

Abstract
Resistance to androgen deprivation therapies leads to metastatic castration-resistant prostate cancer (mCRPC) of adenocarcinoma (AdCa) origin that can transform to emergent aggressive variant prostate cancer (AVPC) which has neuroendocrine (NE)-like features. In this work, we used LuCaP patient-derived xenograft (PDX) tumors, clinically relevant models that reflects and retains key features of the tumor from advanced prostate cancer patients. Here we performed proteome and phosphoproteome characterization of 48 LuCaP PDX tumors and identified over 94,000 peptides and 9,700 phosphopeptides corresponding to 7,738 proteins. We compared 15 NE versus 33 AdCa samples, that included six different PDX tumors for each group in biological replicates and identified 309 unique proteins and 476 unique phosphopeptides that were significantly altered and corresponded to proteins that are known to distinguish these two phenotypes. Assessment of concordance from PDX tumor matched protein and mRNA revealed increased dissonance in transcriptionally regulated proteins in NE and metabolite interconversion enzymes in AdCa. Implications: Overall, our study highlights the importance of protein-based identification when compared to RNA and provides a rich resource of new and feasible targets for clinical assay development and in understanding the underlying biology of these tumors.
Keywords: Proteomics, phosphoproteomics, Neuroendocrine, Adenocarcinoma, biomarkers, surfaceome, secretome, blood proteins, prostate cancer, patient-derived xenograft
Introduction:
Prostate cancer (PCa) is the most diagnosed cancer in men in the United States. Early detection via regular screening of serum prostate-specific antigen (PSA) levels have facilitated PCa diagnosis in organ confined tumors before cancer spread1. If a patient presents with aggressive PCa, a classic upfront therapy involves radiation or surgery with androgen-deprivation therapy (ADT) 2,3. While ADT response is effective initially, tumors progress to a more aggressive disease known as metastatic castration-resistance prostate cancer (mCRPC)3,4. Treatment of mCRPC with adenocarcinoma (AdCa) features consists of hormonal therapies such as enzalutamide, abiraterone acetate, darolutamide, and apalutamide; however, these therapies can often induce novel phenotypes such as aggressive variant prostate cancer (AVPC). AVPC has genetic aberrations, including PTEN and RB1 loss, TP53 mutations, and diminished AR signaling activity. Several definitions of AVPC have been described including treatment-emergent small cell carcinoma, double-negative prostate cancer, amphicrine, or neuroendocrine prostate cancer (NEPC)5,6. Emergence of these drug-resistant phenotypes creates a large unmet medical need to identify new protein or phosphoprotein drug targets for potential biomarker and therapeutic development for this subset of CRPC patients.
Analysis of the genomic aberrations has contributed to the understanding of drug resistance mechanisms in PCa. These include mutations and focal amplifications in the androgen receptor (AR), PIK3CA/B mutations, fusions in BRAF/RAF1, mutations in APC, mutations and amplifications in CTNNB1, focal homozygous deletions in ZBTB16/PLZF, biallelic loss, inactivation and somatic point mutations in BRCA1 and BRCA2 and biallelic loss and point mutations in ATM7. Additional alterations among other biologically relevant genes include point mutations of SPOP, FOXA1, and TP53, copy number alterations in MYC, RB1, PTEN, and CHD1, and E26 transformation-specific (ETS) fusions8–15. The identification of such alterations has paved the way to define pathological categorizations of mCRPC between AR+ (AdCa) and AR- (NE) disease states. These two types of metastatic phenotypes show distinct pathological features, which in most cases are consequences of the implementation of different treatment modalities with no curative therapies available. In solid tumors, prostate cancer nonsynchronous mutational rate is in the lower 25% quartile compared to esophageal and colorectal tumors which are in the higher 75% quartile7,16–22. Furthermore, somatic alterations, such as in the PI3K pathway, are present in 49% (73/150) of the mCRPC afflicted patients, and still half of this population likely will fail to respond if treated with PI3K inhibitors,7,9,23. Overall, this information indicates that the genomic feature of prostate cancer explains some of the tumor progression and therapy responses but dismiss key phenotypic functional expression coming from proteins that may be driving the biology and drug resistance.
Several patient-derived xenograft tumor (PDX) models have been developed in prostate cancer reflecting different clinical subtypes including the typical prostate AdCa and the atypical patterns of progression known as AVPC that includes tumors with NE features. These models have shown to closely reflect the characteristics of the heterogeneity of the patient tumor population, maintaining histopathologic architecture and the genomic footprint of the tumors from which they were derived24–28. The LuCaP series have been extensively characterized, including analyses of genomic alterations, transcriptomic profiles, and single tandem repeats29. However, little is known about the proteomic profiles of these tumors and, specifically, post-translational modifications of these proteins such as phosphorylation.
Recent technological advancements in mass spectrometry (MS) based proteomics have allowed the increase in protein detection, coverage, and quantification30,31. Here, we used Field Asymmetric Ion Mobility Spectrometry (FAIMS)31 technology and performed a global proteome and phosphoproteome analysis of the LuCaP PDX series to elucidate proteome-wide signatures and unique activated pathways between AdCa and AVPC (with an emphasis on NEPC). We have developed the largest proteome and phosphoproteome database of prostate cancer PDX models which provides an extensive list of protein targets for drug development, predicted kinase activities, proteins found in blood (plasma), proteins that are secreted, surface proteins, or proteins within biological pathways. Most importantly, we integrated mRNA sequencing data from the same PDX models with our proteomic data and evaluated the concordance between protein and mRNA. The results revealed dissonance between protein and mRNA expression of some important biological targets that would have been dismissed if only mRNA was analyzed from these tumors. Overall, this large proteomic and phosphoproteomic resource will further our understanding of the underlying cell signaling mechanisms, identification and functional validation of novel drug targets, and future biomarker development in prostate cancer.
Materials and Methods:
LuCaP Patient Derived Xenograft Tumors
A total of 48 PDX tumors from the LuCaP series29 representing 18 LuCaP PDX models; 6 AdCa non-castrated (NCR) mice, 6 AdCa castrated (CR) mice and 6 neuroendocrine-like (2-3 independent tumors samples per model) were used for this analysis. These PDXs were obtained in a frozen pellet that originated from subcutaneously implanted patient-derived advanced prostate cancer from primary tumors and metastatic sites as described previously29. The age and passage of these tumors are Supplementary Table 1 and characterization of these PDX tumors shall be found in Nguyen et al., Prostate 201729.
Cell Preparation and Protein Extraction from PDX Tumors for Proteome and Phosphoproteome Enrichment
Approximately 150 mg of each tumor was processed as previously described32; The lysis buffer contained 7M urea, 2M thiourea, 0.4M Tris pH 8.0, 20% acetonitrile (ACN), 10 mM TCEP, 25 mM chloroacetamide, Thermo Scientific’s Halt protease inhibitor cocktail 1x concentration (originally 100x), and phosphatase inhibitors (HALT from Thermo). After adding 500 μL of the lysis buffer to the tumor pieces, samples were placed on ice, then vortexed and centrifuged at 12,000 x g for 10 min. The samples were then sonicated for 5 seconds using a probe sonicator set at 30% amplitude and kept on ice during the entire sonication process. After sonication, the samples were incubated for 0.5 hrs at 37°C, then at room temperature for 15 min to reduce and alkylate cysteines and centrifuged at 12,000 x g for 10 min at 18°C. Protein concentration was measured using Bradford Assay (Bio-Rad).
We then used 2.5 mg of protein, added 10 μg of 20 μg/mL Lysyl Endopeptidase (WAKO, 125-05061) and incubated the samples @ room temperature for 5-6 hr at pH 7.4. Then adjusted pH to 7.5-8.0 using 1 M Tris, pH 11. Then we added Worthington TPCK-treated trypsin (1 mg/mL) dissolved in 1 mM HCl supplemented with 20 mM of CaCl2 per to prevent autolysis. The trypsin mixture incubated at 4°C for about 1 hour prior to adding to the protein lysate. Samples were diluted 5-fold by adding 10 mM tris, pH 8.0 to dissolve urea <2M followed by trypsin addition at 1:50 trypsin/protein ratio overnight at 37°C.
After incubation, samples were acidified with TFA to pH 3 or less. Two sequential reverse phase extraction methods were used first. Hydrophilic-Lipophilic Balanced (HLB) was used first and then the flow thru from HLB and wash fractions were vacuum dried, resuspended, and cleaned up again using a C18 solid phase extraction method. The peptides were combined from HLB and C18 cleanups and peptide yield was measured using the BCA peptide assay (Thermo Fisher scientific cat# 23275). Sample digestion efficiency prior to mass spectrometry analysis was inspected by evaluating samples before and after enzymatic digestion using SDS-page gels and again after mass spectrometry analysis.
Mass Spectrometry
1 ug of purified peptides was submitted for mass spectrometry analysis (proteome) and 2 mg of peptides were kept and used for enrichment of phosphopeptides using the Sequential Metal Oxide Affinity Chromatography (SMOAC) Kit (Thermo Fisher Cat# A32993, A32992). The quantitative analysis of phosphoserine, phosphotyrosine and phosphothreonine peptides by Quantitative Mass Spectrometry was performed as previously described33,34 with minor modifications in-tandem using the SMOAC assay. The desalted peptide mixture was fractionated online using EASY-spray columns (25 cm 3 75 mm ID, PepMap RSLC C18 2 mm). The gradient was delivered by an easy-nano Liquid Chromatography 1000 ultra-high-pressure liquid chromatography (UHPLC) system (Thermo Scientific). Tandem mass spectrometry (MS/MS) spectra were collected on the FAIMS TRIBRID mass spectrometer (Thermo Scientific). Samples were run in biological replicates, and raw MS files were analyzed using MaxQuant version 1.4.1.235. MS/MS fragmentation spectra were searched using ANDROMEDA against the Uniprot human reference proteome database with canonical and isoform sequences (downloaded August 1st 2021 from http://uniprot.org). N-terminal acetylation, oxidized methionine, and phosphorylated serine, threonine, or tyrosine were set as variable modifications, and carbamidomethyl cysteine (*C) was set as a fixed modification. The false discovery rate was set to 1% using a composite target-reversed decoy database search strategy. Group-specific parameters included max missed cleavages of two and label-free quantitation (LFQ) with an LFQ minimum ratio count of one. Global parameters included match between runs with a match time and alignment time window of 5 and 20 min, respectively, and match unidentified features selected.
