

Abstract
Triple negative breast cancer (TNBC) contains the highest proportion of cancer stem-like cells (CSCs), which display intrinsic resistance to currently available cancer therapies. This therapeutic resistance is partially mediated by an antioxidant defense coordinated by the transcription factor NRF2 and its downstream targets including NQO1. Here, we identified the antioxidant enzymes NQO1 and SOD1 as therapeutic vulnerabilities of ALDH+ epithelial-like CSCs and CD24−/loCD44+/hi mesenchyme-llike CSCs in TNBC. Effective targeting of these CSC states was achieved by utilizing IB-DNQ, a potent and specific NQO1-bioactivatable futile redox cycling molecule, which generated large amounts of reactive oxygen species (ROS) including superoxide and hydrogen peroxide. Furthermore, the CSC killing effect was specifically enhanced by genetic or pharmacological inhibition of SOD1, a copper-containing superoxide dismutase highly expressed in TNBC. Mechanistically, a significant portion of NQO1 resided in the mitochondrial intermembrane space, catalyzing futile redox cycling from IB-DNQ to generate high levels of mitochondrial superoxide, and SOD1 inhibition markedly potentiated this effect resulting in mitochondrial oxidative injury, cytochrome c release, and activation of the caspase 3-mediated apoptotic pathway. Treatment with IB-DNQ alone or together with SOD1 inhibition effectively suppressed tumor growth, metastasis, and tumor-initiating potential in xenograft models of TNBC expressing different levels of NQO1. This futile oxidant-generating strategy, which targets CSCs across the epithelial-mesenchymal continuum, could be a promising therapeutic approach for treating TNBC patients.
INTRODUCTION
Triple negative breast cancer (TNBC) is a very aggressive breast cancer subtype with the least favorable prognosis and few treatment options (1,2). It is also characterized as containing a high proportion of cancer stem-like cells (CSCs) (3–5) that contribute to treatment resistance and tumor recurrence (6–9). Many conventional therapies utilized to treat TNBC such as chemotherapy and radiation actually increase the proportion of CSCs in the residual tumors (10–13), although other studies suggests that a reversible drug-tolerant persister (DTP) cell state also mediates chemotherapy resistance (14–16). This highlights the importance of developing strategies to effectively target CSCs and DTP cells in TNBC to improve clinical outcome. CSCs in solid tumors were initially identified in breast cancer (BC) by virtue of CD24−/loCD44+/hi marker expression (17), and subsequent studies demonstrated that elevated aldehyde dehydrogenase (ALDH) activity also defines a highly tumorigenic cell population in a number of malignancies including BC (18), colon cancer (19,20), prostate cancer (21), and lung adenocarcinoma (22). More recently, we have demonstrated the phenotypic heterogeneity of these CSC populations, with CD24−/loCD44+/hi cells representing more mesenchymal/quiescent and ALDH+ cells more epithelial/proliferative CSC states (23). In fact, there is substantial evidence suggesting that rather than representing a fixed, discrete cell population, CSCs represent a highly plastic tumor cell population across the EMT-MET spectrum (24–27). This plasticity facilitates rapid development of epigenetic resistance to targeted therapies and promotes tumor progression and metastasis (26,28–30). Most attempts to develop CSC targeted therapies have focused on the targeting of CSC self-renewal pathways such as Hedgehog, Notch and Wnt (31,32). While these approaches have shown modest efficacy, their utility is limited by CSC heterogeneity and plasticity (27,33,34).
An alternative approach for targeting CSCs is to exploit their metabolic vulnerabilities. Despite their regulation by multiple signal transduction pathways and networks, CSCs maintain metabolic dependencies characteristic of their EMT/MET states (35–37). We have shown that redox state of the tumor microenvironment regulates CSC E/M state dynamics. The generation of oxidant stress facilitates the transition of EMT-like (CD24−/loCD44+/hi) CSCs, which are largely quiescent and dependent on glycolysis, to a more proliferative epithelial cell state characterized by ALDH expression, which is highly dependent on mitochondrial oxidative phosphorylation (37). These ALDH+ CSCs rely on an antioxidant shield coordinated by NRF2 and its downstream targets including NAD(P)H quinone oxidoreductase 1 (NQO1) to survive oxidative stress (37).
NQO1, a cytosolic enzyme involved in NRF2-regulated antioxidant defenses, has emerged as an attractive target for pro-oxidant-based antitumor strategies (38–40). As a FAD-dependent two-electron reductase, NQO1 is overexpressed in many solid tumors including breast cancer (41), colorectal cancer (42), lung cancer (43), gastric adenocarcinoma (44), hepatocellular carcinoma (45) and cervical cancer (46), and high NQO1 expression is associated with poor patient outcomes (46–49), suggesting an important role of NQO1 in promoting cancer progression.
Although NQO1 plays a cytoprotective role in cancer by detoxifying a variety of ROS-producing quinone toxins, a class of quinone-containing molecules, i.e., β-Lapachone (β-Lap) and deoxynyboquinone (DNQ), are subject to NQO1-catalyzed two-electron reduction into unstable hydroquinone-containing molecules, which react with oxygen via two one-electron oxidation steps back to the original quinones, generating large amounts of ROS via futile redox cycle (50,51). These NQO1-Bioactivatable futile Redox Cycling Compounds (NBRCCs) have the potential to turn this normally cytoprotective enzyme into a therapeutic vulnerability for cancer cells that overexpress NQO1 and under-express catalase (CAT) (40), an enzyme metabolizing hydrogen peroxide.
Despite the role of NBRCCs (i.e., β-Lap) in targeting tumor cells expressing high levels of NQO1 (40,52–55), the potential of NBRCCs in targeting CSCs remains unstudied. Interestingly, a large array of NRF2-regulated antioxidant genes, including NQO1, are markedly elevated in CSCs, especially ALDH+ E-CSCs isolated from patient-derived xenograft (PDX) models of TNBC(37). This increased NQO1 expression in breast CSCs (BCSCs) may serve as a metabolic vulnerability, rendering them susceptible to the ROS generated by NBRCCs. Using a derivative of DNQ, called isobutyl-deoxynyboquinone (IB-DNQ) that exhibited promising efficacy against feline oral squamous cell carcinoma (56), we show here that IB-DNQ is much more potent and specific than β-Lap in killing NQO1-overexpressing TNBC cells with particular efficacy in targeting CSCs. We demonstrate that IB-DNQ-elicited cell death is associated with highly elevated mitochondrial O2•−(mitoROS), which is generated from a portion of NQO1 residing in the mitochondrial intermembrane space (IMS). Furthermore, genetic or pharmacological inhibition of SOD1, but not SOD2 or CAT, synergizes with IB-DNQ to generate mitoROS and abrogate CSCs across the EMT-MET state continuum. By exploiting NQO1 and SOD1 as therapeutic vulnerabilities of CSCs, we demonstrate the utility of this oxidant-generating strategy using IB-DNQ alone or together with SOD1 inhibition to suppresses tumor growth, metastasis, and tumor-initiating potential in xenograft models of TNBC expressing different levels of NQO1.
MATERIALS AND METHODS
Cell lines and culture
SUM149 and SUM159 cells were cultured in Ham’s F-12 (Invitrogen) supplemented with 5% FBS (ThermoFisher Scientific, Pittsburgh, PA), 5 μg/mL insulin, and 1 μg/mL hydrocortisone (both from Sigma, St. Louis, MO), 10 μg/ml gentimycin, and 1% antibiotic-antimycotic (both from Invitrogen). BT20, MDA-MB-231, MDA-MB-468, MDA-MB-157 and MDA-MB-453 were cultured in DMEM-high glucose (Gibco) supplemented with 10% FBS and 1x antibiotic-antimycotic. Vari068, HCC38, HCC70 and HCC1937, and ZR-75–1 were maintained in RPMI1640 medium (ThermoFisher Scientific) supplemented with 10% FBS and 1x antibiotic-antimycotic. MCF7 cells were cultured in Eagle’s Minimum Essential Medium (EMEM, ATCC 30–2003) supplemented with 10% FBS, 10 μg/mL human insulin, and 1x antibiotic-antimycotic. MCF10A is cultured in DMEM/F12 media (50:50, ThermoFisher Scientific) supplemented with 5% horse serum, 1x HEPES, 20 ng/ml EGF, 0.5 mg/ml hydrocortisone, 100 ng/ml cholera toxin, 10 μg/ml insulin and 1x antibiotic-antimycotic. Hs578t were cultured in DMEM (Gibco) supplemented with 10% FBS, 5 μg/mL insulin, and 1x antibiotic-antimycotic. All the cell lines are cultured at 37 °C under 5% CO2 in a humidified chamber and are mycoplasma-free.
MTT and CCK8 assays
For MTT assays, BC cells were seeded at a density of 1000–2000 cells per well in 96-well microplates for overnight and cultured with or without IB-DNQ or beta-lap at various doses alone or with ATN224 (5μM) or Dicumoral (10μM) for 24 hours. After drugs treatment, MTT solution was added to each well and incubated for 3 h. After removing the supernatant, cells in each well were solubilized by adding 150 μL of DMSO and OD absorbance at 590 nm was measured with a microplate reader (Bio-Rad, Hercules, CA, USA). Alternatively, cell viability was analyzed using Cell Counting Kit-8 (CCK8, Beyotime, Shanghai, China) according to the manufacturer’s instructions. For CCK8 assay, cells were seeded at a density of 1000–2000 cells per well in 96-well microplate for overnight. Cells were then treated with various concentrations of IB-DNQ for 24 hours, subsequently 10 μL of CCK-8 reagent was added to each well and then cultured for 2 hours. OD absorbance at 450 nm was analyzed by a microplate reader using wells without cells as blanks. Relative cell survival vs. control cells was plotted.
DNA constructs and lentiviral infection
TRIPZ and SMARTvector Dox-inducible lentiviral shRNA clones against human NQO1, SOD1, SOD2, CAT were obtained from Horizon Discovery with the following clone #: V2THS_235287 (shNQO1, TRIPZ), V3IHSHER_10771835 (shNQO1-1, SMARTvector), V3IHSHER_7177739 (shNQO1-2, SMARTvector), V3IHSHER_8909942 (shNQO1-3, SMARTvector), V3THS_369718 (shSOD1, TRIPZ), V3IHSHER_9961124 (shSOD1-1, SMARTvector), V3IHSHER_9099989 (shSOD1-2, SMARTvector), V3THS_307804 (shSOD2, TRIPZ), V2THS_150247 (shCAT, TRIPZ). Lentiviruses expressing human shNQO1, shSOD1, shSOD2, shCAT or a scrambled sequence (SCR) were packaged at the University of Michigan Vector Core. Vari068, HCC70, SUM149, and SUM159 TNBC cells were infected with lentiviruses in the presence of polybrene (8 μg/mL, Millipore) and the medium containing lentiviruses was replaced with fresh medium after 20 h of lentiviral infection. Puromycin (Invitrogen) selection was performed at a final concentration of 0.5–2.5 μg/mL for 2 weeks to establish Dox-inducible shNQO1, shSOD1, shSOD2, shCAT, or SCR cell lines.
Cell labeling and flow cytometry
To determine the effect of drug treatment on M- and E-BCSCs in basal BC cell lines, SUM149, SUM159, or Vari068 following various treatment were digested by 0.25% Trypsin-EDTA, re-suspended in HF buffer (HBSS plus 2% FBS) at 2×105 cells/100 μl and incubated with antibodies against human CD24 (PE-Cy7-conjugated, 1:100 from BD for SUM149 or 1:50 from Biolegend for SUM159 and Vari068) and CD44 (APC-conjugated, 1:200, from BD) in cold room for 30 minutes. Content of ALDH+ E-BCSCs was determined by Aldefluor assay (StemCell Technologies) per manufacturer’s instructions. After labeling, cells were washed twice, re-suspended in Aldefluor assay buffer containing 1μg/mL of 4′, 6-diamidino-2-phenylindole (DAPI, Sigma) to discriminate live from dead cells. To obtain tumor cells from tumor xenografts, tumors of SUM149 xenografts grown in SCID mice were digested into single cells by collagenase for an hour and briefly shaken every 15 minutes. Cells debris was removed by filtration through a 40μm cell strainer (BD Biosciences) to obtain single cell suspensions. Red blood cells were removed with 0.8% ammonium chloride solution. The dissociated single tumor cells were re-suspended in HF (1xHBSS supplied with 2% FBS) buffer and incubated first with antibody against mouse H2Kd (PE conjugated, 1:100, BD Biosciences) in cold room for 30 minutes. Tumor cells were obtained by gating out mouse H2Kd+ cells. Flow analysis or sorting was performed on a MoFlow XDP or Astrios Cell Sorter (Beckman Coulter) at the University of Michigan Flow Cytometry Core facility.
Analysis of cellular ROS/superoxide and mitochondrial ROS
To measure cellular ROS and mitochondrial superoxide levels, cells were stained with CellROX™ Orange or MitoSOX Red (all from Thermo Fisher Scientific), which generates florescent signals when oxidized by cellular ROS or mitochondrial superoxide in the cells. Cells were incubated with pre-warmed CellROX or MitoSOX Red (diluted in PBS to a final concentration of 2.5 μM) staining solution for 30 min at 37 °C. All subsequent steps were performed in the dark. Cells were washed in PBS, harvested, then analyzed by flow cytometry. Cellular superoxide levels were labeled with the Cellular Superoxide Detection Assay Kit (ab139477, Abcam) following manufacturer’s instructions, and analyzed by flow cytometry. Flow data analysis was performed using the FlowJo software.
Oxygen consumption and ATP assay
Extracellular oxygen consumption from SUM159 breast cancer cells treated with IB-DNQ or mock (DMSO) were measured using a commercially available assay kit following manufacturer’s instructions (Abcam, ab197243). Cellular ATP levels in SUM159 breast cancer cells treated with IB-DNQ or mock (DMSO) were examined with the ATP assay kit from Beyotime Biotechnology (S0026) following manufacturer’s instructions. Fluorescence (for oxygen consumption assay) or luminescence (for ATP assay) intensity was measured under the BioTek SYNERGY H1 microplate reader.
