

Abstract
The lipopolysaccharide (LPS) of the Gram-negative legume symbiont Rhizobium leguminosarum biovar viciae 3841 contains several unique modifications, including the addition of a 27-hydroxyoctacosanoic acid (27OHC28 : 0), also termed the very long chain fatty acid (VLCFA), attached at the 2′ position of lipid A. A transposon mutant that lacks expression of two putative 3-oxo-acyl [acyl-carrier protein] synthase II genes, fabF1 and fabF2, from the VLCFA biosynthetic cluster, was isolated and characterized. MS indicated that the lipid A of the mutant lacked the VLCFA modification, and sodium deoxycholate (DOC)-PAGE of the LPS indicated further structural alterations. The mutant was characteristically sensitive to several stresses that would be experienced in the soil environment, such as desiccation and osmotic stresses. An increase in the excretion of neutral surface polysaccharides was observed in the mutant. This mutant was also altered in its attachment to solid surfaces, and was non-motile, with most of the mutant cells lacking flagella. Despite the pleiotropic effects of the mutation, these mutants were still able to nodulate legumes and fix atmospheric nitrogen. This report emphasizes that a structurally intact VLCFA-containing lipid A is critical to cellular traits that are important for survival in the rhizosphere.
INTRODUCTION
Rhizobium leguminosarum biovar viciae 3841 is a Gram-negative soil bacterium and a nitrogen-fixing symbiont of leguminous plants (Johnston & Beringer, 1975). As with other Gram-negative bacteria, the outer leaflet of the outer membrane of R. leguminosarum is composed primarily of lipopolysaccharide (LPS), which includes three distinct structural components: the lipid A, which is located in the lipid membrane; the core polysaccharide; and the variable O-antigen, which protrudes from the cell surface into the external milieu (Kannenberg et al., 1998).
Structural determinations of the lipid A component from R. leguminosarum and its close relative Rhizobium etli have determined that it contains four unique structural modifications compared with the lipid A from other Gram-negative bacteria. These alterations include a galacturonic acid residue at the 4′ position (Que et al., 2000a, b; Bhat et al., 1994), lack of phosphate groups at the 1 and 4′ positions (Que et al., 2000a, b; Bhat et al., 1994), the presence of a very long chain fatty acid (VLCFA) composed of a 27-hydroxyoctacosanoate chain attached by an acyloxyacyl linkage at the 2′ position (Bhat et al., 1994), and finally the replacement of the reducing-end glucosamine with a 2-deoxy-2-aminogluconate residue (Bhat et al., 1991).
A number of genes have been identified in the genome of R. leguminosarum 3841 that are required to produce the unusual lipid A structure (Basu et al., 2002). In particular, the synthesis and attachment of the VLCFA is thought to require a group of six proteins. The proteins are: an acyl carrier protein (acpXL), a putative dehydratase (fabZ), two putative 3-oxo-acyl [acyl-carrier protein] synthase II enzymes (fabF1 and fabF2), a putative dehydrogenase, and a unique long chain acyltransferase (lpxXL) (Basu et al., 2002).
Mutational analyses of acpXL and lpxXL have confirmed their essential role in the modification of lipid A with the VLCFA in free-living R. leguminosarum (Basu et al., 2002; Ferguson et al., 2005; Vedam et al., 2003, 2006). These studies have also demonstrated the importance of VLCFA during the establishment of the plant symbiosis. Mutation of acpXL or lpxXL delays the establishment of nitrogen-fixing nodules, indicating that the VLCFA plays a role during infection. In addition, Vedam et al. (2006) have found that bacteroids formed by an acpXL mutant are partially restored in the VLCFA. These results indicate that there is a functional replacement for acpXL that is activated in planta, suggesting that VLCFA is in fact essential for symbiosis.
We report here, for the first time, to our knowledge, the isolation of a transposon mutant in the putative 3-oxoacyl [acyl-carrier protein] synthase fabF2 that was discovered during a screen for mutants with defects in their cell envelope. Structural analysis of the lipid A confirms that the mutant lacks the VLCFA. Characterization of the mutant using a desiccation-sensitivity assay determined that it has an increased sensitivity to desiccation stress. In addition, the mutant is altered in biofilm formation and is non-motile. The pleiotropic nature of the mutant emphasizes the importance of the VLCFA in the lipid A structure and suggests that intact lipid A is crucial for survival of R. leguminosarum in the rhizosphere.
METHODS
Strains, media and growth conditions
Strains and plasmids used in this study are listed in Table 1 and primer sequences used are listed in Supplementary Table S1. Escherichia coli strains were cultured using Luria–Bertani (LB) medium (Sambrook et al., 1989), supplemented as necessary with the following concentrations of antibiotics (μg ml−1): gentamicin, 15; ampicillin, 100; spectinomycin, 100; tetracycline, 10. R. leguminosarum cells were cultured using tryptone-yeast (TY) medium, with a standard CaCl2 concentration of 3.5 mM (Beringer, 1974), or Vincent’s minimal medium (VMM) modified with 10 mM mannitol (Vincent, 1970), and supplemented as required with the following concentrations of antibiotics (μg ml−1): gentamicin, 30; neomycin, 100; tetracycline, 5; streptomycin, 500. Motility of the 38EV28 mutant was evaluated by inoculating the wild-type, mutant and complemented strains into yeast extract swimming (YES) medium (Yost et al., 1998) and measuring the swim diameter after incubation for 48 h at 30 °C.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics | Source or reference |
|---|---|---|
| E. coli strains | ||
| TOP 10 | F−
mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(ara-leu)7697 galU galK rpsL (StrR) endA1 nupG |
Invitrogen |
| S17-1 | RP4 tra region, mobilizer strain recA derivative of MM294A with RP4-2 (Tc : : Mu : : Km : : FabF2/F17) integrated into the chromosome |
Simon et al. (1983) |
| DH5α | F−
ϕ80lacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rk, ) phoA supE44 thi-1 gyrA96 relA1 λ− |
Invitrogen |
|
R. leguminosarum
strains | ||
| 3841 | Biovar viciae, JB300 derivative, Smr | Johnston & Beringer (1975) |
| 38EV27 | 3841 fabF2, fabF1 TGN mutant, Smr, Nmr, Gmr | This study |
| 38EV51 | 3841 fabF2/F1 mutant with functional fabF1 expressed on plasmid pEV51, Smr, Nmr, Gmr, Tcr |
This study |
| 38EV28 | 3841 fabF1 mutant, Smr, Gmr | This study |
| Plasmids | ||
| pTGN | Tn5 derivative, Gmr, Ampr promoterless Nmr, gfp | Tang et al. (1999) |
| pCR2.1 TOPO | TOPO TA cloning vector, 4.0 kb, Kmr, Apr | Invitrogen |
| pDG71 | Broad-host-range derivative of pHC41. Constitutively expressed tryptophan promoter and gfp(mut3), Tcr |
Bringhurst et al. (2001) |
| pJQ200SK+ | Suicide vector, P15aori, mob, sacB, Gmr | Quandt & Hynes (1993) |
| pFUS1 | pMP220 derivative with promoterless gusA, Tcr | Reeve et al. (1999) |
| pFUS1P | pFUS1, par cassette inserted from pRR71-H as HindIII fragment | Yost et al. (2004) |
| pCS115 | pWKR56 carrying HindIII fragment from pRIA76, Tcr | Colonna-Romano et al. (1990) |
| pEV35 | pCR2.1 TOPO with 1330 bp PCR fragment containing fabF1, Ampr, Kmr | This study |
| pEV51 | 1330 bp fabF1 fragment cloned from pEV35 into pDG71 with BamHI, Tcr | This study |
| pEV26 | pCR2.1 TOPO with 456 bp PCR fragment containing internal fragment of fabF1, Ampr, Kmr |
This study |
| pEV28 | 456 bp fabF1 fragment cloned from pEV26 into pJQ200SK+ using ApaI and BamHI, Gmr |
This study |
| pNdvB | pFUS1P, ndvB : : gusA, Tcr | This study |
| pAVP | pFUS1, flaA : : gusA, Tcr | Michael Hynes |
| pVNVP | pFUS1P, visN : : gusA, Tcr, par stabilized | Michael Hynes |
| pSVP SUM | pFUS1P, rem : : gusA, Tcr, par stabilized | Michael Hynes |
| pCGR | pRK7813, mcpC : : gusA, Nmr, Tcr | Yost et al. (2004) |
| pDGRP | pFUS1P, mcpD : : gusA, Tcr, par stabilized | Yost et al. (2004) |
Transposon mutagenesis
Mutagenesis was performed using the mini-Tn5 derivative, pTGN (Tang et al., 1999). Biparental matings of the E. coli mobilizer strain S17-1 containing the pTGN vector and R. leguminosarum 3841 were performed at 30 °C for 24 h on TY plates. Transconjugants were selected on VMM with streptomycin and neomycin, and subsequently screened for inability to grow on the solid complex medium TY. We have previously observed that certain mutants with defects in the cell envelope are unable to grow on complex solid media (Gilbert et al., 2007; C. K. Yost and others, unpublished data). Therefore, we used the lack of growth on TY as a screen to enrich for isolation of transposon mutants with defective cell envelopes. Genomic DNA was isolated from transposon mutants unable to grow on TY and used as a PCR template to identify the transposon insertion site. The transposon insertion site was identified using the restriction enzyme site-directed amplification (RESDA) PCR protocol, as described by González-Ballester et al. (2005). Briefly, the specific primer TGN1 (SigmaGenosys Canada), which binds within the GFP gene cassette of the transposon, and the arbitrary degenerate primer DegTaqI, were used in the primary reaction, which was followed by a secondary reaction with the primers TGN2 and Q0 (González-Ballester et al., 2005), and 1 μl of primary reaction as template. Fragments were subsequently cloned into the pCR2.1 TOPO vector according to the manufacturer’s instructions (Invitrogen), and sequenced using the T7 primer binding site available in the pCR 2.1 TOPO vector.
