

Abstract
In eukaryotic cells, DNA damage triggers activation of checkpoint signaling pathways that coordinate cell cycle arrest and repair of damaged DNA. These DNA damage responses serve to maintain genome stability and prevent accumulation of genetic mutations and development of cancer. The p38 MAPK was previously implicated in cellular responses to several types of DNA damage. However, the role of each of the four p38 isoforms and the mechanism for their involvement in DNA damage responses remained poorly understood. In this study, we demonstrate that p38γ, but not the other p38 isoforms, contributes to the survival of UV-treated cells. Deletion of p38γ sensitizes cells to UV exposure, accompanied by prolonged S phase cell cycle arrest and increased rate of apoptosis. Further investigation reveal that p38γ is essential for the optimal activation of the checkpoint signaling caused by UV, and for the efficient repair of UV-induced DNA damage. These findings have established a novel role of p38γ in UV-induced DNA damage responses, and suggested that p38γ contributes to the ability of cells to cope with UV exposure by regulating the checkpoint signaling pathways and the repair of damaged DNA.
Keywords: p38γ, DNA damage, UV, DNA repair, checkpoint signaling
Introduction
p38 is one of the major MAP kinases, which regulates multiple biological processes in mammals (Ono and Han, 2000). It was initially identified as a key mediator of inflammation and stress responses. Later studies also implicated p38 in cell cycle and proliferation, cell differentiation, cellular senescence, and DNA damage responses (Nebreda and Porras, 2000; Johnson and Lapadat, 2002; Han and Sun, 2007).
In response to DNA damage, eukaryotic cells undergo proliferative arrest to allow DNA repair, which is crucial for maintaining genome stability and preventing tumorigenesis (Kastan and Bartek, 2004). The p38 pathway plays an important role in DNA damage responses. p38 is activated by ionizing radiation (Dent et al., 2003), and upon γ-irradiation, activation of MKK6 and p38 is required for dividing cells to arrest at the G2/M phase transition (Wang et al., 2000). Moreover, p38 mediates UV-induced apoptosis by activating p53, through direct phosphorylation of functionally important residues (Ser15, Ser33 and Ser46), and stabilization of the p53 protein (Bulavin et al., 1999; She et al., 2000). Two papers show that p38 mediates UV- or DNA alkylating agent-induced G2/M arrest, and phosphorylation and inactivation of Cdc25B and Cdc25C, two phosphatases that facilitate G2/M progression by removing inhibitory phosphates from Cdk1 (Bulavin et al., 2001; Hirose et al., 2003). A p38 downstream kinase MAPKAPK2 (MK2) may also contribute to the inactivation of Cdc25B and Cdc25C and DNA damage-induced cell cycle arrest (Manke et al., 2005). In addition, a recent study identifies a MAP3K termed TAO, as an upstream activator of p38 during DNA damage responses (Raman et al., 2007).
Like other MAPK pathways, the p38 signaling cascades involves sequential activation of MAP kinase kinase kinases (MAP3Ks), such as MTK, MLK2, MLK3, DLK, ASK and TAK1, and MAP kinase kinases (MKKs), including MKK3, MKK6, and MKK4, which directly activate p38 through phosphorylation in a cell type- and stimulus-dependent manner. Numerous p38 substrates, including protein kinases, transcription factors and cell-cycle regulators, have been identified that mediate a variety of p38 functions (Shi and Gaestel, 2002). The mammalian genomes contain four isoforms of p38, α, β, δ and γ, each encoded by a different gene. These p38 isoforms differ in tissue-specific expression, mode of regulation by upstream stimuli, selectivity for upstream regulatory kinases and phosphatases as well as downstream targets, and specificity for pharmaceutical inhibitors (Han et al., 1994; Jiang et al., 1996; Li et al., 1996; Jiang et al., 1997; Enslen et al., 2000; Tanoue et al., 2001; Shi and Gaestel, 2002). Among these isoforms, only p38α has been shown to be essential for inflammatory and stress responses by genetic analysis in murine models (Kang et al., 2008; Otsuka et al., 2010), while the physiological roles of the other p38 isoforms in inflammation or other cellular functions have been unclear (Beardmore et al., 2005; Sabio et al., 2005). Although previous studies have indicated an important function of p38 in DNA damage responses, the specific role of each p38 isoform in these processes have not been systematically analyzed, and the mechanism underlying the involvement of p38 in DNA damage responses is not fully understood. In the current study, we took advantage of the isogenic cell lines either wild type or null for each of the p38 isoforms, and demonstrate that p38γ, but not other isoforms, contributes to cell survival after UV exposure. Deletion of p38γ leads to increased sensitivity of cells to UV treatment, enhances UV-induced cell cycle arrest and apoptosis, and causes defects in checkpoint signaling and UV lesion repair. These studies have identified p38γ as the major p38 isoform that regulates UV sensitivity, and suggest that p38γ promotes the survival of UV-exposed cells by mediating activation of the DNA damage checkpoint signaling and efficient repair of UV-induced DNA lesions.
Results
Deficiency in p38γ, but not the other isoforms of p38, increases sensitivity of cells to UV treatment
To investigate the role of each individual p38 isoform in UV-induced DNA damage responses, we isolated mouse embryonic fibroblasts (MEFs) from mice harboring floxed alleles of p38α, β, γ, δ (Kang et al., 2008; Otsuka et al., 2010). After immortalization, these cells were transduced with Cre-encoding or control retroviruses to generate isogenic MEF lines either null or wild type for each p38 isoform, respectively (Supplemental Fig. 1). Genomic PCR using allele-specific primers confirmed that Cre had indeed excised the floxed allele of the appropriate p38 isoform, while leaving the loci encoding the other isoform intact (Fig. 1A). After treatment with UV, the survival rate, based on the number of proliferating cells determined by the MTT assay, was comparable between the p38α-, p38β-, or p38δ-null MEF cells and their isogenic wild type counterparts (Fig. 1B). In contrast, the percentage of surviving cells after UV treatment was significantly lower in MEFs that are p38γ deficient, as compared to the isogenic control MEFs (Fig. 1B). The loss of p38γ protein expression after Cre expression in MEFs was confirmed by Western blot analysis (Supplemental Fig. 2). Thus, the loss of p38γ, but not the other p38 isoforms, increases the sensitivity of cells to UV exposure. To further substantiate the involvement of p38γ in UV sensitivity, we treated wild type and p38γ-deficient MEF cells with varying dosages of UV (Fig. 1C). As expected, UV induced a dose-dependent reduction in the survival of both wild type and p38γ-deficient cells. Nevertheless, the UV-induced cell toxicity is significantly more severe in p38γ-deficient cells than in the isogenic control cells at all the dosages. In addition, we measured the percentage of surviving cells at different time points after the UV treatment (Fig. 1D). After treated with 50 J/m2 of UV, the number of proliferating control cells declined within the first 24 h and became steady afterwards, while the surviving p38γ-deficient cells continued to decrease between 24- and 48-h post treatment, suggesting that loss of p38γ sensitizes cells to UV.