Mass Spectrometry Pre-processing of Proteomic and Phosphoproteomic Data
Maxquant imputed peptide level raw intensity files were obtained for each sample after the mass spectrometry runs. At least two peptides had to map to a protein in order to be included in our data analysis for the proteome. We first summed the intensity of duplicated peptides based on the peptide sequences. In considering peptides with missed cleavages, we identified and then summed groups of peptides of variable length but had identical base sequences. We then mapped the peptide sequences to the most likely protein candidate based on algorithms by MaxQuant35. We then averaged the intensity of the peptides that belonged to the same protein. To note, this averaging was only performed for the proteomic data and not the phosphoproteomic data. At this stage, we aggregated all samples and then conducted VSN normalization for each dataset36,37. We omitted one 208.1 NEPC sample due to the unexpected expression of AR, which is normally only detected in AdCa. To nominate statistically significant proteins or phospho-peptides, we utilized reproducibility-optimized test statistic (ROTS)38. FDR adjusted p-values were computed in which we deemed less than 0.05 as statistically significant. This was used as the threshold for differentially represented features in AdCa or NEPC (Supplementary Tables 4, 9). We performed this for both proteomic and phosphoproteomic outputs. Hierarchical clustering was performed using the Cluster 3.0 program with the Pearson correlation and pairwise complete linkage analysis39. Java TreeView was used to visualize clustering results40. Quantitative data for each peptide and phosphopeptide can be found in Supplementary Tables 2 and 7, respectively, and upstream mapping for each phosphopeptide along with kinase substrate enrichment analysis (KSEA) can be found in Supplementary Tables 10, 11.
Proteomic and Transcriptomic Correlation
mRNA data was obtained from the same PDX samples41 in which we averaged, by sample ID, the log2 FPKM data for the mRNA sequencing or the VSN normalized protein abundance data. We only included genes that were detected in both datasets and then conducted Spearman’s correlations between the mRNA expression and protein abundance levels for each gene across all samples. Concordance analysis was done by comparing the hyperabundant proteins from the NE and AdCa groups with the mRNA levels in the transcriptomic data. Concordance indicates that the protein and mRNA fold change is greater than 1.5. Non-concordance was defined as proteins that are greater than 1.5-fold change, with mRNA between −1.5 to 1.5-fold change. Discordance is defined as proteins that are 1.5 fold change hyper abundant, with mRNA is lower than −1.5-fold change. All were statistically significant with an adjusted p-value <0.05.
Protein Annotations and Databases
We analyzed protein functional annotations using Human Proteome Atlas (HPA version 22.0 http://www.proteinatlas.org/) that has categorized blood proteins, secretome42 and surfaceome43. Ontologies were identified using Gene Set Enrichment Analysis (GSEA version 4.2.1). Potential drug targets were further mapped to gene symbol, to PhosphoSitePlus44, Therapeutic Target Database (TTD)45 Genomics of Drug Sensitivity in Cancer (GDSC)46, and HPA to acquire additional information on whether the targets had drug response data, or they were receptors, kinases, or known cancer/FDA-approved/potential drug targets.
Immunohistochemistry on the LuCaP PDX Tissues
The LuCaP UW TMA 103 was constructed from subcutaneous PDX tumors. It contains 39 LuCaP PDX models; 3 tumors per PDX model, 3 punches per PDX model (together 9 cores per model). The antibodies used for IHC were as follows: HOXB 13 (Cell signaling technologies, catalog#90944, 1:50, Antigen retrieval pH6), SYP (Santa Cruz sc-17750, 1:100 Antigen retrieval pH6), AR (BioGenex MU256-0717 1:100 Antigen retrieval pH9), ASCL1 (ABCAM ab-211327, 1:100, Antigen retrieval pH6). The scoring of each TMA was performed as previously published29.
Statistical Analysis
All statistical data were presented after either t-tests or ROTS as described in the figure legends. The tumors collected from the PDX models and different mice yielded samples that represented AdCa-CR (n=18), AdCa-NCR (n=15) and NE (n=15). We applied the statistical tests with the assumption that each sample would have independent proteomic and phosphoproteomic profiles.
Data Availability Statement
The following data can be obtained via the following databases:
LuCaP PDX mRNA sequencing data is available at the Gene Expression Omnibus (GEO) under accession GSE199596.
LuCaP PDX phosphoproteome data is available at ProteomeXchange PDX042859.
LuCaP PDX proteome data is available at ProteomeXchange PXD042867.
Results:
Proteomic and phosphoproteomic platform analysis
We assessed the global proteome and phosphoproteome of 48 PDX tumor samples, which includes 6 different AdCa tumors grown in intact mice (AdCa NCR, n=15), 6 AdCa grown in castrated mice (AdCa CR, n=18) and 6 neuroendocrine (NE, n=15) tumors (Fig 1A-sample collection). These samples were all processed in parallel on the same day and bottom-up proteomics was performed (Fig 1A-sample processing). To evaluate the phosphoproteome, we performed phosphotyrosine (pY), phosphoserine (pS) and phosphothreonine (pT) enrichment using a sequential metal oxide affinity chromatography (SMOAC) assay (Fig 1A-sample processing-II). In parallel, from the pool of peptides prior to phospho-STY enrichment, we used this fraction and evaluated the overall peptide mix, which we defined as the proteome (Fig 1A sample processing-I). The enriched phosphopeptides and total peptides were analyzed using a state-of-art instrument containing a high-field asymmetric waveform ion mobility spectrometry (FAIMS), which is an atmospheric pressure ion mobility technique that separates gas-phase ions by their behavior in strong and weak electric fields. This approach allows better separation and detection of stable peptides (> +2 charge state ions) for confident quantification31 compared to no FAIMS application where the instrument collects +1 charge state ions, which are very unstable and not quantifiable, among other advantageous features (Fig 1A). We used an in-house proteome and phosphoproteome analysis pipeline32 that includes Maxquant35 for peptide/phosphopeptide searches and data processing (Supplementary Fig 1). Using a 1% FDR, we identified a total of 94,517 peptides that mapped to 7,738 non-redundant proteins at the proteome level and a total of 9,722 phosphopeptides that mapped to 3,759 non-redundant phosphoproteins (Fig 1B and 1C, Supplementary Tables 2 and 7) with a phosphosite probability > 0.75. In combination, we identified 8,612 unique master proteins from these samples, where 32.6% of these proteins overlapped between the proteome and phosphoproteome datasets while 9.9% were unique to the phosphoproteome and 57.5% were unique to the proteome (Fig 1D).
Figure 1. Proteomic and Phosphoproteomic Platform and Characterization.