Apoptosis and mitochondrial membrane potential assay
Apoptotic cells in cultured Vari068 or SUM149 BC cells following IB-DNQ, ATN224 alone or combined treatment were determined by labeling with APC-AnnexinV (BD, 1:50) and 4,6-diamidino-2-phenylindole (DAPI) and subjected to flow cytometry to detect AnnexinV-staining cells. To determine mitochondrial membrane potential, Vari068 or SUM149 BC cells following IB-DNQ, ATN224 alone or combined treatment were labeled with PE-TMRM (Invitrogen, 20nM) for 30 minutes at 37 °C. Cells were then washed, incubated with DAPI (Sigma) for 10 min at room temperature. After washing with PBS, cells were subjected to analysis by flow cytometry, using 488nm laser for excitation and a 570 nm emission filter for detection.
TUNEL assay and immunofluorescent staining
Vari068 or SUM149 BC cells were seeded at a density of 2000–5000 cells per slide for 1 days and then treated with vehicle (DMSO), IB-DNQ, ATN224 alone or in combination with IB-DNQ and ATN224 for 20 hours. Cells were fixed with 4% paraformaldehyde in PBS for 30 min at room temperature, rinsed with PBS, and then incubated in permeabilization buffer (0.5% TritonX-100 diluted in PBS) for 15 minutes on ice. Cells were rinsed twice with PBS and incubated with 50 μl of TUNEL reaction mixture per sample. For the negative control, 50 μl of Label Solution was used. Cells were incubated with TUNEL reaction mixture for 60 minutes at 37°C in a humidified atmosphere in the dark and rinsed 3 times with PBS. For immunostaining of ALDH1A3, TUNEL labeled cells were further incubated with antibodies against ALDH1A3 (Invitrogen, Cat# PA5-29188, 1:100) in PBS containing 0.5% Triton X-100 and 5% BSA overnight at 4°C, washed with PBS 3 times, and then incubated with FITC-conjugated goat anti-Rabbit IgG (Jackson Immunoresearch, 1:200) for 1h at room temperature and washed with PBS 3 times. All cell samples were mounted with VECTASHIELD mounting medium with DAPI (Vector Laboratories). Slides were examined under a BX41 microscope (Olympus) using UplanF1 20x-40x/0.5 objective lenses. Images were captured with a DP70 camera with DP Controller version 1.2.1.108 (Olympus).
Confocal microscopy
SUM159 BC cells seeded at a density of 2000–5000 cells per slide were fixed with 4% paraformaldehyde, permeabilized with 0.5% TritonX-100 (diluted in PBS) on ice, and blocked in 5% bovine serum albumin (BSA) buffer (diluted in PBS) for 1 h. Cells were then incubated with primary antibodies against NQO1 (Cat# 62262S, Cell Signaling Technology, 1:100) and Cytochrome c (Cat# 12963S, Cell Signaling Technology, 1:100) in PBS containing 0.5% Triton X-100 and 5% BSA overnight at 4°C. Subsequently, cells were washed three times with PBS, and incubated with Rhodamine-conjugated goat anti mouse IgG (Jackson Immunoresearch, 1:200) and FITC-conjugated goat anti rabbit IgG (Jackson Immunoresearch, 1:200) at room temperature for 1 hour. Nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI) in the dark for 5 min at room temperature and mounted with VECTASHIELD mounting medium (Vector Laboratories). Cells were imaged using a confocal microscope (ZEISS LSM 700, Germany). Colocalization of NQO1 and cytochrome c was analyzed by NIS Elements software.
Cell fractionation assay
To determine NQO1 and SOD1 expression level in mitochondria and cytosol fractions, BC cells treated with vehicle (DMSO) or IB-DNQ, ATN224, or IB-DNQ plus ATN224 were harvested by a cell scraper in ice cold PBS, and cells were centrifuged for 5 minutes at 600g. Freshly prepared 1× Extraction Buffer A (1ml) was added per 1× 107 cells per sample and cells were incubated on ice for 10–15 minutes. Cells were then homogenized on ice using a Dounce homogenizer for 10–30 strokes. Cell homogenates were subject to centrifugation at 600 × g for 10 minutes at 2–8 °C, and supernatants from different cell samples were carefully transferred to a fresh tube and centrifuged at 11,000 g for 10 minutes at 2–8 °C. The resulting supernatant (cytosol) fractions were carefully removed, and the pellet fractions (mitochondria) were each re-suspended in 150–200 μL of CelLytic M Cell Lysis Reagent with Protease Inhibitor Cocktail (1:100 [v/v], Sigma-Aldrich) for further testing.
Transmission electron microscopy (TEM)
Cells were fixed at 4 °C overnight in 3% glutaraldehyde, 3.0% paraformaldehyde in 0.1 M sodium cacodylate buffer, pH7.2, and were processed by the electron microscopy facility at the University of Michigan. Samples in 70nm sections on copper grids were examined using the JEOL JEM-1400+ Transmission Electron Microscope equipped with XR401 sCMOS camera.
Western blotting
Total cell protein was extracted with RIPA buffer (10mM Tris.Cl, 100mM NaCl, 2mM EDTA, 20mM NaF, 5 mM Na3VO4, 0.5% sodium deoxycholate, 0.1% SDS, 1% Triton X-100) supplemented with protease and phosphatase inhibitor cocktail (ThermoFisher Scientific). Cell lysates (20ug per lane) were leaded and subjected to SDS-PAGE with Bolt™ 4–12% Bis-Tris Plus Gel, blotted onto PVDF Membrane (ThermoFisher Scientific), and then blocked with 2% BSA in TBST buffer for 1 hour before incubation with primary antibodies as listed in the key resource table (see Supplementary Information). Membrane was rinsed three times in TBST for 5 minutes each time and incubated with HRP-conjugated rabbit or mouse secondary antibodies (Cell Signaling Technology). Protein bands were visualized using WesternBright Sirius Chemiluminescent Detection Kit (Advansta).
Tumorsphere formation assay
For tumorsphere culture of BC cell lines transduced with shNQO1, shSOD1, shSOD2, shCAT or SCR, 20 live (DAPI−) BC cells were sorted into each well of 96-well ultralow attachment plate (Corning) containing 150 μl of completed human MammoCult medium (StemCell Technologies) supplemented with 4 μg/mL heparin, 1μg/mL hydrocortisone, 1% antibiotic-antimycotic and 20 μg/mL gentamycin (all from Invitrogen) and cultured for 7–10 days in the presence of Dox (0.5μg/ml) together with or without IB-DNQ. Tumorspheres with diameter over 40 μM were counted under an optical microscope with 10x optical lens (EVOS all-in-one digital inverted microscope) and plotted for mean sphere formation per 100 cells. Tumorspheres of parental SUM149, SUM159 or Vari068 BC cells under various drug treatment regimens were cultivated at the condition of 20 cells/well in completed human MammoCult medium for 7–10 days. Tumorspheres settled in these conditions were counted under an optical microscope with 10x optical lens using the EVOS all-in-one digital inverted microscope.
Animal studies and drug treatment
7-week of female SCID mice were purchased from Charles River laboratories and housed in AAALAC-accredited specific pathogen-free rodent facilities at the University of Michigan. Tumor growth was determined by injecting primary breast cancer cells (Vari068: 1×106 cells per site; SUM49: 0.5×106 cells per site) with 30% Matrigel (BD Biosciences) into the 4th mammary fat pads of 7-week-old female SCID mice with 6–8 mice per cohort. Mouse treatments were started when the tumors reached to 3–5mm in diameter using vehicle (20% HPβCD, I.V.), IB-DNQ (10mg/kg), ATN-224 (10mg/kg) or combined treatment (i.v., every two days). Tumor size was measured once a week with a caliper and calculated as tumor volume = Length × Width2/2. Animals were euthanized at the end of treatments. Vari068 tumor xenografts and lung tissue sections were monitored by H&E staining and IHC. For limiting dilution secondary transplantation, SUM149 xenograft tumors were dissociated into single cells, and tumor cells of human origin were obtained by gating out mouse H2Kd+ cells. H2Kd− primary tumor cells sorted by flow cytometry were prepared in 30% Matrigel (BD Biosciences) with 2 or 3 different dilutions and injected bilaterally into the #4 mammary fat pads of 7-week-old female SCID mice with 3–4 mice per dilution. Tumor appearance was monitored for 3 months and frequency of tumor initiating cells was calculated using the ELDA software (Walter + Eliza Hall Bioinformatics, Institute of Medical research). To study the impact of SOD1 KD on IB-DNQ sensitivity in vivo, SUM149 cells harboring shSOD1 and SCR sequence were injected into the 4th mammary fat pads of 7-week-old female SCID mice (1× 106 per site). At the following day, mice (both shSOD1 and SCR) were randomly divided into vehicle or IB-DNQ treatment cohort (n = 6). Water containing Dox (200 μg/ml, Sigma-Aldrich) was administrated to each mouse cohort via bottled water supplied at day 1 after tumor cell implantation. IB-DNQ (10mg/kg) or vehicle (20% HPβCD) treatments were started when the tumors reached to 3–5 mm in diameter with tail vein injection. IB-DNQ or vehicle was given i.v., every two days. Tumor size was measured once a week with a caliper and calculated as tumor volume = Length × Width2 /2. All mouse experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee at the University of Michigan.
Kaplan-Meier analysis
Kaplan-Meier analysis of NQO1, SOD1, SOD2 and CAT antioxidant genes in 4929 breast cancer patient database (including all molecular subtypes) are performed using Kaplan-Meier Plotter (http://kmplot.com/analysis/index.php?p=service&default=true). Selected genes with correlation to patient survival were further analyzed using restricted analysis to different molecular subtypes of BC.
Statistical analysis
All in vitro assays were independently performed at least three times unless otherwise specified. Statistical differences between two groups were calculated using two-tailed student t-test. Statistical differences between three or more groups were calculated with one-way ANOVA with Bonferroni’s correction. Two-way ANOVA was used to analyze the extracellular oxygen consumption or tumor growth of TNBC cells with different treatment at multiple intervals. All analyses were conducted using GraphPad Prism (version 8.0). Unless otherwise indicated, data are presented as the Mean ± SEM (standard error mean), and a two-tailed value of * P <0.05, ** P<0.01, *** P<0.001 and **** P<0.0001 were considered statistically different.
Data availability
Data supporting the findings of this study are available within the article and its supplementary information. Raw data are available from the corresponding authors upon request.
RESULTS
NQO1 and SOD1 are highly expressed in TNBC and are associated with poor patient survival.
To identify potential biomarkers predicting treatment responses to IB-DNQ, we examined the expression of NQO1 in a large panel of patient TNBCs utilizing a tumor tissue array vs. two normal breast tissues (Fig. 1A), which unveiled that NQO1 is overexpressed in over 90% of TNBC tissues vs. that of normal breast. NQO1 overexpression in TNBC provides a therapeutic vulnerability exploitable by NQO1-bioactivatable futile redox cycling from NBRCCs (i.e., IB-DNQ), which produces large amounts of superoxide (O2.−) and downstream ROS (Fig. 1B).
Figure 1. NQO1 and related antioxidant enzyme expression in TNBC tissues, cell lines and CSC populations.
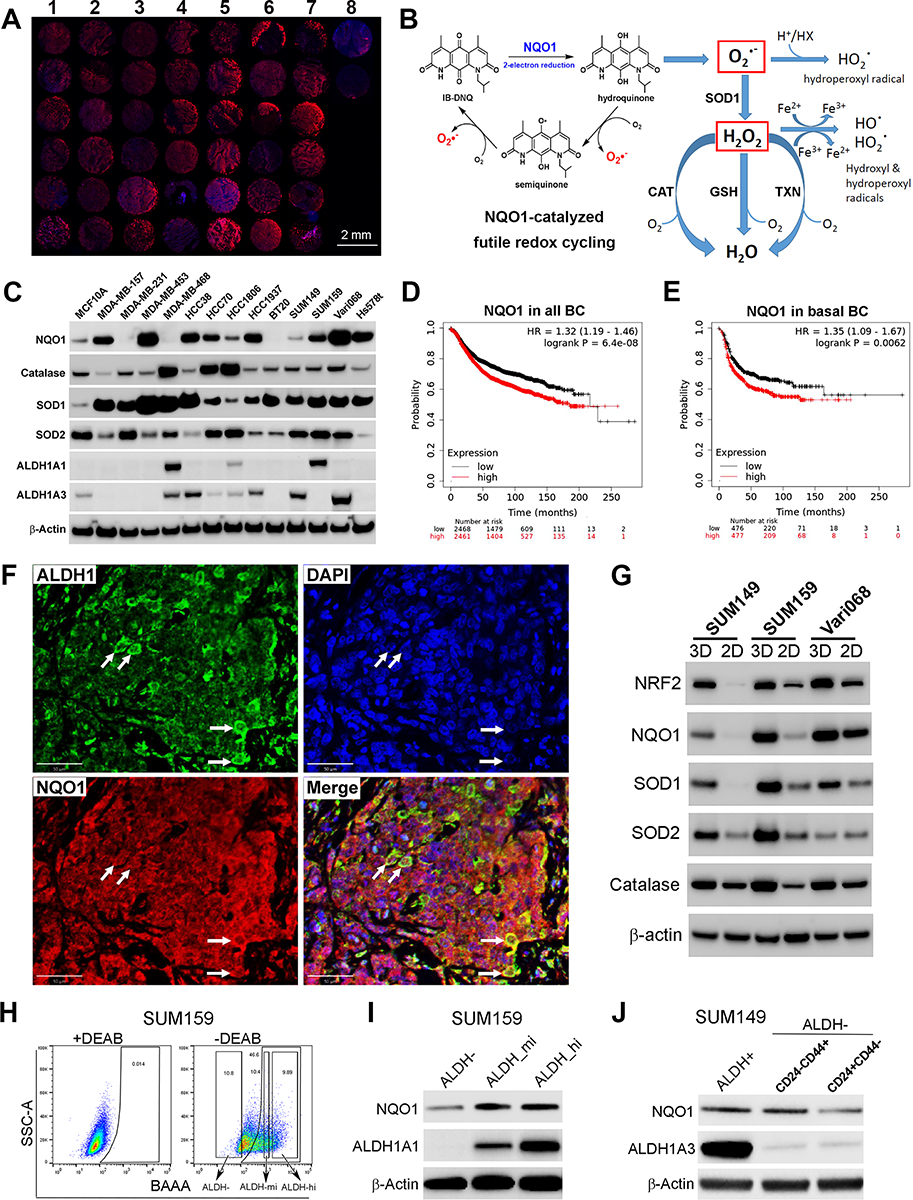
A, Immunofluorescent labeling of NQO1 in a human TNBC tissue array (column1–7) vs. two normal mammary tissues (column 8). Scale bar: 2mm. B, NQO1-catalyzed futile redox cycle from IB-DNQ. C, NQO1 and related antioxidant enzyme expression in 13 TNBC cell lines vs. MCF10A. D-E,NQO1 expression predicts poor RFS in a patient cohort containing all BC subtypes (D) and basal BC (E). F, Double labeling of TNBC tissues using specific antibodies against NQO1 and ALDH1. Bar: 50 μm. G, NQO1 and its related antioxidant enzyme expression in tumorspheres vs. 2D adherent cells. H-I, ALDHhi, ALDHmed and ALDH− bulk tumor cells were sorted based on ALDEFLUOR assay (H) and examined by immunoblotting with ALDH1A1 and NQO1 antibodies (I). J, NQO1 expression in ALDH+ and ALDH−CD24−CD44+ CSCs vs. bulk tumor cells of SUM149.
