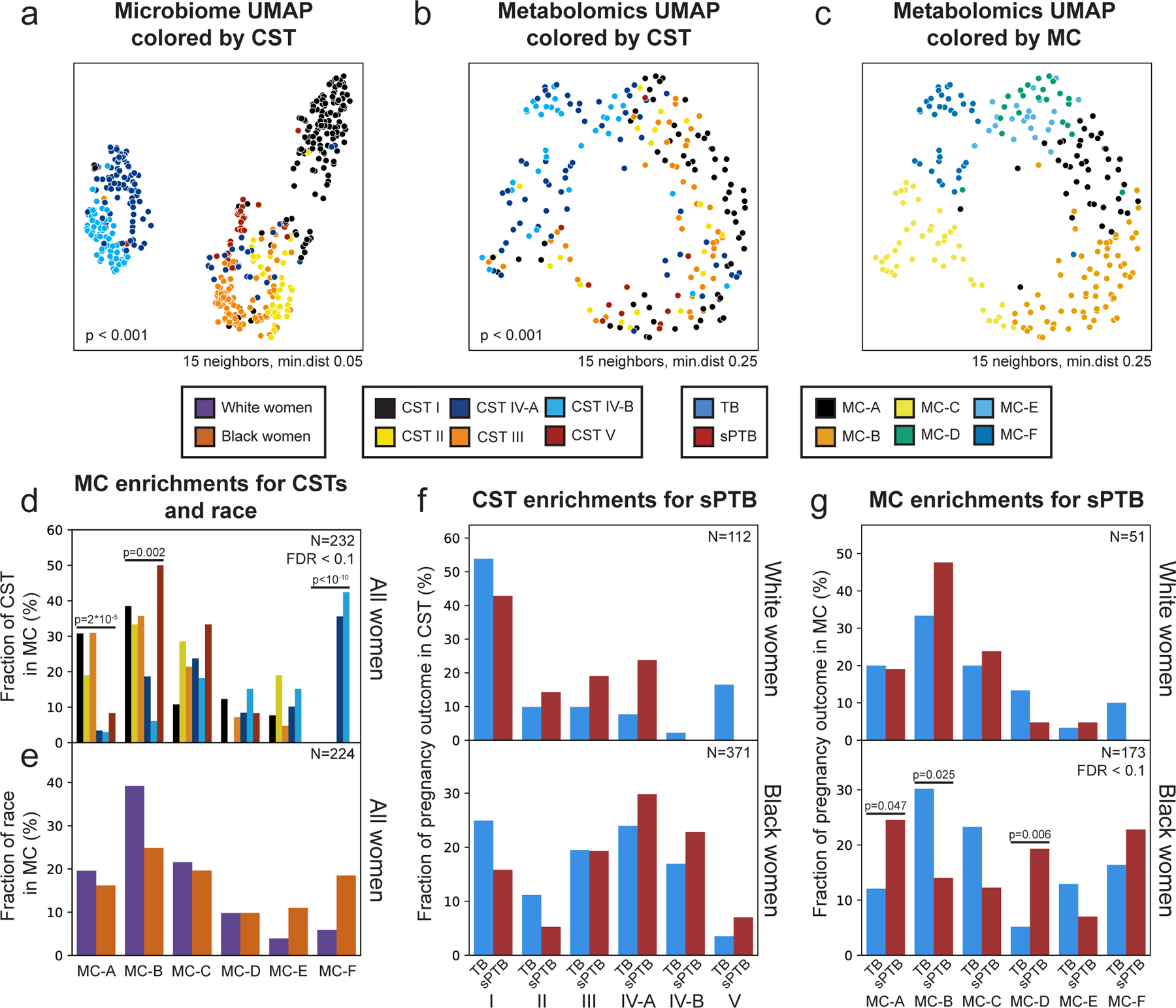
a-c, UMAP ordination of microbiome (a; N=503) and metabolomics data (b,c; N=232), colored by community state types (CSTs; a,b) or de-novo clustering of metabolites data (metabolites clusters [MCs]; Methods; c). The vaginal microbiome and metabolome are significantly separated by CSTs (PERMANOVA p<0.001 for both), yet the separation is less clear in the metabolome. See also Extended Data Fig. 4c,d for similar plots colored by maternal race. d, The fraction of women whose metabolite profiles clustered to each MC, shown for each CST separately. e, Similar to d but shown for Black and White women separately. f, The fraction of White (top) and Black (bottom) women whose microbiomes belonged to each CST, separated by pregnancy outcome. g, Similar to f, for the fraction of women whose metabolomes clustered to each MC. We show a significant association of sPTB with MC-A, B and D among Black women (p=0.047, p=0.025, p=0.006, respectively, q<0.1). Number above horizontal lines in d-g is two-sided Fisher’s exact p, q<0.1.
Metabolite-clusters associate with sPTB
We next performed de-novo k-medoids clustering of the metabolome, revealing six “metabolite-clusters” (MCs A-F; Methods; Fig. 1c, Extended Data Fig. 3; Table S2), which are not as well-separated as the separation of the vaginal microbiome to CSTs. The metabolite sub-pathway most enriched within each MC was polyamine metabolism, dipeptides, dicarboxylated fatty acids, glutamate metabolism, TCA cycle, and dipeptides for MC-A-F, respectively (Fisher’s exact p<0.05 for all). Amino-acid-related metabolites were similarly enriched in MC-A,B,D (p<0.01, q<0.1 for all), and xenobiotics in MC-C (Fisher’s exact p=0.005, q<0.1). While MC-A-D are mostly paired with Lactobacillus dominated CSTs (54%-93%), MC-F is composed entirely of CST-IV, and MC-E is evenly split (50% CST-IV; Fig. 1d, Extended Data Fig. 4a). Reciprocally, we found various enrichments of CSTs in MCs (Extended Data Fig. 4b).
Similar to the strong association between the global microbiome signature and self-identified race in this cohort (PERMANOVA p<0.001; Extended Data Fig. 4c), we saw a significant difference in the metabolome of Black and White women (p<0.001; Extended Data Fig. 4d). However, we found only mild differences between these subgroups in their assignments to MCs (Fig. 1e). Interestingly, while CSTs are only weakly associated with sPTB in White women (Fisher’s exact p=0.047, q=0.21; Fig. 1f, Extended Data Fig. 4e; similar to a previous analysis14), we find that several MCs are significantly associated with sPTB in Black women (p=0.047, p=0.025 and p=0.006, respectively, for MC-A, MC-B, and MC-D; q<0.1 for all; Fig. 1g, Extended Data Fig. 4f). However, we observed no significant associations with early PTB (<32 weeks; q>0.1 for all, Extended Data Fig. 4g). Taken together, our results demonstrate that the metabolome structure in this cohort better captures associations with prematurity in Black women than the microbiome structure.
Multiple metabolites associate with sPTB
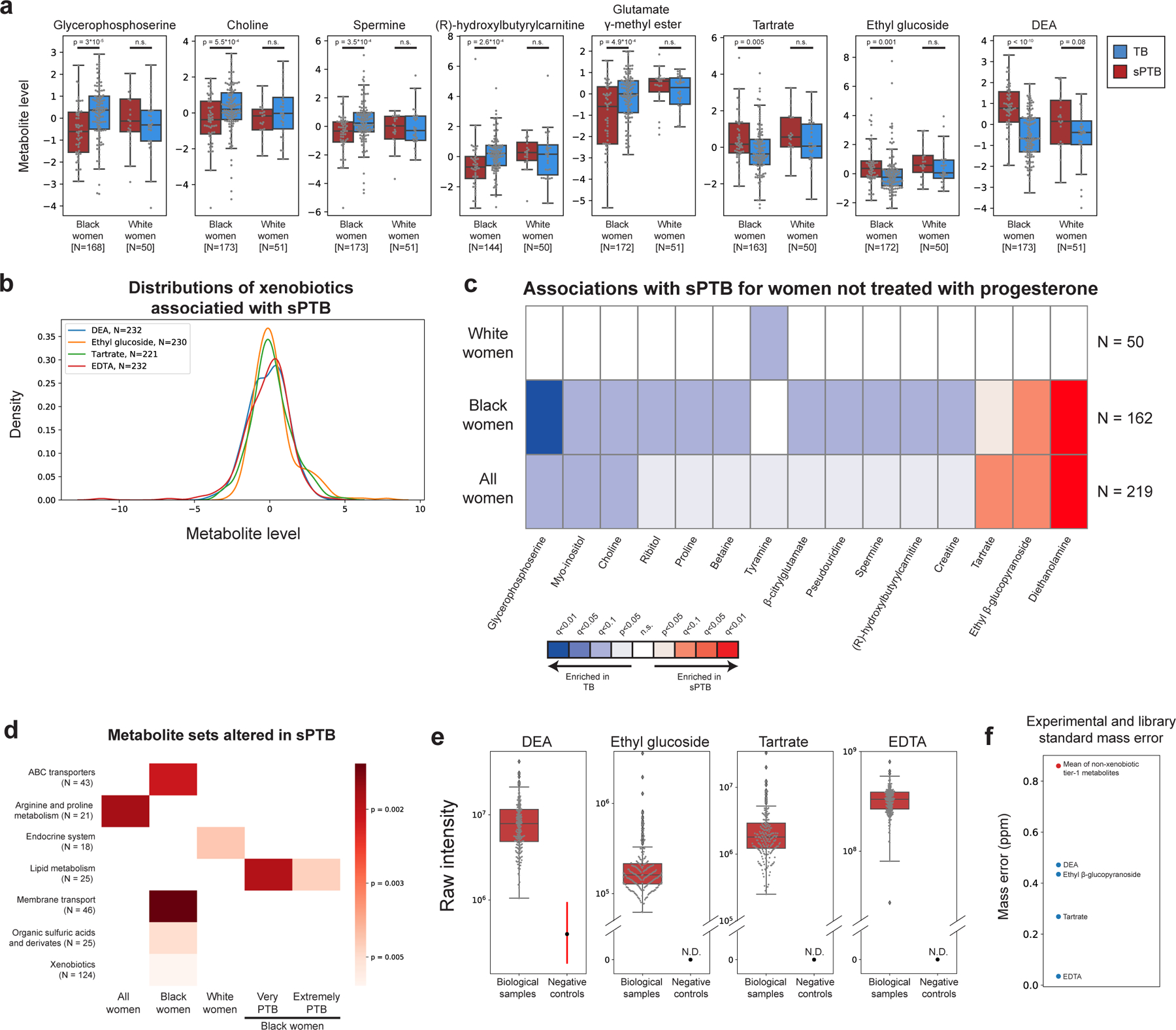
We next investigated associations between sPTB and specific metabolites. We find four metabolites that are significantly associated with sPTB (Mann-Whitney U p<0.05, q<0.1; Fig. 2a, Extended Data Fig. 5a). Three of these, ethyl glucoside (ethyl β-glucopyranoside; p=1.9x10−4, q=0.065); tartrate (p=4.8x10−4, q=0.078); and diethanolamine (DEA; p<10−10, q =5x10−8), all higher in sPTB, appear to be of exogenous source30–36. We confirmed this using AMON37 (Methods), a method that predicts metabolite origins, which predicted that DEA and tartrate were of xenobiotic origin (no prediction could be made for ethyl glucoside; Table S3). Of note, DEA is also associated with MC-A (p=0.006, q=0.014), and MC-D (p=0.04, q=0.07), the MCs we found to be enriched with sPTB (Fig. 1g). Despite their likely exogenous source, these metabolites were detected in >95% of this cohort (Extended Data Fig. 5b).
Figure 2 |. Vaginal metabolites associate with subsequent preterm delivery.
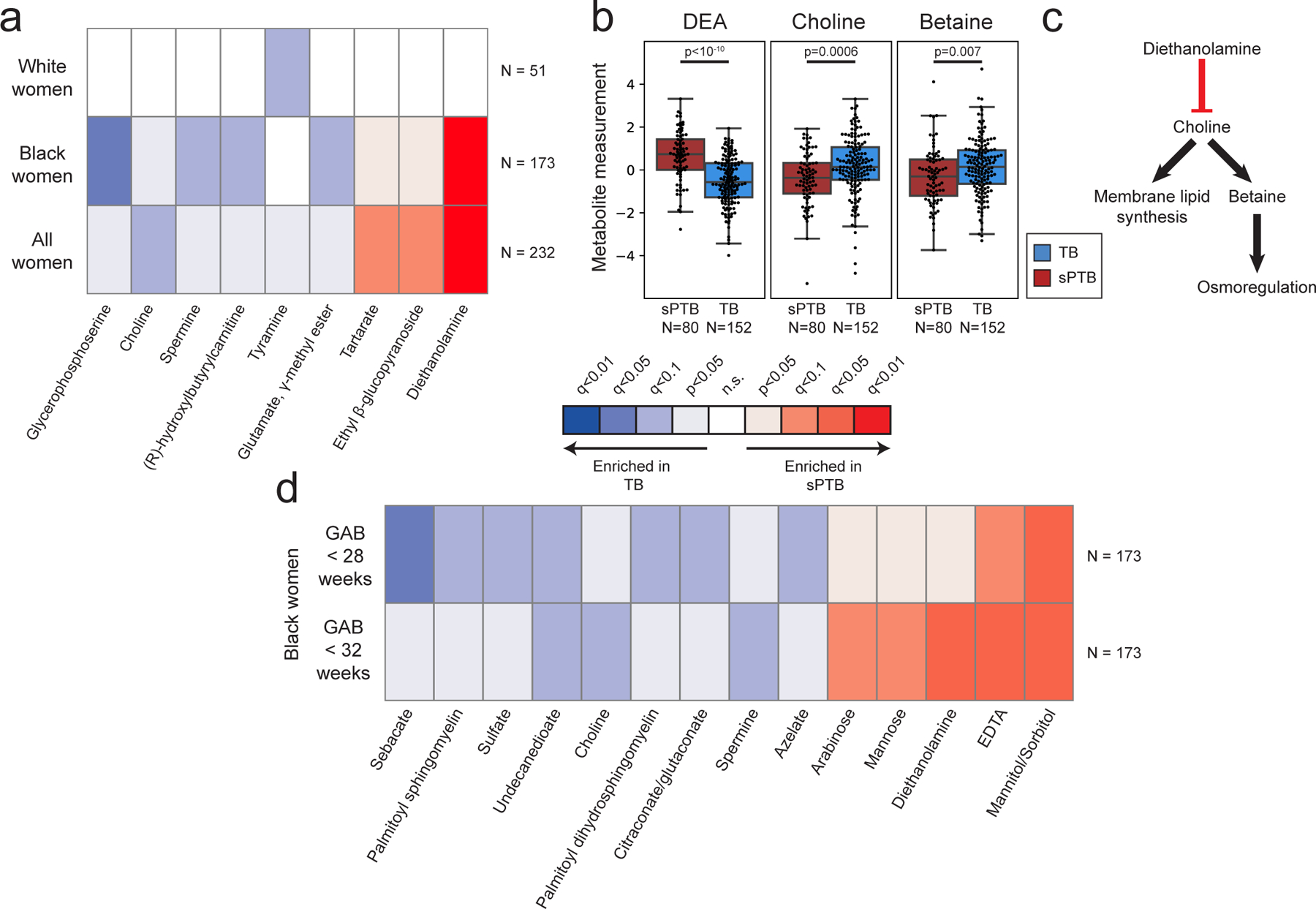
a, Heatmap showing statistically significant associations (Two-sided Mann-Whitney U p<0.05) between specific metabolite measurements and birth outcomes, stratified by maternal race, and colored by significance and direction of association. Only metabolites with at least one association with FDR<0.1 are shown. Metabolites are sorted by their average signed (direction of fold change) log p-value. b, Box and swarm plots (line, median; box, IQR; whiskers, 1.5*IQR) of three metabolites with significant associations with sPTB. p – two-sided Mann-Whitney U. c, Illustration summarizing some of the literature regarding the three metabolites shown in b. Diethanolamine (DEA), which is associated with sPTB, was shown to inhibit choline uptake41. Choline and betaine, both associated with TB, are important for membrane lipid synthesis and osmoregulation38,40. d, Same as a, with stratification by gestational age at birth (GAB), performed among Black women. Middle legend applies to a and d, q<0.1 indicated by bright colors (legend).