Sequence analysis
The transposon insertion site was identified using a blastn search (Altschul et al., 1997) at the Rhizobase database (Kazusa DNA Research Institute; http://bacteria.kazusa.or.jp/rhizobase/). DNA sequencing was performed by the University of Calgary Core DNA Services. DNA sequence data were analysed using 4Peaks software (version 1.7.2; A. Griekspoor and T. Groothuis; http://mekentosj.com/4peaks/). Sequence alignments were performed using clustal w (Thompson et al., 1994). All PCR products that were cloned into pCR2.1 TOPO vectors were sequenced to confirm the nature of the PCR amplicon.
Complementation of fabF2/F1 mutant
To create a fabF2 single mutant, a plasmid expressing fabF1 only was used to complement the polar fabF2/F1 mutant. Primers FabF1F and FabF1R were used to amplify a 1330 bp fragment containing the entire fabF1 gene and upstream region using Pfx50 DNA polymerase (Invitrogen) as recommended by the manufacturer. The PCR product was then cloned into the pCR2.1 TOPO vector as per the manufacturer’s instructions, resulting in pEV35. The vector pEV51 was constructed for complementation by excising the fabF1 fragment from pEV35 with BamHI and ligated into the BamHI site of the vector pDG71. Orientation of the fragment was confirmed by sequencing. The pEV51 vector was then transformed into the mobilizer strain S17-1 and conjugated into the fabF2/F1 mutant. Mutants were selected using tetracycline, and expression of fabF1 was confirmed by RT-PCR.
Mutagenesis of fabF1
Primers FabF2F and FabF1RTR were used to amplify a 456 bp internal fragment of fabF1 using 1 U Taq DNA polymerase (UBI). PCR products were cloned into the pCR2.1 TOPO vector as per the manufacturer’s instructions, resulting in pEV26. The vector pEV28 was constructed for mutagenesis of fabF1 by allelic exchange using the method described by Quandt & Hynes (1993). Briefly, the entire internal fragment of fabF1 was excised using BamHI and ApaI restriction sites in the pCR2.1 TOPO vector and ligated into the same sites in the vector pJQ200SK+, creating pEV28. The pEV28 vector was subsequently transferred to the mobilizer strain S17-1, and biparental matings were performed overnight on VMM supplemented with 0.5 mM proline. Mutants were selected on the basis of gentamicin resistance and further screened for inability to grow on the complex medium TY. Putative mutants were confirmed by PCR.
Standard molecular techniques
Plasmid DNA was isolated using the alkaline lysis method (Sambrook et al., 1989). Restriction endonucleases were purchased from Invitrogen and used according to the manufacturer’s instructions. When necessary, PCR products were isolated from agarose gels using reagents and protocols from the QIAEX II gel extraction kit.
RNA extraction
RNA was extracted using a modification of the method supplied with the Aurum Total RNA Mini kit (Bio-Rad). Briefly, 4 ml samples of overnight cultures were pelleted and resuspended in 650 μl of 500 μg lysozyme ml−1 in Tris-EDTA buffer (10 mM Tris, 1 mM EDTA, pH 7.5) and incubated for 5 min at room temperature. A 350 μl volume of Cell Lysis Solution (Bio-Rad) was then added and the solution was transferred to a tube containing FastPrep RNA matrix (Qbiogene) and vortexed at full speed for 10 min using a 1.5–2 ml vortex adaptor (MoBio Laboratories). Tubes were then centrifuged at 12 000 g for 5 min and the supernatant was transferred to an RNA binding column. The remainder of the extraction used the protocols and reagents supplied with the Aurum Total RNA Mini kit (Bio-Rad).
RT-PCR
Primer sequences for FabZ, FabF2F, FabF2R, FabF1RTF, FabF1RTR, RL2813F and RL2813R, used for RT-PCR, are listed in Supplementary Table S1. Reverse-transcription reactions were carried out according to the protocol of Manzon et al. (2007). Briefly, 10 μl RNA was treated with 1 U DNase I (Fermentas) in a total volume of 16 μl for 30 min at 37 °C. Dnase I was heat-inactivated at 75 °C for 5 min. A 1.2 μl volume of the appropriate 2 μM reverse primer was then added followed by incubation at 70 °C for 10 min. Samples were snap-cooled on ice and 2 μl 10 mM dNTPs were added, followed by incubation at 37 °C for 5 min. A 1 μl volume of RevertAid M-MuLV Reverse Transcriptase (Fermentas) was added and samples were incubated at 42 °C for 50 min, followed by heat-inactivation at 70 °C for 15 min. PCRs contained 1 x reaction buffer, 2 mM MgSO4 (1 mM MgSO4 for FabF2F and FabF1R), 0.2 mM dNTPs, 2 μM forward primer, 0.12 μM reverse primer and 1 U Taq DNA polymerase (UBI). A 2 μl volume from the reverse-transcription reaction was used as template. PCR amplification was performed with a Techne TC312 Thermocycler at 94 °C for 5 min followed by 30 cycles of 95 °C for 30 s, 58 °C for 30 s and 72 °C for 1 min, and a final extension at 72 °C for 5 min. PCR products were subsequently analysed by agarose gel electrophoresis.
Isolation of LPS
LPS was extracted from bacteria using the hot phenol–water extraction procedure described by Westphal & Jann (1965). Dialysed phenol and water phases (3500 molecular weight cut-off tubing) were concentrated, freeze-dried and further purified by ultracentrifugation at 100 000 g for 6 h at 4 °C. LPS recovered from pellets after freeze-drying was purified from any contaminating phospholipids by washing three times with chilled 9 : 1 ethanol : water (v/v).
Lipid A isolation and MS analysis
Lipid A was released from LPS by hydrolysis with 1 % acetic acid at 100 °C for 2 h. Lipid A was separated from oligosaccharides and O-chains by centrifugation for 25 min at 3500 g and 4 °C, washed three times with nanopure water and lyophilized. The lipid A was then extracted with chloroform : methanol : water (2 : 2 : 1.8 by vol.). The organic phase was re-extracted with water and, after concentration under a stream of nitrogen, used for MALDI-MS and compositional analyses.
Lipid A preparations were dissolved in chloroform : methanol, 3 : 1 (v/v). Solutions (5 μg μl−1) were mixed 1 : 1 (v/v) with 0.5 M 2,4,6-trihydroxyaceto-phenone (THAP) matrix and 1 μl was then applied onto a stainless steel MALDI plate. Spectra were acquired in negative reflector mode using an Applied Biosystems 4700 Proteomics Analyser with a TOF/TOF tandem mass spectrometer.
Chemical characterization of lipid A
The compositions of lipid A were determined by preparation of trimethylsilyl (TMS) methyl glycosides. Briefly, samples were subjected to methanolysis with methanolic 2 M HCl at 80 °Cfor 18 h, N-acetylated [3 : 1 : 1 methanol : pyridine : acetic anhydride (by vol.)] at 100 °C for 1 h, and finally trimethylsilylated with Tri-Sil reagent (Pierce) for 30 min at 80 °C, as described by York et al. (1985). The resulting TMS methyl glycosides and fatty acid methyl esters were analysed by combined GC-MS.
Sodium deoxycholate (DOC)-PAGE analysis of LPS
For wild-type, fabF2/F1− and fabF2−, 1 μg extracted LPS was resuspended in 1 μl sample buffer. For fabF2/F1−pCS115, fabF1−, and fabF1−pCS115, cells were scraped off agar plates and resuspended in water. The cell number in each sample was standardized by measuring OD600. Cells were then pelleted and samples for electrophoresis were prepared by resuspending cells of the appropriate strains in Laemmli solubilization buffer (100 μl) and boiling for 10 min (Laemmli, 1970). Contaminating proteins were then removed by incubating samples at 60 °C for 45 min in the presence of 2 mg protease ml−1 (Sigma-Aldrich). LPS molecules were then separated by DOC-PAGE, as described previously (Reuhs et al., 1993) and silver-stained using the Bio-Rad Silver Stain kit. Gels were run three independent times to confirm the observations.