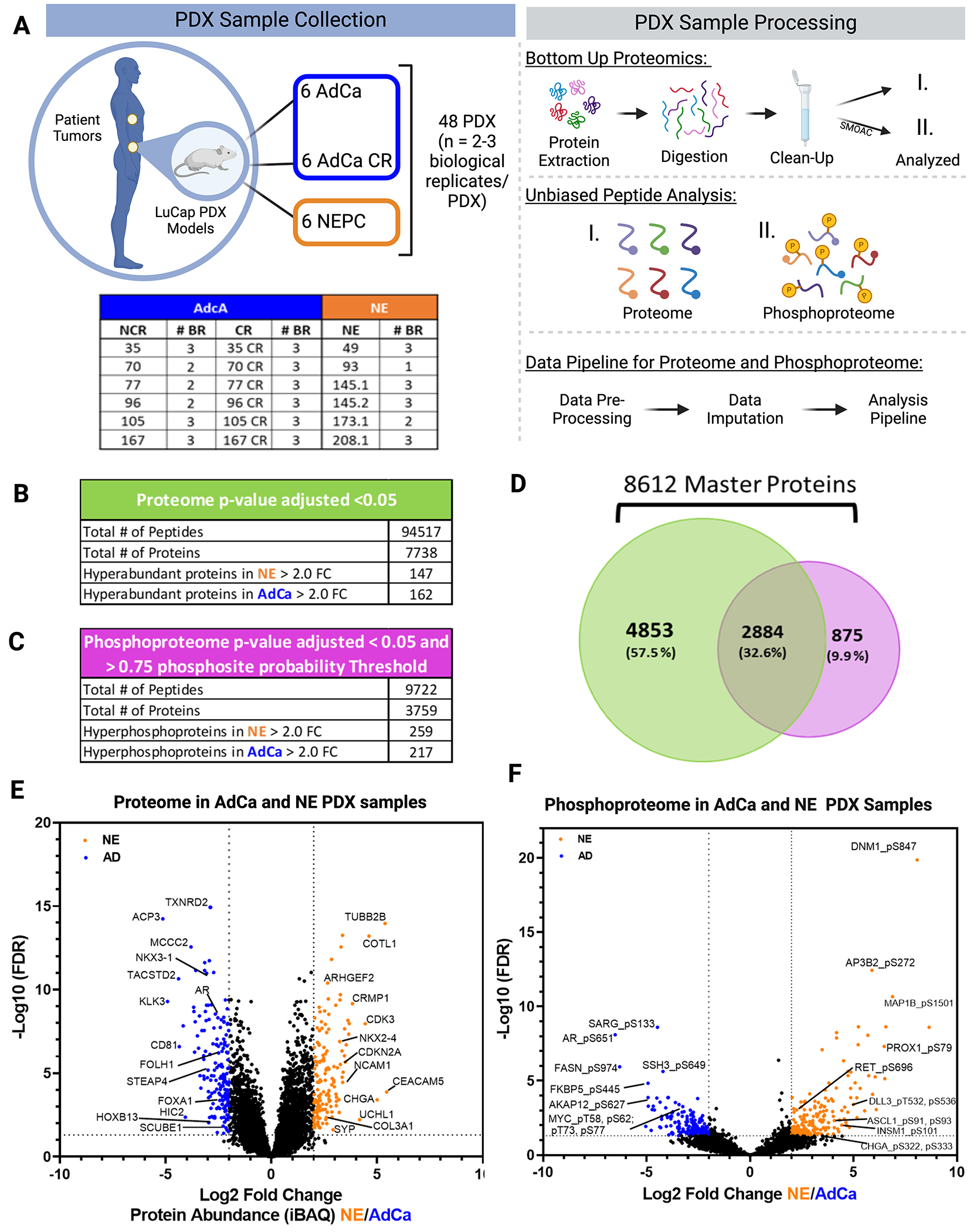
A. The LuCaP series of 48 patient derived xenografts (PDX) tumors depicted in the table, where 33 Adenocarcinoma (AdCa) either castrated and non-castrated (all) tumors are shown in dark blue and 15 neuroendocrine prostate cancer (NEPC) tumors are shown in orange, n=2-3 biological replicates (BR). The PDXs were processed by extracting proteins and an enzymatic digestion was performed using Trypsin and LysC. Peptides were purified by reversed-phase chromatography. The final peptide pool was ran as the proteome (I.) and in parallel from this peptide pool a sequential metal oxide affinity chromatography was performed to enrich for phosphorylated Serine, Threonine and Tyrosine (II.). Finally, raw data was searched, processed, and analyzed. B. Overall proteome results using 1%FDR for protein identification and p-value adjusted < 0.05 log2 fold change significance. C. Overall proteome results using 1%FDR for phosphoprotein identification and p-value adjusted <0.05 log2 fold change significance and >0.75 phosphosite probability threshold. D. Venn diagram of the proteome and phosphoproteome show the total number of 8,612 master proteins identified when both data sets are overlaid. E. Volcano plot depicting the intensity based average quantification (iBAQ) enriched in NE and AdCa. F. Volcano plot of the phosphoprotein enriched in NE vs AdCa. Grayed line in the x-axis and y-axis are the cut off threshold for NE 2-fold change and for AdCa −2-fold change and p value adjusted to (−log10 FDR), respectively in E-F.
To infer protein abundance, we used intensity-based absolute quantification (iBAQ)47. To identify the significantly altered proteins, we performed a variance stabilization normalization (VSN)36 making the sample variances nondependent from their mean intensities using p-value adjusted < 0.05 and a log2 fold change. Using this approach, we identified 147 proteins that were hyper-abundant in the NE group and 162 proteins in the AdCa group (Fig 1E, Supplementary Table 4). Using similar statistical analyses, we identified 259 unique hyperphosphorylated peptides in the NE samples and 217 unique hyperphosphorylated peptides in the AdCa group (Fig 1F, Supplementary Table 9). We performed several comparisons between AdCa grown in castrated mice (CR) versus AdCa grown in intact, or non-castrated, mice (NCR), AdCa-CR versus NE, and AdCa-NCR plus AdCa-CR (pooled AdCa) versus NE. We made all possible comparisons among the different cohorts such as, NCR and CR, CR and NE and AdCa and NE (Supplementary Tables 18–26). However, we observed most of the differences between pooled AdCa (NCR + CR) versus NE, therefore we proceeded with the rest of the analyses comparing NE vs pooled AdCa all.
The LuCaP PDX tumor proteome is consistent with established NE and AdCa gene signatures
To evaluate differences in the overall proteome landscape between NE and the pooled AdCa PDX samples, we performed an unsupervised clustering of all the proteins measured. We observed that there is distinct inter-tumor variability across all PDX samples (Fig 2A). More specifically, there is more variability across non-castrated (NCR) and castrated (CR) AdCa PDX patient-matched pairs than all of the NE samples, which indicates that the relative protein abundance in NE PDX samples are more similar than we might have expected. We then evaluated if this variability is consistent with the top 50 most highly upregulated proteins across all samples and observed that the inter-tumor variability disappears in the AdCa PDX samples but clearly clustered uniquely and distinctly from the NE tumors (Fig 2B).
Figure 2. Proteome Landscape of PDXs in Prostate Cancer.
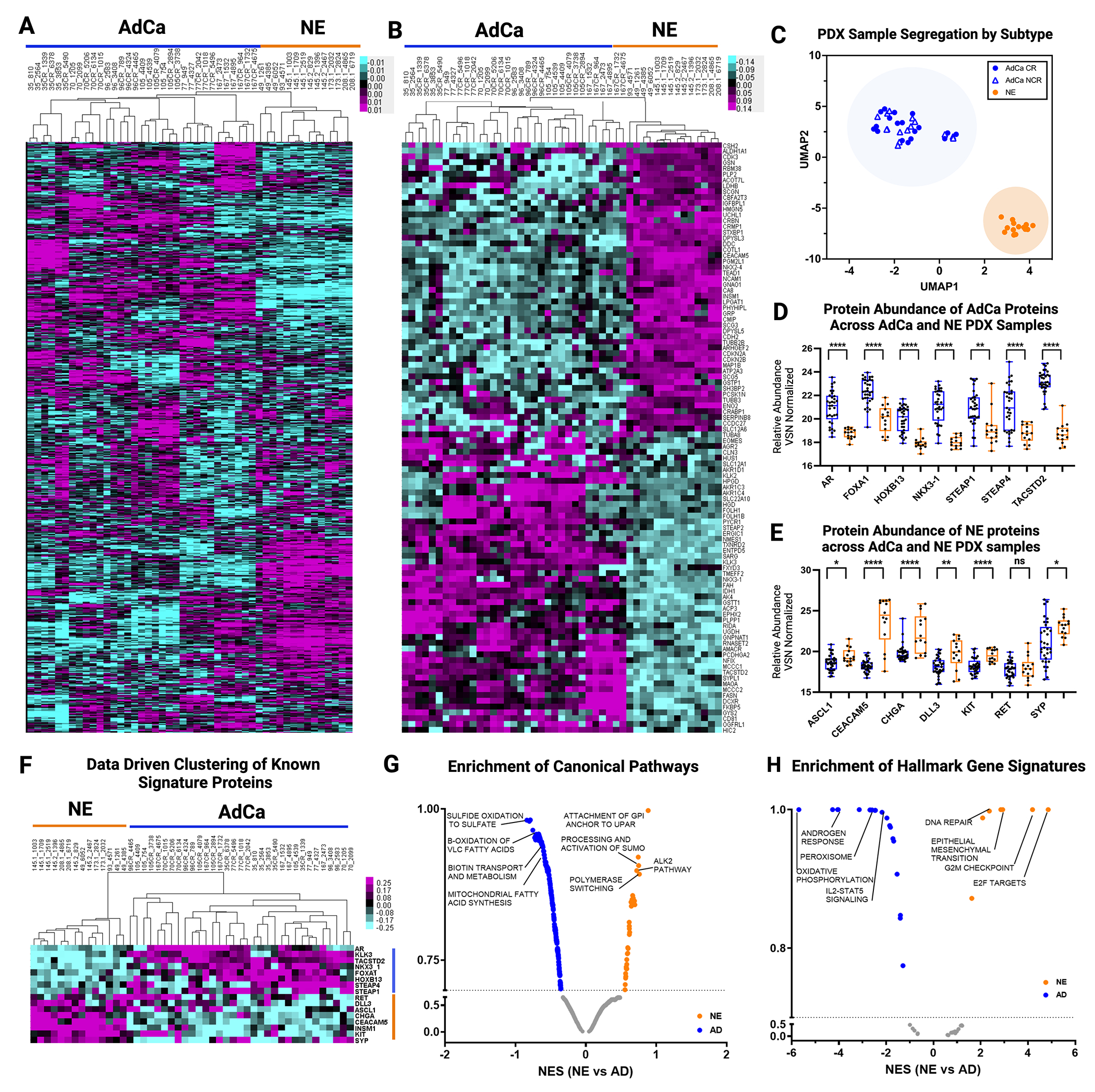
A. Unsupervised clustering data drive 7,738 master proteins 1% FDR. B. Unsupervised clustering of TOP 50 NE and 50 AdCA proteins. C. UMAP analysis of all PDXs. D. Data driven supervised hierarchical clustering of NE and AdCa signatures proteins. E. NE signature proteins. F. AdCa signature protein. G. Pathway Analysis of NE and AdCa highlighting the top 3 pathways on each group. H. Hallmarks in cancer analysis of NE and AdCa highlighting the top 4 pathways on each group (FDR 0.25).
