

Abstract
Objective
Programmed cell death protein 1 (PD-1) checkpoint inhibition and adoptive cellular therapy have had limited success in patients with microsatellite stable colorectal cancer liver metastases (CRLM). We sought to evaluate the effect of interleukin 10 (IL-10) blockade on endogenous T cell and chimeric antigen receptor T (CAR-T) cell antitumour function in CRLM slice cultures.
Design
We created organotypic slice cultures from human CRLM (n=38 patients’ tumours) and tested the antitumour effects of a neutralising antibody against IL-10 (αIL-10) both alone as treatment and in combination with exogenously administered carcinoembryonic antigen (CEA)-specific CAR-T cells. We evaluated slice cultures with single and multiplex immunohistochemistry, in situ hybridisation, single-cell RNA sequencing, reverse-phase protein arrays and time-lapse fluorescent microscopy.
Results
αIL-10 generated a 1.8-fold increase in T cell-mediated carcinoma cell death in human CRLM slice cultures. αIL-10 significantly increased proportions of CD8+ T cells without exhaustion transcription changes, and increased human leukocyte antigen - DR isotype (HLA-DR) expression of macrophages. The antitumour effects of αIL-10 were reversed by major histocompatibility complex class I or II (MHC-I or MHC-II) blockade, confirming the essential role of antigen presenting cells. Interrupting IL-10 signalling also rescued murine CAR-T cell proliferation and cytotoxicity from myeloid cell-mediated immunosuppression. In human CRLM slices, αIL-10 increased CEA-specific CAR-T cell activation and CAR-T cell-mediated cytotoxicity, with nearly 70% carcinoma cell apoptosis across multiple human tumours. Pretreatment with an IL-10 receptor blocking antibody also potentiated CAR-T function.
Conclusion
Neutralising the effects of IL-10 in human CRLM has therapeutic potential as a stand-alone treatment and to augment the function of adoptively transferred CAR-T cells.
INTRODUCTION
Colorectal cancer (CRC) is the third most common cause of cancer-related mortality in the USA.1 For the 25%–30% of patients with CRC who develop liver metastases, the 5-year overall survival (OS) rate is 49% with resection but only 2.2% with palliative chemotherapy. The profound impact of CRC liver metastases (CRLM) is highlighted by the fact that the survival of CRC patients with isolated liver metastases is substantially worse than for those with isolated lung metastases.2
Immune checkpoint inhibition (ICI) with programmed cell death protein 1 (PD-1) blockade is effective in patients with microsatellite instability-high (MSI-H) CRC, but this represents less than 5% of patients with CRC with locally advanced or metastatic disease.3 Among all patients with CRC, objective response rates to checkpoint blockade are seen in less than 5% of patients.4–7 However, tumour-infiltrating lymphocytes correlate with prognosis. The Immunoscore, which grades CD3+ and CD8+ T cell density within CRLM, correlates with better prognosis,8 and high regulatory T cell (Treg cell) infiltrate correlates with shorter OS.9 Both the density and morphology of tumour-associated macrophages (TAMs) correlate with survival in patients with CRLM.10 11 Despite the lack of success of currently approved immunotherapies, modulation of immune cells infiltrating CRLM remains a compelling potential therapeutic approach.
An alternative immunotherapeutic approach is adoptive transfer of chimeric antigen receptor T (CAR-T) cells, but several aspects of the CRLM tumour microenvironment (TME) limit the efficacy of CAR-T cells.12 In a phase I clinical trial of anti-carcinoembryonic antigen (CEA) CAR-T cells for patients with CRLM who had progressed on other therapies, 7 of 10 patients demonstrated stable disease without severe adverse reactions.13 A phase I trial of anti-CEA CAR-T administered via hepatic arterial infusion showed stable disease in one of six patients and tumour necrosis or fibrosis in four of six patients, without grade 3 or 4 adverse events.14 In murine studies of the liver immune TME, myeloid cells suppress antitumour immunity via signal transducer and activator of transcription factor 3 (STAT3)-mediated expression of indoleamine 2,3-deoxygenase (IDO) and programmed death ligand 1 (PD-L1), and CAR-T efficacy can be rescued when murine myeloid cells are depleted.15 16 The effect of the specific organ TME on the immune landscape in a tumour is evidenced by the finding that myeloid cells in liver metastases are more immunosuppressive than those isolated from lung metastases.17 Importantly, the liver-specific myeloid cell functional programme was reversible on transfer into lung tissue, suggesting repolarisation of immunosuppressive liver TAMs as a novel therapeutic approach.
Interleukin 10 (IL-10) has broad immunosuppressive functions through interaction with its cognate receptor (IL-10R) and downstream activation of the STAT3 pathway. Classically, IL-10 inhibits antigen-presenting cells (APCs) leading to T cell inhibition, in addition to directly causing CD4+ T cell anergy.18 A variety of immunosuppressive signals, including IL-10, support a bias towards immune tolerance in the liver, which permits immunological silence towards antigens present in food.19 In non-haematological malignancies including CRC, high serum IL-10 correlates with more advanced stage, worse survival or higher recurrence rate.20 21 In a murine breast cancer model, IL-10 from macrophages suppressed dendritic cell (DC) IL-12 secretion to abrogate CD8+ T cell responses.22 In a murine CRC model, intratumoral injections of a lentiviral vector encoding IL-10-silencing short hairpin RNA (shRNA) reduced tumour growth.23 Conversely, some studies have suggested a beneficial effect of IL-10 including increased CD8+ cell infiltration and increased interferon-gamma release in the TME.24–30 However, clinical trials using pegylated IL-10 treatment have not shown a benefit in patients.31 32 Recent definition of the active structure of the IL-10 ligand/receptor suggests new ways of targeting the IL-10/IL-10R axis in a cell type-specific manner.33
Tumour slice culture (TSC) permits the evaluation of immune cell function in the context of the native three-dimensional (3D) solid tumour structure and cell populations. Our laboratory has demonstrated that the TME remains intact and viable in human TSC for 1–2 weeks34–36 and that antitumour immunity can be enhanced in immune-excluded solid tumours such as pancreatic adenocarcinoma.37 Notably, in TSC, combined CXCR4/PD-1 inhibition improved migration of cytotoxic T cells toward carcinoma cells, an effect mirrored in patients’ tumours on a clinical trial of these therapies.38 Because the effect of IL-10 on antitumour immune function remains controversial, we leveraged the human TSC system to investigate the role of IL-10 in human CRLM. We confirmed the inhibitory effect of IL-10 and demonstrated that IL-10 blockade dramatically enhanced antitumour function of both endogenous and adoptively transferred T cells in a heterogeneous cohort of human CRLM tumours.
MATERIALS AND METHODS
Ethics statement
All investigations were performed according to the principles expressed in the Declaration of Helsinki. Samples for slice culture were procured from patients undergoing hepatic resection for CRLM who provided prior written-informed consent under a research protocol (no. 1852) approved by the University of Washington Institutional Review Board.
Patient and public involvement
Patients were involved in providing voluntary consent for use of their tumour specimens. Patients were not involved in the research design, conduct, reporting or dissemination plans.
The Cancer Genome Atlas (TCGA) analysis
IL10 RNA expression and disease-specific survival (DSS) data from TCGA Pan-Cancer cohort was exported using the UCSC Xena webtool.39 Only colon adenocarcinoma (COAD) primary tumours were included for the survival analysis, and duplicate samples were excluded. Kaplan-Meier DSS curves with 68% CIs were generated using the Python lifelines (v0.26.0) package.
Tumour slice culture
Slice culture of freshly resected human CRLM tumours was performed as previously described.35–37 40 Additional details and treatment antibody data are available in online supplemental table S1. Slice cultures were treated with 20 μg/mL of each treatment antibody.
In situ hybridisation (ISH) and multicolour immunohistochemistry (mIHC)
Detailed description of ISH and mIHC, reagents and antibodies used are available in online supplemental table S2. Slides were imaged with Leica SP8X confocal microscope (Leica Microsystems, Wetzlar, Germany). Areas of nuclear overlap on the images were analysed using IMARIS (Bitplane USA) and Flowjo (Beckton Dickinson).
Reverse phase protein array (RPPA)
Protein microarrays were printed and processed as described previously.41 42 Slides were scanned using Licor Odyssey CLX Scanner (LiCOR). Total signal intensity was normalised to total beta-actin (Sigma, Catalogue No A1978) and quantified using Array-Pro analyzer software package (Media Cybernetics, Maryland, USA).
Terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL)
The Click-iT Plus TUNEL Assay for In Situ Apoptosis Alexa Fluor 488 dye kit (C10617, ThermoFisher) was used according to manufacturer instructions. Slides were imaged using Leica SP8X confocal microscope. After background intensity subtraction, automatic counting of total nuclei and colocalisation was performed in IMARIS using the spots colocalisation function.
Immunohistochemistry (IHC)
Detailed description of IHC steps, reagents used and primary IHC antibodies are available in the online supplemental table S3. The transversely cut slides were imaged at 20x to visualise the entire thickness of the slice. Slides were digitally scanned to NanoZoomer Digital Pathology (Hamamatsu) image viewing software. Positive 3,3’-diaminobenzidine (DAB) cells with haematoxylin-stained nuclei were visually counted using Fiji ImageJ software over at least two high-powered fields (hpf) per slice in at least two different slices per treatment per individual patient tumour.
RNA sequencing
Slices were treated and RNA extracted using Qiagen RNeasy kit according to the manufacturer’s protocol. RNA sequencing was performed using the Northwest Genomics Center core facility with Next Generation Sequencing after samples passed quality controls. Samples were provided as normalised cell counts to housekeeping genes. The data have been deposited and are publicly available.43
Single-cell RNA sequencing (scRNAseq)
ScRNAseq was carried out first by dissociating untreated slices from fresh tumour tissue. Next, slices treated with either control or IL-10 blocking antibody were dissociated. After removal of dead cells and single-cell suspension preparation, 10× droplet sequencing was performed. Details of each steps and the statistical analysis are available in the online supplemental materials. The scRNAseq data have been deposited and are also available.43
Mice, liver metastasis (LM) in vivo model and myeloid cell isolation
C57BL/6 male mice (6–8 weeks old) obtained from Jackson Laboratories were bred and maintained under pathogen-free conditions at Roger Williams Medical Center (RWMC) animal facility. All surgical procedures were approved by RWMC Institutional Animal Care and Use Committee protocol No. 19–2904. To generate LM, mice were anaesthetised and injected with 2.5×106 cells via spleen followed by splenectomy. Liver non-parenchymal cells were isolated from LM, 4 days post-tumour generation, as previously described.15 Immunomagnetic beads against CD11b were used to purify bulk hepatic myeloid cells (Miltenyi Biotech). Further details are available in the online supplemental materials.
