

Abstract
A major challenge in cancer treatment is the development of therapies that target cancer cells with little or no toxicity to normal tissues and cells. Alterations in DNA double strand break (DSB) repair in cancer cells include both elevated and reduced levels of key repair proteins and changes in the relative contributions of the various DSB repair pathways. These differences can result in increased sensitivity to DSB-inducing agents and increased genomic instability. The development of agents that selectively inhibit the DSB repair pathways that cancer cells are more dependent upon will facilitate the design of therapeutic strategies that exploit the differences in DSB repair between normal and cancer cells. Here, we discuss the pathways of DSB repair, alterations in DSB repair in cancer, inhibitors of DSB repair and future directions for cancer therapies that target DSB repair.
Keywords: Homologous recombination, Non-homologous end-joining
Introduction
Cells have evolved a complex network of pathways that function in response to DNA damage. Key components of this response include DNA repair pathways that remove various types of DNA lesions and DNA damage-activated signal transduction pathways that target fundamental cellular processes, including transcription and cell cycle progression. Here, we will focus on the cellular response induced by DNA double strand breaks (DSB)s, considered to be the most lethal form of DNA damage [1]. The cancer predispostion of autosomal recessive human syndromes, such as ataxia telangiectasia, that are characterized by hypersensitivity to DSB-inducing agents indicates the role of the DNA damage response in protecting against the genomic instability induced by DSBs that drives cancer formation and progression [2]. More recently, abnormalities in the DSB response, including defects in DSB repair, have been identified as the underlying cause of hereditary forms of breast cancer [3]. Since genomic instability is a common characteristic of both inherited and sporadic forms of cancer cells, it appears likely that abnormalities in the DNA damage response also contribute to the development and progression of sporadic cancers [1]. However, high-throughput sequencing studies have found that mutation of DNA repair genes occurs infrequently in sporadic cancers [4]. Instead, it has been suggested that genomic instability in sporadic cancers may be mainly due to oncogene-induced DNA replication stress and DNA damage [4]. In addition, oncogenes may also impact the repair and mutagenic consequences of DNA damage by altering the relative activities of DNA repair pathways that repair the same lesion, presumably by epigenetic mechanisms.
DSB repair pathways
The cytotoxicity of DSBs presumably reflects the difficulty of repairing these lesions because, unlike almost all other types of DNA damage that have an intact undamaged template strand to guide the repair, the integrity of both strands of the duplex is lost. Thus, cells that incur more than one DSB have the problem of distinguishing between the previously linked DNA ends and DNA ends from other molecules. The repair of DSBs occurs via two mechanistically distinct groups of pathways, homology-directed pathways and non-homologous end-joining (NHEJ) pathways.
Homology-directed repair
The predominant pathway that repairs replication-associated DSBs is characterized by the invasion of single strand DNA into a homologous duplex [1, 5]. This repair pathway, which is active in late S phase and in the G2 phase of the cell cycle, utilizes the undamaged sister chromatid as the template for repair and so is usually error-free (Fig. 1). Notably, many chemotherapeutic agents block DNA replication, leading to the stalling and/or collapse of replication forks and the generation of lesions that are repaired by homologous recombination (HR) [6]. If the HR pathway is inactivated, there are back-up pathways that can repair DSBs. These pathways, which are described in more detail below, are error-prone, generating deletions and chromosomal translocations (Fig. 1).
Fig. 1. DSB repair in the S and G2 phases of the cell cycle.
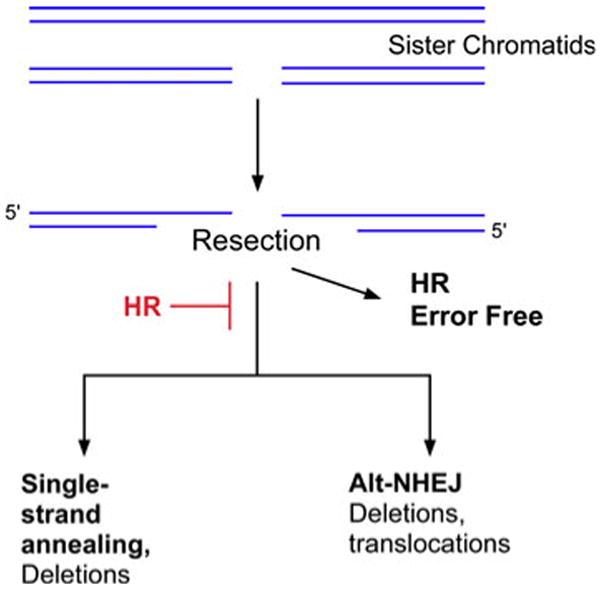
In late S and the G2 phase of the cell cycle, DSBs can be repaired by homologous recombination (HR) using the undamaged sister chromatid. In the initial stage of HR, the ends of the DSBs are resected to generate 3′ single strand regions. If the ends are resected but HR is inactivated, the DSBs can be joined by back-up pathways, single strand annealing and alternative non-homologous end joining. In contrast to the error-free homolgous recombination pathways, the back-up pathways generate genomic rearrangements.
The first step in HR is resection of the DSB in a 5′–3′ manner that involves the human Mre11-Rad50-Nbs1 (MRN) complex and CtIP [7, 8]. The resultant 3′ single strands are bound by RPA, preventing degradation and providing the signal to activate the cell cycle checkpoint kinase ATR [9]. Next, hRad51 is recruited and assembled into a nucleoprotein filament, displacing RPA. This reaction involves several accessory proteins including hRad52, XRCC2, XRCC3 and BRCA2 [10–12]. The recruitment and assembly of hRad51 nucleoprotein filaments can be visualized in cell nuclei as foci formed after IR and in S phase at sites of replication-associated DSBs [13]. Strand invasion by the hRad51 nucleoprotein filament into the adjacent homologous sister chromatid results in formation of a D loop structure. DNA synthesis from the invading 3′ end extends the D loop, increasing its stability [14, 15]. The extension of the D loop also permits capture of the second DNA end, resulting in the formation of a Holliday junction. Resolution of the Holliday junction by a resolvase, such as Gen1 [16], completes the repair, generating two identical sister chromatids [17]. Alternatively, in break-induced replication, the entire sister chromatid is copied following formation of the D loop structure [18].
Homology-directed repair (HR) does occur between homologous chromosomes in G1 cells albeit at a much lower frequency [19] (Fig. 2). These events may generate genetic alterations ranging from gene conversion to loss of heterozygosity (Fig. 2). The presence of repetitive sequences throughout the human genome presents a problem for homology-dependent DSB repair mechanisms. For example, strand invasion can occur with an inappropriate homologous sequence on the sister chromatid, homologous chromosome or even on a non-homologous chromosome, resulting in genomic rearrangements. Finally, a single strand annealing pathway that occurs between repeated sequences on the same chromosome generates intra-chromosomal deletions [20] (Figs. 1, 2).
Fig. 2. DSB repair in the G1 phases of the cell cycle and in non-dividing cells.