As high NQO1/CAT ratio in non-small cell lung cancer (NSCLC) promotes therapeutic efficacy of β-Lap (40), we next examined the expression pattern of NQO1 and related antioxidant enzymes including CAT, SOD1 and SOD2 across 13 TNBC cell lines vs. a nontumorigenic mammary epithelial cell (MaEC) line, MCF10A. NQO1 is overexpressed in a large proportion of TNBC cells, with 5 (Vari068, MDA-MB-453, HCC38, HCC1937 and Hs578t) expressing high levels of NQO1 (NQO1hi), 3 (MDA-MB-157, SUM159 and HCC70) expressing medium levels (NQO1med) and 2 (HCC1806 & SUM149) expressing low levels of NQO1 (NQO1lo) comparable to MCF10A (Fig. 1C). In addition, 3 lines (MDA-MB-231, MDA-MB-468 and BT20) display no detectable NQO1 (NQO1null), presumably due to a *2 mutation (C609T) that promotes NQO1 degradation (57,58).
Surprisingly, CAT, a H2O2 detoxifying enzyme under-expressed in NSCLC but high in benign tissues (40) is expressed in TNBC cells, and 3 TNBC lines have significantly elevated CAT expression (Fig. 1C). However, SOD1, a cytosolic, copper-containing enzyme, is ubiquitously overexpressed across all 13 TNBC cells, while SOD2, a superoxide dismutase localized in the mitochondria, is either unchanged (6/13) or suppressed (7/13) vs. MCF10A (Fig. 1C). These data support the notion that a SOD2 to SOD1 switch occurs during breast oncogenesis (59). We also examined the overall expression of ALDH1A1/A3, two established BCSC markers. Vari068, SUM149, HCC38 and HCC1937 express ALDH1A3 but not ALDH1A1, whereas SUM159, a mesenchymal BC cell line, expresses ALDH1A1 but not ALDH1A3 (Fig. 1C). The identification of NQO1+ TNBC cells expressing ALDH1A1/A3 provides suitable cell models to evaluate the potential of IB-DNQ in targeting ALDH+ CSCs.
As NQO1 and SOD1 are overexpressed in TNBC vs. normal MaEC, we next determined if the expression of these antioxidant enzymes correlate with disease progression across different BC subtypes. Kaplan-Meier analysis of a publicly available 4929-patient BC database (60) revealed that high NQO1 expression correlates with low recurrence-free survival (RFS) in the whole BC cohort (Fig. 1D), and in the luminal A (Supplementary Fig.S1A), luminal B (Supplementary Fig.S1B), HER2+ (Supplementary Fig.S1C) and basal (Fig. 1E) BC cohorts, suggesting a universal role of NQO1 in promoting cancer progression. High SOD1 expression also correlates with low RFS in the whole (Supplementary Fig. S1D) and basal BC (Supplementary Fig. S1E) cohort. In contrast, CAT (Supplementary Fig. S1, F&G) or SOD2 (Supplementary Fig. S1, H&I) expression did not correlate with patient survival in the whole and basal BC cohort. These studies suggest that NQO1 and SOD1, but not CAT or SOD2, may play more significant roles in BC progression.
NQO1 is highly expressed in TNBC CSCs.
ALDH+ CSCs are endowed with elevated NRF2 antioxidant defenses (37). Double labeling immunofluorescence of TNBC tissues using specific antibodies against NQO1 and ALDH1 revealed that NQO1 is highly expressed in ALDH1+ CSCs and bulk tumor cells (Fig. 1F). As tumorspheres enrich for CSCs, we compared the expression of NQO1 and its related antioxidant enzymes (NRF2, CAT, SOD1 and SOD2) in TNBC cells cultured as 3D spheroid vs. 2D adherent cells. NQO1, NRF2, CAT, SOD1 and SOD2 are all overexpressed in spheroid cells of different TNBC lines (Fig. 1G).
To validate if CSCs in different E/M states exhibit elevated NQO1 expression, we utilized SUM159 to sort ALDHhi CSCs as well as ALDHmed and ALDH− bulk BC cells (Fig. 1H) and examined NQO1 expression. NQO1 is elevated in ALDH+ (ALDHhi and ALDHmed) cells vs. ALDH− bulk BC cells (Fig. 1I). Using basal BC cells SUM149 that harbor CSCs in distinct E/M states, we sorted ALDH+ E- and ALDH−CD24−CD44+ M-CSCs and ALDH−CD24+CD44− differentiated cells and examined their relative NQO1 expression. Both E- and M-CSCs from SUM149 display elevated NQO1 expression vs. differentiated BC cells (Fig. 1J). This elevated NQO1 expression in CSCs may render them vulnerable to NQO1-bioactivatable futile redox cycling.
IB-DNQ is more potent and specific than β-lap in killing TNBC cells in a NQO1-dependent fashion.
We compared the potency of IB-DNQ vs. β-Lap, a highly studied NBRCC, in killing Vari068 TNBC cells developed from a primary tumor xenograft (37), which highly expresses NQO1 (Fig. 1C). IB-DNQ kills Vari068 cells (24h treatment) with LD50 (50% lethal dose) of 63 nM, which is 60-fold more potent than β-Lap (LD50 at 3.8 μM). Interestingly, cell lethality elicited by IB-DNQ but not β-Lap is fully rescued by Dicoumarol (DIC, an NQO1 specific inhibitor) (Fig. 2, A–C). Similar studies were conducted in SUM159 (Supplementary Fig. S2, A–C) and MDA-MB-157 (Supplementary Fig. S2, D and E), confirming the high potency and specificity of IB-DNQ vs. β-Lap in targeting TNBC cells overexpressing NQO1. We also tested the potency of IB-DNQ vs. β-Lap in HCC1806 cells that express low level of NQO1 comparable to MCF10A. IB-DNQ kills HCC1806 cells with LD50 of 405 nM, which is significantly more potent than β-Lap (LD50 > 5 μM), and cell lethality elicited by IB-DNQ and β-Lap in HCC1806 cells are both rescued by DIC (Supplementary Fig. S2, F and G).
Figure 2. IB-DNQ is more potent and specific than β-lap in killing TNBC cells and genetic or pharmacological inhibition of SOD1 synergistically enhances IB-DNQ-elicited lethality.
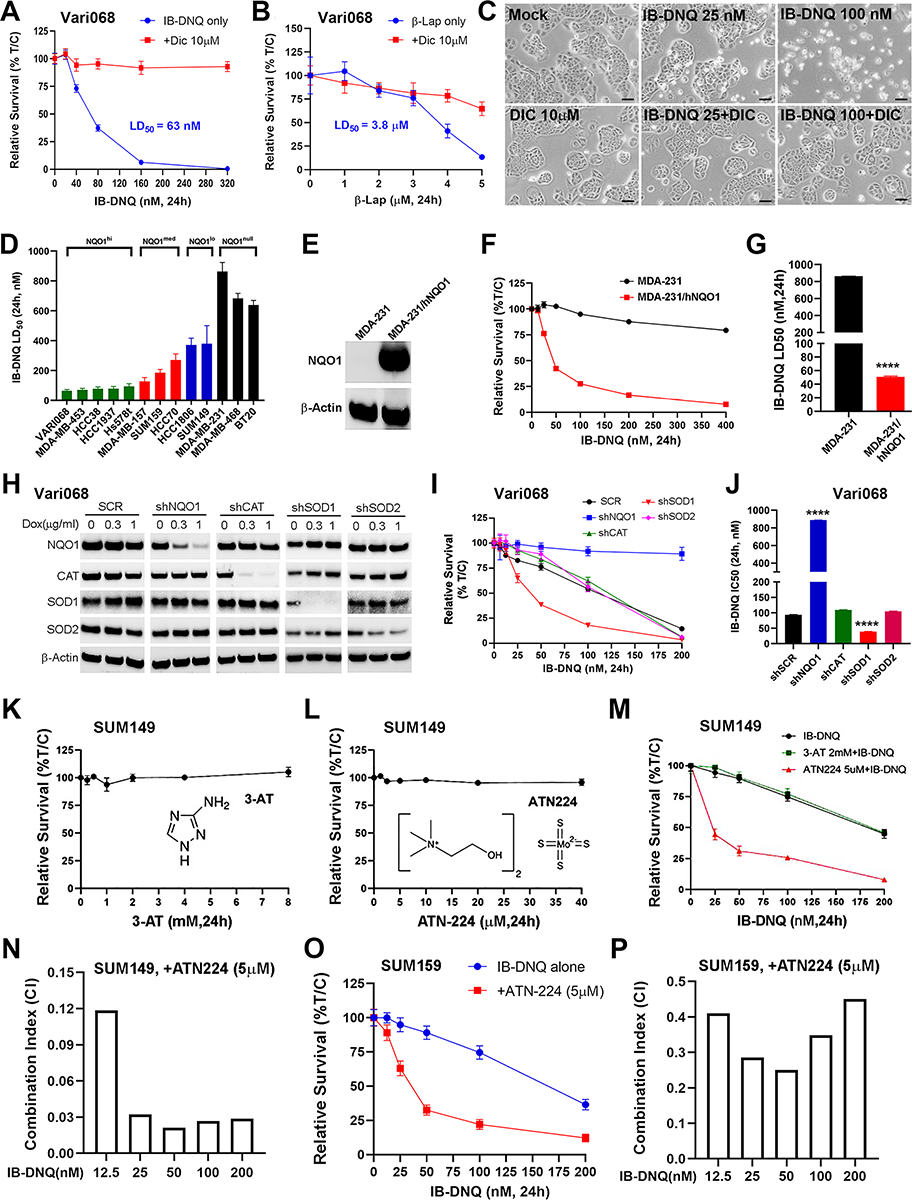
A-B, LD50 of IB-DNQ (A) vs. β-lap (B) in Vari068 BC cells treated with or without DIC (n=6 wells). C, Vari068 cells treated with Mock, DIC (10 μM), IB-DNQ (25 and 100nM), or IB-DNQ+DIC (10μM) for 24h and examined by light microscopy. Scale bar: 50 μm. D, IB-DNQ LD50 in 13 TNBC cells (n=2). E-G, NQO1 expression in NQO− MDA-MB-231 (E) renders these cells sensitive to IB-DNQ (F, n=6 wells) with LD50 < 50nM (G). ****: P<0.0001 vs. NQO− cells (n=2, unpaired student’s t-test). H-J, Dox-inducible KD of NQO1, CAT, SOD1 and SOD2 in Vari068 validated by immunoblotting with respective antibodies (H), and relative survival curve (I, n=6 wells) and IC50 (J) of Vari068 cells with KD of NQO1, CAT, SOD1 or SOD2 vs. SCR cells following IB-DNQ treatment. ****: p< 0.0001 vs. shSCR (n=2, unpaired student’s t-test). K-L, Relative survival of SUM149 treated with 3-AT (K) or ATN224 (L) at different doses for 24h (n=6 wells). M-N, Relative survival of SUM149 cells treated with IB-DNQ alone or together with ATN224 or 3-AT (n= 6 wells, M) and combination index (CI) of ATN224 (5 μM) with IB-DNQ from 12.5–200 nM in SUM149 (N). O-P, Relative survival of SUM159 cells treated with IB-DNQ alone or with ATN224 (n=6 wells, O) and CI of ATN224 with IB-DNQ from 12.5–200 nM in SUM159 (P).
The MTT assay used to measure cell viability is based on mitochondrial dehydrogenase activity, which may be a direct target of NBRCCs. We made a comparison using CCK8 (which involves most of the dehydrogenase activity in the cell) vs. MTT assay on SUM159 BC cells treated with IB-DNQ. This revealed that IB-DNQ at different doses resulted in the same degree of cytotoxicity determined by MTT vs. CCK8 assay (Supplementary Fig. S2H). We further tested the relative survival of SUM159 BC cells treated with IB-DNQ in media containing high (25 mM) vs. low (5.5 mM) concentrations of glucose (Supplementary Fig. S2I). This confirms that the sensitivity of TNBC cells to IB-DNQ is independent of glucose concentrations.
We systemically measured the LD50 of IB-DNQ in 13 TNBC cell lines and found that killing potency of IB-DNQ is correlated with NQO1 protein expression levels (Fig. 2D). NQO1hi Vari068 and MDA-MB-453 display the highest sensitivity, whereas NQO1null cell lines are resistant to IB-DNQ at doses <500 nM. As NQO1null TNBC cells are resistant to IB-DNQ, we next measured LD50 of IB-DNQ in isogenic MDA-MB-231 cells with NQO1+ (expressing WT hNQO1) or NQO1− (expressing NQO1 *2 mutation) gene expression. hNQO1 expression (Fig. 2E) confers high sensitivity of NQO1− cells to IB-DNQ with LD50 < 50nM (Fig. 2F and G).
Genetic or pharmacological inhibition of SOD1 enhances IB-DNQ-elicited cell lethality.