Desiccation, detergent and osmotic stress assays
Desiccation sensitivity assays were performed using the filtration method described by Ophir & Gutnick (1994) and as modified by Gilbert et al. (2007). Detergent sensitivity assays were performed as described by Gilbert et al. (2007). Hyperosmotic stress assays were performed by inoculating strains into TY medium with 69.5 mM NaCl. Wild-type cells will not grow at concentrations higher than 69.5 mM; therefore, this concentration represents the maximum tolerable level of osmotic stress. Cultures were grown at 30 °C for 48 h and then the OD600 was measured. Hypo-osmotic stress tolerance was determined using the low-osmolarity glutamate yeast extract mannitol (GYM) medium described by Dylan et al. (1990).
Quantitative determination of secreted polysaccharides
The method for exopolysaccharide (EPS) quantification was a modification of those described by Ngwai et al. (2006). Briefly, cells were grown to stationary phase in 25 ml VMM minimal media. Cells were pelleted at 7710 g for 20 min at 4 °C, and washed with 25 ml 1 M NaCl, 10 mM EDTA at pH 8.0. The EPS was then precipitated from the combined culture and NaCl–EDTA wash supernatants by the addition of two volumes of ice-cold 2-propanol. The precipitated polysaccharide was then spooled and dried in a sterile Petri dish at 37 °C overnight. Results are reported as the mass of EPS produced per milligram of dry cell mass. The capsular polysaccharide (CPS) was extracted according to the method of Zevenhuizen (1984). Precipitated CPS was then dried overnight at 37 °C and the results reported as the mass of CPS produced per milligram of dry cell mass. The amount of total neutral polysaccharide was determined by combining the remaining ethanol supernatants from the EPS and CPS determinations and quantifying the amount of glucose using the anthrone–sulphuric acid assay as described by Laurentin & Edwards (2003). The amount of reducing sugars in the neutral polysaccharide fraction was determined using the method described by Lever (1972).
Biofilm cultivation
Biofilms were grown in the Calgary Biofilm Device (CBD; Innovotech), using a procedure modified from that previously described by Ceri et al. (1999). First, strains were streaked out twice on the appropriate selective agar medium. Colonies were collected from the second agar subculture using a sterile cotton swab and were suspended in double-distilled water (ddH2O) to match a 1.0 McFarland standard. This standardized bacterial suspension was then diluted 1 : 15 into VMM that contained 1 % mannitol and 500 μg streptomycin ml−1, which, when verified by viable cell counting, provided a starting inoculum of approximately 1 × 107 c.f.u. ml−1. A 22 ml volume of this standardized inoculum was added to the trough of the CBD. The devices were sealed with Parafilm and then incubated at 30 °C for 72 h on a rocking table set to 3.5 rocks per minute.
CBD biofilms were rinsed by placing the peg lid into a microtitre plate that contained 200 μl ddH2O in each well. For viable cell counts, the peg lids were transferred into a second microtitre plate, also containing 200 μl ddH2O in each well, into which biofilms were disrupted using an ultrasonic cleaner as previously described by Ceri et al. (1999). Cells from the disrupted biofilms were serially diluted 10-fold and plated for viable cell counting on VMM + 1 % mannitol agar. Spot plates were grown for 72 h before enumeration.
Confocal laser scanning microscopy (CLSM)
Pegs were broken from the lid of the CBD using a pair of flamed needle-nose pliers. Biofilms, which had been grown on the surface of the peg, were stained with acridine orange (AO) or with Syto-9 in combination with tetramethylrhodamine isothiocyanate-conjugated concanavalin A (TRITC-ConA), as previously described by Harrison et al. (2006, 2007). AO, which fluoresces green, is a nucleic acid intercalator that stains biofilm cells as well as extracellular nucleic acids, and thus may be used a biomass indicator. Syto-9 is also a membrane-permeable nucleic acid intercalator, which fluoresces green, whereas TRITC-ConA is a red fluorophore-conjugated lectin that binds to α-d-mannose and α-d-glucose monomers, as well as certain derivatives of these sugars, with high affinity. In this fashion, Syto-9 and TRITC-ConA can be used in combination to stain bacterial cells and extracellular polysaccharides, respectively. To preserve structure and extracellular biomass, biofilms were fixed with 5 % glutaraldehyde overnight at 4 °C prior to staining with Syto-9 and TRITC-ConA. These fixed biofilms were rinsed twice with ddH2O before mounting for microscopy.
Fluorescently labelled biofilms were placed in two drops of ddH2O on the surface of a glass coverslip. These pegs were examined using a Leica DM IRE2 spectral confocal and multiphoton microscope with a Leica TCS SP2 acoustic optical beam splitter (Leica Microsystems), as previously described (Harrison et al., 2007). For AO-stained samples, biofilms were scanned using 476 nm excitation and fluorescence was measured in the green region of the spectrum. The dual-labelled samples were sequentially scanned, frame-by-frame, first at 488 nm and then at 543 nm. Fluorescence emission was sequentially measured in the green and red regions of the spectrum, respectively. Line averaging ( x 2) was used to capture images with reduced noise. A 63 x water-immersion objective was used in all imaging experiments. Image capture and 2D reconstruction of z-stacks were performed using Leica Confocal Software (Leica Microsystems).
Construction of an ndvB::gusA transcriptional fusion
A 327 bp fragment containing the promoter of ndvB was amplified with the primers ndvBF and ndvBR and the reaction conditions described above for mutagenesis of fabF1. The fragment was subsequently cloned into the pCR2.1 TOPO vector as per the manufacturer’s instructions. The promoter fragment was then excised using KpnI and EcoRI and ligated into the pFUS1P vector containing the gusA reporter gene. The plasmid pNdvB was then conjugated into the wild-type and the fabF1 mutant to measure gene expression as described below.
Motility-related gene fusions
Promoter regions of the flaA, rem and visN genes from R. leguminosarum VF39 were amplified and cloned into the pFUS1 vector (M. F. Hynes and others, unpublished data). flaA, rem, visN, mcpC and mcpD from VF39 are homologous to the following genes in R. leguminosarum 3841: RL0718, RL0727, RL0697, pRL120312 and RL2683, respectively.
β-Glucuronidase (gusA) reporter gene assays
To measure the gene expression of several chemotaxis, flagellar and motility genes, the plasmids pCGR, pDGRP, pAVP, pVNVP and pSVP SUM, with gusA transcriptional fusions to mcpC, mcpD, flaA, visN and rem, respectively, were conjugated into 3841 and 38EV28. The enzyme assays for β-glucuronidase activity were carried out based on the β-galactosidase activity method of Miller (1972), with modifications described by Yost et al. (2004).
Electron microscopy for flagella visualization
Cells from agar cultures were resuspended in distilled water and negatively stained with 1 % uranyl acetate and bacitracin as a wetting agent. Samples on carbon-Formvar-coated grids were examined in a Philips EM 410 electron microscope operating at 60 kV.
RESULTS
The fabF2 gene is part of a three-gene operon
The fabF2 mutant was isolated during a transposon mutagenesis screen intended to identify mutants of R. leguminosarum 3841 with defects in the cell envelope. The transposon insertion site of the fabF2/F1 mutant was mapped to base pair 999 of the putative 3-oxoacyl [acylcarrier protein] synthase fabF2 (Fig. 1). The fabF2 gene is the third of a group of six genes associated with the synthesis of the unique VLCFA component characteristic of rhizobial LPS. Earlier work by Sharypova et al. (2003) suggests that the orthologous gene cluster in Sinorhizobium meliloti is made up of three transcriptional units: (1) acpXL; (2) fabZ, fabF2 and fabF1; and (3) a putative dehydrogenase and lpxXL.
Fig. 1.
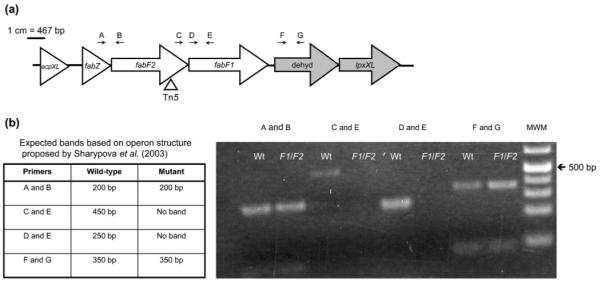
(a) Schematic of fabF2 gene region. dehyd, Putative dehydrogenase. Gene annotations were obtained from the Rhizobase database (Kazusa DNA Research Institute; http://bacteria.kazusa.or.jp/rhizobase/). Shading represents putative operon structures proposed by Sharypova et al. (2003). The triangle indicates the transposon insertion site. Arrows indicate primer binding sites. Refer to Supplementary Table S1 for primer sequences and Methods for amplification conditions used. A, FabZ; B, FabF2R; C, FabF2F; D, FabF1F; E, FabF1R; F, RL2813F; G, RL2813R. (b) RT-PCR of wild-type (Wt) and the fabF2/F1 (F1/F2) mutant to determine operon structure. MWM, molecular weight marker (GeneRuler 1 kb Plus DNA ladder, Fermentas).