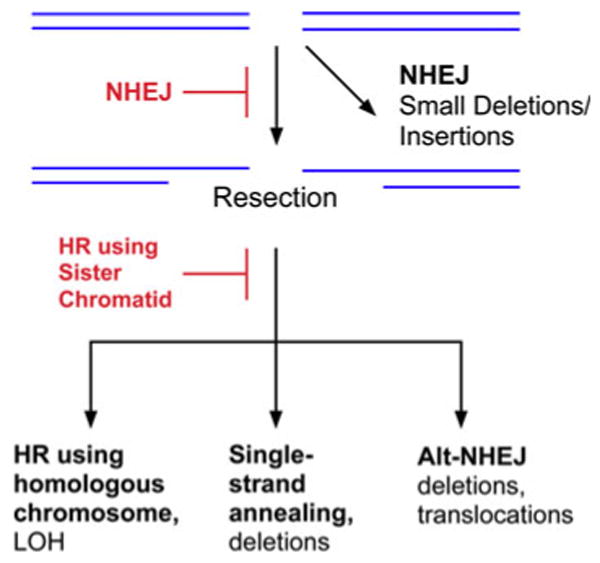
In the G1 phase of the cell cycle and non-dividing cells, the majority of DSBs are repaired by DNA-PK-dependent NHEJ. If this pathway is inactivated, the DSBs can be repaired by homologous recombination (HR) but, in the absence of the sister chromatid, the homologous chromsome will be used to guide the repair. Alternatively, the DSBs can be joined by back-up pathways, single strand annealing and alternative non-homologous end joining.
Non-homologous end-joining
In the repair of DSBs by NHEJ, the DNA ends are brought together in a reaction that is independent of extensive DNA sequence homology and so is prone to introducing errors ranging from small insertions and deletions at the break site to the joining of previously unlinked DNA ends [1, 5, 21]. In addition to repairing DSBs caused by endogenous and exogenous DNA damaging agents, the NHEJ proteins also participate in immunoglobulin gene rearrangements [22]. While the repair of DSBs by NHEJ occurs throughout the cell cycle, NHEJ is the major DSB repair pathway in G0, G1 (Fig. 1) and early S phase [22, 23]. Most DSBs are rapidly repaired by NHEJ, but there is a slower phase that reflects the repair of a subset of DSBs that are either more complex DSB lesions or occur in condensed chromatin [24, 25].
As shown in Fig. 3, the NHEJ pathway is initiated by the Ku70/Ku86 heterodimer, a ring-shaped complex that binds to and encircles DNA ends [26]. This serves to protect the DNA ends from degradation and to recruit the catalytic subunit (cs) of the DNA-dependent protein kinase (DNA-PK) [27–29] to form the activated DNA-PK [30, 31]. The kinase activity of DNA-PK is critical for NHEJ with a key substrate being the DNA-PKcs itself [32]. In addition, DNA-PK phosphorylates Artemis, which binds to DNA-PKcs [33], and activates its endonuclease activity. The key step in NHEJ is the physical juxtaposition of DNA ends. This end-bridging occurs via interactions between DNA-bound DNA-PKcs molecules [34, 35].
Fig. 3.
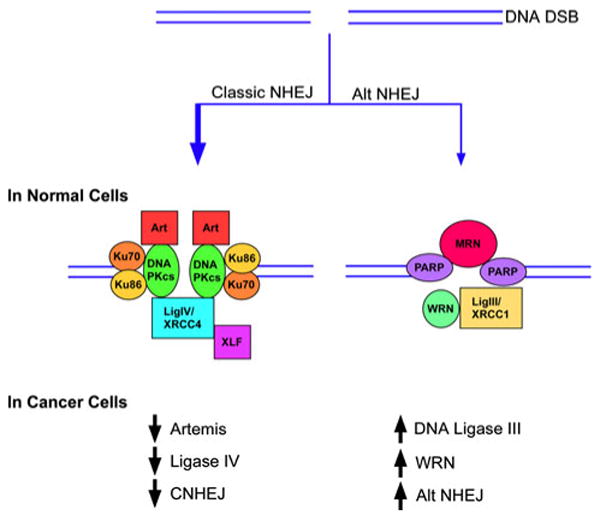
Upper panel. In normal cells, alternative NHEJ pathway (Alt NHEJ) is a minor DSB repair pathway compared with DNA-PK-dependent NHEJ (Classic NHEJ). Lower panel. In cancer cells, the steady state levels of key DNA-PK-dependent NHEJ proteins are reduced whereas the steady state levels of key alternative NHEJ are increased. This results in increased activity of the Alt NHEJ pathway and reduced activity of the DNA-PK-dependent NHEJ (CNHEJ) pathway.
Approximately 10% of endogenous DSBs in mammalian cells have non-ligatable ends [36], whereas a significantly higher fraction of DSBs generated by ionizing radiation are not directly ligatable. When juxtaposed ends can be directly ligated, the repair reaction is completed by DNA ligase IV/XRCC4, which is recruited to DSBs by interactions with DNA-PK [37]. In contrast, there appear to be multiple factors involved in processing non-ligatable ends to generate a ligatable substrate. These include polynucleotide kinase [38], the nucleases FEN-1 [39] and Artemis [36], and the Pol X family members, Pol mu and lambda [40]. As a consequence of these processing reactions, the joining of DSBs by DNA-PK-dependent NHEJ often results in the loss or addition of a few nucleotides at the break site and the presence of short complementary sequences, microhomologies, at the break site that presumably contributed to the alignment of the DNA ends [41, 42]. Another NHEJ factor, XLF or Cernunnos, contributes to the joining of non-complementary DNA ends by interacting with DNA ligase IV/XRCC4 and stimulating the joining of mismatched DNA ends [43].
Although DNA-PK-dependent NHEJ frequently causes small alterations in DNA sequence around the break site, it usually does not join previously unlinked DNA ends [44]. There is, however, increasing evidence for an alternative (Alt) version of NHEJ that results in larger deletions and chromosomal translocations [44, 45] (Figs. 1 and 2). For example, chromosomal abnormalities, including c-myc/IgH translocations are observed in the absence of either Ku or DNA ligase IV/XRCC4 [46–48] and rare aberrant V(D)J coding joins are found in lymphocytes lacking either Ku or DNA-PKcs [49]. Cell-based assays measuring rearrangement of immunoglobulin genes have detected robust end joining by Alt NHEJ that is even detectable in cells with a functional DNA PK-dependent NHEJ pathway [50–52]. The hallmark features of the Alt NHEJ pathway are larger deletions and insertions, longer tracts of microhomology and a much higher frequency of chromosomal translocations compared with DNA PK-dependent NHEJ [44]. A number of DNA repair proteins, including PARP-1, MRN, WRN and DNA ligase IIIα/XRCC1 [53–60], have been implicated in alternative NHEJ, but the mechanisms and regulation of this repair pathway or pathways are poorly defined (Fig. 3).
Since the fidelity of DSB repair by the different pathways described above varies greatly (Figs. 1 and 2), the choice of DSB repair pathway will determine the effect that this type of DNA damage has on genome stability. There is evidence for competition between the major HR and NHEJ pathways, in particular at the DSB binding stage. In addition, regulation of the end resection machinery by cyclin-dependent protein kinases plays a role in limiting HR to the S and G2 phases of the cell cycle [61]. Thus, there are many different potential alterations in DSB repair that could increase genome instability, thereby driving cancer progression. At the present time, our ability to define the DSB repair properties and capacities of tumor samples in biopsies is very limited, making it difficult to design therapeutic strategies that exploit the abnormal DNA repair in tumor cells.
