

Abstract
Background:
Microvasculature dysfunction is a common finding in pathological remodeling of the heart and is thought to play an important role in the pathogenesis of hypertrophic cardiomyopathy (HCM), a disease caused by sarcomere gene mutations. We hypothesized that microvascular dysfunction in HCM was secondary to abnormal microvascular growth and could occur independent of ventricular hypertrophy.
Methods:
We utilized multimodality imaging methods to track the temporality of microvascular dysfunction in HCM mouse models harboring mutations in the sarcomere genes Mybpc3 or Myh6. We performed complementary molecular methods to assess protein quantity, interactions, and post-translational modifications to identify mechanisms regulating this response. We manipulated select molecular pathways in vivo using both genetic and pharmacological methods to validate these mechanisms.
Results:
We found that microvascular dysfunction in our HCM models occurred secondary to reduced myocardial capillary growth during the early postnatal time period and could occur before the onset of myocardial hypertrophy. We discovered that the E3 ubiquitin protein ligase murine double minute 2 (MDM2) dynamically regulates the protein stability of both HIF1α and HIF2α/EPAS1 through canonical and non-canonical mechanisms. The resulting HIF imbalance leads to reduced pro-angiogenic gene expression during a key period of myocardial capillary growth. Reducing MDM2 protein levels by genetic or pharmacological methods normalized HIF protein levels and prevented the development of microvascular dysfunction in both HCM models.
Conclusions:
Our results show that sarcomere mutations induce cardiomyocyte MDM2 signaling during the earliest stages of disease, and this leads to long term changes in the myocardial microenvironment.
Keywords: hypertrophic cardiomyopathy, HCM, microvascular dysfunction, MDM2, HIF1, HIF2, MYBPC3, MYH6, VHL, proteasome, sarcomere mutations, capillary, coronary flow reserve, cardiomyocyte hypertrophy, angiogenesis
INTRODUCTION
Hypertrophic cardiomyopathy (HCM) is a disease characterized by pathological thickening of the left ventricular myocardium which can lead to heart failure and sudden death in humans.1 The prevalence of HCM in humans is ~1:500 and mutations in sarcomere protein genes have been shown to cause this disease.2 The most commonly mutated genes in HCM are cardiac myosin-binding protein C (MYBPC3) and beta myosin heavy chain (MYH7) and mutations in these two genes account for approximately 80% of mutation-positive HCM cases.3, 4 The cause of myocardial hypertrophy in HCM is increased cardiomyocyte growth that occurs during childhood or in adult life in humans. However, pathologic ventricular remodeling in HCM is also associated with changes in the non-cardiomyocyte myocardial cell populations.5 It remains incompletely understood how sarcomere protein mutations that are expressed primarily in the cardiomyocyte cell population lead to changes in the other cell types of the myocardium.
Microvascular dysfunction or disease (MVD), occurs secondary to abnormalities of the capillary beds of multiple different organs such as the brain, kidney, retina, skin, lung, and heart.6–8 MVD has been consistently identified in many different forms of human cardiomyopathy secondary to both ischemic and non-ischemic etiologies.9–14 Mechanistically, MVD often involves an imbalance of pro-angiogenic and anti-angiogenic factors which are critical for capillary formation and maintenance.15 For example, reduced expression of the pro-angiogenic factors such as vascular endothelial growth factor A (VegfA) and angiopoietin 1 and 2 and increased production of the anti-angiogenic factors, angiopoietin 4 and thrombospondin 1, induce capillary rarefaction secondary to pressure overload.16–19 However, the molecular mechanisms regulating the development of MVD in HCM remain poorly defined.
The pre-hypertrophic disease interval is a critical period in the development of HCM.20–24 We hypothesized that MVD develops during this period secondary to alterations in cardiomyocyte signaling that can develop before the onset of hypertrophy. To explore these questions, we utilized multiple sarcomere gene mutant animal models of HCM to assess microvascular growth and dysfunction while exploring the key molecular mechanisms regulating these phenotypes. We then investigated if targeting these pathways was sufficient to prevent the development of MVD in pre-clinical models of HCM.
MATERIALS AND METHODS
All data needed to evaluate the conclusions in the paper are present in the paper and/or the supplemental materials. Additional data related to this paper may be requested from the authors.
Detailed description of the materials and methods can be found in the online-only Data Supplement.
Ethical Considerations
All mice used in the study were housed at American Association for the Accreditation of Laboratory Animal Care (AAALAC) accredited animal facility. The animal experiments were conducted in accordance with the practices defined in the Guide for the Care and Use of Laboratory Animals which were approved and overseen by University of Pittsburgh’s Institutional Animal Care and Use Committee (IACUC).
Statistical Analysis
Experimental data was obtained in a blinded fashion to genotype or treatment group. Estimated sample sizes were calculated using preliminary experimental results. Statistical significance between two groups was calculated using two-tailed unpaired Student’s t-test. If unequal variances were detected using a F test, then a Welch’s t-test was utilized. In the case of more than two groups, a one-way or two-way ANOVA with Tukey’s multiple comparisons test was used for determining statistical significance. If unequal variances were detected using a Brown-Forsythe test, then a Brown-Forsythe and Welch ANOVA with Dunnett’s T3 multiple comparisons test was utilized for one-way ANOVA. All statistical analyses were performed by Prism software version 9 (GraphPad). A p value less than 0.05 was considered statistically significant.
RESULTS
Reduced postnatal capillary formation in Mybpc3−/− myocardium is associated with microvascular dysfunction and tissue hypoxia.
We previously found that deletion of the sarcomere protein Mybpc3 leads to early postnatal left ventricular (LV) hypertrophy.20 Therefore, we wanted to determine if there was evidence of microvascular dysfunction in this model. First, we measured capillary to cardiomyocyte ratios in wild-type (WT) and Mybpc3-null (Mybpc3−/−) LV tissue by staining for the endothelial cell marker CD31. We discovered that capillary density was reduced in Mybpc3−/− LV tissue beginning at postnatal day 7 (P7) (Figure 1A–B, Figure S1A–B). Both male and female Mybpc3−/− mice had similar reductions in LV capillary density (Figure S1C). We then performed immunohistochemistry for an alternative endothelial cell marker, angiopoietin-1 receptor TIE2 (aka TEK), and again found that capillary density was reduced at P7 (Figure 1A–B, Figure S1D). We confirmed that CD31 and TIE2 detect the same cell population in the myocardium (Figure S1E–F). In addition to endothelial cells, pericytes are a critical cell population for proper capillary development.25 Therefore, we performed immunohistochemistry for the pericyte marker neural/glial antigen 2 (NG2) and found that Mybpc3−/− mice also had reduced LV pericytes (Figure 1C–D, Figure S2A).
Figure 1. Reduced postnatal capillary formation in the Mybpc3−/− myocardium is associated with microvascular dysfunction and tissue hypoxia.
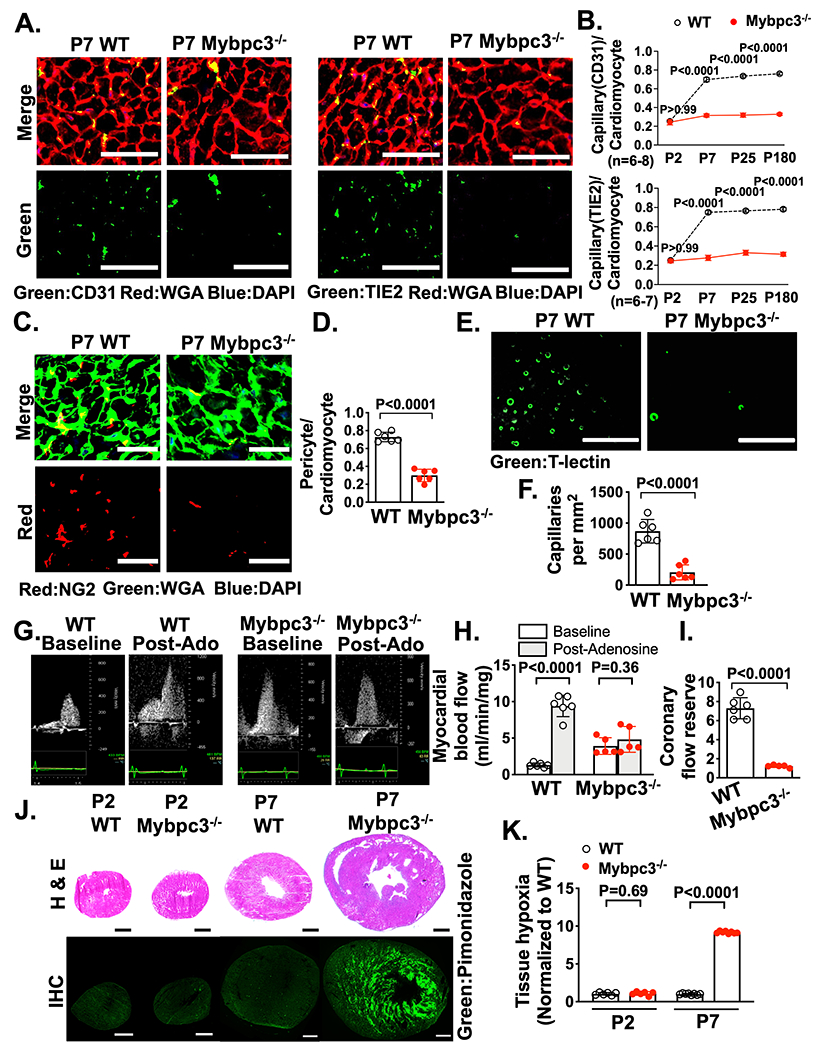
(A) Representative immunohistochemistry images for the endothelial cell markers CD31 [left] or TIE2 [right] (green) co-stained with wheat germ agglutinin (WGA) (red) in left ventricular (LV) tissue cross-sections from postnatal day 7 (P7) wild-type (WT) and Mybpc3−/− mice. Nuclei are blue (DAPI). Scale bars=25 μm. (B) Capillary to cardiomyocyte ratios from WT and Mybpc3−/− (n=6-8/group) LV tissue at P2, P7, P25 or P180. Capillaries were identified with CD31 [top] or TIE2 [bottom]. Minimum 120 cardiomyocytes/sample. (C) Representative immunohistochemistry images for the pericyte marker NG2 (red) co-stained with WGA (green) in LV tissue from P7 WT and Mybpc3−/− mice. Nuclei are blue (DAPI). Scale bars=25 μm. (D) Pericyte to cardiomyocyte ratios from WT (n=6) and Mybpc3−/− (n=6) LV tissue at P7. Minimum 200 cardiomyocytes/sample. (E) Representative fluorescence images for the intravascularly injected endothelial cell stain tomato lectin (T-Lectin) (green) in LV tissue from P7 WT and Mybpc3−/− mice. Scale bars=80 μm. (F) Capillaries per mm2 in WT (n=6) and Mybpc3−/− (n=6) LV tissue at P7. Three cross-sectional images per sample were analyzed. (G) Representative myocardial blood flow velocity tracings using pulsed wave Doppler echocardiography in P60 WT and Mybpc3−/− mice. Myocardial blood flow at baseline and after retro-orbital injection with adenosine (post-adenosine) to induce maximal hyperemia. (H) Myocardial blood flow in P60 WT (n=6) and Mybpc3−/− (n=5) mice. Myocardial blood flow at baseline (white bars) and post-adenosine (light grey bars) were normalized to heart weight. (I) Coronary flow reserve in P60 WT (n=6) and Mybpc3−/− (n=5) mice. Coronary flow reserve is the ratio of myocardial blood flow post-adenosine to myocardial blood flow at baseline. (J) [top] Representative H&E–stained heart cross-sections from P2 or P7 WT and Mybpc3−/− mice. [bottom] Representative immunohistochemistry images of heart cross sections from P2 or P7 WT and Mybpc3−/− mice injected with pimonidazole (hypoxyprobe, green). Scale bars=0.5 mm. (K) Comparison of LV tissue hypoxia in WT and Mybpc3−/− mice at P2 or P7 (n=6-8/group). Green fluorescent intensity for each sample was obtained and then normalized to P2 WT samples. All results are shown as mean±SEM. Student’s or Welch’s t-test were utilized for D, F, H, I and K. Two-way ANOVA with Tukey’s multiple comparison test was for B.