Figure 1. Deletion of p38γ, but not the other p38 isoforms, increases the sensitivity of cells to UV radiation.
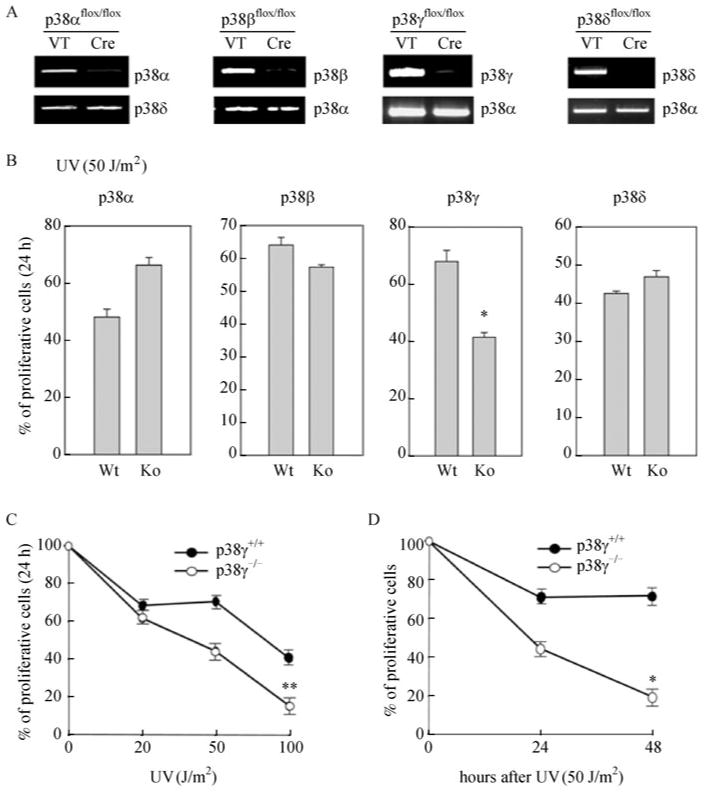
(A) Products of genomic PCR in MEF cells carrying floxed alleles of p38α, β, γ or δ, after transduction with a Cre-encoding (Cre) or control (VT) retrovirus, using allele-specific primers for each indicated p38 isoform. (B) MTTassays with 1 × 104 of MEF cells either wild type (Wt) or null (Ko) for p38α, p38β, p38γ or p38δ, after treatment with 50 J/m2 of UV and recovery for 24 h. (C) MTTassays with 1 × 104 MEF cells either wild type (p38γ+/+) or null (p38γ-/-) for p38γ, after treatment with indicated dosages of UV and recovery for 24 h. (D) MTT assays with 1 × 104 MEF cells either wild type (p38γ+/+) or null (p38γ-/-) for p38γ, after treatment with 50 J/m2 of UV and recovery for indicated periods of time. (B, C, D) % of proliferating cells was calculated by dividing A470 derived from the treated cells by A470 representing 1 × 104 cells on the parallel standard curve. Values are mean ± SD for triplicates. The results are representative of 3 independent repeats. *, P < 0.001; **, P < 0.01, versus the wild type cell line control by Student's t test.
Taken together, these results indicate that p38γ, but not the other p38 isoforms, contributes to cell survival after UV-induced DNA damage.
Deficiency in p38γ prolongs S phase arrest and enhances apoptosis after UV treatment
We next investigated whether the enhanced sensitivity to UV upon loss of p38γ was due to increased cell cycle arrest or cell death. Wild type and p38γ-deficient MEFs had very similar cell cycle profiles under untreated conditions (Fig. 2A, 0 h). After UV-treatment, cells in the S phase gradually accumulated in both wild type and p38γ-deficient populations (Fig. 2A, 6 h and 16 h), indicative of a S-phase cell cycle arrest required for the repair of UV-induced DNA damage. Twenty-four hours after the treatment, the S-phase arrested cells in the wild type control population had already re-entered cell cycle, presumably as a result of successful repair of the UV-induced DNA damage; however, the majority of the cells were still arrested in S phase in the p38γ-deficient population (Fig. 2A, 24 h). Therefore, deletion of p38γ leads to a prolonged S-phase arrest after UV treatment, suggesting that there might be a defect in DNA damage repair in p38γ-deficient cells.
Figure 2. p38γ deficiency leads to prolonged S phase cell cycle arrest and increased apoptosis after UV treatment.
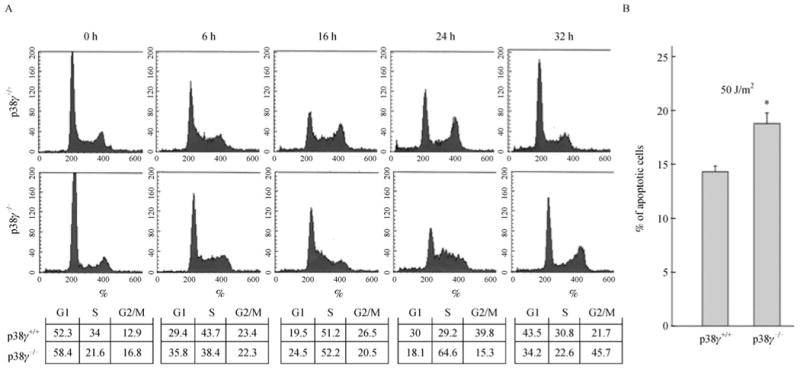
(A) MEF cells either wild type (p38γ+/+) or null (p38γ-/-) for p38γ were treated with 20 J/m2 of UV and allowed to recover for 0, 6, 16, 24 or 32 h. The cell cycle profiles were determined by flow cytometry after fixation in ethanol and staining with propidium iodide. Numbers in the charts indicate the percentages of cells in G1, S or G2/M cell cycle phases in each cell population. (B) MEF cells either wild type (p38γ+/+) or null (p38γ-/-) for p38γ were treated with 50 J/m2 of UV or left untreated, and allowed to recover for 24 h. Both adherent cells and cells in suspension were collected, stained with FITC-conjugated Annexin-V, and analyzed by flow cytometry. Percentage of apoptotic cells was calculated by dividing the percentage of FITC-positive cells in the UV-treated population by that in the untreated population. Values are mean ± SD for triplicates. The results are representative of 2 independent repeats. *P < 0.01 versus the wild type cell line control by Student's t test.