DNA damage-activated cell cycle checkpoints
The potential mutagenic and cytotoxic consequences of unrepaired DNA damage are mitigated by cell cycle checkpoints that prevent the replication and/or segregation of damaged genomes. The Ataxia Telangiactasia Mutated (ATM) protein that is defective in the cancer-prone radiation-sensitive human disease, ataxia telangiectasia, is a central player in the activation of cell cycle checkpoints by DSBs. ATM is recruited to DSBs by the MRN complex [62]. This results in ATM autophosphorylation and its conversion from an inactive dimer to an active monomer [63]. Once activated, ATM phosphorylates MRN and downstream effector proteins to initiate cell cycle checkpoints at the G1/S, intra-S, and G2/M boundaries [2]. The activation of these checkpoints allows increased time for the repair of DNA damage before it is replicated and/or passed onto daughter cells thereby increasing cell survival and preserving genomic integrity. In addition to its role in DSB-activated signal transduction pathways, there is emerging evidence that ATM is involved in the repair of a subset of DSBs in both G0 and G2 cells [24, 64].
Alterations in DSB repair in cancer
Homology-directed repair
Inherited mutations in BRCA1 and BRCA2 predispose these individuals to breast, ovarian and other cancers [65]. Following the classic paradigm of tumor suppressor genes [66], the inheritance of one defective copy of BRCA1 or BRCA2 in the germline is enough to cause cancer predisposition, because it increases the probability of losing the remaining wild-type allele, an event that is consistently observed in tumor cells from these individuals. Cancer susceptibility genes fall into two general classes, “gatekeeper” genes whose altered expression relieves normal controls on cell division, death, or lifespan, promoting the out-growth of cancer cells, and “caretaker genes” whose disruption causes genome instability [67]. Several lines of evidence suggest that BRCA1 and BRCA2 act as caretakers and that loss of these genes lead to spontaneous chromosomal abnormalities. Mouse BRCA2-deficient cells sustain spontaneous chromosomal aberrations that accumulate during cell proliferation [68]. Microscopically, the abnormalities are not restricted to broken chromosomes and chromatids but also include triradial and quadriradial structures, markers of defective mitotic recombination that are hallmarks of the inherited cancer-prone human diseases, Bloom's syndrome (BS) and Fanconi's anemia (FA) [69, 70].
Analysis of the genes involved in FA, which is characterized by cellular hypersensitivity to DNA cross-linking agents, revealed that FANCD2 and BRCA2 are in fact the same gene. Furthermore, while homozygous mutation of FANCD1, FANCN, or FANCJ results in FA, heterozygous mutations in these same genes have been linked to familial breast and ovarian cancer predisposition, highlighting the role of both the BRCA and FA genes as tumor suppressors in the same tissues [71]. In response to DNA damage or replication fork stalling during S phase, the FA core complex is activated and monoubiquitylates FANCD2 and FANCI, leading to their retention in chromatin foci, which colocalize with downstream components of the repair pathway, including FANCD1 (BRCA2), FANCN (PALB2), and FANCJ (BRIP) [72]. BLM is a member of the RecQ helicase family that is involved in both regulating homologous recombinational repair and replication fork regression [73]. Recently, it has been shown that, after treatment of cells with agents that introduce DNA interstrand cross-links, a complex containing BLM associates with the FA core complex to form a 1.5- to 2-MDa supercomplex named BRAFT [74], suggesting that the genomic instability observed in FA, BS and inherited breast cancers may be due to a failure in BRAFT assembly that in turn results in a defect in homologous recombination at stalled replication forks.
Although evidence is emerging that the gross chromosomal alterations observed in BRCA-deficient cells result from inappropriate DSB repair, the exact mechanisms that generate these abnormalities are still not understood. Recent work from several groups shows that, while BRCA1- or BRCA2-deficient rodent cells or human tumors are specifically deficient in HR, NHEJ (and sometimes SSA) remains intact [75]. This suggests that spontaneous or induced DSBs in BRCA-deficient cells are rerouted for repair by error-prone mechanisms, because the preferred mode of (error-free) processing by HR is unavailable (Fig. 1). In accord with this hypothesis, it has been shown that error-prone DSB repair mechanisms predominate in murine BRCA2-deficient cells [76–78], and possibly in BRCA1-deficient cells [79].
The major role of BRCA2 in DSB repair is through control of the hRad51 recombinase, while BRCA1 performs a distinct and more general function as a link between the sensing/signaling and effector components involved in the response to DNA damage, helping to ensure that the response is appropriate for the initiating lesion [80]. Overexpression of hRad51 in a chicken DT40 BRCA1 null mutant rescues defects in proliferation, DNA damage survival, and HR [81]. Furthermore, retrospective analyses of microarray expression data in BRCA1-deficient breast tumors revealed elevated expression of hRad51 and two of its late-acting cofactors, RAD54 and RAD51AP1. Together, these results suggest that upregulation of hRad51 in cells lacking BRCA1 function circumvents the normal requirement for BRCA1 in subnuclear assembly of hRad51 foci [81], Interestingly, while mutations in BRCA genes rarely occur in sporadic breast cancer, hRad51 is frequently upregulated, resulting in increased HR in these cells [82]. It is possible that this dysregulated HR may also lead to inappropriate repair of DSBs.
It is now widely accepted that a variety of tumor cell lines display elevated steady state levels of hRad51 and increased numbers of hRad51 nuclear foci compared with nonmalignant control cell lines [83]. The elevated steady state levels of hRad51 are not caused by gene amplification or changes in protein stability but instead are the result of transcriptional up-regulation [83]. In contrast, decreased levels of Rad51 were observed in multiple cancer cell types under hypoxic conditions but were not associated with the cell cycle distribution or expression of hypoxia-inducible factor [84]. With the accumulating evidence that abnormalities in HR occur frequently in cancer and that these abnormalities are potential therapeutic targets, there is growing interest in the identification of biomarkers that are diagnostic of HR abnormalities. Since alterations in expression of key recombination proteins have been observed, the use of focused microarrays to determine the expression of HR proteins may lead to the identification of diagnostic gene expression patterns for different HR abnormalities. A problem with this approach is that it does not directly measure HR. Recently, Powell and colleagues have developed an ex vivo assay based on formation of BRCA1, hRad51 and FANCD2 foci to detect FA/BRCA pathway defects in breast cancer biopsies [85].
Non-homologous end-joining
The genetic instability caused by deletion of any one of the key components of the main DNA-PK dependent non-homologous end-joining (NHEJ) pathway is characterized by chromosomal translocations [44, 86]. In addition, NHEJ deficiencies animals result in increased rates of neoplastic transformation. For example, DNA-PKcs and Ku70 mutant mice have a high incidence of T-cell lymphomas and Ku70−/− mice have increased rates of fibroblast transformation [87, 88]. In contrast, while fibroblasts from Ku80-null mice show chromosomal instability associated with chromosome aberrations, including breakage, translocations and aneuploidy, the animals have only a slightly earlier onset of cancer compared with wild-type animals. However, p53 inactivation synergizes with Ku80 to promote tumorigenesis such that all the double mutant mice succumb to pro-B-cell lymphoma at an early age [47]. These tumors display a specific set of chromosomal translocations and gene amplifications involving the immunoglobulin heavy chain IgH/Myc locus, reminiscent of Burkitt lymphoma [89]. Similar translocations are also seen in pro-B cell tumors that result from XRCC4- or DNA ligase IV-deficiency in a Trp53-null animals [90]. Since almost all the malignancies observed in NHEJ-deficient mice occur in lymphoid-derived cells, it is possible that they arise as a consequence of the role of NHEJ in immunoglobulin gene rearrangements. However, studies by the Alt laboratory have shown that the cancer predisposition resulting from NHEJ-deficiency occurs in other tissues and cell types [91].
