

Abstract
Pooled CRISPR screens with single-cell RNA-seq readout (Perturb-seq) have emerged as a key technique in functional genomics, but are limited in scale by cost and combinatorial complexity. Here, we modify the design of Perturb-seq by incorporating algorithms applied to random, low-dimensional observations. Compressed Perturb-seq measures multiple random perturbations per cell or multiple cells per droplet and computationally decompresses these measurements by leveraging the sparse structure of regulatory circuits. Applied to 598 genes in the immune response to bacterial lipopolysaccharide, compressed Perturb-seq achieves the same accuracy as conventional Perturb-seq with an order of magnitude cost reduction and greater power to learn genetic interactions. We identify known and novel regulators of immune responses and uncover evolutionarily constrained genes with downstream targets enriched for immune disease heritability, including many missed by existing genome-wide association studies. Our framework enables new scales of interrogation for a foundational method in functional genomics.
Pooled perturbation screens with high-content readouts, ranging from single-cell RNA-seq (Perturb-seq)1–4 to imaging-based spatial profiling5–7, are now enabling systematic studies of the regulatory circuits that underlie diverse cell phenotypes. Perturb-seq has been applied to various model systems, leading to insights about diverse cellular processes including the innate immune response2, in vivo effects of autism risk genes in mice8 and organoids 9,10, and genome-scale effects on aneuploidy, differentiation, and RNA splicing11. Integrating data from population-level genetic screens has also elucidated human disease mechanisms12.
However, due to the large number of genes in the genome, large-scale Perturb-seq screens are still prohibitively expensive and are often limited by the number of available cells, especially for primary cell systems13 and in vivo niches8. In addition, the exponentially larger number of possible genetic interactions makes it impossible to conduct exhaustive combinatorial screens for genetic interactions using existing approaches, so current Perturb-seq studies of genetic interactions are very modest and focused14. Several approaches have been developed to improve the efficiency of scRNA-seq and/or Perturb-seq, including overloading droplets with multiple pre-indexed cells (SciFi-seq15) or pooling multiple guides within cells16. However, pre-indexing requires an additional laborious and complex experimental step, while guide pooling has only been used to study cis and not trans effects of perturbations.
We propose an alternative approach to greatly increase the efficiency and power of Perturb-seq for both single and combinatorial perturbation screens, inspired by theoretical results from compressed sensing17–19 that apply to the sparse and modular nature of regulatory circuits in cells. To elaborate, perturbation effects tend to be sparse in that most perturbations affect only a small number of genes or co-regulated gene programs2,20. In this scenario, rather than assaying each perturbation individually, we can measure a much smaller number of random combinations of perturbations (forming what we call “composite samples”) and accurately learn the effects of individual perturbations from the composite samples using sparsity-promoting algorithms. Moreover, with certain types of composite samples, we can efficiently learn both first-order effects (i.e., from single gene perturbations) and higher-order genetic interaction effects from the same data. We have previously shown that experiments that measure random compositions of the underlying biological dataset can greatly increase the efficiency of measuring expression profiles21 and imaging transcriptomics22.
Here, we develop two experimental strategies to generate composite samples for Perturb-seq screens, and we introduce an inference method, Factorize-Recover for Perturb-seq analysis (FR-Perturb), to learn individual perturbation effects from composite samples. We apply our approach to 598 genes in a human macrophage cell line treated with lipopolysaccharide (LPS). By comparing compressed Perturb-seq to conventional Perturb-seq conducted in the same system, we demonstrate the enhanced efficiency and power of our approach for learning single perturbation effects and second-order genetic interactions. We derive insights into immune regulatory functions and illustrate their connection to human disease mechanisms by integrating data from genome-wide association studies (GWAS) and expression quantitative trait loci (eQTL) studies.
RESULTS
A compressed sensing framework for perturbation screens
In conventional Perturb-seq, each cell in a pool receives one or more genetic perturbations. Each cell is then profiled for the identity of the perturbation(s) and the expression levels of expressed genes. Our goal is to infer the effect sizes of perturbations on the phenotype, which can be the entire gene expression profile ( matrix) or an aggregate multi-gene phenotype2,3,11 such as an expression program or cell state score (length- vector). In both cases, we need samples to learn the effects of perturbations (Fig. 1a) (where sample replicates introduce a constant factor that is subsumed under the big O notation), such that the number of samples scales linearly with the number of perturbations.
Fig. 1. Framework for compressed Perturb-seq.
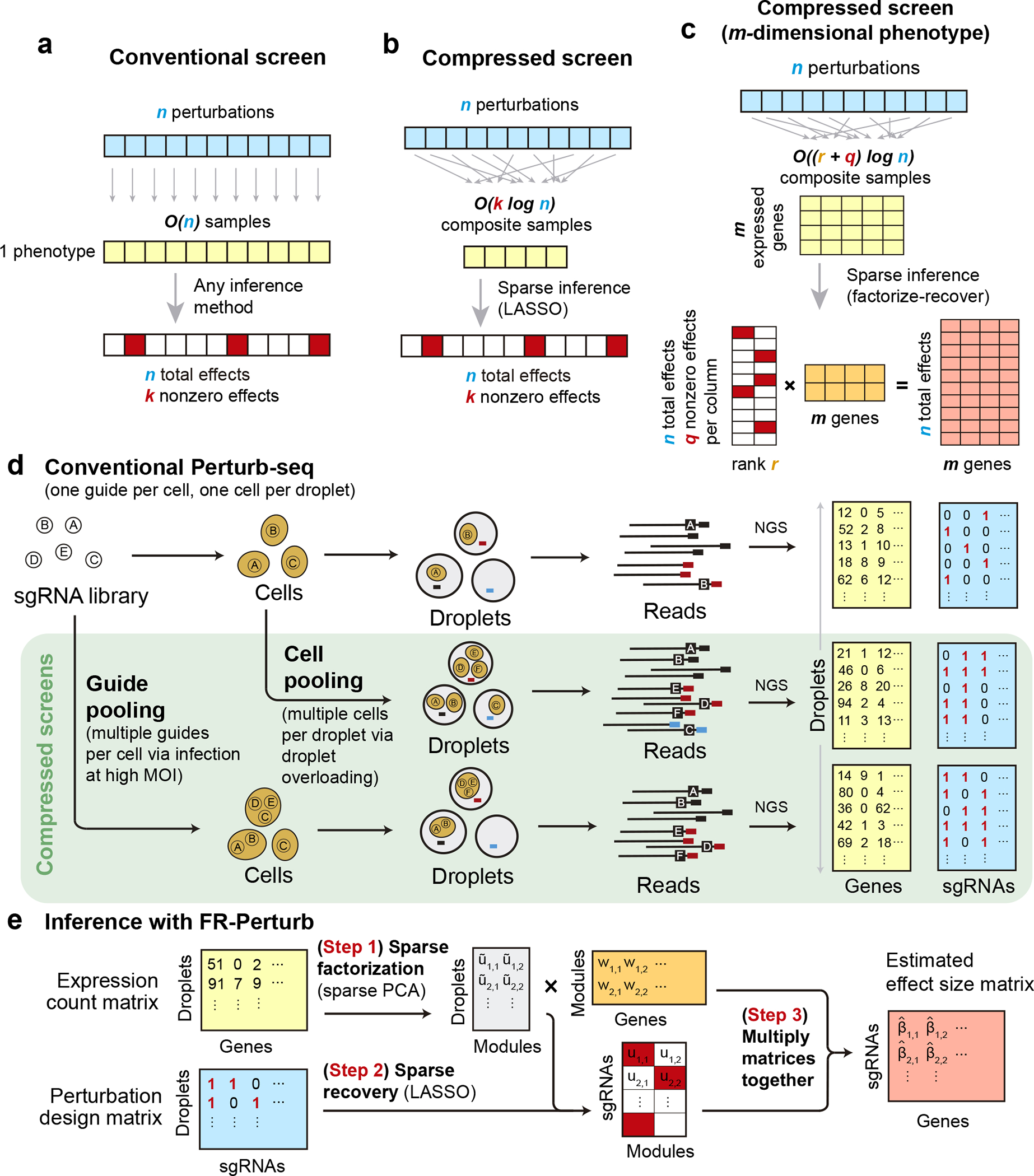
(a) Schematic for conventional perturbation screen with single-valued phenotype. Each sample (yellow) receives a single perturbation (blue). The required number of samples scales linearly with the number of perturbations, as captured by the term. (b) Schematic for compressed perturbation screen with single-valued phenotype. Each “composite” sample (yellow) represents a random combination of perturbations (blue). The required number of samples scales sub-linearly with the number of perturbations given the following: (1) the effects of the perturbations are sparse (i.e., increases more slowly than ), and (2) sparse inference (typically LASSO) is used to infer the effects from the composite sample phenotypes. (c) Schematic for compressed perturbation screen with high-dimensional phenotype, which is the main use case for Perturb-seq. The required number of samples scales sub-linearly with the number of perturbations given the following: (1) the effects of the perturbations are sparse and act on a relatively small number of groups of correlated genes (i.e., and increase more slowly than ), and (2) sparse inference (namely the “factorize-recover” algorithm23) is used to infer the effects from the composite sample phenotypes. (d) Two experimental strategies for generating composite samples for Perturb-seq. Both “cell pooling” and “guide pooling” change one step of the conventional Perturb-seq protocol. The result is a sample whose phenotype corresponds to a random linear combination of the phenotypes of samples from the conventional Perturb-seq screen. (e) Schematic of computational method used to infer perturbation effects from composite sample phenotypes, based on the “factorize-recover” algorithm23.
Based on the theory of compressed sensing17, there exist conditions under which far fewer than samples are sufficient to learn the effects of perturbations. In general, if the perturbation effects are sparse (i.e., relatively few perturbations affect the phenotype), or are sparse in a latent representation (i.e., perturbations tend to affect relatively few latent factors that can be combined to “explain” the phenotype), then we can measure a small number of random composite samples (comprising linear combinations of individual sample phenotypes) and decompress those measurements to infer the effects of individual perturbations. Composite samples can be generated either by randomly pooling perturbations in individual cells, or by randomly pooling cells containing one perturbation each (see below).
The number of required composite samples depends on whether the phenotype is single-valued or high-dimensional. When the phenotype is single-valued (e.g., fitness), composite samples suffice to accurately recover the effects of perturbations18,19, where is the number of nonzero elements among the perturbation effects (Fig. 1b). When most genes do not affect the phenotype, grows more slowly than , and the number of required composite samples scales logarithmically or at worst sub-linearly with the number of perturbations. Meanwhile, when the phenotype is an -dimensional gene expression profile, an efficient approach involves inferring effects on latent expression factors, then reconstructing the effects on individual genes from these factors using the “factorize-recover” algorithm23. This approach requires composite samples, where is the rank of the perturbation effect size matrix (i.e., the maximum number of its linearly independent column vectors), and is the maximum number of nonzero elements in any column of the left matrix of the factorized effect size matrix (Fig. 1c). In our case, is the number of distinct groups of “co-regulated” genes whose expression changes concordantly in response to any perturbation, while is the maximum number of “co-functional” perturbations with nonzero effects on any individual module. Due to the modular nature of gene regulation20,24,25, and are expected to remain small when increases. Indeed, we observed a relatively small number of co-functional and co-regulated gene groups (small and , respectively, relative to ) in previous Perturb-seq screens in various systems2,13. Thus, the number of required composite samples will scale logarithmically or at worst sub-linearly with , leading to much fewer required samples than the conventional approach with large . In simulations, this result held across a wide range of plausible values for and (Extended Data Fig. 1). We provide rough estimates of and from our own screens (see below) in the Supplementary Note, section 1.
Experimentally generating composite samples
We generated composite samples for compressed Perturb-seq by either randomly pooling cells containing one perturbation each in overloaded scRNA-seq droplets15 (“cell-pooling”), or by randomly pooling guides in individual cells via infection with a high multiplicity of infection (MOI)2,16 (“guide-pooling”; Fig. 1d). Under certain assumptions, the resulting expression counts in each droplet from either method represent a random linear combination of log fold-change effect sizes of guides. When cell-pooling, the expression counts in a given droplet are proportional to the average expression counts of the cells in the droplet, which can then be modeled in terms of log fold-change effect sizes of the guides in each cell (Methods). When guide-pooling, the expression counts in a given droplet can be also modeled as the sum of log fold-change effect sizes (Methods), though this requires the non-trivial assumption that the effect sizes of guides tend to combine additively in log expression space when multiple guides are present in the same cell. Although higher-order genetic interaction effects can in theory bias lower-order effect size estimates in guide-pooled data, we note that only a large imbalance in the direction and/or magnitude of higher-order interaction effects across many perturbations will lead to such biases, and that even in this scenario, many of the lower-order effects can still be accurately estimated (Supplementary Note, section 2).
Either of the two methods described above can be used to learn the same underlying perturbation effects, but each has different strengths and limitations (Discussion). Guide-pooling has a key benefit over cell-pooling, in that the generated data can be used to estimate both first-order effects and higher-order genetic interactions (with appropriate sample sizes and explicit interaction terms in the model) (Methods). We illustrate the feasibility of estimating second-order effects from guide-pooled data in later analyses.
FR-Perturb infers effects from compressed Perturb-seq
To infer perturbation effects from the composite samples, we devised a method called FR-Perturb based on the “factorize-recover” algorithm23 (Methods). FR-Perturb first factorizes the expression count matrix with sparse factorization (i.e., sparse PCA), followed by sparse recovery (i.e., LASSO) on the resulting left factor matrix comprising perturbation effects on the latent factors. Finally, it computes perturbation effects on individual genes as the product of the left factor matrix from the recovery step with the right factor matrix (comprising gene weights in each latent factor) from the first factorization step (Fig. 1e; Methods). Because FR-Perturb uses penalized regression, it is not guaranteed to be unbiased. We obtained p-values and false discovery rates (FDR) for all effects by permutation testing (Methods). We evaluated FR-Perturb by comparing it to existing inference methods for Perturb-seq, namely elastic net regression2 and negative binomial regression16, in later analyses.
Compressed Perturb-seq screens of the LPS response
We implemented and evaluated compressed Perturb-seq in the response of THP1 cells (a human monocytic leukemia cell line) to stimulation with LPS when either pooling cells or pooling guides (Fig. 2a,b). In each case, we also performed conventional Perturb-seq, targeting the same genes in the same system for comparison. We selected 598 genes to be perturbed from seven mostly non-overlapping immune response studies (Supplementary Table 1), including genes with roles in the canonical LPS response pathway (34 genes), GWAS for inflammatory bowel disease (79 genes) and infection (106 genes), Mendelian immune diseases from OMIM with keywords for “bacterial infection” (85 genes) and “NF-kappa-B” (102 genes), a previous genome-wide screen for effects on TNF expression in mouse BMDCs26 (93 genes), and genes with large genetic effects in trans on gene expression from an eQTL study in patient-derived macrophages stimulated with LPS27 (79 genes) (Methods, Supplementary Fig. 1). We designed 4 sgRNAs for each gene and 500 each of non-targeting or safe-targeting control sgRNAs, resulting in a total pool of 3,392 sgRNAs (Methods). We introduced the sgRNAs into THP1 cells via a modified CROP-seq vector4 (Methods). After transduction and selection, we treated cells with PMA for 24 hours and grew them for another 48 hours as they differentiated into a macrophage-like state28, then treated them with LPS for three hours before harvesting for scRNA-Seq (Methods). As a baseline, we also collected scRNA-seq data for genetically perturbed cells pre-stimulation (i.e., no LPS treatment) (see Supplementary Note, section 3 and Extended Data Fig. 2 for analysis). For our cell-pooled screen, we used CRISPR-Cas9 to knock out genes2, whereas for our guide-pooled screen we used CRISPRi with dCas9-KRAB to knock down gene expression1 (Fig. 2a) to avoid cellular toxicity due to multiple double-stranded breaks in individual cells29.
Fig. 2. Experimental overview.
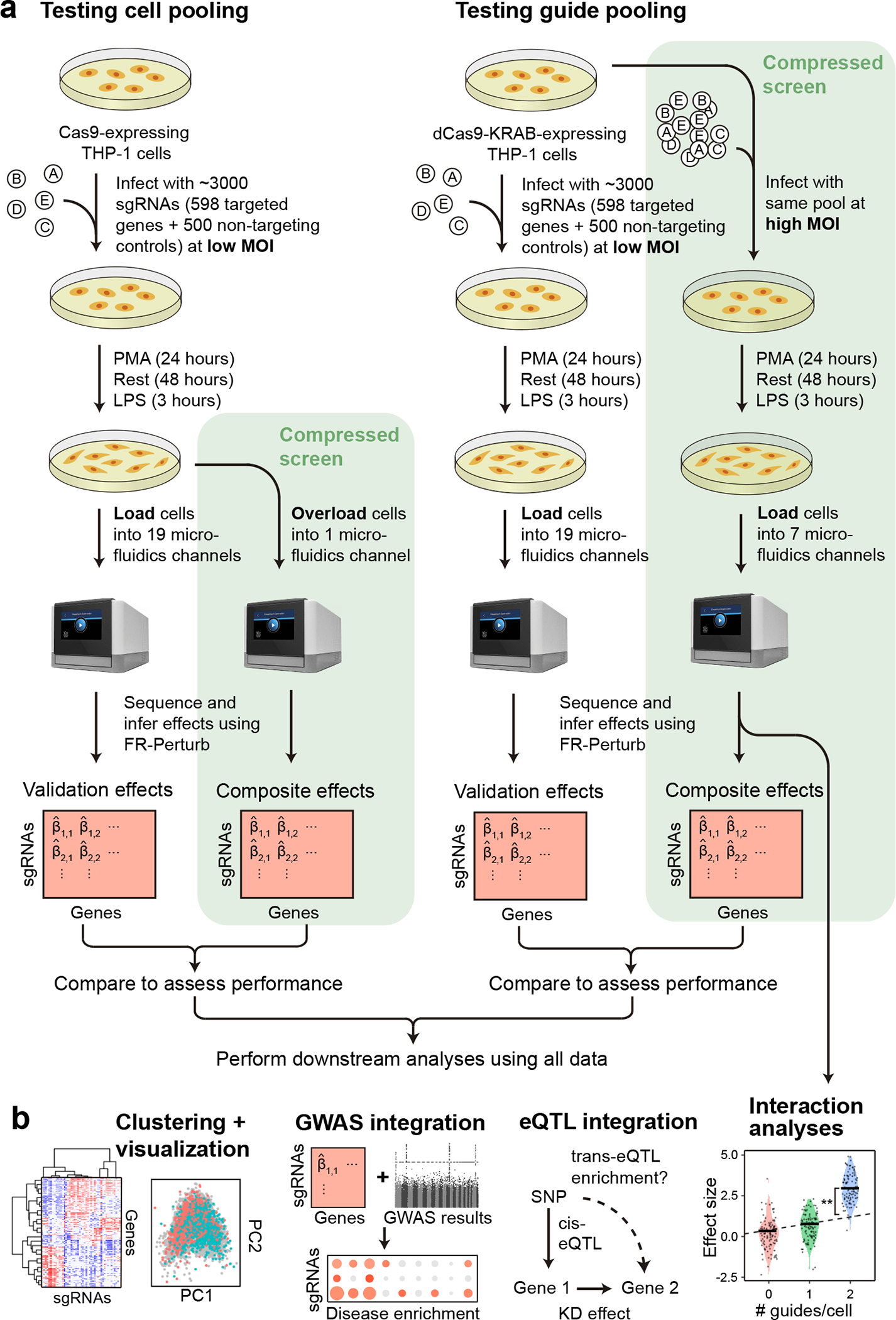
(a) Outline of experiments used to test and validate cell pooling (left) and guide pooling (right). (b) Downstream analyses performed using perturbation effects from all experiments.