UMAP analysis showed that the PDX tumors clustered within their sub-groups (AdCa or NE) despite inter-PDX tumor variability (Fig 2C). Most importantly, individually analyzed proteins that differentiate clinical NEPC (ASCL1, RET, CEACAM5, CHGA, DLL3, SYP, KIT) from CRPC AdCa (AR, FOXA1, HOXB13, NKX3-1, STEAP1, STEAP4, TACSTD2) also were hyper-abundant in their corresponding tumor phenotypes (Fig D-F, Supplementary Table 3). Four of these proteins (AR, HOXB13, SYP, and ASCL1) were validated on LuCaP tissue microarrays and were indeed expressed primarily in the corresponding tumor phenotype, as expected (Supplementary Fig 2). Pathway analysis showed that there are 34 pathways enriched in NE and 267 in AdCa. Canonical pathways enriched in the AdCa PDX tumors consisted of sulfide oxidation to sulfate, β-oxidation of very long chain fatty acids (VLC-FA), (Fig 2G, Supplementary Table 5). In the NE PDX tumor samples, we identified an enrichment of proteins involved in the processing and activation of SUMO, ALK2 pathway, polymerase switching, and attachment of GPI anchor to u-PAR (urokinase-type plasminogen activator) (Fig 2G). We also observed distinct hallmark gene signatures with oxidative phosphorylation, androgen response, among others enriched in the AdCa PDX tumors while E2F targets, G2M checkpoint, and EMT enriched in the NE PDX tumors (Fig 2H, Supplementary Table 6). These results provide evidence that the PDX tumor protein profiles maintain the proteome architecture and footprint similar to the clinical phenotypes previously observed.
The LuCaP PDX tumor phosphoproteome reveals increased inter-patient similarity with established NE and AdCa gene signatures
To evaluate the overall phosphoproteome in the LuCaP PDX samples, we performed a sequential metal oxide phospho-enrichment targeting phosphoserine (pS), phosphothreonine (pT), and phosphotyrosine (pY) residues (Fig 1A. II). Unsupervised hierarchical clustering showed that the AdCa PDX tumors LuCaP 96CR (replicate 10C), 105, 105CR, 167 and 167CR clustered more closely with NE PDX tumors LuCaP 49 (replicate 3A), 145.2, 173.1 and 208.1 while the other NE PDX tumors, LuCaP 49 and 145.1, clustered more closely to AdCa PDX tumors LuCaP 35, 35CR, 70, 70CR, 96, 96CR, 77, and 77CR (Fig 3A). This indicates that the phosphoproteome has more inter-patient cross over of similar phosphoproteins between NE and AdCa PDX tumors than the proteome and the clustering patterns seem to also reflect less interpatient heterogeneity when compared to the proteome. These data provide new insights about the phosphorylation profile of these two mCRPC subgroups where protein phosphorylation reveals more signaling overlap, allowing for the testing of novel drug targets that may treat both AdCa and NE tumors. When clustering the top 50 proteins that mapped to hyper-phosphorylated peptides from each of the AdCa and NE PDX tumors there was distinct segregation between those sub-groups, however, there appeared to be a third group (mainly LuCaP 105, 105CR, 167, and 167CR) that clustered with the AdCa tumors but certain phosphoproteins fell somewhere between the AdCa and NE phenotypes (Fig 3B). While speculative, these tumors may be transitioning from an AdCa to a NE state. Similar to the proteome, we assessed the well accepted clinical NE and AdCa proteins and mapped them to the phosphoproteome data. Interestingly, these phosphopeptides clustered similarly to the NE and AdCa proteins (Fig 2D) even though most of these phosphoresidues have never been analyzed in this context (Fig 3C, Supplementary Table 8). UMAP analyses also showed that these samples segregated within their corresponding sub-groups (Fig 3D), although the AdCa PDX tumor samples were not as tightly clustered as the proteome (Fig 2C), suggesting that a certain group of AdCa PDX tumors may be transitioning toward the NE phenotype. Using the proteins identified from the volcano plot in Figure 1F, we mapped several functional phosphoproteins that have druggable phosphosites (i.e. phosphopeptides that map to phosphoresidues on proteins with known functional activity) that were unique in AdCa (AR_pS651, pS310, pS120; MYC_pT58; RB1_pT373 and SIRT1_pS47) and NE (STMN1_pS38; ADD2_pS697 and pS701; RET_pS696; CDK1_pT161; ARHGEF2_pS956, pS960; MCM2_pS108; EZH2_pT345; USP16_pS552; and E2A_pS379) PDX tumors (Fig 3E, Supplementary Table 9). Kinase Substrate Enrichment Analysis (KSEA) was performed to predict other kinase activity within the phosphoproteome dataset and found ATM and protein kinase C (PKC) were enriched in the AdCa PDX tumors while MAPK activity was enriched in the NE PDX tumors (Fig 3F, Supplementary Tables 10, 11). Further, pathways involved in metabolism of RNA, RNA processing, and PID-HIF TF pathway were enriched in AdCa while chromatin modifying enzymes, neurexins and neuroligins, and transcription regulation by RUNX1 were enriched in NE PDX tumors (Fig 3G, Supplementary Table 12). This confirms that NE PDX tumors are more closely related to a neuronal phenotype, while AdCa is more metabolically defined. Gene set enrichment analysis (GSEA) indicated that androgen response, hypoxia and MYC targets were enriched in AdCa, while G2M checkpoint and E2F targets were enriched in the NE PDX tumors (Fig 3H, Supplementary Table 13), as expected and similar to the proteome data. Therefore, despite some inter-patient cross-over of AdCa and NE PDX tumors from the global phosphoproteome, there is a significant divergence between AdCa and NE PDX tumors when evaluating the most altered phosphopeptides, including some key druggable targets.
Figure 3. Phosphoproteome Landscape of PDXs in Prostate Cancer.
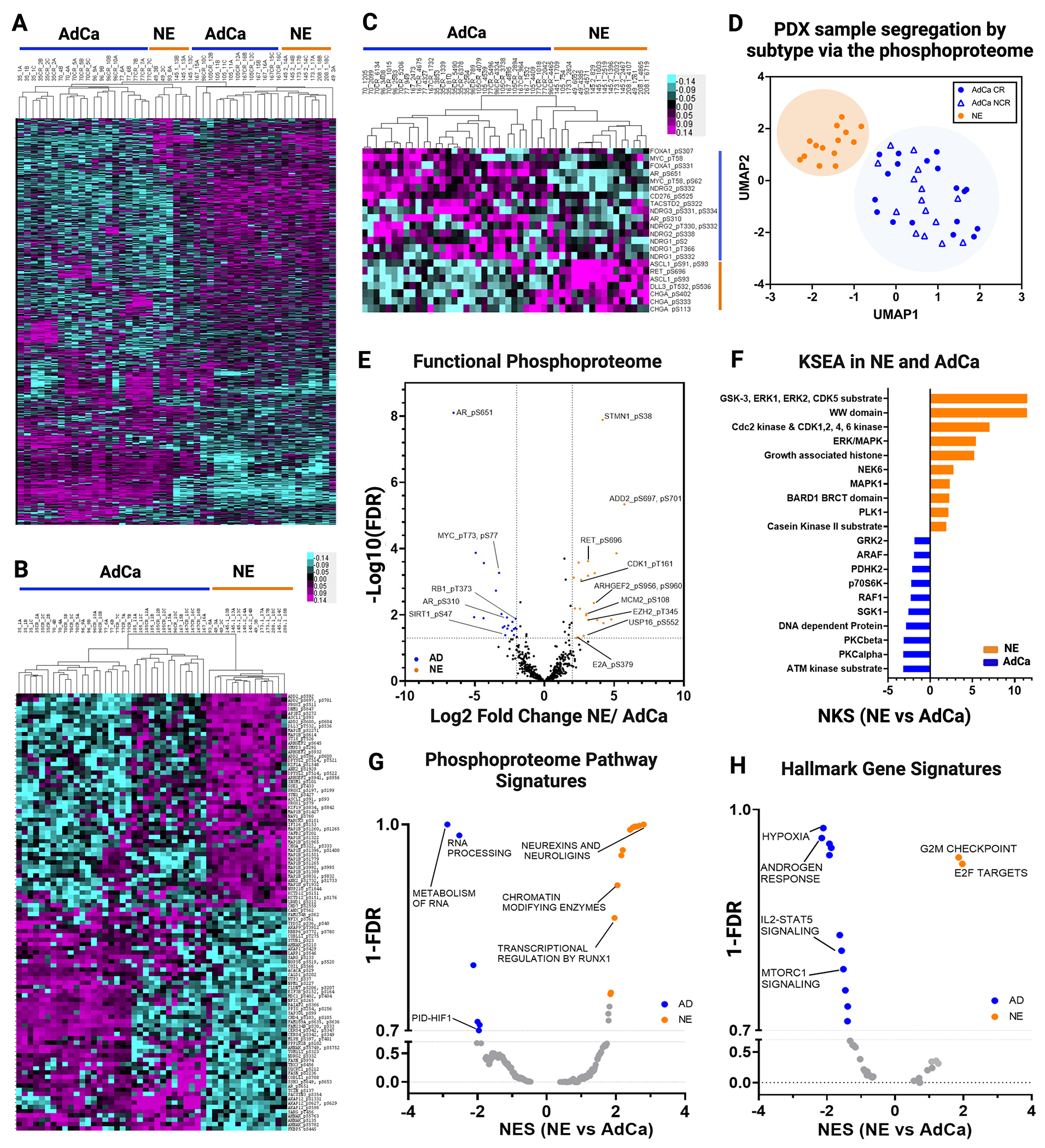
A. Data driven unsupervised clustering of 9,723 phosphopeptides with 1% FDR. B. Unsupervised clustering of TOP 50 NE and 50 AdCA hyper-phosphorylated peptides. C. Unsupervised hierarchical clustering signature protein-phospho-counterpart. D. UMAP analysis of all phosphopeptides. E. Functional phosphoproteome of NE and AD hyperphosphorylated peptides. F. Kinase/substrate enrichment (KSEA) analysis identified unique and known kinases that were predicted from the phosphoproteome (Top 10 hits are showed on each group). G-H. Gene set enrichment analysis (GSEA) was performed to identify canonical pathways (F) and hallmarks in cancer (G) enriched in NE (orange) and in AdCa (blue). NES, normalized enrichment score; orange, hyperphosphorylated in NE and blue hyperphosphorylated in AdCa.