CAR-T generation, proliferation assay and cytotoxicity assay
Using a construct provided by Sorrento and TNK Therapeutics, CAR-T cells were generated from mouse splenocytes as described previously44 and MC38CEA cells were used as target tumour cells. Myeloid cells were isolated from LM tumour-bearing mice livers. CAR-T cells were carboxyfluorescein diacetate succinimidyl ester (CFSE, Life Technologies) labelled as per the manufacturer’s protocol. MC38CEA cells were plated at 2×104 cells/well in 96-well cell culture plate with myeloid cells and CAR-T cells in a 1:1:1 ratio as controls. Cells were treated with 100, 200 or 400 ng/mL of anti-IL-10 antibody (JES5–2A5, BioLegend). Supernatant was collected and analysed for lactate dehydrogenase as per manufacturer’s protocol (Promega). Antibodies used for flow cytometry are available in online supplemental table S4. Cells were collected for analysis using Cytoflex LX (Beckman Coulter), and postacquisition analysis was performed using FlowJo software.
Live imaging
CAR-T cells were stained with CFSE. One million untransduced control or anti-CEA CAR-T cells were added to slices in culture with either IgG antibody control or anti-IL-10 antibody (IL-10) and incubated for 1 day before imaging. Alternatively, the CAR-T cells were thawed and incubated overnight with 20 μg/mL of either control or anti-IL-10R antibody (IL-10R). The pretreated CAR-T cells were then stained, placed directly into the CRLM tissue slice culture media without additional treatment and incubated for 1 day before imaging. Live fluorescent microscopy was performed as previously described on a Leica SP8X confocal microscope.37 40 Image processing and data analysis were performed on Leica LAS X software and IMARIS software. All EpCAM+, CFSE+, SR-FLICA+ cells were counted manually throughout the time imaged at each position.
Flow cytometry
CAR-T cells were stained with CFSE. TSC slices were then treated with control or antigen-specific CAR-T cells with or without IL-10, and following treatment for 1 day in culture the slices were digested to a cell suspension. The cells were then stained with fluorophore-labelled CD69 and CD25 (online supplemental table S4), and flow cytometry was performed. The flow analysis was performed on the LSR2 and Symphony flow cytometry machines (Beckton Dickinson).
Statistical analysis
Statistical significance in the TCGA analysis between the first and fourth IL10 expression quartiles was calculated using the Xena webtool’s implementation of the log-rank test. Data were entered and analysed using Prism (GraphPad, La Jolla, California, USA). For all other experiments, Student’s t-test, paired t-test or one-way analysis of variance with multiple comparisons was used as indicated. Statistical significance was defined as p<0.05. Further details are available in the online supplemental materials.
RESULTS
Patient and tumour characteristics
We generated TSC from 38 individual patients with CRLM (table 1). Microsatellite instability testing was available in 31 cases, and tumours were found to be microsatellite stable (MSS) in all available cases. All tumours treated with anti-CEA CAR-T cells were procured from patients with serum CEA greater than 2 ng/mL.
Table 1.
Characteristics of patients and tumour samples studied (n=38)
| Clinicopathological characteristics | |
| Age, median (range) | 56.5 (38–74) years old |
| Female | 19/38 (50%) |
| Received neoadjuvant chemotherapy prior to hepatic resection | 31/38 (82.6%) |
| Rectosigmoid or rectal location of primary tumour | 17/38 (45.7%) |
| T stage of primary tumour | |
| T1 | 2/38 (5.3%) |
| T2 | 5/38 (13.2%) |
| T3 | 23/38 (60.5%) |
| T4 | 8/38 (21.1%) |
| N stage of primary tumour | |
| N0 | 9/38 (23.7%) |
| N1 | 17/38 (44.7%) |
| N2 | 12/38 (31.5%) |
| Lymphovascular invasion present in primary tumour (unknown for n=11) | 19/27 (70.4%) |
| Perineural invasion present in primary tumour (unknown for n=10) | 10/28 (35.7%) |
| Synchronous metastasis (unknown for n=2) | 23/36 (63.9%) |
| Tumour molecular characteristics | |
| MSS (unknown for n=7) | 31/31 (100%) |
| NRAS wild type (unknown for n=12) | 25/26 (96.2%) |
| KRAS wild type (unknown for n=8) | 21/30 (70%) |
| BRAF wild type (unknown for n=12) | 26/26 (100%) |
MSS, microsatellite stable.
IL-10 blockade induces carcinoma cell death in human CRLM slice cultures refractory to PD-1 blockade
Analysis of TCGA Pan-Cancer dataset showed that elevated IL10 expression correlated with worse 5-year DSS in patients with COAD (online supplemental figure S1). In contrast, transcript levels of PDCD1 and HACVR2, encoding the T-cell coinhibitory receptors PD-1 and T-cell immunoglobulin and mucin domain-3 (TIM-3), respectively, did not correlate with DSS (online supplemental figure S1). To determine the cell types producing and responding to IL-10 in CRLM, we performed single-cell RNA sequencing (scRNAseq) of fresh, untreated CRLM samples from three separate patients. Immune cells, defined by clusters with high expression of PTPRC encoding CD45, comprised an average of 11.1% (range 4.2%–31.4% in the three cases) of all cells. This immune cell population is primarily composed of macrophages, with a cluster high in both CD68 and CD163 expression (5.5% of all cells, 56.7% of immune cells), and CD3 expressing T cells (3.3% of all cells, 33.9% of immune cells) (figure 1A). Of the T cells found within the CRLM tumour, the majority were helper T cells (Th) or cytotoxic T cells, with a smaller proportion of Treg cells (figure 1B). B cells expressing CD79A and DCs combined made up less than 0.3% of all cells and 3.4% of immune cells (figure 1A).
Figure 1.
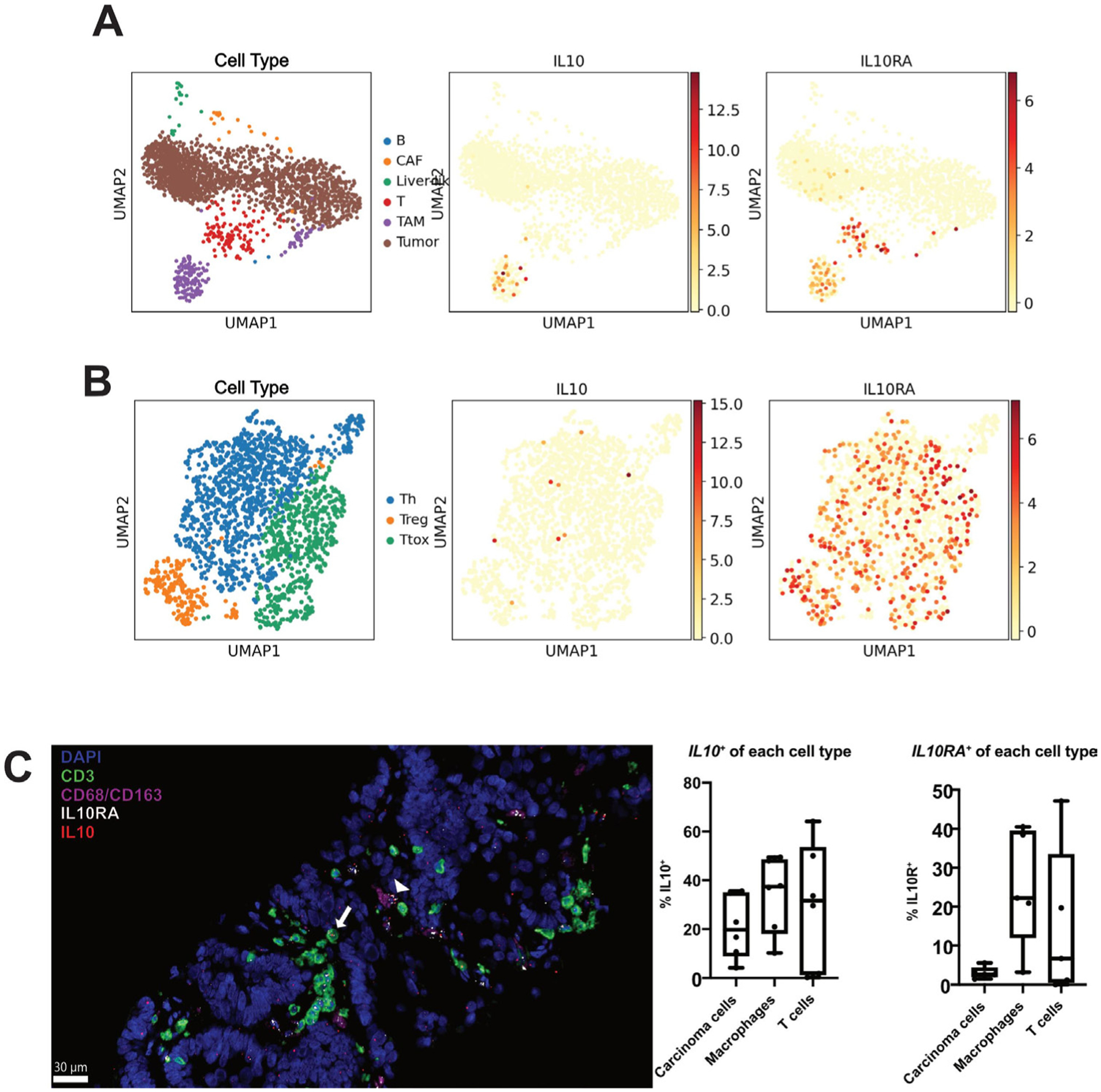
Interleukin 10 (IL-10) is produced by carcinoma cells, macrophages and T cells. (A) UMAP plots of all cell clusters of single cell RNA sequencing (scRNAseq) data from n=3 colorectal cancer liver metastases (CRLM) tumours. TAM, tumour-associated macrophages (purple); CAF, cancer-associated fibroblasts (orange); B cells (blue); liver-like cells (green); T cells (red); tumour cells (brown). IL10 and IL10RA expression highlighted in red/orange. (B) UMAP plots of T cell cluster of scRNAseq data from n=3 CRCLM tumours. Th, helper T cells (blue); Ttox, cytotoxic T cells (green); Treg, regulatory T cells (orange). IL10 and IL10RA expression highlighted in red/orange. (C) Multiplex immunohistochemistry was performed for T cells (CD3, green), macrophages (CD68/CD163, pink) and nuclei (DAPI, blue) with additional in situ hybridisation for IL10 (red) and IL10RA (white). A representative image with IL10+ IL10RA+macrophage (white arrowhead) and IL10+ IL10RA+T cell (white arrow) is shown. Quantification of the proportion of IL10+ and IL10+ cells within each cell type is also shown (n=5 patient tumours). Scale bar=30 μm. Data points represent each human tumour sample.
Analysis of our scRNAseq dataset demonstrated that while detectable RNA expression of IL-10 was predominantly limited to the TAM cluster, there was less, but still detectable, expression of IL10 in the Th cluster, the IL-10 receptor gene (IL10RA) was highly expressed in both T cells and TAMs (figure 1A,B). Given the relatively high expression necessary to reliably identify a transcript via scRNAseq, we next sought to confirm these results using a more sensitive multiplexed in situ hybridisation (mISH) platform. We examined IL10 and IL10RA gene expression in CRLM sections concurrently stained for carcinoma cells (EpCAM), T cells (CD3) and macrophages (CD68/CD163) (n=6). This mISH confirmed expression of IL10 in 30.9% of macrophages, in addition to relatively low levels of transcript expression in 25.6% of T cells and 19.3% of carcinoma cells (figure 1C). As with the single-cell data, via mISH IL10RA was robustly expressed in TAM (25.0%) and T cells (14.9%), with expression rarely detected in carcinoma cells (3.1%, figure 1C).