RT-PCR was used to determine whether R. leguminosarum has an operon structure similar to the putative organization in S. meliloti, and to determine the possible polarity of the transposon mutation (Fig. 1). It was determined that the three genes fabZ, fabF2 and fabF1 are co-transcribed, and that the adjacent downstream dehydrogenase gene is transcribed separately from these three. The predicted ORFs of the fabF2 and fabF1 genes also overlap, suggesting that these two genes are translationally coupled. These results indicate that the transposon mutation is a polar mutation, affecting expression of the fabF1 and fabF2 genes. Both fabF1 and fabF2 single mutants were also isolated and were unable to grow on the complex agar TY (data not shown).
Amino acid sequence analysis of FabF1 and FabF2
The fabF1 and fabF2 genes disrupted in the transposon mutant are both annotated as 3-oxoacyl-[acyl-carrier protein] synthases, which are also referred to as genes encoding putative β-ketoacyl-acyl carrier protein synthases (KAS II). Analysis of the FabF1 and FabF2 amino acid sequences with PSORTb v.2.0 (Gardy et al., 2005) found that both are predicted to be cytoplasmic proteins.
The predicted FabF1 and FabF2 amino acid sequences were aligned with the canonical FabF amino acid sequence from E. coli. Based on this alignment it was determined that the majority of the catalytic triad residues, as well as the other highly conserved active site residues of E. coli FabF, are present in FabF1 but not FabF2 (Supplementary Fig. S1a). A previous structural analysis of FabF from E. coli revealed that it uses a Cys-His-His catalytic triad, with the cysteine residue acting as the nucleophile during the reaction (Huang et al., 1998), and although this hallmark triad is present in FabF1, it is absent from FabF2. FabF2 is highly conserved amongst the Rhizobiales (Vedam et al., 2006). Alignment of the R. leguminosarum 3841 FabF2 with a number of other FabF2 sequences revealed that Cys 98 is highly conserved, and there are a number of conserved histidine residues that could be part of a catalytic triad (Supplementary Fig. S1b).
In E. coli, there are three 3-oxoacyl [acyl-carrier protein] synthase II (KAS) enzymes, designated FabB (KAS I), FabF (KAS II) and FabH (KAS III). FabH is involved in initiating fatty acid biosynthesis by catalysing the initial condensation reaction between malonyl-ACP and acetyl-CoA, whereas FabB and FabF are involved in fatty acid chain elongation by catalysing the condensation of malonyl-ACP to an acyl group on the growing fatty acid chain (reviewed by Heath et al., 2001). In order to determine whether the FabF2 sequence was an orthologue of FabH or FabB, rather than FabF, a neighbour-joining phylogenetic tree was constructed (data not shown). However, based on this analysis it was determined that FabF2 was most closely related to FabF.
Lipid A of the fabF2/F1 mutant lacks the VLCFA
Composition results for the lipid A preparations showed the presence of galacturonic acid (GalA), glucosamine (GlcN) and 2-aminogluconate (GlcNonate). The major fatty acids present in the lipid A from the parent strain (3841) were βOHC14 : 0, βOHC16 : 0, βOHC18 : 0 and 27OHC28 : 0, with smaller amounts of βOHC15 : 0. These fatty acyl components are consistent with the previously reported structure for the lipid A from R. leguminosarum and R. etli strains (Bhat et al., 1994, 1991; Kannenberg et al., 1998). The lipid A from the fabF2/F1 mutant did not contain 27OHC28 : 0 but did contain the other fatty acyl residues found in the parent 3841 lipid A. In addition, the fabF2/F1 mutant lipid A contained C16 : 0, C18 : 0 and C18 : 1. The lipid A from the fabF2 single mutant contained the same fatty acyl components as found in the fabF2/F1 lipid A.
Each lipid A preparation was subjected to MALDI-TOF MS analysis as described in Methods. The spectra are shown in Fig. 2. Proposed structures are consistent with the composition results, the observed ions, as well as the published structures (Kannenberg et al., 1998; Que et al., 2000a, b; Vedam et al., 2003) for R. leguminosarum and R. etli lipid A (Fig. 2). The wild-type (3841) lipid A preparations had compositions and ions that were consistent with structures A, B, C, D, A’, B’ and C’ shown in Fig. 2. Structures A, B and C are penta-acylated lipid A molecules that vary due to the form of the proximal glycosyl residue; i.e. in structure A this residue is GlcNonate, in B it is GlcN, and in C it is GlcNonolactone. The ‘prime’ forms of these molecules were also present, in which the 27-hydroxyl group of 27OHC28 : 0 is esterified by a β-hydroxybutyryl (βOHC4 : 0) residue. During mild acid hydrolytic release of the lipid A from the LPS, the GlcNonate residue can be lactonized and partial release of the fatty acyl residue from O3 of the GlcNonolactone residue occurs through acid-catalysed β-elimination, resulting in structure D. Both the fabF2/F1 mutant and the fabF2 mutant lipid A preparations had compositions and ions that were consistent with structures E, F, G, I, J and K. Structures E, F and G are structures that lack the 27OHC28 : 0 residue from the lipid A. Each of these mutant lipid A preparations also contains compositions and ions consistent with structure H, which is the β-elimination product of structure G, likely produced during the mild acid hydrolytic release of the lipid A from the LPS, as described above for structure D.
Fig. 2.


(a) MALDI-TOF MS spectra of lipid A from the wild-type and the fabF2/F1 and fabF2 mutants. (b) Possible structures of the lipid A from the mutants and the wild-type based on mass values.
The LPS of mutants deficient in VLCFA is altered
The LPS molecules from the wild-type, the fabF2/F1, fabF1 and fabF2 mutants, and the complemented fabF2/F1 and fabF1 mutants, were separated by DOC-PAGE (Fig. 3). PAGE separation of the LPS of the wild-type strain resolved in a ladder pattern that is characteristic of LPS with a different number of O-repeat units in the O-antigen (O-specific polysaccharide) (lane 1) (Carlson, 1984; Hitchcock & Brown, 1983). In contrast, the LPS extracts from the fabF2/F1 and fabF2 mutants contained two intense bands characteristic of the high-molecular-weight LPS and the low-molecular-weight LPS (lanes 2 and 3), and LPS from the fabF1 mutant had one high-molecular-weight band (lane 6). The absence of the ladder pattern in the lanes containing LPS from the mutants suggests alterations to the O-antigen structure. The faster mobility of the low-molecular-weight band of the fabF2/F1 LPS in the gel would indeed be indicative of a loss of the VLCFA from the lipid A, in agreement with compositional results and results of MALDI-MS.
Fig. 3.
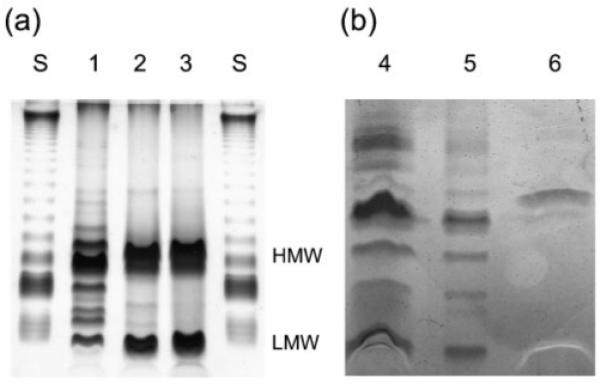
DOC-PAGE analysis of the wild-type and mutant LPS. (a) LPS (1 μg) loaded in 1 μl sample buffer. (b) LPS extracted from 1 ml culture at OD600 0.5, as outlined in Methods. Lanes: S, standard LPS of Salmonella enterica serovar Minnesota (Sigma); 1, wild-type; 2, fabF2/F1−; 3, fabF2−; 4, fabF2/F1−pCS115; 5, fabF1−pCS115; 6, fabF1−. HMW, high-molecular-weight LPS; LMW, low-molecular-weight LPS.
Sensitivity of the fabF2/F1 double mutant, fabF2, and fabF1 single mutants to membrane stressors
The fabF2/F1, fabF2 and fabF1 mutants were unable to grow on the solid complex TY medium; however, the mutants were able to grow in TY broth, although their growth was slowed compared with the wild-type (data not shown). Growth on solid TY could be partially restored by increasing the concentration of Ca2+ from 3.5 to 8.5 mM. These results are similar to those found for an lpxXL mutant in S. meliloti, which is unable to grow on complex medium without additional supplementation with 2.5 mM CaCl2 and 2.5 mM MgSO4 (Ferguson et al., 2005). Divalent cations are known to provide stability to the outer membrane by interacting with LPS molecules (Nikaido & Vaara, 1985). These results suggest that mutations that eliminate the VLCFA from lipid A destabilize the outer membrane and that this can be partially compensated for by the addition of outer-membrane-stabilizing agents.