We further find lower levels of choline in women with subsequent sPTB (p=5.5x10−4, q=0.078; Fig. 2a,b). Choline is an essential nutrient38, and lower choline levels were previously found in cord blood from premature infants39. Choline is also a precursor of betaine40, an osmoregulator which was also negatively associated with sPTB (p=0.007, q=0.29; Fig. 2b). DEA is known to disrupt choline metabolism41, and its dermal administration in mice depleted hepatic choline42,43. We therefore propose that the higher levels of DEA in sPTB may also be linked to lower choline and betaine levels (Fig. 2b,c). DEA was further shown to be carcinogenic44 and teratogenic42 in mice. However, the relative nature of our metabolomic assay precludes quantitative comparison with levels measured in previous studies. Taken together, these results highlight a potential role of several metabolites in prematurity, some of which may arise exogenously from environmental exposures.
Metabolite associations interact with race and sPTB timing
As the metabolome differed between Black and White women, we performed the same association analysis while stratifying by race. Interestingly, we detect five additional metabolites negatively associated with sPTB (Mann–Whitney U p<0.05; q<0.1; Fig 2a, Extended Data Fig. 5a). In Black women, these include glycerophosphoserine (p=3x10−5, q=0.014), previously reported to be altered in preeclampsia45; spermine (p=3.5x10−4, q=0.07), previously shown to be increased in the blood of preterm infants46; hydroxybutyl carnitine (p=2.6x10−4, q=0.065), a ketocarnitine shown to be depleted in the blood of low birth weight full-term neonates47; and glutamate gamma-methyl ester (p=4.9x10−4, q=0.078). Tyramine, a biogenic amine, was significantly lower in samples from White women who delivered preterm (p=2.8x10−4, q=0.065; Fig. 2a). Tyramine was shown to colocalize with synaptic vesicles in the mouse uterine plexus, highlighting a possible role in uterine contractions48. Altogether, these results highlight the potential connection between vaginal metabolites, metabolite levels in other organs, and sPTB.
As several participants in this cohort (N=13, N=11 in Black women) were treated with intravaginal progesterone prior or close to sample collection (at weeks 18–23 of gestation), we performed the same analysis only in women not treated with vaginal progesterone. One association, between glutamate gamma-methyl ester and TB in Black women (Fig. 2a) no longer passed correction for multiple hypothesis testing (p=0.002, q=0.12, Extended Data Fig. 5c). We find, however, an additional seven metabolites to be associated with TB in Black women (p<0.05; q<0.1 for all; Extended Data Fig. 5c). These include proline (p=6x10−4, q=0.082), which comprises about a quarter of the amino acid residues of collagen49, and is integral to the extracellular matrix; spermine, a polyamine important for placental angiogenesis50 which was lower in Black women with subsequent sPTB (p=4x10−4, q=0.08); and betaine (p=9x10−4, q=0.091). N-acetylarginine (p=0.0015, q=0.102), which is produced from proline and is necessary for the synthesis of polyamines such as spermine, was also lower in Black women with subsequent sPTB. Both disordered placental angiogenesis and extracellular matrix remodeling have been associated with sPTB51.
Earlier preterm deliveries are associated with worse outcomes1. Therefore, we next investigated associations between vaginal metabolites and subsequent very and extremely preterm deliveries (gestational age at birth <32 and <28 weeks, respectively). We limited this analysis to Black women, due to their high proportion among such deliveries (21 of 26 and 14 of 15, respectively). We identify 13 metabolites that are associated only with these earlier sPTBs (p<0.05, q<0.1; Fig. 2d). The phospholipids palmitoyl sphingomyelin and palmitoyl dihydro sphingomyelin were both negatively associated with extremely PTB (p=8.7x10−4, q=0.061; p=0.0011, q=0.069; respectively). Citraconate was likewise negatively associated with extremely PTB (p=0.0014, q=0.075), and was previously found to have lower concentrations in placental mitochondria of women with severe preeclampsia52. We also find several sugar or sugar alcohol metabolites to be higher in early PTB, including mannose (p=4x10−4, q=0.052), previously associated with uropathogens such as Escherichia coli53; arabinose (p=9x10−4, q=0.061), previously associated with bacterial vaginosis (BV)54; and mannitol/sorbitol (p=1.7x10−4, q=0.022), previously associated with PTB55. Ethylenediaminetetraacetic acid (EDTA), an additional xenobiotic whose likely-exogenous source56–58 was also confirmed by AMON (Methods; Table S3), was increased in extremely and very PTB (p=8x10−4, q=0.061 and p=1.6x10−4, q=0.044; respectively). EDTA was shown to be cytotoxic in vaginal epithelial cells59, and is teratogenic in rats at non-maternotoxic doses57,60. EDTA was detected in 100% of women in this cohort (Extended Data Fig. 5b), which is expected given its presence in the sample collection buffer, yet this is unlikely to explain these associations. Overall, we find that metabolite associations with sPTB interact with both race and sPTB timing, and detect an additional sPTB-associated xenobiotic.
Functional metabolite sets enriched for sPTB associations
We next checked whether functional groups of metabolites (e.g. KEGG pathways61; Table S4) are enriched for associations with sPTB, even if changes to any specific metabolite are small (Methods). We find significant enrichment in proline and arginine metabolism (p=0.0018, q=0.058; Extended Data Fig. 5d), consistent with our findings regarding proline and N-acetylarginine (Extended Data Fig. 5c). Additionally, and again consistent with the association between tyramine and TB among White women (Fig. 2a), we find an enrichment in metabolites related to the endocrine system among White women (p=0.0045, q=0.077; Extended Data Fig. 5d). We further identify lipid-metabolism-related metabolites to be enriched for associations with early sPTB among Black women (p=0.0019, q=0.032 and p=0.0047, q=0.038 for very and extremely PTB, respectively; Extended Data Fig. 5d), potentially related to other lipid metabolism alterations reported in PTB62. Notably, we identify a global enrichment of xenobiotics associated with sPTB among Black women (p=0.006, q=0.054; Extended Data Fig. 5d), consistent with our finding regarding specific metabolites (Fig. 2).
A network of microbe-metabolite associations in sPTB
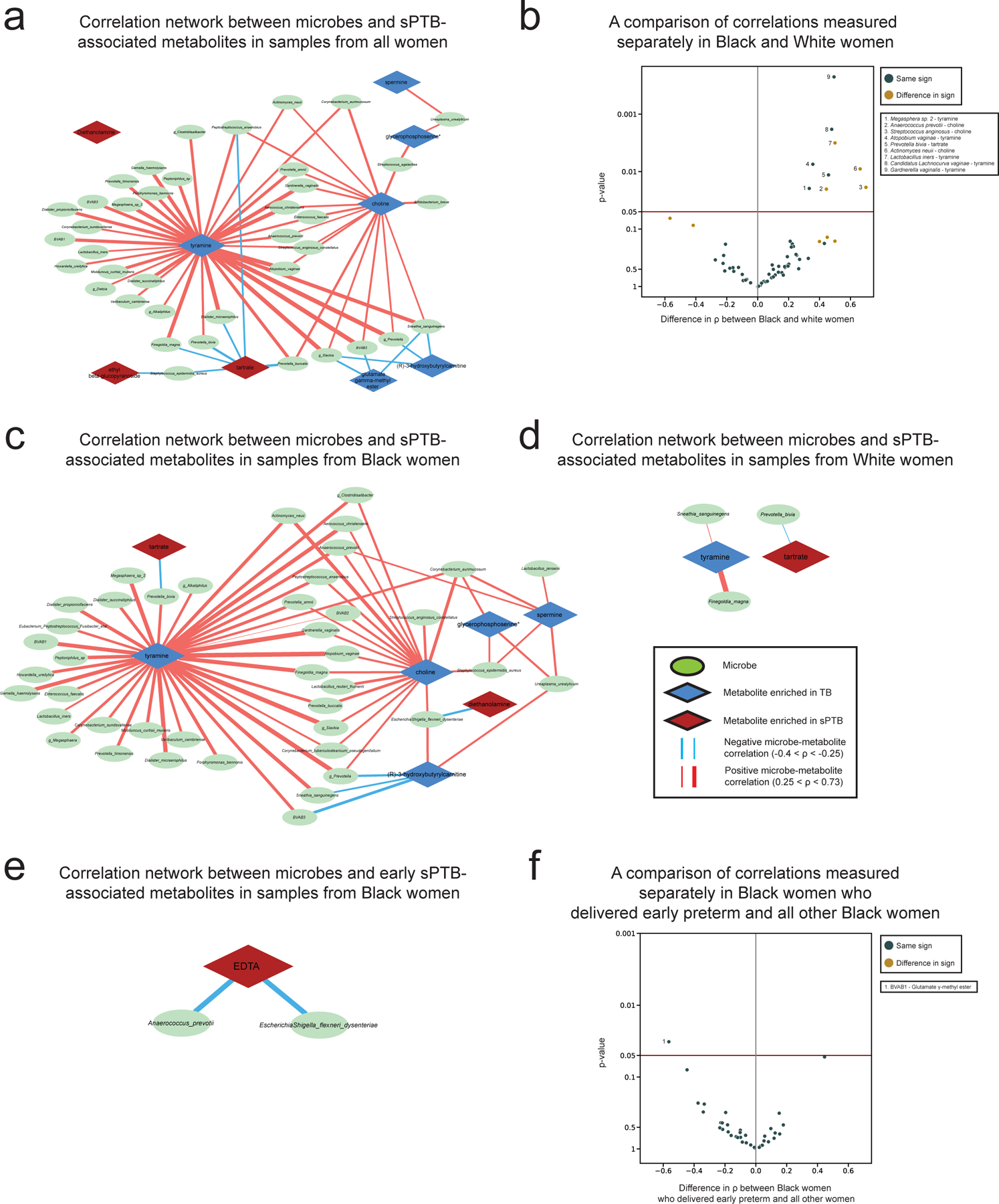
We next investigated the correlations between the estimated absolute abundances of microbial species and sPTB-associated metabolites (Methods). Contrary to metabolite associations with sPTB, we find weak interactions between microbe-metabolite associations and both race and sPTB timing (Supplementary Note 2). Our results replicate multiple known associations, such as between Dialister species or Enterococcus faecalis and tyramine63,64 (Spearman ρ>0.54, p<10−10, q<0.1 for all; Fig. 3a, Extended Data Fig. 6a), as well as evidence for choline metabolism in G. vaginalis65 and Corynebacterium aurimucosum66 (ρ=0.34, p<10−6, q=1.7x10−5 and ρ=0.40, p=4x10−4, q=0.006, respectively). Additionally, higher tyramine concentrations were previously found in BV67, supporting the associations we find with BV-associated microbes (Fig. 3a).
Figure 3 |. Microbe-metabolite correlations and metabolic models suggest sources for sPTB-associated metabolites.
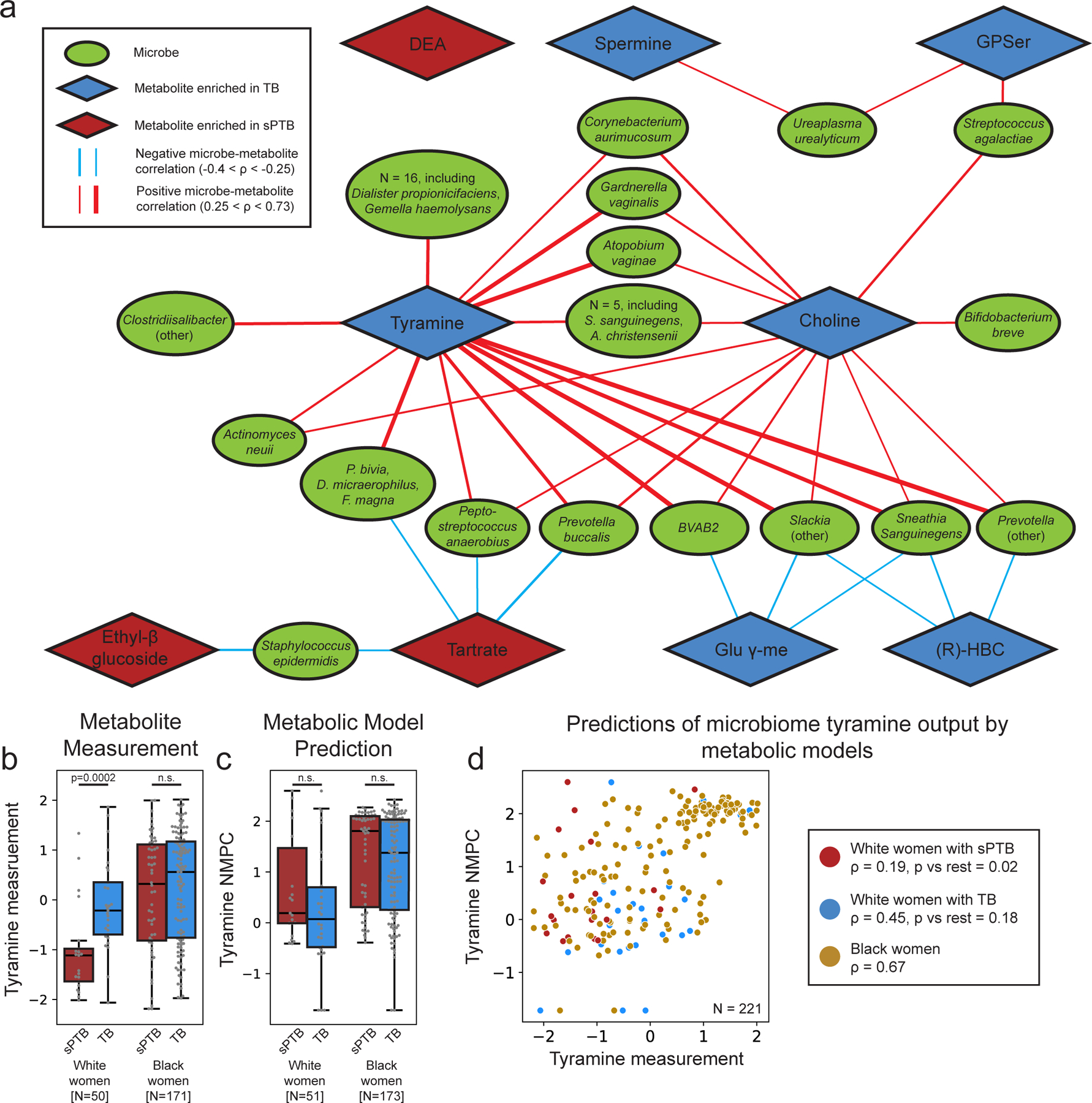
a, A network of microbial correlations with metabolites associated with sPTB. Ellipses, microbial species; blue and red diamonds, metabolites enriched in TB and sPTB, respectively; blue and red edges, negative and positive Spearman correlations with FDR<0.1, |ρ| > 0.25, respectively; edge width, median ρ. See Extended Data Fig. 6a for the same network without grouped nodes. b,c, Box and swarm plots (line, median; box, IQR; whiskers, 1.5*IQR) of tyramine levels, as measured (b) and predicted with metabolic models (Methods; c), comparing preterm and term deliveries and stratifying by maternal self-identified race. White women who delivered preterm had lower measured vaginal levels of tyramine (p=0.0002), yet our metabolic models predict higher, albeit non-statistically significant, microbiome production of tyramine in women who delivered preterm (p=0.18 and p=0.26 for all and White women, respectively). p, Two-sided Mann-Whitney U. d, Tyramine production derived from microbiome metabolic models (NMPC; Methods; y-axis) plotted against measured tyramine levels (x-axis) and colored by race and birth outcome (legend). While our models are generally accurate for tyramine (Spearman ρ=0.62, p<10−10 across all women), the accuracy for White women who delivered preterm was significantly lower (Spearman ρ=0.19, p=0.02 for comparing correlation strength vs the correlation in other women, two-sided Fisher R-to-z transform), suggesting a difference in strains, functional capacity, or a non-microbial interaction not captured by our models.