To validate the roles of NQO1, CAT, SOD1 and SOD2 in regulating IB-DNQ-elicited killing of TNBC cells, we employed the TRIPZ lentiviral vectors to establish doxycycline (Dox) inducible knockdown (KD) of NQO1, CAT, SOD1 and SOD2 in NQO1hi Vari068, NQO1med HCC70, and NQO1lo SUM149 BC cells. Dox-induced KD of NQO1 in Vari068 (Fig. 2, H–J) and HCC70 (Supplementary Fig. S2, J–L) renders these cells resistant to IB-DNQ, whereas the cells expressing a scrambled sequence (SCR) remain sensitive. Surprisingly, Dox-induced KD of CAT in Vari068 and HCC70 had no appreciable effect on IB-DNQ sensitivity. In contrast, Dox-induced KD of SOD1, but not SOD2, markedly enhanced IB-DNQ sensitivity in Vari068 and HCC70. This increased IB-DNQ sensitivity upon KD of SOD1, but not CAT, was also observed in NQO1lo SUM149 cells (Supplementary Fig. S2, M–O). As SOD1 provides major superoxide dismuatase activity in BC cells catalyzing H2O2 generation from O2.− (59), our data suggest the ROS effector responsible for IB-DNQ-elicited killing is actually O2.− rather than H2O2, which is thought to mediate β-Lap-induced tumor cell killing by inducing single-stranded DNA breaks and PARP1 hyperactivation, promoting the depletion of NAD+ and ATP to trigger programmed necrosis (40).
The limited availability of TRIPZ inducible shRNA clones for NQO1 and SOD1 make it hard to assess the specificity of these shRNAs. To address this issue, we obtained SMARTvector Dox-inducible lentiviral shRNA clones for NQO1 and SOD1 and made additional KD cells against NQO1 and SOD1 in SUM159. After Dox induction, SUM159 BC cells expressing 3 different NQO1 shRNAs (shNQO1–1/2/3) all had significantly reduced NQO1 expression, with shNQO1–2 and shNQO1–3 displaying the best KD efficiency (Supplementary Fig. S3A, inset). KD of NQO1 with shNQO1–2 or shNQO1–3 rendered SUM159 BC cells more resistant to IB-DNQ (Supplementary Fig. S3A), consistent with the studies using TRIPZ inducible NQO1 shRNA. Similar studies were done in SUM159 cells using the SMARTvector lentiviral shRNA clones for SOD1 (shSOD1–1/2), which unveiled that Dox-induced KD of SOD1 with shSOD1–2 but not shSOD1–1 significantly reduced SOD1 expression (Supplementary Fig.S3B, inset), leading to significantly increased IB-DNQ sensitivity.
Having genetically validated NQO1 and SOD1 as two therapeutic targets exploitable by IB-DNQ futile redox cycling, we next asked if pharmacological inhibition of SOD1 vs. CAT enhances the killing efficacy of IB-DNQ in NQO1lo TNBC cells. SUM149 treated with a CAT inhibitor 3-AT (Fig. 2K) or SOD1 inhibitor ATN224 alone (Fig. 2L) at different doses for 24h did not significantly affect tumor cell survival. However, treatment with IB-DNQ together with ATN224 but not 3-AT markedly enhanced IB-DNQ-induced killing of SUM149 BC cells (Fig. 2M). Calculation of the combination index (CI) of ATN224 at 5 μM with sublethal doses of IB-DNQ from 12.5–200 nM confirmed that ATN224 generates strong synergy (CI<0.15) with IB-DNQ at each dose examined (Fig. 2N). Similar synergistic effect of ATN224 with IB-DNQ in killing TNBC cells was observed in NQO1lo HCC1806 (Supplementary Fig. S3, C& D) and NQO1med SUM159 (Fig. 2O and P). In contrast, MCF10A treated with IB-DNQ (50–300 nM) plus distinct inhibitors of SOD1, including ATN224 (Supplementary Fig. S3E) or LCS-1 (a SOD1 inhibitor without copper-chelating activity, Supplementary Fig. S3F) failed to generate synergistic killing effect. LCS-1 (1μM) with IB-DNQ from 12.5–200nM also synergistically increased IB-DNQ-elicited killing of SUM149 and SUM159 BC cells (Supplementary Fig. S3, G–I).
IB-DNQ abrogates sphere-forming ability of NQO1-expressing TNBC cells at sub-lethal concentrations, and SOD1 inhibition potentiates this effect.
Using tumorsphere formation at clonal density, an assay widely used to assess CSC activity, we observed that IB-DNQ at 25–50 nM severely suppressed sphere-forming capacity, exemplified by significantly reduced numbers of spheres and smaller sphere size, in Vari068 (Fig. 3A and Supplementary Fig. S4A) and SUM159 (Fig. 3B and Supplementary Fig. S4B) BC cells that overexpress NQO1. In contrast to significantly inhibited tumorsphere formation, Vari068 (Fig. 3C) and SUM159 (Fig. 3D) cells grown at 2D adherent culture containing IB-DNQ at 25–50 nM for 2 weeks only modestly inhibited tumor cell survival.
Figure 3. IB-DNQ treatment preferentially inhibits CSC activity in TNBC cells expressing NQO1.
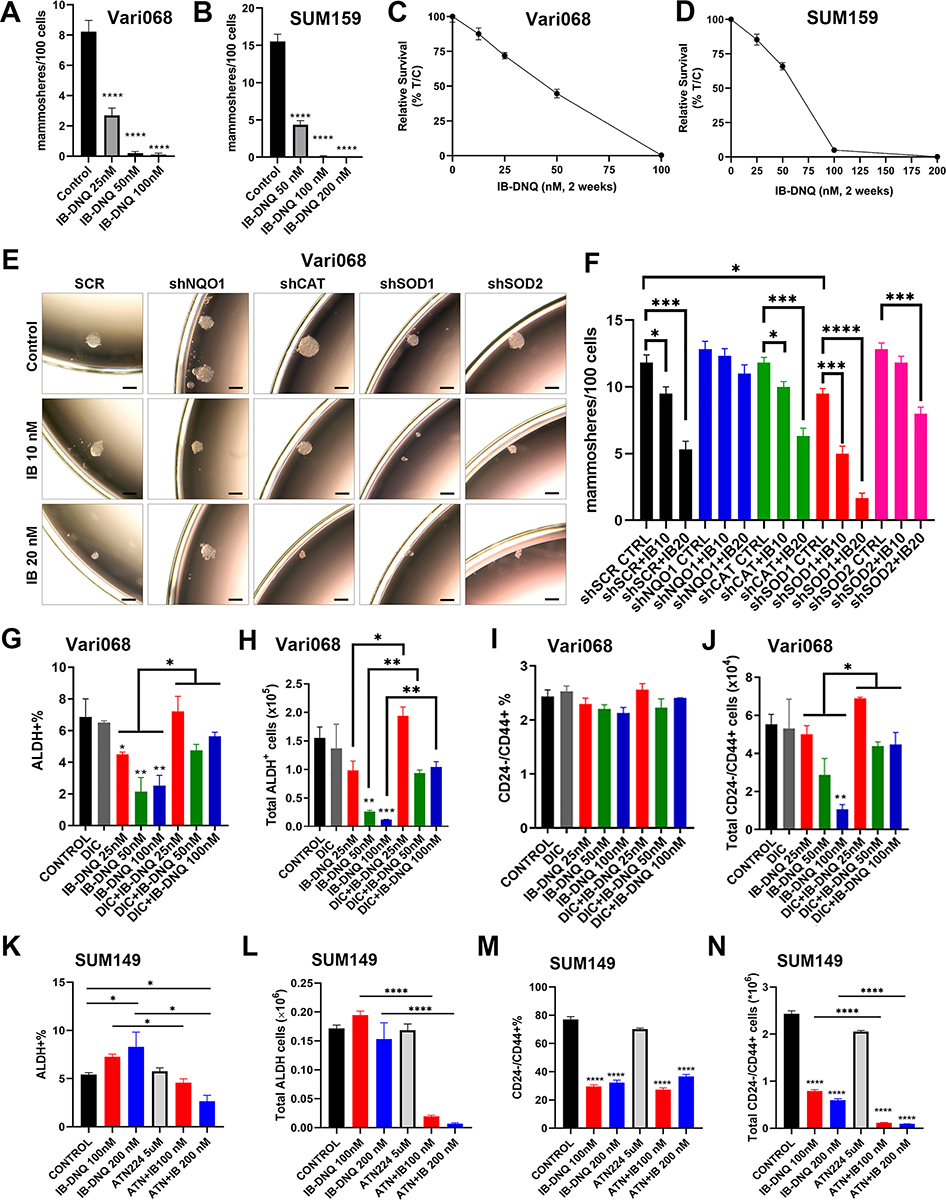
A-D, Tumorsphere formation of Vari068 (A) and SUM159 (C) BC cells in medium containing control (DMSO) or IB-DNQ of various doses, and relative survival of Vari068 (B) and SUM159 (D) cells grown in adherent culture with IB-DNQ for 14 days, n=6 wells. E-F, Sphere formation of Vari068 cells subjected to Dox-induced KD of NQO1, CAT, SOD1 or SOD2 and IB-DNQ treatment (E) and the impact of KD on IB-DNQ suppression of sphere-forming capacity (F). G-N, Vari068 (G-J) and SUM149 (K-N) cells were treated with indicated compounds for 20h and examined for the content and absolute number of ALDH+ and CD24−CD44+ CSCs. *, **, ***, ****: P < 0.05, 0.01, 0.001 or 0.0001 vs. Control or indicated by brackets. n=3, one-way ANOVA for A, C, F, G-J, and K-N.
We next examined if Dox-induced KD of NQO1, CAT, SOD1 or SOD2 alters IB-DNQ suppression of sphere-forming capacity (Fig. 3E and F). Vari068 BC cells expressing a SCR sequence exhibit a dose-dependent inhibition of tumorsphere formation in the presence of IB-DNQ (10 or 20 nM) and Dox (0.5 μg/mL), whereas Dox-induced KD of NQO1 renders Vari068 cells resistant to IB-DNQ. Dox-induced KD of CAT does not alter IB-DNQ inhibition of tumorsphere formation, whereas Dox-induced KD of SOD1, but not SOD2, significantly enhances IB-DNQ suppression of tumorsphere formation. Dox-induced KD of SOD1, but not NQO1, CAT or SOD2, slightly but significantly decreases sphere-forming ability of Vari068 cells, suggesting a role of SOD1 in maintaining CSC activity. As observed in Vari068 cells, NQO1lo SUM149 BC cells expressing a SCR sequence exhibited significantly inhibited sphere-forming activity when treated with IB-DNQ, and Dox-induced KD of CAT did not affect SUM149 sphere-forming ability, nor to significantly alter IB-DNQ inhibition of tumorsphere formation (Supplementary Fig. S4C). In contrast to CAT KD, Dox-induced SOD1 KD severely inhibited SUM149 sphere-forming ability and SOD1 KD together with 20 nM of IB-DNQ completely abrogated SUM149 tumorsphere formation (Supplementary Fig. S4C). SOD2 KD also had an inhibitory effect on SUM149 tumorsphere formation, which failed to enhance IB-DNQ inhibition of sphere-forming ability.
We determined if pharmacological inhibition of SOD1 is synergistic with IB-DNQ in suppressing CSC activity. SUM149 BC cells treated with 2.5 μM of ATN224 or 50 nM of IB-DNQ each exhibited significantly inhibited sphere-forming capacity, and co-treatment with ATN224 and IB-DNQ completely blocked tumorsphere formation (Supplementary Fig. S4D). In SUM159 BC cells, an increased concentration of ATN224 is required to inhibit tumorsphere formation, and ATN224 together wih IB-DNQ generated a strong synergy inhibiting sphere-forming capacity (Supplementary Fig. S4E).
NQO1 and SOD1 maintain ALDH+ E- and CD24−CD4+ M-CSCs respectively, and SOD1 inhibition augments IB-DNQ redox cycling to abrogate both E- and M-CSCs.
IB-DNQ treatment of Vari068 (Fig. 3G and H) and SUM159 (Supplementary Fig. S5, A and B) BC cells dose-dependently decreased the percentage and absolute cell number of ALDH+ CSCs, which were rescued by DIC. Despite that IB-DNQ preferentially targets ALDH+ CSCs in TNBC cells overexpressing NQO1, it failed to reduce the proportion of CD24−CD44+ M-CSC-like cells in Vari068 (Fig. 3I) and SUM159 (Supplementary Fig. S5C), although the absolute number of M-CSC-like cells in Vari068 (Fig. 3J) and SUM159 (Supplementary Fig. S5D) were significantly reduced, which were rescued by DIC. Although Dox-induced KD of NQO1 in Vari068 BC cells renders ALDH+ CSCs (Supplementary Fig. S5, E and F) and bulk tumor cells (Supplementary Fig. S5G) resistant to IB-DNQ, NQO1 KD in Vari068 significantly reduces the proportion and absolute number of ALDH+ CSCs, supporting a role of NQO1 for the maintenance of ALDH+ CSCs (61).
We next tested if SOD1 inhibition augments IB-DNQ redox cycling to abrogate E- and M-CSCs in TNBC expressing low/modest levels of NQO1. IB-DNQ treatment of SUM149 BC cells had a differential effect on distinct CSC states, with the proportion and absolute number of ALDH+ E-CSCs slightly increased (Fig. 3K and L), while the proportion and absolute number of CD24−CD4+ M-CSCs significantly decreased (Fig. 3M and N). These differential responses of E- and M-CSCs to IB-DNQ reflect the fact that modest levels of ROS produced by IB-DNQ in NQO1lo SUM149 BC cells do not harm but promote ALDH+ E-CSCs. In contrast, ROSlo M-CSCs are sensitive to even modestly increased ROS, which induces their transition to a ROShi E-CSC state (37). Although IB-DNQ at sublethal doses is not sufficient to decrease ALDH+ E-CSCs in SUM149, co-treatment with IB-DNQ plus ATN224 significantly reduced the percentage and absolute number of E-CSCs (Fig. 3K and L). ATN224 also potentiates the effect of IB-DNQ in abrogating the absolute number of CD24−CD44+ M-CSCs, despite the percentage of these cells was not further decreased (Fig. 3M and N).