By design, the two compressed screens were substantially smaller than their corresponding conventional screens. In the cell-pooled screen, we analyzed a single channel of droplets (10x Genomics, Methods) overloaded with 250,000 cells, while for the corresponding conventional Perturb-seq screen we analyzed 19 channels at normal loading. We sequenced the library from the overloaded channel to a depth of 4-fold more reads than a conventional channel to account for the larger number of non-empty droplets and greater expected RNA content per droplet. After quality control, there were 32,700 droplets containing at least one sgRNA from the overloaded channel (vs. 4,576 droplets/channel for a total of 86,954 droplets from the conventional screen) (Fig. 3a), with a mean of 1.86 sgRNAs per non-empty droplet (conventional: 1.11) (Fig. 3b) and a mean of 90 droplets containing a guide for each perturbed gene (conventional: 144) (Fig. 3c). We observed 14,987 total genes with measured expression (conventional: 17,552). Thus, the cell-pooled screen had >7 times the number of non-empty droplets per channel compared to the conventional screen; considering library preparation and sequencing costs, it was approximately 8 times cheaper.
Fig. 3. Evaluating cell-pooled Perturb-seq versus conventional Perturb-seq.
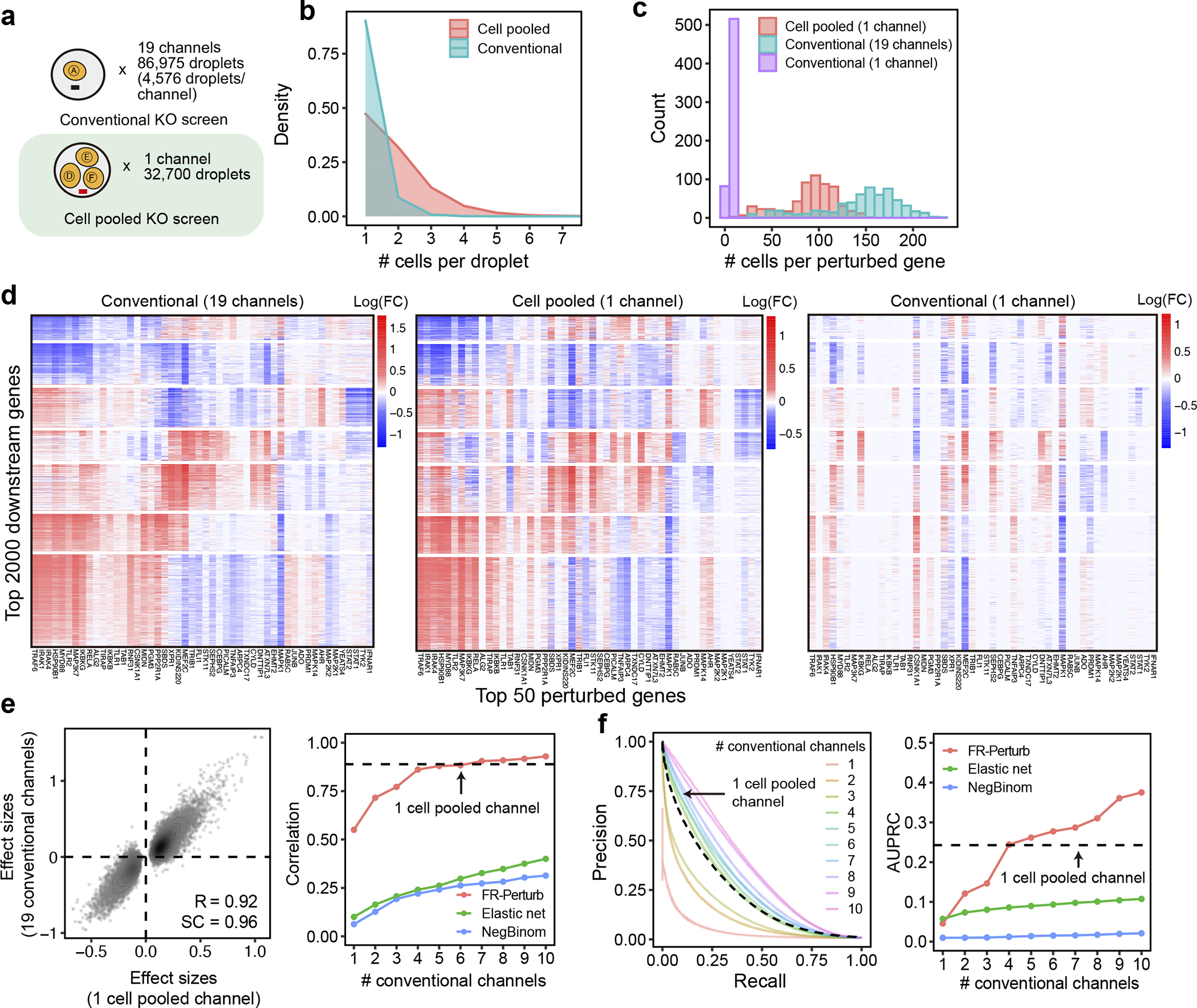
(a) Number of channels and droplets from the conventional validation screen (top) and cell-pooled screen (bottom). (b) Distribution of droplets based on number of cells they contain for the cell-pooled and conventional screens. (c) Distribution of the number of cells containing a guide targeting each perturbed gene in the cell-pooled screen and conventional screen (19 channels = full screen, 1 channel = matching # of channels from cell-pooled screen). (d) Heatmaps of the top effect sizes (inferred with FR-Perturb) from the conventional screen (left), with the same effect sizes shown for the cell pooled screen (middle) and one equivalent channel of the conventional screen (right). X-axis: top 50 perturbed genes, based on their average magnitude of effect on all 17,552 downstream genes. Y-axis: top 2,000 downstream genes, based on the average magnitude of effects of all 598 perturbed genes acting on them. Rows and columns are clustered based on hierarchical clustering in the leftmost plot. For the left plot, all effects with FDR q-value > 0.2 are whited out (q-value threshold relaxed to 0.5 for the middle and right plots). (e) (Left) Scatterplot of all significant effects () from the cell-pooled screen (X-axis) versus the same effects in the conventional screen (Y-axis). Effects represent log-fold changes in expression relative to control cells. R, Pearson’s correlation. SC, sign concordance. (Right) Held-out validation accuracy of top 19,909 effects (Y-axis; Pearson’s correlation with validation dataset) from down-sampled conventional screen (X-axis) and cell-pooled screen (dotted line). The same inference method is used to estimate effects in both the down-sampled conventional data and validation data. The effects from the cell-pooled screen are estimated using FR-Perturb only (see Extended Data Fig. 3d for results using other methods). (f) (Left) Precision-recall curves computed from down-sampled conventional screen and cell-pooled screen (dotted line). True positives = all significant effects () from the held-out validation dataset. The classification threshold being varied (X-axis) is the significance (i.e. p-value) of the effects. All effects displayed are learned using FR-Perturb. (Right) AUPRCs (Y-axis) computed from the down-sampled conventional experiment when varying the number of channels (X-axis).
In the guide-pooled experiment, we infected cells expressing dCas9-KRAB at high MOI (Methods) and profiled a single cell in each droplet across seven channels, while for the corresponding conventional Perturb-seq we infected cells with the same guide library at low MOI and analyzed 19 channels. From the guide-pooled experiment, we obtained 24,192 cells after filtering (conventional: 66,283), where 35% of the cells (8,448) contained three or more guides (Fig. 4a), with 2.50 guides on average per cell (conventional: 1.13) (Fig. 4b) and 101 cells containing a guide for each perturbed gene on average (conventional: 115) (Fig. 4c). We measured expression for 16,268 total genes (conventional: 18,617). The guide-pooled screen was approximately 3 times cheaper than the conventional screen.
Fig. 4. Evaluating guide-pooled Perturb-seq versus conventional Perturb-seq.
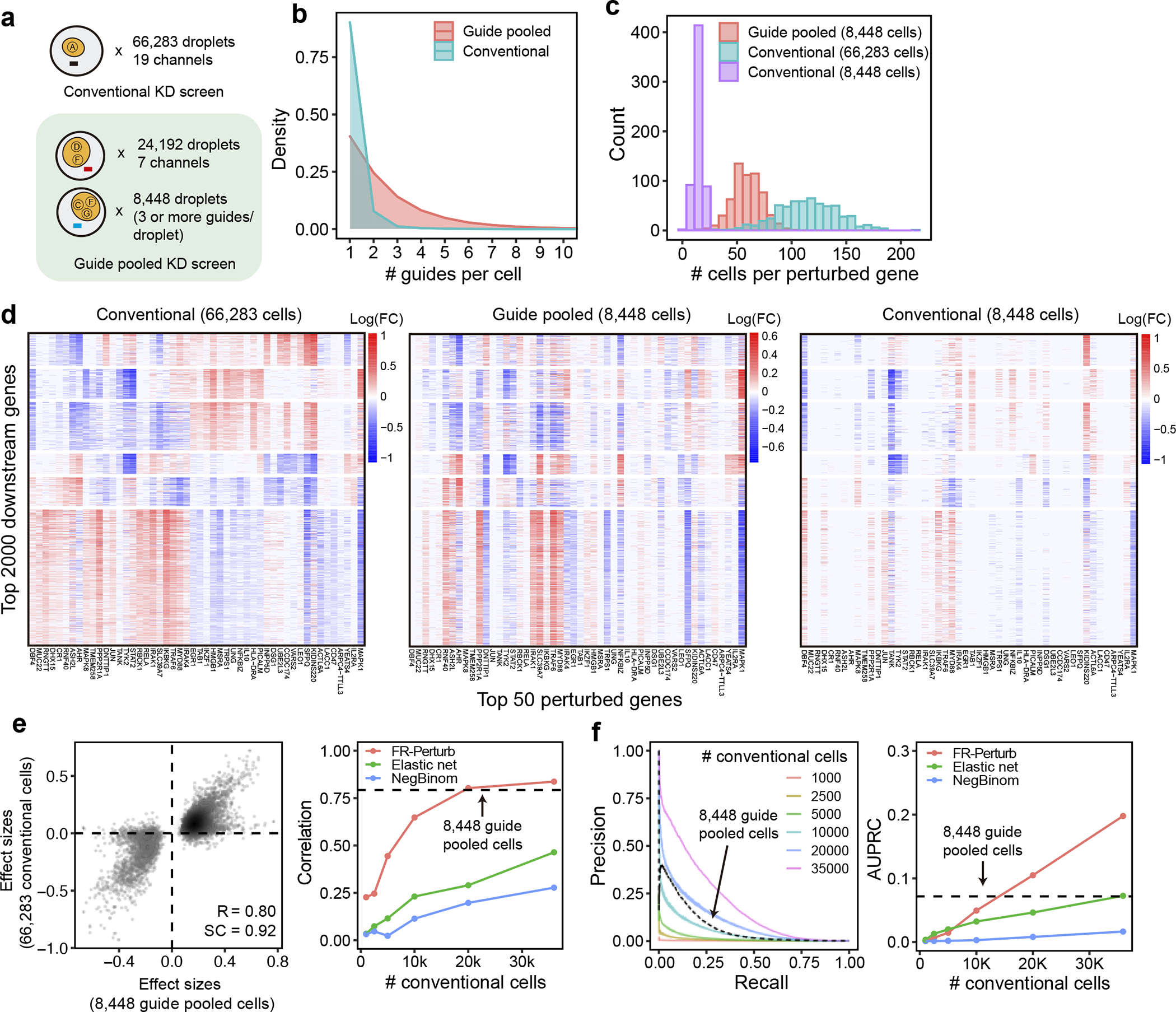
(a) Number of channels and droplets from the conventional validation screen (top) and guide-pooled screen (bottom). We focus our analysis on the subset of 8,448 droplets from the guide-pooled screen with at least 3 guides per droplet. (b) Distribution of cells based on # of guides they contain for the full guide-pooled and conventional screens. In practice, we only directly measure the # of guides/droplet rather than guides/cell, but these quantities are equivalent given 1 cell/droplet. (c-f) See captions for Fig. 3c–f. These analyses were conducted in an identical fashion, with the only difference that the screens are down-sampled based on cell count rather than channel count.
Cell-pooling achieves large efficiency gains
The perturbation effect sizes estimated by Perturb-FR from the cell-pooled Perturb-seq screen (Methods) agreed well with its conventional counterpart. When estimating effects, we included read count, cell cycle, and proportion of mitochondrial reads as covariates2, and we combined sgRNAs targeting the same gene while retaining the subset of sgRNAs for a gene with maximal concordance of effects across random subsets of the data (Methods). The significant effects from the compressed experiment were strongly correlated with the corresponding effects from the conventional experiment (Pearson’s , sign concordance = 0.96, Fig. 3e). Notably, we observed many more significant effects overall in the conventional screen than the cell-pooled screen (216,220 vs. 19,909; FDR q-value < 0.05), but this is expected given that we intentionally generated a larger and more highly powered conventional screen (144 droplets per perturbation, compared to 90 for the cell-pooled screen) to enable data splitting and cross validation analyses (see below).
The cell-pooled experiment yielded substantially more signal per experimental unit (channel) than the conventional one (Fig. 3d–f). First, the global clustering of effects learned from a single cell-pooled channel was much less noisy than from a single conventional channel (adjusted Rand index of 0.53 vs. 0.31 when comparing clusters with those learned from the full conventional screen; Fig. 3d). Moreover, approximately four conventional channels were needed to obtain the same number of significant effects as one cell-pooled channel (Extended Data Fig. 3a). Next, to quantitatively assess the specificity of each approach, we held out half of the conventional data as a validation set, then we down-sampled the remaining half to different numbers of channels and compared the top 19,909 most significant effects learned from the down-sampled data (matching the number of significant effects in the cell-pooled screen) to those in the held-out validation set. We found that 5–6 conventional channels were needed to achieve equivalent validation accuracy (correlation) as one cell-pooled channel (Fig. 3e). The relative efficiency gains of the compressed screen were consistent when varying the number of effects being compared (Extended Data Fig. 3c), when comparing effects on modules rather than on individual genes (Extended Data Fig. 4a), or when evaluating performance based on biological informativeness as reflected by the number of effects with significant heritability enrichment for common diseases (Extended Data Fig. 4b,c). We also assessed the sensitivity of each approach by testing whether the significant effects determined from the validation set were recovered by the down-sampled conventional or cell-pooled screens. We constructed precision-recall curves, calling “true positives” the 79,100 significant effects from the validation dataset and varying the classification threshold by the significance of the effects in the down-sampled conventional or cell-pooled datasets. One cell-pooled channel had comparable AUPRC to 4 conventional channels (Fig. 3f), with consistent efficiency gains when varying the number of true positive effects (Extended Data Fig. 3c).
Moreover, FR-Perturb substantially outperformed the established inference methods we tested: elastic net regression2 and negative binomial regression16. Repeating the same analyses as above with each method (Methods), the concordance between the down-sampled conventional data and validation data, and between cell-pooled and conventional data, was much higher with FR-Perturb than prior methods (Fig. 3e,f, Extended Data Fig. 3d). FR-Perturb also identified more biologically informative effects than prior methods, based on the heritability enrichment of common diseases (Extended Data Fig. 5). By down-sampling the cell-pooled screen, we found that ~1/5 of a cell-pooled channel analyzed with FR-Perturb achieved the same validation accuracy as 10 conventional channels analyzed with existing methods (Extended Data Fig. 3b). We assess the cost savings of cell pooling over the conventional approach while factoring in sequencing costs in the Supplementary Note, section 5.
Guide-pooling achieves large efficiency gains
Guide-pooled Perturb-seq was also concordant with its conventional counterpart, based on a similar evaluation scheme as above. For the guide-pooled screen, we focused on the 8,448 cells with 3 or more guides. This number of guides per cell can be achieved with sequential transduction, as done for 2 of the 7 channels (Methods, Supplementary Fig. 2). We learned perturbation effects from both screens using FR-Perturb, with slight modifications to account for differences in the guide-pooled vs. cell-pooled screens (Methods). The 5,836 significant effects from the guide-pooled cells were strongly correlated with the same effects from the conventional screen (Pearson’s , sign concordance = 0.92) (Fig. 4e). Thus, even if some non-linear effects exist between guides, the overall assumption of additivity holds broadly enough to infer many accurate effects. Analysis of the effects that appear to be visual outliers in the guide-pooled screen (Fig. 4e) showed that they arise from correlated noise rather than genetic interaction effects (Supplementary Note, section 4, Supplementary Fig. 3). As with the cell-pooled screen, the total number of significant effects was much lower in the 8,448 guide-pooled cells vs. the full conventional screen (5,836 vs. 95,526; q-value < 0.05), but this is expected because our conventional screen was by design larger and more highly powered overall to enable down-sampling analyses.