To complement the immunohistochemistry based methods, we then injected fluorescently labeled tomato lectin (T-lectin) which binds to vascular endothelial cells.26 This allowed better visualization of the capillary lumens and confirmed that Mybpc3−/− mice had a significant reduction in LV capillaries (Figure 1E–F, Figure S2B–C, Video S1–2). We confirmed that T-lectin, CD31 and NG2 could detect the same myocardial capillaries (Figure S2D–E). In addition to reduced LV capillaries, we discovered that P7 Mybpc3−/− mice had increased left coronary artery dilation compared to wild type control mice. However, left coronary artery size was similar in both groups of mice after birth at P2 arguing against a difference in embryonic coronary development between the groups (Figure S2F–I). Overall, these results show that LV capillary formation is reduced in the early postnatal period in Mybpc3−/− mice.
Next, we wanted to determine if the reduction of LV capillaries in Mybpc3−/− mice led to microvasculature dysfunction. We measured blood flow in the left coronary artery (LCA) before and after the administration of the vasodilator adenosine to calculate the coronary flow reserve which is dependent on myocardial capillary blood flow (Figure 1G, Figure S2J–L).27 Baseline myocardial blood flow was increased in Mybpc3−/− animals which was partly driven by the increased left coronary artery size in these mice (Figure 1G–H). However, myocardial blood flow minimally increased after adenosine administration (Figure 1H). Therefore, the coronary flow reserve was reduced in Mybpc3−/− animals (Figure 1I). Next, we wanted to determine if the microvascular dysfunction in Mybpc3−/− myocardium was associated with tissue hypoxia. We utilized the tissue hypoxia marker pimonidazole and detected a large increase in LV tissue hypoxia in Mybpc3−/− heart tissue (Figure 1J–K). Overall, these results show that the reduction of myocardial capillary formation in Mybpc3−/− mice leads to microvascular dysfunction and LV tissue hypoxia.
Dynamic changes in HIF1α and HIF2α occur during the early postnatal period in the Mybpc3−/− myocardium.
Since we detected tissue hypoxia in the Mybpc3−/− myocardium, we measured the protein levels of the hypoxia-regulated transcriptions factors, HIF1α and HIF2α/EPAS1.28 We detected no differences in the protein levels at P2 (Figure 2A+B). In contrast, there was a reduction of HIF1α protein and an increase of HIF2α in the Mybpc3−/− LV tissue at P7 (Figure 2A+B). However, by P25, the HIF1α and HIF2α LV protein levels were similar between WT and Mybpc3−/− mice (Figure 2A+B). We compared protein levels across the different ages and discovered that myocardial HIF1α protein levels do not increase at P7 in the Mybpc3−/− LV tissue when compared to WT LV tissue (Figure 2C). In contrast, HIF2α protein levels increase at P7 in Mybpc3−/− LV tissue and then return to control levels by P25 (Figure 2D). We then performed immunohistochemistry to confirm that the changes in HIF1α and HIF2α protein levels were cardiomyocyte-specific (Figure 2E–G). Overall, these results show that during the early postnatal time period when myocardial capillary formation is reduced in the Mybpc3−/− mice, there are dynamic changes in cardiomyocyte HIF1α and HIF2α protein levels.
Figure 2. Dynamic changes in HIF1⍺ and HIF2⍺ occur during the early postnatal period in the Mybpc3−/− myocardium.
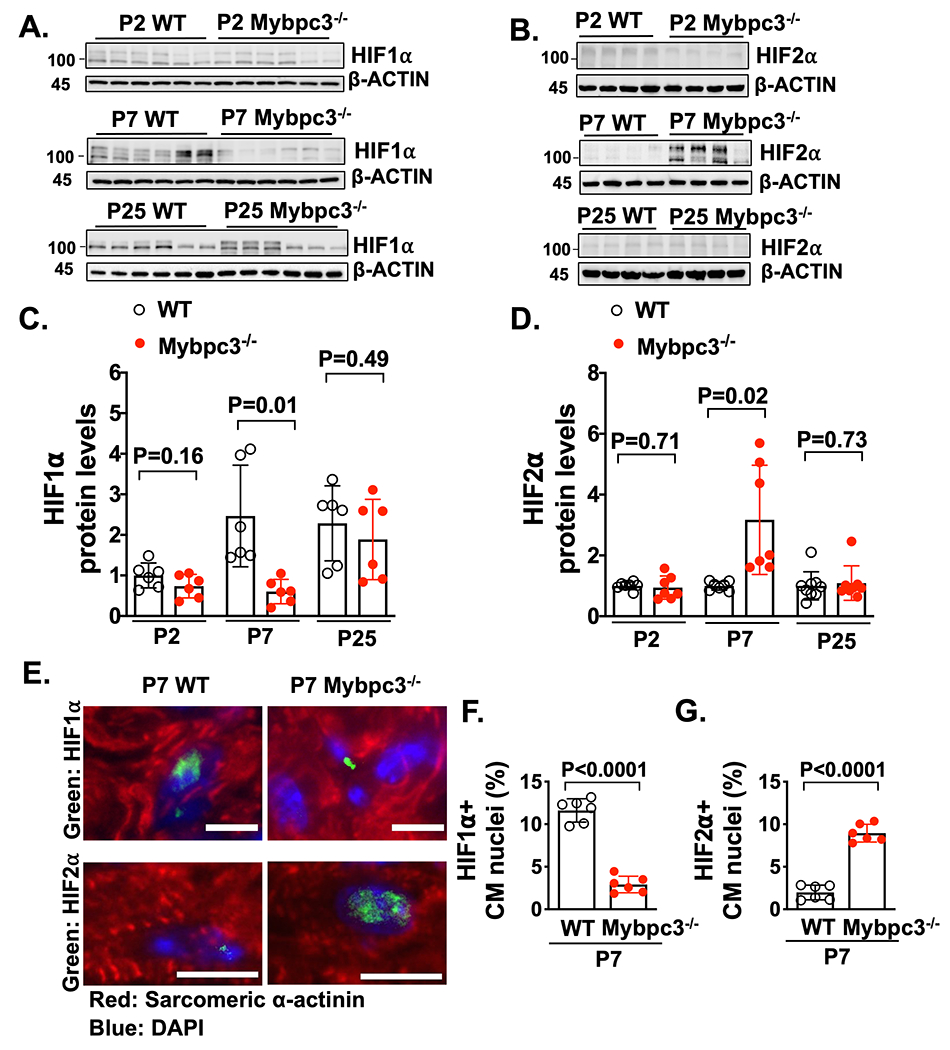
Immunoblots for hypoxia inducible factor 1α (HIF1α) (A) and hypoxia inducible factor 2α (HIF2α) (B) in LV tissue lysates from wild-type (WT) and Mybpc3−/− mice at postnatal day 2 (P2), P7 or P25. (C) HIF1α protein quantification from WT (n=6) and Mybpc3−/− (n=6) LV tissue lysates from P2, P7 or P25 mice normalized to β-actin protein expression and relative to P2 WT mice. (D) HIF2α protein quantification from WT (n=7-9) and Mybpc3−/− (n=7-8) LV tissue from P2, P7 or P25 mice normalized to β-actin and relative to P2 WT mice. (E) Representative immunohistochemistry images for HIF1α (top, green) and HIF2α (bottom, green) co-stained with sarcomeric α-actinin (red), in LV tissue from P7 WT and Mybpc3−/− mice. Nuclei are blue (DAPI). Scale bars=5um HIF1, 10 μm HIF2. (F) HIF1α positive CM nuclei (% of total nuclei) in LV tissue from P7 WT (n=6) and Mybpc3−/− (n=6) mice. Minimum 100 nuclei/sample. (G) HIF2α positive CM nuclei (% of total nuclei) in LV tissue from P7 WT (n=6) and Mybpc3−/− (n=6) mice. Minimum 100 nuclei/sample. All results are shown as mean±SEM. Student’s or Welch’s t-test were utilized for C, D, F and G.
The non-canonical degradation of HIF1⍺ in the Mybpc3−/− myocardium is regulated by cardiomyocyte MDM2.
We identified a reduction in Hif1α protein in the Mybpc3−/− myocardium at P7. However, it was unclear if the reduction of HIF1α protein was secondary to a reduction in gene expression or increased protein degradation. We found that despite the reduction of HIF1α protein at P7, there was increased Hif1α mRNA in Mybpc3−/− LV tissue (Figure 3A). Therefore, we hypothesized that there was increased degradation of HIF1α in the Mybpc3−/− myocardium. We administered the proteasomal inhibitor bortezomib (BTZ) and this normalized HIF1α protein (Figure 3B–C). We then performed an in situ proximity ligation assay (PLA) to measure the amount of ubiquitinated HIF1α in the groups of mice that had been administered BTZ. We discovered a robust increase in ubiquitinated HIF1α in the Mybpc3−/− LV tissue (Figure 3D–E, Figure S3A). These data show that the reduction of HIF1α in Mybpc3−/− LV tissue is secondary to increased ubiquitination and proteasomal degradation of HIF1α.
Figure 3. The non-canonical degradation of HIF1⍺ in the Mybpc3−/− myocardium is regulated by cardiomyocyte MDM2.