As a copper-chelating inhibitor of SOD1, ATN224 may have off target effects by inhibiting other copper-containing proteins, confounding a specific role of SOD1 inhibition in synergizing IB-DNQ redox cycling targeting CSCs. To address this issue, we examined the efficacy of IB-DNQ targeting ALDH+ E- and CD24−CD4+ M-CSCs in SUM149 BC cells expressing Dox-inducible shSOD1 or SCR sequence. In Dox-treated SUM149 BC cells expressing a SCR sequence, distinct CSC states exhibited differential responses to IB-DNQ, with the percentage and absolute number of ALDH+ E-CSCs increased (Supplementary Fig. 5, H and I), but CD24−CD4+ M-CSCs significantly decreased in a dose-dependent fashion (Supplementary Fig. S5, J and K). Although IB-DNQ at sublethal doses failed to abrogate but promote ALDH+ E-CSCs in SUM149 expressing SCR sequence, Dox-induced KD of SOD1 markedly enhanced the efficacy of IB-DNQ to reduce the percentage (Supplementary Fig. S5H) and total number (Supplementary Fig. S5I) of ALDH+ CSCs. Dox-induced SOD1 KD also potentiates the effect of IB-DNQ in abrogating the absolute number of CD24−CD4+ M-CSCs (Supplementary Fig. S5K). Notably, Dox-induced KD of SOD1 in SUM149 selectively decreased the proportion and absolute number of CD24−CD4+ M- (Supplementary Fig. S5, J and K) but not ALDH+ E- (Supplementary Fig. S5, H and I) CSCs, suggesting a specific role of SOD1 in maintaining M-CSCs. In SUM159 BC cells, co-treatment with IB-DNQ and ATN224 also synergistically enhances the depletion of ALDH+ CSCs, and this effect was rescued by DIC (Supplementary Fig. S5, L and M).
IB-DNQ triggers apoptosis of ALDH+ CSCs and SOD1 inhibition potentiates this effect in TNBC expressing low/modest levels of NQO1.
To explore the nature of cell death elicited by IB-DNQ, we first treated NQO1hi Vari068 TNBC cells with sublethal doses of IB-DNQ for 20h, which were then labeled with Annexin V and DAPI to assess the cells undergoing early (DAPI−AnnexinV+) or late (DAPI+AnnexinV+) apoptosis (Supplementary Fig. 6A). IB-DNQ induces both early and late apoptosis of Vari068 BC cells in a dose-dependent manner (Fig. 4A). To confirm if IB-DNQ directly induces apoptosis of ALDH+ CSCs, we examined IB-DNQ-treated Vari068 cells by ALDH1A3 and TUNEL (apoptotic marker) dual immunofluorescence. Vari068 BC cells treated with IB-DNQ (0.1 μM) vs. vehicle exhibit significantly increased TUNEL+ labeling in ALDH1A3+ CSCs (arrows) (Fig. 4B), confirming that IB-DNQ at sublethal doses eradicates ALDH+ CSCs in TNBC overexpressing NQO1 through apoptotic death.
Figure 4. IB-DNQ induces apoptosis of Vari068 ALDH+ CSCs and ATN224 enhances IB-DNQ-mediated apoptosis in SUM149 BC cells expressing low/modest levels of NQO1.
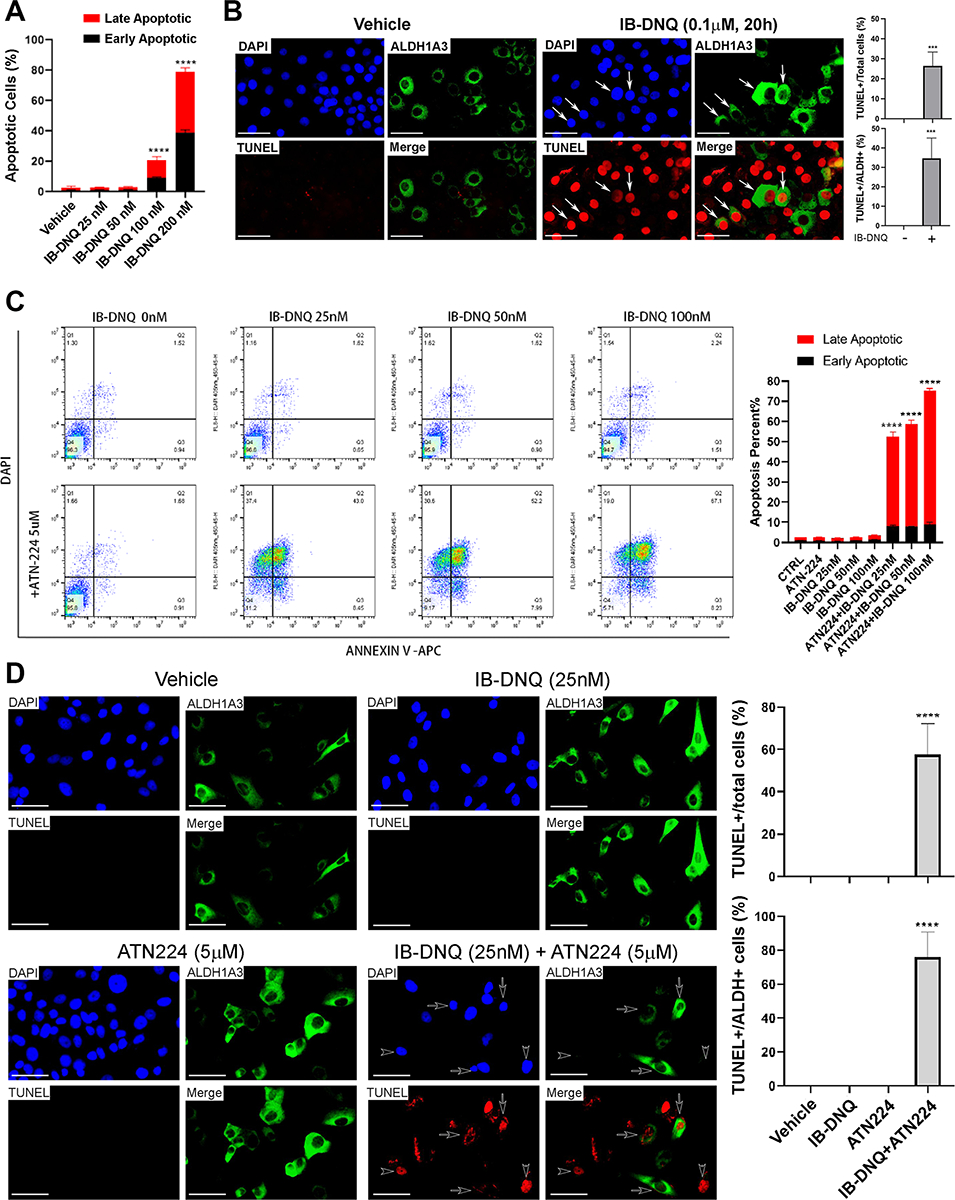
A-B, Vari068 cells treated with IB-DNQ for 20h were examined by flow cytometry to assess early and late apoptotic cells (A), or fixed and examined by ALDH1A3 and TUNEL dual labeling fluorescent microscopy (B) and % of TUNEL+ over total or ALDH1A3+ cells were scored in 3 different areas. C-D, SUM149 cells treated with IB-DNQ alone or together with ATN224 (5 μM) for 20h were examined by flow cytometry to detect early and late apoptotic cells (C), or fixed and examined by ALDH1A3 and TUNEL dual labeling (D), and % of TUNEL+ over total or ALDH1A3+ cells were scored in 3 different areas. ****: P < 0.0001 vs. Vehicle. n=3, one-way ANOVA for A and C and unpaired student’s t-test for B and D. Scale bar: 50 μm.
To verify if IB-DNQ alone or together with ATN224 triggers apoptotic death in NQO1lo TNBC cells, we treated SUM149 cells with 25–100 nM of IB-DNQ alone or together with ATN224 (5 μM) for 20h. Following treatment, SUM149 cells were harvested, labeled with Annexin V-APC and DAPI, and analyzed by flow cytometry (Fig. 4C, left panel). Although IB-DNQ (25–100 nM) or ATN224 (5μM) alone did not generate detectable apoptosis, addition of ATN224 plus 25–100 nM of IB-DNQ markedly induced early and late apoptosis of SUM149 BC cells (Fig. 4C, right panel). Treatment with a sublethal dose of IB-DNQ in combination with ATN224 synergistically induced TUNEL+ apoptotic death in ALDH+ CSCs (arrows) and ALDH− bulk tumor cells (arrowheads, Fig. 4D).
IB-DNQ plus ATN224 synergistically enhance mitoROS production and loss of mitochondrial membrane potential.
As NQO1-mediated redox cycling induces tumor cell killing via oxidant stress, we next measured the levels of cellular ROS by CellROX labeling and mitochondrial superoxide (mitoROS) by MitoSOX labeling with flow cytometry. IB-DNQ significantly increased total ROS (Fig. 5, A–C) and mitoROS (Fig. 5, D–F) in a dose-dependent fashion in Vari068, SUM159 and SUM149 BC cells. Similar to increased levels of total ROS and mitoROS, IB-DNQ significantly increased the levels of cellular superoxide in SUM159 (Supplementary Fig. 6B) and SUM149 (Supplementary Fig. 6C) BC cells. When these cells were treated with sublethal doses of IB-DNQ plus ATN224 (5μM), cellular ROS (Fig. 5, A–C), superoxide (Supplementary Fig. S6, B & C) and mitoROS (Fig. 5, D–F and Supplementary Fig. S6, D–F) were markedly increased vs. the cells treated with IB-DNQ alone, and this increase is more prominent in NQO1lo SUM149 BC cells.
Figure 5. IB-DNQ in combination with ATN224 synergistically enhances total ROS and mitoROS production resulting in loss of mitochondrial membrane potential.
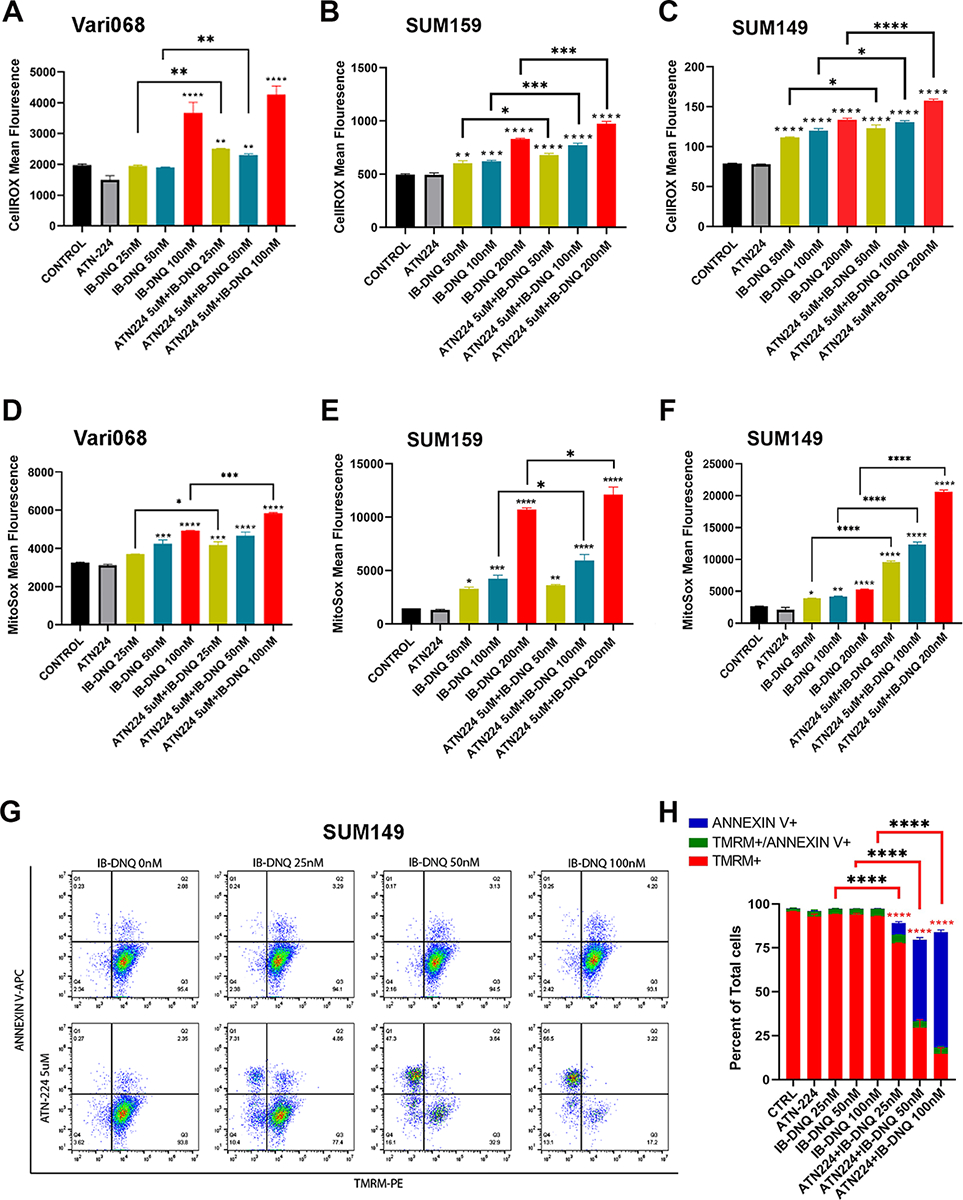
A-F, Vari068, SUM159 and SUM149 cells were treated with Control (DMSO), ATN224 (5 μM), IB-DNQ alone or IB-BNQ+ATN224 for 20h and examined by CellROX (A-C) or MitoSOX (D-F) labeling followed by flow cytometry. G-H, SUM149 cells treated with IB-DNQ (0–100 nM) alone or together with 5 μM of ATN224 for 20h were stained with AnnexinV-APC plus TMRM-PE and examined by flow cytometry (G), and % of TMRM+, TMRM+AnnexinV+ and AnnexinV+ cells was plotted (H). *, **, ***, ****: P< 0.05, 0.01, 0.001 or 0.0001 vs. Control or indicated by brackets, n=3, one-way ANOVA for A-F and H.
To further explore if increased mitoROS promotes mitochondrial oxidative damage, triggering the intrinsic apoptosis pathway, we measured mitochondrial membrane potential (by TMRM labeling) and apoptosis (by Annexin V labeling) in SUM149 cells treated with sublethal doses of IB-DNQ alone, or together with ATN224 (Fig. 5G). This revealed that IB-DNQ (25–100 nM) or ATN224 (5 μM) alone is not sufficient to induce significant loss of mitochondrial membrane potential and apoptosis. However, in the presence of ATN224, IB-DNQ induces the loss of mitochondrial membrane potential and increases apoptosis in a dose-dependent fashion (Fig. 5H).
NQO1 resides in the mitochondrial IMS driving IB-DNQ futile redox cycling, leading to excessive levels of mitoROS and mitochondrial oxidative damage.