LuCaP PDX tumor proteomic and transcriptomic integration reveals dissonance between mRNA and protein targets
It has been established that mRNA expression has low to moderate correlation to protein expression with a 40-50% concordance48–50, which might misguide potential nomination of novel targets if evaluated at the mRNA level only. Previous work on primary prostate cancer tissues observed a weak correlation between mRNA and the corresponding protein expression via mass spectrometry (median Spearman’s ρ=0.21)51. We proceeded to expand upon this and assess the concordance of mRNA and protein on the LuCaP PDX series derived from metastatic CRPC tumors to evaluate potential discrepancy and nominate targets confidently for a clinical assay or biomarker development. Importantly, these PDX models provide consistent, reproducible material that will allow us to test and evaluate these discrepancies functionally. The LuCaP PDX mRNA data41 was analyzed against the LuCaP PDX proteomic data collected in this manuscript. We analyzed and compared the proteins that were statistically significant and hyper-abundant (>1.5-fold change) in the NE (336 proteins) and AdCa (360 proteins) PDX tumors with the corresponding matching mRNA transcripts that were statistically significant (matching proteins and mRNA transcripts). We then plotted each proteins’ relative intensity based average quantification (iBAQ) log2 fold change against the correspondent transcript FPKM log2 fold change, the overall linear regression correlation was low with a statistical significance r2=0.2359 as expected from previously published work51 (Supplementary Fig 3). Next, we performed concordance analysis where we focused only on the hyper-abundant proteins and their matching mRNA expression level counterparts. While all the proteins analyzed here were statistically significant and hyper-abundant, we observed that only 54% (NE) and 59% (AdCa) of the matching proteins/mRNA transcripts were concordant (C; mRNA and protein are upregulated and hyper-abundant, respectively) while more than 40% in NE and 35% in AdCa proteins were non-concordant either discordant level I (DC.I; mRNA is not altered/changed significantly and protein is hyper-abundant) or discordant level II (DC.II; mRNA is significantly downregulated while the protein is hyper-abundant) (Fig 4A). These data strongly indicate that we are missing important drug targets and tumor biology within these two PDX sub-groups if we were to focus only on mRNA changes. We then analyzed the directionality of the proteins versus the mRNA counterpart within the subgroups and the overall dynamic range of mRNA expression was greater in the concordant group than the relative abundance in protein expression (Fig 4B, right side) compared to the non-concordant groups (DC.I and DC.II) (Fig 4B, left side and middle). We then performed two sub analyses focusing on the AdCa and NE PDX tumors alone to show the overall distribution of the mRNA FPKM vs protein iBAQ fold changes (Fig 4C and 4D). After evaluation of the protein class analysis (Fig 4E), we observed that NE hyper-abundant proteins, which were classified as discordant Level I (DC.I) and discordant level II (DC.II), were mainly categorized as transcriptional regulators (such as NKX2-4, SMARCD1, ATF2, ZBTB21, MYEF2, and more), chromatin binding proteins (CENPH), and DNA metabolic proteins (TIPIN). This indicates that the mRNA transcripts of these proteins were not changed, or the expression was downregulated while the protein was hyper-abundant (Fig 4D). These targets, have relevance in the biology of NEPC and would have been missed if only the mRNA transcripts were analyzed.
Figure 4. Proteomic and Transcriptomic Data Integration Reveals Dissonance of Targetable Proteins.
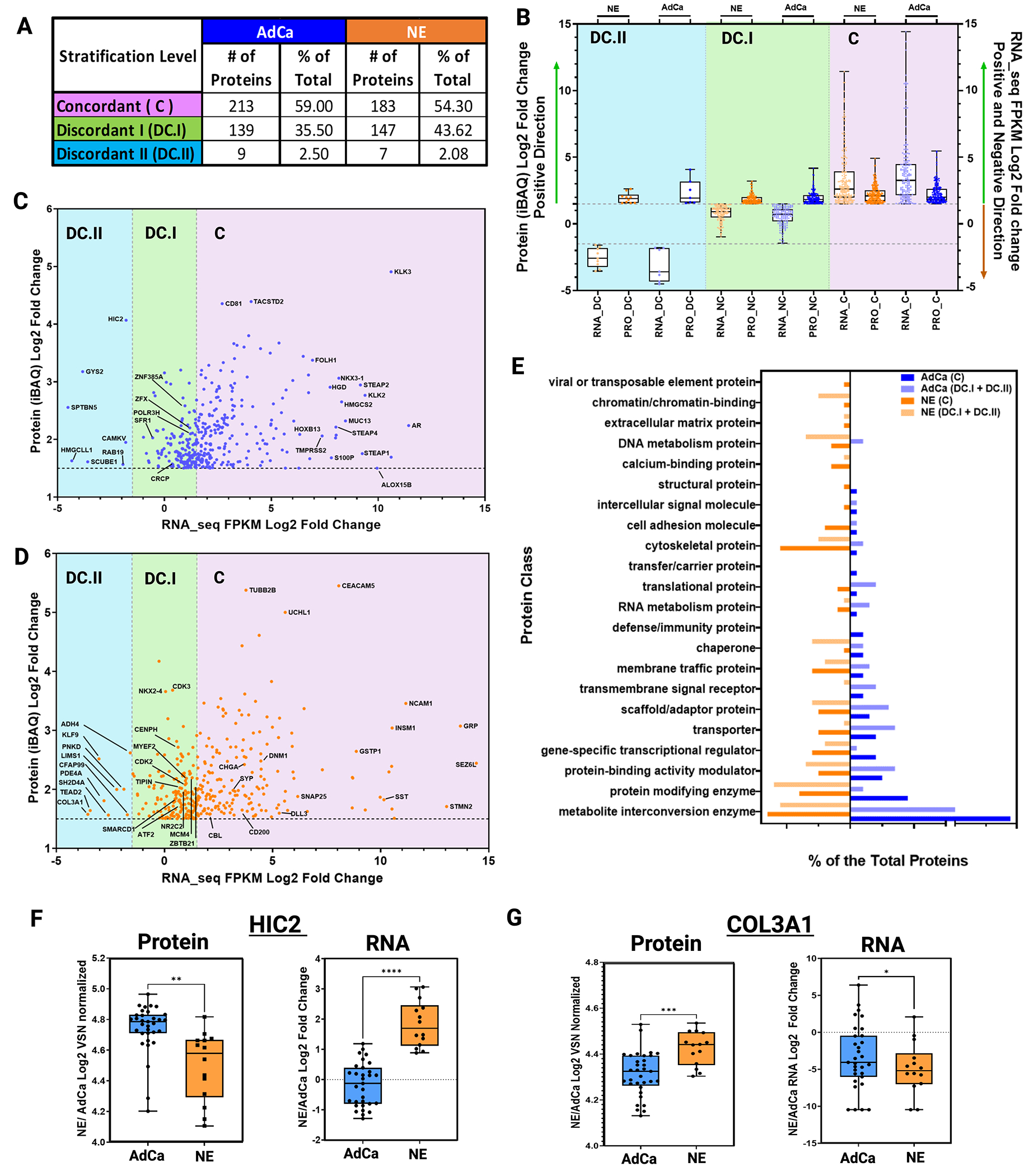
A. Table shows the three main stratification levels of protein and mRNA expression agreements, concordant (C); discordant I (DC.I); discordant II (DC.II) and the total number of hyper-abundant proteins in AdCa (n=361) and in NE (n=337) including percent distribution of total, respectively. B. Protein and mRNA Log2 fold change evaluating only the hyper-abundant protein in NE (337 proteins) and AdCa (361 proteins) and simultaneously evaluating the direction of the mRNA expression of those proteins that are stratified as concordant (C; mRNA and protein are upregulated and hyper-abundant), discordant I (DC.I; mRNA is not altered significantly and protein is hyper-abundant) and discordant II (DC.II; mRNA is significantly downregulated while the protein is hyper-abundant). C-D. AdCa and NE hyper-abundant proteins iBAQ (VSN normalized and ROTS p-value adjusted <0.05 significances) mRNA FPKM (ROTS normalized and p-value adjusted <0.05) Log2 fold change highlighting proteins of interest. E. GO protein class analysis of the NE and AdCa concordant and non-concordant plus discordant proteins. Box plots of protein log 2-fold change VSN normalized and mRNA log 2-fold change of n=33 AdCa and n=14 NE evaluating the overall expression in F. HIC-2 and G. COL3A1. Data are represented as mean ± SEM and. ∗∗p < 0.01, ∗∗∗p < 0.001, two-tailed Welch-corrected.
To identify if the proteins that are discordant level I (DC.I) and discordant level II (DC.II) share any common characteristics, we performed a gene ontology protein class analysis (Fig 4E). From this, we identified that the proteins involved in chromatin binding, DNA metabolism, chaperon, and protein modifying enzymes were over-represented in the NE discordant level I and discordant level II groups (DC.I + DC.II) compared to AdCa; while translational proteins, transporter, scaffold proteins, and metabolite interconversion enzymes were greater in the AdCa DC.I and DC.II groups. This data also shows that there was greater dissonance in the NE DC.I and DC.II than in AdCa PDX tumors indicating that these targets would have been disregarded if only mRNA would have been analyzed.