The guide-pooled screen was substantially more efficient than the conventional screen per experimental unit (cell), and FR-Perturb provided more accurate effect sizes than established methods. Around 2.5x more conventionally studied cells were needed to obtain the same number of significant effects as guide-pooled cells (Extended Data Fig. 3e). Globally, the effect size patterns learned from the same number of cells (8,448 cells) were much less noisy in the guide-pooled screen than in the conventional screen (adjusted Rand index of 0.45 vs. 0.35 when comparing clusters with those learned from the full conventional screen; Fig. 4d). Approximately twice as many conventional cells were required to learn effect sizes at the same correlation (Fig. 4e) or to attain the same AUPRC (Fig. 4f) as guide-pooled cells when comparing to a held-out validation set. This relative efficiency gain was consistent when varying the number of compared effects (Extended Data Fig. 3g) or when comparing effects on modules rather than on individual genes (Extended Data Fig. 4a) Moreover, the effect sizes inferred by FR-Perturb had substantially better validation accuracy than those from the two established inference methods in both the guide-pooled and conventional data (Fig. 4e,f, Extended Data Fig. 3h). Around 3,200 guide-pooled cells analyzed with FR-Perturb achieved the same validation accuracy as 36,000 conventional cells analyzed with existing approaches (Fig. 2f), leading to an approximately 10-fold cell count and cost reduction over existing experimental and computational approaches (Supplementary Note, section 5).
Guide pooling is the more impactful compression approach
We conducted a detailed comparison of the strengths and limitations of cell-pooling and guide-pooling relative to each other (Supplementary Note, section 6–7, Supplementary Fig. 4). Notably, the performance of cell-pooling does not scale with the number of cells per droplet, and the overall efficiency gains of cell-pooling stem from obtaining more non-empty droplets per channel (Extended Data Fig. 6). On the other hand, the performance of guide-pooling does scale with the number of guides per cell, with the best performance attained by cells with 4 or more guides (Extended Data Fig. 6). This suggests that guide-pooling has the potential to achieve even higher efficiency with a greater degree of overloading than we attained in our experiment.
The effectiveness of compressed Perturb-seq has important implications for existing Perturb-seq screens, each of which already has some overloaded droplets (cell-pooling) and multi-guide expressing cells (guide-pooling) by chance or by design1,2,13. While these cells/droplets are often discarded, our results suggest that these cells/droplets can contain even more signal than the single-guide/single-cell containing ones and thus should be retained. To illustrate this, we used FR-Perturb to analyze a Perturb-seq knock-out screen of 1,130 genes in mouse bone marrow derived dendritic cells30 (BMDCs). In this screen, 519,535 droplets containing a single cell were obtained, of which 33% contained more than one guide by chance. By stratifying cells by the number of guides and comparing the learned effect sizes from FR-Perturb with a held-out validation subset of the data with single guide perturbations, we show that the accuracy of the effect sizes scales with the number of guides per cell and is highest in cells containing three guides (Extended Data Fig. 7a). Thus, by retaining all cells with more than one guide, the sample size of the experiment could effectively be doubled compared to the conventional approach that discards these cells (Extended Data Fig. 7b).
Regulatory circuitry of the LPS response
We next leveraged the overall concordance of all perturbation data (conventional and compressed, knock-out (KO) and knock-down (KD)) to investigate the underlying regulatory circuitry of the LPS response. To maximize power, we merged droplets from the compressed and conventional screens together, then re-estimated all effects. There were 251,792 significant effects in the combined conventional and cell-pooled KO screen (131,161 effects in the combined conventional and guide-pooled KD), an increase of 16% (KD: 37%) over the conventional screen alone. We focused all subsequent analyses on effects from these combined screens.
Overall, the KO and KD screens were concordant, with most of the significant effects (FDR q-value < 0.05) attributed to relatively few (~5%) of the perturbations, each with widespread effects on many genes (Fig. 5a). As expected, there were substantially more significant effects in the KO compared to the KD screen (251,792 vs. 131,161 effects), consistent with larger effects of KO on the target gene’s activity31. Effects significant in both screens were highly correlated between the screens (; sign consistency = 0.99; Supplementary Fig. 5a–d). The perturbations did not lead to new global cell states, such that profiles from perturbed (one or more targeting guides) and unperturbed (control guide) cells spanned the same low-dimensional space (Fig. 5c). Thus, while many perturbations had significant and widespread effects, they did not yield radically altered phenotypic states, consistent with previous studies of this cellular response2.
Fig. 5. Analysis of knock-out and knock-down perturbation effects in the LPS response.

(a) Distribution of perturbed genes based on their number of significant effects () on downstream genes. (b) Distribution of downstream genes based on how many perturbed genes significantly affect their expression. (c) PCA of perturbed and control cells based on the expression of the top 2,000 most variable genes. Control cells (grey) contain a non-targeting guide only. Perturbed cells (red/blue) contain a guide for one of the following genes. Red: IKBKB, IKBKG, IRAK1, IRAK4, MAP2K1, MAP3K7, MAPK14, MYD88, RELA, TIRAP, TLR1, TLR2, TRAF6. Blue: CISH, CYLD, STAT3, TNFAIP3, TRIB1, ZFP36. Numbers in parentheses indicate percent variance explained by PCs. (d) Heatmaps of perturbation effect sizes (inferred with FR-Perturb) from the knock-out (left) and knock-down (right) screens. Rows: top 50 perturbed genes based on their average magnitude of effects on all downstream genes. Columns: top 2,000 downstream genes based on the average magnitude of effects of all perturbed genes acting on them. Rows and columns are clustered using Leiden clustering. Clusters are labelled based on their GO enrichment terms. All effects with are whited out. (e) (Left) Correlation of knock-out effect sizes (y-axis) between all pairs of perturbed genes (x-axis). Top and bottom gene pairs are labelled. (Top right) Graph of all perturbed genes that physically interact with XPR1 and/or KIDINS220, based on AP-MS data from Bioplex 3.046. Edges represent physical interaction. (Bottom right) Mean effects of perturbed genes from top right on P1-P4. (f) Analysis of genetic interaction effects. (Left) Effect sizes relative to control (y-axis) of cells containing 0, 1, or 2 guides (x-axis) within each perturbation module (lines connecting three dots). Modules with significant effects () are highlighted in color and labeled, with the expected effect of cells containing two guides in the module represented with a dotted line. Error bars represent standard errors obtained from bootstrapping. (Right plots) Violin plots of the mean effects of individual cells containing 0, 1, or 2 guides in the three perturbation modules with significant interaction effects. Dotted line represents the expected effect of cells with 2 guides. Two-sided p-values are computed from permutation testing.
We organized the perturbations and genes by clustering their effect size profiles (Methods), observing four broad co-regulated programs of downstream genes with correlated responses across the perturbations, and three broad co-functional modules of perturbations with correlated effects on downstream genes (Fig. 5d).
The four major co-regulated programs were present in both the KO and KD screens (Fig. 5d), spanning key aspects of the response to LPS: inflammation (P1; cytokine, chemotaxis and LPS response genes; Supplementary Fig. 5e,f); macrophage differentiation (P2; immune cell activation, differentiation, and cell adhesion genes); antiviral response (P3; type I interferon response genes); and ECM and developmental genes (P4) (Supplementary Table 2). Inflammation (P1) and the antiviral response (P3) are known to be regulated by LPS signaling through AP1/NF-kB and IRF3, respectively32, and were mostly anti-correlated in their responses to perturbation in our screen, consistent with reports that downregulation of the inflammatory response can lead to upregulation of type I interferon response33,34. Inflammatory signaling is known to lead to macrophage differentiation35, but almost all perturbations with significant effects on inflammation (P1) (in any direction) down-regulated macrophage differentiation (P2). This suggests that additional factors beyond inflammatory signaling mediate macrophage differentiation in response to LPS36.
Of the three major co-functional modules, KO/KD of the first module (M1) resulted in strong down-regulation of inflammation and macrophage differentiation (P1–2) and upregulation of the antiviral response and ECM/developmental genes (P3–4) (Fig. 5d). M1 was mainly composed of core TLR/LPS response genes and genes directly up- or downstream of the pathway32, including MYD88, IRAK1, IRAK4, RELA, TRAF6, TIRAP, IKBKB, IKBKG, TAB1, TANK, TLR1, TLR2, MAPK14, MAP3K7, FOS, JUNB, and CHUK. Given the known function of these genes, we expect that their KO/KD will lead to down-regulation of inflammation and macrophage differentiation (P1–2), as we indeed observed. Other genes in M1 previously shown to down-regulate TNF and the inflammatory response when knocked out26 included two LUBAC complex proteins (RBCK1 and RNF31), genes in the OST complex (DAD1, TMEM258) and ER transport (HSP90B1, SEC61A1, ALG2), and other genes with diverse functions (MIDN, AHR, PPP2R1A, ASH2L). M1 also included two additional ER transport genes not previously implicated in immune pathways (RAB5C, PGM3), highlighting the important role of N-glycosylation and trafficking in macrophage activation37.
KO/KD of the second co-functional module (M2) primarily resulted in strong downregulation of the antiviral program (P3), with weak/mixed effects on other programs. M2 comprised four genes known to be core components of the type 1 interferon response38 – STAT1, STAT2, TYK2, and IFNAR1 – for which downregulation of the antiviral program in response to their perturbation is expected.
KO/KD of the third and final co-functional module (M3) resulted in upregulation of inflammation (P1), downregulation of macrophage differentiation and the antiviral response (P2–3), and mixed effects on ECM/development (P4). M3 included many genes with known inhibitory effects on inflammation, including ZFP36, an RNA-binding protein that destabilizes TNF mRNA39, enzymes CYLD and TNFAIP3 involved in deubiquitination of NF-kB pathway proteins40,41, pseudokinase TRIB1 and ubiquitin ligase RFWD2 which are involved in degradation of JUN42,43, and RELA-homolog DNTTIP126. Other genes in M3 included transcription factors (MEF2C, FLI, and EGR1), chromatin modifiers (EHMT2, ATXN7L3), and kinases (CSNK1A1, STK11).
Interestingly, two of the M3 genes with particularly strong effects on all programs did not have prior immune annotations – XPR1, a retrovirus receptor involved in phosphate export, and KIDINS220, a transmembrane scaffold protein previously reported in neurons44. In the KO screen, this pair of genes had the fourth highest correlation of downstream effects among all perturbation pairs (Fig. 5e), following IRAK1/IRAK4, IRAK1/TRAF6, and IRAK4/TRAF6 which are all known to form a physical LPS signaling complex32. XPR1 and KIDINS220 have recently been shown to form a complex that is required for normal regulation phosphate efflux in certain cancer cells45. Furthermore, in AP-MS data46, XPR1 and KIDINS220 physically associate with each other and TNF receptor TNFRSF1A. Knockout of TNFRSF1A in our screen results in effects opposite to XPR1/KIDINS220 KO (Fig. 5e), suggesting a possible inhibitory effect of this complex on TNFRSF1A.
We experimentally validated several of the novel results described in this section, namely the effects of RAB5C, PGM3, XPR1, and KIDINS220 KO on the inflammatory response in LPS-stimulated THP1 cells, as measured by the secretion of IL6 (Methods). We found that RAB5C and PGM3 KO both led to a modest decrease (~0.85-fold) in IL6 secretion (consistent with our finding that KO of these genes led to downregulation of the P1 program), while XPR1 and KIDINS220 knock-out both led to a substantial increase (~2.6-fold) in IL6 secretion (consistent with our previous finding that KO of these genes led to upregulation of P1; Extended Data Fig. 8).
Guide pooling reveals second-order genetic interactions
Genetic interactions (non-additive effects) between two or more genes can in principle be inferred from cells containing two or more guides, which are generated by chance when transducing cells at low or high MOI (Fig. 4b). Here, guide-pooling can provide increased efficiency compared to the conventional approach, like in the first-order case (Supplementary Note, section 9).
We first attempted to estimate second-order interaction effects and their p-values from the guide-pooled screen and corresponding conventional KD screen by adding interaction terms to the perturbation design matrix (Methods). However, although we could generate point estimates of second-order effects2, none of these effects was significant in either screen due to insufficient power (Supplementary Fig. 6a), even with a lax significance threshold .
To increase power, we aggregated perturbations into modules defined by GO annotations (Supplementary Table 3a) and learned the overall impact of second-order interactions within and between each module on each gene program (Methods). Here, we define an interaction effect as the deviation from the sum of first-order effects for cells that contain any two perturbations from either the same module (intra-module interactions) or two different modules (inter-module interactions) (Methods). To ensure adequately sized groupings, we aggregated perturbations into 490 (possibly overlapping) modules each with at least 20 genes, such that any pair of perturbations in each module was represented in an average of 87 cells in the guide-pooled screen (conventional: 30 cells) (Supplementary Fig. 6b). We also constructed 30 non-overlapping modules by clustering the original 490 modules (Methods), resulting in module pairs among which we could compute inter-module interactions. To increase power, we grouped downstream genes by their program (P1–4) membership (Fig. 5d), computing mean effects on these four programs rather than on individual genes. The results from this analysis represent the extent of intra- and inter-module interactions on each key program.
We detected three co-functional modules with significant intra-module interaction effects on at least one program from the guide-pooled screen (Fig. 5f, Supplementary Table 3b), while we detected no significant interactions from the substantially larger conventional screen (even at ) (Supplementary Fig. 6c, Supplementary Table 3c). Two of the significant interaction effects – with genes for regulation of chromosome organization and antigen processing – had insignificant first-order effects on the antiviral program (P3), while having significant positive second-order effects. The third, TNFa signaling, had a significant negative first-order effect on the inflammatory/LPS program (P1) and significant positive second-order effect . This effect is consistent with the reported non-linear relationship between gene dosage and TNF signaling activity when comparing heterozygous versus homozygous KO mice for either TNF47 or the TNF receptor TNFRSF1A48. Interestingly, we did not observe any significant inter-module interactions from either screen (Supplementary Fig. 6d, Supplementary Table 3d,e), which may suggest that perturbations in different modules are less likely to interact with each other49,50.
Integrating Perturb-seq with genome-wide association studies
Because dysregulation of innate immune responses plays a key role in many human diseases51, we next asked whether the perturbation effects learned from our in vitro screens can help identify disease-relevant genes and processes. In vitro screens may be especially helpful for this aim given that many of the perturbed genes from our screens are under strong selective constraint in human populations (Supplementary Fig. 7a), making them challenging to directly connect to disease through genome-wide association studies52 (GWAS) due to fewer common variants in or around the gene53,54. To investigate this, we obtained summary statistics from GWAS of 64 distinct human diseases and traits (Supplementary Table 4a), including autoimmune diseases and blood traits, as well as non-immune traits/diseases (e.g. height, BMI, schizophrenia, type 2 diabetes). Using sc-linker55, we computed the overall heritability enrichment of these 64 traits/diseases in SNPs in/around genes comprising perturbation modules M1–3 (Methods). We observed significant heritability enrichment for M3 (genes that suppress the LPS response) for two blood traits (lymphocyte and neutrophil percentage), but did not observe significant enrichment for M1 (positive regulators of the LPS response) or M2 (genes involved in the antiviral response) for any traits (Supplementary Fig. 7b).
Instead, we hypothesized that if a perturbed gene is important for disease, then disease heritability may be enriched near the downstream genes it affects12,56. To test this hypothesis, we constructed two “perturbation signatures” for each perturbed gene that include all genes that are significantly upregulated (“negative” targets) or downregulated (“positive” targets) by its KO/KD. We retained signatures with at least 100 genes, resulting in a total of 1,634 perturbation signatures from both the KO and KD screens. We also constructed signatures corresponding to the gene programs P1–4 (Fig. 5d). As above, we used sc-linker to test for disease heritability enrichment for each signature/phenotype pair (Methods).
23 signatures associated with 16 perturbed genes had significant heritability enrichment scores for at least two phenotypes (p-value < 0.001). Meanwhile, 7 phenotypes that reflect immune or blood traits (inflammatory bowel disease, eczema, rheumatoid arthritis, asthma, primary biliary cirrhosis, and eosinophil percentage) had significant scores for at least two perturbation signatures (Fig. 6a; Supplementary Fig. 7c,d; Supplementary Table 4b,c). As an important negative control, no non-immune/blood traits had any significant enrichment. Most of the significant signatures (15/23) were from the KO screen, suggesting that the expression effects from KO are more suited for this analysis (either because they are more disease-relevant or more powered due to capturing more effects). Among the downstream programs P1–4, we observed significant enrichment from only P2 on three immune traits: inflammatory bowel disease, eczema, and primary biliary cirrhosis (Supplementary Fig. 7b).
Fig. 6. Integration of population-genetic screens with Perturb-seq.

(a) Heritability enrichment scores of signatures comprising genes significantly modulated by perturbations (rows) across human traits (columns), computed using sc-linker55. “Pos” indicates the set of genes whose expression changes in the same direction as the perturbed gene (i.e., downregulated by the perturbation), with the opposite applying to “neg”. Displayed are all perturbation signatures and traits with at least 2 significant () effects. Non-significant scores are greyed out. (Barplot) Probability of loss-of-function intolerance54 (pLI) of the corresponding perturbed gene. (b) Schematic of eQTL integration analysis, aiming to test whether trans-regulatory relationships learned from Perturb-seq are also present in eQTL studies. For all gene pairs in which gene exerts an effect on gene (i.e., has a significant knock-down effect in our Perturb-seq screen), we would expect that gene and gene are enriched for cis-by-trans eQTLs. (c) Using data from an eQTL study closely matching our cell type and treatment27, shown is the probability of observing significant cis-by-trans eQTLs among the top 15 perturbed genes from our knock-down screen and their affected downstream genes (red) compared to random downstream genes (grey). (d) Enrichment of significant cis-by-trans eQTLs among various sources of gene-gene pairs: significant KO/KD effects (representing significant gene-gene effects from our KO and KD screens, respectively), curated transcription factor (TF) and target gene pairs65, and the top 1,000/10,000 most co-expressed gene pairs (based on correlation of expression across samples) from the eQTL dataset. Enrichment is computed relative to random trans genes for each cis gene, then averaged over all cis genes. (e) Selective constraint on trans genes from (D) plus all significant cis-by-trans eQTLs from the Fairfax dataset. Each point represents a cis gene, while the x-axis represents the proportion of the trans genes for each cis gene that are under selective constraint (determined as having a pLI > 0.5). Boxplots represent the median and first/third quartile of points, while the bounds of the whiskers represent 1.5*IQR.