The sensitivity of the fabF2/F1, fabF2 and fabF1 mutants to detergents was determined to confirm that the outer membrane structure was compromised (Table 2). The mutants were hypersensitive to the anionic detergents deoxycholate and sarcosyl. Also, both biofilm and planktonic cells were hypersensitive to doxycycline, a hydrophobic antibiotic (data not shown). All of these results suggest that the outer membrane integrity is compromised in the fabF2/F1, fabF2 and fabF1 mutants.
Table 2.
Sensitivity of the VLCFA mutants to detergents, desiccation and osmotic stress
| Strain | VMM+DOC* | VMM+SARC* | Desiccation tolerance† |
Hyperosmotic stress‡ |
Hypo-osmotic stress‡ |
|---|---|---|---|---|---|
| Wild-type | 125.0±4.45 | 123±3.42 | 42±1.8 | 79.4±1.42 | 57.0±8.08 |
| fabF2/F1 − | 12.9±4.72§ | 9.6±2.07§ | 3.46±1.68§ | 41.2±7.26§ | 10.7±0.970§ |
| fabF2/F1−+pCS115 | 110.0±7.67 | 110.2±7.73 | 15.7±5.42 | 90.7±4.57 | 50.8±2.72 |
| fabF2 − | 16.1±9.75§ | 17.6±2.56§ | nd | 30.1±5.21§ | 26.2±2.79§ |
| fabF1 − | 10.5±0.502§ | 10.9±1.05§ | nd | 47.1±2.08§ | nd |
| fabF1−+pCS115 | 94.8±4.64 | 74.5±6.82 | nd | 87.9±6.57 | nd |
Strains were grown in VMM broth or VMM supplemented with 0.075 % DOC or 0.050 % sarcosyl (SARC) for 2 days. OD600 values were measured. Percentage growth was calculated as described in Methods. Values are the mean (±sd) from three independent trials.
Desiccation sensitivity assays were performed as described in Methods. Percentage survival values presented are the mean (±sd) percentage survival from three independent trials.
Strains were grown in TY supplemented with 69.5 mM NaCl (hyperosmotic) or GYM medium (hypo-osmotic) for 2 days, and the growth was compared with that of strains grown in TY medium. OD600 values were measured. Percentage growth values are the mean and sd from three independent trials.
Difference between the wild-type and mutant is statistically significant at P <0.001 (Student’s t test).
Lack of VLCFA-containing lipid A contributes to an increased sensitivity to desiccation and osmotic stress
To quantify the fabF2/F1 mutant’s sensitivity to desiccation, the ability of the mutant to survive drying at ambient temperature and humidity for 16 h was determined (Table 2). We found that the mutant had more than an eightfold reduction in percentage survival following desiccation, compared with the wild-type. This suggests that structurally intact LPS is important in protecting cells against desiccation stress.
In addition to desiccation, the mutant was sensitive to hyper- and hypo-osmotic stress (Table 2). These results agree with data for acpXL and lpxXL mutants of S. meliloti and R. leguminosarum, which were also sensitive to hyperosmotic stress (Ferguson et al., 2005; Vedam et al., 2003). The fabF2 and fabF1 single mutants were also sensitive to osmotic stress, further confirming the results for the transposon mutant.
Excretion of neutral polysaccharides is increased in a mutant lacking the VLCFA of lipid A
In rhizobia, hypo-osmotic stress tolerance is associated with the production of periplasmic cyclic β-(1,2)-glucans (Dylan et al., 1990). Analysis of the polysaccharide content of the transposon mutant showed that it excreted 74.48 ± 16.74 μg neutral polysaccharide (mg dry cell weight)−1 onto the cell surface. Both the wild-type and the complemented mutant strain fabF2/F1 + pCS115 had approximately fivefold lower amounts of neutral polysaccharides on their cell surface, with values of 15.18 ± 2.3 and 16.69 ± 5.33 μg neutral polysaccharide (mg dry cell weight)−1, respectively. No changes were observed in the amount of CPS or EPS in the transposon mutant (data not shown). Two distinct forms of neutral polysaccharides have been described for R. leguminosarum: a low-molecular-weight cyclic β-(1,2)-glucan (Zevenhuizen et al., 1990) and a high-molecular-weight glucomannan (Laus et al., 2006). To determine whether one or both of the neutral polysaccharides were elevated in the fabF2/F1 mutant, the amount of reducing sugar present in the neutral polysaccharide fraction was determined using the method of Lever (1972). The cyclic β-(1,2)-glucan molecule contains no terminal glucose residues, and therefore no reducing sugars, whereas the glucomannan is described as having terminal mannose and some terminal glucose residues, both of which are reducing sugars (Laus et al., 2006). Therefore, if the increased amount of neutral polysaccharide in the mutant was due to an increase in the amount of glucomannan, there would be an expected increase in the amount of reducing sugars. However, no difference was found in the amount of reducing sugars between the fabF2/F1 mutant [1.35 ± 0.209 μg reducing sugar (mg dry cell weight)−1] and the wild-type [1.50±0.244 μg reducing sugar (mg dry cell weight)−1]. Consequently, the increased amount of neutral polysaccharide in the mutant is suggestive of an increase in the amount of cyclic β-(1,2)-glucan.
The suggested increase in the cell surface-associated cyclic β-(1,2)-glucan could be caused by one of two mechanisms: (1) the fabF2/F1 mutant synthesizes more cyclic β-(1,2)-glucan than the wild-type; (2) the cyclic β-(1,2)-glucan is distributed in the mutant differently from in the wild-type. In R. leguminosarum, cyclic β-(1,2)-glucans are found in the periplasm, within the cell capsule, and also associated with the more diffuse EPS (Breedveld & Miller, 1994). It could be that alterations to the outer membrane permeability caused by the fabF2/F1 mutation result in leakage of β-(1,2)-glucans from the periplasm to the cell surface.
In R. leguminosarum, cyclic β-(1,2)-glucans are synthesized from UDP-glucose by the cytoplasmic membrane protein NdvB. To determine whether the possible increase in secreted β-(1,2)-glucan was due to an increase in biosynthesis, the activity of the ndvB promoter was measured in the wild-type and the fabF2/F1 mutant. There was no significant difference in the expression of the ndvB promoter between the wild-type and the fabF2/F1 mutant, with expression activities of 2038 ± 306 and 2023 ± 248 Miller units, respectively. These observations agree with those of Breedveld & Miller (1994), who found that cells grown in conditions such as high salt that increase the permeability of the outer membrane excrete large amounts of β-(1,2)-glucan, without a corresponding increase in ndvB expression. Taken together, these results suggest that the VLCFA-deficient lipid A structure observed in the fabF2/F1 mutant increases the permeability of the outer membrane to small, neutral molecules, such as the β-(1,2)-glucans, and the predicted decrease in periplasmic β-(1,2)-glucan may therefore result in increased sensitivity to hypo-osmotic stress.
The fabF2/F1 mutant is altered in biofilm formation
Biofilm formation by the fabF2/F1 mutant strain was analysed to determine whether the pleiotropic changes in mutant cell structure might affect the attachment of the mutant to solid surfaces. When assessed by viable cell counting, both the wild-type and the mutant strain formed biofilms with similar mean cell densities of 5.3 ± 0.5 and 5.7 ± 0.3 c.f.u. per peg in the CBD, respectively. However, the nature of how the adherent bacteria were organized on the plastic surfaces was substantially different between the wild-type and the mutant.
Staining of biofilms with AO and examination by CLSM (Fig. 4) indicated that the wild-type strain had formed loosely organized microcolonies at the air–liquid-surface interface, and single cells, which were attached to the surface by their poles, were scattered across the CBD peg. In contrast, the fabF2/F1 mutant formed large, tightly organized microcolonies at the air–liquid-surface interface, yet by comparison, single cells were sparingly attached to the remainder of the peg surface. Unlike the polar attachment of wild-type cells, the fabF2/F1 cells were attached to the plastic by their sides.
Fig. 4.
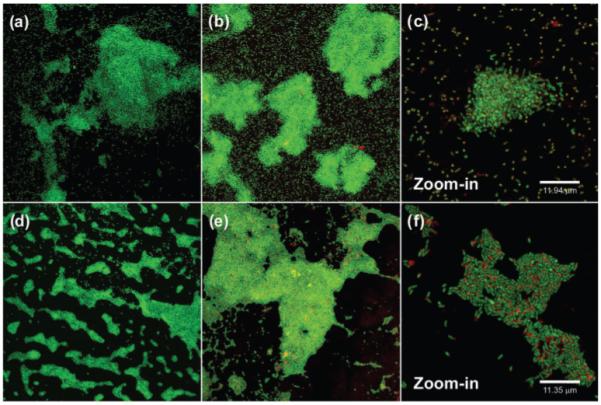
CLSM of biofilms produced by wild-type, 3841 (a–c) and the fabF2/F1 mutant (d–f). Biofilms were stained with AO (a, d) or Syto-9 (green) and TRITC-ConA (red) (b, c, e, f).