To illustrate an example of extreme discordance, such as in DC.II, patterns between protein and mRNA, we show two examples of proteins that are highly abundant in AdCa (HIC2, Fig 4F) and NE (COL3A1, Fig 4G) but the mRNA expression levels are very low. HIC ZBTB Transcriptional Repressor 2 (HIC2), a protein that enables protein C-terminus binding activity, predicted to be involved in regulation of transcription by RNA polymerase. In prostate cancer, HIC2 protein expression was shown to be increased in tumors compared to benign hyperplasia and normal tissue with a Gleason score greater than 7 and grade 352. Collagen Type III Alpha 1 Chain (COL3A1) is a protein that is found in most soft connective tissues along with type I collagen53,54. It is involved in regulation of cortical development, and it is the major ligand of ADGRG1 in the developing brain. Moreover, COL3A1 binding to ADGRG1 inhibits neuronal migration and activates the RhoA pathway by coupling ADGRG1 to GNA13 and possibly GNA1255. In prostate cancer, COL3A1 is suppressed by miR-29b in DU145, and after treatment with the miR-29b inhibitor COL3A1 expression is increased as well as the cells’ invasiveness54. Therefore, in the case of these two proteins and their vital role in prostate cancer development or metastases, they would have been missed if only mRNA was used to analyze differential changes, impeding opportunities toward the possible development of therapies against these proteins.
Systematic analysis of the functional proteome and phosphoproteome for actionable targets
We further investigated the hyper-abundant proteins and phosphoproteins from the NE and AdCa sub-groups to identify key biomarkers and drug targets. We used the following databases for our query: the secretome (found in the extracellular matrix)42 and blood proteins (found in blood plasma) from the Human Proteome Atlas, the surfaceome43 and drug analysis from the Therapeutic Target Database45, and Genomics of Drug Sensitivity in Cancer (GDSC)46. We analyzed the proteins that were hyper-abundant (Fig 5A, 5B, Supplementary Tables 16, 17, 27) and hyper-phosphorylated (Fig 5C, 5D, Supplementary Tables 14, 15, 28) in AdCa and NE PDXs and searched them against these databases. We included the concordance stratification level on the proteome and for the phosphoproteome, concordance was only possible by mapping the protein concordance level from the proteome to the phosphorylated peptide counterpart (Fig 5C and 5D). Of the 82 and 70 hyper-abundant proteins in AdCa and NE, respectively, 44% (36 proteins) and 36% (25 proteins) have drug targets in different stages of development, while over 50% of the remaining proteins would be categorized as novel targets for drug development (Fig 5E and 5F).
Figure 5. Functional Proteome and Phosphoproteome Characterization.
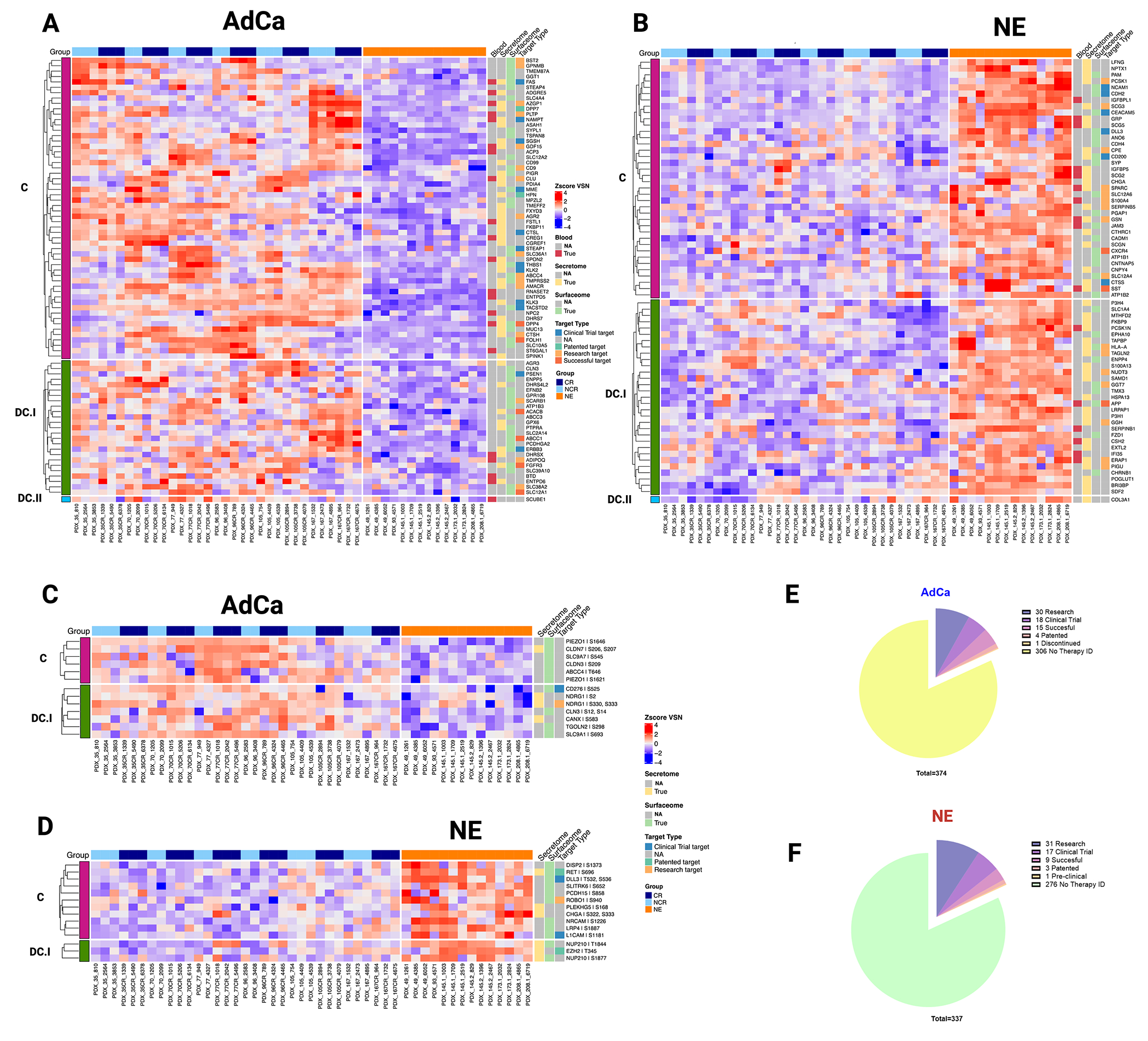
Heatmap data illustrates z-score VSN normalized protein hyper-abundance expression for AdCa (n=82) (A.) and NE (n=70) (B.) Heatmap data illustrates z-score VSN normalized proteins hyper-phosphorylated expression for AdCa (n=13) (C.) and NE (n= 14) (D.) Concordance level was defined by using the master protein counterpart and clustered based on this concordance from the proteome. Pie charts illustrate the therapeutic target distribution identified across the hyper-abundant proteins in AdCa (E.) with a total number 337 and NE (F.) with a total of 374 analyzed. Functional proteins color coding that are identified as Blood (red), Secreted (yellow) Surface (light green) and therapy target type such as clinical trial in blue, patented in green, research target in light orange, successful in terracotta and no available data identified as NA in gray.
At the phosphoproteome level, 16% (2 proteins) and 36% (5 proteins) of the hyperphosphorylated proteins in AdCa and NE, respectively, have drug therapies developed in different stages, while the remaining have not been investigated as potential therapeutic targets. Our data identified several proteins and phosphoproteins that would be viable biomarkers and cell surface proteins that could be used for future biologics such as CAR-T cell therapy, antibody drug conjugates (ADCs) and radio-ligand targeted therapy development. Overall, our data demonstrated that there is a strong mRNA/protein concordance for certain protein targets, but other protein targets may have been overlooked or misinterpreted based on mRNA data alone. These new protein targets could provide new insights into prostate cancer biology, identification and development of biomarkers, and drug targets.
Discussion:
The continuous emergence of therapeutic resistance to current treatment modalities in mCRPC necessitates identification of novel prognostic and predictive biomarkers as well as specific targets for personalized patient stratification and development of new effective therapies. Different prostate cancer PDX models have been developed and evaluated at the transcriptome26,28,56,57, proteome28, genome26,29, copy number variation26,29 and at the single-cell level26 to understand the underlying mechanisms of resistance to therapies. Although these models have elucidated some very interesting biology, protein post translational modifications, such as phosphorylation, have not been fully explored. In this work, we expanded beyond the proteome to include the phosphoproteome and reported one of the largest global proteomic and phosphoproteomic databases capable of differentiating NE from AdCa in the LuCaP PDX tumor models. With this approach, we combined both the proteome and phosphoproteome and we were able to measure over 8,600 proteins and demonstrated inter-PDX tumor variability. Furthermore, comparison of individual AdCa matched tumor pairs grown in non-castrated (NCR) versus castrated (CR) mice showed that their proteome and phosphoproteome signals were mainly unchanged and the differences between the pairs were not significant (Fig 2A, B; Fig 3A, B; Supplementary Tables 18–26). Interestingly, unsupervised clustering analysis of the phosphoproteome revealed that select neuroendocrine PDXs (LuCaP 49, 93, 145.1) clustered with the majority of the AdCa PDXs, while select AdCa PDXs (LuCaP 105, 105CR, 167, and 167 CR) clustered more closely with the majority of the NE PDXs. However, when selecting the top 50 most phosphorylated proteins from both NE and AdCa, the separation between these two groups was more apparent highlighting that the differences between these groups are within the most hyperphosphorylated proteins. In addition, detected phosphorylated proteins such as AR, MYC, NDRG1, NDRG2 FOXA1, TACSTD2 in AdCa PDXs, and ASCL1, RET, DLL3, CHGA in NE PDXs clustered with their known protein/gene signature counterpart (Fig 3D). The characterization of the unsupervised and supervised clustering of these phosphorylated peptides at a global scale is relatively new in prostate cancer since phosphoprotein signatures have not been established or defined to date. However, we can clearly observe that the detected phosphorylated proteins fall within their respective disease phenotype.