Leveraging our human TSC experimental system, we next aimed to examine the effects of IL-10 and PD-1 blockade in CRLM. Consistent with clinical trial results,45 anti-PD-1 did not increase tumour apoptosis compared with control in four individual human CRLM slice cultures (figure 2A,B). Given the inverse correlation between IL10 expression and DSS, we posited that intratumoral IL-10 suppresses effective antitumour immune responses. We first confirmed that addition of an IL-10 neutralising monoclonal antibody (αIL-10) significantly reduced IL-10 protein from 8.5 pg/mL to 0.39 pg/mL in supernatant from CRLM slice cultures (n=5, online supplemental figure S2A). In the majority of 32 unique CRLM patients’ TSCs, αIL-10 caused dramatic tumour necrosis (figure 2A), leading to 1.8-fold increased apoptosis compared with control (median 51.3% vs 29.1% cleaved Caspase-3+ cells, p<0.0001; figure 2C). Cleaved Caspase-3 colocalised with EpCAM, an epithelial marker, confirming that apoptosis occurred predominantly in carcinoma cells, which comprise the bulk of cells within CRLM (online supplemental figure S2B). We saw a similar magnitude of αIL-10-induced tumour apoptosis when evaluating αIL-10 versus control-treated tumour slices for cleaved Caspase-3 protein expression via RPPA (mean relative signal intensity 2.1 vs 1.1 a.u., n=3 cases, p=0.015; figure 2D) and TUNEL (median 38.8% vs 20.3% TUNEL+ of total nuclei, n=4 cases, p<0.001; figure 2E). Together, these results demonstrate the ability of IL-10 blockade to induce tumour-specific apoptosis in a human CRLM TSC model.
Figure 2.
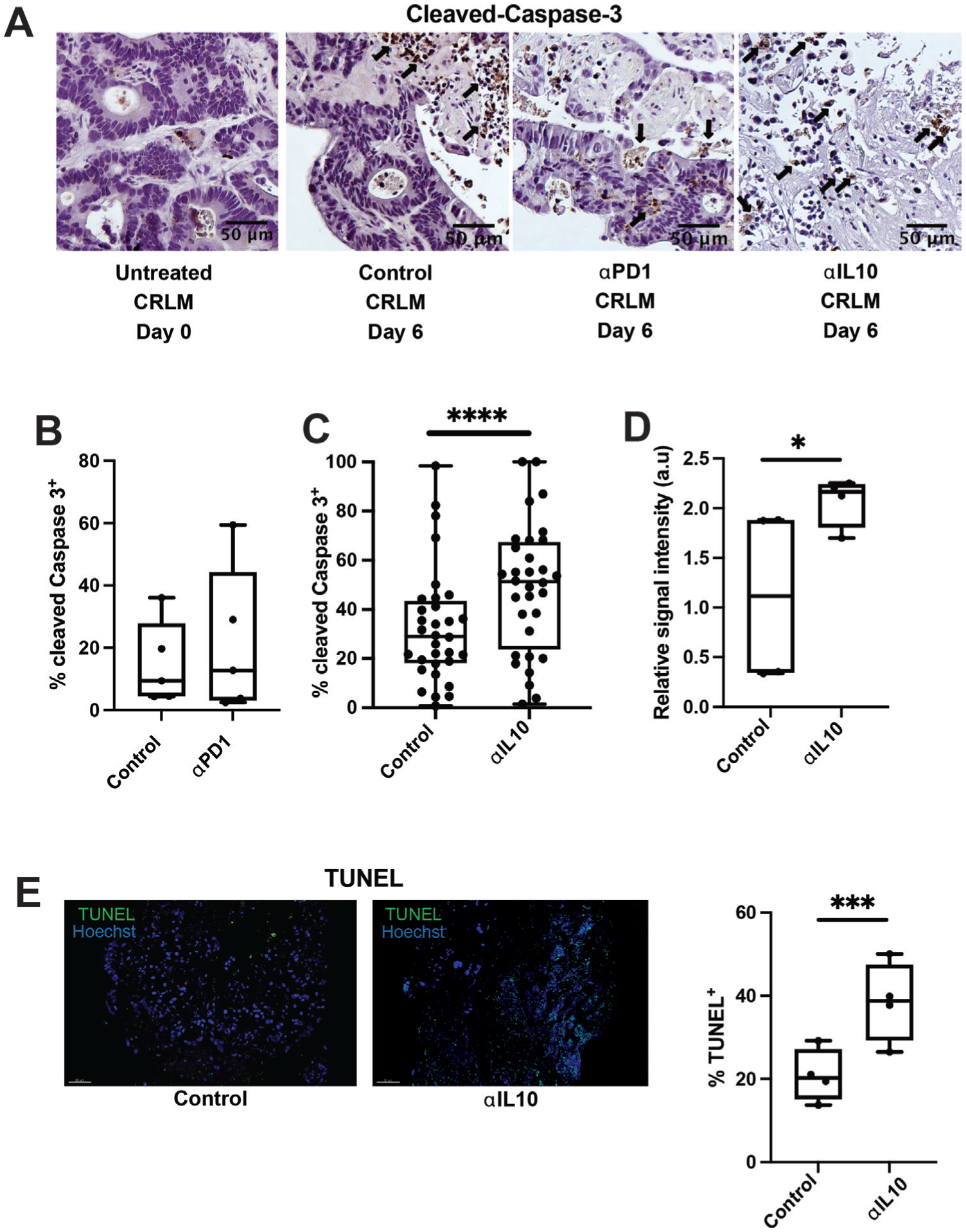
Interleukin 10 (IL-10) blockade facilitates significantly increased apoptosis of carcinoma cells. (A) Immunohistochemistry (IHC) for cleaved Caspase-3 was performed on tumour slice culture (TSC) after 6 days of treatment with immunomodulating agents. Representative images at 20x magnification from day 0, and then day 6 of antibody control, anti-programmed cell death protein-1 (anti-PD-1) antibody and αIL-10 treated slices. Black arrows denote apoptotic cells positive for cleaved Caspase-3 (brown). Scale bar=50 μm. (B) Quantification of percentage of cleaved Caspase-3+ cells in slices treated with control or anti-PD-1 antibody (n=4 separate human cases with at least two slices per case). (C) Quantification of percentage of cleaved Caspase-3+ cells in slices treated with control or αIL-10 (n=32 separate human cases with at least two slices per case). Data points represent each human tumour sample which consists of counts over multiple high-powered fields (hpf) in multiple slices to account for intratumoral heterogeneity. Student’s t-test. ****p<0.0001. (D) Reverse phase protein array with relative signal intensity of total cleaved Caspase-3 in colorectal cancer liver metastases TSC after 6 days in culture with αIL-10 (n=4 cases). Student’s t-test. *p<0.05. (E) Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining with fluorescent microscopy demonstrating TUNEL+ cells (green) after 6 days of αIL-10 (n=4 separate human cases). Scale bar=30 μm. Data points represent each human tumour sample which consists of counts over multiple hpf in multiple slices to account for intratumoral heterogeneity. Student’s t-test. ***p<0.001.
IL-10 blockade-mediated cell death in human CRLM is a result of immune activation
Given the ability of IL-10 blockade to specifically increase tumour cell death in our human CRLM TSC model, we hypothesised that IL-10 blockade in CRLM promotes a shift towards an inflammatory antitumour immune response in the TME. To further explore this hypothesis, we first quantified the presence of immune cells within each TSC treated with control antibody or IL-10 blockade. We found that the frequency of CD163+ macrophages was not significantly different (p=0.17) between control and αIL-10 treated tumours (figure 3A), but there was an increase in the number of HLA-DR+ cells (median 19.5% vs 11.5%, n=5 cases, p=0.02; figure 3B).
Figure 3.