We note that xenobiotics positively associated with sPTB have significantly weaker correlations with vaginal microbes than those observed for the rest of the metabolites (Mann-Whitney U p=0.024). DEA, for example, shows only weak correlations with all vaginal microbes (ρ<0.23, q>0.1 for all microbes). This observation provides further support for an exogenous source for these metabolites.
We find the strongest and most numerous correlations for tyramine (35 associations, Spearman 0.27<ρ<0.73; Fig. 3a) which was higher in TB among White women (Fig. 2a). Eight out of the 35 tyramine-correlated microbes are also correlated with choline, which was enriched in TB across all women (Fig. 2a). Interestingly, many of the species positively correlated with TB-associated metabolites, including Atopobium vaginae, G. vaginalis, several Prevotella species, BVAB, and many others, were previously reported to be associated with negative outcomes, such as BV68, preterm birth13–15,17 and other adverse pregnancy69 and neonatal70 outcomes. We find a similarly paradoxical negative correlation between Staphylococcus epidermidis, previously associated with BV71 and late-onset sepsis in preterm neonates72, and both tartrate and ethyl glucoside (ρ=-0.28, p=6.9x10−4, q=0.009; ρ=-0.26, p=0.0015, q=0.016, respectively; Fig. 3a), which were positively associated with sPTB. Therefore, even as many of these associations were known, our results also suggest complex interactions between suboptimal vaginal microbes, sPTB-associated metabolites, and health outcomes.
Metabolic models support microbiome production of tyramine
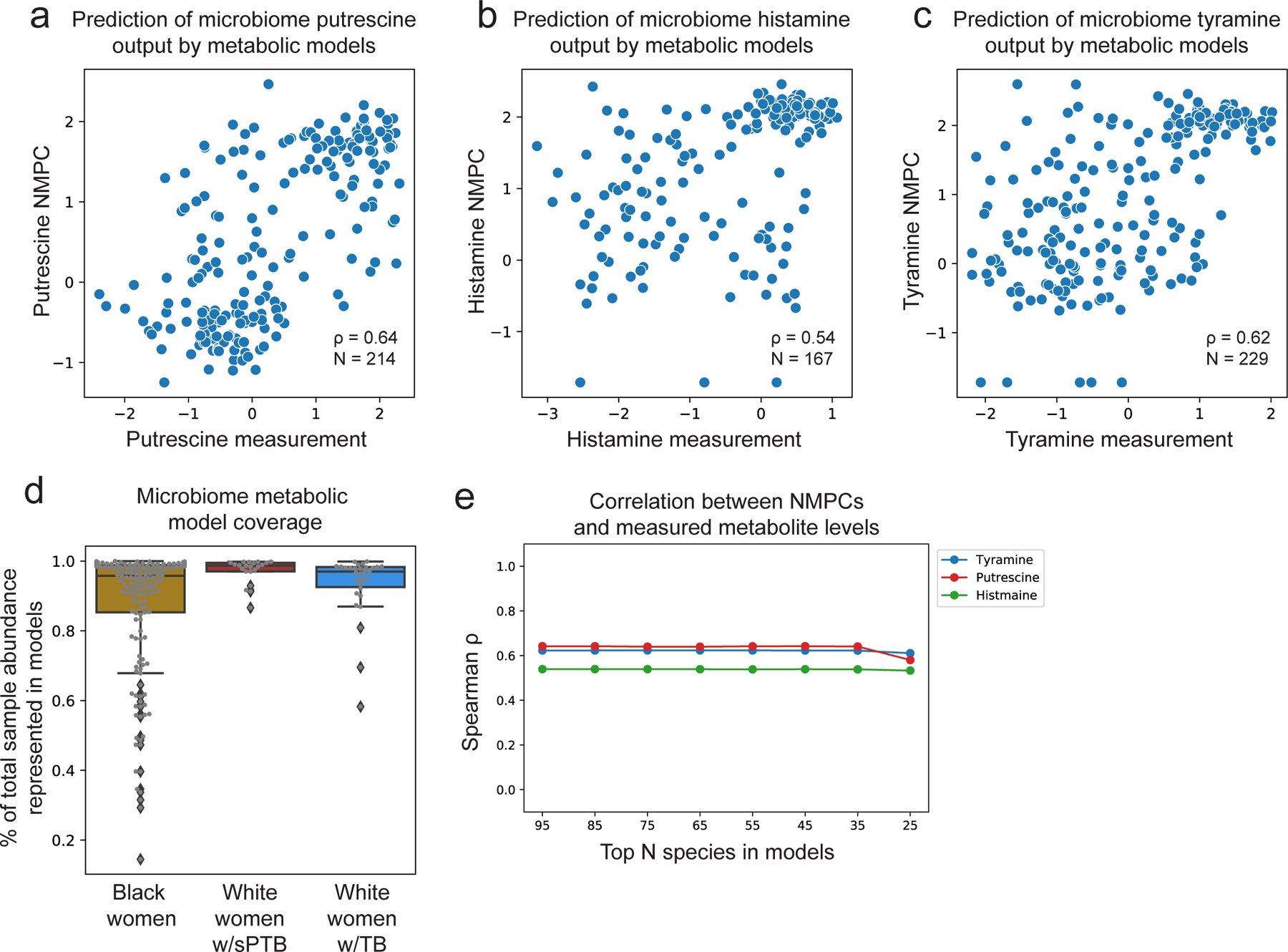
To gain some mechanistic insight into the correlations we found, we used community-level metabolic models73, which integrate genetic and biochemical knowledge to predict the metabolic output of each microbiome sample (community net maximal production capacity73 [NMPC]; Methods). Our models show accurate predictions for several metabolites known to be produced by the vaginal microbiome63,74, such as putrescine and histamine (Spearman ρ=0.64 between NMPCs and metabolomic measurements, N=214, p<10−10 and ρ=0.54, N=167, p<10−10, respectively; Extended Data Fig. 7a,b).
Two sPTB-associated metabolites, tyramine and choline, were represented in our models. As our models predicted that choline was not affected by the vaginal microbiome (NMPCs of 0 for all women), we focused on tyramine, which previous studies suggest is produced by vaginal microbes63,74. Following genomic curation (Methods), the predictions of our models were highly accurate (Spearman ρ=0.62, N=229, p<10−10; Extended Data Fig. 7c). Interestingly we find that, among White women, while the measured levels of tyramine were enriched in TB (Mann-Whitney U p=2.8x10−4; Fig. 3b), its predicted microbiome output was not, and was even somewhat higher in sPTB (p=0.26; Fig. 3c). This stems from lower accuracy in tyramine predictions in White women who delivered preterm (Spearman ρ=0.19 versus ρ=0.65, p=0.02 for difference in ρ’s; Fig. 3d).
This difference in accuracy could not be explained by the representation of microbes in the metabolic models, which was in fact lower in Black women (Mann-Whitney p=0.05, Extended Data Fig. 7d), likely due to the generally higher vaginal microbial diversity in this population75. Furthermore, tyramine prediction accuracy was not sensitive to constraints on metabolite uptakes or to the representation of low abundance taxa (Methods; Table S5; Extended Data Fig. 7e). As these analyses suggest that lower tyramine prediction accuracy in White women with sPTB is not the result of a modeling artifact, the different accuracy could stem from a difference in strains, functional capacity, or a non-microbial effect. Either phenomenon also has the potential to explain the aforementioned paradoxical microbial associations with tyramine (Fig. 3a). The possibility of a microbial difference or a host effect is also supported by AMON37, which predicts that tyramine is either microbial or host derived (Table S3). Overall, our results demonstrate the utility of metabolic models in studying microbiome-metabolome interactions, and raise intriguing hypotheses for further investigation.
Early prediction of sPTB risk using the vaginal metabolome
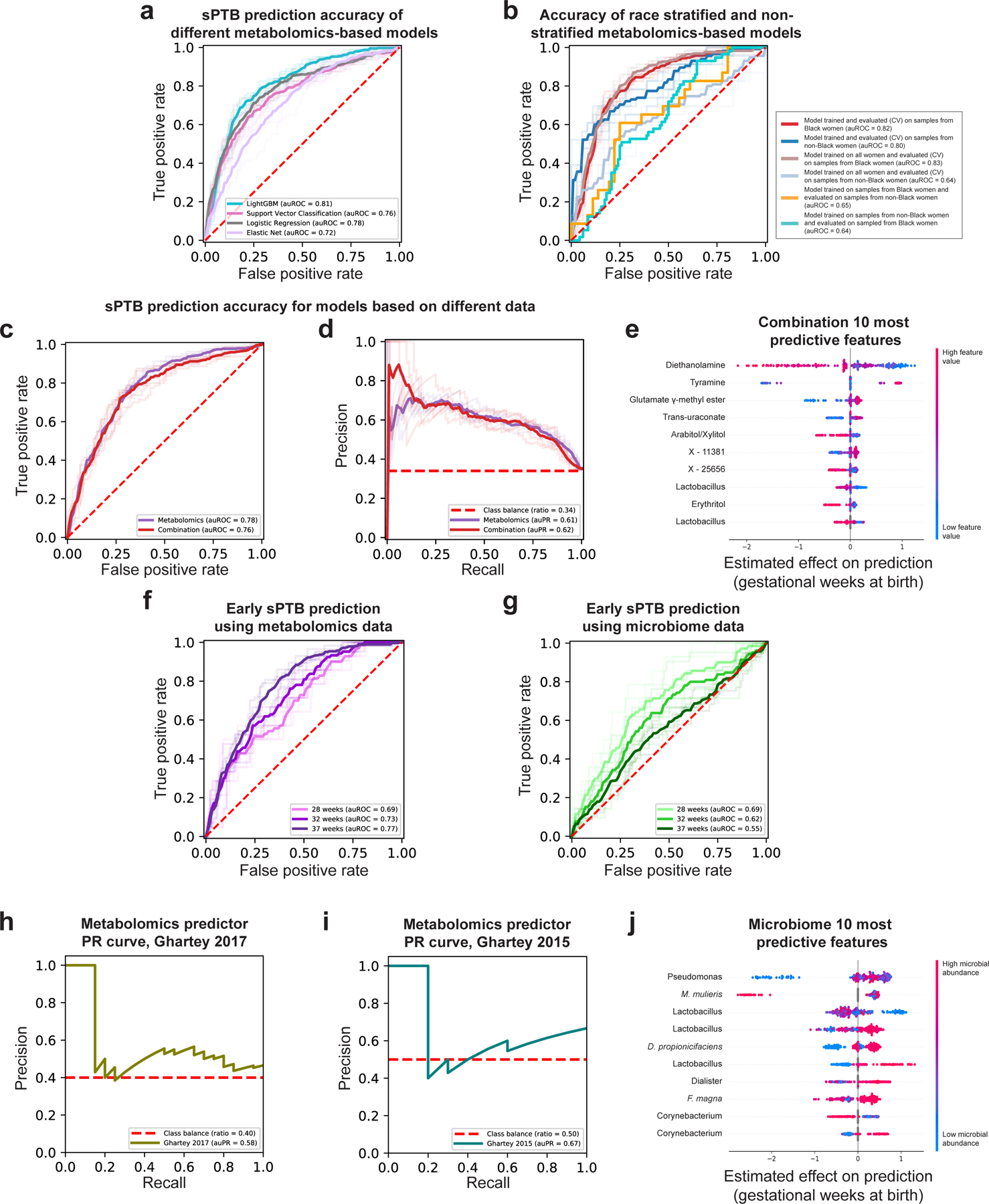
Early diagnosis of pregnancies with high risk for prematurity is crucial for the development of prevention and intervention strategies. We therefore explored whether we can use clinical, microbiome or metabolome data, collected ~3 months prior to delivery (mean±std of 14.5±4.2 weeks), to predict subsequent sPTB. We used boosted decision trees, which were superior to alternative models (Extended Data Fig. 8a). For microbiome- and metabolome-based models, we trained composite predictors, such that a separate model was used for White and Black women. Despite the smaller effective sample size for each model, this resulted in better performance (Extended Data Fig. 8b). We evaluated all models on held-out samples using nested cross-validation without test-data leakage (Methods).
Our models using clinical (age, BMI, race, PTB history and nulliparity) and microbial abundances data obtained limited accuracy (area under receiver operating characteristic [auROC]=0.59, area under precision-recall curve [auPR]=0.46 for clinical data; auROC=0.55, auPR=0.41 for microbiome data; p=0.12 for difference between the models; Methods; Fig. 4a,b). Notably, using metabolomics data, we were able to generate a model with superior accuracy (auROC=0.78, auPR=0.61; p<10−10 for comparison with either clinical or microbiome models; Methods; Fig. 4a,b). Lastly, a model combining clinical, microbiome, and metabolomics data obtained similar accuracy to the metabolome-based model (auROC=0.76, auPR=0.62; p=0.44 vs. metabolome-based model; Extended Data Fig. 8c,d), with metabolites as the most prominent contributors to the model (Extended Data Fig. 8e). This suggests that metabolite measurements are a sufficient representation of information contained in these three data types.