Single cells in glutaraldehyde-fixed biofilms of the wild-type strain stained with Syto-9+TRITC-ConA had a yellow hue, suggesting that lectin binding was distributed across the cell surfaces; furthermore, many cells also had a concentration of lectin binding at a single pole of the cell (Fig. 4). However, the mutant cells appeared green, with the lectin binding in relatively large abundance at a single pole of the cell. Asymmetrical distribution of cell-surface polysaccharides is important for the normal polar adhesion of R. leguminosarum to surfaces (Laus et al., 2006). The increased longitudinal attachment of the fabF2/F1 mutant to the polystyrene surface suggests that this process is disrupted by alterations to the VLCFA of LPS; however, a putative mechanism underlying this phenomenon is not clear from the present work. Similar experiments with the complemented strain fabF2/F1 + pCS115 showed that it had a biofilm and cell structure similar to the wild-type (data not shown).
Mutations in the fabF2 and fabF1 genes result in loss of motility
Alterations in LPS structure have been observed to cause motility-related defects (Priefer, 1989; Garcia-de los Santos & Brom, 1997; Hynes & McGregor, 1990). The mutant 38EV28 was used to assess the effect of a fabF1 mutation on the motility of R. leguminosarum 3841. The fabF2/F1 transposon mutant was also non-motile; however, motility was not restored in the complemented strain and therefore further investigation was limited to the fabF1 mutant. The fabF1 mutant was non-motile, whereas the wild-type had a swimming diameter of 7.8 ± 0.84 mm when inoculated into YES medium for 48 h. Complementation of the fabF1 mutant with pCS115 restored motility, with a swimming diameter of 5.0 ± 1.7 mm. In addition, phase-contrast microscopy was used to confirm that there was no observable swimming in the mutant strain.
A non-motile phenotype has also been described for ndvA and ndvB mutants under hypo-osmotic conditions. To see whether the non-motile phenotype of the fabF1 mutant was related to the observed alterations in cyclic β-(1,2)-glucan, the ability of the mutant to swim in YES medium supplemented with 50 mM NaCl was determined. The mutant was still unable to swim even in the presence of additional salt, while both the wild-type and complemented strains swam normally (data not shown), suggesting that the alterations to the lipid A structure directly cause the observed defects in motility rather than the sensitivity of the mutant to hypo-osmotic conditions. Electron microscopy was used to determine whether or not the mutant strain was flagellated (Fig. 5). The large amount of dark background material on the grids is EPS. We found that while some of the mutant cells had flagella, the majority of the cells were not flagellated. Based on the swimming and phase-contrast microscopy, it is likely that the small number of flagella produced are non-functional.
Fig. 5.
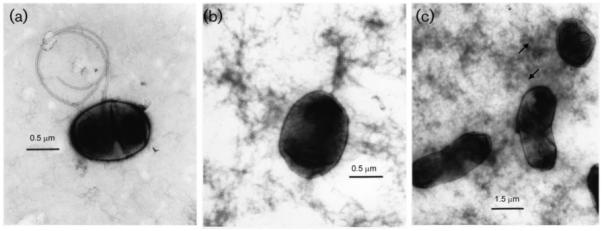
Electron microscopy of cells stained with 1 % uranyl acetate to determine the presence of flagella. (a) Flagellated wild-type cell, (b) fabF2/F1 mutant without flagella, (c) flagellated fabF1−pCS115.
To further confirm the non-motile phenotype of the fabF1 mutant, the expression of several motility-related genes was determined using gusA transcriptional fusions (Table 3). From these experiments it was found that the class III motility genes flaA, mcpC and mcpD were all down-regulated two- to threefold in the fabF1 mutant compared with the wild-type. Also, the class IA visN gene and the class IB regulator rem were significantly downregulated. In S. meliloti, the motility, chemotaxis and flagellar genes are arranged in a three-tiered hierarchy of expression, with the VisNR global regulator at the top followed by the Rem regulator, which regulates the expression of the class II and class III genes (Rotter et al., 2006). The data we have presented suggest that the fabF1 mutation affects motility gene expression at the highest levels of regulation. The lack of flagella in the mutant suggests a feedback mechanism whereby motility genes are downregulated due to the inability of the mutant to effectively assemble flagella.
Table 3.
Expression of motility-related genes in the fabF1 mutant, 38EV28
| Strain |
gusA activity* (Miller units) for gene fusion: |
|||||
|---|---|---|---|---|---|---|
| No plasmid | mcpC (pCGR) | mcpD (pDGRP) | flaA (pAVP) | visN (pVNVP) | rem (pSVP SUM) | |
| Wild-type | 160±8.98 | 2.39×103±449 | 2.22×103±557 | 6.54×103±781 | 7.29×103±881 | 1.35×104±1.27×103 |
| fabF1 − | 120±15.9 | 549±132† | 795±136† | 2.89×103±667† | 2.36×103±237† | 9.36×103±347† |
Strains were grown to late-exponential phase in TY broth and specific activity was determined as described in Methods. Data presented are the mean (±sd) of three independent trials.
Difference between the wild-type and mutant is statistically significant at P <0.006 (Student’s t test).
DISCUSSION
This study describes the isolation and characterization of mutations within the putative 3-oxoacyl [acyl-carrier protein] synthase II genes fabF1 and fabF2 from R. leguminosarum 3841. This research provides experimental evidence that FabF1 and FabF2 are required for synthesis of the VLCFA component of lipid A, and that osmotic and detergent stress tolerance, phenotypes previously observed in VLCFA-deficient lipid A acpXL and lpxXL mutants of R. leguminosarum and S. meliloti, are also observed in the fabF2 and fabF1 mutants. In addition, we have attributed the activities of FabF1 and FabF2 to several new processes that are presumably important for the persistence of free-living cells in the rhizosphere, including desiccation tolerance, biofilm formation, and motility.
Several researchers have speculated that LPS plays a role in desiccation tolerance (Cytryn et al., 2007; Vriezen et al., 2007). Recent microarray data from Bradyrhizobium japonicum has identified several genes related to synthesis of LPS that are upregulated in response to desiccation (Cytryn et al., 2007). Lloret et al. (1995) found that structural changes to LPS occur when cells are stressed by conditions that mimic desiccation stress, but that similar changes are not seen when the same cells are osmotically stressed, suggesting that cells may alter LPS structure as a specific adaptation to desiccation. The mutagenesis results provided here are the first direct evidence linking LPS structure to desiccation tolerance in rhizobia. The DOC-PAGE analyses of the LPS from the various fabF2/F1 mutants suggest that changes may have occurred to the O-antigen of the mutant. Notably, studies with Salmonella enterica serovar Typhimurium have found that cells lacking the O-antigen have increased sensitivity to drying, which Garmiri et al. (2008) suggested was due to loss of the ability to maintain a hydrated protective layer around the cell. Therefore, it is possible that the desiccation sensitivity of the mutant is related to alterations in the polysaccharide component of the LPS and may not necessarily be directly linked to the altered lipid A structure. Future research will be directed to determining the specific contributions of the lipid A and polysaccharide components of the LPS to desiccation tolerance in rhizobia.
The interaction of the fabF2/F1 mutant with plastic surfaces was altered compared with wild-type cells, and this affected the structure of R. leguminosarum bv viciae biofilms. The wild-type R. leguminosarum cells showed polar attachment to the neutrally charged, polystyrene surface of the CBD, whereas the fabF2/F1 mutant cells attached to the plastic surface longitudinally. Staining of the mutant and wild-type biofilms with a fluorescent lectin (TRITC-ConA) suggests that relative to wild-type cells, a mannose- and/or glucose-rich polysaccharide accumulates at the poles of the fabF2/F1 mutant. Since the fabF2/F1 mutant does not appear to produce more glucomannan than wild-type cells it remains to be determined whether the suggested increase in cyclic β-(1,2)-glucan excretion in the fabF2/F1 mutant is contributing to the observed alterations in lectin binding or whether the glucomannan is distributed differently on the surface of the mutant compared with wild-type biofilm cells. Furthermore, an explanation of why this polar polysaccharide does not anchor cells to surfaces is not clear from the present study. Taken together, our data suggest that the VLCFA modification of LPS is directly or indirectly involved in the normal, asymmetrical localization of polysaccharides in cells and that alterations to LPS affect how R. leguminosarum naturally organizes onto surfaces. It will be interesting to determine whether the observed difference in polysaccharide localization between mutant and wild-type biofilm cells is present on planktonic cells. Laus et al. (2006) have recently proposed that a unipolar, high-molecular-weight glucomannan in R. leguminosarum RBL5523 plays a role in bacterial cell docking at the host plant root surface via lectins. Therefore, future research will also be directed towards determining whether the fabF2/F1 mutant is altered in attachment to plant roots.