Most of the significant signatures (17/23) were from genes in core LPS and TLR signaling pathways that fall into perturbation module M1 (even though M1 did not exhibit any direct heritability enrichment itself; Supplementary Fig. 7b): TRAF6 (positive), TLR7 (positive), TLR2 (positive), TLR1 (positive), TIRAP (positive), TAB1 (positive), MYD88 (positive), MAP3K7 (positive), IRAK4 (positive), IRAK1 (positive), and IKBKG (positive). Other significant signatures include HSP90B1 (positive), an ER transport gene important for innate immunity57 that is co-functional with the core LPS genes (Fig. 5d); FADD (negative), a pro-apoptotic gene downstream of LPS signaling that serves for negative feedback32; MYC (negative), an oncogene with known immunosuppressive effects58,59; and poorly characterized pseudogene HLA-L. The two remaining significant signatures are for genes whose functions are not previously associated with the immune system, including APLP1 (an amyloid beta precursor-like gene primarily involved in brain function that interestingly contains a missense variant associated with severe influenza60) and GPAA1 (involved in anchoring proteins to the cell membrane). Thus, by leveraging gene-gene links learned from our screens, we were able to identify disease-relevant genes that we were underpowered to detect through direct heritability analyses (Discussion).
To complement our results that focus on common diseases and variants, we also computed the enrichment of Mendelian immune disease genes among the same signatures derived from our screens from above. We found significant enrichment in a similar number of signatures, particularly those with strong effects on the antiviral response (Supplementary Note, section 10, Supplementary Fig. 8).
Perturbation effects do not concord with trans-eQTLs
Trans-genetic gene regulation (i.e., regulation of gene expression distal to the given SNP) has been proposed as a primary mediator of genetic effects on human disease61. Trans-genetic gene regulation can be studied through either population-level genetic data (via expression quantitative trait loci (eQTL) studies62,63), or through experimental perturbation of gene expression12, such as the screens conducted in our study. Although both types of data can in principle be used to learn the same trans effects, their consistency with each other has not been empirically evaluated.
We therefore compared gene-gene regulatory links between our Perturb-seq screen and a trans-eQTL analysis in primary patient-derived monocytes treated with LPS27 , closely matching our cell line. For validation, we repeated this analysis using a much larger trans-eQTL dataset (eQTLGen; ), although in a model system less similar to ours (whole blood samples). We define a gene-gene regulatory link in eQTL studies based on cis-by-trans colocalization, where a cis-eQTL for gene is also a trans-eQTL for gene via a (presumed) trans-regulatory effect of gene on gene (Fig. 6b). Here, we assume that a perturbation of a cis-eQTL on the expression of gene is analogous to the experimental KD in our system. We used coloc64 to compute the posterior probability of cis-by-trans colocalization, while accounting for linkage disequilibrium between SNPs (Methods). To determine whether the regulatory links learned for a given perturbed gene from Perturb-seq are reflected in the eQTL analysis, we compared the proportion of downstream genes of gene in Perturb-seq that colocalize with gene in the eQTL study, , with the proportion of random expressed genes that colocalize with (Methods).
Surprisingly, was slightly lower than for individual perturbed genes (Fig. 6c, Supplementary Table 5), as well as when aggregating across all perturbed genes (Fig. 6d). Moreover, we observed no relationship between either the significance or magnitude of the effect of gene on gene and (Supplementary Fig. 9a). We observed similar negative results when obtaining gene-gene links from our KO data or from a curated list of transcription factor-target gene pairs65 (Fig. 6d). Using an alternative way of quantifying gene-gene links in eQTL studies that does not make assumptions about the number of causal variants (i.e., bivariate Haseman-Elston regression to estimate genetic correlation of expression66; Methods) yielded similar results (Supplementary Fig. 9b,c). We observed similar negative results when taking cis-by-trans eQTLs from eQTLGen (Supplementary Fig. 10).
Conversely, we did observe significant enrichment of cis-by-trans eQTLs in gene pairs co-expressed in the same eQTL study (Fig. 6d), as has been observed in other trans-eQTL studies62. Notably, co-expression in eQTL datasets is dominated by environmental effects rather than genetic effects67. Thus, given that the two effects are independent across samples, we would not ordinarily expect the most strongly co-expressed genes to be enriched for cis-by-trans eQTLs, suggesting that they may be confounded in part by unmodelled technical artifacts or inter-cellular heterogeneity (Supplementary Note, section 11). We also observed that the level of negative selection on the trans gene mirrored the patterns of cis-by-trans eQTL enrichment (or lack thereof) we observed in the previous analyses (Fig. 6e), suggesting that our power to detect cis-by-trans eQTLs was affected by selection-induced depletion of SNPs affecting the trans genes54,68 (Supplementary Note, section 12).
DISCUSSION
Here, we evaluated a new approach for conducting Perturb-seq based on generating composite samples, which involves either overloading microfluidics chips to generate droplets containing multiple cells (cell pooling), or infecting cells at high MOI so that each cell contains multiple guides (guide pooling). We also propose a new method, FR-Perturb, to estimate perturbation effect sizes from composite samples, which increases power by estimating sparsity-constrained effects on latent gene expression factors rather than on individual genes. We tested our approach by perturbing 598 immune-related genes in a human macrophage cell line. We found that our experimental approaches of cell pooling and guide pooling, combined with the use of FR-Perturb to infer effect sizes, lead to large cost reductions over conventional Perturb-seq while maintaining the same accuracy. Guide pooling also significantly increases power to detect genetic interaction effects and reduces the number of cells needed for screening.
In our study, we report that cell-pooling led to a 4–20 fold cost reduction, while guide-pooling led to a 10-fold cost reduction, over existing approaches (Supplementary Note, section 5). Both these approaches reduce costs due to RNA library preparation without altering the sequencing step of scRNA-seq. Thus, they can in principle be paired with approaches that increase the efficiency of sequencing via new technologies69 or targeted sequencing70, resulting in further improvements to the efficiency of Perturb-seq. Concurrent results also demonstrate the power of compressed screening with bio-chemical perturbations in high-fidelity cellular model systems (Mead et al.71, companion manuscript).
Inference with FR-Perturb leads to substantially improved out-of-sample validation accuracy over conventional gene-by-gene methods (e.g., elastic net, negative binomial regression) in both conventionally generated data and compressed data. FR-Perturb is thus useful for inferring effects in any type of Perturb-seq screen, even conventional screens that do not adopt our proposed experimental changes. The improved performance of FR-Perturb in both conventional and compressed settings likely stems from perturbation effect sizes being inferred on latent gene expression factors that aggregate many co-expressed genes, thereby denoising the expression counts of individual genes which are especially noisy/sparse in single-cell data. However, the performance of FR-Perturb is likely to suffer when inferring effects for perturbations that cannot be well-approximated by these factors (due to idiosyncratic effects of the perturbations21).
Cell pooling and guide pooling are complementary approaches with different strengths and limitations. Unlike cell pooling, guide pooling has the drawbacks that it requires that nonlinear interaction effects do not systematically bias phenotypes (though not all interaction effects will impart bias; Supplementary Note, section 2), and it potentially suffers from cellular toxicity caused by multiple viruses infecting each cell and/or multiple double stranded breaks. Meanwhile, unlike guide pooling, cell pooling has the drawbacks that it requires increased sequencing depth per channel to account for more non-empty droplets, and it loses per-droplet signal due to dilution of effect sizes (Supplementary Note, section 8). Due to the latter fact, cell pooling requires many more cells than guide pooling to achieve the same performance, which can be prohibitive in certain settings where cell count is limited8,13. Because guide pooling performs best with high guide number per cell (4 or more), whereas cell pooling does not perform well with high cell count per droplet, we posit that guide pooling (but not cell pooling) can be readily scaled up to very compressed designs (in which case the use of knock-down over knock-out and Cas12/13 over Cas9 may be desirable to avoid cellular toxicity), likely leading to even larger efficiency gains than we observed in our screens. To aid in the design of future experiments, we also conducted simulations showing the performance of compressed Perturb-seq when varying factors such as sequencing depth and guide efficiency, finding that is it is robust in many different scenarios (Supplementary Note, section 13, Extended Data Fig. 9, Supplementary Fig. 11).
An additional key advantage of guide pooling over cell pooling is that guide pooling naturally allows for the study of higher-order interaction effects. In our study, we were underpowered (even with guide pooling) to detect second-order interaction effects between individual gene pairs. However, we detected significant intra-module interaction effects from the guide-pooled but not conventional screen, serving as a proof-of-concept that such signal can be detected in the guide-pooled screen, and may be further probed in more powered future experiments. The efficiency gains brought about from guide pooling can in theory counteract the exponential growth of gene combinations (given that various assumptions are satisfied), potentially making it the only tractable way to systematically study higher-order interaction effects (Supplementary Note, section 9). To aid in the design of future experiments, we conducted simulations showing the number of cells needed to learn second-order interaction effects at various levels of guide pooling, finding that guide pooling can dramatically reduce the number of cells needed to learn a given number of second-order interaction effects (Supplementary Note, section 14, Extended Data Fig. 10).
By integrating data from GWAS, our screens highlighted perturbed genes with downstream genes enriched for disease heritability. Many of these perturbed genes are under strong selective constraint and would require up to millions of samples to detect in GWAS72. Thus, our analysis represents a potential way to circumvent the issue of negative selection removing GWAS signal from some large-effect disease-relevant genes, a key challenge for biological interpretation of common-variant GWAS.
Gene-gene effects learned from our Perturb-seq screens were not enriched for cis-by-trans eQTLs in a closely matched cell type and treatment. Many possible explanations exist for this observation, including (1) insufficient power to detect trans-eQTLs in the eQTL dataset, (2) biological differences between our cell line and primary monocytes used in the eQTL study, (3) large differences in the magnitude of perturbation between experimental KO/KD and eQTLs, and (4) confounders in the eQTL dataset (Supplementary Note, section 11). Explanation (1) can in theory be addressed with larger trans-eQTL studies62, though we observed similar negative results when replicating our results in a large trans-eQTL dataset (eQTLGen). Such studies often suffer from issues with confounding/intercellular heterogeneity, as evidenced by very low reported out-of-sample replication accuracy and substantial overlap (>50%) of detected trans-eQTLs with variants known to influence cell type proportion62. Meanwhile, single-cell eQTL studies73 can potentially address explanation (4), though such studies suffer from low power relative to sample size (~1,000 significant trans-eQTL effects detected from ~1.2 million cells73 versus ~200,000 trans perturbation effects detected from ~100,000 cells in our screen). We propose that our compressed screen is a powerful tool to learn trans-effects on gene expression, while additional work is needed to fully reconcile the differences between population-level genetic screens and experimental perturbation screens.
Methods
EXPERIMENTAL PROCEDURES
Cell culture and stimulation
THP-1 cells (ATCC, TIB202) were cultured in RPMI medium (ATCC, 30–2001) supplemented with 10% FBS (ATCC, 30–2020) and 0.05mM 2-mercaptoethanol (Sigma Aldrich, M7522). Cells were maintained between 0.8 and 2 million cells per milliliter.
Cell lines for knockout (KO) and knockdown (KD) screens were engineered with lentiviral vectors containing Cas9 (pxpr311) and dCas9-KRAB (pxpr121), respectively. Viruses were prepared using a previously published protocol (https://portals.broadinstitute.org/gpp/public/dir/download?dirpath=protocols/production&filename=TRC%20shRNA%20sgRNA%20ORF%20Low%20Throughput%20Viral%20Production%20201506.pdf) and concentrated by centrifugation in a column with a cut size of 100kDa (MilliporeSigma UFC903096). Cells were transduced by spinfection as previously described (https://portals.broadinstitute.org/gpp/public/resources/protocols).
THP-1 cell lines were infected with sgRNA libraries (described below) at a multiplicity of infection (MOI) specific for each guide-pooled experiment. 12 hours after spinfection, cells and media were diluted 1:10 and cells were allowed to recover for 48h. Cells were selected with puromycin (2 g/mL) for four days. The selected cells were differentiated into macrophages by stimulation in 20ng/mL phorbol 12-myristate 13-acetate (Sigma Aldrich, P8139–1mg) for 24 hours. Cells were then allowed to rest in normal culture medium for 48 hours before stimulation in medium containing 100ng/mL LPS (MilliporeSigma, L4391–1mg) for 3 hours.
Guide library production and validation
sgRNAs for the perturbed panel of genes (described below) were designed using the Crispr-Pick tool from the Broad Institute. Four distinct sgRNAs were designed for each perturbed gene. In addition, 500 non-targeting sgRNAs and 500 safe-targeting sgRNAs (i.e., guides targeting intergenic regions of the genome) were included. Oligonucleotide libraries were synthesized by Twist Biosciences, then amplified and inserted into a CROP-Seq vector4 with sgOpti scaffold (Addgene #106280) via Gibson assembly. Cloned libraries for KO, KD, and control sgRNAs (non-targeting and safe-targeting) were sequence-validated as previously described (https://portals.broadinstitute.org/gpp/public/dir/download?dirpath=protocols/production&filename=cloning_of_oligos_for_sgRNA_shRNA_nov2019.pdf). Viral libraries were produced as described above (without concentration), and an MOI was determined by transfecting cells with scaled dilutions of the virus covering a 100-fold dynamic range and quantifying survival rate after selection.
Conventional Perturb-Seq, cell-pooling, and guide-pooling (scRNAseq & dialout library production)
For conventional screens, the infected (MOI 0.25) and stimulated THP-1 cell suspension was prepared for droplet generation according to the manufacturer’s suggested protocol (10x Genomics, CG00053 Rev C). Channels aiming to recover 5,000–10,000 cells were loaded on the 10x Chromium Controller and the protocol was followed according to the manual for Chromium Next GEM Single Cell 3ʹ Reagent Kits v3.1 (CG000315 Rev C).
For cell-pooling (MOI 0.25), the standard 10x single cell 3’ RNAseq protocol (Chromium Next GEM Single Cell 3’ GEM, Library & Gel Bead Kit v3.1 PN-1000121) was run according to manufacturer’s recommendation, except the concentration of cells was increased to co-encapsulate multiple cells per droplet (250,000 cells loaded per channel).
For guide-pooling, cells were infected at an MOI of 10 before selection and stimulation, or were left to rest for 2 days after initial infection before infecting a second time at an MOI of 10 before selection and stimulation (Supplementary Fig. 2). High MOI cells were loaded into droplets as in the conventional screens.
After the generation of double-stranded cDNA, part of the whole transcriptome amplification (WTA) product was set aside for targeted amplification to recover the perturbation barcode. 10ng of WTA from each channel were input into 8 cycles of PCR (primer 1 CTACACGACGCTCTTCCGATCT; primer 2 GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTTGTGGAAAGGACGAAACACC). The sample underwent a 1x AMPure XP Reagent SPRI clean (Beckman Coulter A63881) and was amplified for another 9 cycles with 8bp indexed PCR primers and purified with a 0.7x SPRI clean (primer 1 AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTC, primer 2 CAAGCAGAAGACGGCATACGAGATGTCGAGCAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT).
Guide effect validation screens
For guide effect validation, two guides (out of four) were chosen for 6 targets: MYD88, STAT1, RAB5C, PGM3, XPR1 and KIDINS220, as well as two of the non-targeting controls. RAB5C, PGM3, XPR1, and KIDINS220 represent novel regulators of the inflammatory response, while MYD88 and STAT1 were included as positive controls. The two guides for each target were selected by computing the pairwise correlation of effect sizes of the four individual guides on all genes, then taking the pair with the highest correlation. Single guides were cloned into the CROP-seq vector as previously detailed. 2 million cells were infected for each guide. Cells were then selected with 4ug/mL puromycin for 2 days, then expanded in culture for 10 days. Cells infected with the first guide targeting XPR1 all died, so that condition was removed from the validation experiment. THP1s were differentiated into macrophages using PMA as in the main screen. 3 wells of a 24 well plate were seeded for each guide, with 250,000 cells per well. After 24 hours in PMA, the medium was changed for fresh medium, and cells recovered for 2 days. Cells were then stimulated with 250uL of medium containing LPS (100ng/mL) for 8 hours, then medium was collected, spun at 1,000g for 2 minutes to remove cell debris, and stored at −80C. 2 extra wells of cells infected with non-targeting guides received fresh medium as a non-stimulated control. ELISAs were conducted following the manufacturer protocol (https://www.abcam.com/ps/products/178/ab178013/documents/Human-IL-6-ELISA-kit-protocol-book-v4a-ab178013%20(website).pdf)
COMPUTATIONAL PROCEDURES
Selecting genes to be perturbed
A set of perturbed genes was compiled from several sources (Supplementary Table 1). These included: a manually curated list of 35 canonical LPS response genes; the top 100 genes from a previous genome-wide CRISPR screen for regulation of TNF expression after LPS stimulation26; 100 genes identified as being a cis eQTL target of SNPs that were (in total) associated with trans eQTL effects for at least 4 downstream genes in primary monocytes treated with LPS27; 95 genes near high confidence variants in IBD GWAS loci74; 108 genes associated with Mendelian disorders identified by search for “bacterial infection” in the Online Mendelian Inheritance in Man (OMIM) database75 and 115 Mendelian genes similarly identified by “NF-kappa-b” search; and 173 genes reported in studies identified by a GWAS Catalog76 search for “infection” with diseases/traits related to liver disease and HIV-1 infection excluded.
The (perhaps surprisingly small) intersections between gene lists from these sources is depicted in Supplementary Fig. 1. The final list of 598 perturbed genes was obtained by intersecting genes expressed in THP-1 cells with the combined list of 758 genes from all sources.
Generating expression and perturbation design matrix
Starting with raw Illumina BCL files from the sequencing output, the “cellranger mkfastq” command with default parameters (from the 10x CellRanger tool v6.0.1; https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest) was used to generate FASTQ files. The “cellranger count” command with default parameters was used to align the expression reads to the GRCh38 build of the human transcriptome and generate a gene expression count matrix (see below for details on normalization of expression counts).