In addition to cell cycle arrest, UV also induces apoptosis in cells. Based on staining with fluorescence-conjugated Annexin V, UV induced a higher percentage of apoptotic cells in the p38γ-deficient MEFs than in the isogenic control population (Fig. 2B). Thus, our findings indicate that the increased UV sensitivity upon loss of p38γ is resulted from both prolonged S phase cell cycle arrest and enhanced apoptosis after UV treatment.
p38γ is required for activation of the checkpoint signaling pathway in responses to UV
The UV-induced DNA damage response, including cell cycle arrest and DNA damage repair, is coordinated via the checkpoint signal transduction cascades involving multiple protein kinases, phosphatases, adaptor proteins, and transcription factors (Latonen and Laiho, 2005). UV radiation triggers phosphorylation and activation of the phosphatidylinositol 3-kinase-like kinases ATR and its downstream target the Chk1 protein kinase, and the p53 tumor suppressor protein (Abraham, 2001; Latonen and Laiho, 2005; Cimprich and Cortez, 2008). These proteins collectively mediate the cell cycle arrest and facilitate the repair of UV-induced lesions on DNA. In addition, NBS1, a subunit of the Mre11/Rad50/NBS1 (MRN) protein complex, is also activated through phosphorylation upon UV treatment, and contributes to the activation of ATR and downstream signaling of ATR (Olson et al., 2007). Therefore, we analyzed the effect of p38γ status on the UV-induced checkpoint signaling pathway.
As expected, UV induced phosphorylation of Chk1 at Ser345 in wild type MEF cells a dose-dependent manner; however, the induction of Chk1 phosphorylation was greatly decreased at every dosage of UV in p38γ-deficient cells (Fig. 3A). We performed a time-course analysis of Chk1 phosphorylation in these cells. UV induced robust Chk1 phosphorylation at Ser345 at about 30 min post treatment, after which the amount of phosphorylated Chk1 gradually declined (Fig. 3B). The level of Chk1-S345 phosphorylation was much weaker in p38γ-deficient cells than in the isogenic wild type cells at each time point examined. To confirm the specificity of the effect of p38γ on Chk1 activation, wild type p38γ was introduced back into the p38γ-deficient MEFs via recombinant retroviruses. Restoration of p38γ expression in p38γ-deficient cells led to a marked increase in UV-induced Chk1-S345 phosphorylation as compared to the cells transduced with a vector control (Fig. 3C), indicating that reduced Chk1 phosphorylation in p38γ-deficient MEFs was indeed a specific result of the loss of p38γ.
Figure 3. p38γ deficiency attenuates activation of the checkpoint signaling pathway after UV treatment.
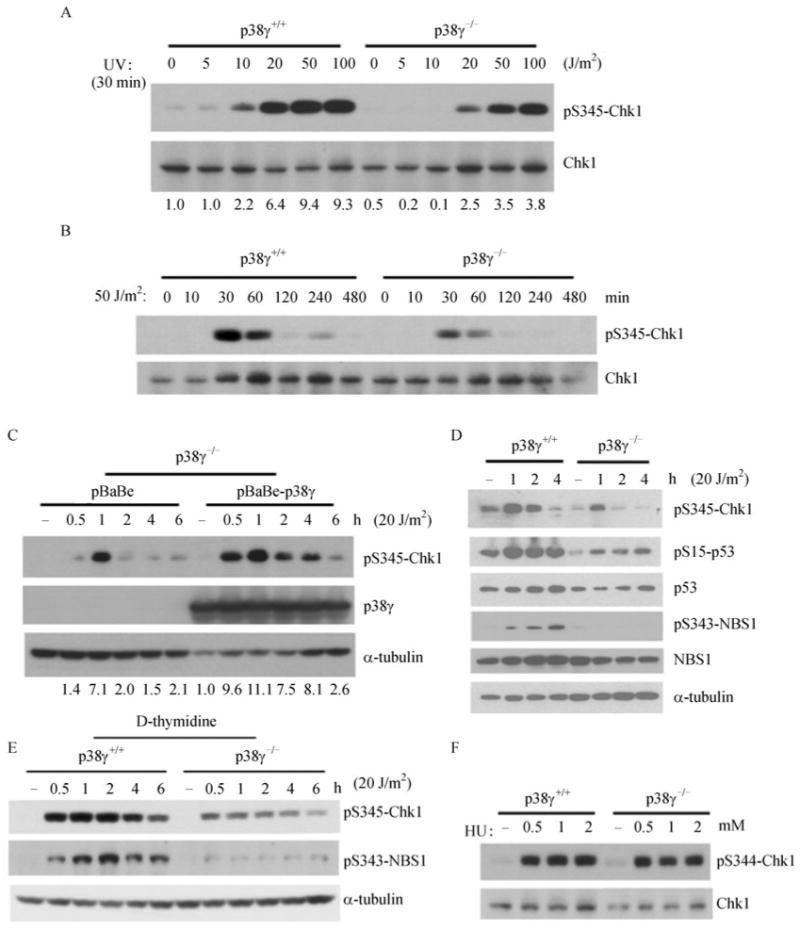
(A) Western blot analysis of MEF cells either wild type (p38γ+/+) or null (p38γ-/-) for p38γ after treatment with indicated dosages of UV and recovery in complete medium for 30 min, detecting phospho-Chk1-S345 and Chk1. Numbers represent relative levels of the phospho-Chk1-S345 signals. (B) Western blot analysis of MEF cells either wild type (p38γ+/+) or null (p38γ-/-) for p38γ after treatment with 50 J/m2 of UV and recovery in complete medium for indicated periods of time, detecting phospho-Chk1-S345 and Chk1. (C) Western blot analysis of p38γ-null MEF cells (p38γ-/-) transduced with a retroviral p38γ expression vector pBabeHygro-p38γ (pBabe-p38γ) or the pBabeHygro vector control (pBabe), after treatment with 20 J/m2 of UV and recovery in complete medium for indicated periods of time, detecting phospho-Chk1-S345, p38γ and α-tubulin. Numbers represent relative levels of the phospho-Chk1-S345 signals. (D) Western blot analysis of MEF cells either wild type (p38γ+/+) or null (p38γ-/-) for p38γ after treatment with 20 J/m2 of UVor none (−) and recovery in complete medium for 1, 2 or 4 h, detecting phospho-Chk1-S345, phospho-p53-S15, p53, phospho-NBS1-S343, NBS1 and α-tubulin. (E) Western blot analysis of wild type (p38γ+/+) and p38γ-null (p38γ-/-) MEF cells arrested in S phase by double thymidine block, after treatment with 20 J/m2 of UV or none (−) and recovery in complete medium for 0.5, 1, 2, 4 or 6 h, detecting phospho-Chk1-S345, phospho-NBS1-S343 and α-tubulin. (F) Western blot analysis of wild type (p38γ+/+) and p38γ-null (p38γ-/-) MEF cells after treatment with indicated concentrations of hydroxyurea (HU) or vehicle control (−) for 30 min, detecting phospho-Chk1-S345 and Chk1.