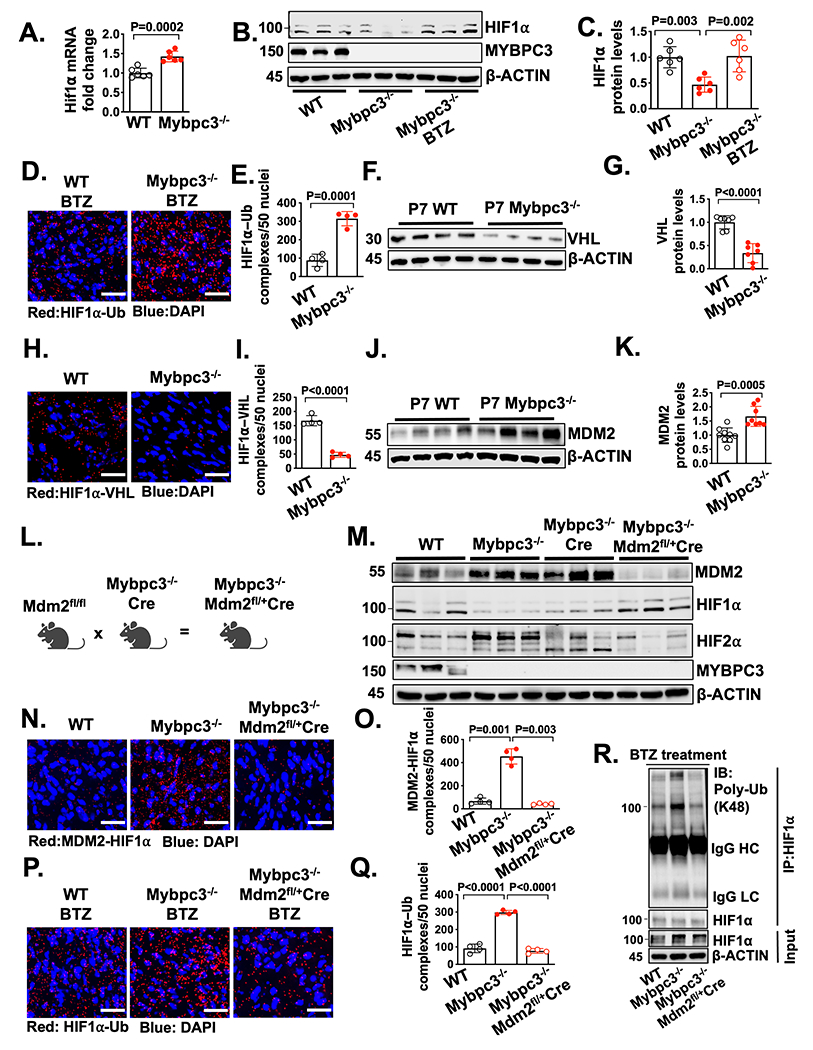
(A) Hif1α gene expression in LV tissue RNA from P7 WT (n=6) and Mybpc3−/− (n=6) mice. Hif1α gene expression was normalized to Rpl32 and fold change relative to WT. (B+C) Immunoblots and protein quantification for HIF1α in LV tissue lysates from P7 WT (n=6), Mybpc3−/− (n=6) and bortezomib (BTZ) injected Mybpc3−/− (n=6) mice normalized to β-actin and relative to WT. (D+E) Representative in situ proximity ligation assay (PLA) images and quantification for Ubiquitin (Ub) modified HIF1α in LV tissue from BTZ injected P7 WT (n=4) and Mybpc3−/− (n=4) mice. HIF1α-Ub complexes are red and nuclei are blue (DAPI). Three non-overlapping LV images/sample. Scale bars=25 μm. (F+G) Immunoblot and quantification for Von Hippel-Lindau (VHL) in LV tissue lysate from P7 WT (n=7) and Mybpc3−/− (n=7) mice normalized to β-actin and relative to WT. (H+I) Representative in situ PLA images and quantification for HIF1α and VHL protein complexes in LV tissue from P7 WT (n=4) and Mybpc3−/− (n=4) mice. HIF1α-VHL complexes are shown in red and nuclei are blue (DAPI). Three non-overlapping LV images/sample. Scale bars=25 μm. (J+K) Immunoblot and quantification for murine double minute 2 (MDM2) in LV tissue from P7 WT (n=9) and Mybpc3−/− (n=8) mice normalized to β-actin and relative to WT. (L) Schematic of cardiomyocyte selective reduction of MDM2 in Mybpc3−/− mice generated by crossing MDM2fl/fl , Mybpc3−/−, and Myh6:Cre mouse lines to create Mybpc3−/−MDM2fl/+/Myh6:Cre. (M) Immunoblot for MDM2, HIF1α, HIF2α, MYPBC3 and β-actin in LV tissue from P7 WT, Mybpc3−/−, Mybpc3−/−/Myh6:Cre and Mybpc3−/−MDM2fl/+/Myh6:Cre mice. (N+O) Representative in situ PLA images and quantification for MDM2 and HIF1α protein complexes in LV tissue from P7 WT (n=4), Mybpc3−/− (n=4) and Mybpc3−/−MDM2fl/+/Myh6:Cre (n=4) mice. MDM2-HIF1α complexes are shown in red and nuclei are blue (DAPI). Three non-overlapping LV images/sample. Scale bars=25 μm. (P+Q) Representative in situ PLA images and quantification for Ub modified HIF1α in LV tissue from BTZ injected P7 WT (n=4), Mybpc3−/− (n=4) and Mybpc3−/−MDM2fl/+/Myh6:Cre (n=4) mice. HIF1α-Ub complexes are red and nuclei are blue (DAPI). Three non-overlapping LV images/sample. Scale bars=25 μm. (R) Immunoprecipitation (IP) for HIF1α was performed on LV tissue lysates from BTZ injected P7 WT, Mybpc3−/− and Mybpc3−/−MDM2fl/+/Myh6:Cre mice and then immunoblots were performed for K48-linked ubiquitin (Poly-Ub) and HIF1α. The input LV tissue lysates also underwent immunoblotting for HIF1α and β-actin. All results are shown as mean±SEM. Student’s t-test was utilized for A, E, G, I and K. One-way ANOVA with Tukey’s or Dunnett’s T3 multiple comparison test were utilized for C, O and Q.
The canonical HIF1α ubiquitination and proteasomal degradation pathway require oxygen-dependent prolyl hydroxyl domain (PHD) enzymes to hydroxylate HIF1α which then allows the E3 ligase, Von Hippel-Lindau (VHL), to ubiquitinate HIF1α.29, 30 To determine if the canonical HIF1α degradation pathway was regulating HIF1α degradation we first measured VHL protein levels. Surprisingly, VHL protein levels were reduced in Mybpc3−/− (Figure 3F–G). We then measured in situ HIF1α-VHL protein complexes and found that HIF1α-VHL complexes were significantly reduced in Mybpc3−/− LV tissue (Figure 3H–I, Figure S3B). In addition, Phd gene expression was unchanged and the Pan-PHD inhibitor, molidustat, did not increase HIF1α protein levels in Mybpc3−/− mice (Figure S3C–F). This data shows that the canonical oxygen-dependent PHD-VHL degradation pathway is not regulating the increased proteasomal degradation of HIF1α in the Mybpc3−/− LV tissue.
In addition to the oxygen-dependent canonical HIF degradation pathway, oxygen-independent non-canonical HIF degradation pathways have been identified.31, 32 The E3 ligase murine double minute 2 (MDM2) has been shown to ubiquitinate HIF1α.33 Therefore, we wanted to determine if MDM2 was regulating the ubiquitination of HIF1α in Mybpc3−/− cardiomyocytes. First, we measured MDM2 protein levels and discovered that MDM2 protein levels were increased in Mybpc3−/− LV tissue during the same time point (P7) when HIF1α protein was decreased (Figure 3J–K). Next, we selectively reduced cardiomyocyte MDM2 levels in vivo using a transgenic strategy that utilized a cardiomyocyte selective Cre line (Myh6:Cre) (Figure 3L). This strategy successfully reduced MDM2 protein levels (Figure 3M, Figure S3M). Selective reduction of cardiomyocyte MDM2 led to the normalization of HIF1α and HIF2α protein levels in Mybpc3−/− LV tissue (Figure 3M, Figure S3M).
We previously discovered that Mybpc3−/− cardiomyocytes develop increased DNA damage and increased gene expression of MDM2 at P25.21 Therefore, we measured MDM2 mRNA levels at P7 to determine if increased MDM2 gene expression was the reason for the elevated MDM2 protein levels at P7 in Mybpc3−/− mice. Interestingly, we found that MDM2 mRNA was reduced in the Mybpc3−/− mice at P7 but was increased at P25 (Figure S3G). It was previously shown that MDM2 can serve as a negative regulator of its own gene expression through inhibiting p53.34 Indeed, we detected a rebound in MDM2 mRNA levels when cardiomyocyte MDM2 protein levels were reduced in Mybpc3−/− mice at P7 (Figure S3H). Likewise, MDM2 mRNA increased at P25 when levels of MDM2 protein declined (Figure S3G+I). These results suggested that cardiomyocyte MDM2 gene expression was not the primary cause of increased MDM2 protein levels at P7 and suggested that MDM2 protein stability was altered in the Mybpc3−/− mice. Indeed, we found that MDM2 protein ubiquitination was reduced in P7 Mybpc3−/− LV tissue (Figure S3J+K). In addition, we found that MDM2 and MDM4 (aka MDMX) protein complexes were reduced in Mybpc3 LV tissue (Figure S3L). It was previously shown that MDM4 can stimulate MDM2 autoubiquitination and degradation.35 Importantly, when MDM2 protein levels decrease at P25 in the Mybpc3−/− mice (Fig S3I), this is associated with a normalization of myocardial levels of HIF1α and HIF2α (Fig 2A+B). Overall, these results suggest that MDM2 protein is increased at P7 in the Mybpc3−/− myocardium secondary changes in its protein stability.
Next, we performed measured in-situ HIF1α-MDM2 protein complexes and discovered that they were robustly increased in Mybpc3−/− LV tissue and greatly reduced by selective cardiomyocyte reduction of MDM2 (Figure 3N–O, Figure S3N). We then measured in situ HIF1α ubiquitination in LV tissue and found that a reduction of cardiomyocyte MDM2 led to a corresponding reduction in HIF1α ubiquitination (Figure 3P–Q). We confirmed our in-situ PLA results by performing immunoprecipitation of LV tissue lysates using a HIF1α antibody and then immunoblotting with a K48 ubiquitin selective antibody (Figure 3R). Finally, the E3 ligase function of MDM2 is a well-described inducer of p53 protein degradation.36 Therefore, we wanted to determine if reducing cardiomyocyte MDM2 led to a surge of p53 protein levels. However, this did not occur, and LV p53 protein levels decreased in parallel with a reduction of cardiomyocyte MDM2 protein (Figure S3O). Overall, these results show that cardiomyocyte MDM2 is facilitating the non-canonical ubiquitination and degradation of HIF1α in Mybpc3−/− LV tissue.
Increased HIF2α in the Mybpc3−/− myocardium occurs secondary to MDM2 facilitated degradation of VHL.
Although we identified the molecular mechanisms regulating the increased degradation of HIF1α in Mybpc3−/− cardiomyocytes, it remained unclear why HIF2α protein levels were increased. We hypothesized that the reduced VHL protein levels may be contributing to the increased levels of HIF2α (Figure 3F). We first measured Vhl gene expression and detected no difference between the groups (Figure 4A). This result suggested the reduction of VHL protein in Mybpc3−/− LV tissue was secondary to increased proteasomal degradation. Indeed, we found that VHL protein levels rebounded to normal levels when a proteasome inhibitor was administered to the Mybpc3−/− mice (Figure 4B–C). Next, we performed a coimmunoprecipitation assay to determine if MDM2 bound to VHL and discovered that there was increased VHL binding to MDM2 in Mybpc3−/− LV tissue lysates (Figure 4D). In situ MDM2-VHL protein complexes were also increased in Mybpc3−/− LV tissue and were reduced with the selective reduction of cardiomyocyte MDM2 (Figure 4E–F, Figure S4A). Next, we discovered that in situ VHL ubiquitination was increased in Mybpc3−/− LV tissue and this increase was eliminated with reduction of cardiomyocyte MDM2. (Figure 4G–H, Figure S4B). We confirmed our in-situ PLA results by performing immunoprecipitation of LV tissue lysates using a VHL antibody and then immunoblotting with a K48 ubiquitin selective antibody (Figure 4I). In contrast to VHL ubiquitination, the neddylation of VHL was unchanged (Figure S4C–D). Taken together, these results show that MDM2 regulates the ubiquitination and degradation of VHL in Mybpc3−/− cardiomyocytes.
Figure 4. Increased HIF2⍺ in the Mybpc3−/− myocardium occurs secondary to MDM2 facilitated degradation of VHL.