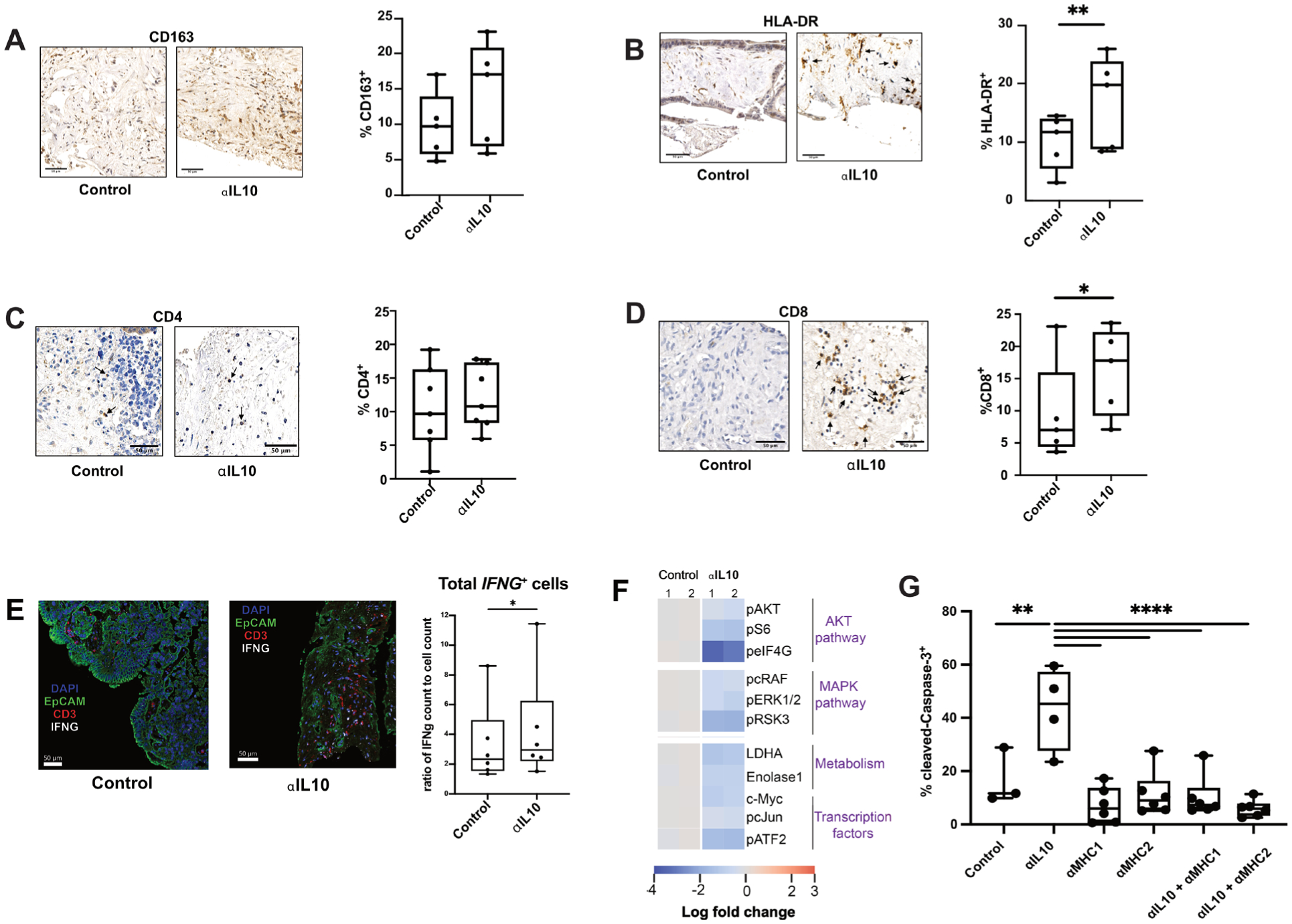
Increased CD8+ T-cell population and inflammatory immune microenvironment occurs after interleukin 10 (IL-10) blockade. (A) Representative images at 20x magnification from IHC staining for CD163 of fixed TSC treated with control or αIL-10 after 4 days and quantification by cell count of CD163+ cells (n=5 separate human tumours). Scale bar=50 μm. Data points represent each human tumour sample which consists of counts over multiple hpf in multiple slices to account for intratumoral heterogeneity. Not significant by Student’s t-test. (B) Representative images at 20x magnification from IHC of fixed TSC treated with control or αIL-10 after 4 days, with black arrows denoting HLA-DR+ cells, and quantification by cell count of HLA-DR+ cells (n=5 separate human tumours). Scale bar=50 μm. Data points represent each human tumour sample which consists of counts over multiple hpf in multiple slices to account for intratumoral heterogeneity. Student’s t-test. *p<0.05. (C) Representative images at 20x magnification from IHC of fixed TSC treated with control or αIL-10 after 4 days, with black arrows denoting CD4+ cells, and quantification by cell count of CD4+ cells (n=7 separate human tumours). Scale bar=50 μm. Data points represent each human tumour sample which consists of counts over multiple hpf in multiple slices to account for intratumoral heterogeneity. (D) Representative images at 20x magnification from IHC of fixed TSC treated with control or αIL-10 after 4 days, with black arrows denoting CD8+ cells. Quantification by cell count of CD8+ cells (n=5 separate human tumours). Scale bar=50 μm. Data points represent each human tumour sample which consists of counts over multiple hpf in multiple slices to account for intratumoral heterogeneity. Student’s t-test. *p<0.05. (E) Multiplex IHC was performed for T cells (CD3, red), carcinoma cells (EpCAM, green) and nuclei (DAPI, blue) with additional in situ hybridisation for IFNG (white). Quantification of proportion of IFNG+ per total number of cells by ISH and by IHC are also shown (n=6 patient tumours). Scale bar=50 μm. Data points represent each human tumour sample. Student’s t-test. *p<0.05. (F) RPPA data from slices treated with antibody control or IL-10 blockade showing downregulation of various molecular pathways. (G) Quantification of percentage of cleaved Caspase-3+ cells following αIL-10 with or without MHC blockade shows that while αIL-10 alone increases apoptotic cell proportion, the effect is reversible with MHC blockade back to below baseline levels of cell death. Representative data with data points representing a slice from one human tumour experiment, repeated in a total of three human tumour samples with similar results. Error bars display SEM. ANOVA with multiple comparisons. **p<0.01, ****p<0.0001.
While αIL-10 did not alter the CD4+ cell frequency (figure 3C), αIL-10 doubled the frequency of intratumoral CD8+ cells compared with control (median 17.8% vs 7.0%, n=5, p=0.02; figure 3D). Similarly, αIL-10 increased gene expression of the immunostimulatory cytokine interferon-gamma, IFNG, when normalised to total cell number on days 2–5 (median 2.9% vs 2.3%, respectively, p<0.05, n=6; figure 3E) and a non-statistically significant increase in total granzyme B (mean 2.3% granzyme B positive tissue area vs 1.2%, n=6, p=0.13; online supplemental figure S2D). Although PD-1 does serve as a marker of T-cell exhaustion following prolonged (greater than 1 week) antigen stimulation,46 we tested its expression after 2–5 days in our TSC model to identify early activated T cells potentially undergoing clonal expansion.47 48 We found a non-statistically significant increase in CD3+ T-cell expression of PD-1 in αIL-10-treated slices (p=0.08; online supplemental figure S2D), suggesting a shift towards an activated or clonally expanding phenotype.
To further understand the effects of IL-10 blockade on the immune TME, we additionally performed scRNAseq of αIL-10-and control-treated human CRLM TSCs from two individual patients at three consecutive time points (days 1, 2 and 3). We captured a total of approximately 7300 cells across a total of six biological replicates. While not significant, we found that activation signature (CD69, FASLG, CD27, CD28, TNFRSF9, TNFRSF4, LAMP1, EOMES, TBX21) scores for both CD4+ and CD8+ T cells were higher in αIL-10-treated samples when compared with controls at each of the three examined time points (online supplemental figure S3A,B). Importantly, no such trend was observed for a T cell exhaustion signature (PDCD1, CTLA4, HAVCR2, TIGIT, LAG3) across treated and untreated samples (online supplemental figure S3C). We additionally observed a non-significant upregulation in MHC-II presentation Reactome pathway genes in TAMs (online supplemental figure S3D), which closely mirrored our previous IHC findings (figure 3A,B). As HLA-DR and CD163 colocalised on sequential IHC staining (n=4; online supplemental figure S2C), these results together strongly suggest that IL-10 blockade increases macrophage MHC-II expression and thereby macrophage activation. These findings thereby suggest that the antitumour immune response induced by IL-10 blockade is mediated by an increase in the frequency of CD8+ cytotoxic T cell and concurrent immunostimulatory polarisation of TAMs.
We next sought to confirm the role of both direct CD8+ cytotoxic T cell activity and indirect CD4+ helper T cell and TAM activity in the αIL-10 antitumour response. Towards this, we treated CRLM tumours with αIL-10 in the presence or absence of antibody-mediated MHC class I or class II blockade. Confirming our hypothesis, we found that blockade of MHC-I, required for cytotoxic T cell antigen recognition, reversed the effects of IL-10 blockade on carcinoma cell apoptosis. In addition, blockade of MHC-II, required for CD4+ T cell and TAM antigen presentation, similarly completely and independently abrogated the effects of αIL-10 (figure 3G). That is, these results directly demonstrate that both the MHC-I and MHC-II pathways are necessary, though not necessarily sufficient, for the IL-10 blockade-mediated antitumour immune response. In sum, our results demonstrate that IL-10 blockade in human CRLM TSC induces and requires a classical antitumour immune response involving the activation of cytotoxic T cells and APCs.
Murine myeloid cell-derived IL-10 reversibly suppresses CAR-T cell proliferation
We previously demonstrated in a murine model of CRLM that myeloid cells suppress inflammation and inhibit CAR-T cell function in a STAT-3-dependent manner.15 16 Based on our present findings on the effect of ⍺IL-10 on native immune cells in the CRLM TME, we hypothesised that tumour-associated myeloid cells were suppressing CAR-T cells by secreting IL-10 and causing downstream IL-10R/STAT3 signalling in adjacent CAR-T cells. By flow cytometry, we found 37.4% of murine myeloid cells expressed IL-10, while few T cells produced IL-10 (figure 4A). In addition, multiplexed immunofluorescent imaging confirmed the presence of IL-10+CD11b+GR1+ myeloid-derived suppressor cells and IL-10RA+CD3+ T cells (figure 4B). Addition of myeloid cells to coculture of anti-CEA CAR-T cells and CEA-expressing carcinoma cells significantly reduced CAR-T cell proliferation from 50.3% to 29.3% (figure 4C). However, when the CAR-T cell, carcinoma cell and myeloid cell cocultures were treated with αIL-10, CAR-T proliferation was restored in a dose-dependent fashion (39.7%–45.9% proliferation, n=4, p<0.01; figure 4C). Myeloid cells also possess the ability to inhibit the cytotoxic activity of CAR-T cells. Importantly, as with its effect on CAR-T proliferation, addition of αIL-10 to the coculture system reversed the suppressive effects of myeloid cells on tumour cytotoxicity (p<0.001; figure 4D). Therefore, our results demonstrate that myeloid cell suppression of anti-CEA CAR-T cell proliferation and cytotoxicity can be reversed by antibody neutralisation of IL-10.
Figure 4.
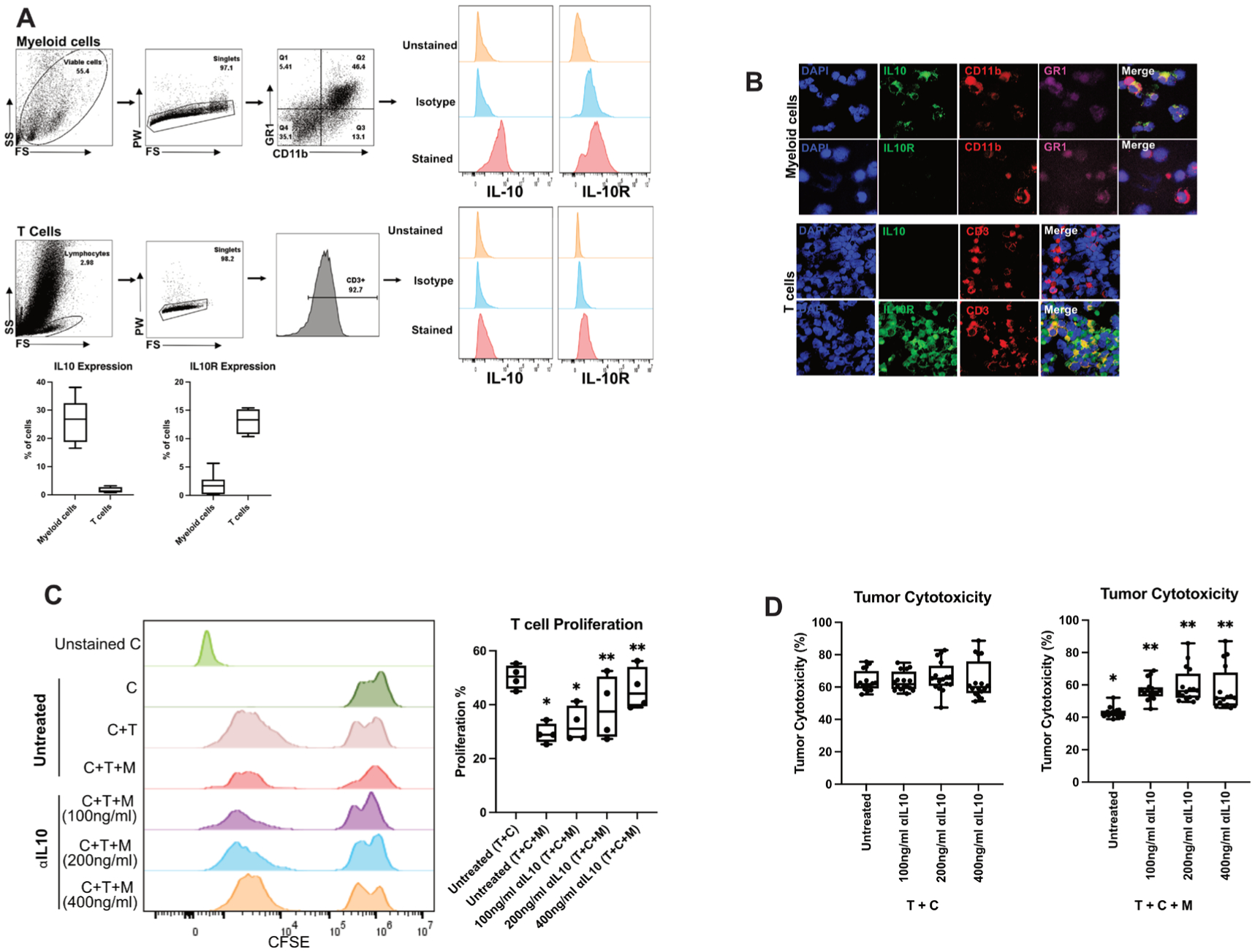
Treatment with interleukin 10 (IL-10) antibody reverses murine myeloid cell suppressive activity of chimeric antigen receptor T (CAR-T) cells. (A) Phenotypic characterisation of CAR-T cells and CD11b+ cells isolated 4 days post-tumour generation was performed as per the gating strategy. Representative flow cytometry plots showing expression of IL-10 and IL-10R for myeloid cells (n=7) and CAR-T (n=4). (B) Immunofluorescence performed on CAR-T and myeloid cells confirmed expression of IL-10 (green) in myeloid cells and IL-10R (green) on CAR-T cells. (C) Carboxyfluorescein diacetate succinimidyl ester (CFSE)-labelled CAR-T (C) cells were cocultured with myeloid cells (M) isolated from liver metastases (LM) tumour-bearing mice and MC38CEA tumour cells (T) in 1:1:1 ratio. Cells were treated with 100 ng/mL or 200 ng/mL or 400 ng/mL anti-IL-10 antibody for 24 hours. *p<0.005 versus untreated (T+C), **p<0.01 versus untreated (T+C+M). (D) The in vitro cytotoxic activity of CAR-T (C) cells was evaluated by incubating MC38CEA (T) cells with myeloid cells (M) treated with at a 1:1:1 ratio as shown in C. Corresponding tumour and CAR-T controls were used for the assay. *p<0.0001 versus T+C, **p<0.001 versus untreated T+C+M. All data were analysed by one-way analysis of variance and Tukey’s test or two-sided Student’s t-test.