In addition to Chk1, p38γ also appeared to be important for the activation of the other components of the checkpoint signaling pathway. Loss of p38γ led to abrogation of phosphorylation of NBS1 at Ser343 and phosphorylation of p53 at Ser15 (Fig. 3D), both of which have been shown to be direct substrate sites of ATM/ATR and are crucial for activation of these proteins, respectively (Tibbetts et al., 1999; Gatei et al., 2000; Lim et al., 2000; Wu et al., 2000; Zhao et al., 2000).
In order to rule out the influence from possible changes in the cell cycle profile as a result of loss of p38γ, we analyzed the checkpoint activation in MEF cells arrested in early S phase by double thymidine block before the UV treatment. Again, at every time point upon release from the UV treatment, the induction of phosphorylation of Chk1 at Ser345 and NBS1 at Ser343 was greatly diminished in p38γ-deficient cells as compared to the wild type control cells (Fig. 3E). This observation indicates that the weakened checkpoint signaling is likely to be a specific consequence of the loss of p38γ function, rather than an indirect effect of altered cell cycle phase distribution in p38γ-/- cells. Furthermore, in contrast to UV that mainly induces DNA damage in the form of adducts between adjacent DNA bases, hydroxyurea, a DNA synthesis inhibitor that causes stalled DNA replication forks and accumulation of single-stranded DNA by depleting deoxyribonumceotides, triggered comparable levels of Chk1 activation in both control and p38γ-/- MEF cells (Fig. 3F), suggesting that p38γ is specifically required for cellular responses to only certain types of DNA damage.
Another direct substrate of ATM/ATR is the variant histone H2AX. Upon DNA damage, H2AX is rapidly phosphorylated at Ser139 by these kinases on chromatin regions surrounding the damaged sites (van Attikum and Gasser, 2009). Using immunofluorescence analysis, we found that the Ser139-phosphorylated form of H2AX (γH2AX) was accumulated rapidly and robustly on DNA damage foci in UV-treated wild type MEFs, but to a much less extent in the p38γ-deficient cells (Fig. 4A and 4B). Taken together, our results demonstrate that p38γ is critical for the efficient induction of the checkpoint signaling pathway in UV-treated cells.
Figure 4. p38γ deficiency leads to reduced localization of γ-H2AX on DNA damage foci.
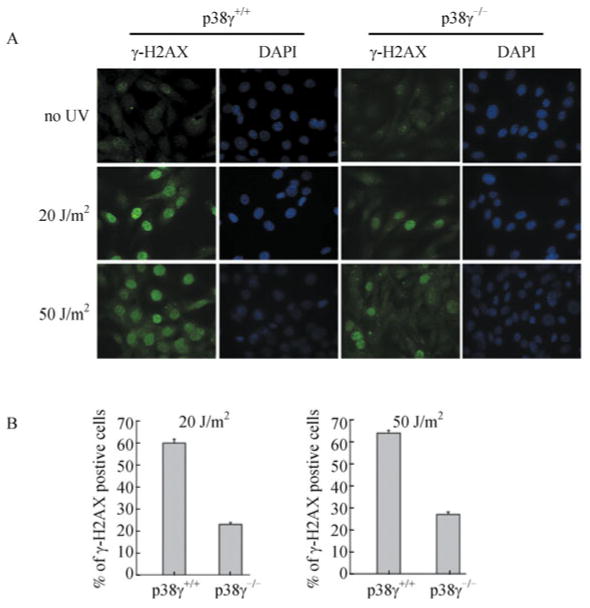
(A) Immunofluorescent staining of wild type (p38γ+/+) and p38γ-null (p38γ-/-) MEF cells, after treatment with 20 J/m2 or 50 J/m2 of UV or none (no UV) and recovery in complete medium for 30 min. The cells were stained with an anti-γ-H2AX antibody followed by a FITC-conjugated secondary antibody, and mounted with DAPI-containing medium. Cells were photographed under microscope using filters detecting FITC (γ-H2AX) and DAPI. (B) Quantification of the percentage of cells with more than 5 γ-H2AX positive foci in wild type (p38γ+/+) and p38γ-null (p38γ-/-) MEF cell lines after treatment with 20 or 50 J/m2 of UV and recovery in complete medium for 30 min. At least 200 cells were counted in at least 5 randomly chosen fields. Values are mean ± SD for triplicates.