The findings described above suggest that IB-DNQ may directly target mitochondria in ALDH+ CSCs to induce apoptosis. Notably, SOD1 localizes in the cytoplasm and mitochondrial IMS, where it provides major dismutase activity to protect mitochondrial integrity of BC cells (59). We hypothesize that a portion of NQO1 localizes in the mitochondria, where it coresides with SOD1 in the mitochondrial IMS. Therefore, SOD1 inhibition, by augmenting mitoROS production from IB-DNQ, potentiates mitochondrial oxidative injury to activate mitochondrial apoptotic pathway.
To determine the subcellular localization of NQO1, we purified mitochondrial and cytosol fractions from NQO1med (SUM159) and NQO1lo (SUM149 and HCC1806) TNBC cells and examined NQO1 and SOD1 expression in each fraction. In addition to cytosolic localization, a significant portion of NQO1 and SOD1 are both localized to the mitochondria fraction (Fig. 6A). In contrast, the mitochondrial protein SDHA (succinate dehydrogenase complex subunit A) is exclusively localized to the mitochondria fraction, while S phase kinase-associated protein 1 (Skp1), a cytosolic protein, is exclusively localized in the cytosol fraction (Fig. 6A). We also performed cell fractionation using a panel of NQO1hi BC cells, including Vari068, MDA-MB-436, MDA-MB-453, MCF7 and ZR-75–1. This revealed that, in addition to cytosolic NQO1, a significant portion of NQO1 is localized to the mitochondrial fraction in each BC cell line examined (Fig. 6B). In contrast to NQO1, two mitochondrial proteins SDHA and VDAC (voltage-dependent anion channel) are exclusively localized to the mitochondria fraction, while Skp1 is exclusively localized in the cytosol fraction. Therefore, NQO1-mediated futile redox cycling may occur not only in the cytosol but also in the mitochondria.
Figure 6. NQO1 residing in the mitochondrial IMS drives IB-DNQ redox cycling, promoting mitochondrial oxidative damage, Cyto c release, and activation of Casp3-mediated mitochondrial apoptotic pathway.
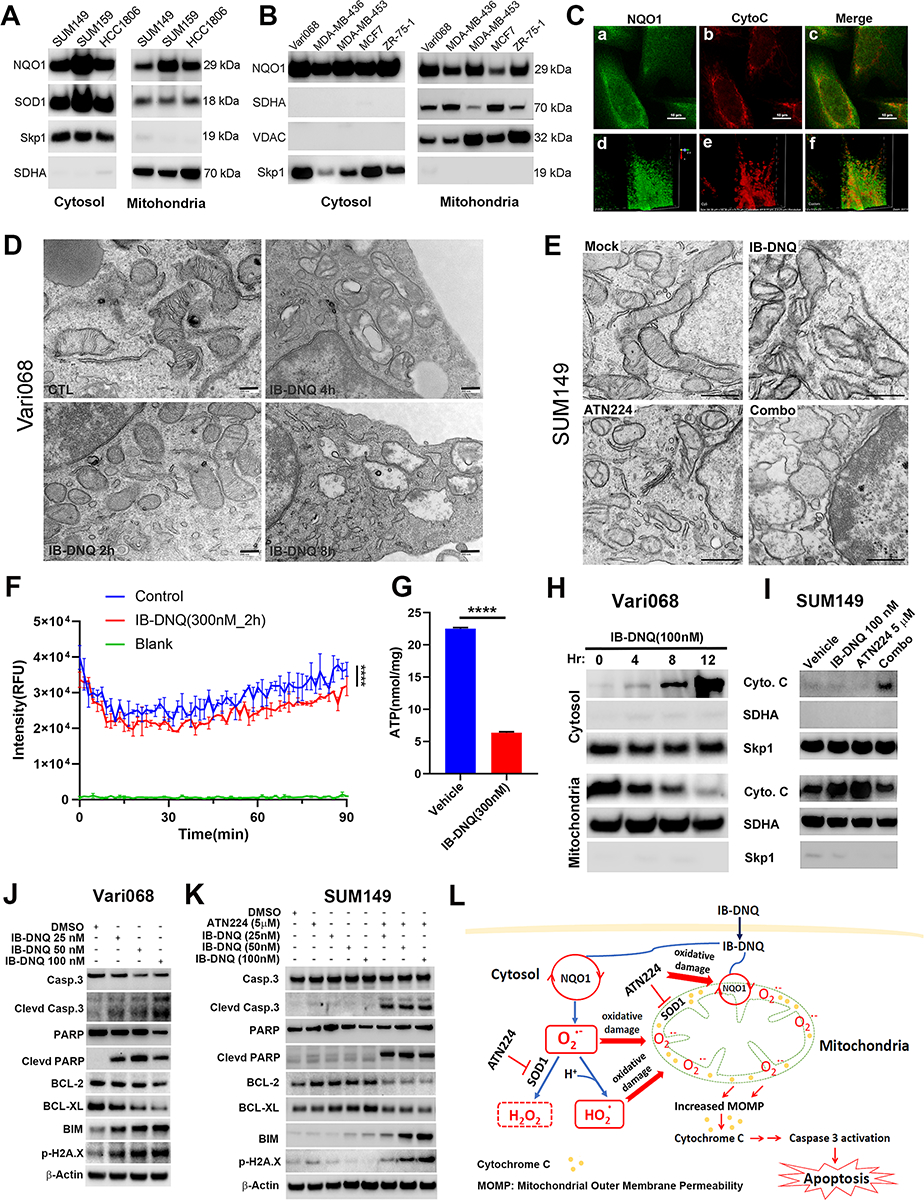
A-B, NQO1med and NQO1lo (A) and NQO1hi (B) TNBC cells were examined for NQO1, SOD1, SDHA and Skp1 expression in the mitochondrial and cytosol fraction. C, SUM159 cells labeled with specific antibodies against NQO1 and Cyto c were examined by confocal microscopy. Scale bar: 120 μm. D-E, Vari08 and SUM149 cells treated with 100 nM of IB-DNQ for 0–8h (D) or with vehicle, IB-DNQ (100 nM), ATN224 (5 μM), IB-DNQ+ATN224 for 16h (E) were processed and examined by TEM. Bars: 200 (D) and 500 (E) nm. F, SUM159 cells were treated with vehicle or IB-DNQ for 2h and measured for oxygen consumption based on fluorescent intensity (RFU) of an oxygen-bleaching fluorescent dye in the media covered by mineral oil at 1.5 min intervals for 90 min. ****: p<0.0001 (n=3, two-way ANOVA). G, ATP levels in the lysates of SUM159 cells treated with vehicle or IB-DNQ for 2h were measured and normalized by protein concentrations. ****: p<0.0001 with unpaired student’s t-test (n=3). H-I, Vari068 and SUM149 cells treated with IB-DNQ (100 nM) for 0–12h (H) or IB-DNQ and ATN224 alone or in combination for 16h (I) were used to isolate mitochondrial and cytosol fractions and examine Cyto c, SDHA and Skp1 expression. J-K, Lysates of Vari068 (J) and SUM149 (K) cells treated with IB-DNQ alone or IB-DNQ plus ATN224 for 20h were subjected to immunoblotting to examine the expression of apoptosis-related proteins. L, A model illustrating the mechanisms of action for IB-DNQ mediated pro-oxidant therapy targeting TNBC cells including CSCs.
To assess co-localization of NQO1 and cytochrome c (Cyto c), a protein known to reside in the mitochondrial IMS, we performed confocal microscopy of SUM159 BC cells subjected to dual-color immunofluorescent staining with NQO1 and Cyto c specific antibodies. This revealed that a significant portion of NQO1 colocalizes with Cyto c in the mitochondrial IMS (Fig. 6C, a–c). Further volume assay of NQO1 and Cyto c labeling from a specific angle using Volume Viewer of the NIS Elements software confirmed that NQO1 colocalizes with Cyto c in the mitochondria (Fig. 6C, d–f).
To test if IB-DNQ futile redox cycling generates mitochondrial oxidative damage, we treated NQO1hi Vari068 BC cells with 100 nM of IB-DNQ for 0, 2, 4, and 8 hours and determined the effect of IB-DNQ on mitochondrial morphology using transmission electron microscopy (TEM). Compared to control (DMSO) treated cells, which display typical mitochondrial morphology with intact cristae, Vari068 cells treated with IB-DNQ exhibit gradual loss of cristae and dilated/vacuolized mitochondria (Fig. 6D), indicative of mitochondrial oxidative damage. Similar TEM studies were performed on NQO1lo SUM149 BC cells treated with mock (DMSO), IB-DNQ (100 nM), ATN224 (5 μM), or IB-DNQ and ATN224 for 16h. Co-treatment with IB-DNQ and ATN224 generates a synergy in producing mitochondrial oxidative damage, manifested by loss of cristae and dilated/vacuolized mitochondria, while treatment with IB-DNQ alone has no effect (Fig. 6E). Inhibition of SOD1 with ATN224 produces mild mitochondrial oxidative damage manifested by some degree of cristae loss and mitochondrial vacuolization (Fig. 6E), consistent with a previous study (59).
Given a role of IB-DNQ redox cycling in targeting mitochondria to produce oxidative stress in TNBC cells, we next determined if IB-DNQ impairs mitochondrial oxidative metabolism and ATP biosynthesis. Treatment of SUM159 BC cells with IB-DNQ (at 300 nM) for 2 hours did not affect cell survival (Supplementary Fig. S7A), but significantly inhibited extracellular oxygen consumption (Fig. 6F) and intracellular ATP levels (Fig. 6G), suggesting a role of IB-DNQ in suppressing mitochondrial respiration and ATP production.
NQO1-mediated futile redox cycling from IB-DNQ results in the release of mitochondrial Cyto c and activation of Casp3-mediated apoptosis.
To test if IB-DNQ promotes Cyto c release from mitochondrial IMS, initiating the mitochondrial apoptotic pathway (62), we utilized 100 nM of IB-DNQ to treat NQO1hi Vari068 BC cells for 0–12h, and conducted cell fractionation to isolate the mitochondrial and cytosol fraction at each time point. IB-DNQ gradually induces the loss of Cyto c from the mitochondria, accompanied by increased Cyto c in the cytosol (Fig. 6H). Interestingly, compared to vehicle treated cells, treatment of NQO1lo SUM149 cells with IB-DNQ at 100 nM or ATN224 at 5 μM for 16h significantly increased mitochondrial Cyto c protein expression (Fig. 6I). This suggests that IB-DNQ or ATN224 treatment is sufficient to increase mitochondrial oxidative stress in SUM149, leading to mitochondrial antioxidant responses by boosting Cyto c expression, as Cyto c itself serves as an antioxidant protein (63). However, when SUM149 cells were treated with IB-DNQ plus ATN224, the enhanced mitochondrial Cyto c expression was lost (Fig. 6I), accompanied by significantly increased Cyto c in the cytosol fraction (Fig. 6I).
We investigated the downstream molecular mechanisms associated with Cyto c release. Treatment with IB-DNQ dose-dependently induced the cleavage of caspase 3 (Casp3), accompanied by the cleavage of its substrate poly (ADP-ribose) polymerase (PARP), and decreased BCL-2 and BCL-XL (antiapoptotic) but increased BIM and p-H2A.X (proapoptotic) expression in NQO1hi Vari068 BC cells (Fig. 6J). In NQO1lo SUM149 BC cells, treatment with IB-DNQ (100 nM) or ATN224 (5 μM) each had no effect on Casp3 activation, PARP cleavage, and BCL-2/BCL-XL or BIM/p-H2A.X expression. However, co-treatment with IB-DNQ and ATN224 synergistically enhanced Casp3 activation and PARP cleavage, associated with reduced BCL-2/BCL-XL, but increased BIM and p-H2A.X expression (Fig. 6K). IB-DNQ treatment of SUM159 BC cells also triggered Casp3 activation and PARP cleavage, associated with reduced BCL-2 and increased BIM expression, and treatment with ATN224+IB-DNQ significantly augmented the efficacy of IB-DNQ in promoting Casp3 and PARP cleavage, the reduction of BCL-2 and induction of BIM expression (Supplementary Fig. S7B). Inhibition of Casp3 with Z-DEVD-FMK in SUM159 BC cells significantly rescued IB-DNQ-induced cell lethality, whereas ferrostatin-1, an inhibitor of ferroptosis, failed to significantly rescue this lethality (Supplementary Fig. S7C). Parallel with the rescue of IB-DNQ-induced cell lethality, Z-DEVD-FMK partially but significantly rescued the decreased proportion of ALDH+ CSCs in SUM159 treated with IB-DNQ (Supplementary Fig. S7D). These studies suggest a novel mechanism of action for NQO1-mediated futile redox cycling from IB-DNQ in killing TNBC cells including CSCs (Fig. 6L), where a significant portion of NQO1 and SOD1 both reside in the mitochondrial IMS, and SOD1 inhibition synergizes with IB-DNQ redox cycling to abrogate CSCs and bulk tumor cells by augmenting mitochondrial oxidative injury, resulting in Cyto c release and activation of Casp3-mediated apoptosis.
IB-DNQ alone or together with SOD1 inhibition inhibits tumor growth, metastasis, and tumor-initiating potential of TNBC xenografts.