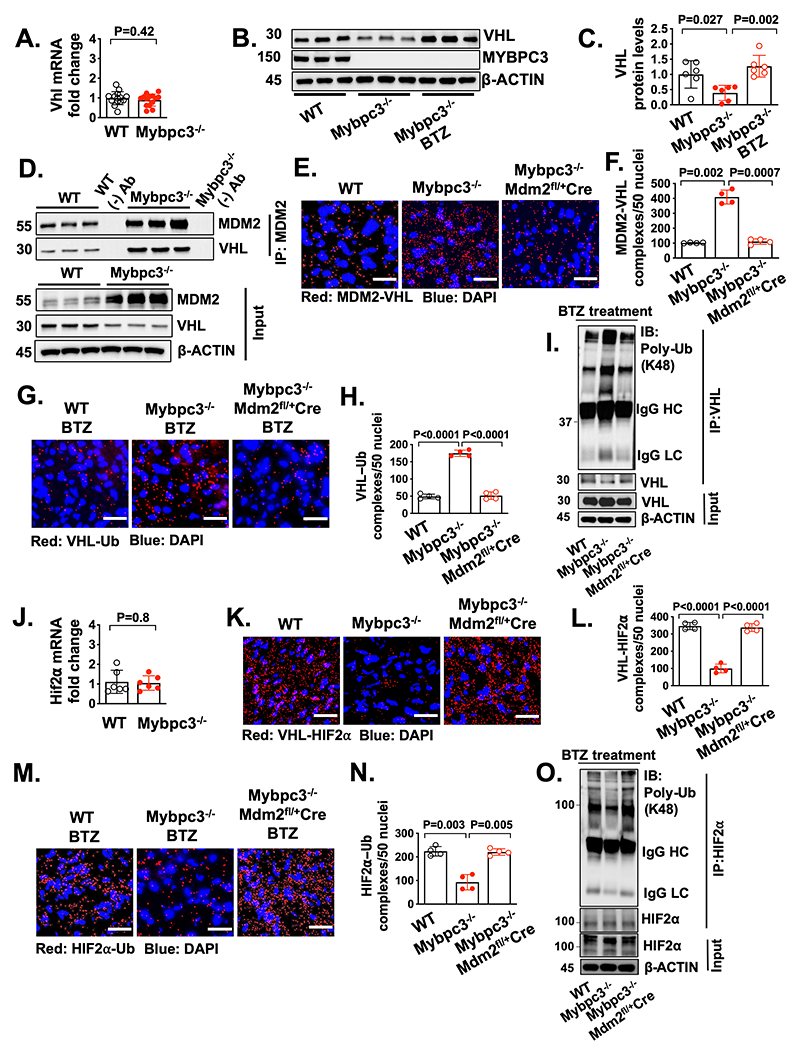
(A) Von Hippel-Lindau (Vhl) gene expression in LV tissue RNA from P7 WT (n=14) and Mybpc3−/− (n=14) mice. Vhl gene expression was normalized to Rpl32 expression and fold change is relative to WT. (B+C) Immunoblots and protein quantification for VHL in LV tissue from P7 WT (n=6), Mybpc3−/− (n=6) and BTZ injected Mybpc3−/− (n=6) mice normalized to β-actin and relative to WT. (D) Coimmunoprecipitation (Co-IP) for MDM2 was performed in LV tissue lysates from P7 WT and Mybpc3−/− mice and then immunoblots were performed for VHL and MDM2. The third WT and Mybpc3−/− sample also underwent bead only precipitation without the MDM2 antibody as a negative control experiment (WT (−) Ab and Mybpc3−/− (−) Ab). The input LV tissue lysates also underwent immunoblotting for MDM2, VHL, and β-actin. (E+F) Representative in situ proximity ligation assay (PLA) images and quantification for MDM2 and VHL protein complexes in LV tissue from P7 WT (n=4), Mybpc3−/− (n=4) and Mybpc3−/−MDM2fl/+/Myh6:Cre (n=4) mice. MDM2-VHL complexes are red and nuclei are blue (DAPI). Three non-overlapping LV images/sample. Scale bars=25 μm. (G+H) Representative in situ PLA images and quantification for ubiquitin (Ub) modified VHL in LV tissue from BTZ-treated P7 WT (n=4), Mybpc3−/− (n=4) and Mybpc3−/−MDM2fl/+/Myh6:Cre (n=4) mice. VHL-Ub complexes are red and nuclei are blue (DAPI). Three non-overlapping LV images/sample. Scale bars=25 μm. (I) Immunoprecipitation (IP) for VHL was performed on LV tissue lysates from BTZ injected P7 WT, Mybpc3−/− and Mybpc3−/−MDM2fl/+/Myh6:Cre mice and then immunoblots were performed K48-linked ubiquitin (Poly-Ub) and VHL. The input LV tissue lysates also underwent immunoblotting for VHL and β-actin. (J) Hypoxia inducible factor (Hif) 2α gene expression in LV tissue RNA from P7 WT (n=6) and Mybpc3−/− (n=6) mice. Hif2α gene expression was normalized to Rpl32 and fold change is relative to WT. (K+L) Representative in situ PLA images for VHL and HIF2α protein complexes in LV tissue from P7 WT (n=4), Mybpc3−/− (n=4) and Mybpc3−/−MDM2fl/+/Myh6:Cre (n=4) mice. VHL-HIF2α complexes are red and nuclei are blue (DAPI). Three non-overlapping LV images/sample. Scale bars=25 μm. (M+N) Representative in situ PLA images and quantification for Ub modified HIF2α in LV tissue from BTZ injected P7 WT (n=4), Mybpc3−/− (n=4) and Mybpc3−/−MDM2fl/+/Myh6:Cre (n=4) mice. HIF2α-Ub complexes are red and nuclei are blue (DAPI). Three non-overlapping LV images/sample. Scale bars=25 μm. (O) Immunoprecipitation (IP) for HIF2α was performed on LV tissue lysates from BTZ injected P7 WT, Mybpc3−/− and Mybpc3−/−MDM2fl/+/Myh6:Cre mice and then immunoblots for K48-linked ubiquitin (Poly-Ub) and HIF2α. The input LV tissue lysates also underwent immunoblotting for HIF2α and β-actin. All results are shown as mean±SEM. Student’s t-test was utilized for A and J. One-way ANOVA with Tukey’s or Dunnett’s T3 multiple comparison test were utilized for C, F, H, L and N.
We identified a novel role for cardiomyocyte MDM2 in the regulation of VHL ubiquitination and VHL protein stability. Therefore, we hypothesized that the increase in HIF2α protein levels in Mybpc3−/− LV tissue was related to the changes we identified in VHL. First, we confirmed that Hif2α expression was unchanged in the groups (Figure 4J). However, in situ VHL-HIF2α protein complexes were greatly reduced in the Mybpc3−/− LV tissue but were normalized with the reduction of MDM2 (Figure 4K–L, Figure S4E). Importantly, we confirmed that there were minimal MDM2 and HIF2α protein complexes in LV tissue (Figure S4F–G). We then measured in situ HIF2α ubiquitination and found that HIF2α ubiquitination was reduced in Mybpc3−/− LV tissue and was normalized by the cardiomyocyte selective reduction of MDM2 (Figure 4M–N, Figure S4H). Again, we confirmed our in-situ PLA results by performing immunoprecipitation of LV tissue lysates using a HIF2α antibody and then immunoblotting with a K48 ubiquitin selective antibody (Figure 4O). Overall, these results show that cardiomyocyte MDM2 facilitates the degradation of VHL which leads to dynamic increases in HIF2α protein levels.
Reduction of cardiomyocyte MDM2 in Mybpc3−/− mice increases myocardial capillary formation and pro-angiogenic gene expression
We identified a role for cardiomyocyte MDM2 in regulating the protein stability of both HIF1α and HIF2α. Therefore, we wanted to determine if selective reduction of cardiomyocyte MDM2 would impact capillary formation in Mybpc3−/− mice. Indeed, we found that the reduction of cardiomyocyte MDM2 led to increased LV capillaries in Mybpc3−/− mice (Figure 5A–B). Likewise, pericytes were also increased with selective reduction of cardiomyocyte MDM2 in Mybpc3−/− mice (Figure S5A–B). We wanted to determine if the reduction of LV capillary density in the Mybpc3−/− myocardium was associated with reduced expression of angiogenic genes. Indeed, we found that Mybpc3−/− LV tissue had reduced expression of multiple pro-angiogenic genes (Figure 5C). Importantly, the selective cardiomyocyte reduction of MDM2 could reverse these gene expression changes (Figure 5C).
Figure 5. Reduction of cardiomyocyte MDM2 in Mybpc3−/− mice increases myocardial capillary formation and pro-angiogenic gene expression.
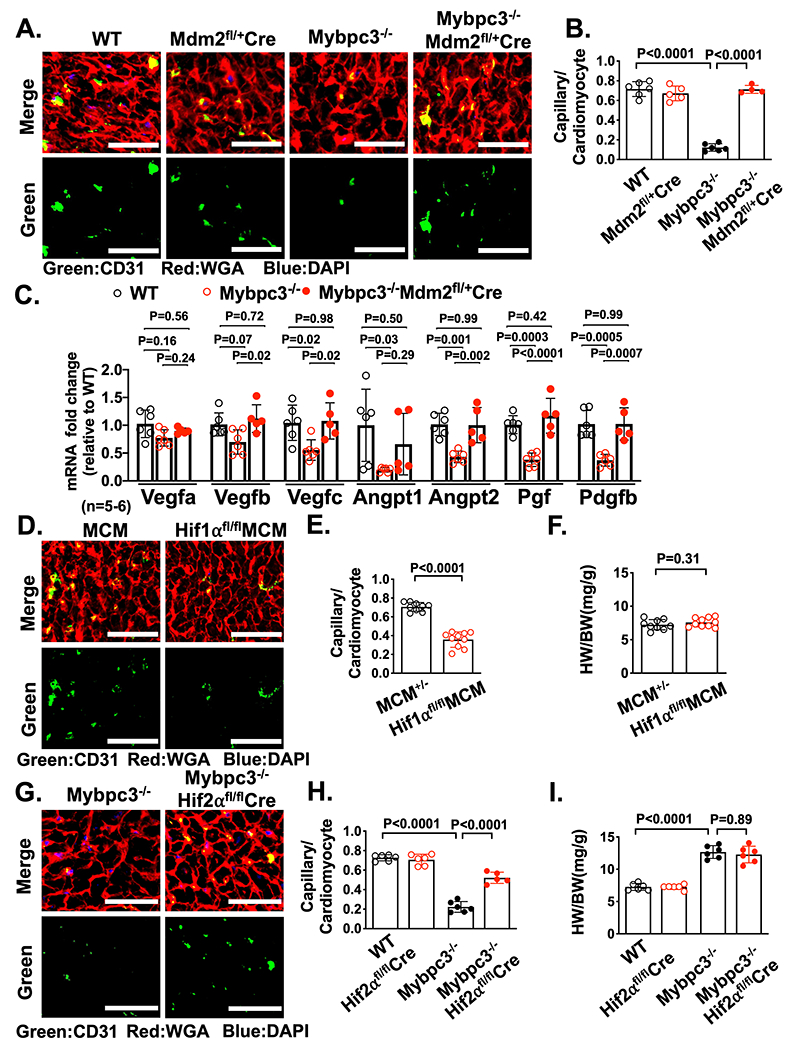
(A) Representative immunohistochemistry images for CD31 (green) co-stained with WGA (red) in LV tissue from P7 WT, Mdm2fl/+/Myh6:Cre, Mybpc3−/− and Mybpc3−/−MDM2fl/+/Myh6:Cre mice. Nuclei are blue (DAPI). Scale bars=30 μm. (B) Capillary to cardiomyocyte ratios in LV tissue from P7 WT (n=6), Mdm2fl/+/Myh6:Cre (n=5), Mybpc3−/− (n=6) and Mybpc3−/− MDM2fl/+/Myh6:Cre (n=4) mice. Minimum 100 cardiomyocytes/sample. (C) Pro-angiogenic gene expression (Vascular endothelial growth factor a [Vegfa], Vegfb, Vegfc, Angiopoietin 1 [Angpt1], Angpt2, Placental growth factor [Pgf], Platelet derived growth factor subunit b [Pdgfb]) in LV tissue RNA from P7 WT (n=6), Mybpc3−/− (n=6) and Mybpc3−/− MDM2fl/+/Myh6:Cre (n=5) mice. The genes of interest were normalized to Rpl32 and fold changes are relative to WT. (D) Representative immunohistochemistry images for CD31 (green) co-stained with WGA (red) in LV tissue from P7 Myh6:MerCreMer (MCM) and HIF1αfl/flMCM mice injected with tamoxifen at P1 and P4. Nuclei are blue (DAPI). Scale bars=50 μm. (E) Capillary to cardiomyocyte ratios in LV tissue from P7 MCM(n=9) and HIF1αfl/flMCM (n=10) injected with tamoxifen at P1 and P4. Minimum 200 cardiomyocytes/sample. (F) Heart weight (mg) to body weight (g) ratios (HW/BW) from P7 MCM (n=9) and HIF1αfl/fl MCM (n=10) mice injected with tamoxifen at P1 and P4. (G) Representative immunohistochemistry images for CD31 (green) co-stained with WGA (red) in LV tissue from P7 Mybpc3−/− and Mybpc3−/−HIF2αfl/fl/Myh6:Cre mice. Nuclei are blue (DAPI). Scale bars=50 μm. (H) Capillary to cardiomyocyte ratios in LV tissue from P7 WT (n=6), HIF2αfl/fl/Myh6:Cre (n=6), Mybpc3−/− (n=6) and Mybpc3−/−HIF2αfl/fl/Myh6:Cre (n=5) mice. Minimum 200 cardiomyocytes/sample. (I) Heart weight (mg) to body weight (g) ratios (HW/BW) from P7 WT (n=6), HIF2αfl/fl/Myh6:Cre (n=6), Mybpc3−/− (n=6) and Mybpc3−/−HIF2αfl/fl/Myh6:Cre (n=6) mice. All results are shown as mean±SEM. Student’s t-test was utilized for E and F. One-way ANOVA with Tukey or Dunnett’s T3 multiple comparison test were utilized for C. Two-way ANOVA with Tukey’s multiple comparison test was utilized for B, H and I.