p38γ is required for the nucleotide excision repair of UV-induced DNA damage
The major types of UV-induced DNA damage are cyclobutane-type pyrimidine dimmers (CDPs) and (6-4)-photoproducts (6-4PPs) in which adjacent DNA bases are cross-linked (Latonen and Laiho, 2005). These DNA adducts are repaired by nucleotide excision repair (NER) in mammalian cells. The prolonged S phase cell cycle arrest in p38γ-deficient cells after UV treatment (Fig. 2A) suggests that these cells may have defects in the repair of UV-induced DNA damage. To test this possibility, we compared the ability of wild type and p38γ-null MEF cells to repair a UV-damaged luciferase reporter gene, using a host cell reactivation assay in which DNA repair was measured by the restoration of luciferase expression upon transfection of the in vitro-damaged reporter plasmid into cells (Jia et al., 1999; Hu et al., 2004). We initially validated this reporter assay using a patient-derived cell line (XP12R0) deficient in XPA, an essential component of NER, and its isogenic cell line (GM15876A) that had been complemented with a wild type human XPA gene (Bomgarden et al., 2006). As expected, the repair of UV-damaged luciferase reporter was greatly improved in the XPA-complemented cell line as compared to the XPA-deficient cell line (Fig. 5A). More importantly, the efficiency in repairing the UV-induced DNA damage was significantly reduced in p38γ-/- MEFs than in the wild type control cells (Fig. 5B), indicating that the repair of UV-damage in cells is impaired by the loss of p38γ. Since the majority of the damages induced by UV in vitro are DNA adducts (Jia et al., 1999; Hu et al., 2004), this finding reveals a critical role of p38γ in NER, and has thus provided a molecular mechanism underlying the prolonged S phase arrest and increased UV sensitivity in p38γ-deficient cells.
Figure 5. p38γ is required for the efficient repair of UV-induced DNA damage in cells.
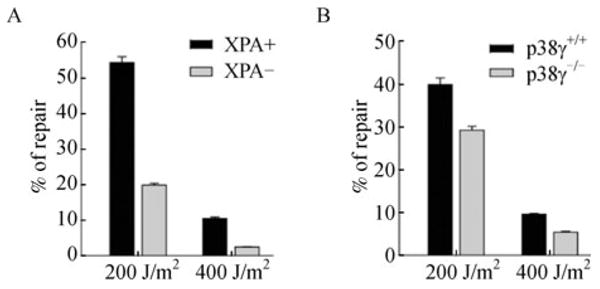
(A) Cell reactivation assay in XPA-complemented GM15876A cells (XPA+, black bars) and XPA-deficient XP12R0 cells (XPA−, grey bars). (B) Cell reactivation assay in wild type (p38γ+/+, black bars) and p38γ-null (p38γ-/-, grey bars) MEF cells. (A, B) Firefly luciferase reporter plasmid pGL3 treated with 200 or 400 J/m2 of UV or left untreated was co-transfected with un-damaged Renilla luciferase reporter plasmid pRL-TK into XPA+ or XPA− (A), and p38γ+/+ or p38γ-/- (B) cells. Cells were lysed and luciferase activity was measured 24 h after transfection. Percentage of repair was calculated by dividing the firefly luciferase activity normalized to Renilla luciferase activity obtained with UV-treated pGL3 by that with untreated pGL3. Values are mean ± SD for triplicates.
Discussion
Although the p38 MAP kinase has been implicated in cellular responses to DNA damage, the role of each individual p38 isoform and the mechanism underlying their role in DNA damage are poorly understood. In this study, we showed that p38γ, but not the other isoforms, appeared to be critical for the survival of cells upon UV-induced DNA damage. Deletion of p38γ led to reduction in the number of proliferating cells after UV treatment, as a result of both prolonged cell cycle arrest in S phase and increased apoptosis. Further investigation revealed that p38γ was essential for the optimal activation of the DNA damage checkpoint signaling and efficient repair of UV-induced DNA damage. These findings have established a novel role of p38γ in mediating UV-induced DNA damage responses that are important for cells to cope with the UV exposure.
DNA damage triggers highly coordinated responses in cells including repair of the damaged DNA and cell cycle arrest that temporarily halts DNA replication and cell division until the repair is completed. These responses are coordinated by the checkpoint signaling pathway (Abraham, 2001; Latonen and Laiho, 2005; Cimprich and Cortez, 2008). It has been reported in previous studies that p38 MAPK contributes to DNA damage responses. Upon induction by DNA damage agents such as UV, the p38α isoform stimulates the activity of p53 through direct phosphorylation of the Ser33 residue and stabilization of the p53 protein (Bulavin et al., 1999; She et al., 2000). Our data indicate that the p38γ isoform contributes to the activation of the checkpoint signaling, including the activating phosphorylation of NBS1, Chk1, p53 and γH2AX, in response to UV exposure, suggesting that p38γ acts either upstream of these checkpoint proteins or in a positive feedback loop in checkpoint signaling. The difference between our findings and previous results may reflect a functional diversity between different p38 isoforms. It is equally possible that the same p38 isoforms contribute to DNA damage response through regulation at multiple levels.
It has been reported that an active checkpoint signaling pathway is crucial for the efficient repair of DNA damages. In addition to the proteins that recognize DNA damage and thus trigger the checkpoint signaling and repair, p53 directly participates in DNA damage repair by regulating the transcription of, and by direct interaction with, the components of the repair machineries (Cline and Hanawalt, 2003; Latonen and Laiho, 2005). Therefore, the reduced efficiency in repairing UV-induced DNA damage in p38γ-deficient cells is possibly due to the requirement of p38γ in UV-induced checkpoint signaling, including the activation of p53. However, a more direct effect of p38γ on NER cannot be ruled out. In a recent study using SB203580, a chemical inhibitor of p38, it was shown that p38 augments NER by promoting chromatin relaxation and ubiquitin-mediated proteolysis of DDB2, a subunit of the UV-damaged DNA binding (UV-DDB) complex, which needs to be degraded before the recruitment of XPC to the damage site (Zhao et al., 2008). SB203580 mainly targets p38α and p38β, but it also inhibits the other p38 isoforms with lower affinity. Thus, it remains to be determined whether the p38γ is the major, or at least one of the, p38 isoform(s) contributing to these processes, and whether these regulations occur through the effect of p38 on the checkpoint signaling.
Interestingly, although an active checkpoint pathway mediates both cell cycle arrest and DNA damage repair, dampened checkpoint signaling in p38γ-deficient cells was accompanied only by reduction in repair, but not by impairment in cell cycle arrest. On the contrary, a prolonged S phase arrest was observed as compared to the wild type control cells, most likely due to the difficulty in repairing the DNA damages in the absence of p38γ. These results suggest that p38γ only contributes to certain, but not all, aspects of the checkpoint signaling. As a result, loss of p38γ may have spared the activation of some redundant signaling components that are sufficient to mediate cell cycle arrest. Alternatively, the severity of reduction in the strength of the checkpoint signaling in p38γ-deficient cells may be only enough to disrupt DNA damage repair, but not the cell cycle arrest.