While the genome is typically constant, the proteome is quite different from cell to cell, both spatially and temporally. Proteins have over 400-post translational modifications, so the diversity generated from a single protein is larger than that of the corresponding gene58,59. Many attempts have been made to correlate mRNA with proteins35,49,50. This has uncovered that for a given amount of protein to be translated, it will depend on the gene classification that is transcribed such as a metabolic gene (required for survival) versus a transcription factor (will be turned on/off or degraded as needed for different biological responses), the current cell state, and the post-translational modification that is driving the signal. Based off of this information, we proceeded to integrate the transcriptomic41 data to the current proteome data in this manuscript. There were two main goals of this work; 1) to investigate concordance/discordance of mRNA and protein expression and 2) to identify protein-based biomarkers that can lead to the identification or development of therapeutic targets. We initially performed a traditional Spearman’s correlation that did not identify any significant targets due to small sample size (Supplementary Fig 3). However, we were able to evaluate the overall dissonance between mRNA and proteins by comparing the directionality of the differential protein expression (focusing on the hyper-abundant) and mRNA (focusing on any directionality: upregulated, downregulated, or not changed) expression of NE vs pooled AdCa PDXs. Interestingly, several of the proteins known to be involved in prostate cancer biology showed discordance between protein and mRNA levels, a finding that may have future clinical disease management implications.
NEPC has been defined as a disease that is highly transcriptionally active regulating proteins involved in DNA replication (for example DNA polymerase, thymidine kinase, dihydrofolate reductase and cdc6), and chromosomal regulation60 while prostate cancer AdCa that expresses AR and without neuroendocrine features is highly metabolically61 regulated. Furthermore, it has been shown that mRNA from transcription factors have increased average decay rates compared with other mRNA transcripts and are enriched for “fast-decaying” mRNA transcripts with half-lives <2 hours48. On the other hand, mRNAs related to biosynthetic proteins have decreased decay rates and are deficient in fast-decaying mRNAs48,49. This discordance can be explained further by comparing half-lives between proteins and mRNA as well as the timing of data collection. As an example, there are proteins involved in RNA metabolism that would have been missed since the mRNA molecules would have degraded much faster (2-10 hours) than its respective protein (10-46 hours)62. Therefore, the identification of discordance observed between mRNA and protein expression levels in these two CRPC disease states (NE vs AdCa) might be explained mainly by the nature of the proteins expressed in that tumor type and/or the timing of data collection. There are some current algorithms that implement different variables (time and space), including protein isoforms, that could potentially increase the probability of mRNA/protein predictability, but these are still a way off from true implementation63. Thus, prediction of protein abundance or activity in either AdCa or NE tumors based on mRNA transcript levels alone may be misleading and nominating biomarkers or subsequently designing clinical assays based on the most stable molecule, such as proteins, and evaluating if these proteins are either metabolically or transcriptionally involved will be highly recommended for assay development decision making.
The limitation of this study, as well as other studies similar to this that uses mouse models, is that there is an 80% gene homology between human and mouse protein-coding genes implying that some of our identified peptides may map to the mouse proteome64. Importantly, our study showed that the analyzed LuCaP PDX models have a high degree of similarity with known adenocarcinoma and neuroendocrine-like phenotypes and recapitulates many of the underlying mechanisms present in human CRPC. Confirmation and follow up studies, such as western blot analysis, evaluating the expression of the protein of interest identified from mouse models is recommended using human tissue or cell lines. Our work also revealed a very interesting observation: the global phosphoproteome data intersected between AdCa and NE phenotypes indicating a hierarchy of signaling that is somewhat consistent or maintained across these phenotypes. Importantly, these phosphoproteins could help identify new druggable targets that overlaps across prostate cancer phenotypes. Unfortunately to date, the use of phosphoprotein analyses and signature generation is currently hindered by the ability to generate robust phosphoprotein data from small tissue amounts in clinical samples.
In conclusion, we generated the largest proteome and phosphoproteome resource database on clinically relevant and widely used CRPC LuCaP PDX models. Our analysis showed that the overall proteome maintained its fidelity with known CRPC AdCa (AR+) and NE (AR-) markers. We found proteins that are known to be overexpressed and hyper-phosphorylated such AR, RET, ASCL1, DLL3, KIT, CECAM, PSMA/FOHL1 and novel proteins specifically with important functional characteristics for biomarker or drug development, such as surface localization, secreted to the extracellular matrix and/or found in blood plasma. Furthermore, our analyses showed that there is discordance between multiple proteins and their mRNA counterpart that is more dominantly found in transcriptionally regulated proteins compared to metabolic proteins. Future follow up studies where both mRNA and proteins are collected at the same time and measured in parallel, will be highly recommended to rule in/out any temporal changes that might affect the mRNA levels to the protein expression. Moving forward, we encourage multi-omic level analysis, including incorporating the proteome, as a vital element for biomarker and drug development and for effective personalized medicine.
Supplementary Material
Acknowledgements
ZES is supported by the Department of Defense Prostate Cancer Research Program E01 W81XWH-20-1-0070-P00002 and Molecular, Genetic, and Cellular Targets of Cancer Training Program. The establishment and characterization of the LuCaP PDX models were supported by the Pacific Northwest Prostate Cancer SPORE P50 CA97186, the P01 NIH grant CA163227, and the Institute for Prostate Cancer Research. We would like to thank the Comparative Medicine Animal Caregivers for assistance with the LuCaP PDX maintenance and the patients and their families who generously donated tissues that made this research possible. JMD is supported by the NIH R01CA269801 and the Department of Defense Prostate Cancer Research Program W81XWH-18-1-0542. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations:
- AdCa
adenocarcinoma
- ADT
androgen deprivation therapy
- AVPC
aggressive variant prostate cancer
- CR
castrate resistant
- FAIMS
Field Asymmetric Ion Mobility Spectrometry
- KSEA
kinase substrate enrichment analysis
- mCRPC
metastatic castration-resistant prostate cancer
- MS
mass spectrometry
- NCR
non-castrate resistant
- NE
neuroendocrine
- NEPC
neuroendocrine prostate cancer
- PCa
prostate cancer
- PDX
patient derived xenograft
- PSA
prostate specific antigen
- ROTS
reproducibility-optimized test statistic
- SMOAC
Sequential Metal Oxide Affinity Chromatography
- VSN
variance stabilization normalization
Footnotes
Conflict of Interest Statement
ZES has no conflicts relevant to this work, however she works as a consultant to Astrin Bioscience as a senior scientist. EC is a consultant of DotQuant, and obtained research support form AbbVie, Genentech, Bayer Pharmaceuticals, Forma Therapeutics, KronoBio, Foghorn, MacroGenics, Gilead, Janssen Research and GSK. JMD has no conflicts relevant to this work. However, he holds equity in and serves as Chief Scientific Officer of Astrin Biosciences. This interest has been reviewed and managed by the University of Minnesota in accordance with its Conflict-of-Interest policies. None of these companies contributed to or directed any of the research reported in this article. The remaining authors declare no potential conflicts of interest.