We next investigated whether reduction of capillary density in the Mybpc3−/− myocardium was secondary to the reduction of HIF1α or the increase of HIF2α protein at P7. Selective reduction of cardiomyocyte HIF1α during the P2 to P7 postnatal time period in WT mice led to a reduction in LV capillary formation (Figure 5D–E, Figure S5C). In contrast, the reduction of cardiomyocyte HIF2α in Mybpc3−/− mice resulted in a partial increase of capillary density in Mybpc3−/− mice but had no impact on WT LV capillary density (Figure 5G–H, Figure S5D). These results show that the transient imbalance of both HIF1α and HIF2α are contributing to the capillary formation abnormality in Mybpc3−/− mice.
Mybpc3−/− mice rapidly develop LV hypertrophy and dysfunction in the early postnatal time period.20 Therefore, we measured cardiac structure and function in Mybpc3−/− mice with reduction of cardiomyocyte MDM2. We discovered that selective reduction of cardiomyocyte MDM2 did reduce LV hypertrophy and improve systolic function in Mybpc3−/− mice during the early postnatal time period (Figure S5E–G). In contrast, targeted elimination of cardiomyocyte HIF1α and HIF2α did not cause significant changes in cardiac structure or function in the early postnatal time period (Figure S5H–M). These results show that the MDM2-induced HIF imbalance selectively alters LV capillary formation during the early postnatal time period in Mybpc3−/− mice.
MDM2 regulates capillary formation before the development of ventricular hypertrophy in Myh6R404Q/WT mice.
We discovered that Mybpc3−/− mice have reduced LV capillary formation during the early postnatal time period and that MDM2 plays a critical role in this process. However, it remained unclear if LV capillary formation defects could occur independently of LVH and if other sarcomere gene mutations had shared pathophysiological responses to the Mybpc3−/− mice. To address these questions, we utilized genetic editing to create a murine model with a point mutation in the Myh6 gene (Myh6R404Q/WT) (Figure S6A+B) to model the human MYH7 R403Q mutation, which is a well described cause of HCM in humans.37 The murine MYH6 protein is targeted because it is the dominant myosin heavy chain protein in the adult mouse LV versus MYH7 in the adult human LV and this strategy was successfully utilized in the past with homologous recombination methods.38 In contrast to Mybpc3−/− mice that rapidly develop LVH by postnatal day 7, heterozygote Myh6R404Q/WT mice had normal LV wall thickness through postnatal day 25 but developed LVH by postnatal day 60 without systolic dysfunction (Figure 6A–C, Figure S6C). Male and female Myh6R404Q/WT mice developed similar levels of LVH by P60 but male mice had slightly more LVH by 6 months of age (Figure S6D–G). Similar to the development of myocardial hypertrophy, the heterozygote Myh6R404Q/WT mice developed cardiomyocyte hypertrophy by P60 (Figure 6D–E). Therefore, this HCM model of adult onset LVH allowed us to determine if sarcomere protein dysfunction impacted myocardial capillary formation before the development of left ventricular hypertrophy.
Figure 6. MDM2 regulates capillary formation in Myh6R404Q/WT mice before the development of ventricular hypertrophy.
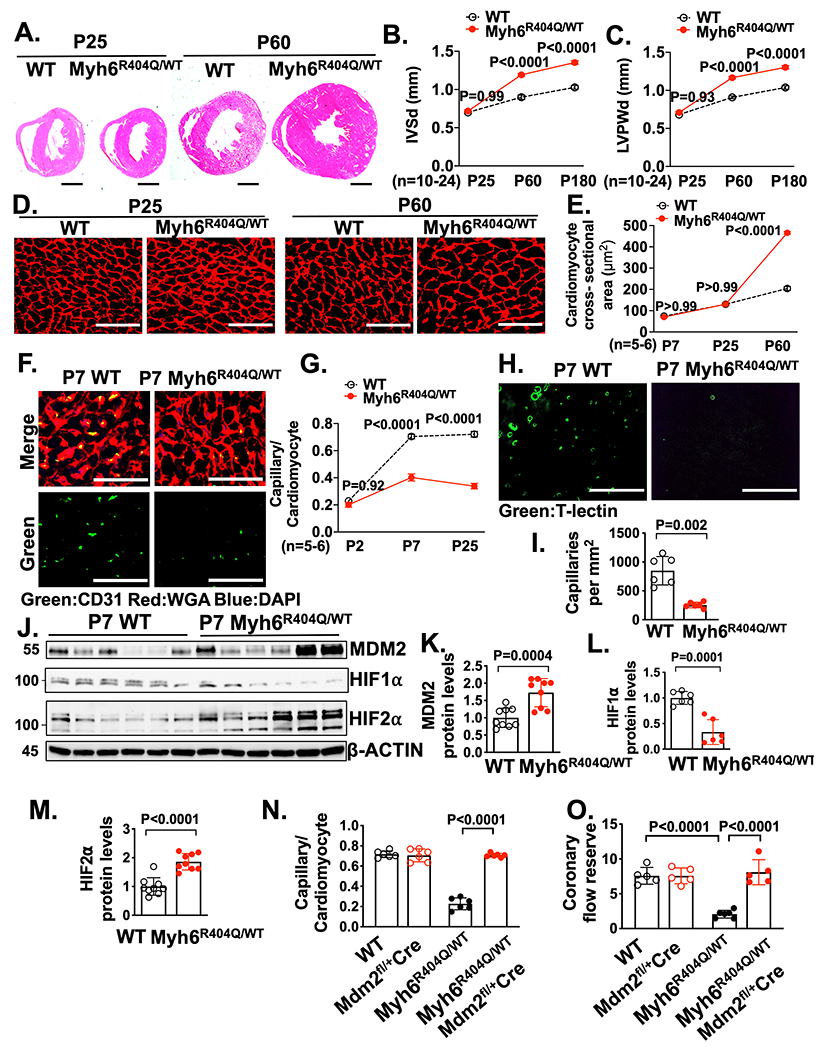
(A) Representative H&E–stained heart cross-sections of P25 and P60 WT and Myh6R404Q/WT mice. Scale bars=1 mm. Echocardiography assessment of (B) interventricular septal thickness at end diastole (IVSd) and (C) LV posterior wall thickness at end diastole (LVPWd) in WT (n=10-11), and Myh6R404Q/WT (n=19-24) mice at P25, P60 or P180. (D) Representative images of WGA (red) stained LV tissue from WT and Myh6R404Q/WT mice at P25 or P60. Scale bar=75 μm. (E) Cardiomyocyte cross-sectional areas from WGA stained LV tissue from WT (n=5-6) and Myh6R404Q/WT (n=5-6) mice at P7, P25 or P60. Minimum 50 cardiomyocytes/sample. (F) Representative immunohistochemistry images for CD31 (green) co-stained with WGA (red) in LV tissue from P7 WT and Myh6R404Q/WT mice. Nuclei are blue (DAPI). Scale bars=50 μm. (G) Capillary to cardiomyocyte ratios in LV tissue from WT (n=5-6) and Myh6R404Q/WT (n=5-6) mice at P2, P7 or P25. Minimum 200 cardiomyocytes/sample. (H) Representative fluorescence images for the intravascularly injected endothelial cell stain tomato lectin (T-lectin) (green) in LV tissue from P7 WT and Myh6R404Q/WT mice. Scale bars=80 μm. (I) Capillaries per mm2 in LV tissue from P7 WT (n=6) and Myh6R404Q/WT (n=6) mice. Three cross-sectional images per sample were analyzed. (J-M) Immunoblots and quantification for MDM2, HIF1α, and HIF2α in LV tissue lysates from P7 WT (n=6-9) and Myh6R404Q/WT (n=6-9) mice normalized to β-actin and relative to WT. (N) Capillary to cardiomyocyte ratios were calculated from LV tissue in P7 WT (n=6), MDM2fl/+/Myh6:Cre (n=6), Myh6R404Q/WT (n=6) and Myh6R404Q/WTMDM2fl/+/Myh6:Cre (n=6) mice. Minimum 200 cardiomyocytes/sample. (O) Coronary flow reserve in P25 WT (n=5), Mdm2fl/+/Myh6:Cre (n=5), Myh6R404Q/WT (n=6) and Myh6R404Q/WTMdm2fl/+/Myh6:Cre (n=5) mice. All results are shown as mean±SEM. Student’s t-test or Welch’s t-test were utilized for I, K, L and M. Two-way ANOVA with Tukey multiple comparison test was utilized for B, C, E, G, N and O.
We analyzed LV capillary density in the Myh6R404Q/WT mice and discovered that capillary formation was reduced (Figure 6F–I, and Video 3–4). Similar to Mybpc3−/− mice, both male and female Myh6R404Q/WT mice had similar reductions in capillary density (Figure S6H). However, in contrast to Mybpc3−/− mice, left coronary artery size of Myh6R404Q/WT mice was similar to WT mice at P7 (Figure S6I–J). We then wanted to determine if the changes in LV capillary formation in Myh6R404Q/WT mice were related to changes in MDM2-HIF signaling. We found that Myh6R404Q/WT myocardium had an increase in MDM2 protein (Figure 6J–K). Similar to Mybpc3−/− mice,21 the increase in MDM2 in the Myh6R404Q/WT mice was associated with increased cardiomyocyte DNA damage (Figure S6K–L). Importantly, we found no evidence of increased endothelial cell DNA damage (Figure S6M–N). We then measured HIF1α and HIF2α protein levels and discovered that P7 Myh6R404Q/WT myocardium had reduced HIF1α and increased HIF2α protein levels (Figure 6J, 6L–M). In addition, VHL protein levels were also reduced in P7 Myh6R404Q/WT myocardium (Figure S6O–P). Next, we wanted to determine if selective reduction of cardiomyocyte MDM2 would increase capillary density and prevent microvascular dysfunction in Myh6R404Q/WT mice in the pre-LVH period. Indeed, we found that genetic reduction of cardiomyocyte MDM2 in Myh6R404Q/WT mice led to increased LV capillary formation and normalization of coronary flow reserve (Figure 6N–O, Figure S7A–C). In contrast to the Mybpc3−/− model that had LVH at P7, we did not detect evidence of myocardial tissue hypoxia in the Myh6R404Q/WT model at P7 (Figure S7D). Therefore, despite the absence of myocardial tissue hypoxia in the Myh6R404Q/WT model, the abnormalities in the microvasculature and molecular alterations in MDM2, HIF1/2 and VHL were conserved with the Mybpc3−/− model. Overall, these results show that myocardial capillary formation precedes the development of ventricular hypertrophy in Myh6R404Q/WT mice and microvascular dysfunction can be prevented by genetic reduction of cardiomyocyte MDM2.