Our results showed that p38γ deficiency led to reduction in checkpoint signaling induced by UV, but not that by hydroxyurea, indicating that the role of p38γ is limited to UV-induced DNA damage responses. UV induces the formation of DNA adducts that cause distortions in the DNA helix and halt RNA polymerase elongation along DNA, resulting in stalled DNA replication forks. Hydroxyurea also stalls DNA replication by inhibiting ribonucleotide reductase, thus causing depletion of deoxyribonucleotides in cells. In both cases, stalled replication forks lead to accumulation of single-stranded DNA and activation of the ATR-Chk1 checkpoint pathway. The differential effect of p38γ on UV- and hydroxyurea-induced DNA damage responses suggest that p38γ is likely involved in sensing DNA adducts formed upon UV treatment, rather than other types of DNA damages.
Materials and Methods
Cell lines and cell culture
Mouse embryonic fibroblast (MEF) cells and LinX-E retroviral packaging cells (Sun et al., 1998) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum, glutamine and antibiotics. MEF cells isolated from 14.5 dpc embryos using a published protocol (Serrano et al., 1997), and immortalized with a recombinant retrovirus encoding SV40 large T antigen. The immortalized MEF cells isolated from mice harboring floxed alleles of p38α, β, γ or δ (Kang et al., 2008; Otsuka et al., 2010) were transduced with Cre-encoding or control retroviruses to generate isogenic MEF lines either null or wild type for each p38 isoform, respectively. XPA-complemented (GM15876A) and XPA-deficient (XP12R0) cell lines were purchased from the Cornell Cell Repository and maintained in DMEM supplemented with 10% fetal calf serum, glutamine and antibiotics.
Genotyping of mice and isogenic MEF cell lines
Genotypes of mice carrying floxed alleles of p38α, β, γ or δ, and isogenic MEF lines either null or wild type for each p38 isoform were determined by genomic PCR using allele-specific primers, as described previously (Kang et al., 2008; Otsuka et al., 2010).
Retroviral gene transduction
Retroviral gene transduction was carried out as previously described, using ecotropic packaging cell line LinX-E (Sun et al., 1998). Transduced cells were selected with 1 μg/mL of puromycin, 80 μg/mL of hygromycin B, or 400 μg/mL of G418.
UV sensitivity assay
MTT assay was performed to measure the sensitivity of MEF cells to UV treatment. MEF cells were seeded into 96-well plates at a density of 1 × 104 cells/well, and treated with indicated dosages of UV. After recovery in complete medium for an indicated period of time, cells were washed, incubated with 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and then dissolved in DMSO. Absorbance at 470 nm was quantified by spectrophotometer. In each MTT assay, a parallel standard curve was established by plotting the A470 reading vs number of cells ranging from 100 to 2 × 105. The percentage of surviving cells was calculated by dividing the A470 value derived from the treated cells by the A470 value representing 1 × 104 cells on the standard curve. Each experimental point was performed in triplicates.
Cell cycle analysis
MEF cells were seeded into 6-well plates at a density of 2 × 105 cells/well. After exposure to 20 J/m2 of UV or being left untreated, cells were allowed to recover in complete medium for an indicated period time before they were harvested by trypsinization, washed with phosphate-buffered saline (PBS), and fixed in 100% ethanol for overnight. The fixed cells were then spun down, washed with PBS, resuspended in PBS containing 1% of fetal calf serum and 20 μg/mL of propidium iodide, and analyzed by flow cytometry.
Apoptotic assay
MEF cells were seeded into 6-well plates at a density of 2 × 105 cells/well. After exposure to 50 J/m2 of UV, cells were allowed to recover in complete medium for 24 h. Adherent cells were harvested by trypsinization and combined with cells collected from suspension, after which the cells were collected by centrifugation, resuspended in complete medium, washed with PBS, and then resuspended in 500 μL of 1 × Annexin-V binding buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, and 2.5 mM CaCl2). Five microliter of FITC-conjugated Annexin-V (BD Pharmingen, 51-65874X) and 2 μL of 1 mg/mL propidium iodide were added and incubated with cells at room temperature for 15 min. The percentage of FITC-positive apoptotic cells were determined by flow cytometry. Each experimental point was performed in triplicates.
Synchronization of cells by double thymidine block
To synchronize cells at the G1/S boundary, cells were treated with 2.5 mM of thymidine for 16 h, released into complete medium without thymidine for 9 h, and treated again with 2.5 mM of thymidine for 16 h. Cells were then released into medium without thymidine for 1 h to allow entry into early S phase, and treated with 20 J/m2 of UV. After recovery in complete medium for an indicated period of time, cells were lysed and subjected to Western blot analysis.
Cell reactivation assay
Cell reactivation assay was performed to determine the capability of different cell line in repairing UV-induced DNA damage, following published protocols (Jia et al., 1999; Hu et al., 2004) with minor modifications. One ml of 0.1 μg/mL of the Firefly luciferase reporter plasmid pGL3 (Promega) was irradiated with 0, 200 or 400 J/m2 of UV in a 6-cm plate. 0.8 μg of treated pGL3 was co-transfected together with 16 ng of un-damaged Renilla luciferase reporter plasmid pRL-TK (Promega), into 5 × 104 of indicated cells seeded in 24-well plates using 2 μL of Lipofectamine 2000. Cells were lysed and luciferase activity was measured 24 h after transfection using Dual Luciferase Assay System (Promega). After normalization of the Firefly luciferase activity to the Renilla luciferase activity, % of repaired DNA was calculated by dividing the normalized activity obtained with pGL3 treated with 200 or 400 J/m2 of UV by that with pGL3 treated with 0 J/m2 of UV. Each experimental point was performed in triplicates.
Western blot analysis
MEF cells were seeded into 6-well plates at a density of 2 × 105 cells/well, treated with indicated dosages of UV, recovered in complete medium for indicated period of time, and then lysed in 80 μL of 1 × Laemmli sample buffer. After sonication, 20 μL of lysates were resolve on 4%–20% SDS-PAGE gel, transferred to nitrocellulose membrane, and subjected to Western blot using appropriate antibodies. Antibodies against phospho-Chk1-S345, Chk1, phospho-p53-S15, phospho-NBS1-S343, and NBS1 were purchased from Cell Signaling; antibody against p38γ was purchased from R&D Systems; antibody against p53 (CM5) was purchased from Novocastra; and antibody against α-tubulin (DM1A) was purchased from Sigma. After incubation with proper secondary antibodies, signals were detected using enhanced chemiluminescence.