To generate the droplet by perturbation design matrix, paired-end reads (in FASTQ format) containing a droplet barcode and UMI on read 1 and sgRNA sequence on read 2 were aligned using Bowtie2 as follows. Read 2 reads were aligned to a reference constructed from the labeled sgRNA sequences using the --local option with default parameters, which performs local read alignment. Then, using a custom script, droplet barcodes were matched to the mapped guides for each paired-end read. A guide was called as “present” in a droplet if there were at least 5 UMIs for each droplet barcode / guide barcode pairs.
Inference using FR-Perturb
From the sequencing output of each of our Perturb-seq experiments, two matrices were directly generated (see above):
raw gene expression count matrix , where is the number of droplets and is the number of sequenced genes.
perturbation design matrix , where is the number of droplets and is the total number of perturbed genes. Here, represents a binary indicator variable for whether droplet contains a guide targeting gene (we discuss below how we collapse multiple guides for the same gene). also includes two additional columns corresponding to the presence of a non-targeting control guide and safe-targeting guide, respectively. Cells containing a non-targeting guide are treated as “control” cells (see below), while cells containing a safe-targeting guide are used to test for general effects of genome-targeting guides.
From these data, a effect size matrix is estimated, where represents the log fold change of the expression of gene relative to control expression when gene is perturbed. Two slightly different versions of FR-Perturb were formulated to learn from and generated from cell and guide pooling, respectively, as follows.
Version 1: Composition in expression space (for cell pooling).
This scenario arises from cell pooling. The relationship between , and in a given droplet is modeled as:
| #(1) |
where is a vector of length corresponding to the expression counts of all genes in droplet is the number of guides contained in droplet (used as a proxy for the number of cells in the droplet), is a binary scalar indicating whether cell contains a guide for gene is a vector of length indicating the expected control expression counts of all genes, and is a vector of length indicating the fold-change of expression relative to control expression for cells containing a guide for gene (with representing the log fold-change). Note that the symbol here is used to distinguish fold-changes from log fold-changes, since the latter units are more commonly used to report effect sizes on gene expression. Conceptually, this model reflects the fact that expected expression measured in a droplet containing cells is the average of the expected expression counts of the individual cells in the droplet (where the latter quantity can be expressed as for cells containing guide ).
In practice, it is advantageous to model the measured expression in each droplet as the geometric rather than arithmetic mean of expression of the constituent cells. Simulations with real cells show that the arithmetic versus geometric mean of expression across multiple cells are very similar (Supplementary Fig. 12a), but modeling expression counts in a droplet as the latter enables us to perform inference in the space of log fold-changes rather than fold-changes. The former is symmetric around zero (whereas the latter is not) and thus leads to balanced inference of up- versus down-regulation.
Thus, Equation (1) is rewritten as follows:
| #(2) |
Equation (2) can be expressed simply in matrix form as , where each row of , equals , and is with rows normalized to sum 1. In order to infer , is transformed into by taking the of all gene expression counts and subtracting from each row of (where represents the average of all genes in cells containing only non-targeting control guides). A pseudocount of 1 is included because the sparse nature of gene expression counts prevents directly taking their logarithm.
Next, the factorize-recover algorithm is applied to and to infer . In the first “factorize” step of factorize-recover, sparse factorization is applied to alone using sparse PCA, which produces left factor matrix and right factor matrix . is a hyperparameter that controls the rank of . In the second “recover” step, sparse recovery is used to learn matrix from the following regression model: , using LASSO applied to each column of (so that one column of is learned at a time). By multiplying by obtained from the factorize step, a matrix is obtained, which is an estimate of .
In practice, the magnitude of elements of was strongly correlated with overall expression level of the downstream gene in control cells. This correlation changed (but was not removed) when varying the arbitrary pseudocount of 1 and/or scale factor of 10,000, suggesting that it was an artifact arising from log-transforming lowly expressed gene expression counts77. Indeed, simulations show that the magnitude of effects estimated with FR-Perturb had a negative bias that scaled with the expression level of the downstream gene, with the largest biases observed for the most lowly-expressed genes (Supplementary Fig. 12c).
This bias was removed with the following heuristic correction. First, LOESS was used to fit a curve to the plot of effect size magnitude vs. expression level in control cells for all entries of . Next, all effect sizes were scaled based on the ratio of their fitted effect size magnitude from LOESS and the fitted effect size magnitude of genes with the highest expression counts (log(average TP10K) > 2). This procedure removes the global relationship between effect size magnitude and expression level of the downstream gene, while preserving heterogeneity in the average magnitude of effect sizes on individual downstream genes. In simulations, this procedure produced much less biased effect size estimates than when not scaling (Supplementary Fig. 12b,c).
Version 2: Composition in log fold change effect size space (for guide pooling)
For guide pooling data, the relationship between , , and in a given droplet is modeled as:
| (3) |
The only difference between Equation (2) and Equation (3) is the absence of the normalizing factor in front of the second term of the right side of Equation (3). Inference to learn is performed as in Version 1, with the only difference that the rows of are not normalized to have a sum of 1.
Covariates
Covariates corresponding to the proportion of mitochondrial reads, the total read count per cell, and cell cycle state (as determined by the CellCycleScoring function from the Seurat R package78) were accounted for when estimating effect sizes using FR-Perturb, by regressing the covariates out of the expression matrix according to the linear model . Here, represents the normalized expression matrix (where is the number of cells and is the number of sequenced genes), represents the covariate matrix including an intercept term (where represents number of covariates with all covariates centered to mean 0), and represents the fitted matrix of covariate effects on gene expression. All downstream inference was performed on the residual matrix .
Hyperparameters for FR-Perturb
The spams R package79 was used to perform the steps of factorize-recover, including sparse PCA and LASSO. Three hyperparameters are set in FR-Perturb: the rank of , a tuning parameter for sparse PCA during the factorize step (which is the solution of so that ), and a tuning parameter for LASSO during the recover step (which is the solution of so that ). These were set based on maximizing cross-validation as , and . Analysis results were not especially sensitive to different values of , and (Supplementary Fig. 12d–f).
Permutation testing for significance
Permutation testing was used to obtain two-tailed p-values for elements of . To generate an empirical null distribution for each element of , samples were permuted (i.e., rows of ) and was re-inferred using FR-Perturb for each permutation. Permuting rows of has no impact on the factorize step, since this step does not involve (and the alternative approach of permuting rows of does not affect the individual factors). Thus, only the recover step was performed and was estimated for each permutation, followed by multiplying the null by obtained from the factorize step to obtain the null estimate. In addition, to reduce computational cost, only 500 permutations total were performed. For entries of that had p-value = 0 based on these 500 permutations, a skew-t distribution was fit to the empirical null distribution for each entry using the selm function from the sn R package, and p-values were then re-computed for these entries from the fitted distribution. False discovery q-values were computed using the Benjamini-Hochberg procedure applied to the p-values for all entries of .
Inference using negative binomial regression
Using the glmGamPoi R package80, was inferred by separately running differential expression analysis for each perturbation (i.e., column of ), where the two groups being compared were droplets containing only non-targeting control guides and droplets containing a guide for the perturbed gene of interest. For droplets containing multiple guides, other guides present in the droplet were ignored when forming these groups. Analytic p-values and false discovery q-values were obtained for all effect sizes from the method output.
Inference using elastic net
Using the spams R package79, the same elastic net inference procedure proposed in Dixit et al.2 was used to infer from the following models: for version 1, and for version 2 from above with and (where elastic net finds the solution to for each column of ), matching the values used in Dixit et al. Other values for the parameters yielded similar results (Supplementary Fig. 12g). P-values for all effect sizes were obtained by permuting the rows of a total of 10 times and re-estimating to generate a null distribution across all values of , matching the procedure used in Dixit et al.
Selecting optimal guide combination for each gene
Four distinct sgRNAs were generated for each perturbed gene. When inferring effect sizes, guides were aggregated by perturbed gene to increase sample size and simplify downstream analyses. When generating the perturbation design matrix , a cell containing any guide for the gene was labelled as receiving a perturbation for the gene. However, sgRNAs have varying efficiency at KO or KD their target gene, and including guides that do not work will add noise to the effect size inference. To retain only sgRNAs that had measurable effects on their target gene, we retained guides with concordant effect size estimates across random sample-wise splits of the data (i.e., the subset of guides to the same gene showing maximal concordance).
Specifically, let represent the index of a given perturbed gene, so that corresponds to the column of that indicates which cells received perturbation , and corresponds to the column of that indicates the effects sizes on all genes’ expression from perturbing gene . For each , 15 different version of were generated, corresponding to all possible subsets of the 4 guides. For each version, any cell receiving a guide within the given subset of guides is labelled as containing a perturbation for the gene, while the remaining guides are ignored. Only in was modified and the remaining columns were kept the same. Next, the dataset of interest was randomly split in half by samples (cells). FR-Perturb was used to infer effect sizes for all perturbed genes within each half. Then, the of was computed between the two halves (restricting to only effects with an FDR q-value < 0.2), and the specific guide subset that produced that highest was retained. The same procedure was repeated for each to learn the optimal guide combination for each perturbed gene.
Simulations
Perturb-seq datasets were simulated at various levels of overloading using real expression counts and perturbation effect sizes estimated from our data.
Simulating cell-pooled data.
To simulate expression data for droplets containing cells each, the expression of cells (each containing 1 guide) were first simulated by randomly sampling control cells from our experiment, and scaling their expression counts by the fold change effect sizes of a given perturbed gene (estimated from our conventional knock-out Perturb-seq screen). A 10% probability of receiving a control guide (i.e., no change in expression) was simulated to match the proportion of control guides in the real data. Next, the expression counts of cells were randomly averaged at a time to create cell-pooled data.
Simulating guide-pooled data.
To simulate expression data for cells containing guides each, perturbed genes were randomly selected for each cell, and the expression of a randomly selected control cell was then scaled by the product of the fold change effect sizes of the perturbed genes. As before, a 10% probability of receiving a control guide was simulated.
Clustering and dimensionality reduction
For Fig. 5c, dimensionality reduction was performed using PCA on the expression counts of all cells, where the expression values of each gene are scaled and centered to mean 0 and variance 1.
The rows and columns of Fig. 5d were clustered using Leiden clustering81. First, the Euclidian distance between all pairs of genes was calculated by their perturbation effect sizes, and the FindNeighbors function from the Seurat R package78 was used to compute a shared nearest neighbor graph from these distances (), followed by the FindClusters function to perform Leiden clustering on the graph with resolution parameter = 0.5, selected by visual inspection of the resulting clusters. GO enrichment analysis of the genes in the resulting clusters was performed with the ClusterProfiler package82 with gene sets obtained from the C2 (curated gene sets) and C5 (ontology gene sets) collections of the MSigDB83.
Learning second-order effects for individual perturbation pairs
Second-order interaction effects on gene expression in cell with multiple guides were modeled as:
Here, is a vector of length corresponding to the log expression counts of all genes in droplet and are binary scalars indicating whether cell contains a guide for gene and/or gene is a vector of length indicating the expected control expression counts of all genes, is a vector of length indicating the first-order effect size of guide on the expression of genes, and is a vector of length indicating the second-order effect size of guides and on the expression of genes. In matrix form, the above can be represented as:
where each row of equals is an indicator matrix for whether each cell contains any of perturbation pairs, and is a matrix of second-order interaction effects. is known from estimating first-order effects previously, which enables the following equation to be written:
where . Finally, is estimated using FR-Perturb in the exact same manner as To reduce the large size of , only perturbation pairs that were present in a minimum of 5 cells were included.
When estimating the significance of entries of , the uncertainty in both and must be accounted for, since the latter depends on the former. Thus, when generating a null distribution for the entries of , the rows of both and were permuted and was re-estimated for each permutation.
Learning second-order effects for perturbation modules
Intra-modular interactions.
A second-order intra-modular interaction effect was estimated for each co-functional perturbation module (i.e., group of perturbed genes) on each co-regulated gene program (i.e., group of downstream genes) as follows. For each pair of and , cells were partitioned into three sets:
Control set. Cells containing only non-targeting control guides or guides for genes without significant effects on . The latter group of guides is included to increase sample size, and all these guides are collectively referred to as “control guides”.
First-order set. Cells with exactly one guide in , with remaining guides in the cells falling into the “control guide” set.
Second-order set. Cells with exactly two guides in , with remaining guides in the cells falling into the “control guide” set.
A mean expression value for was computed for each set ( and respectively) as the average standardized expression of all genes in among the cells in the set, with covariates corresponding to read count per cell, percent mitochondrial reads, cell cycle state, and number of guides per cell regressed out of the expression matrix, and expression standardized to mean 0 and variance 1. The effect size of the first-order set was computed as and the interaction effect size of the second-order set as . P-values for all interaction effects were computed by permuting the set membership labels of all the cells and recomputing , and for the permuted sets. Standard errors for all interaction effects were computed via bootstrapping, by resampling cells from each of the sets without changing their labels.
Inter-modular interactions.
Inter-modular interaction effects were computed using a similar approach as above. The 490 total modules were first reduced into 30 disjoint modules using Leiden clustering of a shared nearest neighbor graph defined based on the number of genes shared between gene sets. For two co-functional modules and , the first order effects and were computed in the same manner as above. The second-order set was defined as cells with at least one guide from each of and , with the remaining guides in the cell falling into the “control guide” category, as defined above. The mean expression of the second-order group is . The interaction effect is defined as and p-values and standard errors were estimated using permutation testing and bootstrapping, respectively.
Heritability analyses
Sc-linker55 was used as previously described to compute a disease heritability enrichment score for each gene set constructed from the KO and KD perturbation effect sizes or perturbation modules and gene programs. Using sc-linker, SNPs were first linked to genes using a combination of histone marks from the Epigenomics Roadmap84 and the activity-by-contact strategy85, then an enrichment score was computed for the SNPs based on the heritability enrichment of the SNPs obtained from stratified LD score regression (S-LDSC86,87).
More specifically, for each gene set , a set of weights between 0 and 1 was constructed for each SNP based on the confidence of them influencing any gene in , following the procedure described in Jagadeesh et al.55 using activity-by-contact scores88 and the Epigenomics Roadmap histone marks84 for whole blood samples. For gene sets defined from membership in perturbation modules (M1–3) or gene programs (P1–4) (Supplementary Table 2), modules/programs were merged between the KO and KD screens. For gene sets defined based on perturbation effects, each gene was weighted by the effect size of the perturbation on the gene, normalized to lie between 0 and 1. A set of weights was also constructed, representing the confidence of the SNP influencing any gene across the genome. Next, heritability enrichment estimates and were computed for each and , respectively, using S-LDSC86,87. Here, (where represents the squared effect size of SNP on the phenotype and represents the total number of SNPs), and . Conceptually, represents the fraction of the total genetic effect on the phenotype attributed to SNPs in , while represents the effective fraction of SNPs that are contained in . Thus, the ratio is essentially the average effect size magnitude on the phenotype for SNPs in . Finally, the enrichment score for was computed as . Subtracting controls for the baseline level of heritability enrichment for SNPs that influence any gene (since most SNPs do not influence any genes). P-values were obtained for the null hypothesis using a block jackknife procedure86.
eQTL analyses
Raw genetic data for 432 European individuals and gene expression data for primary monocytes from these individuals profiled 2 hours after treatment with LPS was obtained from Fairfax et al.27. For each cis-trans gene pair, plink89 was used to compute marginal association statistics of all SNPs within 1 megabase of the promoter of the cis gene with the expression of both the cis gene and trans gene. All our analyses were restricted to cis genes with at least one significant cis-eQTL in the Fairfax dataset. Next, coloc64 was applied to the association statistics to estimate the posterior probability (with the default prior) that the cis and trans gene have a shared eQTL within 1 megabase of the cis gene, setting a posterior probability threshold of 0.75 to determine significant colocalization (varying this threshold does not change downstream results, Supplementary Fig. 9d). The posterior probability that each cis gene colocalizes with random trans genes was also computed. For all analyses, the top 20 principal components of the gene expression matrix were included as covariates, matching the covariates included by Fairfax et al. in their trans-eQTLs analysis and selected based on the fact that they maximize the number of significant trans-eQTLs in Fairfax et al. By restricting the cis gene to having a significant eQTL and comparing our effects to random genes while keeping the cis gene the same, we control for differences in power for detecting cis-by-trans eQTLs that arise from differential levels of selective constraint on the cis gene. In particular, the cis genes selected to be perturbed in our screens include many genes under selective constraint (Supplementary Fig. 7a), for which we have decreased power to detect cis-by-trans eQTLs compared to random cis genes.
Bivariate Haseman-Elston regression as implemented in the GCTA software tool66 was also used to compute the genetic correlation between the expression of the cis gene and the trans gene when restricting to the region 1 megabase around the promoter of the cis gene. Again, the top 20 principal components of the gene expression matrix were included as covariates. The method outputs a genetic correlation estimate and standard error estimate for each cis-trans gene pair. In order to obtain a combined genetic correlation estimate for all downstream genes of a given perturbed gene, all estimates were first squared and then combined using inverse variance weighing. The variance of was estimated from using the Delta method: .
Extended Data
Extended Data Figure 1. Performance of compressed Perturb-seq in simulations with different effect size structure.

Effect sizes were simulated for 100 perturbations on 10,000 genes by separately simulating factor matrices, comprising a (1) 100 perturbation x module “activity” matrix and (2) module x 10,000 gene “dictionary” matrix, then multiplying the matrices together to obtain the final effect size matrix. Entries for both factor matrices were drawn from N(0, 1). The latent dimensionality (corresponding to r in the main text) of the final matrix was set by varying the number of modules (i.e. columns of the activity matrix or rows of the dictionary matrix). The perturbation sparsity (corresponding to q in the main text) was set by randomly setting a given proportion of entries in the module activity matrix to zero. Samples were generated by taking random rows (or sums of random combinations of rows) of the perturbation-by-gene effect size matrix, with the number of rows represented per sample set to 1 for conventional samples or 5 for composite samples. Noise from N(0, 9) was added to all samples to generate phenotypes with 10% signal and 90% noise for the 1 perturbation/sample scenario (plausible for single-cell expression data). Unless otherwise specified, inference was performed using the Factorize-Recover algorithm. (a) Correlation of inferred vs. true effects (Y-axis) when varying the latent dimensionality r of the perturbation effect size matrix (X-axis). q was fixed at 0.1 (left) or 1 (right). (b) Correlation of inferred vs. true effects (Y-axis) when varying the perturbation sparsity q (i.e. the proportion of nonzero entries in the module activity matrix; X-axis). r was fixed at 10 (left) or 50 (right).