Studies with an acpXL mutant of R. leguminosarum 3841 found that the mutant is able to fix nitrogen, but is delayed in the formation of nitrogen-fixing nodules (Vedam et al., 2003). Similar to the acpXL mutant, the VLCFA-deficient fabF2/F1 mutant is able to form functional nitrogen-fixing nodules (data not shown). Analysis of the lipid A structure of the bacteroids from the acpXL mutant showed that they are restored in their ability to produce the VLCFA in planta (Vedam et al., 2006). Vedam et al. (2006) identified a region of the symbiotic plasmid pRL10 that contains a putative acp gene, which they speculate is able to compensate for lack of acpXL expression during symbiosis. Notably, the gene region of this acp does not contain additional fabF-like genes, suggesting that R. leguminosarum 3841 cannot use alternative genes to compensate for fabF1 and fabF2 mutations during symbiosis. It will be interesting to determine whether the LPS of the fabF2/F1 mutant bacteroids contains the VLCFA modification on the lipid A.
A blastp search of published bacterial genomes using the amino acid sequences of the VLCFA biosynthetic proteins suggests that the unique 27-hydroxyoctacosanoate VLCFA modification is limited to the Rhizobiales and to a number of intracellular pathogens, Legionella spp., Brucella spp. and Bartonella spp. (Vedam et al., 2006). However, a specific explanation of why only these Gram-negative bacteria require a VLCFA remains elusive. In this study we have identified a role for the VLCFA in several key phenotypes linked to free-living survival of rhizobia, including desiccation tolerance, biofilm formation and motility. These results indicate that in addition to its possible role during symbiosis, the VLCFA modification is likely to play an important role in the survival of R. leguminosarum 3841 in the rhizosphere.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Silvia Wehmeier for help with the DOC-PAGE protocol, Michael Hynes, University of Calgary, for the gift of the pAVP, pVNVP and pSVP SUM fusions, and Raymond Turner for insightful comments and access to instrumentation. This research has been supported by Natural Sciences and Engineering Research Council (NSERC) grants to C. K. Y. and S. F. K. E. M. V. was supported by a Postgraduate Scholarship from NSERC. Analysis of the lipid A was supported, in part, by National Institutes of Health Grant GM39583 to R. W. C., and Department of Energy Grant DEFG02-98ER20307 to the Complex Carbohydrate Research Center. J. J. H. was supported by a Canada Graduate Scholarship from NSERC as well as by a PhD studentship from the Alberta Heritage Foundation for Medical Research. CLSM was facilitated by a grant from the Canadian Foundation for Innovation to H. C. at the University of Calgary. We gratefully acknowledge Judy Sholdice and Caitlin Ward for technical assistance with the EM analysis.
Abbreviations
- AO
acridine orange
- CBD
Calgary Biofilm Device
- CLSM
confocal laser scanning microscopy
- CPS
capsular polysaccharide
- ddH2O
double-distilled H2O
- DOC
sodium deoxycholate
- EPS
exopolysaccharide
- TRITC-ConA
tetramethylrhodamine isothiocyanate-conjugated concanavalin A
- VLCFA
very long chain fatty acid
REFERENCES
- Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ. Gapped blast and psi-blast: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu SS, Karbarz MJ, Raetz CRH. Expression cloning and characterization of the C28 acyltransferase of lipid A biosynthesis in Rhizobium leguminosarum. J Biol Chem. 2002;277:28959–28971. doi: 10.1074/jbc.M204525200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beringer JE. R factor transfer in Rhizobium leguminosarum. J Gen Microbiol. 1974;84:188–198. doi: 10.1099/00221287-84-1-188. [DOI] [PubMed] [Google Scholar]
- Bhat UR, Mayer H, Yokota A, Hollingsworth RI, Carlson RW. Occurrence of lipid A variants with 27-hydroxyoctacosanoic acid in lipopolysaccharides from members of the family Rhizobiaceae. J Bacteriol. 1991;173:2155–2159. doi: 10.1128/jb.173.7.2155-2159.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat UR, Forsberg LS, Carlson RW. Structure of lipid A component of Rhizobium leguminosarum bv phaseoli lipopolysaccharide – unique nonphosphorylated lipid A containing 2-amino-8-deoxygluconate, galacturonate, and glucosamine. J Biol Chem. 1994;269:14402–14410. [PubMed] [Google Scholar]
- Breedveld MW, Miller KJ. Cyclic β-glucans of members of the family Rhizobiaceae. Microbiol Rev. 1994;58:145–161. doi: 10.1128/mr.58.2.145-161.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringhurst RM, Cardon ZG, Gage DJ. Galactosides in the rhizosphere: utilization by Sinorhizobium meliloti and development of a biosensor. Proc Natl Acad Sci U S A. 2001;98:4540–4545. doi: 10.1073/pnas.071375898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson RW. Heterogeneity of Rhizobium lipopolysaccharides. J Bacteriol. 1984;158:1012–1017. doi: 10.1128/jb.158.3.1012-1017.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceri H, Olson ME, Stremick CA, Read RR, Morck D, Buret A. The Calgary Biofilm Device: new technology for rapid determination of antibiotic susceptibilities of bacterial biofilms. J Clin Microbiol. 1999;37:1771–1776. doi: 10.1128/jcm.37.6.1771-1776.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna-Romano S, Arnold W, Schluter A, Boistard P, Puhler A, Priefer UB. An Fnr-like protein encoded in Rhizobium leguminosarum biovar viciae shows structural and functional homology to Rhizobium meliloti FixK. Mol Gen Genet. 1990;223:138–147. doi: 10.1007/BF00315806. [DOI] [PubMed] [Google Scholar]
- Cytryn EJ, Sangurdekar DP, Streeter JG, Franck WL, Chang WS, Stacey G, Emerich DW, Joshi T, Xu D, Sadowsky MJ. Transcriptional and physiological responses of Bradyrhizobium japonicum to desiccation-induced stress. J Bacteriol. 2007;189:6751–6762. doi: 10.1128/JB.00533-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dylan T, Helinski DR, Ditta GS. Hypoosmotic adaptation in Rhizobium meliloti requires β-(1→2)-glucan. J Bacteriol. 1990;172:1400–1408. doi: 10.1128/jb.172.3.1400-1408.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson GP, Datta A, Carlson RW, Walker GC. Importance of unusually modified lipid A in Sinorhizobium stress resistance and legume symbiosis. Mol Microbiol. 2005;56:68–80. doi: 10.1111/j.1365-2958.2005.04536.x. [DOI] [PubMed] [Google Scholar]
- Garcia-de los Santos A, Brom S. Characterization of two plasmid-borne lpsβ loci of Rhizobium etli required for lipopolysaccharide synthesis and for optimal interaction with plants. Mol Plant Microbe Interact. 1997;10:891–902. doi: 10.1094/MPMI.1997.10.7.891. [DOI] [PubMed] [Google Scholar]
- Gardy JL, Laird MR, Chen F, Rey S, Walsh CJ, Ester M, Brinkman FSL. PSORTb v.2.0: expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis. Bioinformatics. 2005;21:617–623. doi: 10.1093/bioinformatics/bti057. [DOI] [PubMed] [Google Scholar]
- Garmiri P, Coles K, Humphrey T, Cogan T. Role of outer membrane lipopolysaccharides in the protection of Salmonella enterica serovar Typhimurium from desiccation damage. FEMS Microbiol Lett. 2008;281:155–159. doi: 10.1111/j.1574-6968.2008.01093.x. [DOI] [PubMed] [Google Scholar]
- Gilbert KB, Vanderlinde EM, Yost CK. Mutagenesis of the carboxy terminal protease CtpA decreases desiccation tolerance in Rhizobium leguminosarum. FEMS Microbiol Lett. 2007;272:65–74. doi: 10.1111/j.1574-6968.2007.00735.x. [DOI] [PubMed] [Google Scholar]
- González-Ballester D, de Montaigu A, Galvan A, Fernandez E. Restriction enzyme site-directed amplification PCR: a tool to identify regions flanking a marker DNA. Anal Biochem. 2005;340:330–335. doi: 10.1016/j.ab.2005.01.031. [DOI] [PubMed] [Google Scholar]
- Harrison JJ, Ceri H, Yerly J, Stremick CA, Hu Y, Martinuzzi R, Turner RJ. The use of microscopy and three-dimensional visualization to evaluate the structure of microbial biofilms cultivated in the Calgary Biofilm Device. Biol Proced Online. 2006;8:194–215. doi: 10.1251/bpo127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JJ, Ceri H, Yerly J, Rabiei M, Hu Y, Martinuzzi R, Turner RJ. Metal ions may suppress or enhance cellular differentiation in Candida albicans and Candida tropicalis biofilms. Appl Environ Microbiol. 2007;73:4940–4949. doi: 10.1128/AEM.02711-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath RJ, White SW, Rock CO. Lipid biosynthesis as a target for antibacterial agents. Prog Lipid Res. 2001;40:467–497. doi: 10.1016/s0163-7827(01)00012-1. [DOI] [PubMed] [Google Scholar]