In humans, there is emerging circumstantial evidence that defects in DNA-PK-dependent NHEJ result in cancer predisposition. One of the five individuals identified with mutant LIG4 alleles [92, 93] had leukemia. A more recent study provided evidence that LIG4 polymorphisms might influence the risk of acute lymphoblastic leukemia in children [94]. Finally, there is also evidence showing that the steady state levels of key factors in DNA-PK-dependent NHEJ are frequently reduced in cancer cell lines [55, 91] (Fig. 3). Specifically, it has been shown that DNA ligase IV is reduced in colon, cervical and breast cancer cell lines [95] and both DNA ligase IV and Artemis are reduced in chronic myeloid leukemia (CML) cell lines [55]. Notably, primary CML cells and cell lines established from CML patients expressing BCR-ABL1 have elevated levels of ROS and increased endogenous DSBs [96, 97]. Furthermore, repair of DSBs by NHEJ in CML cells is characterized by large deletions around the break-point junction and joining of DNA ends at regions of DNA sequence microhomology [98]. This abnormal and error-prone DSB repair is not only due to the reduced activity of the DNA-PK-dependent NHEJ pathway [55] but also the increased activity of the Alt NHEJ pathway, resulting from elevated steady state levels of WRN and DNA ligase IIIα [55] (Fig. 3). Together these studies suggest that Alt NHEJ is upregulated in a variety of cancers and is likely to contribute to the deletions and translocations that drive cancer progression. Importantly, knockdown of DNA ligase IIIα reduces DSB repair by NHEJ in CML but not normal myeloid cells, indicating that the upregulated Alt NHEJ pathway in cancer cells is a potential therapeutic target [55].
Use of DNA repair inhibitors in cancers with DSB repair defects
Cancer therapy has until recently been focused on creating cytotoxicity through DNA damaging agents, such as ionizing radiation, cis platinum and temozolomide [99]. Although differences in the DNA damage response between normal and cancer cells presumably underlie the ability of these agents to preferentially kill cancer cells, their use is often limited by normal tissue toxicity. Since abnormalities in the DNA damage response of cancer cells are becoming more clearly defined, there is growing interest in the development of small molecules that will selectively target the abnormal DNA repair in cancer cells with the hope that these compounds either alone or in combination with DNA damaging agents will effectively kill cancer cells, while minimizing damage to normal cells.
DSB-activated cell cycle checkpoints
Since ATM is the predominant kinase responsible for the activation of multiple cell cycle checkpoints following DSB induction and ATM-deficient cells are exquisitely sensitive to ionizing radiation [2], inhibitors of ATM should potentiate the cytotoxicity of ionizing radiation and chemotherapeutic drugs that cause DSBs. Initial studies with caffeine and LY294002 that inhibit a broad range of protein kinases, including ATM, provided support for this idea [100, 101] and prompted the identification of a more specific ATM inhibitor, KU-55933, from a library of LY294002 derivatives [101]. As expected, KU-55933 efficiently sensitizes tumor cells to ionizing radiation and DSB-inducing chemotherapeutic agents, such as camptothecin and etoposide [101]. A potential problem with using ATM inhibitors as cancer therapeutics is that they may also sensitize normal tissues to DNA damage. In this scenario, the inhibitor of the DNA damage response will not preferentially enhance killing of the cancer cell and so there will be no therapeutic gain. Since cancer cells are presumed to have abnormalities in the DNA damage response, a subset of cancers with a particular DNA repair abnormality may be uniquely sensitive to ATM inhibition. In support of this idea, ATM was identified in a high-throughput siRNA screen for gene products whose knockdown is lethal to cells with a defective FA pathway [102]. Since the FA pathway is known to be disrupted in several types of sporadic cancers [102], inhibition of ATM may be an effective therapeutic strategy in this subset of cancers.
HR
As mentioned above, the key strand exchange protein in HR, hRad51, is overexpressed in a variety of tumors, and elevated hRad51 expression is correlated with a poor prognosis [103]. Although the DSB repair function of hRad51 protects normal cells from acquiring genetic changes that drive tumor development, overexpressed hRad51 in tumors contributes to their resistance to chemotherapy agents such as cisplatin [103–105], indicating that hRad51 is a potential target for antitumor drugs. Hine and colleagues exploited the overexpression of Rad51 in cancer cells to design a hRad51 promoter-based anticancer therapy. They cloned 2,931 bp of upstream regulatory sequences, the first noncoding exon of the Rad51, and the sequence encoding the first 12 amino acids of the hRad51 open reading frame (ORF) into a luciferase plasmid construct. The resultant plasmid was transfected into normal and cancer cells and luciferase activity was analyzed by flow cytometry. They found that the difference in promoter activity between normal and cancer cells increases to an average of 840-fold with a maximum difference of 12,500-fold. Based on this dramatic difference in promoter activity between normal and cancer cells, they designed a therapeutic strategy in which the hRad51 promoter was fused to a sequence encoding diphtheria toxin A (DTA), and the resultant plasmid was transfected into a variety of cancer cell types, including fibrosarcoma, breast and cervical cancer cells, and normal breast epithelial cells and fibroblasts. Notably, the cancer but not normal cells were killed by the plasmid, presumably as a consequence of increased expression of DTA. While the strategy described above has the problem of specifically introducing a nucleic acid into the cancer cells, these results suggest that therapies based on the hRad51 promoter could be highly tumor specific and may open new avenues for targeting a broad range of cancers [103].
A small molecule inhibitor of the MRN complex, Mirin, has been identified by high throughput screening of a small molecule library using a X. laevis extract assay. As expected, Mirin inhibits MRN-dependent activation of ATM and homology-dependent repair of DSBs [106]. Since HR factors such as MRN and hRad51 are required for cell viability, there is a concern that small molecule inhibitors of these essential proteins will be cytotoxic for normal as well as cancer cells and so will not have utility as anti-cancer agents. Factors such as hRad52, XRCC2 and XRCC3 that contribute to hRad51 strand exchange, but are not essential, may be alternative therapeutic targets [5]. It has been suggested that, while there appears to some functional redundancy among these factors, abnormalities in HR may make cancer cells more dependent upon one or more of these factors [107]. While these accessory factors do not have enzymatic activities, their activity appears to be mediated by protein–protein interactions, so it may be possible to design high throughput screens for small molecules that block protein–protein interactions. Although this is a relatively unexplored approach compared with targeting enzyme active sites, it has been used successfully to identify a small molecule that prevents an inhibitory protein binding to wild-type p53, thereby restoring the tumor suppressor function of p53 [108].
PARP inhibitors
The abundant nuclear protein Poly(ADP-ribose) polymerase (PARP-1) binds avidly to DNA single strand breaks, an event that activates PARP-1 polymerase activity [109]. Activated PARP-1 utilizes NAD to synthesize poly (ADP-ribose) polymers that are attached to PARP-1 itself and to other nuclear proteins. Poly (ADP-ribosylated) PARP-1 serves as a recruitment factor for DNA ligase IIIα/XRCC1 and other factors involved in the repair of DNA single strand breaks [110]. Although there are other PARP family members, PARP-1 is the predominant enzyme that synthesizes poly (ADP-ribose) in response to DNA damage [111]. The replication of DNA containing single strand breaks cause DSBs, and so preventing the repair of DNA single strand breaks by inhibiting PARP-1 results in an increase in DSBs. Since these replication-associated DSBs would normally be repaired by HR, cells that are defective in HR are hypersensitive to PARP inhibitors. Based on this rationale, potent and specific inhibitors of PARP were developed as therapeutic agents for inherited forms of breast and ovarian cancer as the PARP inhibitors should be cytotoxic for brca mutant tumors but not normal tissues with a functional BRCA allele [112, 113].