IL-10 blockade facilitates CAR-T-mediated tumour cell death
Based on our results in murine CRLM, we hypothesised that αIL-10 would also enhance cytotoxicity of CEA-specific CAR-T cells in human CRLM. We previously demonstrated the ability of intravital confocal microscopy to demonstrate early effects of T cell-targeted therapies in human PDA slice cultures.37 We first confirmed adequate migration of CAR-T cells into the tumour slice after 1 day (figure 5A). We then used live imaging to assess CAR-T function in the presence of αIL-10 or control. CEA-specific CAR-T cells demonstrated low (mean 30.1%) basal migration to target cells (defined by EpCAM+ cell with CFSE+ CAR-T cell within 20 μm), which was enhanced with addition of αIL-10 (mean 82.5%, n=3 separate CRLM tumours, p<0.001; figure 5B). Additionally, there was lower antitumour activity in the control or antigen-specific CAR-T cells treated with control when compared with αIL-10 (67.2% vs 15.6%–26.6% SR-FLICA+ EpCAM+ cells, n=3 cases, p<0.03; figure 5C). To determine if the increase in cancer cell death was mediated by CAR-T cells and not another cell type, we specifically evaluated the subset of carcinoma cells adjacent to a CAR-T cell. We found 58.2% of EpCAM+ cells adjacent to anti-CEA CAR-T cells were SR-FLICA+ when treated additionally with αIL-10, while this was true of only 18.7% of EpCAM+ cells treated with anti-CEA CAR-T cells in the absence αIL-10 (p=0.007, n=3 separate human CRLM tumours; figure 5D). Additionally, there was no difference in the proportion of SR-FLICA+ EpCAM+ cells not adjacent to a CFSE+ CAR-T cell. Together, these results demonstrate that IL-10 inhibits tumour-specific CAR-T function in both human and murine CRLM.
Figure 5.
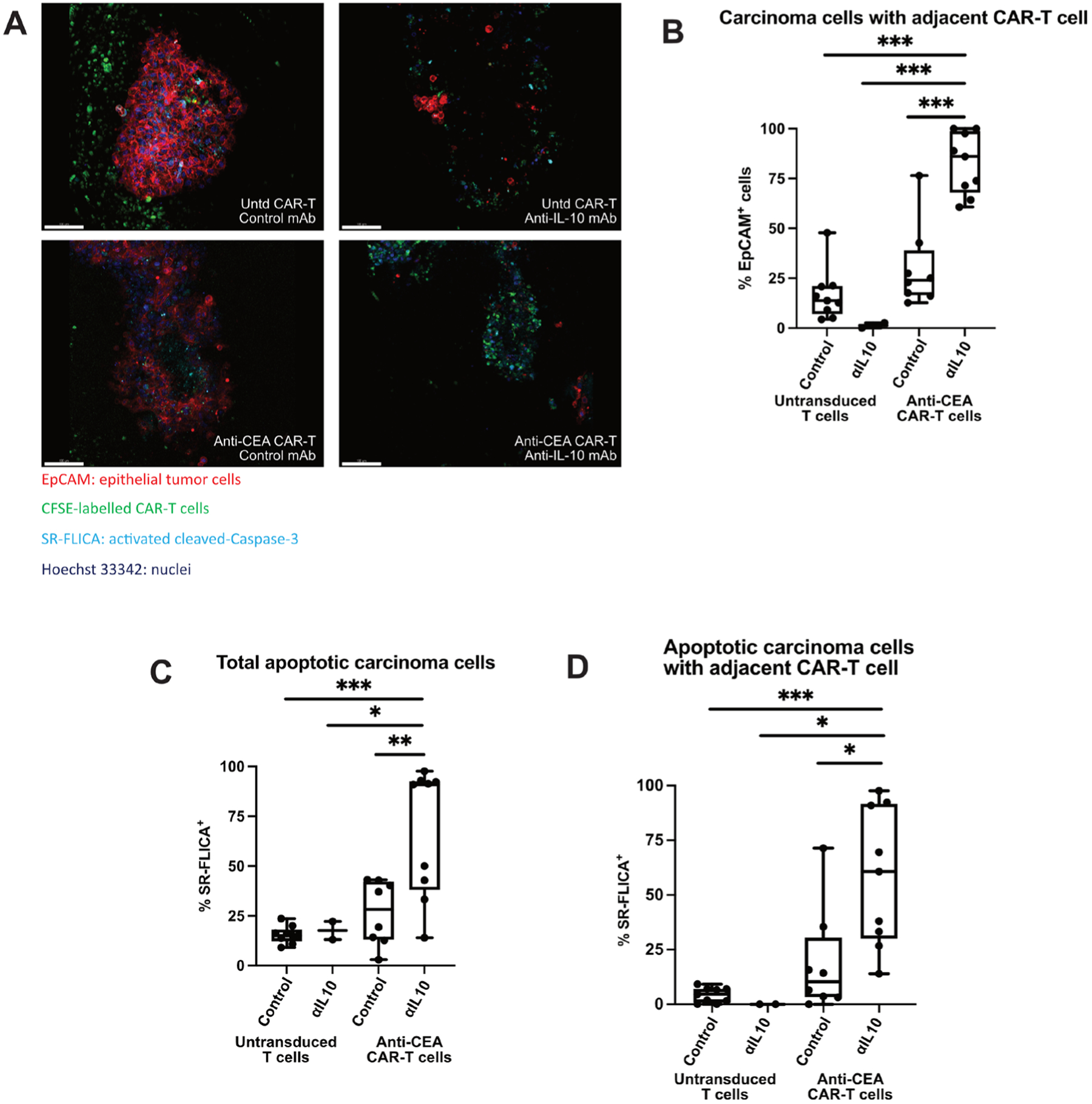
Interleukin 10 (IL-10) blockade enhances chimeric antigen receptor T (CAR-T) cell migration and tumour apoptosis. (A) Live staining and fluorescent microscopy of colorectal cancer liver metastases (CRLM) tumour slice culture (TSC) treated with untransduced CAR-T with control, untransduced CAR-T with αIL-10, anti-carcinoembryonic antigen (CEA) CAR-T with control, and anti-CEA CAR-T with αIL-10 show CFSE+ CAR-T cells (green) within the tumour and near epithelial carcinoma cells (red) when antigen-specific CAR-T are used in combination with αIL-10. Apoptotic cells are shown when activated cleaved Caspases are present and positive for the SR-FLICA reagent (cyan). Tumour slices were imaged after 1 day of treatment. Scale bar=100 μm. (B) Quantification of the carcinoma cells with a CAR-T cell nearby (defined as within 20 μm) with untransduced control CAR-T cells and anti-CEA CAR-T cells treated concurrently with or without αIL-10. Data points represent each high-powered fields (hpf) area imaged, with 3 hpf imaged in each tumour (n=3 separate human tumour samples). Analysis of variance with multiple comparisons. ***p<0.001. (C,D) Quantification of the percentage of total carcinoma cells that are SR-FLICA+, and SR-FLICA+ carcinoma cells with a CAR-T cell nearby (defined as within 20 μm) after untransduced control CAR-T and anti-CEA CAR-T cells are treated concurrently with or without αIL-10. Data points represent each hpf area imaged, with 3 hpf imaged in each tumour (n=3 separate human tumour samples). Analysis of variance with multiple comparisons. *p<0.05, **p<0.01, ***p<0.001.
To better characterise the downstream effects of IL-10 blockade on antigen-specific CAR-T cells in human CRLM, we performed RNA sequencing of the treated human tumour slices after 1 day. We specifically found that genes associated with cytolytic functions (GNLY, GZMA, PRF1), inflammation (IFNG, GBP5,49 NOS2) and T cell migration/chemotaxis (CXCL9, CXCL10, CXCL11,50 CCL7, CCL8) were relatively upregulated after antigen-specific CAR-T cells were treated with αIL-10 (figure 6A). We then performed a gene ontology analysis of upregulated genes in those samples treated with anti-CEA CAR-T cells and αIL-10 versus anti-CEA CAR-T cells and control IgG. Transcriptional pathways including cellular response to interferon-gamma, inflammatory response, cellular response to IL-1, lymphocyte chemotaxis and T cell migration were all upregulated in the antigen-specific CAR-T cells treated with αIL-10 compared with the same CAR-T cells treated with control antibody (figure 6B). Additionally, flow cytometry performed on the CAR-T cells after coculture in slice culture demonstrated increased expression of the activation markers CD25 and CD69 (figure 6C; online supplemental figure S5).
Figure 6.