Figure 4 |. Metabolomics-based prediction of subsequent spontaneous preterm birth.
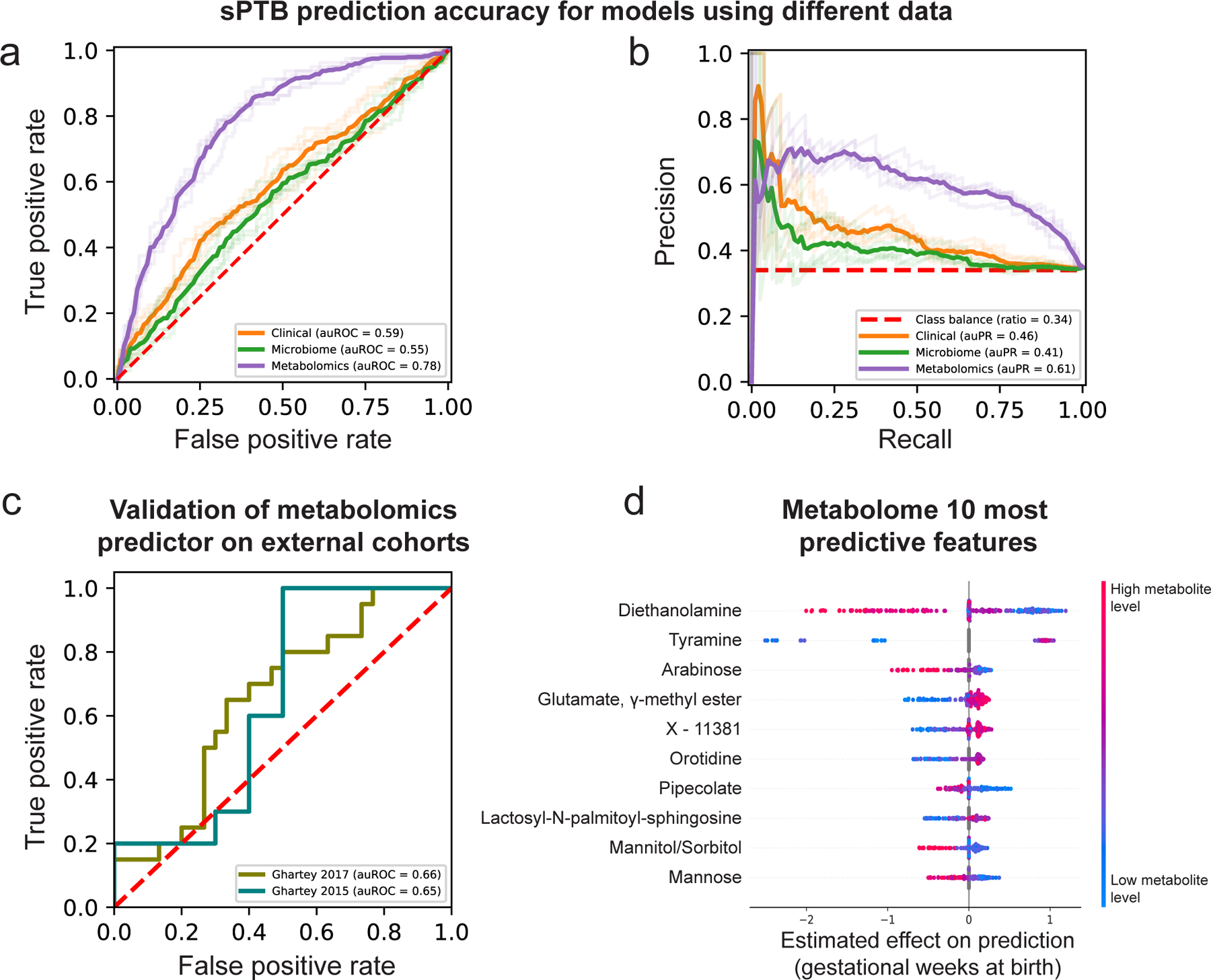
a,b, Receiver operating characteristic (ROC, a) and precision-recall (PR, b) curves comparing sPTB prediction accuracy for models based on clinical (auROC=0.59, auPR=0.46), microbiome (auROC=0.55, auPR=0.41) and metabolomics (auROC=0.78, auPR=0.61) data (legend), evaluated in nested cross-validation (Methods). N=232 for all. Shaded lines show results from five independent and outer 10-fold cross-validation draws (Methods). c, ROC curve evaluating the performance of our metabolomics-based predictor on two external cohorts. Despite a challenging replication setting, with different inclusion criteria, measured metabolites, and batch effects, our predictor obtains relatively accurate predictions without retraining (auROC=0.66, auROC=0.65, for the Ghartey 2017 [N=50] and 2015 [N=20] cohorts, respectively; Methods). d, Effect on total prediction (SHAP-based83; x-axis) for the 10 most predictive metabolites in our metabolomics-based predictor, sorted with descending importance. Each dot represents a specific sample, with the color corresponding to the relative level of the metabolite in the sample compared to all other samples.
Our metabolome-based model is superior or similar in accuracy to several previously-published models, such as those using amniotic fluid metabolomics (auROC=0.65–0.70, N=24)76, maternal serum metabolome and clinical data (auROC=0.73, N=164)77, maternal urine and plasma metabolome (auROC=0.69–0.79, N=146)78, blood cell-free RNA measurements (auROC=0.81, N=38)79, or vaginal protein biomarkers (auROC=0.86, N=150, sPTB N=11)80, many of which have small sample sizes, lack demographic diversity, or focus on high-risk cohorts. Overall, our results demonstrate the promising utility of vaginal metabolites as early and accurate biomarkers of sPTB.
We next evaluated the same models, without retraining, for predicting extremely and very PTB in Black women from the same held-out data (i.e., only the ground-truth classification of outcome changed). Interestingly, while the metabolome-based model shows a slight decrease in accuracy (auROC of 0.69 and 0.73 for extremely and very PTB, respectively, compared to auROC=0.77 for sPTB in Black women; p=4.3x10−4 and p=0.001, respectively; Extended Data Fig. 8f), our microbiome-based model shows increasing accuracy (auROC of 0.69 and 0.62, respectively, compared to auROC=0.55; p=0.031 and p=0.49, respectively; Extended Data Fig. 8g). These results may reflect the potentially increased involvement of the vaginal microbiome in earlier sPTBs1.
Metabolome-based predictor replicates in external cohorts
To test the generalizability of our metabolome-based model, we validated its accuracy in two independent cohorts (Methods): a case-control study of 20 women (10 PTB), mostly (75%) White, at high-risk for PTB, with samples collected at 24–28 weeks of gestation (“Ghartey 2015”)81; and a case-control study of 50 women (20 PTB), mostly (88%) Black, presenting with symptoms of preterm labor and no PTB history, with samples collected at 22–34 weeks of gestation (“Ghartey 2017”)55.
This validation is extremely challenging: due to the different inclusion criteria and population structure; due to significant batch effects in metabolomics measurements across different studies82; and finally, as data was generated 4–6 years earlier, only a small fraction of metabolites used by our predictor were measured (34% and 39%). To emphasize this, only one and two (for Ghartey 2015 and 2017, respectively) of the 10 associations we detected between vaginal metabolites and sPTB (Fig. 2a) could be examined in these cohorts (Methods), of which none were significant (Mann-Whitney p>0.05). These sPTB-associated metabolites are likely important features for prediction, making generalization across these cohorts difficult. Despite this challenging setting, our metabolome-based predictor, trained only on the 232 samples profiled here, without any retraining or adaptation, provided relatively accurate predictions in both external cohorts (auROC=0.65, auPR=0.67 and auROC=0.66, auPR=0.58 for Ghartey 2015 and 2017, respectively; Fig. 4c, Extended Data Fig. 8h,i). These results demonstrate the robustness of the vaginal metabolome and of our predictive approach to study-specific biases.
Model interpretation reveals other contributing features
To obtain insights into the features used by the models, we assessed the contribution of each feature towards the prediction for each sample using SHAP83 (Table S6). As expected, six of the ten most predictive metabolites, namely DEA, tyramine, arabinose, glutamate gamma-methyl ester, mannitol/sorbitol and mannose, were also identified in our association analysis, with a similar direction of association (Fig. 2, 4d). We additionally find that high pipecolate levels and low levels of lactosyl-N-palmitoyl-sphingosine and orotidine contribute to sPTB predictions. Of these, pipecolate was shown to be elevated in women with BV84.
A similar analysis of our microbiome-based predictor also captures previously-detected associations between vaginal microbes and sPTB, including those of M. mulieris14 and Finegoldia magna85, and of Lactobacillus14 and Dialister species15 (Extended Data Fig. 8j). These results highlight the interpretability of our models and their ability to model complex non-linear interactions, enabling us to expose associations not detected by univariate analyses.
Discussion
In this study, we measured the second trimester vaginal metabolome of 232 pregnant women. We show that it is associated with the vaginal microbiome, and that metabolite signatures are enriched for sPTB among Black women. We identify multiple metabolites that are associated with sPTB, across the cohort and separately for Black and White women. Our results highlight exogenous metabolites with strong associations with sPTB, which we suggest constitute important risk factors. We further uncover intriguing interactions between TB-associated metabolites and potentially suboptimal microbes, and propose a difference in the vaginal metabolism of tyramine in White women who delivered preterm. Finally, we demonstrate that metabolome-based models can predict subsequent sPTB weeks to months in advance, potentially paving the way for early diagnostics.
We detected several sPTB-associated xenobiotics, DEA, ethyl glucoside, tartrate, and EDTA, which prior literature and a functional analysis37 suggest are of exogenous source. DEA, a chemical with no known natural source86, commonly used in drilling and metalworking fluids35, and to which reproductive-aged women are highly exposed87; and ethyl glucoside, present in alcohol-containing products31; are both precursors or ingredients in hygienic and cosmetic products30,33. Tartrate and EDTA are used as food additives32,58 and are also common in hygienic and cosmetic products32,57. While we have not identified the sources of these metabolites, the fact that all are documented in hygienic and cosmetic products raises concern that some of these products may increase the risk for sPTB. Our results coincide with recent studies raising concerns regarding environmental exposures in pregnancy88,89, and identify these chemicals in the reproductive tract. Further study is warranted to identify the sources of these metabolites and to disentangle their effects on the host, microbiome, and pregnancy outcomes, so that policy recommendations can be made regarding their use in various products and during pregnancy.
The cohort we analyzed includes a majority of Black women, offering an opportunity to study PTB in women who are disproportionately burdened by PTB and other adverse pregnancy outcomes, while also represented in small numbers in many studies. However, we urge caution in drawing conclusions from differences in associations between Black and White women, as maternal self-identified race represents a complex array of preexisting differences, disparities, and clinical covariates at the time of sampling. Nevertheless, we note that the enrichment of sPTB associations among the xenobiotic metabolite set in Black women may potentially reflect disparities in environmental and exogenous exposures90,91, consistent with reports that Black women have greater exposures to endocrine disrupting chemicals through personal care products92,93 and with studies that identified exogenous chemicals as possible drivers of PTB94,95. Metabolomic exposure patterns could contribute to the association between racial disparities in prematurity rates and racial differences in the vaginal microbiome96.
We used community-scale metabolic models to investigate microbial tyramine metabolism, which have important limitations. Model curation is an ongoing effort, and thus models may not be tailored to each sample or may lack representation of niche-specific metabolic capabilities. Another limitation stems from the resolution of 16S rRNA amplicon sequencing, which identifies taxa at the species or genus level, precluding strain-specific modeling. Despite these limitations, our models accurately predicted several metabolites, and offered insights regarding potential sources of tyramine.
Our predictive modeling approach has several noteworthy limitations: (1) Our use of a case-control cohort enriched for PTB limits our ability to assess population-level predictive value, and further validation is required in prospective studies. (2) As this cohort was focused on sPTB, we are unable to assess if our models are specific to sPTB or are detecting a general risk for adverse pregnancy outcomes. (3) The use of race in our models, while common throughout medicine97, is controversial and creates issues in implementation98. This was driven by differences in both sample size and the vaginal metabolome itself between Black and White women in this cohort, and resulted in an overall increased accuracy. (4) Finally, there is additional unexplored potential in using even earlier samples for prediction. A larger sample size, and combination with other sources of data, such as maternal urine or serum metabolomics, vaginal metagenomics, or cell free RNA measurements, could further improve prediction accuracy.
Our results demonstrate the utility of vaginal metabolites as early biomarkers of PTB, and identify xenobiotic metabolites as potentially modifiable sPTB risk factors, which may also disproportionately affect Black women. The strong associations we observe motivate the investigation of the vaginal microbiome and metabolome in the context of other adverse pregnancy outcomes such as preeclampsia, indicated preterm birth and bacterial vaginosis.
Methods
Study design and cohort description
We analyzed banked samples from the previously collected and described Motherhood & Microbiome (M&M) cohort (NCT02030106)14. This cohort was approved by the Institutional Review Board at the University of Pennsylvania (IRB #818914) and the University of Maryland School of Medicine (HP-00045398), and all participants provided written informed consent. The M&M cohort recruited 2,000 women with a singleton pregnancy prior to 20 weeks of gestation. Women were followed to delivery, and spontaneous preterm birth was defined as delivery before 37 weeks of gestation with a presentation of cervical dilation and/or premature rupture of membranes. Of these, the vaginal microbiota of 503 women was previously characterized via 16S rRNA gene amplicon sequencing (V3-V4 region) of vaginal swabs collected between 20 to 24 weeks of gestation, and total bacterial load was assessed using the TaqMan® BactQuant assay14. For this study, out of women with available microbiome data, all available samples were selected from women who delivered preterm (N=80), in addition to samples from 152 controls who delivered at term. The selected cervicovaginal samples were replicates of those used for 16S rRNA gene sequencing, collected using a double shaft dacron swab. Cervicovaginal swabs were either self-collected or collected by a research coordinator during a study visit14.
Statistics and reproducibility
No data was excluded from analysis in the present study. No statistical method was used to predetermine sample size. As the study was observational, there was no allocation or randomization. The study included all available samples who delivered preterm (N=80), and no statistical methods were used to predetermine sample sizes; our sample size is similar to those reported in previous publications25,26. Samples were randomly distributed across metabolomics batches and metabolomics analysis was performed by Metabolon Inc. (Durham, NC, USA), who were blinded to the outcome assessment of each sample. Two-sided Mann-Whitney U tests (SciPy 1.5.2) and logistic regression (Statsmodels 0.12.1) were used to identify associations between metabolite levels and sPTB. Two-sided Fisher’s exact tests (R stats 3.6.1) were used to identify associations between MCs, CSTs, race, and sPTB. PERMANOVA tests (scikit-bio 0.5.6) were used to identify associations between the microbiome, metabolome, and CST, race, and metabolomics batches. Metabolite set enrichment analysis (Methods) was used to identify associations between metabolite sets and sPTB. Spearman correlations were used to measure the agreement between metabolite levels and NMPCs and between metabolite levels and microbial abundances. Fisher-R-to-z transform was used to compare correlations measured within subgroups. Evaluation of machine learning models was performed using scikit-learn 0.24.2. pandas 1.1.5 and NumPy 1.18.5 were used for data processing. Robust assessment of generalization error of predictive models was achieved via nested cross-validation.