As expected, PARP inhibitors increased the cytotoxicity of a range of anti-cancer agents including temozolomide and ionizing radiation that cause DNA single strand breaks [114, 115], and both brca1- and brca2-mutant cell lines were hypersensitive to PARP inhibitors in cell culture and mouse xenograft assays [116]. These results formed the basis for a phase I clinical trial, which demonstrated that the PARP inhibitor AZD2281 (Olaparib) exhibited antitumor activity in patients with ovarian and breast tumors resulting from either BRCA1 or BRCA2 mutations [117]. This phase I study was conducted in patients with advanced solid tumors (n = 60). The patient population was enriched for BRCA1 or BRCA2 mutation carriers (n = 23), including three breast cancer patients with inherited BRCA2 mutations, in order to assess an objective antitumor effect of the PARP inhibitor in BRCA-deficient tumors. Within this group, treatment with Olaparib resulted in a confirmed partial response rate of 39% (9/23) with a sustained response in one patient for more than 76 weeks. As expected, no responses were observed in the tumors of non-BRCA mutation carriers. Olaparib treatment was evaluated further in two separate phase II studies for BRCA-associated breast and ovarian cancer [118, 119]. These studies recruited BRCA1 or BRCA2 mutation carriers with advanced breast cancer who had progressed following at least one previous cycle of chemotherapy. Twenty-seven patients were treated in each cohort and the activity of Olaparib as a single agent was confirmed. In the 400-mg group, an objective response rate of 41% (11/27) was seen with a progression-free survival of 5.7 months. Response was lower in the 100-mg group suggesting that the extent of PARP inhibition may be important. These proof of concept studies were the first to report single agent activity for a PARP inhibitor in BRCA-related breast cancer. Although a potentially exciting breakthrough in breast cancer treatment, the results of these small nonrandomized trials will require confirmation in larger phase III trials. A phase II trial investigating the single agent activity of the PARP inhibitor AG014699 (Pfizer GRD, La Jolla, California, USA) and a phase I trial investigating the PARP inhibitor ABT-888 (Abbott, North Chicago, Illinois, USA) in BRCA-associated breast and ovarian cancers are ongoing. Since these trials employ different modes of administration, scheduling, specificity and potency of PARP inhibition, it will be interesting to see if the responses differ from those observed with Olaparib. The promising results obtained with PARP inhibitors as a single agent in the treatment of hereditary breast cancer have prompted the design of trials to determine whether combining PARP inhibitors with other cancer therapeutics improves the outcome in other forms of cancer.
NHEJ
In addition to DNA damage-activated cell cycle checkpoints and HR, there is evidence that the NHEJ pathway may be a valid target for the development of more effective cancer treatments. Cells deficient in Ku70/80 or the catalytic subunit of DNA-PK (DNA-PKcs) are sensitive to DSBs induced by IR or chemotherapeutic agents [120]. In addition, DNA-PK is upregulated in some cancers, suggesting that it may be an important factor for tumor growth and survival. Indeed, upregulation of DNA-PK activity has been shown to impair apoptosis in B-cell chronic lymphocytic leukemia [121, 122]. In initial studies, the broad-spectrum phosphoinositide-3-kinase-related protein kinase (PIKK) inhibitors, wortmannin and LY294002 [123], that inhibit DNA-PK and other PIKKs such as ATM and ATR, were found to sensitize tumor cells to chemotherapeutic agents, prompting the development of more specific PIKK inhibitors. Treatment with a flavone-based DNA-PK inhibitor IC87361 led to tumor regression [124]. Similarly, a highly potent and selective DNA-PK inhibitor NU7441 with an IC50 of 13 nM caused sensitization of tumor cells to radiation and chemotherapeutic agents both in culture and in mouse xenograft models [125].
DNA ligase inhibitors
DNA joining events are required for the completion of almost all DNA repair pathways and for DNA replication [126]. In human cells, there are three genes that encode DNA ligases with different but partially overlapping cellular functions [126]. Thus, inhibitors of DNA ligases are predicted to be cytotoxic and to sensitize cells to a variety of DNA damaging agents depending upon the specificity of the inhibitor for the three human DNA ligases. Using computer-aided drug design based on the structure of human DNA ligase I complexed with nicked DNA, a series of small molecule inhibitors of human DNA ligases, which exhibit different specificities for the three human DNA ligases in vitro, have been identified [95, 127]. In cell culture assays, a subset of these compounds were cytotoxic, killing both normal and cancer cells. Interestingly, the cytotoxic ligase inhibitors inhibit both DNA ligases I and III whereas a DNA ligase I-specific inhibitor was cytostatic [95]. It is possible that DNA ligase III substitutes for DNA ligase I, the replicative DNA ligase, and so cytotoxicity is observed only when both enzymes are inhibited. Since expression of phosphorylation site mutants of human DNA ligase I induces cell senescence [128], it is possible that the cytostatic effect of the DNA ligase I-specific inhibitor is due to induction of cellular senescence. Alternatively, the cytotoxic effects of the DNA ligase inhibitors may be a consequence of inhibition of DNA repair and/or DNA ligase III-dependent mitochondrial DNA metabolism [129].
Notably, subtoxic concentrations of the ligase inhibitors preferentially sensitized cancer cells to DNA alkylating agents and ionizing radiation [95]. The molecular mechanisms that underlie the ability of the DNA ligase inhibitors to enhance the killing of cancer cells by DNA damaging agents are not known but presumably reflect differences in the DNA damage response between cancer and normal cells. In theory, DNA ligase inhibitors constitute an extremely versatile group of agents that, depending on their specificity for the three human DNA ligases, can be used to target a variety of DNA repair pathways that would be chosen based on the DNA damaging agent. For example, a DNA ligase IV-specific inhibitor is predicted to sensitize cells with a functional DNA-PK-dependent NHEJ pathway to ionizing radiation. The situation with inhibitors of DNA ligases I and III is more complex because these enzymes participate in multiple nuclear DNA repair pathways, and DNA ligase III also functions in mitochondrial DNA metabolism [126].
Future directions in cancer therapeutics
A wide variety of DNA damaging agents are used as cancer therapeutics. It is generally assumed that differences in the DNA damage response between normal and cancer cells underlies the utility of DNA damaging agents in cancer treatment. However, abnormalities in the DNA damage response in cancer cells are poorly defined. An understanding of the alterations in the DNA damage response of cancer cells would permit the design of novel therapeutic strategies involving the use of inhibitors of the DNA damage response alone or in combination with DNA damaging agents that selectively target the altered DNA damage response in cancer cells. The development of PARP inhibitors that selectively target the defect in DSB repair in breast tumors in women with herditary breast cancer is the first example of this therapeutic strategy [116]. This has prompted interest in defining and then targeting DNA repair abnormalities in sporadic cancers. Although there are a growing number of publications that describe DNA repair abnormalities in cancer cell lines, there is a need for the development of assays and identification of biomarkers that can be used to identify DNA repair abnormalities in clinical samples. In addition, the indentification and characterization of a wider repertoire of small molecules that target DSB repair proteins will not only increase our ability to probe DSB repair abnormalities in cancer cells but also to develop different combinatorial therapeutic strategies that selectively target the DSB repair abnormality in cancer cells.
Acknowledgments
Work in the Tomkinson laboratory is supported by grants from the National Institutes for Health (R01 GM47251, R01 GM57479, R01 ES12512 and P01 CA92584). Work in the Rassool Laboratory is supported by grants from the Leukemia Lymphoma Society (LLS 6085-07), NIH/NCI 5R01CA125635-02, State of Maryland 08072925 and V Foundation.