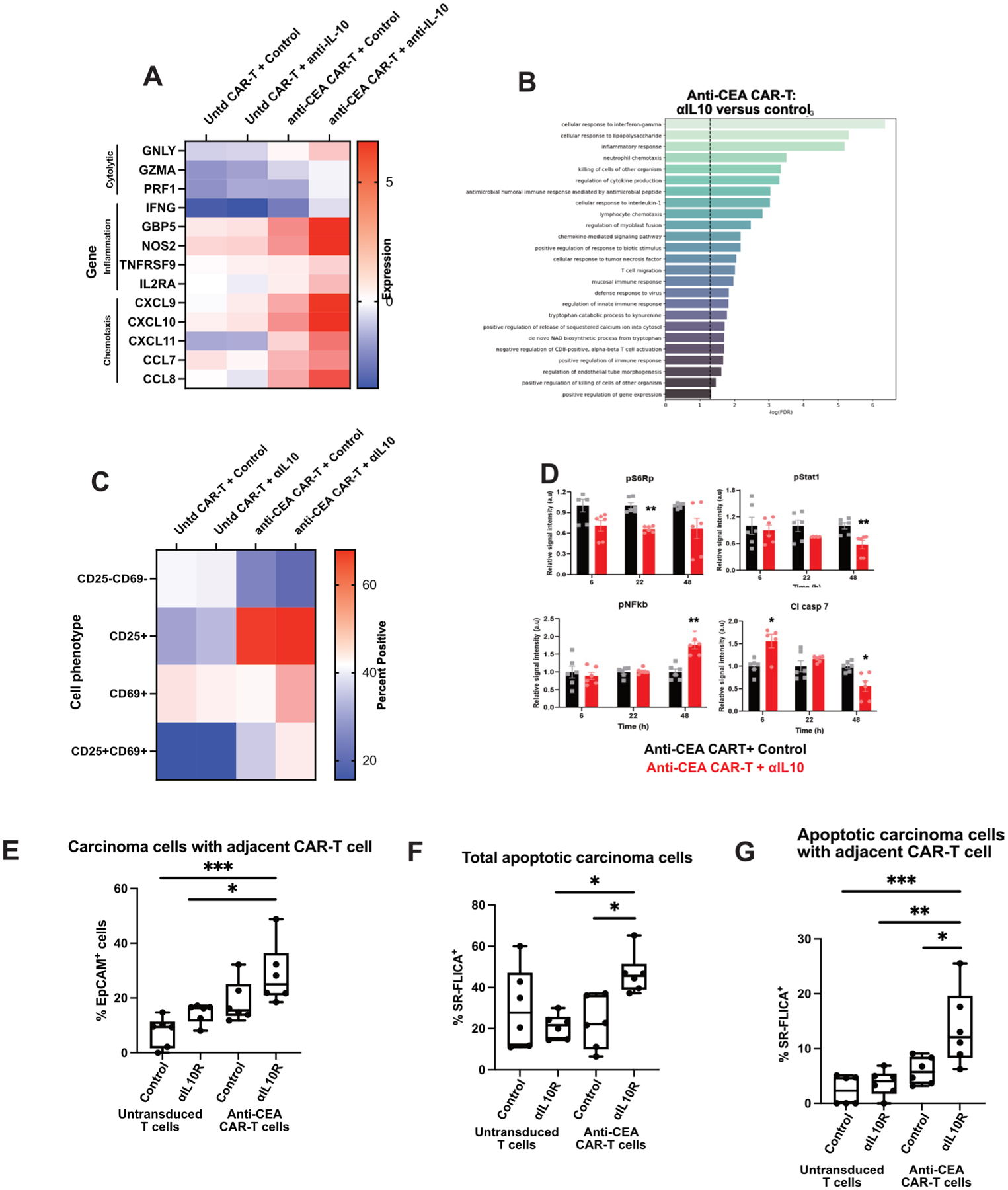
Interleukin 10 (IL-10) blockade increases antigen-specific chimeric antigen receptor T (CAR-T) cell activation through a mechanism that requires intact IL-10R on CAR-T cells. Human colorectal cancer liver metastases (CRLM) slices were treated with control or carcinoembryonic antigen (CEA)-specific CAR-T cells, plus control or αIL-10, for 1 day. (A) Tumour slices were analysed by bulk RNA sequencing with heatmap demonstrating expression of genes related to T cell cytolytic functioning, activation and migration/chemotaxis for each treatment group (n=2 tumours). Dark red denotes high expression of the listed gene and dark blue denotes comparably low expression normalised to housekeeping genes. (B) Gene expression pathways increased with anti-CEA CAR-T cells are treated with αIL-10 versus control. Untd, untransduced control CAR-T cells. (C) Representative case of flow cytometry performed on CAR-T cells after treatment with control or IL-10 blocking antibody demonstrating expression of CD25 and CD69 (n=2 tumours). (D) Reverse phase protein array showing expression of pS6, Stat1, nuclear factor kappa B (NF-κB) and cleaved Caspase-7 protein of slices treated with anti-CEA CAR-T cells and either control or αIL-10 at 6, 22 and 48 hours. After 6 hours, cleaved Caspase-7 expression is increased in the slices treated with the CAR-T in addition to αIL-10, and at 22 hours pS6 expression is decreased. After 48 hours, Stat1 and cleaved Caspase-7 are decreased with an increase in NF-κB. Each data point represents a replicate from n=2 cases. Student’s t-test. *p<0.05; **p<0.01. (E–G) Following treatment of either untransduced control or anti-CEA CAR-T cells with either control or αIL-10R, the CAR-T cells alone were incubated with tumour slice culture and live imaging was performed. Again, quantification of the carcinoma cells with a CAR-T cell adjacent (defined as within 20 μm), total SR-FLICA+ carcinoma cells and SR-FLICA+ carcinoma cells with a CAR-T cell adjacent. Data points represent each high-powered fields (hpf) area imaged, with 3 hpf imaged in each tumour (n=3 separate human tumour samples). Analysis of variance with multiple comparisons. *p<0.05, **p<0.01, ***p<0.001.
To determine the molecular mechanisms within the CAR-T cells, we employed RPPA to assess changes in the state and activity of >20 proteins including major oncogenic signalling pathways (RAF/MEK/ERK, PI3K/AKT, RTK signalling and STATs) in slices treated with anti-CEA CAR-T cells and αIL-10. We found a decrease in pS6 ribosomal protein expression within 24 hours followed by a decrease in STAT1 at 48 hours (figure 6D). Cleaved Caspase-7 increased in the anti-CEA CAR-T and αIL-10-treated slices in 6 hours and was then low at 48 hours. At 48 hours, we noted an increase in nuclear factor kappa B (NF-κB) expression. NF-κB is unlikely to be upregulated in dying tumour cells and therefore this change likely represents CAR-T cells as the next most abundant cell type, and high NF-κB activity has been demonstrated in antitumour T-cell responses.51–53 The time course of the increase in cleaved Caspase is consistent with our findings of increased cell death in about within 1 day as previously described on fluorescence imaging, and the subsequent decrease may be due to early significant cell death leading to less Caspase activity at later time points. In addition, we noted high NF-κB activity after 24–48 hours, consistent with our findings of increased antitumour activity of the antigen-specific CAR-T cells within 2 days.
Given the rapid effect of αIL-10 on CAR-T function, we hypothesised that this was due to relief of direct suppressive effects of IL-10 on CAR-T cells, rather than from secondary effects attributable to modulation of myeloid cells. To test this hypothesis, we pretreated the CAR-T cells with an antagonistic anti-IL-10 receptor alpha (IL-10R) antibody before adding them alone to the CRLM TSC. CAR-T cells pretreated with αIL-10R showed a trend towards being in closer proximity to carcinoma cells compared with control treated CAR-T (figure 6E). Furthermore, pretreatment with αIL-10R increased CAR-T cytotoxicity, as evidenced by increased total carcinoma cell apoptosis, and apoptosis with an adjacent CAR-T cell (p<0.05; figure 6F,G). These data demonstrate that IL-10 blockade activates CAR-T cell function in part through direct inhibition of IL-10R signalling in CAR-T cells.
DISCUSSION
Defining tumour-specific mechanisms of immune evasion is critical to developing new immunotherapies against ICI-resistant cancers, and testing these strategies in accurate preclinical models is essential to future clinical success. Using a unique human tumour-based model of metastatic CRC, we demonstrate that the immunosuppressive cytokine IL-10 is a pivotal mechanism of immune evasion in human CRLM. In a heterogeneous cohort of 38 unique patients’ tumours, IL-10 blockade nearly doubled tumour apoptosis (figure 2C–E) through an immune-mediated mechanism dependent on both MHC class I and class II antigen presentation (figure 3G). The relevance of IL-10 in CRLM to antigen-specific T-cell function was further underscored by CAR-T cell experiments, in which αIL-10 dramatically increased proliferation and cytotoxicity of CEA-specific CAR-T cells in a murine cell coculture systems (figure 4) and human CRLM slice cultures (figures 5 and 6).
Combined, our data identify IL-10 as a major immune checkpoint to overcome in human CRLM and support a model in which IL-10 in the CRLM TME maintains MHC class II-low TAMs with poor antigen presenting capacity, resulting in ineffective cytotoxicity of tumour antigen-specific T cells. We found that blocking the effect of IL-10 in the TME results in reduced protein kinase B (AKT) and mitogen-activated protein kinase (MAPK) pathway phosphorylation, followed by increased MHC-II-dependent antigen presentation by macrophage, leading to CD8+ T cell proliferation and activation, resulting in tumour cell death. It is perhaps due to either the relatively short duration of treatment (1 week) or to the presence of and help from CD4+ T cells on CD8+ T cells and the short culture period that despite T-cell activation, we do not see transcriptomic evidence of T cell exhaustion.54 To test the effects of IL-10 and IL-10 blockade on antigen-specific T cells within the human CRLM environment, we modified the TSC system to include adoptive transfer of control or CEA-specific CAR-T cells. Although CAR-T recognise surface antigen through a different mechanism than the native T-cell receptor/peptide/MHC complex, adoptive CAR-T transfer was the most feasible approach within our heterogeneous human tumours. Within this modified system, we found that antagonism of IL-10 ligand or receptor caused rapid activation of antigen-specific CAR-T cells in the TME, with concurrent transcriptional and proteomic changes reflective of cytotoxic function (increased expression of GBP5, NOS2, CXCL9, CXCL10 and CCL8) and T-cell activation (increased NF-κB expression and increased CD25+CD69+ population). Overall, these data suggest direct IL-10 effects via IL-10R signalling in both the myeloid and T-cell compartments in CRLM, inhibition of which leads to rapid changes in gene and protein expression in IL-10-target cells that culminate in immune-mediated tumour killing.