Immuno-fluorescence staining of γ-H2AX
MEF cells were seeded on cover slips placed in 12-well plates at a density of 1 × 105 cells/well, treated with 20 or 50 J/m2 of UV, recovered in complete medium for 30 min, washed with PBS, and fixed with 4% of paraformaldehyde in PBS for 45 min at RT. The cells were then washed 3 times with PBS for 5 min each, permeabilized in PBS containing 0.1% Triton X-100 and 1% BSA for 30 min at RT. The cover slips were blocked with 3% BSA in PBS for 1 h at RT, incubated with an anti-γ-H2AX antibody (Cell Signaling) at a 1:200 dilution in 3% BSA/PBS for 2 h at RT, washed 3 times with 0.1% Tween-20 in Tris-buffered saline (TBST), and incubated with a FITC-conjugated anti-rabbit IgG antibody (Vector) at a 1:500 dilution in 3% BSA/PBS for 30 min at RT. After washing 3 times with TBST, the cover slips were mounted in VECTASHIELD mounting medium with DAPI (Vector). The cells were photographed and the percentage of cells with more than 5 γ-H2AX-positive foci was determined under a fluorescence microscope. Each experimental point was performed in triplicates. At least 200 cells were counted in at least 5 randomly chosen fields.
Supplementary Material
Acknowledgments
This work is supported by grants from National Institute of Health, USA (CA106768, CA131231 and RR025744 to P.S., AI041637 and AI068896 to J.H., and CA102361 and CA140972 to XW), and an international collaborative grant from National Science Foundation in China (30828019 to P.S. and J.H.). The Scripps manuscript number is 20721.
Abbreviations
- 6-4PP
(6-4)-photoproducts
- ATM
ataxia telangiectasia mutated
- ATR
ataxia telangiectasia and Rad3 related
- BSA
bovine serum albumin
- Cdk1
cyclin-dependent protein kinase 1
- CDP
cyclobutane-type pyrimidine dimmer
- DDB2
damage-specific DNA binding protein 2
- DMEM
Dulbecco's modified Eagle's medium
- DMSO
dimethyl sulfoxide
- dpc
days post coitum
- FITC
fluorescein isothiocyanate
- MAPK
mitogen-activated protein kinase
- MAP3K
mitogen-activated protein kinase kinase kinase
- MKK3 (4, 6)
mitogen-activated protein kinase kinase 3 (4, 6)
- MAPKAPK2 (MK2)
mitogen-activated protein kinase-activated protein kinase 2
- MEF
mouse embryonic fibroblast
- MRN
Mre11-Rad50-Nbs1
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NBS1
Nijmegen breakage syndrome 1
- NER
nucleotide excision repair
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- RT
room temperature
- SV40
Simian virus 40
- TBST
Tris-buffered saline containing Tween-20
- UV-DDB
UV-damaged DNA binding
- XPA
xeroderma pigmentosum, complementation group A
- XPC
xeroderma pigmentosum, complementation group C
Contributor Information
Jiahuai Han, Email: jhan@scripps.edu.
Peiqing Sun, Email: pqsun@scripps.edu.
References
- Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- Beardmore VA, Hinton HJ, Eftychi C, Apostolaki M, Armaka M, Darragh J, McIlrath J, Carr JM, Armit LJ, Clacher C, et al. Generation and characterization of p38beta (MAPK11) gene-targeted mice. Mol Cell Biol. 2005;25:10454–10464. doi: 10.1128/MCB.25.23.10454-10464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomgarden RD, Lupardus PJ, Soni DV, Yee MC, Ford JM, Cimprich KA. Opposing effects of the UV lesion repair protein XPA and UV bypass polymerase eta on ATR checkpoint signaling. EMBO J. 2006;25:2605–2614. doi: 10.1038/sj.emboj.7601123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulavin DV, Higashimoto Y, Popoff IJ, Gaarde WA, Basrur V, Potapova O, Appella E, Fornace AJ., Jr Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature. 2001;411:102–107. doi: 10.1038/35075107. [DOI] [PubMed] [Google Scholar]
- Bulavin DV, Saito S, Hollander MC, Sakaguchi K, Anderson CW, Appella E, Fornace AJ., Jr Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J. 1999;18:6845–6854. doi: 10.1093/emboj/18.23.6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline SD, Hanawalt PC. Who's on first in the cellular response to DNA damage? Nat Rev Mol Cell Biol. 2003;4:361–372. doi: 10.1038/nrm1101. [DOI] [PubMed] [Google Scholar]
- Dent P, Yacoub A, Fisher PB, Hagan MP, Grant S. MAPK pathways in radiation responses. Oncogene. 2003;22:5885–5896. doi: 10.1038/sj.onc.1206701. [DOI] [PubMed] [Google Scholar]
- Enslen H, Brancho DM, Davis RJ. Molecular determinants that mediate selective activation of p38 MAP kinase isoforms. EMBO J. 2000;19:1301–1311. doi: 10.1093/emboj/19.6.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatei M, Young D, Cerosaletti KM, Desai-Mehta A, Spring K, Kozlov S, Lavin MF, Gatti RA, Concannon P, Khanna K. ATM-dependent phosphorylation of nibrin in response to radiation exposure. Nat Genet. 2000;25:115–119. doi: 10.1038/75508. [DOI] [PubMed] [Google Scholar]
- Han J, Lee JD, Bibbs L, Ulevitch RJ. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. 1994;265:808–811. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- Han J, Sun P. The pathways to tumor suppression via route p38. Trends Biochem Sci. 2007;32:364–371. doi: 10.1016/j.tibs.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Hirose Y, Katayama M, Stokoe D, Haas-Kogan DA, Berger MS, Pieper RO. The p38 mitogen-activated protein kinase pathway links the DNA mismatch repair system to the G2 checkpoint and to resistance to chemotherapeutic DNA-methylating agents. Mol Cell Biol. 2003;23:8306–8315. doi: 10.1128/MCB.23.22.8306-8315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Feng Z, Tang MS. Nickel (II) enhances benzo [a]pyrene diol epoxide-induced mutagenesis through inhibition of nucleotide excision repair in human cells: a possible mechanism for nickel (II)-induced carcinogenesis. Carcinogenesis. 2004;25:455–462. doi: 10.1093/carcin/bgh012. [DOI] [PubMed] [Google Scholar]