Extended Data Figure 2. Analysis of pre-stimulated cells.
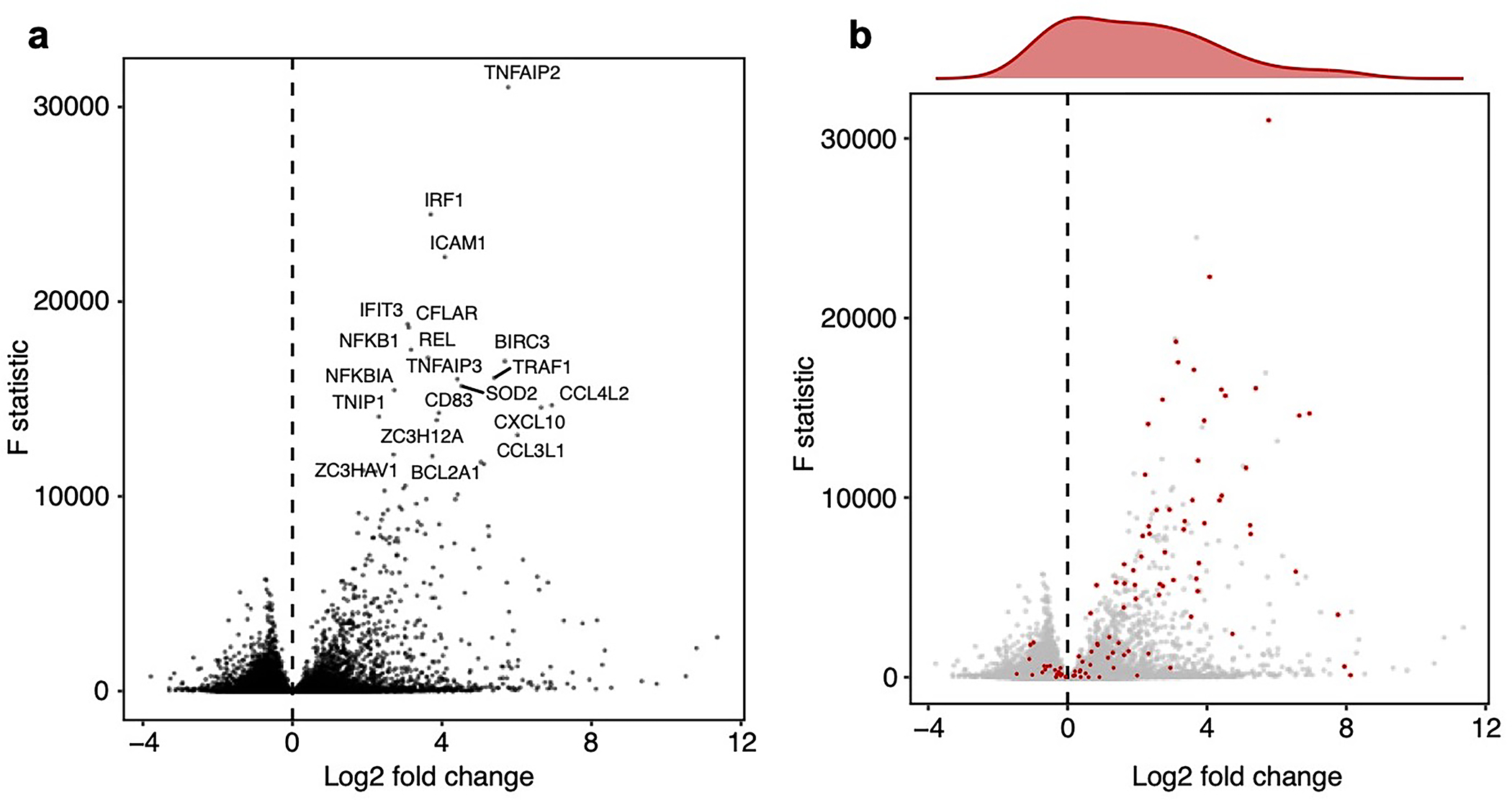
Volcano plots showing the log2 fold changes (x-axis) and F statistics (y-axis) of all genes from differential expression analysis of pre-stimulated vs. LPS-stimulated cells. (a) Top 20 most significantly differentially expressed genes are labeled. (b) Same data as a, but instead the top 100 genes (based on the number of perturbations that significantly modulate them) are highlighted in red. Density plot shows the distribution of log2 fold changes of these 100 genes.
Extended Data Figure 3. Additional analyses comparing compressed versus conventional screens.
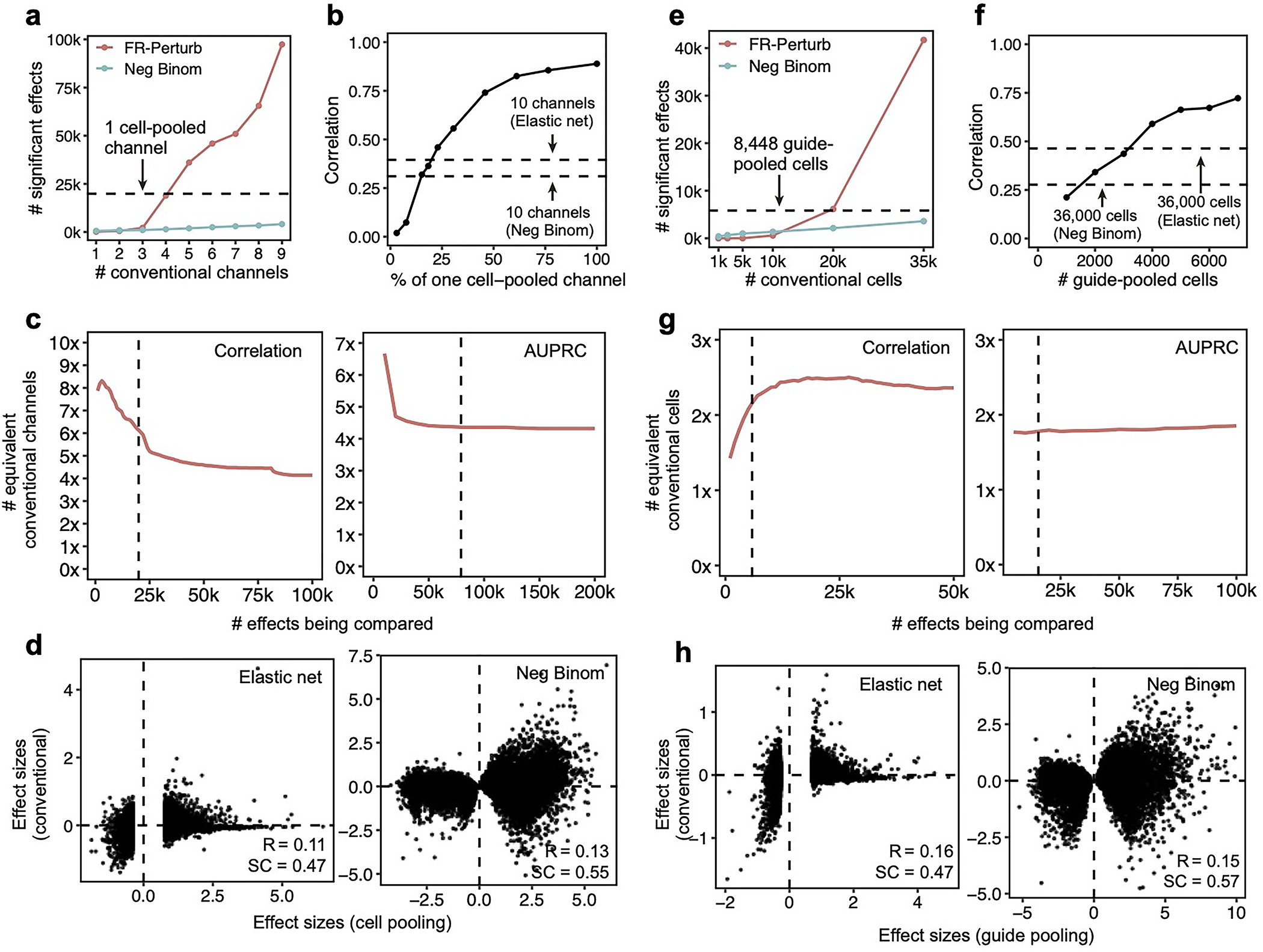
(a) Number of significant effects () detected by FR-Perturb and negative binomial regression (y-axis) as a function of number of channels (x-axis) from the conventional knock-out screen. We do not include the number of significant effects from elastic net due to its extremely large magnitude (>1,000,000), which is inconsistent with the performance of elastic net in held-out validation analyses. (b) Sample size in terms of percentage of a single cell-pooled channel by droplet count (x-axis) versus out-of-sample validation accuracy (y-axis). Validation accuracy of 10 channels analyzed with elastic net or negative binomial regression is indicated with dotted lines. (c) Performance of cell-pooled versus conventional screen (y-axis) while varying the number of effects being compared (x-axis). Performance is quantified as the number of conventional channels needed to obtain the same correlation (left) or AUPRC (right) as one cell-pooled channel. Dotted line represents the cutoffs used in Fig. 3e–f. (d) Scatterplots of top 19,909 estimated effects from the cell-pooled screen (x-axis) versus the same effects in the conventional screen (y-axis) when estimating effects using elastic net regression (left) or negative binomial regression (right). R = Pearson’s correlation, SC = sign concordance. (e-h) Same as a-d, but showing results from the guide-pooled screen (restricting to cells with 3 or more guides) and corresponding conventional screen.
Extended Data Figure 4. Additional analyses comparing compressed versus conventional screens.

(a) Same as Fig. 3e (left) and 4e (right), but correlation (Y-axis) is computed based on perturbation effects on gene modules rather than effects on individual genes. FR-Perturb produces module dictionaries that are correlated but not identical when applied to different datasets, which precludes the direct comparison of perturbation effects on modules in different datasets. Thus, to enable this comparison, the module dictionary was fixed to be the one obtained from the held-out validation dataset for all results above. We note that overall lower correlation is observed in this figure than Fig. 3e and 4e because we compared all perturbation’s effects on all modules rather than only significant effects on genes. (b) Same as Fig. 3e, but performance is assessed based on the number of gene sets constructed from the perturbation effects with significant GWAS heritability enrichment estimated using sc-linker ( for at least two traits out of 63 total; same threshold used as Fig. 6a; see Methods and section “Integrating Perturb-seq with genome-wide association studies” in the main text). P-values are two-sided and obtained from sc-linker. (c) Individual heritability enrichment estimates for all significant gene sets and traits from the full knock-out screen (combined cell-pooled and conventional screens, leftmost plot). The same effects are shown for gene sets constructed from perturbation effects estimated from 1 conventional channel, 1 cell-pooled channel, and 4 conventional channels. Effects with are greyed out.
Extended Data Figure 5. Additional analyses comparing inference methods.
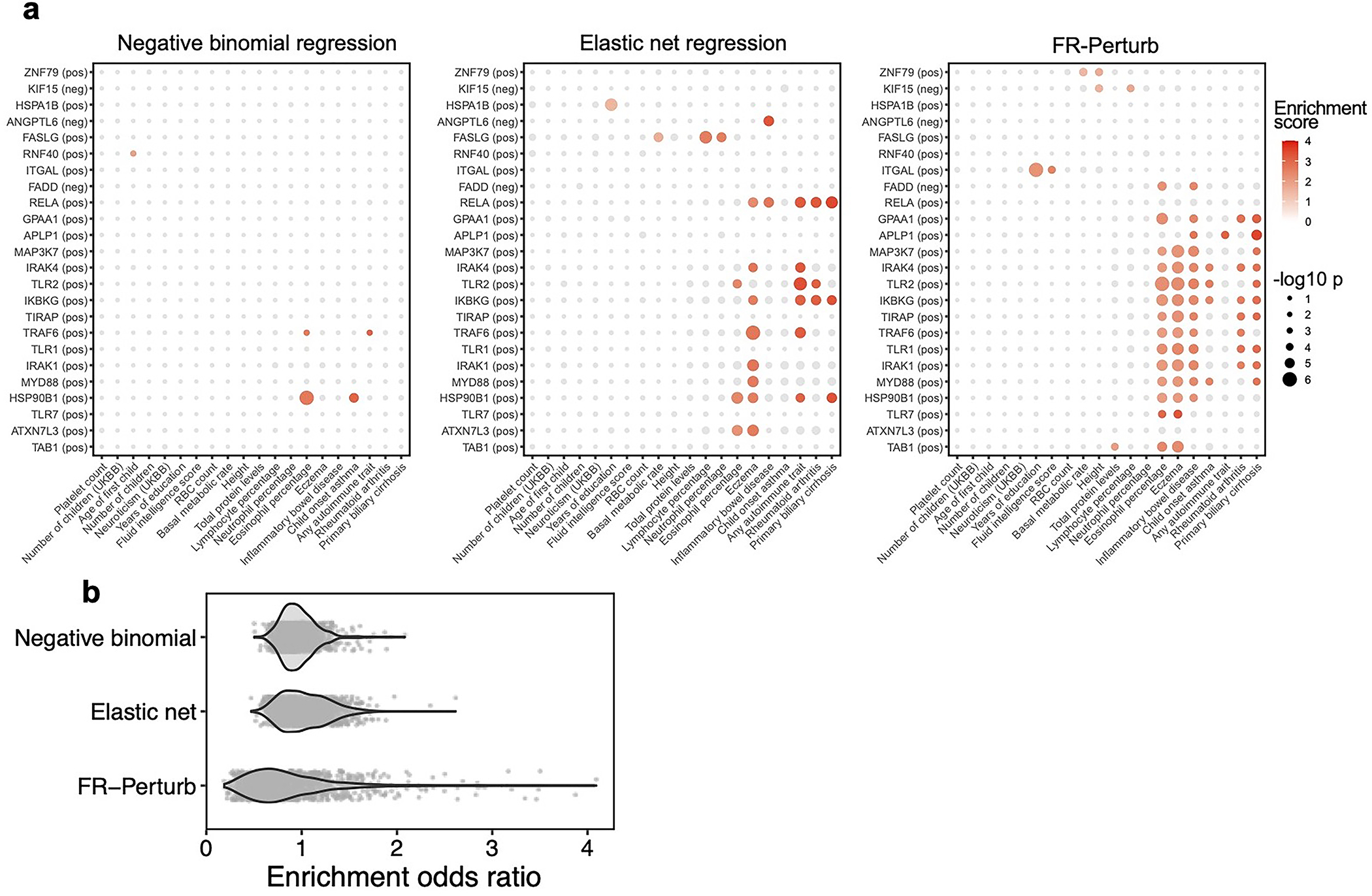
(A) Heritability enrichment estimates and p-values (estimated using sc-linker; Methods) for gene sets and traits that are significant in at least one of the three inference methods. Gene sets were constructed in the same manner as in Figure 6a (see section “Integrating Perturb-seq with genome-wide association studies” in the main text). Significance is determined as having two or more effects with (same threshold used as in Figure 6a). Greyed out points correspond to p-value > 0.001. Gene sets are constructed from the conventional knock-out screen. (b) Odds ratios for enrichment of evolutionarily constrained genes (pLI > 0.9) in all gene sets (comprising the top 500 upregulated or downregulated genes from each perturbation) estimated from the three inference methods. Each point represents a gene set.
Extended Data Figure 6. Relationship between degree of overloading and performance.
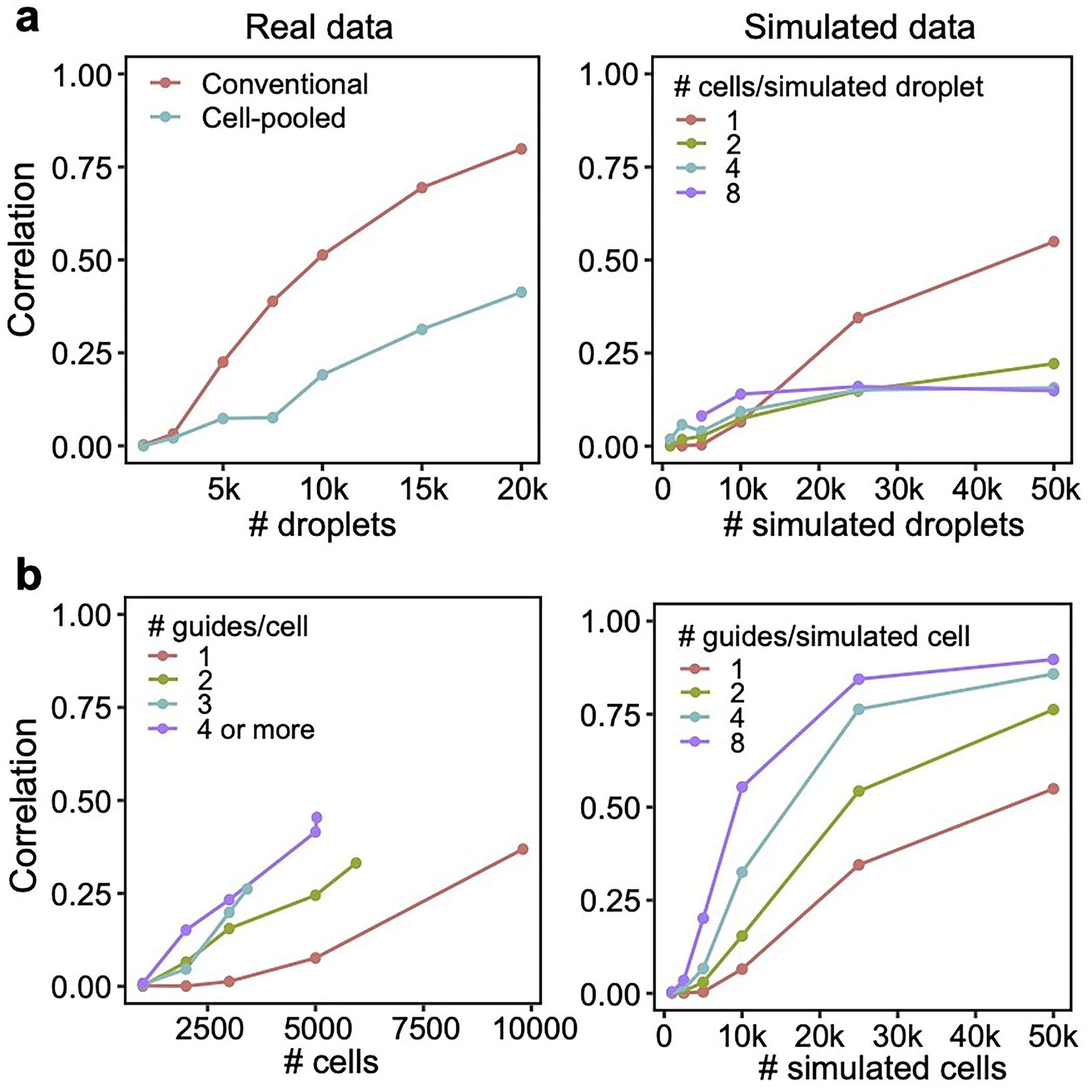
(a) Down-sampling droplets from cell-pooled and conventional screens. (Left) Correlation of top 10,000 estimated effects with held-out validation data (y-axis) when varying droplet count (x-axis). (Right) Correlation of top 10,000 estimated effects with true effects in simulations of cell-pooled data with varying numbers of cells/droplet. (b) Down-sampling cells from guide-pooled screen stratified by # guides/cell. (Left) Correlation of top 10,000 estimated effects with held-out validation data (y-axis) when varying cell count (x-axis). (Right) Correlation of top 10,000 estimated effects with true effects in simulations of guide-pooled data.