References
- 1.Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27:247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 2.O'Driscoll M, Jeggo PA. The role of double-strand break repair—insights from human genetics. Nat Rev Genet. 2006;7:45–54. doi: 10.1038/nrg1746. [DOI] [PubMed] [Google Scholar]
- 3.Futreal PA, Liu Q, Shattuck-Eidens D, Cochran C, Harshman K, Tavtigian S, Bennett LM, Haugen-Strano A, Swensen J, Miki Y, et al. BRCA1 mutations in primary breast and ovarian carcinomas. Science. 1994;266:120–122. doi: 10.1126/science.7939630. [DOI] [PubMed] [Google Scholar]
- 4.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability—an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- 5.Hartlerode AJ, Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J. 2009;423:157–168. doi: 10.1042/BJ20090942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keller KL, Overbeck-Carrick TL, Beck DJ. Survival and induction of SOS in Escherichia coli treated with cisplatin, UV-irradiation, or mitomycin C are dependent on the function of the RecBC and RecFOR pathways of homologous recombination. Mutat Res. 2001;486:21–29. doi: 10.1016/s0921-8777(01)00077-5. [DOI] [PubMed] [Google Scholar]
- 7.Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huertas P. DNA resection in eukaryotes: deciding how to fix the break. Nat Struct Mol Biol. 17:11–16. doi: 10.1038/nsmb.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu Y, Sugiyama T, Kowalczykowski SC. DNA annealing mediated by Rad52 and Rad59 proteins. J Biol Chem. 2006;281:15441–15449. doi: 10.1074/jbc.M601827200. [DOI] [PubMed] [Google Scholar]
- 10.Petalcorin MI, Sandall J, Wigley DB, Boulton SJ. CeBRCA-2 stimulates D-loop formation by RAD-51 and promotes DNA single-strand annealing. J Mol Biol. 2006;361:231–242. doi: 10.1016/j.jmb.2006.06.020. [DOI] [PubMed] [Google Scholar]
- 11.Liu N, Lamerdin JE, Tebbs RS, Schild D, Tucker JD, Shen MR, Brookman KW, Siciliano MJ, Walter CA, Fan W, Narayana LS, Zhou ZQ, Adamson AW, Sorensen KJ, Chen DJ, Jones NJ, Thompson LH. XRCC2 and XRCC3, new human Rad51-family members, promote chromosome stability and protect against DNA cross-links and other damages. Mol Cell. 1998;1:783–793. doi: 10.1016/s1097-2765(00)80078-7. [DOI] [PubMed] [Google Scholar]
- 12.Sonoda E, Zhao GY, Kohzaki M, Dhar PK, Kikuchi K, Redon C, Pilch DR, Bonner WM, Nakano A, Watanabe M, Nakayama T, Takeda S, Takami Y. Collaborative roles of gammaH2AX and the Rad51 paralog Xrcc3 in homologous recombinational repair. DNA Repair (Amst) 2007;6:280–292. doi: 10.1016/j.dnarep.2006.10.025. [DOI] [PubMed] [Google Scholar]
- 13.Tarsounas M, Davies D, West SC. BRCA2-dependent and independent formation of RAD51 nuclear foci. Oncogene. 2003;22:1115–1123. doi: 10.1038/sj.onc.1206263. [DOI] [PubMed] [Google Scholar]
- 14.McIlwraith MJ, Vaisman A, Liu Y, Fanning E, Woodgate R, West SC. Human DNA polymerase eta promotes DNA synthesis from strand invasion intermediates of homologous recombination. Mol Cell. 2005;20:783–792. doi: 10.1016/j.molcel.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 15.Burgers PM. Polymerase dynamics at the eukaryotic DNA replication fork. J Biol Chem. 2009;284:4041–4045. doi: 10.1074/jbc.R800062200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ip SC, Rass U, Blanco MG, Flynn HR, Skehel JM, West SC. Identification of Holliday junction resolvases from humans and yeast. Nature. 2008;456:357–361. doi: 10.1038/nature07470. [DOI] [PubMed] [Google Scholar]
- 17.Mimitou EP, Symington LS. Nucleases and helicases take center stage in homologous recombination. Trends Biochem Sci. 2009;34:264–272. doi: 10.1016/j.tibs.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 18.Kaye JA, Melo JA, Cheung SK, Vaze MB, Haber JE, Toczyski DP. DNA breaks promote genomic instability by impeding proper chromosome segregation. Curr Biol. 2004;14:2096–2106. doi: 10.1016/j.cub.2004.10.051. [DOI] [PubMed] [Google Scholar]
- 19.Stark JM, Jasin M. Extensive loss of heterozygosity is suppressed during homologous repair of chromosomal breaks. Mol Cell Biol. 2003;23:733–743. doi: 10.1128/MCB.23.2.733-743.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ivanov EL, Sugawara N, Fishman-Lobell J, Haber JE. Genetic requirements for the single-strand annealing pathway of double-strand break repair in Saccharomyces cerevisiae. Genetics. 1996;142:693–704. doi: 10.1093/genetics/142.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lieber MR. The mechanism of human nonhomologous DNA end joining. J Biol Chem. 2008;283:1–5. doi: 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- 22.Lieber MR, Yu K, Raghavan SC. Roles of nonhomologous DNA end joining, V(D)J recombination, and class switch recombination in chromosomal translocations. DNA Repair (Amst) 2006;5:1234–1245. doi: 10.1016/j.dnarep.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 23.Lieber MR, Ma Y, Pannicke U, Schwarz K. Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol. 2003;4:712–720. doi: 10.1038/nrm1202. [DOI] [PubMed] [Google Scholar]
- 24.Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, Reis C, Dahm K, Fricke A, Krempler A, Parker AR, Jackson SP, Gennery A, Jeggo PA, Lobrich M. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol Cell. 2004;16:715–724. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 25.Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Lobrich M, Jeggo PA. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell. 2008;31:167–177. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 26.Walker JR, Corpina RA, Goldberg J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature. 2001;412:607–614. doi: 10.1038/35088000. [DOI] [PubMed] [Google Scholar]
- 27.Falzon M, Fewell JW, Kuff EL. EBP-80, a transcription factor closely resembling the human autoantigen Ku, recognizes single- to double-strand transitions in DNA. J Biol Chem. 1993;268:10546–10552. [PubMed] [Google Scholar]
- 28.Gottlieb TM, Jackson SP. The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell. 1993;72:131–142. doi: 10.1016/0092-8674(93)90057-w. [DOI] [PubMed] [Google Scholar]
- 29.Mimori T, Hardin JA. Mechanism of interaction between Ku protein and DNA. J Biol Chem. 1986;261:10375–10379. [PubMed] [Google Scholar]
- 30.Calsou P, Frit P, Humbert O, Muller C, Chen DJ, Salles B. The DNA-dependent protein kinase catalytic activity regulates DNA end processing by means of Ku entry into DNA. J Biol Chem. 1999;274:7848–7856. doi: 10.1074/jbc.274.12.7848. [DOI] [PubMed] [Google Scholar]
- 31.Singleton BK, Torres-Arzayus MI, Rottinghaus ST, Taccioli GE, Jeggo PA. The C terminus of Ku80 activates the DNA-dependent protein kinase catalytic subunit. Mol Cell Biol. 1999;19:3267–3277. doi: 10.1128/mcb.19.5.3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lees-Miller SP, Chen YR, Anderson CW. Human cells contain a DNA-activated protein kinase that phosphorylates simian virus 40 T antigen, mouse p53, and the human Ku autoantigen. Mol Cell Biol. 1990;10:6472–6481. doi: 10.1128/mcb.10.12.6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 2002;108:781–794. doi: 10.1016/s0092-8674(02)00671-2. [DOI] [PubMed] [Google Scholar]