Our results provide a human cancer context for the large body of literature detailing the direct inhibitory effects of IL-10 on both APC and T-cell function.18 In the context of liver-mediated tolerance,19 we hypothesised that IL-10 might also inhibit APC and T-cell function in the liver TME. Our findings corroborate what others have demonstrated in the primary murine TME. A murine model of breast cancer demonstrated that macrophage-derived IL-10 suppressed DC production of IL-12, which was necessary for maximal CD8+ T cell function and chemotherapy response.22 In a flank tumour model of lung cancer, tumour growth was enhanced in transgenic mice overexpressing Il10, which was reversible with antibody-mediated IL-10 blockade.55 Tumour monocyte-derived IL-10 was directly inhibitory on T-cell proliferation in an orthotopic murine model of CRC.56 In the clinical context, a meta-analysis of 1788 patients spanning 10 different cancer types demonstrated correlation between serum IL-10 concentration and worse survival.21 Together, these studies describe an immunosuppressive, protumoral effect of IL-10.
However, evidence also suggests that IL-10 paradoxically activates antigen-experienced T cells and induces immune-mediated tumour regression. PEGylated IL-10 (pegilodecakin, AM0010) has been shown to induce CD8+ T cell-dependent tumour regression in a murine flank model of squamous cell carcinoma.26 A phase I trial of pegilodecakin in advanced solid cancers demonstrated regression in 5/24 patients.27 In responding patients, activated T cells expanded in both the peripheral blood and tumour and peripheral clonal T cell expansion coincided with delayed tumour response.28 IL-10 in vitro increased granzyme B effector molecule expression in activated human peripheral blood CD8+ T cells, and increased cytotoxicity of tumour-specific CAR-T cells cocultured with target human leukaemia cells.57 An IL-10 fusion protein, IL-10-Fc, expanded and rescued terminally exhausted T cells in murine melanoma and CRC through metabolic reprogramming.58 Together, these data provide an argument for potential immune-activating, antitumour effects of IL-10. However, two clinical trials of pegilodecakin in pancreatic cancer34 and non-small cell lung cancer35 did not meet prespecified endpoints.
The key to the IL-10 paradox likely relates to context, including stoichiometry, kinetics of ligand/receptor interactions and organ-specific immune environmental factors. Notably, the 3D structure of the active IL-10/IL-10RA/IL-10RB hexamer complex was recently defined. This informed design of IL-10 variants with varying receptor subtype affinity, allowing for cell type-specific induction of myeloid but not T cell IL-10R signalling.33 A limitation of the current study is the cell type-indiscriminate effects of αIL-10. The requirement of both MHC class I and class II for maximal αIL-10 effect (figure 3G) demonstrates that both endogenous T cells and APC are required for optimal response, suggesting that more precise myeloid cell targeting could further enhance immune responses. Development of novel IL-10R antagonists and additional RNA-based methods of gene knockdown will enhance the TSC platform and help address these next-order questions.
Another limitation of the TSC model is the absence of circulating blood or draining lymph node components. In vivo murine models are a potential strategy to investigate the effects of systemic or regional lymphocytes, but must be interpreted with caution, given the limitations of carcinogen-derived, transplanted flank models, which are often more immunogenic than human cancer counterparts.59 60 For example, one study in IL-10−/− mice with MC38 murine colon carcinoma cells demonstrated that IL-10 inhibited inflammatory cytokines and resulted in reduced tumour growth.61 Specifically, the MC38 mouse model has been shown to represent MSI-high CRC,62 which clinically in humans correlates with high tumour mutation burden (TMB) and response to ICI. However, most patients with metastatic CRC have MSS tumours with low TMB and do not respond to ICI; thus, modelling and designing new immunotherapeutic approaches for patients with MSS CRC is a major clinical need. The fact that αIL-10 is nevertheless able to elicit immune-mediated tumour regression is notable, given the complete reliance on immune cells present in a 6 mm diameter×250 μm-thick tumour slice. Our findings build on the foundation of profiled intratumoral T cells8 63 and myeloid cells,10 confirming the presence of T cells and APC capable of together mounting an antitumour response. Future work will investigate whether IL-10 blockade causes clonal CD8+ and/or CD4+ T-cell expansion and parallel studies will define the baseline and αIL-10-induced polarisation of TAMs. In CRLM slices, macrophages were a major source of IL-10 (figure 1B), yet IL-10 blockade significantly increased MHC class II expression (figure 3B; online supplemental figure S3D). IL-10 blockade may therefore represent a novel approach to repolarising inhibitory macrophages in CRLM.
Given the safety of CAR-T therapy in CRLM in human clinical trials,13 14 strategies to enhance adoptive cellular therapy are also needed. Here, we found that αIL-10 significantly increased CAR-T migration, proliferation and cytotoxicity in CRLM (figures 4–6). Our findings suggest that ‘arming’ CAR-T cells against IL-10, for example, through transgenic expression of a neutralising IL-10 antibody, may achieve the dual benefit of enhancing CAR-T function and serving as a novel delivery vehicle of IL-10 blockade. Interestingly, IL-10R blockade prior to adoptive transfer leads to similar results, indicating a direct mechanistic effect of IL-10 on the CAR-T cells (figure 6E–G). Future work will define molecular mechanisms underlying the rapid (<24 hours) response of CAR-T cells to αIL-10, such as downstream changes in STAT3 phosphorylation and expression. Given the paradoxical effects of IL-10 observed in leukaemia-specific CAR-T cells in vitro,57 it will be important to define in which contexts, for example, liver metastases, optimal T-cell activation results from blockade rather than additional IL-10/IL-10R signalling.
The controversial role of IL-10 in cancer underscores the importance of studying this cytokine in accurate preclinical models that faithfully recapitulate human cancer. Here, we present evidence that IL-10 suppresses antitumour immunity in human CRLM and that a neutralising antibody against IL-10 can reverse this effect to induce dramatic immune-mediated tumour death. We previously demonstrated that TSC accurately recapitulates tumour and stromal elements of multiple GI carcinomas35 37 and predicts patient responses to both standard chemotherapies34 and novel immunotherapy combinations.38 Despite limitations of human TSC, it is nevertheless notable that we observed a response in over two-thirds of patients’ tumours, despite variability in tumour mutational status and preoperative chemotherapy response. We conclude that IL-10 is a major immune checkpoint to overcome in CRLM. Future work will investigate the relevance of IL-10 blockade to other primary and secondary liver tumours, and other immune-infiltrated tumours resistant to ICI.
Supplementary Material
WHAT IS ALREADY KNOWN ON THIS TOPIC
Despite evidence that the immune system plays a role in tumour development and progression, immunotherapy has yet to demonstrate substantial benefit for patients with metastatic microsatellite stable (MSS) colorectal carcinoma. Although interleukin 10 (IL-10) is primarily believed to be an immunosuppressive cytokine, its role in advanced gastrointestinal malignancies remains unclear. A human tumour slice culture model serves as an accurate predictor of therapeutic drug effects on the tumour microenvironment (TME). In mouse models of colorectal cancer liver metastases (CRLM), myeloid cells are immunosuppressive and inhibit activity of anti-carcinoembryonic antigen chimeric antigen receptor T (CAR-T) cells.
WHAT THIS STUDY ADDS
IL-10 is produced by macrophages, T cells and tumour cells in CRLM. IL-10 blockade overcomes the immunosuppressive TME in a model of human CRLM and reactivates endogenous antitumour immunity, generating nearly twofold greater carcinoma cell death. IL-10 blockade dramatically enhances CAR-T cell cytotoxicity in vitro and in human CRLM slice cultures.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
IL-10 represents a major immunosuppressive signal to overcome in patients with CRLM. Targeting IL-10/IL-10 receptor signalling has great potential to be incorporated into systemic therapy for patients with immune checkpoint blockade-resistant, MSS CRLM. CAR-T cells, armed against IL-10 suppression, may pave the way for more effective adoptive cell therapies for patients with MSS CRLM refractory to currently available surgical and medical therapies.
Acknowledgements
This research was also supported by the Experimental Histopathology Shared Resource of the Fred Hutch/University of Washington Cancer Consortium (P30 CA015704). We acknowledge support from the National Institutes of Health (S10 OD016240) to the W. M. Keck Center for Advanced Studies in Neural Signaling and the assistance of Keck Center manager, Dr Nathaniel Peters. We thank Stella Shin and CB Lim for assistance with RPPA samples. We also thank Dr Jeffrey Schlom for the generous gift of MC38CEA cell line. The authors also thank Sorrento and TNK Therapeutics for generously preparing and providing the anti-CEA CAR-T cells and CEA-Fc detection reagent.
Funding
KMS received funding from the Fibrolamellar Cancer Foundation and Cancer Research Institute (FCF/CRI Postdoctoral Fellowship). TSG received funding from the American Cancer Society (RSG-19-197-01). TSK received funding from the UW/Fred Hutchinson Cancer Center Support Grant (CCSG), P30 CA015704, New Investigator Award. QT received funding from USAMRAA (CA150370P1) and NIH (NCI R01 CA190122). VGP and SCK received funding from the US Army Medical Research Acquisition Activity (USAMRAA; CA150370P2). RSY received funding from USAMRAA (CA150370). VGP received funding from Seattle Translational Tumor Research, Brotman Baty Institute for Precision Medicine, and Merck Investigator Studies Program.
Competing interests
VGP is a member of the scientific advisory board for TriSalus Life Sciences. He has served as a consultant for Merck & Company in 2018, GlaxoSmithKline in 2019, Imvax in 2019, Takeda in 2020 and Umoja in 2022. He has research funding from AstraZeneca, Ipsen, Merck, OncoResponse and NGM. The funders had no role in the conceptualisation, design, data collection, analysis, decision to publish or preparation of the manuscript. The anti-CEA CAR vector was provided by TNK Therapeutics, who had no role in the conceptualisation, design, data collection, analysis or preparation of the manuscript. SCK served as an advisor for TNK Therapeutics until December 2019, is currently Chief Medical Officer of TriSalus Life Sciences and SAB member of Nkarta, Takeda and Sentibio, and has received research funding from Takeda and Nkata.
Footnotes
Patient and public involvement Patients and/or the public were not involved in the design, conduct, reporting or dissemination plans of this research.
Additional supplemental material is published online only. To view, please visit the journal online (http://dx.doi.org/10.1136/gutjnl-2021-325808).
Ethics approval This study involves human participants and was approved by University of Washington Institutional Review Board (protocol no. 1852). Participants gave informed consent to participate in the study before taking part.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as supplementary information. Data from our RNA sequencing and single-cell RNAseq have been deposited and are available at https://doi.org/10.5061/dryad.0cfxpnw54.