Chemical inhibition of MDM2 prevents microvascular dysfunction in two distinct HCM models
We discovered that cardiomyocyte MDM2 regulates the development of microvascular dysfunction in both the Mybpc3−/− and Myh6R404Q/WT mouse lines. Therefore, we wanted to determine if chemically targeting MDM2 could prevent the development of microvascular dysfunction in these models. We utilized MD-224, a chemical proteolysis targeting chimeric (PROTAC) compound, to selectively degrade MDM2 in the early postnatal time period (Figure 7A). Chemical targeting of MDM2 led to the normalization of HIF1α and HIF2α levels in both Mybpc3−/− (Figure 7B) and Myh6R404Q/WT mice (Figure 7C). Likewise, chemical MDM2 degradation led to an increase in LV capillary density in both Mybpc3−/− and Myh6R404Q/WT mice (Figure 7D–E).
Figure 7. Chemical inhibition of MDM2 prevents microvascular dysfunction in two distinct HCM models.
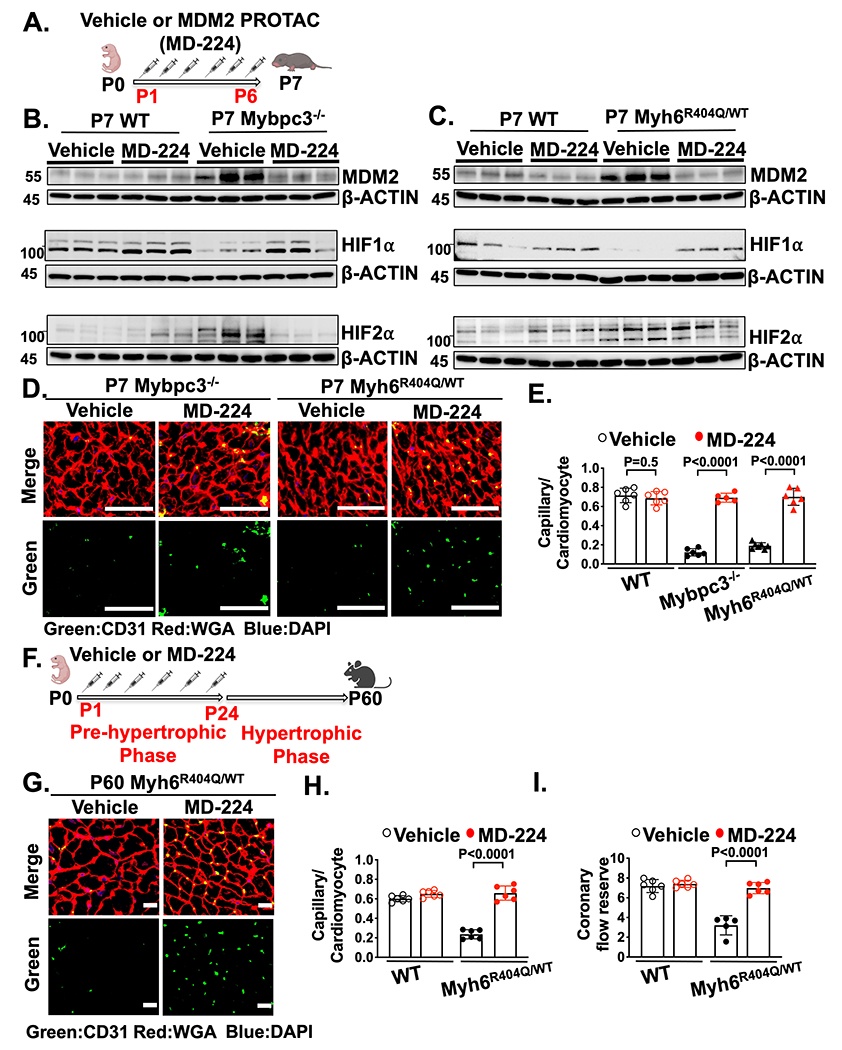
(A) Schematic of injections of vehicle or MDM2 PROTAC (MD-224) from P1 to P6. (B) Immunoblots for MDM2, HIF1α and HIF2α in LV tissue from P7 WT or Mybpc3−/− injected with vehicle or MD-224. (C) Immunoblots for MDM2, HIF1α and HIF2α in LV tissue from P7 WT and Myh6R404Q/WT injected with vehicle or MD-224. (D) Representative immunohistochemistry images for CD31 (green) co-stained with WGA (red) in LV tissue from P7 Mybpc3−/− and Myh6R404Q/WT injected with vehicle or MD-224 from P1-P6. Nuclei are blue (DAPI). Scale bars=50 μm. (E) Capillary to cardiomyocyte ratios in LV tissue from P7 WT vehicle (n=6), WT MD-224 (n=6), Mybpc3−/− vehicle (n=6), Mybpc3−/− MD-224 (n=5), Myh6R404Q/WT vehicle (n=7) and Myh6R404Q/WT MD-224 (n=6) mice. All groups injected from P1-P6 with either vehicle or MD-224. Minimum 200 cardiomyocytes/sample. (F) Schematic of WT or Myh6R404Q/WT mice injected with vehicle or MDM2 PROTAC (MD-224) from P1 to P24 and then analyzed at P60. (G) Representative immunohistochemistry images for CD31 (green) co-stained with WGA (red) in LV tissue from P60 Myh6R404Q/WT mice injected with vehicle or MD-224 from P1 to P24. Nuclei are blue (DAPI). Scale bars=15 μm. (H) Capillary to cardiomyocyte ratios in LV tissue from P60 WT vehicle (n=6), WT MD-224 (n=6), Myh6R404Q/WT vehicle (n=6) and Myh6R404Q/WT MD-224 (n=6) mice. All groups injected from P1 to P24 with vehicle or MD-224. Minimum 140 cardiomyocytes/sample. (I) Coronary flow reserve was calculated in P60 WT vehicle (n=6), WT MD-224 (n=6), Myh6R404Q/WT vehicle (n=5), and Myh6R404Q/WT MD-224 (n=6) mice. All results are shown as mean±SEM. Student’s t-test or Welch’s t-test were utilized for E, H, and I.
Similar to genetic reduction of MDM2, chemical targeting of MDM2 using MD-224 reduced myocardial hypertrophy and improved LV systolic function in Mybpc3−/− mice (Figure S8A–F). Next, we wanted to determine if transient targeting of MDM2 during the pre-hypertrophic period of Myh6R404Q/WT mice would lead to persistent improvement in LV capillary density and coronary flow reserve in the adult animal (Figure 7F). We found that transient chemical targeting of MDM2 during the early postnatal time period led to a persistent increase in LV capillary density and normalized coronary flow reserve in adult Myh6R404Q/WT mice (Figure 7G–I, Figure S8G). In addition, transient chemical targeting of MDM2 prevented the development of LVH in the Myh6 Myh6R404Q/WT model at P60 (Figure S8H–M). In summary, these findings show that transient chemical targeting of MDM2 prevents the HIF imbalance and myocardial capillary dysfunction in two unrelated models of HCM.
DISCUSSION
Myocardial microvascular dysfunction (MVD) has been identified in multiple different forms of human cardiomyopathy and is often thought to occur as a consequence of the pathological myocardial remodeling process.39 Myocardial MVD has been demonstrated in humans with hypertrophic cardiomyopathy (HCM) and is thought to be a significant contributor to disease pathogenesis.40–42 However, it remains unclear when MVD develops in HCM and the molecular mechanisms regulating this pathological process. Utilizing two distinct animal models of HCM, we discovered that early postnatal myocardial capillary formation is impaired, and this leads to MVD in adult animals. The reduction in myocardial capillary formation in HCM is caused by the E3 ligase MDM2 inducing an imbalance of cardiomyocyte HIF1α and HIF2α protein levels during the early postnatal time period. Importantly, myocardial MVD can precede the development of myocardial hypertrophy in HCM and selective targeting of MDM2 prevents myocardial MVD from developing. This study identified a key regulatory mechanism controlling the development of myocardial MVD and provides unique insights into molecular pathways contributing to the pathophysiology of HCM (Figure S9).
We have identified a unique role for MDM2 as a critical regulator of MVD in HCM by reducing myocardial angiogenesis in the early postnatal heart. MDM2 has not been previously shown to regulate MVD in the heart or other organ systems. In contrast, increased expression of MDM2 has been shown to play a pro-angiogenic role in malignant tissues.43–45 The divergent pro and anti-angiogenic properties of MDM2 appear to be partially explained by the acute versus chronic overexpression of the protein. For example, the chronic overexpression of MDM2 in malignant tissues can lead to increased p53 degradation which facilitates pro-angiogenic gene expression.43 In contrast, the acute increase in cardiomyocyte MDM2 in our HCM models led to an anti-angiogenic role for this protein through its role in simultaneously regulating the protein stability of HIF1α and HIF2α.
We found that MDM2 concurrently regulated the non-canonical degradation of HIF1α and the canonical degradation of HIF2α leading to an imbalance in these proteins. Previously it was shown that MDM2 can regulate HIF1α ubiquitination in cancer cell lines31, but our data indicates this non-canonical mechanism of HIF1α degradation is important in vivo in a non-malignant context. In addition, we discovered that in vivo cardiomyocyte MDM2 can bind and regulate the ubiquitination of VHL leading to a reduction in the canonical degradation of HIF2α. Our results identified MDM2 as a specific regulator of HIF and VHL protein stability in the early stages of HCM and reinforces the increasing evidence that the ubiquitin-proteosome system is an important regulator in HCM.46–49 Interestingly, it was previously shown that MDM2 can also regulate VHL neddylation in cancer cell lines.50 However, in contrast to the effect that MDM2 has on VHL ubiquitination, we found no evidence that MDM2 dynamically regulated VHL neddylation in vivo in our disease model. Interestingly, we discovered that HIF1α and HIF2α have opposing roles in regulating myocardial capillary formation in the postnatal mammalian heart, with HIF1α serving a pro-angiogenic role and HIF2α serving an anti-angiogenic role. This is in contrast to tumor angiogenesis, where HIF1α and HIF2α are more often reported to have synergistic roles in promoting angiogenesis.51 Our data shows that the regulation of organ angiogenesis by HIF1α and HIF2α is highly dependent on cell type and tissue environment. This study adds to the growing data that cardiomyocyte HIF signaling has distinct roles in regulating myocardial growth and maturation depending on the developmental time point when they are expressed.52, 53
Another important result from this study is that MVD can develop before cardiomyocyte hypertrophy. Using our Myh6R404Q HCM model that develops LVH in the adolescent period of mouse development, we identified early postnatal capillary defects before the development of LVH. These results suggest that myocardial MVD and myocardial hypertrophy in HCM can occur through distinct molecular mechanisms. Supporting this conclusion are the results from our targeted cardiomyocyte deletion of the MDM2 target proteins HIF1α and HIF2α. Selective reduction of cardiomyocyte HIF1α and HIF2α had opposing effects on myocardial capillary growth but had no measurable impact on myocardial hypertrophic growth in the early postnatal time period. Likewise, we found that targeting MDM2 activity in the early postnatal time period in the Myh6R404Q model prevented myocardial MVD well before the onset of left ventricular hypertrophy. Our results are further supported by clinical studies in human carriers of pathogenic sarcomere gene mutations which identified evidence of MVD even before the development of myocardial hypertrophy.54–56 Although we have defined a unique role of MDM2 in regulating myocardial HIF signaling and capillary growth in two distinct HCM models, it remains unknown how alterations in MDM2 signaling lead to changes in cardiomyocyte hypertrophy in HCM.