- 34.Yaneva M, Kowalewski T, Lieber MR. Interaction of DNA-dependent protein kinase with DNA and with Ku: biochemical and atomic-force microscopy studies. EMBO J. 1997;16:5098–5112. doi: 10.1093/emboj/16.16.5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DeFazio LG, Stansel RM, Griffith JD, Chu G. Synapsis of DNA ends by DNA-dependent protein kinase. EMBO J. 2002;21:3192–3200. doi: 10.1093/emboj/cdf299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen L, Trujillo K, Sung P, Tomkinson AE. Interactions of the DNA ligase IV-XRCC4 complex with DNA ends and the DNA-dependent protein kinase. J Biol Chem. 2000;275:26196–26205. doi: 10.1074/jbc.M000491200. [DOI] [PubMed] [Google Scholar]
- 37.Lobrich M, Jeggo PA. The two edges of the ATM sword: co-operation between repair and checkpoint functions. Radiother Oncol. 2005;76:112–118. doi: 10.1016/j.radonc.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 38.Chappell C, Hanakahi LA, Karimi-Busheri F, Weinfeld M, West SC. Involvement of human polynucleotide kinase in double-strand break repair by non-homologous end joining. EMBO J. 2002;21:2827–2832. doi: 10.1093/emboj/21.11.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu X, Wilson TE, Lieber MR. A role for FEN-1 in nonhomologous DNA end joining: the order of strand annealing and nucleolytic processing events. Proc Natl Acad Sci USA. 1999;96:1303–1308. doi: 10.1073/pnas.96.4.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma Y, Lu H, Tippin B, Goodman MF, Shimazaki N, Koiwai O, Hsieh CL, Schwarz K, Lieber MR. A biochemically defined system for mammalian nonhomologous DNA end joining. Mol Cell. 2004;16:701–713. doi: 10.1016/j.molcel.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 41.Roth DB, Porter TN, Wilson JH. Mechanisms of non-homologous recombination in mammalian cells. Mol Cell Biol. 1985;5:2599–2607. doi: 10.1128/mcb.5.10.2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roth DB, Wilson JH. Nonhomologous recombination in mammalian cells: role for short sequence homologies in the joining reaction. Mol Cell Biol. 1986;6:4295–4304. doi: 10.1128/mcb.6.12.4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. 2006;124:301–313. doi: 10.1016/j.cell.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 44.Nussenzweig A, Nussenzweig MC. A backup DNA repair pathway moves to the forefront. Cell. 2007;131:223–225. doi: 10.1016/j.cell.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 45.Iliakis G. Backup pathways of NHEJ in cells of higher eukaryotes: cell cycle dependence. Radiother Oncol. 2009;92:310–315. doi: 10.1016/j.radonc.2009.06.024. [DOI] [PubMed] [Google Scholar]
- 46.Jankovic M, Nussenzweig A, Nussenzweig MC. Antigen receptor diversification and chromosome translocations. Nat Immunol. 2007;8:801–808. doi: 10.1038/ni1498. [DOI] [PubMed] [Google Scholar]
- 47.Difilippantonio MJ, Zhu J, Chen HT, Meffre E, Nussenzweig MC, Max EE, Ried T, Nussenzweig A. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature. 2000;404:510–514. doi: 10.1038/35006670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu C, Mills KD, Ferguson DO, Lee C, Manis J, Fleming J, Gao Y, Morton CC, Alt FW. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell. 2002;109:811–821. doi: 10.1016/s0092-8674(02)00770-5. [DOI] [PubMed] [Google Scholar]
- 49.Bogue MA, Wang C, Zhu C, Roth DB. V(D)J recombination in Ku86-deficient mice: distinct effects on coding, signal, and hybrid joint formation. Immunity. 1997;7:37–47. doi: 10.1016/s1074-7613(00)80508-7. [DOI] [PubMed] [Google Scholar]
- 50.Corneo B, Wendland RL, Deriano L, Cui X, Klein IA, Wong SY, Arnal S, Holub AJ, Weller GR, Pancake BA, Shah S, Brandt VL, Meek K, Roth DB. Rag mutations reveal robust alternative end joining. Nature. 2007;449:483–486. doi: 10.1038/nature06168. [DOI] [PubMed] [Google Scholar]
- 51.Soulas-Sprauel P, Rivera-Munoz P, Malivert L, Le Guyader G, Abramowski V, Revy P, de Villartay JP. V(D)J and immunoglobulin class switch recombinations: a paradigm to study the regulation of DNA end-joining. Oncogene. 2007;26:7780–7791. doi: 10.1038/sj.onc.1210875. [DOI] [PubMed] [Google Scholar]
- 52.Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, Gumaste S, Geyer M, Zarrin AA, Manis JP, Rajewsky K, Alt FW. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449:478–482. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- 53.Audebert M, Salles B, Calsou P. Effect of double-strand break DNA sequence on the PARP-1 NHEJ pathway. Biochem Biophys Res Commun. 2008;369:982–988. doi: 10.1016/j.bbrc.2007.11.132. [DOI] [PubMed] [Google Scholar]
- 54.Wang M, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34:6170–6182. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sallmyr A, Tomkinson AE, Rassool FV. Up-regulation of WRN and DNA ligase IIIalpha in chronic myeloid leukemia: consequences for the repair of DNA double-strand breaks. Blood. 2008;112:1413–1423. doi: 10.1182/blood-2007-07-104257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deriano L, Stracker TH, Baker A, Petrini JH, Roth DB. Roles for NBS1 in alternative nonhomologous end-joining of V(D)J recombination intermediates. Mol Cell. 2009;34:13–25. doi: 10.1016/j.molcel.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang H, Rosidi B, Perrault R, Wang M, Zhang L, Windhofer F, Iliakis G. DNA ligase III as a candidate component of backup pathways of nonhomologous end joining. Cancer Res. 2005;65:4020–4030. doi: 10.1158/0008-5472.CAN-04-3055. [DOI] [PubMed] [Google Scholar]
- 58.Dinkelmann M, Spehalski E, Stoneham T, Buis J, Wu Y, Sekiguchi JM, Ferguson DO. Multiple functions of MRN in end-joining pathways during isotype class switching. Nat Struct Mol Biol. 2009;16:808–813. doi: 10.1038/nsmb.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xie A, Kwok A, Scully R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat Struct Mol Biol. 2009;16:814–818. doi: 10.1038/nsmb.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rass E, Grabarz A, Plo I, Gautier J, Bertrand P, Lopez BS. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat Struct Mol Biol. 2009;16:819–824. doi: 10.1038/nsmb.1641. [DOI] [PubMed] [Google Scholar]