Metabolomics profiling and preprocessing
Metabolite levels were measured from vaginal swabs by Metabolon Inc. (Durham, NC, USA), using an untargeted LC MS/MS platform99. See Supplementary Note 1 and Extended Data Fig. 2 for discussion of batch processing of the samples. We note that swab lot number, sterile swabs for blank processing, and sample collector (coordinator or self-collection) are not available. While this limits analysis of potential batch effects, we find batch confounding (e.g., swab lot associated with sPTB) unlikely as samples were collected prior to delivery and outcome determination.
Following a methanol-based small molecule extraction, samples were divided into 5 µl aliquots and each was resuspended in an appropriate extraction solvent and separated via one of four chromatography techniques. Each chromatographic method was optimized for the extraction of hydrophobic, basic, or polar compounds. The chromatographic method used for the quantification of each metabolite is provided in Table S4. Isotopically labeled or halogenated standards were added to all aliquots at fixed concentrations prior to extraction in order to serve as retention time markers. Following extraction, compounds were subjected to electrospray ionization and measured via tandem mass spectrometry by a Q-Exactive Hybrid Quadrupole-Orbitrap high resolution mass spectrometer. Data dependent acquisition mode was used to generate fragmentation spectra of high intensity m/z peaks detected during the first round of mass spectrometry. m/z peaks were identified and annotated by Metabolon Inc. (Durham, NC, USA) using proprietary software and comparisons to their database of retention indices and fragment ion spectra. The areas under annotated m/z peaks were taken as metabolite measurements. A comprehensive overview of all chromatographic and mass spectrometry parameters is available in Table S7. Process blanks (negative controls) were run with each metabolomic plate, and metabolites were considered present only if they are detected with levels that are at least 3 times higher than these controls. Detected levels of the xenobiotics highlighted in this study, in vaginal samples and negative controls, are shown in Extended Data Fig. 5e, demonstrating the same. See also Extended Data Fig. 5f for the mass error of these xenobiotics, showing high identification quality compared to other non-xenobiotic metabolites.
While the majority of named metabolites (N=556) were tier 1 identified by Metabolon via fragmentation spectra matches to experimentally measured library standards, only tier 2 assignments are available for independent identification due to the proprietary nature of the Metabolon platform. Metabolite measurements were volume normalized to the volume of buffer used, which may not necessarily account for differences in the original tissue. This was followed by robust standardization27 of the log (base 10) transformed values (subtracting the median and dividing by the standard deviation calculated while clipping the top and bottom 5% of outliers). The Shapiro-Wilk test was used to determine that log (base 10) transformed values deviated from normality for the majority of metabolites (389 of 635 named metabolites). For this reason, non-parametric tests were used in subsequent metabolomic analyses.
Microbiome data processing
All microbiome-based analyses were done using data previously processed with DADA2100 and SpeciateIT14, available from Supplementary Data 2 of ref. 14. A single exception to this are predictive models, which were trained on 97%-clustered OTUs using the USEARCH pipeline101. We obtained raw sequences from the database of Genotypes and Phenotypes (dbGaP) under study accession: phs001739.v1.p1. Primers were aligned to reads and then trimmed, followed by end merging and quality filtering (-fastq_maxee 1.0). The filtered reads were then pooled together, dereplicated, clustered with a 97% threshold, and chimera filtered with the UPARSE algorithm to produce the OTU count matrix.
Global microbiome and metabolome structure
PERMANOVA analysis was performed using Bray-Curtis distance for microbiome data and the Canberra distance for metabolites data, which is robust to outliers and sensitive to differences in common features. We determined the optimal number of clusters by comparing the within cluster sum of square error and the gap statistic for clustering solutions with K between 1 and 15 (Extended Data Fig. 3a,b). To check the robustness and consistency of these clusters, we performed 100 random selections of 209 (90%) of the 232 samples, recreating clusters de novo with the same procedure for each random subset. Many of the resulting subsets (36) had over 95% of samples assigned to the same metabolite cluster as the original assignment (Table S2), with an average assignment accuracy of 86% across all random subsets (Extended Data Fig. 3g), demonstrating that our metabolite clusters are indeed consistent. Uniform Manifold Approximation and Projection (UMAP)102 was performed using the Python umap-learn package103, with n_neighbors=15 and min_dist=0.05 for microbiome data and n_neighbors=15 and min_dist=0.25 for metabolomics data. To further describe each metabolomics cluster, Fisher’s exact test was used to identify metabolite super and sub pathways enriched among metabolites associated with each cluster (p<0.05).
Differential abundance testing and metabolite set enrichment analysis
Differential abundance tests between metabolite levels were done using the two-sided Mann-Whitney U test for metabolites which were present in at least half of the cases. All associations with early PTB were calculated using only samples from Black women, due to their high proportion among these deliveries (21 of 26 for childbirths <32 weeks of gestation and 14 of 15 for childbirth <28 weeks). To identify functional sets of metabolites that were perturbed between sPTB and TB, we compared, for each set, the Mann-Whitney p values for differential abundance between PTB and sPTB for metabolites within the set to the same p values for metabolites outside the sets, using an additional Mann-Whitney U test. We calculated significance by comparing the p value of the latter test to 10,000 similar p values calculated on random permutations of sPTB and TB labels. For functional sets, we used definitions of super and sub-pathways provided by Metabolon, as well as KEGG61 pathways. FDR correction was performed separately for each metabolite set type.
Prediction of metabolite origins using AMON
AMON37 is a method that uses functional annotations according to the KEGG database61 to predict metabolite origins for all metabolites which could be matched to a KEGG entry (N=334 of 635 named metabolites). We used PICRUSt2103 to generate functional profiles for each sample, and then applied AMON37 to predict whether metabolites that had matching entries in the KEGG Database are products of human or microbial metabolism. When both were false, we interpreted the metabolite to be a xenobiotic.
Microbe-metabolite correlations
To identify associations between microbes and metabolites we estimated microbial absolute abundance by multiplying the relative abundances of each taxon by the total 16S rRNA copy number for the sample, obtained using the TaqMan qPCR-based panel14,104,105, and calculated Spearman correlations with the levels of metabolites we found to be associated with sPTB. Across all correlation network analyses (Fig. 3a, Extended Data Fig. 6a,c,d,e) we included correlations with at least 22% of paired measurements, corresponding to 50 samples of 232 for Fig. 3a. All correlation measurements used available data without imputation, and correction for multiple testing was performed via the Benjamini-Hochberg FDR method. To determine whether edges in our network were influenced by race (Extended Data Fig. 6b) or by the severity of sPTB (Extended Data Fig. 6f), we used a two-sided Fisher R-to-z transform to compare these correlations in Black women to the same correlations in White women as well as compare these correlations in Black women who delivered prior to 32 weeks to the same correlations in all other Black women.
Creating and interrogating vaginal microbiome models
Microbiome metabolic modeling was done using Microbiome Modeling Toolbox (COBRA toolbox commit: 71c117305231f77a0292856e292b95ab32040711)73,106, using models from AGORA2107. All computations were performed in MATLAB version 2019a (Mathworks, Inc.), using the IBM CPLEX (IBM, Inc.) 12.10.0 solver.
For each sample, tailored microbiome models were created through the compartmentalization technique108: metabolic reconstructions of species present in the sample are merged into a shared compartment, and input and output compartments are added. The shared compartment enables microbes to share metabolites while input and output compartments are present to enable compounds intake and secretion. Coupling constraints are added as in refs. 109,110 to ensure a dependency between relative abundances and each species network fluxes. Finally, sample-specific microbiome biomass objective functions, composed by the sum of each microbial biomass multiplied by the corresponding relative abundance value, are added to each microbiome model.
To interrogate the secretion potential of each sample-specific microbiome model, we computed Net Maximal Production Capacities (NMPCs) using the pipeline mgPipe.m of the Microbiome Modeling Toolbox73 (Table S8). NMPC calculation accounts for maximal microbiome compound production and uptake rates and aims at predicting the overall contribution of microbiomes to the metabolism of specific compounds73. To later assess prediction accuracy, we computed Spearman correlations between NMPCs and the corresponding metabolite measurements without imputation.
To support and improve the accuracy of our tyramine predictions, we validated the presence of the TDC gene, coding for tyrosine decarboxylase. For each species represented in our metabolic models (N=95), we used Prodigal111 to predict open reading frames in up to 200 randomly selected Refseq112 assemblies, and searched them for evidence of TDC using the hmmsearch function of Hmmer3.3.2113 and a profile hmm for TDC114 (NCBI HMM accession: TIGR03811.1). We then curated our metabolic models, making sure that the corresponding reaction exists in models for which at least one assembly contained the corresponding gene.
To compile the metabolic models, we matched between the species detected in the microbiome samples and those present in AGORA2107 (Table S9). To increase the representativeness of our models, we added three representatives for abundant vaginal species without a corresponding AGORA2 model that were present with >5% relative abundance in at least 20 samples (listed in Table S9). The only species that passed this threshold which was not included in our models was Ca. L. vaginae (BVAB1), for which no suitable AGORA model was available. To generate species level models, we combined metabolic models from available strains using the function createPanModels.m of the Microbiome Modeling Toolbox73. Altogether, our microbiome metabolic models included 95 different species, with an average of 20 species in each sample. As the vaginal microbiome has a very skewed distribution28, this resulted in a median [IQR] of 96.7% [88.4–98.8%] of the total abundance across samples represented by our models (Extended Data Fig. 7d).
As a test of the sensitivity of our models to the lack of representation of low abundance microbes, we performed simulations where we iteratively removed the 10 least abundant species from consideration by our models, and evaluated the accuracy of our models in predicting the well-modeled metabolites tyramine, putrescine, and histamine. As expected, as our models account for the abundance of each microbe and as the vaginal microbiome has a skewed distribution, our models were not sensitive to the representation of low abundance microbes (Extended Data Fig. 7e), even when removing 70 out of 95 models.
Metabolic modeling requires environmental conditions such as media and carbon source availability115. We therefore formulated a “general vaginal media” (Table S10), as the union of all metabolites present in at least 50 samples to which a corresponding metabolite was identified in AGORA, assuming them to be present in an unlimited (i.e., very high) concentration. This vaginal media was applied to each microbiome model input compartment in the form of constraints on metabolite uptake reactions, constraining uptake of compounds not present in the environment to zero. Uptake of specific gut-related dietary compounds, automatically performed in mgPipe, was disabled acknowledging the different metabolic environment in the vagina, and essential metabolites required for achieving microbiome growth, together with their respective flux value, were detected and added to the vaginal media using the fastFVA and findMIIS functions of the COBRA Toolbox106. A comparison of the “general” media to subgroup-specific media, defined as metabolites present in 75% of samples from Black and White women separately, with uptake fluxes constrained to the mean value across the subgroup; and to a person-specific media, in which uptake fluxes were constrained for each sample separately, showed similar accuracy with respect to tyramine predictions (Table S5).
Training, testing, and validation of sPTB classifiers
We constructed predictive models separately using the clinical (age, race, parity status, history of sPTB, and BMI), microbiome, and metabolomics data, as well as a combination model consisting of all of these data types combined. As race had very strong interactions with microbiome and metabolomics data, we trained a composite predictor for microbiome, metabolomics, and combination models, whereas a separate model was trained for Black women. Despite the smaller sample size for each model, this empirically improved prediction performance (Extended Data Fig. 8b). Microbiome-based models used absolute abundances, calculated from USEARCH-processed OTUs as described above. In cases where qPCR based total load was not available (N=14), it was imputed to the mean total load using only training samples.
Samples were split into training and test sets using 10-fold cross validation (“outer folds”), block-stratified for deciles of gestational age at birth (GAB), and for microbiome, metabolomics, and combined models, also stratified for race. To account for stochasticity in the division to 10 folds, we repeated this process 5 times. Train-test sterility was strictly maintained. To tune the optimal set of hyperparameters (including parameters for feature engineering and selection), and to obtain a robust estimate of the generalization error, we used nested cross-validation. In this extension of the training-test-validation framework, the training set was further split to 5 folds (“inner folds”), on which we used 1,000 iterations of a random set of hyperparameters (Table S11). Once more, to account for stochasticity, we repeated this process 5 times. We selected the best hyperparameter set as the model with the top average auROC score out of the top 5 most accurate models based on average R2 for sPTB classification, based on performance on the inner folds. We then used these hyperparameters to train a model on the entire training data for the outer fold, and evaluated it on the held-out test data. Of note, in this framework, hyperparameters are selected using strictly the training data of each outer 10-fold cross-validation fold, and are evaluated just once on the test set. Our prediction pipeline included standardization and imputation (for metabolomics data), optional PCA transformation, and feature selection using sparsity, SHAP83 feature importance, information gain and/or Spearman correlation, followed by prediction using LightGBM116, with all steps performed strictly using training data. The selected models were then evaluated, without retraining, on classification of extremely (GAB < 28 weeks) or very (GAB < 32 weeks) PTB on the outer fold. Benchmark analyses (Extended Data Fig. 8a,b) were done using 10-fold cross-validation, repeated 5 times. We assessed the significance of the difference in auROC between two models by computing z-scores of the normal distributions of auROCs117.
To obtain a final model for interpretation and validation, we trained new composite models on the entire cohort (N=232), using the hyperparameters selected for each of the outer folds (50 models), and picked the model with the best auROC on the same cohort (training fit). The final parameter set for each model is listed in Table S12. For validation on external vaginal metabolome datasets, we note that information on maternal race at the subject level was not available to us. We therefore applied the metabolomics model used for non-Black women, without retraining or adaptation, to metabolomics data from the Ghartey 201581 cohort, as this cohort contained mostly White women; and similarly applied the metabolomics model used for Black women to metabolomics data from the Ghartey 201755 cohort. For validation of associations of metabolites with sPTB (Fig. 2a) in these cohorts, we note that of the 10 metabolites in Fig. 2a, only the six that apply to all and White women can be validated in the Ghartey 2015 cohort, of which only one was measured; and only the nine that apply to all and Black women can be validated in the Ghartey 2017 cohort, of which only two were measured.
Extended Data
Extended Data Figure 1 |. Prevalence and super pathway of assayed metabolites.