REFERENCES
- 1.Siegel RL, Miller KD, Fedewa SA, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin 2017;67:177–93. [DOI] [PubMed] [Google Scholar]
- 2.Engstrand J, Nilsson H, Strömberg C, et al. Colorectal cancer liver metastases - a population-based study on incidence, management and survival. BMC Cancer 2018;18:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koopman M, Kortman GAM, Mekenkamp L, et al. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br J Cancer 2009;100:266–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Topalian SL, Hodi FS, Brahmer JR, Safety BJR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brahmer JR, Tykodi SS, Chow LQM, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee JJ, Sun W, Bahary N. Phase 2 study of pembrolizumab in combination with azacitidine in subjects with metastatic colorectal cancer. Journal of Clinical Oncology 2017;35:3054–54. [Google Scholar]
- 7.Segal NH, Kemeny NE, Cercek A. Non-randomized phase II study to assess the efficacy of pembrolizumab (Pem) plus radiotherapy (RT) or ablation in mismatch repair proficient (pMMR) metastatic colorectal cancer (mCRC) patients. Journal of Clinical Oncology 2016;34:3539–39. [Google Scholar]
- 8.Wang Y, Lin H-C, Huang M-Y, et al. The immunoscore system predicts prognosis after liver metastasectomy in colorectal cancer liver metastases. Cancer Immunol Immunother 2018;67:435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katz SC, Bamboat ZM, Maker AV, et al. Regulatory T cell infiltration predicts outcome following resection of colorectal cancer liver metastases. Ann Surg Oncol 2013;20:946–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donadon M, Torzilli G, Cortese N, et al. Macrophage morphology correlates with single-cell diversity and prognosis in colorectal liver metastasis. J Exp Med 2020;217. doi: 10.1084/jem.20191847. [Epub ahead of print: 02 11 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cavnar MJ, Turcotte S, Katz SC, et al. Tumor-Associated macrophage infiltration in colorectal cancer liver metastases is associated with better outcome. Ann Surg Oncol 2017;24:1835–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin K-T, Chen B, Liu Y-Y, et al. Monoclonal antibodies and chimeric antigen receptor (CAR) T cells in the treatment of colorectal cancer. Cancer Cell Int 2021;21:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang C, Wang Z, Yang Z, et al. Phase I Escalating-Dose Trial of CAR-T Therapy Targeting CEA+ Metastatic Colorectal Cancers. Mol Ther 2017;25:1248–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katz SC, Burga RA, McCormack E, et al. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T-cell therapy for CEA+ liver metastases. Clin Cancer Res 2015;21:3149–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thorn M, Guha P, Cunetta M, et al. Tumor-Associated GM-CSF overexpression induces immunoinhibitory molecules via STAT3 in myeloid-suppressor cells infiltrating liver metastases. Cancer Gene Ther 2016;23:188–98. [DOI] [PubMed] [Google Scholar]
- 16.Burga RA, Thorn M, Point GR. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol Immunother 2015;64:817–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guha P, Gardell J, Rabinowitz B, et al. Monocytic and granulocytic myeloid-derived suppressor cell plasticity and differentiation are organ-specific. Oncogene 2021;40:693–704. [DOI] [PubMed] [Google Scholar]
- 18.Saraiva M, Vieira P, O’Garra A. Biology and therapeutic potential of interleukin-10. J Exp Med 2020;217. doi: 10.1084/jem.20190418. [Epub ahead of print: 06 01 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crispe IN. Hepatic T cells and liver tolerance. Nat Rev Immunol 2003;3:51–62. [DOI] [PubMed] [Google Scholar]
- 20.Li B, Wang F, Ma C, et al. Predictive value of IL-18 and IL-10 in the prognosis of patients with colorectal cancer. Oncol Lett 2019;18:713–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao S, Wu D, Wu P, et al. Serum IL-10 predicts worse outcome in cancer patients: a meta-analysis. PLoS One 2015;10:e0139598–e98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruffell B, Chang-Strachan D, Chan V, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014;26:623–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rossowska J, Anger N, Szczygieł A, et al. Reprogramming the murine colon cancer microenvironment using lentivectors encoding shRNA against IL-10 as a component of a potent DC-based chemoimmunotherapy. J Exp Clin Cancer Res 2018;37:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mocellin S, Panelli MC, Wang E, et al. The dual role of IL-10. Trends Immunol 2003;24:36–43. [DOI] [PubMed] [Google Scholar]
- 25.Mannino MH, Zhu Z, Xiao H, et al. The paradoxical role of IL-10 in immunity and cancer. Cancer Lett 2015;367:103–7. [DOI] [PubMed] [Google Scholar]
- 26.Mumm JB, Emmerich J, Zhang X, et al. Il-10 elicits IFNγ-dependent tumor immune surveillance. Cancer Cell 2011;20:781–96. [DOI] [PubMed] [Google Scholar]
- 27.Naing A, Papadopoulos KP, Autio KA, et al. Safety, antitumor activity, and immune activation of PEGylated recombinant human interleukin-10 (AM0010) in patients with advanced solid tumors. J Clin Oncol 2016;34:3562–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Naing A, Infante JR, Papadopoulos KP, et al. PEGylated IL-10 (Pegilodecakin) Induces Systemic Immune Activation, CD8+ T Cell Invigoration and Polyclonal T Cell Expansion in Cancer Patients. Cancer Cell 2018;34:775–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan IH, Wu V, Bilardello M, et al. PEG-rIL-10 treatment decreases FoxP3(+) Tregs despite upregulation of intratumoral IDO. Oncoimmunology 2016;5:e1197458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emmerich J, Mumm JB, Chan IH, et al. IL-10 directly activates and expands tumor-resident CD8(+) T cells without de novo infiltration from secondary lymphoid organs. Cancer Res 2012;72:3570–81. [DOI] [PubMed] [Google Scholar]
- 31.Lilly Announces phase 3 study in patients with metastatic pancreatic cancer did not meet primary endpoint of overall survival 2019. Available: https://investor.lilly.com/news-releases/news-release-details/lilly-announces-phase-3-study-patients-metastatic-pancreatic [Accessed 1/7/20 2019].
- 32.Spigel D, Jotte R, Nemunaitis J, et al. Randomized phase 2 studies of checkpoint inhibitors alone or in combination with Pegilodecakin in patients with metastatic NSCLC (cypress 1 and cypress 2). J Thorac Oncol 2021;16:327–33. [DOI] [PubMed] [Google Scholar]
- 33.Saxton RA, Tsutsumi N, Su LL, et al. Structure-Based decoupling of the pro-and anti-inflammatory functions of interleukin-10. Science 2021;371. doi: 10.1126/science.abc8433. [Epub ahead of print: 19 03 2021]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jabbari N, Kenerson HL, Lausted C, et al. Modulation of immune checkpoints by chemotherapy in human colorectal liver metastases. Cell Rep Med 2020;1:100160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang X, Seo YD, Chang JH, et al. Long-Lived pancreatic ductal adenocarcinoma slice cultures enable precise study of the immune microenvironment. Oncoimmunology 2017;6:e1333210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang X, Seo YD, Sullivan KM. Establishment of slice cultures as a tool to study the cancer immune microenvironment. Methods Mol Biol 1884;2019:283–95. [DOI] [PubMed] [Google Scholar]
- 37.Seo YD, Jiang X, Sullivan KM, et al. Mobilization of CD8+ T Cells via CXCR4 Blockade Facilitates PD-1 Checkpoint Therapy in Human Pancreatic Cancer. Clin Cancer Res 2019;25:clincanres.0081.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bockorny B, Semenisty V, Macarulla T, et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the combat trial. Nat Med 2020;26:878–85. [DOI] [PubMed] [Google Scholar]
- 39.Goldman M, Craft B, Hastie M. The UCSC Xena platform for public and private cancer genomics data visualization and interpretation. bioRxiv 2019. [Google Scholar]
- 40.Kenerson HL, Sullivan KM, Labadie KP, et al. Protocol for tissue slice cultures from human solid tumors to study therapeutic response. STAR Protoc 2021;2:100574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luckert K, Gujral TS, Chan M, et al. A dual array-based approach to assess the abundance and posttranslational modification state of signaling proteins. Sci Signal 2012;5:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gujral TS, Karp RL, Finski A, et al. Profiling phospho-signaling networks in breast cancer using reverse-phase protein arrays. Oncogene 2013;32:3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lausted C, Sullivan KM, Guha P. Colorectal cancer interleukin-10 blockade scRNA-seq. Dryad Digital Repository 2022. [Google Scholar]
- 44.Burga RA, Thorn M, Point GR, et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol Immunother 2015;64:817–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Le DT, Uram JN, Wang H, et al. Pd-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015;372:2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015;15:486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simon S, Vignard V, Florenceau L, et al. Pd-1 expression conditions T cell avidity within an antigen-specific repertoire. Oncoimmunology 2016;5:e1104448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gros A, Robbins PF, Yao X, et al. Pd-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J Clin Invest 2014;124:2246–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tretina K, Park E-S, Maminska A, et al. Interferon-Induced guanylate-binding proteins: guardians of host defense in health and disease. J Exp Med 2019;216:482–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tokunaga R, Zhang W, Naseem M, et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation - A target for novel cancer therapy. Cancer Treat Rev 2018;63:40–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hopewell EL, Zhao W, Fulp WJ, et al. Lung tumor NF-κB signaling promotes T cell-mediated immune surveillance. J Clin Invest 2013;123:2509–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gerondakis S, Siebenlist U. Roles of the NF-kappaB pathway in lymphocyte development and function. Cold Spring Harb Perspect Biol 2010;2:a000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Philipson BI, O’Connor RS, May MJ, et al. 4–1Bb costimulation promotes CAR T cell survival through noncanonical NF-κB signaling. Sci Signal 2020;13. doi: 10.1126/scisignal.aay8248. [Epub ahead of print: 31 03 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Borst J, Ahrends T, Bąbała N, et al. CD4+ T cell help in cancer immunology and immunotherapy. Nat Rev Immunol 2018;18:635–47. [DOI] [PubMed] [Google Scholar]
- 55.Hagenbaugh A, Sharma S, Dubinett SM, et al. Altered immune responses in interleukin 10 transgenic mice. J Exp Med 1997;185:2101–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jung K, Heishi T, Khan OF, et al. Ly6Clo monocytes drive immunosuppression and confer resistance to anti-VEGFR2 cancer therapy. J Clin Invest 2017;127:3039–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gorby C, Sotolongo Bellón J, Wilmes S, et al. Engineered IL-10 variants elicit potent immunomodulatory effects at low ligand doses. Sci Signal 2020;13. doi: 10.1126/scisignal.abc0653. [Epub ahead of print: 15 09 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guo Y, Xie Y-Q, Gao M, et al. Metabolic reprogramming of terminally exhausted CD8+ T cells by IL-10 enhances anti-tumor immunity. Nat Immunol 2021;22:746–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guerin MV, Finisguerra V, Van den Eynde BJ, et al. Preclinical murine tumor models: a structural and functional perspective. Elife 2020;9. doi: 10.7554/eLife.50740. [Epub ahead of print: 28 Jan 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhong W, Myers JS, Wang F, et al. Comparison of the molecular and cellular phenotypes of common mouse syngeneic models with human tumors. BMC Genomics 2020;21:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tanikawa T, Wilke CM, Kryczek I, et al. Interleukin-10 ablation promotes tumor development, growth, and metastasis. Cancer Res 2012;72:420–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Efremova M, Rieder D, Klepsch V, et al. Targeting immune checkpoints potentiates immunoediting and changes the dynamics of tumor evolution. Nat Commun 2018;9:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Van den Eynde M, Mlecnik B, Bindea G, et al. The link between the Multiverse of immune microenvironments in metastases and the survival of colorectal cancer patients. Cancer Cell 2018;34:1012–26. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as supplementary information. Data from our RNA sequencing and single-cell RNAseq have been deposited and are available at https://doi.org/10.5061/dryad.0cfxpnw54.