Extended Data Figure 7. Additional signal in cells containing multiple guides in a conventional 1,130 gene Perturb-seq screen in mouse BMDCs.
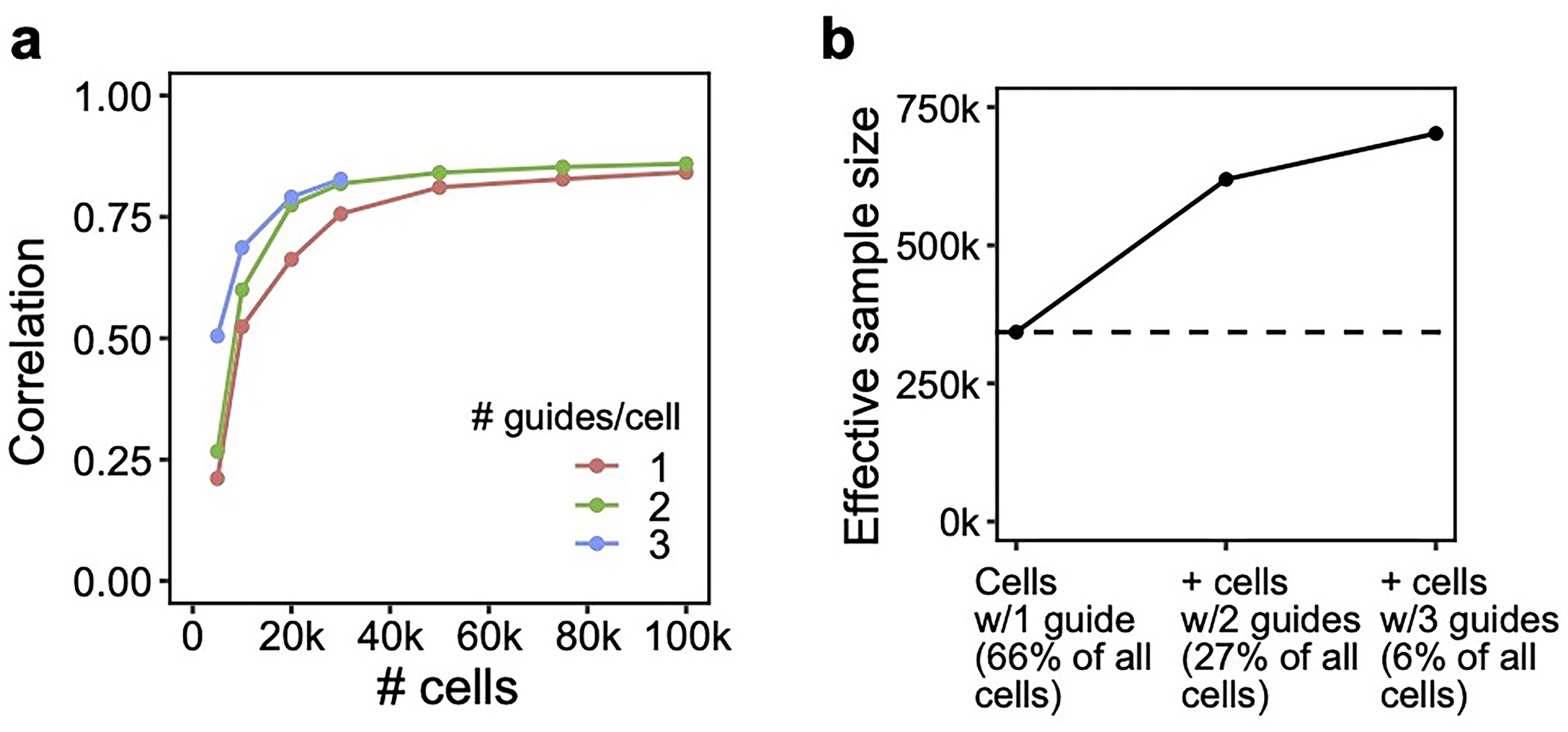
These cells would normally be discarded before analysis. (a) Correlation of top 10,000 estimated effects with held-out validation (y-axis) when varying cell count (x-axis). (b) Increase in effective sample size in cells (y-axis) when including cells containing 2 or 3 guides (x-axis). Effective sample size for cells with 2 or 3 guides is computed as the number of single-guide containing cells needed to achieve the same held-out validation accuracy (from a).
Extended Data Figure 8. Experimental validation of six regulators of the inflammatory response.

RAB5C, PGM3, XPR1, and KIDINS220 represent novel regulators of the inflammatory response, while MYD88 and STAT1 were included as positive controls. (a) IL6 concentration (as measured by ELISA) in LPS-stimulated THP1 cells infected with single guides. Two guides were included for each target (excluding XPR1, which only has one guide due to all cells receiving the other guide dying). Individual bars represent guides, while individual points represent experimental replicates. (b) Left: Log fold changes of IL6 protein in cells receiving perturbations (averaged across the two guides for each target) relative to non-targeting controls. Right: Mean log fold change of expression of genes in P1 (inflammatory program, see Fig. 5d).
Extended Data Figure 9. Additional simulations.
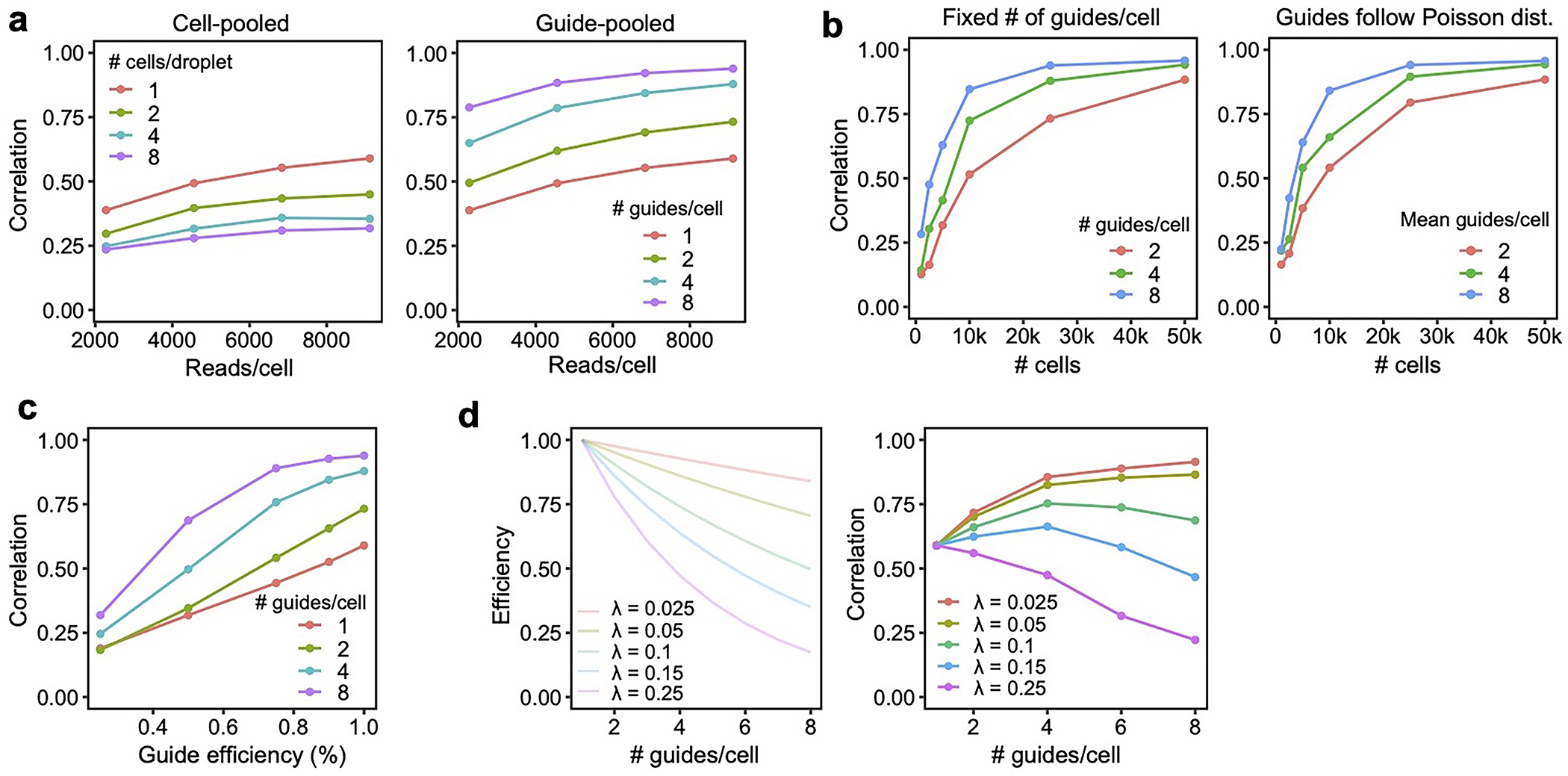
(a) Performance of cell/guide pooling when varying sequencing depth (X-axis). Y-axis: correlation of the top 10,000 most significant effects with the true effects. (b) Performance of guide pooling when simulating cells with a fixed number of guides per cell (left; matching the simulation in Extended Data Fig. 6) or when simulating cells with number of guides following a zero-truncated Poisson distribution with mean guides/cell matching the left plot. (c) Performance of guide pooling vs. the efficiency of all guides (x-axis). Guide efficiency is simulated as the proportion of guides that had the intended effect on their target. For example, for a guide efficiency of 0.8, 20% of guides were randomly selected to have no downstream effects. (d) Performance of guide pooling when efficiency within cells decays as a function of the number of guides per cell. Left: 5 different simulated decay scenarios, where the efficiency per and is the number of guides in the cell. Right: Performance of guide pooling across different # of guides/cell for these 5 scenarios.
Extended Data Figure 10. Theoretical number of cells needed to learn pairwise interactions at different levels of guide pooling.

Number of total perturbations (x-axis) vs. number of cells needed to learn second-order interaction effects between all pairs of perturbation (y-axis), based on the formula , where is the number of cells, is the number of perturbations, and the number of guides per cell.
Supplementary Material
Acknowledgements
We thank Atray Dixit for early discussions on efficient screens and Orit Rozenblatt-Rosen for discussions and help. BC was supported by the Broad Fellows program and a Merkin Institute Fellowship at the Broad Institute. DY was supported by the NSF Graduate Research Fellowship Program (Grant #1745303). AG was supported by R01 HG012133 and R01 HG006399. AR was supported by an NHGRI Center of Excellence in Genome Science grant (CEGS; RM1HG006193; AR), the Howard Hughes Medical Institute, and the Klarman Cell Observatory and Klarman Incubator at the Broad Institute. AR was a Howard Hughes Medical Institute Investigator when this study was initiated. KKD is funded by R00HG012203, P30 CA008748, and the Josie Robertson Investigators Program.
Footnotes
Conflict of Interest statement
AR is a co-founder and equity holder of Celsius Therapeutics, an equity holder in Immunitas, and was a scientific advisory board member of ThermoFisher Scientific, Syros Pharmaceuticals, Neogene Therapeutics and Asimov until July 31, 2020. AR, BE, and KGS are employees of Genentech from August 1, 2020, March 10, 2022, and November 16, 2020, respectively. AR and KGS have equity in Roche. BC and AR are co-inventors on patents filed by the Broad Institute relating to Perturb-seq and compressed sensing methods of this paper. The remaining authors declare no competing interests.
Code Availability
Software implementing FR-Perturb can be found at https://github.com/douglasyao/FR-Perturb91.
Data Availability
Raw and processed data for all Perturb-seq screens (including all perturbation effect sizes estimated with FR-Perturb) were deposited in NCBI’s Gene Expression Omnibus under accession number GSE22132190. SNP-to-gene links (for running sc-linker) can be found at https://github.com/kkdey/GSSG. GWAS summary statistics can be found at https://data.broadinstitute.org/alkesgroup/sumstats_formatted/. eQTLGen data can be found at https://www.eqtlgen.org/phase1.html. Genotypes and expression data from the Fairfax et al.27 study can be found at the European Genome-phenome Archive (https://ega-archive.org/) under study ID EGAS00000000109, though approval is needed to obtain raw data. Gene sets from the Molecular Signatures Database used to run enrichment analysis can be found at https://www.gsea-msigdb.org/gsea/msigdb/collections.jsp.