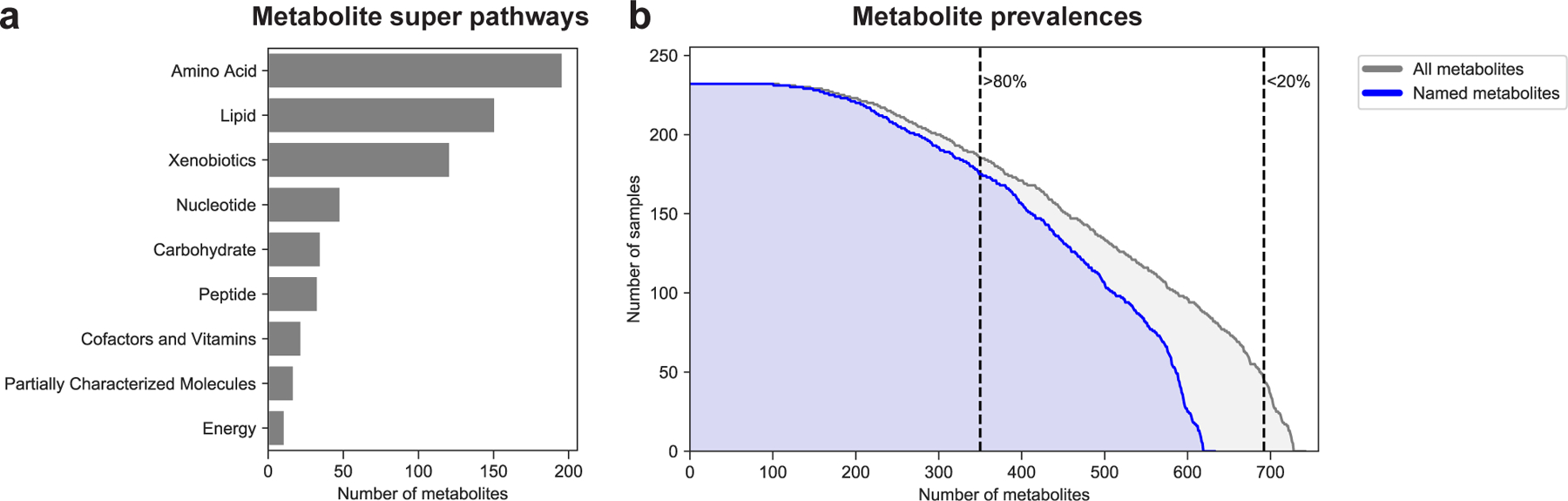
a, Distribution of metabolite super pathways among assayed metabolites. Metabolite super pathway assignments were provided by Metabolon Inc. (Durham, NC, USA). b, Distribution of metabolite prevalences across samples. Gray distribution reflects prevalences of all metabolites (N=745). Blue distribution only reflects prevalences of named metabolites (N=635). Dashed lines distinguish metabolites prevalent in more than 80% (N=352) and more than 20% of samples (N=694).
Extended Data Figure 2 |. Robustness of analyses to metabolomics batch effects.
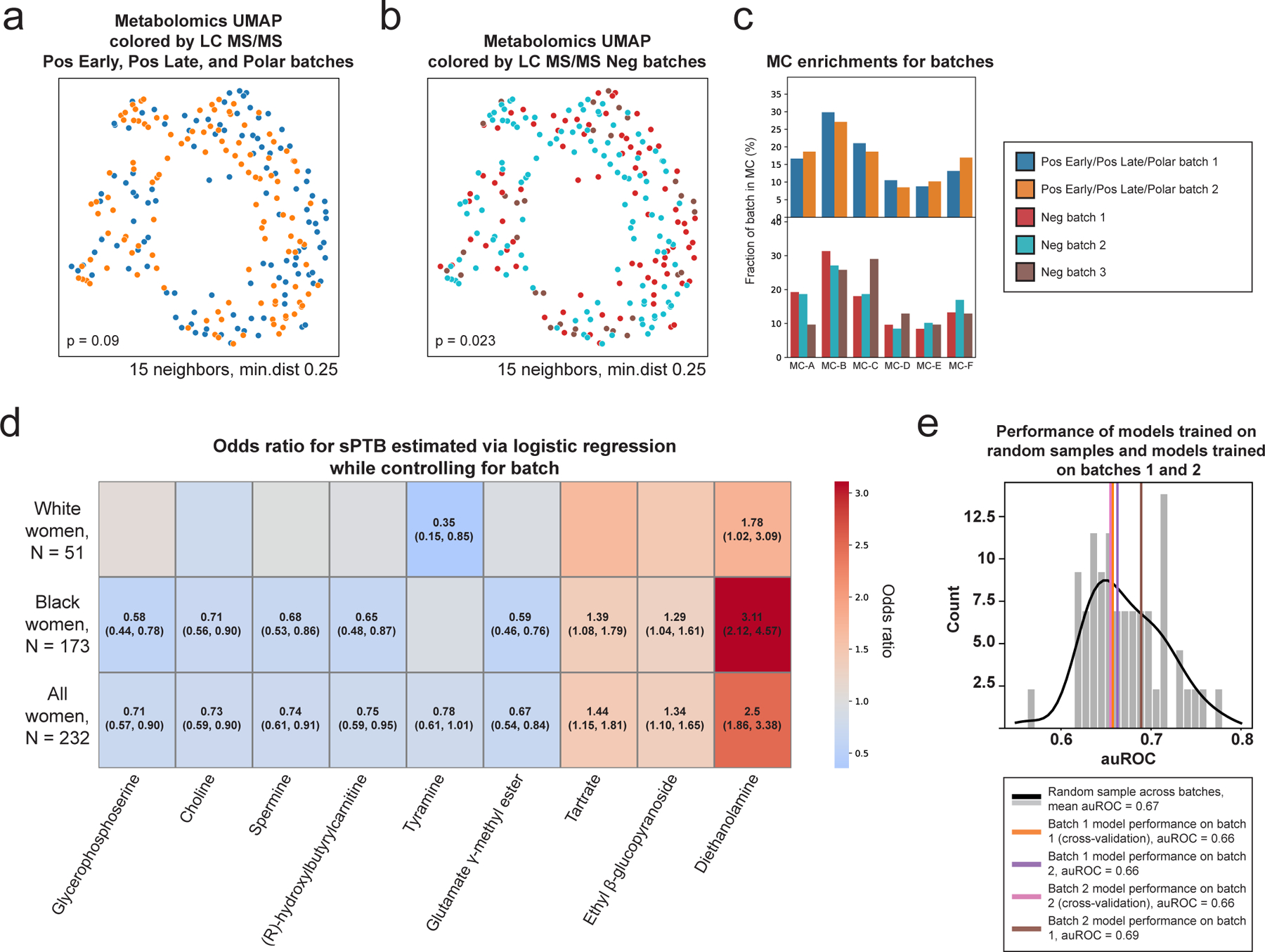
a,b, UMAP ordination of metabolomics data (N=232), same as Fig. 1b, colored by Pos Early, Pos Late, and Polar platform batches (a; 2 batches) and by Neg platform batches (b; 3 batches). See Table S4 for which metabolites were measured by each platform. Limited batch effect is noted, which is statistically significant only for the 3 batches (PERMANOVA p=0.09 and p=0.023 for 2 and 3 batches, respectively). c, The fraction of samples from each batch (y-axis; top, Pos Early, Pos Late, and Polar platform batches; bottom, Neg platform batches) whose metabolite profiles clustered to each metabolite cluster (MC; x-axis), shown for each MC separately. No significant batch effect was detected in MC assignments (Two-sided Fisher’s exact p > 0.05 for all without FDR correction). d, Heatmap showing odds ratio for sPTB (color bar) for each metabolite from Fig. 2a (x-axis) using a logistic regression model adjusting for batch (according to the appropriate platform for the metabolite, Table S4), stratified by maternal race (y-axis). The exact odds ratio and confidence interval are written in the cell for all statistically significant associations (FDR < 0.1). e, sPTB classification accuracy (auROC, x-axis) for a prediction model similar to those used for the entire cohort (Fig. 4, Methods), that is: trained and evaluated in cross validation on batch 1 (N=114; orange; auROC=0.66; one-sided permutation p=0.44 for lower accuracy than random draw); trained on batch 1 (N=114) and evaluated on batch 2 (N=118; violet; auROC=0.66; p=0.46); trained and evaluated in cross validation on batch 2 (N=118; magenta; auROC=0.66; p=0.44); and trained on batch 2 (N=118) and evaluated on batch 1 (N=114; brown; auROC=0.69; p=0.66). Gray histogram (black line, KDE) shows accuracy of models evaluated in cross-validation on random samples (N=116) from this cohort (mean auROC=0.67). This analysis demonstrates that a prediction model trained on one of the two batches generalizes well to the other batch, and that both accuracies are to be expected given the limited sample size.
Extended Data Figure 3 |. Characteristics of metabolite clusters.
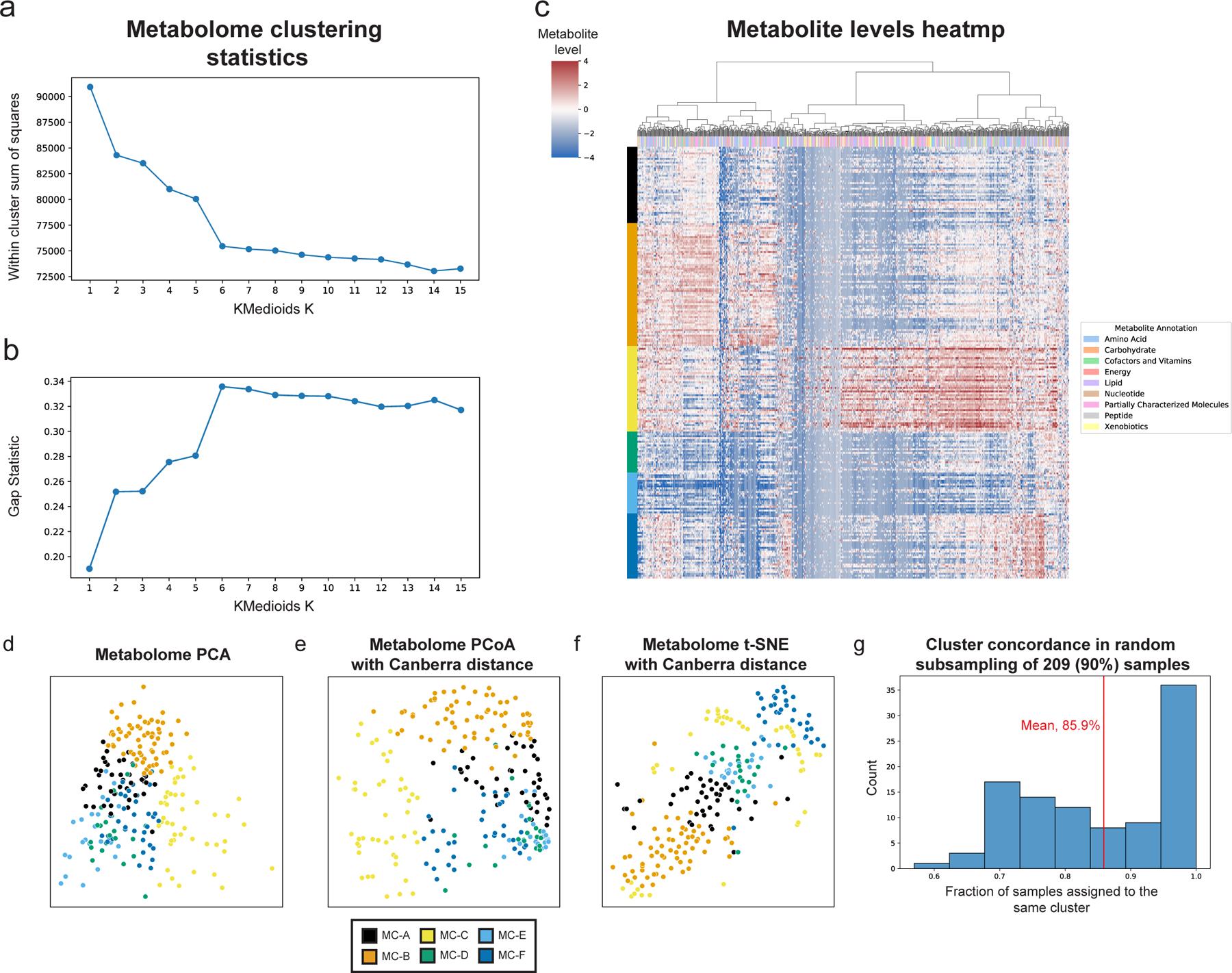
a,b, Within cluster sum of squared distances (a) and gap statistic (b) for k-medoids clustering using Canberra distances with k from 1 to 15. A shoulder (a) and peak (b) are visible for k=6. c, Heatmap showing metabolite levels for each subject (rows) and metabolite (columns). Subjects are sorted by their assigned metabolites cluster (MC) and metabolites are clustered hierarchically using Canberra distance and Ward linkage. The color above each column reflects metabolite annotations (legend to the right). d-f, Same as Fig. 1c, using PCA (d), Canberra distance-based PCoA (e) and t-SNE (f). g, Histogram of consistency of MC assignment, defined as the fraction of samples assigned to the same MC (x-axis) in 100 iterations in which we randomly selected 90% (209 women) of the cohort, and generated 6 metabolite clusters de novo. The analysis shows that many of the iterations (36 iterations, 36%) had over 95% consistency, with an overall mean consistency of 86%.
Extended Data Figure 4 |. Metabolite clusters correspond to CSTs.