In contrast to this study which is focused on how a lifelong genetic hypertrophic stimulus leads to MVD, acquired forms of LVH secondary to hypertension or aortic stenosis typically occur during adulthood. MVD in acquired forms of LVH is thought to be from capillary regression leading to microvascular rarefaction. Therefore, it would be assumed that MVD in genetic and acquired forms of hypertrophy may have divergent mechanisms. However, capillary rarefaction in pressure overload was also shown to occur before the development of LVH.18 Likewise, alterations in p53 activity were previously identified to regulate capillary regression in a pressure overload LVH model.16 Therefore, it will be important to determine how dynamic changes in MDM2 signaling impacts capillary rarefaction in acquired forms of LVH.
Our results show that MVD can be prevented by partial genetic or chemical reduction of MDM2 in two different mouse models of HCM. Importantly, we found that partial chemical or genetic reduction of MDM2 in our models did not lead to an increase in myocardial p53 protein levels. In contrast, previous studies have shown that complete ablation of cardiomyocyte MDM2 during the embryonic or adult period leads to cardiomyopathy and lethality secondary to elevated myocardial p53 levels.57, 58 Likewise, the overexpression of a negative regulator of Mdm2 mRNA stability, ZFP36L2, was found to facilitate the development of peripartum cardiomyopathy secondary to increased cardiomyocyte p53 activity leading to a reduction of mTorc1.59 The divergent phenotypic responses in models with partial versus complete ablation of MDM2 is at least somewhat explained by these divergent p53 responses. However, the type and timing of the pathogenic stimulus may also be an important contributor.
Our data demonstrating that MDM2 regulates a critical stage of HCM disease development further reinforces that the pre or early LVH period of HCM is important in establishing the long term disease. We show that targeting MDM2 during this key period can modify long-term HCM pathological remodeling. Therefore, it may be possible to target MDM2 or related pathways during the earliest stages of HCM disease development and positively impact long-term remodeling while avoiding the potential negative consequences of chronic MDM2 inhibition. The pre-LVH period in mouse HCM models is measured in days to months post-birth but in humans this period is typically years in duration. Therefore, the window for targeting disease modifying pathways in human HCM may be much larger. Supporting the strategy of early disease modification in HCM was a recent clinical trial administering an angiotensin receptor blocker during the earliest stages of hypertrophic growth which successfully modified pathological structural remodeling in HCM patients.60
In conclusion, our results show that sarcomere mutations cause abnormal cardiomyocyte MDM2 signaling which can occur even before the development of cardiomyocyte hypertrophy. Importantly, the early postnatal pathological changes in cardiomyocyte MDM2 signaling led to persistent changes in the myocardial microenvironment of the adult animals. This study reinforces the importance of targeting HCM during the earliest periods of disease development in order to successfully prevent the long-term pathological remodeling of the myocardium in this disease.
Supplementary Material
CLINICAL PERSPECTIVE.
What is New?
We utilized two distinct murine models of hypertrophic cardiomyopathy (HCM) and discovered that microvascular dysfunction in these models is secondary to reduced myocardial capillary formation.
We discovered that the protein murine double minute 2 (MDM2) regulated this process through modulation of cardiomyocyte HIF signaling.
Targeting MDM2 either genetically or pharmacologically increased myocardial capillary formation in both HCM models and prevented microvascular dysfunction.
What are the Clinical Implications?
Microvascular dysfunction can develop early in the pathogenesis of HCM and results from abnormalities in myocardial capillary formation, not regression.
Targeting the MDM2 signaling pathway during the earliest stages of HCM may be able to reduce the long term sequela of MVD such as chronic angina and adverse ventricular remodeling.
Sarcomere mutations can impact different stages of myocardial development and therapeutic strategies designed to exploit these developmental responses may successfully modify disease progression.
Acknowledgements
We would like to acknowledge Chunming Bi and Zhaohui Kou (Mouse Embryo Services Core, University of Pittsburgh, Department of Immunology) for microinjection of zygotes to produce the Myh6R404Q founder mice.
Sources of Funding
This work was supported by grants from the National Institutes of Health (HL136824 and HL160890 to J.R.B.). University of Pittsburgh Small Animal Ultrasonography Core supported by NIH 1S10OD023684-01A1.
Non-standard Abbreviations and Acronyms
- CD31
Cluster of differentiation 31
- CFR
Coronary flow reserve
- HIF2α/EPAS1
Endothelial PAS domain-containing protein 1
- HCM
Hypertrophic cardiomyopathy
- HIF1α
Hypoxia-inducible factor 1 alpha
- IVSd
Interventricular septal thickness at end diastole
- LCA
Left coronary artery
- LVH
Left ventricular hypertrophy
- LVPWd
Left ventricular posterior wall thickness at end diastole
- LVIDd
Left ventricular internal diameter at end diastole
- MBF
Myocardial blood flow
- MDM2
Murine double minute 2
- MVD
Microvascular dysfunction
- MYBPC3
Cardiac myosin binding protein 3
- MYH6
Myosin heavy chain 6
- TIE2/TEK
Tyrosine kinase with immunoglobulin-like loops and epidermal growth factor homology domains-2
- Ub
Ubiquitin
- VHL
Von Hippel-Lindau
Footnotes
Disclosures
None.
REFERENCES:
- 1.Maron BJ, Bonow RO, Cannon RO, Leon MB and Epstein SE. Hypertrophic cardiomyopathy. Interrelations of clinical manifestations, pathophysiology, and therapy (2). N Engl J Med. 1987;316:844–52. [DOI] [PubMed] [Google Scholar]
- 2.McNally EM, Barefield DY and Puckelwartz MJ. The genetic landscape of cardiomyopathy and its role in heart failure. Cell Metab. 2015;21:174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marian AJ and Braunwald E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ Res. 2017;121:749–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bos JM, Towbin JA and Ackerman MJ. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009;54:201–11. [DOI] [PubMed] [Google Scholar]
- 5.Teekakirikul P, Eminaga S, Toka O, Alcalai R, Wang L, Wakimoto H, Nayor M, Konno T, Gorham JM, Wolf CM, Kim JB, Schmitt JP, Molkentin JD, Norris RA, Tager AM, Hoffman SR, Markwald RR, Seidman CE and Seidman JG. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. J Clin Invest. 2010;120:3520–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Del Buono MG, Montone RA, Camilli M, Carbone S, Narula J, Lavie CJ, Niccoli G and Crea F. Coronary Microvascular Dysfunction Across the Spectrum of Cardiovascular Diseases: JACC State-of-the-Art Review. J Am Coll Cardiol. 2021;78:1352–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koton S, Schneider ALC, Windham BG, Mosley TH, Gottesman RF and Coresh J. Microvascular Brain Disease Progression and Risk of Stroke: The ARIC Study. Stroke. 2020;51:3264–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kang DH, Kanellis J, Hugo C, Truong L, Anderson S, Kerjaschki D, Schreiner GF and Johnson RJ. Role of the microvascular endothelium in progressive renal disease. J Am Soc Nephrol. 2002;13:806–816. [DOI] [PubMed] [Google Scholar]
- 9.Gulati A, Ismail TF, Ali A, Hsu LY, Gonçalves C, Ismail NA, Krishnathasan K, Davendralingam N, Ferreira P, Halliday BP, Jones DA, Wage R, Newsome S, Gatehouse P, Firmin D, Jabbour A, Assomull RG, Mathur A, Pennell DJ, Arai AE and Prasad SK. Microvascular Dysfunction in Dilated Cardiomyopathy: A Quantitative Stress Perfusion Cardiovascular Magnetic Resonance Study. JACC Cardiovasc Imaging. 2019;12:1699–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jia G, Hill MA and Sowers JR. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ Res. 2018;122:624–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crea F, Camici PG and Bairey Merz CN. Coronary microvascular dysfunction: an update. Eur Heart J. 2014;35:1101–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shah SJ, Lam CSP, Svedlund S, Saraste A, Hage C, Tan RS, Beussink-Nelson L, Ljung Faxén U, Fermer ML, Broberg MA, Gan LM and Lund LH. Prevalence and correlates of coronary microvascular dysfunction in heart failure with preserved ejection fraction: PROMIS-HFpEF. Eur Heart J. 2018;39:3439–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dorbala S, Vangala D, Bruyere J, Quarta C, Kruger J, Padera R, Foster C, Hanley M, Di Carli MF and Falk R. Coronary microvascular dysfunction is related to abnormalities in myocardial structure and function in cardiac amyloidosis. JACC Heart Fail. 2014;2:358–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh A, Greenwood JP, Berry C, Dawson DK, Hogrefe K, Kelly DJ, Dhakshinamurthy V, Lang CC, Khoo JP, Sprigings D, Steeds RP, Jerosch-Herold M, Neubauer S, Prendergast B, Williams B, Zhang R, Hudson I, Squire IB, Ford I, Samani NJ and McCann GP. Comparison of exercise testing and CMR measured myocardial perfusion reserve for predicting outcome in asymptomatic aortic stenosis: the PRognostic Importance of MIcrovascular Dysfunction in Aortic Stenosis (PRIMID AS) Study. Eur Heart J. 2017;38:1222–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van der Laan AM, Piek JJ and van Royen N. Targeting angiogenesis to restore the microcirculation after reperfused MI. Nat Rev Cardiol. 2009;6:515–23. [DOI] [PubMed] [Google Scholar]
- 16.Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M, Shimizu I, Asahara T, Hamada H, Tomita S, Molkentin JD, Zou Y and Komuro I. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–8. [DOI] [PubMed] [Google Scholar]
- 17.Heineke J, Auger-Messier M, Xu J, Oka T, Sargent MA, York A, Klevitsky R, Vaikunth S, Duncan SA, Aronow BJ, Robbins J, Crombleholme TM, Cromblehol TM and Molkentin JD. Cardiomyocyte GATA4 functions as a stress-responsive regulator of angiogenesis in the murine heart. J Clin Invest. 2007;117:3198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perrino C, Naga Prasad SV, Mao L, Noma T, Yan Z, Kim HS, Smithies O and Rockman HA. Intermittent pressure overload triggers hypertrophy-independent cardiac dysfunction and vascular rarefaction. J Clin Invest. 2006;116:1547–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dittrich GM, Froese N, Wang X, Kroeger H, Wang H, Szaroszyk M, Malek-Mohammadi M, Cordero J, Keles M, Korf-Klingebiel M, Wollert KC, Geffers R, Mayr M, Conway SJ, Dobreva G, Bauersachs J and Heineke J. Fibroblast GATA-4 and GATA-6 promote myocardial adaptation to pressure overload by enhancing cardiac angiogenesis. Basic Res Cardiol. 2021;116:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nixon BR, Williams AF, Glennon MS, de Feria AE, Sebag SC, Baldwin HS and Becker JR. Alterations in sarcomere function modify the hyperplastic to hypertrophic transition phase of mammalian cardiomyocyte development. JCI Insight. 2017;2:e90656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pal S, Nixon BR, Glennon MS, Shridhar P, Satterfield SL, Su YR and Becker JR. Replication Stress Response Modifies Sarcomeric Cardiomyopathy Remodeling. J Am Heart Assoc. 2021;10:e021768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cannon L, Yu ZY, Marciniec T, Waardenberg AJ, Iismaa SE, Nikolova-Krstevski V, Neist E, Ohanian M, Qiu MR, Rainer S, Harvey RP, Feneley MP, Graham RM and Fatkin D. Irreversible triggers for hypertrophic cardiomyopathy are established in the early postnatal period. J Am Coll Cardiol. 2015;65:560–9. [DOI] [PubMed] [Google Scholar]