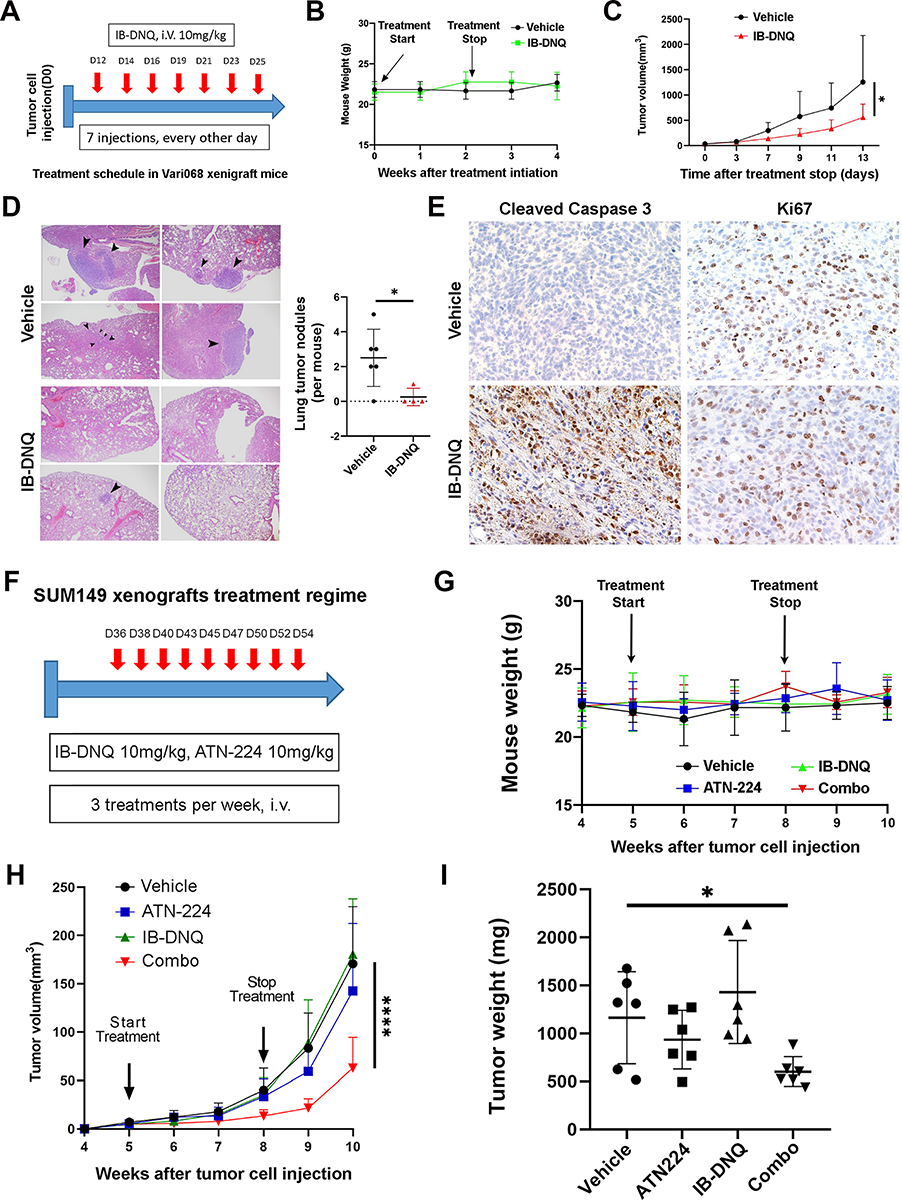
To test the feasibility of utilizing IB-DNQ as an antitumor therapy for TNBC cells overexpressing NQO1, we injected Vari068 BC cells into the #4 mammary fat pad (MFP) of SCID mice and treated tumor bearing mice with vehicle (20% HPβCD) or IB-DNQ (10 mg/kg, I.V.) for 2 weeks as outlined in Fig. 7A. This treatment regime had no significant effect on mouse body weight (Fig. 7B), confirming low toxicity of IB-DNQ. Monitoring tumor growth after the last treatment for two weeks indicated that IB-DNQ significantly inhibited primary tumor growth (Fig. 7C), and examination of lung histology from each group of mice revealed that IB-DNQ treatment markedly suppressed lung metastatic tumor nodule formation (Fig. 7D). Further immunohistochemical staining revealed that IB-DNQ induced Casp3 cleavage while Ki67 staining was not significantly changed (Fig. 7E), suggesting that IB-DNQ suppresses tumor growth and metastasis formation by promoting apoptotic death.
Figure 7. IB-DNQ alone or together with SOD1 inhibition suppresses tumor growth, metastasis, and tumor-initiating potential of TNBC xenografts.
A-C, Vari068 mammary tumor xenograft mice were randomized and treated with vehicle or IB-DNQ as indicated in (A). Mouse body weight was examined weekly for 4 weeks after treatment initiation (B) and mammary tumor growth following the last treatment was monitored for 2 weeks (C). *: p<0.05 (n=6). D, Lung histological sections from each group of mice were harvested to examine metastatic tumor nodule formation. *: p<0.05. E, staining of tumor sections treated with IB-DNQ vs. vehicle using antibodies against cleaved Casp3 or Ki67. F-H, SUM149 mammary tumor xenograft mice were randomized into 4 groups and treated with the regime indicated in (F), and mouse body weight (G) and mammary tumor growth (H) were examined weekly for 6 weeks. ****: p<0.0001(vs. Vehicle or IB-DNQ, n=6). I, Tumor weight for each group of mice at the end of tumor monitoring. *: p<0.05 (Vehicle vs. Combo, n=6). C & H: two-way ANOVA; D & I: two-tailed student’s t-test.
To further validate the effectiveness of IB-DNQ alone or together with SOD1 inhibition on tumor growth and tumor-initiating potential in xenograft models of TNBC expressing low/modest levels of NQO1, we injected SUM149 BC cells into the #4 MFP of SCID mice and treated tumor bearing mice by I.V. with vehicle (20% HPβCD), IB-DNQ (10 mg/kg), ATN224 (10 mg/kg), and IB-DNQ (10 mg/kg)+ATN224 (10 mg/kg) for 3 weeks as outlined in Fig. 7F. Single or combo therapy with IB-DNQ and ATN224 had no significant effect on mouse body weight (Fig. 7G), suggesting that combination of IB-DNQ and ATN224 did not generate added toxicity. Monitoring tumor growth following treatment initiation for 5 weeks indicates that IB-DNQ alone did not significantly inhibit primary tumor growth, while ATN224 modestly inhibited the growth of SUM149 BC cells. In contrast to IB-DNQ or ATN224 alone, combo treatment with IB-DNQ+ATN224 generated a synergistic effect in suppressing mammary tumor growth (Fig. 7H). Examination of tumor weight in each group of mice at the end of tumor monitoring confirmed that treatment with IB-DNQ plus ATN224 significantly reduced residual tumor weight (Fig. 7I). Further limiting dilution transplantation studies in secondary SCID mice using dissociated H2Kd− live tumor cells from residual tumors revealed that treatment with ATN224 but not IB-DNQ was able to decrease tumor initiating potential, and treatment with ATN224 plus IB-DNQ had the most significant effect in reducing tumor initiating potential of residual tumor cells (Table 1).
Table 1.
Limiting dilution transplantation in secondary SCID mouse
| Tumors/injection sites | TIC frequency | p-value (vs. vehicle) | |||
|---|---|---|---|---|---|
| cells injected | 50,000 | 5,000 | 500 | ||
| Vehicle | 8/8 | 8/8 | 6/8 | 1/152 | |
| IB-DNQ | 8/8 | 8/8 | 2/8 | 1/546 | 0.055 |
| ATN-224 | 8/8 | 5/8 | 3/8 | 1/1586 | 0.0002 |
| Combo | 5/8 | 1/6 | 0/6 | 1/20391 | 2.6e-15 |
As a copper-chelating inhibitor of SOD1, ATN224 may inhibit other copper/zinc proteins that are involved in angiogenesis (64–66) and mitochondrial oxidative phosphorylation (67), confounding a specific role of SOD1 inhibition in synergizing IB-DNQ redox cycling to suppress tumor growth and CSC activity. To address this issue, we injected SUM149 BC cells expressing Dox-inducible KD (shSOD1) or scrambled control (SCR) into the #4 MFP of female SCID mice, and mice bearing mammary tumors expressing shSOD1 or SCR sequence were each randomized into two groups and subject to treatment with Dox (200 μg/ml in water) plus vehicle (20% HPβCD, I.V.) or IB-DNQ (10 mg/kg, I.V.) for 3 weeks as indicated in the treatment regime (Supplementary Fig. S8A). Consistent with the study performed with parent SUM149 BC cells (Fig. 7H), mice bearing tumor cells expressing the SCR sequence did not exhibit significant tumor growth retardation after IB-DNQ treatment (Supplementary Fig. S8B). In contrast to the modest effect of ATN224 in suppressing SUM149 tumor growth (Fig. 7H), Dox-induced KD of SOD1 significantly inhibited tumor growth, and IB-DNQ treatment further enhanced tumor growth retardation of SOD1 KD cells (Supplementary Fig. S8B). Together, these studies suggests that IB-DNQ redox cycling serves as a feasible approach targeting TNBC overexpressing NQO1, and SOD1 inhibition greatly augments antitumor efficacy of this pro-oxidant strategy in TNBC cells expressing low or modest levels of NQO1. Furthermore, this approach is able to target CSCs which are resistant to conventional therapies.
DISCUSSION
In this study, we examined the antitumor efficacy and mechanism of action of IB-DNQ alone or in combination with SOD1 inhibition in abrogating CSCs in TNBC expressing different levels of NQO1. We demonstrate that a significant portion of NQO1 and SOD1 are localized in the mitochondria, and genetic or pharmacological inhibition of SOD1 synergizes with NQO1-bioactivated redox cycling from IB-DNQ to elicit CSC lethality by augmenting mitochondrial oxidative injury, leading to Cyto c release and activation of Casp3-mediated mitochondrial apoptotic pathway.
Through the study, we identified two antioxidant enzymes NQO1 and SOD1 as therapeutic vulnerabilities of ALDH+ E- and CD24−/loCD44+/hi M-CSCs in TNBC. Dox-inducible KD of NQO1 in NQO1hi Vari068 preferentially decreases ALDH+ E-CSCs whereas Dox-inducible KD of SOD1 in NQO1lo SUM149 selectively abrogates CD24−/loCD44+/hi M-like CSCs. Of note, targeting NQO1 with sublethal doses of IB-DNQ preferentially abrogates ALDH+ E-CSCs in TNBC expressing high levels of NQO1 (i.e., Vari068 and SUM159). However, for TNBC cells expressing modest/low levels of NQO1 (i.e., SUM149), the efficacy of IB-DNQ targeting ALDH+ E- but not CD24−CD44+ M-CSCs is significantly decreased due to the lower sensitivity of E (vs. M) CSCs to the modest levels of ROS elicited by IB-DNQ. We show that SOD1 is highly expressed in all TNBC cell lines examined, and genetic or pharmacological inhibition of SOD1, but not CAT or SOD2, markedly enhances IB-DNQ-elicited mitoROS production and killing of CSCs in both E and M states. Thus, SOD1 overexpression provides a therapeutic window for CSCs in TNBC with modest/low levels of NQO1 expression.
We confirmed in vitro and in TNBC xenograft models that combined treatment with IB-DNQ and SOD1 inhibition renders both BCSCs and bulk tumor cells more sensitive to IB-DNQ-elicited lethality. As CSCs can be generated from bulk tumor cells under chemotherapeutic stress through the “dedifferentiation” process, simultaneously targeting CSCs and bulk differentiated tumor cells by IB-DNQ-based futile redox cycling will be a promising therapeutic approach for advanced BC such as TNBC. Our data in TNBC xenograft models and a recent study in feline oral squamous cell carcinoma (56) suggest that IB-DNQ is well-tolerated, consistent with other studies showing that β-Lap produced only mild toxicity (anemia and possible methemoglobinemia) in a phase 1 clinical trial (52). In addition, the SOD1 inhibitor ATN224 is orally available and was well-tolerated in a phase 1 study of patients with advanced solid cancers (68), and a derivative of ATN224, tetrathiomolybdate (TM) is FDA approved to treat Wilson’s disease. These preclinical and clinical results suggest that combination therapy using IB-DNQ and ATN224 or TM will be well-tolerated in TNBC patients.
Previous studies from our group and others have demonstrated that proliferative ALDH+ CSCs are highly reliant on mitochondrial oxidative metabolism (36,37,69), providing a rationale for targeting mitochondrial vulnerabilities to eradicate this CSC population. We show here that targeting NQO1 by IB-DNQ at sublethal doses is sufficient to trigger apoptosis of ALDH1A3+ E-CSCs in TNBC overexpressing NQO1, and that pharmacological inhibition of SOD1, by enhancing mitoROS production, greatly enhances IB-DNQ-induced loss of mitochondrial membrane potential and apoptosis of ALDH1A3+ CSCs. These data strongly suggest that IB-DNQ redox cycling from NQO1 directly targets mitochondria in ALDH+ CSCs, triggering the intrinsic apoptotic pathway.
As increased ROS stress promotes the conversion of CD24−/loCD44+/hi M-CSCs to more proliferative ALDH+ E-CSCs (37), by targeting two elevated antioxidant enzymes NQO1 and SOD1 via IB-DNQ mitochondrial futile redox cycling, this study provides a novel therapeutic strategy effectively targeting CSCs including ALDH+ E- and CD24−/loCD44+/hi M-CSCs in TNBC. By promoting the conversion of CD24−/loCD44+/hi M-CSCs to ALDH+ E-CSCs and effectively inducing apoptosis in the later, our pro-oxidant strategy described in this study is able to target CSCs across the EMT-MET continuum. Since NQO1 and SOD1 are overexpressed in many other malignancies, this CSC targeting strategy may have wide applicability.
A limitation of this study is the use of xenograft models of TNBC established in immunodeficient SCID mice to validate the antitumor efficacy of targeting NQO1 and SOD1 through IB-DNQ mitochondrial futile redox cycling. This animal model is appropriate to assess the efficacy of this strategy directly on CSCs and bulk tumor cells. However, other NBRCC such as β-Lap has been shown to play a role in promoting host antitumor immunity (55). As the host immune system plays important roles in regulating CSCs and therapeutic responses, future studies are necessary to assess if and how this futile-oxidant-generating strategy using IB-DNQ alone or together with SOD1 inhibitors eradicates CSCs and bulk tumor cells in immunocompetent mouse models. This limitation should be considered when interpreting the results and translating to patients.
Supplementary Material
Significance:
Combining NQO1-bioactivatable futile oxidant generators with SOD1 inhibition eliminates breast cancer stem cells, providing a therapeutic strategy that may have wide applicability as NQO1 and SOD1 are overexpressed in several cancers.
Acknowledgments
This research was supported by NIH R35 CA197585, R01 CA28298-01A1 and BCRF-22-173 to M.S.W., NIH R01 DE026836 and R35 CA283859 to P.J.H., National Science Foundation of China (No. 82373084 and 82103385), Shenzhen High-Level Hospital Construction Fund, Peking University Shenzhen Hospital Scientific Research Fund (KYQD2023256), Shenzhen San-Ming Project (SZSM201612010), and Shenzhen Key Medical Discipline Construction Fund (SZXK017).
Footnotes
Competing interests: The authors declare no competing interests.