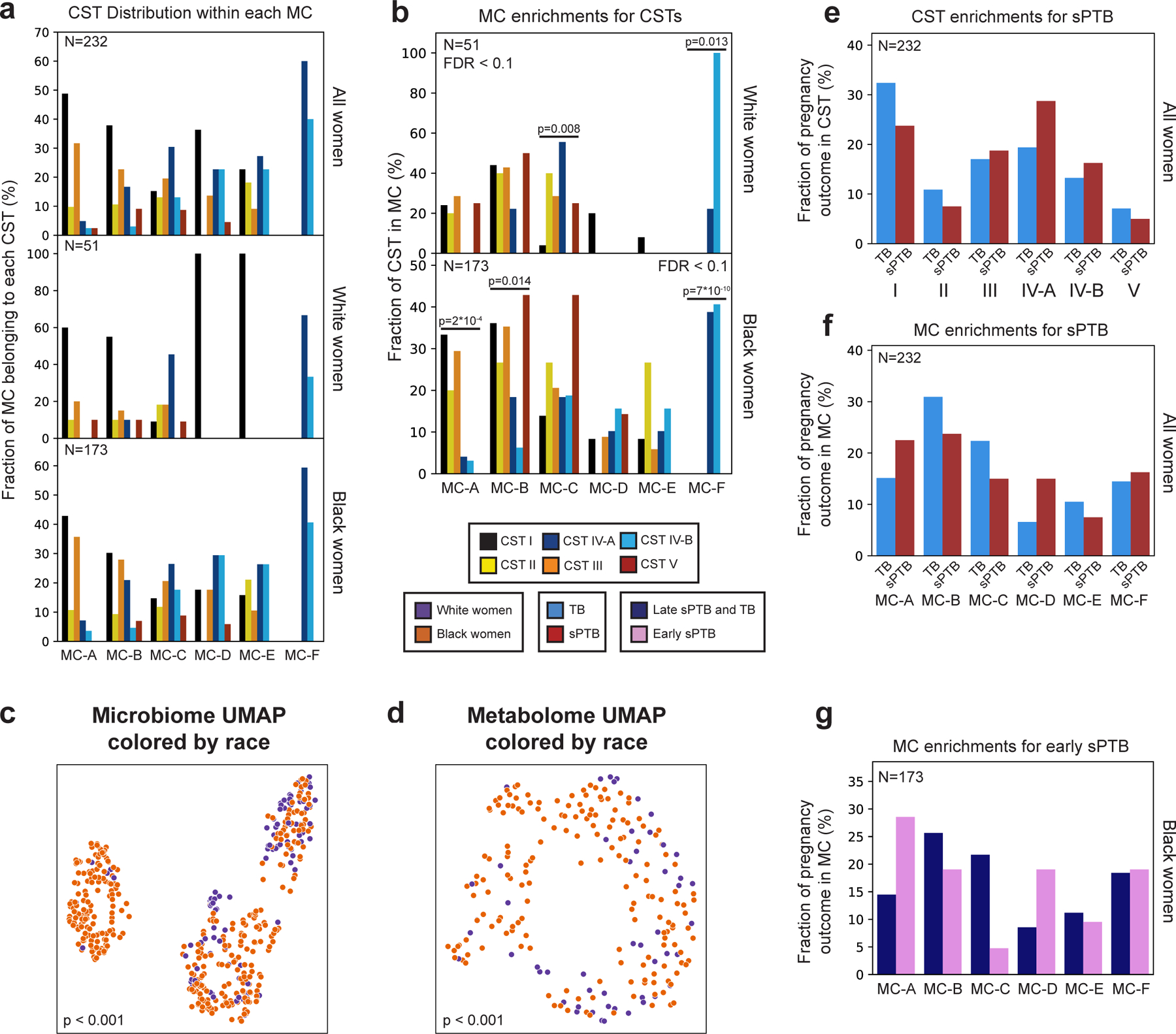
a, Distribution of CSTs within each metabolite cluster, for all (top; N=232), White (middle; N=51) and Black (bottom; N=173) women. Each group of bars corresponds to a single metabolite cluster and bars within a group sum to 100%. b, Same as Fig. 1d, stratified by race. p - two-sided Fisher’s exact p-values, q<0.1. c,d, Same as Fig. 1b,c, colored by maternal race. p - PERMANOVA. e,f, Same as Fig. 1f,g, performed for all women combined. g, Same as Fig. 1g, for association with early sPTB (gestational age at birth < 32).
Extended Data Figure 5 |. Metabolites altered in sPTB.
a, Box and swarm plots (line, median; box, IQR; whiskers, 1.5*IQR) of the levels of metabolites associated with sPTB, comparing preterm and term deliveries and stratifying by maternal self-identified race. p – two-sided Mann Whitney U. b, Distribution (kernel density estimation) of four xenobiotics associated with sPTB or early sPTB across this cohort. Samples with no metabolite detected are excluded. c, Same as Fig. 2a, for women not treated with progesterone. d, Heatmap showing metabolite sets altered in sPTB in various subsets of this cohort. Colors correspond to two-sided p-value of metabolite set enrichment analysis (Methods). Only associations with FDR<0.1 are shown. e, Raw intensity levels measured across samples for the same four xenobiotics as in b, compared to measures from plate negative process controls. Box mid-line, median; box, IQR; whiskers, 1.5*IQR; vertical line, min:max range; dot, mean; N.D., not detected. N=232 for Diethanolamine; N=230 for ethyl glucoside; N=221 for tartrate; N=232 for EDTA. f, Mass error for spectral matching (y-axis) for the same xenobiotics, compared to the mean mass error for all non-xenobiotic, tier 1 metabolites, showing that the four xenobiotic metabolites had very good identification quality.
Extended Data Figure 6 |. Networks of microbial correlations with PTB-associated metabolites.
a, Same as Fig 3a, but with each microbial taxa represented as an individual node. b, Volcano plot where every point represents a microbe-metabolite association. X-axis displays the difference between spearman ρ’s calculated separately among Black and White women. Y-axis displays the significance of the difference, using the two-sided Fisher’s R-to-z transform. Horizontal maroon line designates p=0.05. Gold points indicate associations where there is a difference in sign between the correlations among Black and White women. c,d, Same as a, for associations only among Black (c) and White (d) women. e, Same as a, for metabolites associated with extremely or very PTB among Black women. f, Same as b, for difference in associations between Black women who delivered extremely or very preterm and the rest of the Black women in the cohort.
Extended Data Figure 7 |. Metabolic models provide accurate predictions of putrescine, histamine, and tyramine.
a,b,c, Putrescine (a), histamine (b), and tyramine (c) predictions derived from microbiome metabolic models (NMPC; Methods; y-axis) plotted against measured metabolite levels (x-axis), showing good accuracy for all (Spearman ρ=0.64; ρ=0.54; and ρ=0.62, respectively, p<10−10 for all). d, Model coverage (y-axis; line, median; box, IQR; whiskers, 1.5*IQR), described as the fraction of total sample abundance represented by metabolic models, for each subgroup separately. Samples from White women had higher model coverage compared to samples from Black women, despite the lower accuracy for tyramine prediction in the former group. N=173 for Black women; N=21 for White women with sPTB; N=30 for White women with TB. e, Spearman ρ between metabolic model predictions (NMPCs) and metabolite measurements (y-axis) for models that only contain a maximum of N most abundant species (x-axis). As our metabolic models account for the abundance of each microbe, and as the vaginal microbiome has a skewed distribution, our models are robust to lack of representation of low-abundance microbes.
Extended Data Figure 8 |. Performance and features of prediction models for sPTB.
a, Receiver operating characteristic (ROC) curve comparing the performance of different sPTB prediction algorithms on metabolomics data. LightGBM (auROC=0.81) outperforms logistic regression (auROC=0.78, p=0.017 for auROC comparison against LightGBM), support vector classification (auROC=0.76, p=2.9x10−4) and elastic net (auROC=0.72, p=0.004). b, ROC curve comparing the performance of a composite model stratified for race against a model trained on all samples. A model trained on samples from all women achieves the same accuracy as a model trained only on samples from Black women when evaluated in 10-fold cross-validation on sPTB prediction for Black women (auROC of 0.83 and 0.82, respectively). However, a model trained on samples from all women significantly underperforms a model trained only on samples from women who do not identify as Black when evaluated in 10-fold cross-validation on the same subgroup (auROC of 0.64 vs. 0.80, p=4x10−7 for auROC comparison). Demonstrating that a different model is learned on each subgroup, models trained separately on each subgroup do not generalize as well to the other subgroup (auROC of 0.64 and 0.65). c,d, ROC (c) and precision-recall (PR; d) curves, evaluated in nested cross-validation, comparing sPTB prediction accuracy for models based on metabolomics data alone (auROC=0.78, auPR=0.61), and on metabolomics data combined with microbiome and clinical data (“combination”; auROC=0.76, auPR=0.62; p=0.44). e, SHAP83-based effect on total prediction (x-axis) for the top 10 features used in our combination models, sorted with descending importance. Each dot represents a sample, with the color corresponding to the metabolite level in the sample compared to all samples. f,g, ROC curves for the same metabolome-based (f) and microbiome-based (g) models as in Fig. 4a,b, when prediction is evaluated for extremely (<28 weeks of gestation) and very (<32 weeks) PTB. The microbiome-based models show increasing accuracy for predicting extremely and very PTB (auROC of 0.69 and 0.62, respectively, compared to auROC of 0.55 for all sPTB, p=0.03 and p=0.49, respectively). h,i, PR curve for sPTB prediction on two external cohorts, obtained using our metabolome-based predictor without retraining or adaptation. j, Same as (e) for the microbiome-based model. Shaded lines in a-d,f,g show results from five independent 10-fold cross validation draws (Methods). p-values for comparisons between ROC curves are based on the two-sided test described in ref. 117.
Supplementary Material
Acknowledgments
We thank Michal A. Elovitz, Jacques Ravel, Kristin D. Gerson, Pawel Gajer, and Lauren Anton for initiating, collecting, and sharing samples, and for assistance in funding acquisition. We thank them, members of the Korem group, Liat Shenhav, David Zeevi, Noam Bar and Ronald Wapner for useful discussions. The M&M cohort was funded by the National Institute of Nursing Research (NINR; R01NR014784). One of the datasets used was obtained from the database of Genotypes and Phenotypes (dbGaP) through dbGaP accession number phs001739.v1.p1. The current study was supported by NINR (R01NR014784), the Center for Precision Medicine at the University of Pennsylvania, the Vagelos Award provided by Columbia University Precision Medicine Initiative, the Program for Mathematical Genomics at Columbia University, and the CIFAR Azrieli Global Scholarship in the Humans & the Microbiome Program. W.F.K was supported by NIH T32GM007367 and F30HD108886. I.T. and A.H. were supported by grants from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No 757922) awarded to I.T.
Footnotes
Code availability
Scripts to reproduce the analysis are available in a GitHub repository: https://github.com/korem-lab/PTB_Metabs_2021. The mgPipe pipeline is available within the COBRA toolbox (https://github.com/opencobra/cobratoolbox).
Competing interests
M.L. and T.K. are inventors on a provisional patent application related to this work. Other authors declare no conflict of interests.
Data availability
The 16S rRNA gene amplicon sequencing data and the associated samples and subjects’ metadata analyzed in this study are publicly available in the database of Genotypes and Phenotypes (dbGaP) under accession number phs001739.v1.p1 as well as in Supplementary Data 2 of ref. 14. Raw metabolomics data is available in Table S1. Mass spectral data is available from MetaboLights under accession number MTBLS702 (https://www.ebi.ac.uk/metabolights/MTBLS702 ). Additional information regarding xenobiotics is provided in Table S13. The KEGG Database is available at https://www.genome.jp/kegg/ and the AGORA models are available at vmh.life.
References
- 1.Goldenberg RL, Culhane JF, Iams JD & Romero R Epidemiology and causes of preterm birth. Lancet 371, 75–84 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Howson CP, Kinney MV, McDougall L & Lawn JE Born too soon: preterm birth matters. Reprod. Health 10, 1–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martin JA, Hamilton BE & Osterman MJK Births in the United States, 2018. NCHS Data Brief Hyattsville MD Natl. Cent. Health Stat 1–8 (2019). [PubMed]
- 4.Braveman P et al. Explaining the Black-White Disparity in Preterm Birth: A Consensus Statement From a Multi-Disciplinary Scientific Work Group Convened by the March of Dimes. Front. Reprod. Health 3, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meertens LJ et al. Prediction models for the risk of spontaneous preterm birth based on maternal characteristics: a systematic review and independent external validation. Acta Obstet. Gynecol. Scand 97, 907–920 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conde‐Agudelo A, Papageorghiou AT, Kennedy SH & Villar J Novel biomarkers for the prediction of the spontaneous preterm birth phenotype: a systematic review and meta‐analysis. BJOG Int. J. Obstet. Gynaecol 118, 1042–1054 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Zeevi D et al. Personalized nutrition by prediction of glycemic responses. Cell 163, 1079–1094 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Qin N et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 513, 59–64 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Qin J et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Wirbel J et al. Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat. Med 25, 679–689 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomas AM et al. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat. Med 25, 667–678 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown RG et al. Vaginal dysbiosis increases risk of preterm fetal membrane rupture, neonatal sepsis and is exacerbated by erythromycin. BMC Med 16, 9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Callahan BJ et al. Replication and refinement of a vaginal microbial signature of preterm birth in two racially distinct cohorts of US women. Proc. Natl. Acad. Sci 114, 9966–9971 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elovitz MA et al. Cervicovaginal microbiota and local immune response modulate the risk of spontaneous preterm delivery. Nat. Commun 10, 1305 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fettweis JM et al. The vaginal microbiome and preterm birth. Nat. Med 25, 1012–1021 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DiGiulio DB et al. Temporal and spatial variation of the human microbiota during pregnancy. Proc. Natl. Acad. Sci. U. S. A 112, 11060–11065 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Romero R et al. The vaginal microbiota of pregnant women who subsequently have spontaneous preterm labor and delivery and those with a normal delivery at term. Microbiome 2, 18 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bayar E, Bennett PR, Chan D, Sykes L & MacIntyre DA The pregnancy microbiome and preterm birth. Semin. Immunopathol 42, 487–499 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thaiss CA et al. Microbiota Diurnal Rhythmicity Programs Host Transcriptome Oscillations. Cell 167, 1495–1510.e12 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Yoshimoto S et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Koeth RA et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med 19, 576–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levy M et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 163, 1428–1443 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yachida S et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med 25, 968–976 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Lloyd-Price J et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flaviani F et al. Cervicovaginal microbiota and metabolome predict preterm birth risk in an ethnically diverse cohort. JCI Insight 6, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pruski P et al. Direct on-swab metabolic profiling of vaginal microbiome host interactions during pregnancy and preterm birth. Nat. Commun 12, 5967 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bar N et al. A reference map of potential determinants for the human serum metabolome. Nature 588, 135–140 (2020). [DOI] [PubMed] [Google Scholar]
- 28.Ravel J et al. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci 108, 4680–4687 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stafford GP et al. Spontaneous Preterm Birth Is Associated with Differential Expression of Vaginal Metabolites by Lactobacilli-Dominated Microflora. Front. Physiol 8, 615 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fiume MM et al. Safety assessment of decyl glucoside and other alkyl glucosides as used in cosmetics. Int. J. Toxicol 32, 22S–48S (2013). [DOI] [PubMed] [Google Scholar]
- 31.Waters B et al. A validated method for the separation of ethyl glucoside isomers by gas chromatography-tandem mass spectrometry and quantitation in human whole blood and urine. J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci 1188, 123074 (2021). [DOI] [PubMed] [Google Scholar]
- 32.Kassaian J-M Tartaric Acid. in Ullmann’s Encyclopedia of Industrial Chemistry 671–677 (American Cancer Society, 2000). doi: 10.1002/14356007.a26_163. [DOI] [Google Scholar]
- 33.Fiume MM et al. Safety Assessment of Diethanolamine and Its Salts as Used in Cosmetics. Int. J. Toxicol 36, 89S–110S (2017). [DOI] [PubMed] [Google Scholar]
- 34.Final Report on the Safety Assessment of Cocamide DEA, Lauramide DEA, Linoleamide DEA, and Oleamide DEA. J. Am. Coll. Toxicol 5, 415–454 (1986). [Google Scholar]
- 35.Mirer F Updated epidemiology of workers exposed to metalworking fluids provides sufficient evidence for carcinogenicity. Appl. Occup. Environ. Hyg 18, 902–912 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Shariq L et al. Irrigation of wheat with select hydraulic fracturing chemicals: Evaluating plant uptake and growth impacts. Environ. Pollut. Barking Essex 1987 273, 116402 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Shaffer M et al. AMON: annotation of metabolite origins via networks to integrate microbiome and metabolome data. BMC Bioinformatics 20, 614 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zeisel SH & da Costa K-A Choline: an essential nutrient for public health. Nutr. Rev 67, 615–623 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bernhard W et al. Choline concentrations are lower in postnatal plasma of preterm infants than in cord plasma. Eur. J. Nutr 54, 733–741 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Ueland PM Choline and betaine in health and disease. J. Inherit. Metab. Dis 34, 3–15 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Kirman CR, Hughes B, Becker RA & Hays SM Derivation of a No-significant-risk-level (NSRL) for dermal exposures to diethanolamine. Regul. Toxicol. Pharmacol 76, 137–151 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Craciunescu CN, Wu R & Zeisel SH Diethanolamine alters neurogenesis and induces apoptosis in fetal mouse hippocampus. FASEB J 20, 1635–1640 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lehman-McKeeman LD et al. Diethanolamine induces hepatic choline deficiency in mice. Toxicol. Sci. Off. J. Soc. Toxicol 67, 38–45 (2002). [DOI] [PubMed] [Google Scholar]