- 23.Gedicke-Hornung C, Behrens-Gawlik V, Reischmann S, Geertz B, Stimpel D, Weinberger F, Schlossarek S, Precigout G, Braren I, Eschenhagen T, Mearini G, Lorain S, Voit T, Dreyfus PA, Garcia L and Carrier L. Rescue of cardiomyopathy through U7snRNA-mediated exon skipping in Mybpc3-targeted knock-in mice. EMBO Mol Med. 2013;5:1128–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang J, Wakimoto H, Seidman JG and Seidman CE. Allele-specific silencing of mutant Myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science. 2013;342:111–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Armulik A, Genove G and Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215. [DOI] [PubMed] [Google Scholar]
- 26.Robertson RT, Levine ST, Haynes SM, Gutierrez P, Baratta JL, Tan Z and Longmuir KJ. Use of labeled tomato lectin for imaging vasculature structures. Histochem Cell Biol. 2015;143:225–34. [DOI] [PubMed] [Google Scholar]
- 27.Teng B, Tilley SL, Ledent C and Mustafa SJ. In vivo assessment of coronary flow and cardiac function after bolus adenosine injection in adenosine receptor knockout mice. Physiol Rep. 2016;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Semenza GL. Hypoxia-inducible factor 1 and cardiovascular disease. Annu Rev Physiol. 2014;76:39–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schofield CJ and Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343–54. [DOI] [PubMed] [Google Scholar]
- 30.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS and Kaelin WG. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–8. [DOI] [PubMed] [Google Scholar]
- 31.Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, Dillehay LE, Madan A, Semenza GL and Bedi A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000;14:34–44. [PMC free article] [PubMed] [Google Scholar]
- 32.Nieminen AL, Qanungo S, Schneider EA, Jiang BH and Agani FH. Mdm2 and HIF-1alpha interaction in tumor cells during hypoxia. J Cell Physiol. 2005;204:364–9. [DOI] [PubMed] [Google Scholar]
- 33.Joshi S, Singh AR and Durden DL. MDM2 regulates hypoxic hypoxia-inducible factor 1α stability in an E3 ligase, proteasome, and PTEN-phosphatidylinositol 3-kinase-AKT-dependent manner. J Biol Chem. 2014;289:22785–22797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu X, Bayle JH, Olson D and Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7:1126–32. [DOI] [PubMed] [Google Scholar]
- 35.Linares LK, Hengstermann A, Ciechanover A, Müller S and Scheffner M. HdmX stimulates Hdm2-mediated ubiquitination and degradation of p53. Proc Natl Acad Sci U S A. 2003;100:12009–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kubbutat MH, Jones SN and Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. [DOI] [PubMed] [Google Scholar]
- 37.Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE and Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006. [DOI] [PubMed] [Google Scholar]
- 38.Geisterfer-Lowrance AA, Christe M, Conner DA, Ingwall JS, Schoen FJ, Seidman CE and Seidman JG. A mouse model of familial hypertrophic cardiomyopathy. Science. 1996;272:731–4. [DOI] [PubMed] [Google Scholar]
- 39.Camici PG, d’Amati G and Rimoldi O. Coronary microvascular dysfunction: mechanisms and functional assessment. Nat Rev Cardiol. 2015;12:48–62. [DOI] [PubMed] [Google Scholar]
- 40.Cecchi F, Olivotto I, Gistri R, Lorenzoni R, Chiriatti G and Camici PG. Coronary microvascular dysfunction and prognosis in hypertrophic cardiomyopathy. N Engl J Med. 2003;349:1027–35. [DOI] [PubMed] [Google Scholar]
- 41.Olivotto I, Cecchi F, Gistri R, Lorenzoni R, Chiriatti G, Girolami F, Torricelli F and Camici PG. Relevance of coronary microvascular flow impairment to long-term remodeling and systolic dysfunction in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2006;47:1043–8. [DOI] [PubMed] [Google Scholar]
- 42.Olivotto I, Girolami F, Sciagrà R, Ackerman MJ, Sotgia B, Bos JM, Nistri S, Sgalambro A, Grifoni C, Torricelli F, Camici PG and Cecchi F. Microvascular function is selectively impaired in patients with hypertrophic cardiomyopathy and sarcomere myofilament gene mutations. J Am Coll Cardiol. 2011;58:839–48. [DOI] [PubMed] [Google Scholar]
- 43.Patterson DM, Gao D, Trahan DN, Johnson BA, Ludwig A, Barbieri E, Chen Z, Diaz-Miron J, Vassilev L, Shohet JM and Kim ES. Effect of MDM2 and vascular endothelial growth factor inhibition on tumor angiogenesis and metastasis in neuroblastoma. Angiogenesis. 2011;14:255–66. [DOI] [PubMed] [Google Scholar]
- 44.Venkatesan T, Alaseem A, Chinnaiyan A, Dhandayuthapani S, Kanagasabai T, Alhazzani K, Dondapati P, Alobid S, Natarajan U, Schwartz R and Rathinavelu A. MDM2 Overexpression Modulates the Angiogenesis-Related Gene Expression Profile of Prostate Cancer Cells. Cells. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hou H, Sun D and Zhang X. The role of MDM2 amplification and overexpression in therapeutic resistance of malignant tumors. Cancer Cell Int. 2019;19:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vignier N, Schlossarek S, Fraysse B, Mearini G, Kramer E, Pointu H, Mougenot N, Guiard J, Reimer R, Hohenberg H, Schwartz K, Vernet M, Eschenhagen T and Carrier L. Nonsense-mediated mRNA decay and ubiquitin-proteasome system regulate cardiac myosin-binding protein C mutant levels in cardiomyopathic mice. Circ Res. 2009;105:239–48. [DOI] [PubMed] [Google Scholar]
- 47.Predmore JM, Wang P, Davis F, Bartolone S, Westfall MV, Dyke DB, Pagani F, Powell SR and Day SM. Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation. 2010;121:997–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schlossarek S, Schuermann F, Geertz B, Mearini G, Eschenhagen T and Carrier L. Adrenergic stress reveals septal hypertrophy and proteasome impairment in heterozygous Mybpc3-targeted knock-in mice. J Muscle Res Cell Motil. 2012;33:5–15. [DOI] [PubMed] [Google Scholar]
- 49.Schlossarek S, Englmann DR, Sultan KR, Sauer M, Eschenhagen T and Carrier L. Defective proteolytic systems in Mybpc3-targeted mice with cardiac hypertrophy. Basic Res Cardiol. 2012;107:235. [DOI] [PubMed] [Google Scholar]
- 50.Wolf ER, Mabry AR, Damania B and Mayo LD. Mdm2-mediated neddylation of pVHL blocks the induction of antiangiogenic factors. Oncogene. 2020;39:5228–5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lv X, Li J, Zhang C, Hu T, Li S, He S, Yan H, Tan Y, Lei M, Wen M and Zuo J. The role of hypoxia-inducible factors in tumor angiogenesis and cell metabolism. Genes Dis. 2017;4:19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kimura W, Nakada Y and Sadek HA. Hypoxia-induced myocardial regeneration. J Appl Physiol (1985). 2017;123:1676–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Menendez-Montes I, Escobar B, Palacios B, Gomez MJ, Izquierdo-Garcia JL, Flores L, Jimenez-Borreguero LJ, Aragones J, Ruiz-Cabello J, Torres M and Martin-Puig S. Myocardial VHL-HIF Signaling Controls an Embryonic Metabolic Switch Essential for Cardiac Maturation. Dev Cell. 2016;39:724–739. [DOI] [PubMed] [Google Scholar]
- 54.Camaioni C, Knott KD, Augusto JB, Seraphim A, Rosmini S, Ricci F, Boubertakh R, Xue H, Hughes R, Captur G, Lopes LR, Brown LAE, Manisty C, Petersen SE, Plein S, Kellman P, Mohiddin SA and Moon JC. Inline perfusion mapping provides insights into the disease mechanism in hypertrophic cardiomyopathy. Heart. 2020;106:824–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hughes RK, Camaioni C, Augusto JB, Knott K, Quinn E, Captur G, Seraphim A, Joy G, Syrris P, Elliott PM, Mohiddin S, Kellman P, Xue H, Lopes LR and Moon JC. Myocardial Perfusion Defects in Hypertrophic Cardiomyopathy Mutation Carriers. J Am Heart Assoc. 2021;10:e020227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tesic M, Beleslin B, Giga V, Jovanovic I, Marinkovic J, Trifunovic D, Petrovic O, Dobric M, Aleksandric S, Juricic S, Boskovic N, Tomasevic M, Ristic A, Orlic D, Stojkovic S, Vukcevic V, Stankovic G, Ostojic M and Djordjevic Dikic A. Prognostic Value of Transthoracic Doppler Echocardiography Coronary Flow Velocity Reserve in Patients With Asymmetric Hypertrophic Cardiomyopathy. J Am Heart Assoc. 2021;10:e021936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grier JD, Xiong S, Elizondo-Fraire AC, Parant JM and Lozano G. Tissue-specific differences of p53 inhibition by Mdm2 and Mdm4. Mol Cell Biol. 2006;26:192–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hauck L, Stanley-Hasnain S, Fung A, Grothe D, Rao V, Mak TW and Billia F. Cardiac-specific ablation of the E3 ubiquitin ligase Mdm2 leads to oxidative stress, broad mitochondrial deficiency and early death. PLoS One. 2017;12:e0189861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kouzu H, Tatekoshi Y, Chang HC, Shapiro JS, McGee WA, De Jesus A, Ben-Sahra I, Arany Z, Leor J, Chen C, Blackshear PJ and Ardehali H. ZFP36L2 suppresses mTORc1 through a P53-dependent pathway to prevent peripartum cardiomyopathy in mice. J Clin Invest. 2022;132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ho CY, Day SM, Axelsson A, Russell MW, Zahka K, Lever HM, Pereira AC, Colan SD, Margossian R, Murphy AM, Canter C, Bach RG, Wheeler MT, Rossano JW, Owens AT, Bundgaard H, Benson L, Mestroni L, Taylor MRG, Patel AR, Wilmot I, Thrush P, Vargas JD, Soslow JH, Becker JR, Seidman CE, Lakdawala NK, Cirino AL, Burns KM, McMurray JJV, MacRae CA, Solomon SD, Orav EJ, Braunwald E and Investigators V. Valsartan in early-stage hypertrophic cardiomyopathy: a randomized phase 2 trial. Nat Med. 2021;27:1818–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Haeussler M, Schonig K, Eckert H, Eschstruth A, Mianne J, Renaud JB, Schneider-Maunoury S, Shkumatava A, Teboul L, Kent J, Joly JS and Concordet JP. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pelletier S, Gingras S and Green DR. Mouse genome engineering via CRISPR-Cas9 for study of immune function. Immunity. 2015;42:18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nicolas N and Roux E. 3D Imaging and Quantitative Characterization of Mouse Capillary Coronary Network Architecture. Biology (Basel). 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Robertson RT, Levine ST, Haynes SM, Gutierrez P, Baratta JL, Tan Z and Longmuir KJ. Use of labeled tomato lectin for imaging vasculature structures. Histochem Cell Biol. 2015;143:225–34. [DOI] [PubMed] [Google Scholar]
- 65.Chang WT, Fisch S, Chen M, Qiu Y, Cheng S and Liao R. Ultrasound based assessment of coronary artery flow and coronary flow reserve using the pressure overload model in mice. J Vis Exp. 2015:e52598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Teng B, Tilley SL, Ledent C and Mustafa SJ. In vivo assessment of coronary flow and cardiac function after bolus adenosine injection in adenosine receptor knockout mice. Physiol Rep. 2016;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG and Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3:995–1000. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.