REFERENCES
- 1.Garrido-Castro AC, Lin NU, Polyak K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov 2019;9:176–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loizides S, Constantinidou A. Triple negative breast cancer: Immunogenicity, tumor microenvironment, and immunotherapy. Front Genet 2022;13:1095839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer research 2009;69:1302–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abdoli Shadbad M, Hosseinkhani N, Asadzadeh Z, Derakhshani A, Karim Ahangar N, Hemmat N, et al. A Systematic Review to Clarify the Prognostic Values of CD44 and CD44(+)CD24(−) Phenotype in Triple-Negative Breast Cancer Patients: Lessons Learned and The Road Ahead. Front Oncol 2021;11:689839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olsson M, Larsson P, Johansson J, Sah VR, Parris TZ. Cancer stem cells are prevalent in the basal-like 2 and mesenchymal triple-negative breast cancer subtypes in vitro. Front Cell Dev Biol 2023;11:1237673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shafee N, Smith CR, Wei S, Kim Y, Mills GB, Hortobagyi GN, et al. Cancer stem cells contribute to cisplatin resistance in Brca1/p53-mediated mouse mammary tumors. Cancer research 2008;68:3243–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. Journal of the National Cancer Institute 2008;100:672–9 [DOI] [PubMed] [Google Scholar]
- 8.Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009;458:780–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Croker AK, Allan AL. Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDHhiCD44(+) human breast cancer cells. Breast cancer research and treatment 2012;133:75–87 [DOI] [PubMed] [Google Scholar]
- 10.Tanei T, Morimoto K, Shimazu K, Kim SJ, Tanji Y, Taguchi T, et al. Association of breast cancer stem cells identified by aldehyde dehydrogenase 1 expression with resistance to sequential Paclitaxel and epirubicin-based chemotherapy for breast cancers. Clinical cancer research : an official journal of the American Association for Cancer Research 2009;15:4234–41 [DOI] [PubMed] [Google Scholar]
- 11.Phillips TM, McBride WH, Pajonk F. The response of CD24(−/low)/CD44+ breast cancer-initiating cells to radiation. Journal of the National Cancer Institute 2006;98:1777–85 [DOI] [PubMed] [Google Scholar]
- 12.Lu H, Samanta D, Xiang L, Zhang H, Hu H, Chen I, et al. Chemotherapy triggers HIF-1-dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proceedings of the National Academy of Sciences of the United States of America 2015;112:E4600–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu H, Xie Y, Tran L, Lan J, Yang Y, Murugan NL, et al. Chemotherapy-induced S100A10 recruits KDM6A to facilitate OCT4-mediated breast cancer stemness. J Clin Invest 2020;130:4607–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Echeverria GV, Ge Z, Seth S, Zhang X, Jeter-Jones S, Zhou X, et al. Resistance to neoadjuvant chemotherapy in triple-negative breast cancer mediated by a reversible drug-tolerant state. Sci Transl Med 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dhanyamraju PK, Schell TD, Amin S, Robertson GP. Drug-Tolerant Persister Cells in Cancer Therapy Resistance. Cancer research 2022;82:2503–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pu Y, Li L, Peng H, Liu L, Heymann D, Robert C, et al. Drug-tolerant persister cells in cancer: the cutting edges and future directions. Nat Rev Clin Oncol 2023;20:799–813 [DOI] [PubMed] [Google Scholar]
- 17.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America 2003;100:3983–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell stem cell 2007;1:555–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang EH, Hynes MJ, Zhang T, Ginestier C, Dontu G, Appelman H, et al. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer research 2009;69:3382–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carpentino JE, Hynes MJ, Appelman HD, Zheng T, Steindler DA, Scott EW, et al. Aldehyde dehydrogenase-expressing colon stem cells contribute to tumorigenesis in the transition from colitis to cancer. Cancer research 2009;69:8208–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van den Hoogen C, van der Horst G, Cheung H, Buijs JT, Lippitt JM, Guzman-Ramirez N, et al. High aldehyde dehydrogenase activity identifies tumor-initiating and metastasis-initiating cells in human prostate cancer. Cancer research 2010;70:5163–73 [DOI] [PubMed] [Google Scholar]
- 22.Sullivan JP, Spinola M, Dodge M, Raso MG, Behrens C, Gao B, et al. Aldehyde dehydrogenase activity selects for lung adenocarcinoma stem cells dependent on notch signaling. Cancer research 2010;70:9937–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, et al. Breast Cancer Stem Cells Transition between Epithelial and Mesenchymal States Reflective of their Normal Counterparts. Stem cell reports 2014;2:78–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jolly MK, Somarelli JA, Sheth M, Biddle A, Tripathi SC, Armstrong AJ, et al. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol Ther 2019;194:161–84 [DOI] [PubMed] [Google Scholar]
- 25.Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S, et al. Identification of the tumour transition states occurring during EMT. Nature 2018;556:463–8 [DOI] [PubMed] [Google Scholar]
- 26.Luo M, Brooks M, Wicha MS. Epithelial-mesenchymal plasticity of breast cancer stem cells: implications for metastasis and therapeutic resistance. Current pharmaceutical design 2015;21:1301–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brooks MD, Burness ML, Wicha MS. Therapeutic Implications of Cellular Heterogeneity and Plasticity in Breast Cancer. Cell stem cell 2015;17:260–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gupta PB, Pastushenko I, Skibinski A, Blanpain C, Kuperwasser C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell stem cell 2019;24:65–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu W, Kang Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev Cell 2019;49:361–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quintanal-Villalonga A, Chan JM, Yu HA, Pe’er D, Sawyers CL, Sen T, et al. Lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nat Rev Clin Oncol 2020;17:360–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol 2015;12:445–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther 2020;5:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dashzeveg NK, Taftaf R, Ramos EK, Torre-Healy L, Chumakova A, Silver DJ, et al. New Advances and Challenges of Targeting Cancer Stem Cells. Cancer research 2017;77:5222–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thankamony AP, Saxena K, Murali R, Jolly MK, Nair R. Cancer Stem Cell Plasticity - A Deadly Deal. Front Mol Biosci 2020;7:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luo M, Wicha MS. Targeting Cancer Stem Cell Redox Metabolism to Enhance Therapy Responses. Seminars in Radiation Oncology 2019;29:42–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo M, Wicha MS. Metabolic plasticity of cancer stem cells. Oncotarget 2015;6:35141–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo M, Shang L, Brooks MD, Jiagge E, Zhu Y, Buschhaus JM, et al. Targeting Breast Cancer Stem Cell State Equilibrium through Modulation of Redox Signaling. Cell metabolism 2018;28:69–86 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang K, Chen D, Ma K, Wu X, Hao H, Jiang S. NAD(P)H:Quinone Oxidoreductase 1 (NQO1) as a Therapeutic and Diagnostic Target in Cancer. J Med Chem 2018;61:6983–7003 [DOI] [PubMed] [Google Scholar]
- 39.Starcher CL, Pay SL, Singh N, Yeh IJ, Bhandare SB, Su X, et al. Targeting Base Excision Repair in Cancer: NQO1-Bioactivatable Drugs Improve Tumor Selectivity and Reduce Treatment Toxicity Through Radiosensitization of Human Cancer. Front Oncol 2020;10:1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang X, Motea EA, Moore ZR, Yao J, Dong Y, Chakrabarti G, et al. Leveraging an NQO1 Bioactivatable Drug for Tumor-Selective Use of Poly(ADP-ribose) Polymerase Inhibitors. Cancer Cell 2016;30:940–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang Y, Zhang Y, Wu Q, Cui X, Lin Z, Liu S, et al. Clinical implications of high NQO1 expression in breast cancers. J Exp Clin Cancer Res 2014;33:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mikami K, Naito M, Ishiguro T, Yano H, Tomida A, Yamada T, et al. Immunological quantitation of DT-diaphorase in carcinoma cell lines and clinical colon cancers: advanced tumors express greater levels of DT-diaphorase. Jpn J Cancer Res 1998;89:910–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cui X, Jin T, Wang X, Jin G, Li Z, Lin L. NAD(P)H:quinone oxidoreductase-1 overexpression predicts poor prognosis in small cell lung cancer. Oncol Rep 2014;32:2589–95 [DOI] [PubMed] [Google Scholar]
- 44.Lin L, Qin Y, Jin T, Liu S, Zhang S, Shen X, et al. Significance of NQO1 overexpression for prognostic evaluation of gastric adenocarcinoma. Exp Mol Pathol 2014;96:200–5 [DOI] [PubMed] [Google Scholar]
- 45.Lin L, Sun J, Tan Y, Li Z, Kong F, Shen Y, et al. Prognostic implication of NQO1 overexpression in hepatocellular carcinoma. Hum Pathol 2017;69:31–7 [DOI] [PubMed] [Google Scholar]
- 46.Ma Y, Kong J, Yan G, Ren X, Jin D, Jin T, et al. NQO1 overexpression is associated with poor prognosis in squamous cell carcinoma of the uterine cervix. BMC Cancer 2014;14:414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li R, Bianchet MA, Talalay P, Amzel LM. The three-dimensional structure of NAD(P)H:quinone reductase, a flavoprotein involved in cancer chemoprotection and chemotherapy: mechanism of the two-electron reduction. Proceedings of the National Academy of Sciences of the United States of America 1995;92:8846–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oh ET, Kim JW, Kim JM, Kim SJ, Lee JS, Hong SS, et al. NQO1 inhibits proteasome-mediated degradation of HIF-1alpha. Nat Commun 2016;7:13593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Z, Zhang Y, Jin T, Men J, Lin Z, Qi P, et al. NQO1 protein expression predicts poor prognosis of non-small cell lung cancers. BMC Cancer 2015;15:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parkinson EI, Hergenrother PJ. Deoxynyboquinones as NQO1-Activated Cancer Therapeutics. Acc Chem Res 2015;48:2715–23 [DOI] [PubMed] [Google Scholar]
- 51.Pink JJ, Planchon SM, Tagliarino C, Varnes ME, Siegel D, Boothman DA. NAD(P)H:Quinone oxidoreductase activity is the principal determinant of beta-lapachone cytotoxicity. J Biol Chem 2000;275:5416–24 [DOI] [PubMed] [Google Scholar]
- 52.Gerber DE, Beg MS, Fattah F, Frankel AE, Fatunde O, Arriaga Y, et al. Phase 1 study of ARQ 761, a beta-lapachone analogue that promotes NQO1-mediated programmed cancer cell necrosis. Br J Cancer 2018;119:928–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beg MS, Huang X, Silvers MA, Gerber DE, Bolluyt J, Sarode V, et al. Using a novel NQO1 bioactivatable drug, beta-lapachone (ARQ761), to enhance chemotherapeutic effects by metabolic modulation in pancreatic cancer. J Surg Oncol 2017;116:83–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Motea EA, Huang X, Singh N, Kilgore JA, Williams NS, Xie XJ, et al. NQO1-dependent, Tumor-selective Radiosensitization of Non-small Cell Lung Cancers. Clinical cancer research : an official journal of the American Association for Cancer Research 2019;25:2601–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li X, Liu Z, Zhang A, Han C, Shen A, Jiang L, et al. NQO1 targeting prodrug triggers innate sensing to overcome checkpoint blockade resistance. Nat Commun 2019;10:3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lundberg AP, Boudreau MW, Selting KA, Chatkewitz LE, Samuelson J, Francis JM, et al. Utilizing feline oral squamous cell carcinoma patients to develop NQO1-targeted therapy. Neoplasia 2021;23:811–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Traver RD, Siegel D, Beall HD, Phillips RM, Gibson NW, Franklin WA, et al. Characterization of a polymorphism in NAD(P)H: quinone oxidoreductase (DT-diaphorase). Br J Cancer 1997;75:69–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsvetkov P, Adamovich Y, Elliott E, Shaul Y. E3 ligase STUB1/CHIP regulates NAD(P)H:quinone oxidoreductase 1 (NQO1) accumulation in aged brain, a process impaired in certain Alzheimer disease patients. J Biol Chem 2011;286:8839–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Papa L, Hahn M, Marsh EL, Evans BS, Germain D. SOD2 to SOD1 switch in breast cancer. J Biol Chem 2014;289:5412–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gyorffy B Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput Struct Biotechnol J 2021;19:4101–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Madajewski B, Boatman MA, Chakrabarti G, Boothman DA, Bey EA. Depleting Tumor-NQO1 Potentiates Anoikis and Inhibits Growth of NSCLC. Mol Cancer Res 2016;14:14–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol 2020;21:85–100 [DOI] [PubMed] [Google Scholar]
- 63.Korshunov SS, Krasnikov BF, Pereverzev MO, Skulachev VP. The antioxidant functions of cytochrome c. FEBS Lett 1999;462:192–8 [DOI] [PubMed] [Google Scholar]
- 64.Pan Q, Kleer CG, van Golen KL, Irani J, Bottema KM, Bias C, et al. Copper deficiency induced by tetrathiomolybdate suppresses tumor growth and angiogenesis. Cancer research 2002;62:4854–9 [PubMed] [Google Scholar]
- 65.Hassouneh B, Islam M, Nagel T, Pan Q, Merajver SD, Teknos TN. Tetrathiomolybdate promotes tumor necrosis and prevents distant metastases by suppressing angiogenesis in head and neck cancer. Mol Cancer Ther 2007;6:1039–45 [DOI] [PubMed] [Google Scholar]
- 66.Lowndes SA, Harris AL. Copper chelation as an antiangiogenic therapy. Oncol Res 2004;14:529–39 [DOI] [PubMed] [Google Scholar]
- 67.Ramchandani D, Berisa M, Tavarez DA, Li Z, Miele M, Bai Y, et al. Copper depletion modulates mitochondrial oxidative phosphorylation to impair triple negative breast cancer metastasis. Nat Commun 2021;12:7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lowndes SA, Adams A, Timms A, Fisher N, Smythe J, Watt SM, et al. Phase I study of copper-binding agent ATN-224 in patients with advanced solid tumors. Clinical cancer research : an official journal of the American Association for Cancer Research 2008;14:7526–34 [DOI] [PubMed] [Google Scholar]
- 69.Lamb R, Bonuccelli G, Ozsvari B, Peiris-Pages M, Fiorillo M, Smith DL, et al. Mitochondrial mass, a new metabolic biomarker for stem-like cancer cells: Understanding WNT/FGF-driven anabolic signaling. Oncotarget 2015;6:30453–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting the findings of this study are available within the article and its supplementary information. Raw data are available from the corresponding authors upon request.