- 44.National Toxicology Program. NTP Toxicology and Carcinogenesis Studies of Diethanolamine (CAS No. 111–42-2) in F344/N Rats and B6C3F1 Mice (Dermal Studies). Natl. Toxicol. Program Tech. Rep. Ser 478, 1–212 (1999). [PubMed] [Google Scholar]
- 45.Korkes HA et al. Lipidomic assessment of plasma and placenta of women with early-onset preeclampsia. PloS One 9, e110747 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Casti A et al. Pattern of human blood spermidine and spermine in prematurity. Clin. Chim. Acta Int. J. Clin. Chem 147, 223–232 (1985). [DOI] [PubMed] [Google Scholar]
- 47.Vidarsdottir H et al. Does metabolomic profile differ with regard to birth weight? Pediatr. Res 89, 1144–1151 (2021). [DOI] [PubMed] [Google Scholar]
- 48.Obayomi SB & Baluch DP Tyramine Localization Closely Corelates to Circular Vesicles Within the Mouse Uterine Horn Using Correlational Fluorescence and Scanning Electron Microscopy. Microsc. Microanal 26, 1348–1349 (2020). [Google Scholar]
- 49.Albaugh VL, Mukherjee K & Barbul A Proline Precursors and Collagen Synthesis: Biochemical Challenges of Nutrient Supplementation and Wound Healing. J. Nutr 147, 2011–2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu G, Bazer FW, Cudd TA, Meininger CJ & Spencer TE Maternal nutrition and fetal development. J. Nutr 134, 2169–2172 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Strauss JF Extracellular matrix dynamics and fetal membrane rupture. Reprod. Sci. Thousand Oaks Calif 20, 140–153 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou X et al. Impaired mitochondrial fusion, autophagy, biogenesis and dysregulated lipid metabolism is associated with preeclampsia. Exp. Cell Res 359, 195–204 (2017). [DOI] [PubMed] [Google Scholar]
- 53.Sauer MM et al. Binding of the Bacterial Adhesin FimH to Its Natural, Multivalent High-Mannose Type Glycan Targets. J. Am. Chem. Soc 141, 936–944 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Benito R, Vazquez JA, Berron S, Fenoll A & Saez-Nieto JAY 1986. A Modified Scheme for Biotyping Gardnerella Vaginalis. J. Med. Microbiol 21, 357–359. [DOI] [PubMed] [Google Scholar]
- 55.Ghartey J, Anglim L, Romero J, Brown A & Elovitz MA Women with Symptomatic Preterm Birth Have a Distinct Cervicovaginal Metabolome. Am. J. Perinatol 34, 1078–1083 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Fashemi B, Delaney ML, Onderdonk AB & Fichorova RN Effects of feminine hygiene products on the vaginal mucosal biome. Microb. Ecol. Health Dis 24, 10.3402/mehd.v24i0.19703 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lanigan RS & Yamarik TA Final report on the safety assessment of EDTA, calcium disodium EDTA, diammonium EDTA, dipotassium EDTA, disodium EDTA, TEA-EDTA, tetrasodium EDTA, tripotassium EDTA, trisodium EDTA, HEDTA, and trisodium HEDTA. Int. J. Toxicol 21, 95–142 (2002). [DOI] [PubMed] [Google Scholar]
- 58.Evstatiev R et al. The food additive EDTA aggravates colitis and colon carcinogenesis in mouse models. Sci. Rep 11, 5188 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Youn H, Hong K, Yoo J-W & Lee CH ICAM-1 expression in vaginal cells as a potential biomarker for inflammatory response. Biomark. Biochem. Indic. Expo. Response Susceptibility Chem 13, 257–269 (2008). [DOI] [PubMed] [Google Scholar]
- 60.Brownie CF et al. Teratogenic effect of calcium edetate (CaEDTA) in rats and the protective effect of zinc. Toxicol. Appl. Pharmacol 82, 426–443 (1986). [DOI] [PubMed] [Google Scholar]
- 61.Kanehisa M & Goto S KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Catov JM et al. Early pregnancy lipid concentrations and spontaneous preterm birth. Am. J. Obstet. Gynecol 197, 610.e1–610.e7 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Nelson TM et al. Vaginal biogenic amines: biomarkers of bacterial vaginosis or precursors to vaginal dysbiosis? Front. Physiol 6, 253 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bargossi E et al. The Capability of Tyramine Production and Correlation between Phenotypic and Genetic Characteristics of Enterococcus faecium and Enterococcus faecalis Strains. Front. Microbiol 6, 1371 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cornejo OE, Hickey RJ, Suzuki H & Forney LJ Focusing the diversity of Gardnerella vaginalis through the lens of ecotypes. Evol. Appl 11, 312–324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res 45, D158–D169 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wolrath H, Forsum U, Larsson P-G & Borén H Analysis of bacterial vaginosis-related amines in vaginal fluid by gas chromatography and mass spectrometry. J. Clin. Microbiol 39, 4026–4031 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ravel J et al. Daily temporal dynamics of vaginal microbiota before, during and after episodes of bacterial vaginosis. Microbiome 1, 29 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Al-Memar M et al. The association between vaginal bacterial composition and miscarriage: a nested case–control study. BJOG Int. J. Obstet. Gynaecol 127, 264–274 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mann C, Dertinger S, Hartmann G, Schurz R & Simma B Actinomyces neuii and neonatal sepsis. Infection 30, 178–180 (2002). [DOI] [PubMed] [Google Scholar]
- 71.Holst E, Wathne B, Hovelius B & Mårdh PA Bacterial vaginosis: microbiological and clinical findings. Eur. J. Clin. Microbiol 6, 536–541 (1987). [DOI] [PubMed] [Google Scholar]
- 72.Moles L et al. Staphylococcus epidermidis in feedings and feces of preterm neonates. PloS One 15, e0227823 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baldini F et al. The Microbiome Modeling Toolbox: from microbial interactions to personalized microbial communities. Bioinformatics 35, 2332–2334 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen KC, Forsyth PS, Buchanan TM & Holmes KK Amine content of vaginal fluid from untreated and treated patients with nonspecific vaginitis. J. Clin. Invest 63, 828–835 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Serrano MG et al. Racioethnic diversity in the dynamics of the vaginal microbiome during pregnancy. Nat. Med 25, 1001–1011 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baraldi E et al. Untargeted Metabolomic Analysis of Amniotic Fluid in the Prediction of Preterm Delivery and Bronchopulmonary Dysplasia. PloS One 11, e0164211 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Souza RT et al. Trace biomarkers associated with spontaneous preterm birth from the maternal serum metabolome of asymptomatic nulliparous women - parallel case-control studies from the SCOPE cohort. Sci. Rep 9, 13701 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Aung MT et al. Prediction and associations of preterm birth and its subtypes with eicosanoid enzymatic pathways and inflammatory markers. Sci. Rep 9, 17049 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ngo TTM et al. Noninvasive blood tests for fetal development predict gestational age and preterm delivery. Science 360, 1133–1136 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Leow SM et al. Preterm birth prediction in asymptomatic women at mid-gestation using a panel of novel protein biomarkers: the Prediction of PreTerm Labor (PPeTaL) study. Am. J. Obstet. Gynecol. MFM 2, 100084 (2020). [DOI] [PubMed] [Google Scholar]
- 81.Ghartey J, Bastek JA, Brown AG, Anglim L & Elovitz MA Women with preterm birth have a distinct cervicovaginal metabolome. Am. J. Obstet. Gynecol 212, 776.e1–12 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brunius C, Shi L & Landberg R Large-scale untargeted LC-MS metabolomics data correction using between-batch feature alignment and cluster-based within-batch signal intensity drift correction. Metabolomics 12, 173 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lundberg SM & Lee S-I A unified approach to interpreting model predictions in Proceedings of the 31st International Conference on Neural Information Processing Systems 4768–4777 (Curran Associates Inc., 2017). [Google Scholar]
- 84.Srinivasan S et al. Metabolic Signatures of Bacterial Vaginosis. mBio 6, e00204–15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Freitas AC, Bocking A, Hill JE, Money DM, & VOGUE Research Group. Increased richness and diversity of the vaginal microbiota and spontaneous preterm birth. Microbiome 6, 117 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Howard PH Handbook of Environmental Fate and Exposure Data For Organic Chemicals (CRC Press, 1990). [Google Scholar]
- 87.Wambaugh JF et al. High Throughput Heuristics for Prioritizing Human Exposure to Environmental Chemicals. Environ. Sci. Technol 48, 12760–12767 (2014). [DOI] [PubMed] [Google Scholar]
- 88.Wang A et al. Suspect Screening, Prioritization, and Confirmation of Environmental Chemicals in Maternal-Newborn Pairs from San Francisco. Environ. Sci. Technol 55, 5037–5049 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Woodruff TJ, Zota AR & Schwartz JM Environmental chemicals in pregnant women in the United States: NHANES 2003–2004. Environ. Health Perspect 119, 878–885 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bullard RD Race and Environmental Justice in the United States. Yale J. Int. Law 18, 319 (1993). [Google Scholar]
- 91.Morello-Frosch R & Lopez R The riskscape and the color line: examining the role of segregation in environmental health disparities. Environ. Res 102, 181–196 (2006). [DOI] [PubMed] [Google Scholar]
- 92.Helm JS, Nishioka M, Brody JG, Rudel RA & Dodson RE Measurement of endocrine disrupting and asthma-associated chemicals in hair products used by Black women. Environ. Res 165, 448–458 (2018). [DOI] [PubMed] [Google Scholar]
- 93.James-Todd T, Senie R & Terry MB Racial/ethnic differences in hormonally-active hair product use: a plausible risk factor for health disparities. J. Immigr. Minor. Health 14, 506–511 (2012). [DOI] [PubMed] [Google Scholar]
- 94.Longnecker MP, Klebanoff MA, Zhou H & Brock JW Association between maternal serum concentration of the DDT metabolite DDE and preterm and small-for-gestational-age babies at birth. Lancet Lond. Engl 358, 110–114 (2001). [DOI] [PubMed] [Google Scholar]
- 95.Ferguson KK et al. Environmental phthalate exposure and preterm birth in the PROTECT birth cohort. Environ. Int 132, 105099 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fettweis JM et al. Differences in vaginal microbiome in African American women versus women of European ancestry. Microbiol. Read. Engl 160, 2272–2282 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vyas DA, Eisenstein LG & Jones DS Hidden in Plain Sight - Reconsidering the Use of Race Correction in Clinical Algorithms. N. Engl. J. Med 383, 874–882 (2020). [DOI] [PubMed] [Google Scholar]
- 98.Cooper RS, Kaufman JS & Ward R Race and Genomics. N. Engl. J. Med 348, 1166–1170 (2003). [DOI] [PubMed] [Google Scholar]
- 99.Ford L et al. Precision of a Clinical Metabolomics Profiling Platform for Use in the Identification of Inborn Errors of Metabolism. J. Appl. Lab. Med 5, 342–356 (2020). [DOI] [PubMed] [Google Scholar]
- 100.Callahan BJ et al. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Edgar RC Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010). [DOI] [PubMed] [Google Scholar]
- 102.McInnes L, Healy J & Melville J UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. ArXiv180203426 Cs Stat (2020).
- 103.Douglas GM et al. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol 38, 685–688 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liu CM et al. BactQuant: An enhanced broad-coverage bacterial quantitative real-time PCR assay. BMC Microbiol 12, 56 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jian C, Luukkonen P, Yki-Järvinen H, Salonen A & Korpela K Quantitative PCR provides a simple and accessible method for quantitative microbiota profiling. PLOS ONE 15, e0227285 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Heirendt L et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v. 3.0. Nat. Protoc 14, 639–702 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Heinken A et al. AGORA2: Large scale reconstruction of the microbiome highlights wide-spread drug-metabolising capacities. bioRxiv 2020.11.09.375451 (2020) doi: 10.1101/2020.11.09.375451. [DOI]
- 108.Klitgord N & Segrè D Environments that induce synthetic microbial ecosystems. PLoS Comput Biol 6, e1001002 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Heinken A, Sahoo S, Fleming RMT & Thiele I Systems-level characterization of a host-microbe metabolic symbiosis in the mammalian gut. Gut Microbes 4, 28–40 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Baldini F et al. Parkinson’s disease-associated alterations of the gut microbiome predict disease-relevant changes in metabolic functions. BMC Biol 18, 62 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hyatt D et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.O’Leary NA et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44, D733–745 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Eddy SR Accelerated profile HMM searches. PLoS Comput. Biol 7, e1002195 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Connil N et al. Identification of the Enterococcus faecalis Tyrosine Decarboxylase Operon Involved in Tyramine Production. Appl. Environ. Microbiol 68, 3537–3544 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Orth JD, Thiele I & Palsson BØ What is flux balance analysis? Nat. Biotechnol 28, 245–248 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ke G et al. LightGBM: a highly efficient gradient boosting decision tree in Proceedings of the 31st International Conference on Neural Information Processing Systems 3149–3157 (Curran Associates Inc., 2017). [Google Scholar]
- 117.Hanley JA & McNeil BJ A method of comparing the areas under receiver operating characteristic curves derived from the same cases. Radiology 148, 839–843 (1983). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The 16S rRNA gene amplicon sequencing data and the associated samples and subjects’ metadata analyzed in this study are publicly available in the database of Genotypes and Phenotypes (dbGaP) under accession number phs001739.v1.p1 as well as in Supplementary Data 2 of ref. 14. Raw metabolomics data is available in Table S1. Mass spectral data is available from MetaboLights under accession number MTBLS702 (https://www.ebi.ac.uk/metabolights/MTBLS702 ). Additional information regarding xenobiotics is provided in Table S13. The KEGG Database is available at https://www.genome.jp/kegg/ and the AGORA models are available at vmh.life.