- Hitchcock PJ, Brown TM. Morphological heterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained polyacrylamide gels. J Bacteriol. 1983;154:269–277. doi: 10.1128/jb.154.1.269-277.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WJ, Jia J, Edwards P, Dehesh K, Schneider G, Lindqvist Y. Crystal structure of β-ketoacyl-acyl carrier protein synthase II from E. coli reveals the molecular architecture of condensing enzymes. EMBO J. 1998;17:1183–1191. doi: 10.1093/emboj/17.5.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes MF, McGregor NF. Two plasmids other than the nodulation plasmid are necessary for formation of nitrogen-fixing nodules by Rhizobium leguminosarum. Mol Microbiol. 1990;4:567–574. doi: 10.1111/j.1365-2958.1990.tb00625.x. [DOI] [PubMed] [Google Scholar]
- Johnston AWB, Beringer JE. Identification of Rhizobium strains in pea root nodules using genetic markers. J Gen Microbiol. 1975;87:343–350. doi: 10.1099/00221287-87-2-343. [DOI] [PubMed] [Google Scholar]
- Kannenberg EL, Reuhs BL, Forsberg SL, Carlson RW. Lipopolysaccharides and K-antigens: their structures, biosynthesis, and function. In: Spaink HP, Kondorosi A, Hooykaas PJJ, editors. The Rhizobiaceae: Molecular Biology of Model Plant-Associated Bacteria. Kluwer Academic Publishers; Dordrecht, The Netherlands: 1998. pp. 119–154. [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Laurentin A, Edwards C. A microtiter modification of the anthrone–sulfuric acid colorimetric assay for glucose-based carbohydrates. Anal Biochem. 2003;315:143–145. doi: 10.1016/s0003-2697(02)00704-2. [DOI] [PubMed] [Google Scholar]
- Laus MC, Logman TJ, Lamers GE, Van Brussel AN, Carlson RW, Kijne JW. A novel polar surface polysaccharide from Rhizobium leguminosarum binds host plant lectin. Mol Microbiol. 2006;59:1704–1713. doi: 10.1111/j.1365-2958.2006.05057.x. [DOI] [PubMed] [Google Scholar]
- Lever M. A new reaction for colorimetric determination of carbohydrates. Anal Biochem. 1972;47:273–279. doi: 10.1016/0003-2697(72)90301-6. [DOI] [PubMed] [Google Scholar]
- Lloret J, Bolanos L, Lucas MM, Peart JM, Brewin NJ, Bonilla I, Rivilla R. Ionic stress and osmotic pressure induce different alterations in the lipopolysaccharide of a Rhizobium meliloti strain. Appl Environ Microbiol. 1995;61:3701–3704. doi: 10.1128/aem.61.10.3701-3704.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzon RG, Neuls TM, Manzon LA. Molecular cloning, tissue distribution, and developmental expression of lamprey transthyretins. Gen Comp Endocrinol. 2007;151:55–65. doi: 10.1016/j.ygcen.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1972. [Google Scholar]
- Ngwai YB, Adachi Y, Ogawa Y, Hara H. Characterization of biofilm-forming abilities of antibiotic-resistant Salmonella typhimurium DT104 on hydrophobic abiotic surfaces. J Microbiol Immunol Infect. 2006;39:278–291. [PubMed] [Google Scholar]
- Nikaido H, Vaara M. Molecular basis of bacterial outer membrane permeability. Microbiol Rev. 1985;49:1–32. doi: 10.1128/mr.49.1.1-32.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ophir T, Gutnick D. A role for exopolysaccharides in the protection of microorganisms from desiccation. Appl Environ Microbiol. 1994;60:740–745. doi: 10.1128/aem.60.2.740-745.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priefer UB. Genes involved in lipopolysaccharide production and symbiosis are clustered on the chromosome of Rhizobium leguminosarum biovar viciae VF39. J Bacteriol. 1989;171:6161–6168. doi: 10.1128/jb.171.11.6161-6168.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quandt J, Hynes MF. Versatile suicide vectors which allow direct selection for gene replacement in Gram-negative bacteria. Gene. 1993;127:15–21. doi: 10.1016/0378-1119(93)90611-6. [DOI] [PubMed] [Google Scholar]
- Que NLS, Ribeiro AA, Raetz CRH. Two-dimensional NMR spectroscopy and structures of six lipid A species from Rhizobium etli CE3 – detection of an acyloxyacyl residue in each component and origin of the aminogluconate moiety. J Biol Chem. 2000a;275:28017–28027. doi: 10.1074/jbc.M004009200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que NLS, Lin SH, Cotter RJ, Raetz CRH. Purification and mass spectrometry of six lipid A species from the bacterial endosymbiont Rhizobium etli – demonstration of a conserved distal unit and a variable proximal portion. J Biol Chem. 2000b;275:28006–28016. doi: 10.1074/jbc.M004008200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve WG, Tiwari RP, Worsley PS, Dilworth MJ, Glenn AR, Howieson JG. Constructs for insertional mutagenesis, transcriptional signal localization and gene regulation studies in root nodule and other bacteria. Microbiology. 1999;145:1307–1316. doi: 10.1099/13500872-145-6-1307. [DOI] [PubMed] [Google Scholar]
- Reuhs BL, Carlson RW, Kim JS. Rhizobium fredii and Rhizobium meliloti produce 3-deoxy-d-manno-2-octulosonic acid-containing polysaccharides that are structurally analogous to group II K antigens (capsular polysaccharides) found in Escherichia coli. J Bacteriol. 1993;175:3570–3580. doi: 10.1128/jb.175.11.3570-3580.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotter C, Muhlbacher S, Salamon D, Schmitt R, Scharf B. Rem, a new transcriptional activator of motility and chemotaxis in Sinorhizobium meliloti. J Bacteriol. 2006;188:6932–6942. doi: 10.1128/JB.01902-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: a Laboratory Manual. 2nd edn. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- Sharypova LA, Niehaus K, Scheidle H, Holst O, Becker A. Sinorhizobium meliloti acpXL mutant lacks the C28 hydroxylated fatty acid moiety of lipid A and does not express a slow migrating form of lipopolysaccharide. J Biol Chem. 2003;278:12946–12954. doi: 10.1074/jbc.M209389200. [DOI] [PubMed] [Google Scholar]
- Simon R, Priefer U, Puhler A. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Biotechnology. 1983;1:784–791. [Google Scholar]
- Tang X, Lu BF, Pan SQ. A bifunctional transposon mini-Tn5-gfp-km which can be used to select for promoter fusions and report gene expression levels in Agrobacterium tumefaciens. FEMS Microbiol Lett. 1999;179:37–42. doi: 10.1111/j.1574-6968.1999.tb08704.x. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. clustal w – improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedam V, Kannenberg EL, Haynes JG, Sherrier DJ, Datta A, Carlson RW. A Rhizobium leguminosarum AcpXL mutant produces lipopolysaccharide lacking 27-hydroxyoctacosanoic acid. J Bacteriol. 2003;185:1841–1850. doi: 10.1128/JB.185.6.1841-1850.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedam V, Kannenberg E, Datta A, Brown D, Haynes-Gann JG, Sherrier DJ, Carlson RW. The pea nodule environment restores the ability of a Rhizobium leguminosarum lipopolysaccharide acpXL mutant to add 27-hydroxyoctacosanoic acid to its lipid A. J Bacteriol. 2006;188:2126–2133. doi: 10.1128/JB.188.6.2126-2133.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent VM. A Manual for the Practical Study of Root-Nodule Bacteria. Blackwell; Oxford, UK: 1970. IBP Handbook no. 15. [Google Scholar]
- Vriezen JAC, de Bruijn FJ, Nusslein K. Responses of rhizobia to desiccation in relation to osmotic stress, oxygen, and temperature. Appl Environ Microbiol. 2007;73:3451–3459. doi: 10.1128/AEM.02991-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal O, Jann K. Bacterial lipopolysaccharides. Methods Carbohydr Chem. 1965;5:83–91. [Google Scholar]
- York WS, Darvill AG, McNeil M, Stevenson TT, Albersheim P. Isolation and characterization of plant cell walls and cell wall components. Methods Enzymol. 1985;118:3–40. [Google Scholar]
- Yost CK, Rochepeau P, Hynes MF. Rhizobium leguminosarum contains a group of genes that appear to code for methyl-accepting chemotaxis proteins. Microbiology. 1998;144:1945–1956. doi: 10.1099/00221287-144-7-1945. [DOI] [PubMed] [Google Scholar]
- Yost CK, Del Bel KL, Quandt J, Hynes MF. Rhizobium leguminosarum methyl-accepting chemotaxis protein genes are downregulated in the pea nodule. Arch Microbiol. 2004;182:505–513. doi: 10.1007/s00203-004-0736-7. [DOI] [PubMed] [Google Scholar]
- Zevenhuizen L. Gel-forming capsular polysaccharide of fast-growing rhizobia: occurrence and rheological properties. Appl Microbiol Biotechnol. 1984;20:393–399. [Google Scholar]
- Zevenhuizen LP, van Veldhuizen A, Fokkens RH. Re-examination of cellular cyclic beta-1,2-glucans of Rhizobiaceae: distribution of ring sizes and degrees of glycerol-1-phosphate substitution. Antonie Van Leeuwenhoek. 1990;57:173–178. doi: 10.1007/BF00403952. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.