- Jia L, Wang XW, Harris CC. Hepatitis B virus X protein inhibits nucleotide excision repair. Int J Cancer. 1999;80:875–879. doi: 10.1002/(sici)1097-0215(19990315)80:6<875::aid-ijc13>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Chen C, Li Z, Guo W, Gegner JA, Lin S, Han J. Characterization of the structure and function of a new mitogen-activated protein kinase (p38beta) J Biol Chem. 1996;271:17920–17926. doi: 10.1074/jbc.271.30.17920. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Gram H, Zhao M, New L, Gu J, Feng L, Di Padova F, Ulevitch RJ, Han J. Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinases, p38delta. J Biol Chem. 1997;272:30122–30128. doi: 10.1074/jbc.272.48.30122. [DOI] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- Kang YJ, Chen J, Otsuka M, Mols J, Ren S, Wang Y, Han J. Macrophage deletion of p38alpha partially impairs lipopolysaccharide-induced cellular activation. J Immunol. 2008;180:5075–5082. doi: 10.4049/jimmunol.180.7.5075. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- Latonen L, Laiho M. Cellular UV damage responses—functions of tumor suppressor p53. Biochim Biophys Acta. 2005;1755:71–89. doi: 10.1016/j.bbcan.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Li Z, Jiang Y, Ulevitch RJ, Han J. The primary structure of p38 gamma: a new member of p38 group of MAP kinases. Biochem Biophys Res Commun. 1996;228:334–340. doi: 10.1006/bbrc.1996.1662. [DOI] [PubMed] [Google Scholar]
- Lim DS, Kim ST, Xu B, Maser RS, Lin J, Petrini JH, Kastan MB. ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature. 2000;404:613–617. doi: 10.1038/35007091. [DOI] [PubMed] [Google Scholar]
- Manke IA, Nguyen A, Lim D, Stewart MQ, Elia AE, Yaffe MB. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol Cell. 2005;17:37–48. doi: 10.1016/j.molcel.2004.11.021. [DOI] [PubMed] [Google Scholar]
- Nebreda AR, Porras A. p38 MAP kinases: beyond the stress response. Trends Biochem Sci. 2000;25:257–260. doi: 10.1016/s0968-0004(00)01595-4. [DOI] [PubMed] [Google Scholar]
- Olson E, Nievera CJ, Lee AY, Chen L, Wu X. The Mre11-Rad50-Nbs1 complex acts both upstream and downstream of ataxia telangiectasia mutated and Rad3-related protein (ATR) to regulate the S-phase checkpoint following UV treatment. J Biol Chem. 2007;282:22939–22952. doi: 10.1074/jbc.M702162200. [DOI] [PubMed] [Google Scholar]
- Ono K, Han J. The p38 signal transduction pathway: activation and function. Cell Signal. 2000;12:1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- Otsuka M, Kang YJ, Ren J, Jiang H, Wang Y, Omata M, Han J. Distinct effects of p38alpha deletion in myeloid lineage and gut epithelia in mouse models of inflammatory bowel disease. Gastroenterology. 2010;138:1255–1265. doi: 10.1053/j.gastro.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman M, Earnest S, Zhang K, Zhao Y, Cobb MH. TAO kinases mediate activation of p38 in response to DNA damage. EMBO J. 2007;26:2005–2014. doi: 10.1038/sj.emboj.7601668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabio G, Arthur JS, Kuma Y, Peggie M, Carr J, Murray-Tait V, Centeno F, Goedert M, Morrice NA, Cuenda A. p38gamma regulates the localisation of SAP97 in the cytoskeleton by modulating its interaction with GKAP. EMBO J. 2005;24:1134–1145. doi: 10.1038/sj.emboj.7600578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- She QB, Chen N, Dong Z. ERKs and p38 kinase phosphorylate p53 protein at serine 15 in response to UV radiation. J Biol Chem. 2000;275:20444–20449. doi: 10.1074/jbc.M001020200. [DOI] [PubMed] [Google Scholar]
- Shi Y, Gaestel M. In the cellular garden of forking paths: how p38 MAPKs signal for downstream assistance. Biol Chem. 2002;383:1519–1536. doi: 10.1515/BC.2002.173. [DOI] [PubMed] [Google Scholar]
- Sun P, Dong P, Dai K, Hannon GJ, Beach D. p53-independent role of MDM2 in TGF-beta1 resistance. Science. 1998;282:2270–2272. doi: 10.1126/science.282.5397.2270. [DOI] [PubMed] [Google Scholar]
- Tanoue T, Yamamoto T, Maeda R, Nishida E. A novel MAPK phosphatase MKP-7 acts preferentially on JNK/SAPK and p38 alpha and beta MAPKs. J Biol Chem. 2001;276:26629–26639. doi: 10.1074/jbc.M101981200. [DOI] [PubMed] [Google Scholar]
- Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, Taya Y, Prives C, Abraham RT. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999;13:152–157. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Attikum H, Gasser SM. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 2009;19:207–217. doi: 10.1016/j.tcb.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Wang X, McGowan CH, Zhao M, He L, Downey JS, Fearns C, Wang Y, Huang S, Han J. Involvement of the MKK6-p38gamma cascade in gamma-radiation-induced cell cycle arrest. Mol Cell Biol. 2000;20:4543–4552. doi: 10.1128/mcb.20.13.4543-4552.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Ranganathan V, Weisman DS, Heine WF, Ciccone DN, O'Neill TB, Crick KE, Pierce KA, Lane WS, Rathbun G, et al. ATM phosphorylation of Nijmegen breakage syndrome protein is required in a DNA damage response. Nature. 2000;405:477–482. doi: 10.1038/35013089. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Barakat BM, Qin S, Ray A, El-Mahdy MA, Wani G, Arafa S, Mir SN, Wang QE, Wani AA. The p38 mitogen-activated protein kinase augments nucleotide excision repair by mediating DDB2 degradation and chromatin relaxation. J Biol Chem. 2008;283:32553–32561. doi: 10.1074/jbc.M803963200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Weng YC, Yuan SS, Lin YT, Hsu HC, Lin SC, Gerbino E, Song MH, Zdzienicka MZ, Gatti RA, et al. Functional link between ataxia-telangiectasia and Nijmegen breakage syndrome gene products. Nature. 2000;405:473–477. doi: 10.1038/35013083. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.