References
- 1.Adamson B, Norman TM, Jost M, Cho MY, Nuñez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, et al. (2016). A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 167, 1867–1882.e21. 10.1016/j.cell.2016.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, et al. (2016). Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167, 1853–1866.e17. 10.1016/j.cell.2016.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jaitin DA, Weiner A, Yofe I, Lara-Astiaso D, Keren-Shaul H, David E, Salame TM, Tanay A, Oudenaarden A van, and Amit, I. (2016). Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell 167, 1883–1896.e15. 10.1016/j.cell.2016.11.039. [DOI] [PubMed] [Google Scholar]
- 4.Datlinger P, Rendeiro AF, Schmidl C, Krausgruber T, Traxler P, Klughammer J, Schuster LC, Kuchler A, Alpar D, and Bock C (2017). Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 14, 297–301. 10.1038/nmeth.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen KH, Boettiger AN, Moffitt JR, Wang S, and Zhuang X (2015). Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090. 10.1126/science.aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Codeluppi S, Borm LE, Zeisel A, La Manno G, van Lunteren JA, Svensson CI, and Linnarsson S (2018). Spatial organization of the somatosensory cortex revealed by osmFISH. Nat. Methods 15, 932–935. 10.1038/s41592-018-0175-z. [DOI] [PubMed] [Google Scholar]
- 7.Wang X, Allen WE, Wright MA, Sylwestrak EL, Samusik N, Vesuna S, Evans K, Liu C, Ramakrishnan C, Liu J, et al. (2018). Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 361, eaat5691. 10.1126/science.aat5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jin X, Simmons SK, Guo A, Shetty AS, Ko M, Nguyen L, Jokhi V, Robinson E, Oyler P, Curry N, et al. (2020). In vivo Perturb-Seq reveals neuronal and glial abnormalities associated with autism risk genes. Science 370. 10.1126/science.aaz6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fleck JS, Jansen SMJ, Wollny D, Zenk F, Seimiya M, Jain A, Okamoto R, Santel M, He Z, Camp JG, et al. (2022). Inferring and perturbing cell fate regulomes in human brain organoids. Nature, 1–8. 10.1038/s41586-022-05279-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paulsen B, Velasco S, Kedaigle AJ, Pigoni M, Quadrato G, Deo AJ, Adiconis X, Uzquiano A, Sartore R, Yang SM, et al. (2022). Autism genes converge on asynchronous development of shared neuron classes. Nature 602, 268–273. 10.1038/s41586-021-04358-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Replogle JM, Saunders RA, Pogson AN, Hussmann JA, Lenail A, Guna A, Mascibroda L, Wagner EJ, Adelman K, Lithwick-Yanai G, et al. (2022). Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq. Cell 185, 2559–2575.e28. 10.1016/j.cell.2022.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freimer JW, Shaked O, Naqvi S, Sinnott-Armstrong N, Kathiria A, Garrido CM, Chen AF, Cortez JT, Greenleaf WJ, Pritchard JK, et al. (2022). Systematic discovery and perturbation of regulatory genes in human T cells reveals the architecture of immune networks. Nat. Genet. 54, 1133–1144. 10.1038/s41588-022-01106-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frangieh CJ, Melms JC, Thakore PI, Geiger-Schuller KR, Ho P, Luoma AM, Cleary B, Jerby-Arnon L, Malu S, Cuoco MS, et al. (2021). Multimodal pooled Perturb-CITE-seq screens in patient models define mechanisms of cancer immune evasion. Nat. Genet. 53, 332–341. 10.1038/s41588-021-00779-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Norman TM, Horlbeck MA, Replogle JM, Ge AY, Xu A, Jost M, Gilbert LA, and Weissman JS (2019). Exploring genetic interaction manifolds constructed from rich single-cell phenotypes. Science 365, 786–793. 10.1126/science.aax4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Datlinger P, Rendeiro AF, Boenke T, Senekowitsch M, Krausgruber T, Barreca D, and Bock C (2021). Ultra-high-throughput single-cell RNA sequencing and perturbation screening with combinatorial fluidic indexing. Nat. Methods 18, 635–642. 10.1038/s41592-021-01153-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gasperini M, Hill AJ, McFaline-Figueroa JL, Martin B, Kim S, Zhang MD, Jackson D, Leith A, Schreiber J, Noble WS, et al. (2019). A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens. Cell 176, 377–390.e19. 10.1016/j.cell.2018.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Candes EJ, and Wakin MB (2008). An Introduction To Compressive Sampling. IEEE Signal Process. Mag. 25, 21–30. 10.1109/MSP.2007.914731. [DOI] [Google Scholar]
- 18.Candes EJ, Romberg J, and Tao T (2006). Robust uncertainty principles: exact signal reconstruction from highly incomplete frequency information. IEEE Trans. Inf. Theory 52, 489–509. 10.1109/TIT.2005.862083. [DOI] [Google Scholar]
- 19.Donoho DL (2006). Compressed sensing. IEEE Trans. Inf. Theory 52, 1289–1306. 10.1109/TIT.2006.871582. [DOI] [Google Scholar]
- 20.Petti S, Reddy G, and Desai MM (2023). Inferring sparse structure in genotype-phenotype maps. 2022.09.27.509675. 10.1101/2022.09.27.509675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cleary B, Cong L, Cheung A, Lander ES, and Regev A (2017). Efficient Generation of Transcriptomic Profiles by Random Composite Measurements. Cell 171, 1424–1436.e18. 10.1016/j.cell.2017.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cleary B, Simonton B, Bezney J, Murray E, Alam S, Sinha A, Habibi E, Marshall J, Lander ES, Chen F, et al. (2021). Compressed sensing for highly efficient imaging transcriptomics. Nat. Biotechnol, 1–7. 10.1038/s41587-021-00883-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharan V, Tai KS, Bailis P, and Valiant G (2019). Compressed Factorization: Fast and Accurate Low-Rank Factorization of Compressively-Sensed Data. In Proceedings of the 36th International Conference on Machine Learning (PMLR), pp. 5690–5700. [Google Scholar]
- 24.Yeung KY, and Ruzzo WL (2001). Principal component analysis for clustering gene expression data. Bioinformatics 17, 763–774. 10.1093/bioinformatics/17.9.763. [DOI] [PubMed] [Google Scholar]
- 25.Brunet J-P, Tamayo P, Golub TR, and Mesirov JP (2004). Metagenes and molecular pattern discovery using matrix factorization. Proc. Natl. Acad. Sci. 101, 4164–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parnas O, Jovanovic M, Eisenhaure TM, Herbst RH, Dixit A, Ye CJ, Przybylski D, Platt RJ, Tirosh I, Sanjana NE, et al. (2015). A Genome-wide CRISPR Screen in Primary Immune Cells to Dissect Regulatory Networks. Cell 162, 675–686. 10.1016/j.cell.2015.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fairfax BP, Humburg P, Makino S, Naranbhai V, Wong D, Lau E, Jostins L, Plant K, Andrews R, McGee C, et al. (2014). Innate Immune Activity Conditions the Effect of Regulatory Variants upon Monocyte Gene Expression. Science 343, 1246949. 10.1126/science.1246949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chanput W, Mes JJ, and Wichers HJ (2014). THP-1 cell line: An in vitro cell model for immune modulation approach. Int. Immunopharmacol. 23, 37–45. 10.1016/j.intimp.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 29.Aguirre AJ, Meyers RM, Weir BA, Vazquez F, Zhang C-Z, Ben-David U, Cook A, Ha G, Harrington WF, Doshi MB, et al. (2016). Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov. 6, 914–929. 10.1158/2159-8290.CD-16-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geiger-Schuller K, Eraslan B, Kuksenko O, Dey KK, Jagadeesh KA, Thakore PI, Karayel O, Yung AR, Rajagopalan A, Meireles AM, et al. (2023). Systematically characterizing the roles of E3-ligase family members in inflammatory responses with massively parallel Perturb-seq. 2023.01.23.525198. 10.1101/2023.01.23.525198. [DOI] [Google Scholar]
- 31.Rosenbluh J, Xu H, Harrington W, Gill S, Wang X, Vazquez F, Root DE, Tsherniak A, and Hahn WC (2017). Complementary information derived from CRISPR Cas9 mediated gene deletion and suppression. Nat. Commun. 8, 15403. 10.1038/ncomms15403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brubaker SW, Bonham KS, Zanoni I, and Kagan JC (2015). Innate Immune Pattern Recognition: A Cell Biological Perspective. Annu. Rev. Immunol. 33, 257–290. 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palucka AK, Blanck J-P, Bennett L, Pascual V, and Banchereau J (2005). Cross-regulation of TNF and IFN-α in autoimmune diseases. Proc. Natl. Acad. Sci. 102, 3372–3377. 10.1073/pnas.0408506102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mavragani CP, Niewold TB, Moutsopoulos NM, Pillemer SR, Wahl SM, and Crow MK (2007). Augmented interferon-alpha pathway activation in patients with Sjögren’s syndrome treated with etanercept. Arthritis Rheum. 56, 3995–4004. 10.1002/art.23062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dorrington MG, and Fraser IDC (2019). NF-κB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front. Immunol. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang N, Liang H, and Zen K (2014). Molecular Mechanisms That Influence the Macrophage M1–M2 Polarization Balance. Front. Immunol. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Komura T, Sakai Y, Honda M, Takamura T, Wada T, and Kaneko S (2013). ER stress induced impaired TLR signaling and macrophage differentiation of human monocytes. Cell. Immunol. 282, 44–52. 10.1016/j.cellimm.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 38.Platanias LC (2005). Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5, 375–386. 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 39.Carballo E, Lai WS, and Blackshear PJ (1998). Feedback Inhibition of Macrophage Tumor Necrosis Factor-α Production by Tristetraprolin. Science 281, 1001–1005. 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- 40.Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, and Mosialos G (2003). CYLD is a deubiquitinating enzyme that negatively regulates NF-κB activation by TNFR family members. Nature 424, 793–796. 10.1038/nature01803. [DOI] [PubMed] [Google Scholar]
- 41.Shembade N, Ma A, and Harhaj EW (2010). Inhibition of NF-κB Signaling by A20 Through Disruption of Ubiquitin Enzyme Complexes. Science 327, 1135–1139. 10.1126/science.1182364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wertz IE, O’Rourke KM, Zhang Z, Dornan D, Arnott D, Deshaies RJ, and Dixit VM (2004). Human De-Etiolated-1 Regulates c-Jun by Assembling a CUL4A Ubiquitin Ligase. Science 303, 1371–1374. 10.1126/science.1093549. [DOI] [PubMed] [Google Scholar]
- 43.Kiss-Toth E, Bagstaff SM, Sung HY, Jozsa V, Dempsey C, Caunt JC, Oxley KM, Wyllie DH, Polgar T, Harte M, et al. (2004). Human Tribbles, a Protein Family Controlling Mitogen-activated Protein Kinase Cascades *. J. Biol. Chem. 279, 42703–42708. 10.1074/jbc.M407732200. [DOI] [PubMed] [Google Scholar]
- 44.Scholz-Starke J, and Cesca F (2016). Stepping Out of the Shade: Control of Neuronal Activity by the Scaffold Protein Kidins220/ARMS. Front. Cell. Neurosci. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bondeson DP, Paolella BR, Asfaw A, Rothberg MV, Skipper TA, Langan C, Mesa G, Gonzalez A, Surface LE, Ito K, et al. (2022). Phosphate dysregulation via the XPR1–KIDINS220 protein complex is a therapeutic vulnerability in ovarian cancer. Nat. Cancer 3, 681–695. 10.1038/s43018-022-00360-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huttlin EL, Bruckner RJ, Navarrete-Perea J, Cannon JR, Baltier K, Gebreab F, Gygi MP, Thornock A, Zarraga G, Tam S, et al. (2021). Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell 184, 3022–3040.e28. 10.1016/j.cell.2021.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Amiot F, Boussadia O, Cases S, Fitting C, Lebastard M, Cavaillon J-M, Milon G, and Dautry F (1997). Mice heterozygous for a deletion of the tumor necrosis factor-α and lymphotoxin-α genes: biological importance of a nonlinear response of tumor necrosis factor-α to gene dosage. Eur. J. Immunol. 27, 1035–1042. 10.1002/eji.1830270434. [DOI] [PubMed] [Google Scholar]
- 48.Simon A, Park H, Maddipati R, Lobito AA, Bulua AC, Jackson AJ, Chae JJ, Ettinger R, de Koning HD, Cruz AC, et al. (2010). Concerted action of wild-type and mutant TNF receptors enhances inflammation in TNF receptor 1-associated periodic fever syndrome. Proc. Natl. Acad. Sci. 107, 9801–9806. 10.1073/pnas.0914118107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Segrè D, DeLuna A, Church GM, and Kishony R (2005). Modular epistasis in yeast metabolism. Nat. Genet. 37, 77–83. 10.1038/ng1489. [DOI] [PubMed] [Google Scholar]
- 50.Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JLY, Toufighi K, Mostafavi S, et al. (2010). The Genetic Landscape of a Cell. Science 327, 425–431. 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lang KS, Burow A, Kurrer M, Lang PA, and Recher M (2007). The role of the innate immune response in autoimmune disease. J. Autoimmun. 29, 206–212. 10.1016/j.jaut.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 52.O’Connor LJ, Schoech AP, Hormozdiari F, Gazal S, Patterson N, and Price AL (2019). Extreme Polygenicity of Complex Traits Is Explained by Negative Selection. Am. J. Hum. Genet. 105, 456–476. 10.1016/j.ajhg.2019.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jagadeesh KA, Dey KK, Montoro DT, Mohan R, Gazal S, Engreitz JM, Xavier RJ, Price AL, and Regev A (2022). Identifying disease-critical cell types and cellular processes by integrating single-cell RNA-sequencing and human genetics. Nat. Genet. 54, 1479–1492. 10.1038/s41588-022-01187-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morris JA, Daniloski Z, Domingo J, Barry T, Ziosi M, Glinos DA, Hao S, Mimitou EP, Smibert P, Roeder K, et al. (2021). Discovery of target genes and pathways of blood trait loci using pooled CRISPR screens and single cell RNA sequencing. 2021.04.07.438882. 10.1101/2021.04.07.438882. [DOI] [Google Scholar]
- 57.Graustein A, Misch EA, Musvosvi M, Shey M, Shah J, Wells R, Hanekom W, Hatherill M, Scriba T, and Hawn T (2016). HSP90B1 Regulates TLR-dependent Monocyte Signaling and its Common Variants are Associated with BCG-specific T-cell Responses and Protection from Pediatric TB Disease. J. Immunol. 196, 200.18–200.18. [Google Scholar]
- 58.Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, Gouw AM, Baylot V, Gütgemann I, Eilers M, et al. (2016). MYC regulates the antitumor immune response through CD47 and PD-L1. Science 352, 227–231. 10.1126/science.aac9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Swigart LB, Littlewood TD, and Evan GI (2017). Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 171, 1301–1315.e14. 10.1016/j.cell.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Garcia-Etxebarria K, Bracho MA, Galán JC, Pumarola T, Castilla J, Lejarazu R.O. de, Rodríguez-Dominguez M, Quintela I, Bonet N, Garcia-Garcerà M, et al. (2015). No Major Host Genetic Risk Factor Contributed to A(H1N1)2009 Influenza Severity. PLOS ONE 10, e0135983. 10.1371/journal.pone.0135983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu X, Li YI, and Pritchard JK (2019). Trans Effects on Gene Expression Can Drive Omnigenic Inheritance. Cell 177, 1022–1034.e6. 10.1016/j.cell.2019.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Võsa U, Claringbould A, Westra H-J, Bonder MJ, Deelen P, Zeng B, Kirsten H, Saha A, Kreuzhuber R, Yazar S, et al. (2021). Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 53, 1300–1310. 10.1038/s41588-021-00913-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Westra H-J, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, Christiansen MW, Fairfax BP, Schramm K, Powell JE, et al. (2013). Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243. 10.1038/ng.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, and Plagnol V (2014). Bayesian Test for Colocalisation between Pairs of Genetic Association Studies Using Summary Statistics. PLOS Genet. 10, e1004383. 10.1371/journal.pgen.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Han H, Cho J-W, Lee S, Yun A, Kim H, Bae D, Yang S, Kim CY, Lee M, Kim E, et al. (2018). TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 46, D380–D386. 10.1093/nar/gkx1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang J, Lee SH, Goddard ME, and Visscher PM (2011). GCTA: A Tool for Genome-wide Complex Trait Analysis. Am. J. Hum. Genet. 88, 76–82. 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lukowski SW, Lloyd-Jones LR, Holloway A, Kirsten H, Hemani G, Yang J, Small K, Zhao J, Metspalu A, Dermitzakis ET, et al. (2017). Genetic correlations reveal the shared genetic architecture of transcription in human peripheral blood. Nat. Commun. 8, 483. 10.1038/s41467-017-00473-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Umans BD, Battle A, and Gilad Y (2021). Where Are the Disease-Associated eQTLs? Trends Genet. 37, 109–124. 10.1016/j.tig.2020.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Simmons SK, Lithwick-Yanai G, Adiconis X, Oberstrass F, Iremadze N, Geiger-Schuller K, Thakore PI, Frangieh CJ, Barad O, Almogy G, et al. (2022). Mostly natural sequencing-by-synthesis for scRNA-seq using Ultima sequencing. Nat. Biotechnol, 1–8. 10.1038/s41587-022-01452-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schraivogel D, Gschwind AR, Milbank JH, Leonce DR, Jakob P, Mathur L, Korbel JO, Merten CA, Velten L, and Steinmetz LM (2020). Targeted Perturb-seq enables genome-scale genetic screens in single cells. Nat. Methods 17, 629–635. 10.1038/s41592-020-0837-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mead BE, Kummerlowe C, Liu N, Kattan WE, Cheng T, Cheah JH, Soule CK, Peters J, Lowder KE, Blainey PC, et al. (2023). Compressed phenotypic screens for complex multicellular models and high-content assays. 2023.01.23.525189. 10.1101/2023.01.23.525189. [DOI] [Google Scholar]
- 72.O’Connor LJ (2021). The distribution of common-variant effect sizes. Nat. Genet. 53, 1243–1249. 10.1038/s41588-021-00901-3. [DOI] [PubMed] [Google Scholar]
- 73.Yazar S, Alquicira-Hernandez J, Wing K, Senabouth A, Gordon MG, Andersen S, Lu Q, Rowson A, Taylor TRP, Clarke L, et al. (2022). Single-cell eQTL mapping identifies cell type–specific genetic control of autoimmune disease. Science. 10.1126/science.abf3041. [DOI] [PubMed] [Google Scholar]
Methods-only References
- 74.Huang H, Fang M, Jostins L, Umićević Mirkov M, Boucher G, Anderson CA, Andersen V, Cleynen I, Cortes A, Crins F, et al. (2017). Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 547, 173–178. 10.1038/nature22969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Amberger JS, Bocchini CA, Scott AF, and Hamosh A (2019). OMIM.org: leveraging knowledge across phenotype–gene relationships. Nucleic Acids Res. 47, D1038–D1043. 10.1093/nar/gky1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, et al. (2019). The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012. 10.1093/nar/gky1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.O’Hara R, and Kotze J (2010). Do not log-transform count data. Nat. Preced, 1–1. 10.1038/npre.2010.4136.1. [DOI] [Google Scholar]
- 78.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mairal J, Bach F, Ponce J, and Sapiro G (2010). Online Learning for Matrix Factorization and Sparse Coding. J. Mach. Learn. Res. 11, 19–60. [Google Scholar]
- 80.Ahlmann-Eltze C, and Huber W (2020). glmGamPoi: fitting Gamma-Poisson generalized linear models on single cell count data. Bioinformatics 36, 5701–5702. 10.1093/bioinformatics/btaa1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Traag VA, Waltman L, and van Eck NJ (2019). From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 9, 5233. 10.1038/s41598-019-41695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yu G, Wang L-G, Han Y, and He Q-Y (2012). clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 16, 284–287. 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fulco CP, Nasser J, Jones TR, Munson G, Bergman DT, Subramanian V, Grossman SR, Anyoha R, Doughty BR, Patwardhan TA, et al. (2019). Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 51, 1664–1669. 10.1038/s41588-019-0538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh P-R, Anttila V, Xu H, Zang C, Farh K, et al. (2015). Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235. 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gazal S, Finucane HK, Furlotte NA, Loh P-R, Palamara PF, Liu X, Schoech A, Bulik-Sullivan B, Neale BM, Gusev A, et al. (2017). Linkage disequilibrium–dependent architecture of human complex traits shows action of negative selection. Nat. Genet. 49, 1421–1427. 10.1038/ng.3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nasser J, Bergman DT, Fulco CP, Guckelberger P, Doughty BR, Patwardhan TA, Jones TR, Nguyen TH, Ulirsch JC, Lekschas F, et al. (2021). Genome-wide enhancer maps link risk variants to disease genes. Nature 593, 238–243. 10.1038/s41586-021-03446-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al. (2007). PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yao D, Binan L, Bezney J, Simonton B, Freedman J, Frangieh C, Gusev A, Regev A, Cleary B Compressed Perturb-seq: highly efficient screens for regulatory circuits using random composite perturbations. Gene Expression Omnibus https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE221321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yao D, Binan L, Bezney J, Simonton B, Freedman J, Frangieh C, Gusev A, Regev A, Cleary B Factorize-Recover for Perturb-seq analysis (FR-Perturb). Github. https://github.com/douglasyao/FR-Perturb. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed data for all Perturb-seq screens (including all perturbation effect sizes estimated with FR-Perturb) were deposited in NCBI’s Gene Expression Omnibus under accession number GSE22132190. SNP-to-gene links (for running sc-linker) can be found at https://github.com/kkdey/GSSG. GWAS summary statistics can be found at https://data.broadinstitute.org/alkesgroup/sumstats_formatted/. eQTLGen data can be found at https://www.eqtlgen.org/phase1.html. Genotypes and expression data from the Fairfax et al.27 study can be found at the European Genome-phenome Archive (https://ega-archive.org/) under study ID EGAS00000000109, though approval is needed to obtain raw data. Gene sets from the Molecular Signatures Database used to run enrichment analysis can be found at https://www.gsea-msigdb.org/gsea/msigdb/collections.jsp.