References
- 1.Stephan C, Rittenhouse H, Hu X, Cammann H & Jung K Prostate-Specific Antigen (PSA) Screening and New Biomarkers for Prostate Cancer (PCa). EJIFCC 25, 55–78 (2014). [PMC free article] [PubMed] [Google Scholar]
- 2.Watson PA, Arora VK & Sawyers CL Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer 15, 701–711, doi: 10.1038/nrc4016 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wadosky KM & Koochekpour S Androgen receptor splice variants and prostate cancer: From bench to bedside. Oncotarget 8, 18550–18576, doi: 10.18632/oncotarget.14537 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antonarakis ES, Armstrong AJ, Dehm SM & Luo J Androgen receptor variant-driven prostate cancer: clinical implications and therapeutic targeting. Prostate Cancer Prostatic Dis 19, 231–241, doi: 10.1038/pcan.2016.17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beltran H et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med 22, 298–305, doi: 10.1038/nm.4045 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abida W et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U S A 116, 11428–11436, doi: 10.1073/pnas.1902651116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robinson D et al. Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228, doi: 10.1016/j.cell.2015.05.001 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baca SC et al. Punctuated evolution of prostate cancer genomes. Cell 153, 666–677, doi: 10.1016/j.cell.2013.03.021 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taylor BS et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22, doi: 10.1016/j.ccr.2010.05.026 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berger MF et al. The genomic complexity of primary human prostate cancer. Nature 470, 214–220, doi: 10.1038/nature09744 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barbieri CE et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet 44, 685–689, doi: 10.1038/ng.2279 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tomlins SA et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 448, 595–599, doi: 10.1038/nature06024 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Pflueger D et al. Discovery of non-ETS gene fusions in human prostate cancer using next-generation RNA sequencing. Genome Res 21, 56–67, doi: 10.1101/gr.110684.110 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang XS et al. Characterization of KRAS rearrangements in metastatic prostate cancer. Cancer Discov 1, 35–43, doi: 10.1158/2159-8274.CD-10-0022 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cooper CS et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nat Genet 47, 367–372, doi: 10.1038/ng.3221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lam HM et al. Characterization of an Abiraterone Ultraresponsive Phenotype in Castration-Resistant Prostate Cancer Patient-Derived Xenografts. Clin Cancer Res 23, 2301–2312, doi: 10.1158/1078-0432.CCR-16-2054 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen B et al. Pan-cancer Analysis of CDK12 Alterations Identifies a Subset of Prostate Cancers with Distinct Genomic and Clinical Characteristics. Eur Urol 78, 671–679, doi: 10.1016/j.eururo.2020.03.024 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nickols NG et al. MEK-ERK signaling is a therapeutic target in metastatic castration resistant prostate cancer. Prostate Cancer Prostatic Dis 22, 531–538, doi: 10.1038/s41391-019-0134-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pascal LE et al. Lineage relationship of prostate cancer cell types based on gene expression. BMC Med Genomics 4, 46, doi: 10.1186/1755-8794-4-46 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vogelstein B et al. Cancer genome landscapes. Science 339, 1546–1558, doi: 10.1126/science.1235122 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Armenia J et al. The long tail of oncogenic drivers in prostate cancer. Nat Genet 50, 645–651, doi: 10.1038/s41588-018-0078-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garraway LA & Lander ES Lessons from the cancer genome. Cell 153, 17–37, doi: 10.1016/j.cell.2013.03.002 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Schwartz S et al. Feedback suppression of PI3Kalpha signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kbeta. Cancer Cell 27, 109–122, doi: 10.1016/j.ccell.2014.11.008 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin D et al. High fidelity patient-derived xenografts for accelerating prostate cancer discovery and drug development. Cancer Res 74, 1272–1283, doi: 10.1158/0008-5472.CAN-13-2921-T (2014). [DOI] [PubMed] [Google Scholar]
- 25.Shi M, Wang Y, Lin D & Wang Y Patient-derived xenograft models of neuroendocrine prostate cancer. Cancer Lett 525, 160–169, doi: 10.1016/j.canlet.2021.11.004 (2022). [DOI] [PubMed] [Google Scholar]
- 26.Risbridger GP et al. The MURAL collection of prostate cancer patient-derived xenografts enables discovery through preclinical models of uro-oncology. Nat Commun 12, 5049, doi: 10.1038/s41467-021-25175-5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palanisamy N et al. The MD Anderson Prostate Cancer Patient-derived Xenograft Series (MDA PCa PDX) Captures the Molecular Landscape of Prostate Cancer and Facilitates Marker-driven Therapy Development. Clin Cancer Res 26, 4933–4946, doi: 10.1158/1078-0432.CCR-20-0479 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flores-Morales A et al. Proteogenomic Characterization of Patient-Derived Xenografts Highlights the Role of REST in Neuroendocrine Differentiation of Castration-Resistant Prostate Cancer. Clin Cancer Res 25, 595–608, doi: 10.1158/1078-0432.CCR-18-0729 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Nguyen HM et al. LuCaP Prostate Cancer Patient-Derived Xenografts Reflect the Molecular Heterogeneity of Advanced Disease an--d Serve as Models for Evaluating Cancer Therapeutics. Prostate 77, 654–671, doi: 10.1002/pros.23313 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hebert AS et al. Comprehensive Single-Shot Proteomics with FAIMS on a Hybrid Orbitrap Mass Spectrometer. Anal Chem 90, 9529–9537, doi: 10.1021/acs.analchem.8b02233 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swearingen KE & Moritz RL High-field asymmetric waveform ion mobility spectrometry for mass spectrometry-based proteomics. Expert Rev Proteomics 9, 505–517, doi: 10.1586/epr.12.50 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drake JM et al. Phosphoproteome Integration Reveals Patient-Specific Networks in Prostate Cancer. Cell 166, 1041–1054, doi: 10.1016/j.cell.2016.07.007 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng LC, Li Z, Graeber TG, Graham NA & Drake JM Phosphopeptide Enrichment Coupled with Label-free Quantitative Mass Spectrometry to Investigate the Phosphoproteome in Prostate Cancer. J Vis Exp, doi: 10.3791/57996 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nita-Lazar A, Saito-Benz H & White FM Quantitative phosphoproteomics by mass spectrometry: past, present, and future. Proteomics 8, 4433–4443, doi: 10.1002/pmic.200800231 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cox J & Mann M MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26, 1367–1372, doi: 10.1038/nbt.1511 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Huber W, von Heydebreck A, Sultmann H, Poustka A & Vingron M Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 18 Suppl 1, S96–104, doi: 10.1093/bioinformatics/18.suppl_1.s96 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Valikangas T, Suomi T & Elo LL A systematic evaluation of normalization methods in quantitative label-free proteomics. Brief Bioinform 19, 1–11, doi: 10.1093/bib/bbw095 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suomi T, Seyednasrollah F, Jaakkola MK, Faux T & Elo LL ROTS: An R package for reproducibility-optimized statistical testing. PLoS Comput Biol 13, e1005562, doi: 10.1371/journal.pcbi.1005562 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eisen MB, Spellman PT, Brown PO & Botstein D Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A 95, 14863–14868 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saldanha AJ Java Treeview--extensible visualization of microarray data. Bioinformatics 20, 3246–3248, doi: 10.1093/bioinformatics/bth349 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Coleman IM et al. Therapeutic Implications for Intrinsic Phenotype Classification of Metastatic Castration-Resistant Prostate Cancer. Clin Cancer Res 28, 3127–3140, doi: 10.1158/1078-0432.CCR-21-4289 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uhlen M et al. The human secretome. Sci Signal 12, doi: 10.1126/scisignal.aaz0274 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Bausch-Fluck D et al. The in silico human surfaceome. Proc Natl Acad Sci U S A 115, E10988–E10997, doi: 10.1073/pnas.1808790115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hornbeck PV et al. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43, D512–520, doi: 10.1093/nar/gku1267 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou Y et al. Therapeutic target database update 2022: facilitating drug discovery with enriched comparative data of targeted agents. Nucleic Acids Res 50, D1398–D1407, doi: 10.1093/nar/gkab953 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang W et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res 41, D955–961, doi: 10.1093/nar/gks1111 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schwanhausser B et al. Global quantification of mammalian gene expression control. Nature 473, 337–342, doi: 10.1038/nature10098 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Yang E et al. Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res 13, 1863–1872, doi: 10.1101/gr.1272403 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu Y, Beyer A & Aebersold R On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 165, 535–550, doi: 10.1016/j.cell.2016.03.014 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Maier T, Guell M & Serrano L Correlation of mRNA and protein in complex biological samples. FEBS Lett 583, 3966–3973, doi: 10.1016/j.febslet.2009.10.036 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Sinha A et al. The Proteogenomic Landscape of Curable Prostate Cancer. Cancer Cell 35, 414–427 e416, doi: 10.1016/j.ccell.2019.02.005 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gaballah MSA et al. Small extracellular vesicle-associated miR-6068 promotes aggressive phenotypes of prostate cancer through miR-6068/HIC2/SIRT1 axis. Am J Cancer Res 12, 4015–4027 (2022). [PMC free article] [PubMed] [Google Scholar]
- 53.Jia D et al. Stromal FOXF2 suppresses prostate cancer progression and metastasis by enhancing antitumor immunity. Nat Commun 13, 6828, doi: 10.1038/s41467-022-34665-z (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ivanovic RF et al. miR-29b enhances prostate cancer cell invasion independently of MMP-2 expression. Cancer Cell Int 18, 18, doi: 10.1186/s12935-018-0516-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Engqvist H et al. Immunohistochemical validation of COL3A1, GPR158 and PITHD1 as prognostic biomarkers in early-stage ovarian carcinomas. BMC Cancer 19, 928, doi: 10.1186/s12885-019-6084-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iglesias-Gato D et al. The Proteome of Prostate Cancer Bone Metastasis Reveals Heterogeneity with Prognostic Implications. Clin Cancer Res 24, 5433–5444, doi: 10.1158/1078-0432.CCR-18-1229 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Iglesias-Gato D et al. The Proteome of Primary Prostate Cancer. Eur Urol 69, 942–952, doi: 10.1016/j.eururo.2015.10.053 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Ramazi S & Zahiri J Posttranslational modifications in proteins: resources, tools and prediction methods. Database (Oxford) 2021, doi: 10.1093/database/baab012 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Conibear AC Deciphering protein post-translational modifications using chemical biology tools. Nat Rev Chem 4, 674–695, doi: 10.1038/s41570-020-00223-8 (2020). [DOI] [PubMed] [Google Scholar]
- 60.Xia H et al. A Novel Gene Signature Associated With “E2F Target” Pathway for Predicting the Prognosis of Prostate Cancer. Front Mol Biosci 9, 838654, doi: 10.3389/fmolb.2022.838654 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ahmad F, Cherukuri MK & Choyke PL Metabolic reprogramming in prostate cancer. Br J Cancer 125, 1185–1196, doi: 10.1038/s41416-021-01435-5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moss Bendtsen K, Jensen MH, Krishna S & Semsey S The role of mRNA and protein stability in the function of coupled positive and negative feedback systems in eukaryotic cells. Sci Rep 5, 13910, doi: 10.1038/srep13910 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Magnusson R et al. RNA-sequencing and mass-spectrometry proteomic time-series analysis of T-cell differentiation identified multiple splice variants models that predicted validated protein biomarkers in inflammatory diseases. Front Mol Biosci 9, 916128, doi: 10.3389/fmolb.2022.916128 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Breschi A, Gingeras TR & Guigo R Comparative transcriptomics in human and mouse. Nat Rev Genet 18, 425–440, doi: 10.1038/nrg.2017.19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The following data can be obtained via the following databases:
LuCaP PDX mRNA sequencing data is available at the Gene Expression Omnibus (GEO) under accession GSE199596.
LuCaP PDX phosphoproteome data is available at ProteomeXchange PDX042859.
LuCaP PDX proteome data is available at ProteomeXchange PXD042867.