- 61.Huertas P, Cortes-Ledesma F, Sartori AA, Aguilera A, Jackson SP. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature. 2008;455:689–692. doi: 10.1038/nature07215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–611. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- 63.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 64.Beucher A, Birraux J, Tchouandong L, Barton O, Shibata A, Conrad S, Goodarzi AA, Krempler A, Jeggo PA, Lobrich M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009;28:3413–3427. doi: 10.1038/emboj.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marshall M, Solomon S. Hereditary breast-ovarian cancer: clinical findings and medical management. Plast Surg Nurs. 2007;27:124–127. doi: 10.1097/01.PSN.0000290280.48197.e7. [DOI] [PubMed] [Google Scholar]
- 66.Knudson A. Alfred Knudson and his two-hit hypothesis (Interview by Ezzie Hutchinson) Lancet Oncol. 2001;2:642–645. doi: 10.1016/s1470-2045(01)00524-1. [DOI] [PubMed] [Google Scholar]
- 67.Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–627. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- 68.Patael-Karasik Y, Daniely M, Gotlieb WH, Ben-Baruch G, Schiby J, Barakai G, Goldman B, Aviram A, Friedman E. Comparative genomic hybridization in inherited and sporadic ovarian tumors in Israel. Cancer Genet Cytogenet. 2000;121:26–32. doi: 10.1016/s0165-4608(00)00224-7. [DOI] [PubMed] [Google Scholar]
- 69.Hickson ID, Davies SL, Li JL, Levitt NC, Mohaghegh P, North PS, Wu L. Role of the Bloom's syndrome helicase in maintenance of genome stability. Biochem Soc Trans. 2001;29:201–204. doi: 10.1042/0300-5127:0290201. [DOI] [PubMed] [Google Scholar]
- 70.D'Andrea AD, Grompe M. The Fanconi anaemia/BRCA pathway. Nat Rev Cancer. 2003;3:23–34. doi: 10.1038/nrc970. [DOI] [PubMed] [Google Scholar]
- 71.Niedernhofer LJ, Lalai AS, Hoeijmakers JH. Fanconi anemia (cross)linked to DNA repair. Cell. 2005;123:1191–1198. doi: 10.1016/j.cell.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 72.Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- 73.Wu L, Hickson ID. DNA helicases required for homologous recombination and repair of damaged replication forks. Annu Rev Genet. 2006;40:279–306. doi: 10.1146/annurev.genet.40.110405.090636. [DOI] [PubMed] [Google Scholar]
- 74.Meetei AR, Sechi S, Wallisch M, Yang D, Young MK, Joenje H, Hoatlin ME, Wang W. A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol Cell Biol. 2003;23:3417–3426. doi: 10.1128/MCB.23.10.3417-3426.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O'Connor MJ, Tutt AN, Zdzienicka MZ, Smith GC, Ashworth A. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly (ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–8115. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 76.Tutt A, Bertwistle D, Valentine J, Gabriel A, Swift S, Ross G, Griffin C, Thacker J, Ashworth A. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 2001;20:4704–4716. doi: 10.1093/emboj/20.17.4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xia F, Taghian DG, DeFrank JS, Zeng ZC, Willers H, Iliakis G, Powell SN. Deficiency of human BRCA2 leads to impaired homologous recombination but maintains normal nonhomologous end joining. Proc Natl Acad Sci USA. 2001;98:8644–8649. doi: 10.1073/pnas.151253498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7:263–272. doi: 10.1016/s1097-2765(01)00174-5. [DOI] [PubMed] [Google Scholar]
- 79.Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- 80.Nagaraju G, Scully R. Minding the gap: the underground functions of BRCA1 and BRCA2 at stalled replication forks. DNA Repair (Amst) 2007;6:1018–1031. doi: 10.1016/j.dnarep.2007.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martin RW, Orelli BJ, Yamazoe M, Minn AJ, Takeda S, Bishop DK. RAD51 up-regulation bypasses BRCA1 function and is a common feature of BRCA1-deficient breast tumors. Cancer Res. 2007;67:9658–9665. doi: 10.1158/0008-5472.CAN-07-0290. [DOI] [PubMed] [Google Scholar]
- 82.Mao Z, Jiang Y, Liu X, Seluanov A, Gorbunova V. DNA repair by homologous recombination, but not by nonhomologous end joining, is elevated in breast cancer cells. Neoplasia. 2009;11:683–691. doi: 10.1593/neo.09312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Raderschall E, Stout K, Freier S, Suckow V, Schweiger S, Haaf T. Elevated levels of Rad51 recombination protein in tumor cells. Cancer Res. 2002;62:219–225. [PubMed] [Google Scholar]
- 84.Bindra RS, Schaffer PJ, Meng A, Woo J, Maseide K, Roth ME, Lizardi P, Hedley DW, Bristow RG, Glazer PM. Down-regulation of Rad51 and decreased homologous recombination in hypoxic cancer cells. Mol Cell Biol. 2004;24:8504–8518. doi: 10.1128/MCB.24.19.8504-8518.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Willers H, Taghian AG, Luo CM, Treszezamsky A, Sgroi DC, Powell SN. Utility of DNA repair protein foci for the detection of putative BRCA1 pathway defects in breast cancer biopsies. Mol Cancer Res. 2009;7:1304–1309. doi: 10.1158/1541-7786.MCR-09-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ferguson DO, Sekiguchi JM, Chang S, Frank KM, Gao Y, DePinho RA, Alt FW. The nonhomologous end-joining pathway of DNA repair is required for genomic stability and the suppression of translocations. Proc Natl Acad Sci USA. 2000;97:6630–6633. doi: 10.1073/pnas.110152897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kurimasa A, Ouyang H, Dong LJ, Wang S, Li X, Cordon-Cardo C, Chen DJ, Li GC. Catalytic subunit of DNA-dependent protein kinase: impact on lymphocyte development and tumorigenesis. Proc Natl Acad Sci USA. 1999;96:1403–1408. doi: 10.1073/pnas.96.4.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li GC, Ouyang H, Li X, Nagasawa H, Little JB, Chen DJ, Ling CC, Fuks Z, Cordon-Cardo C. Ku70: a candidate tumor suppressor gene for murine T cell lymphoma. Mol Cell. 1998;2:1–8. doi: 10.1016/s1097-2765(00)80108-2. [DOI] [PubMed] [Google Scholar]
- 89.Lovisa F, Mussolin L, Corral L, Pillon M, Cazzaniga G, Biondi A, Rosolen A. IGH and IGK gene rearrangements as PCR targets for pediatric Burkitt's lymphoma and mature B-ALL MRD analysis. Lab Invest. 2009;89:1182–1186. doi: 10.1038/labinvest.2009.81. [DOI] [PubMed] [Google Scholar]
- 90.Gao Y, Ferguson DO, Xie W, Manis JP, Sekiguchi J, Frank KM, Chaudhuri J, Horner J, DePinho RA, Alt FW. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature. 2000;404:897–900. doi: 10.1038/35009138. [DOI] [PubMed] [Google Scholar]
- 91.Sharpless NE, Ferguson DO, O'Hagan RC, Castrillon DH, Lee C, Farazi PA, Alson S, Fleming J, Morton CC, Frank K, Chin L, Alt FW, DePinho RA. Impaired nonhomologous end-joining provokes soft tissue sarcomas harboring chromosomal translocations, amplifications, and deletions. Mol Cell. 2001;8:1187–1196. doi: 10.1016/s1097-2765(01)00425-7. [DOI] [PubMed] [Google Scholar]
- 92.Riballo E, Critchlow SE, Teo SH, Doherty AJ, Priestley A, Broughton B, Kysela B, Beamish H, Plowman N, Arlett CF, Lehmann AR, Jackson SP, Jeggo PA. Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Curr Biol. 1999;9:699–702. doi: 10.1016/s0960-9822(99)80311-x. [DOI] [PubMed] [Google Scholar]
- 93.O'Driscoll M, Cerosaletti KM, Girard PM, Dai Y, Stumm M, Kysela B, Hirsch B, Gennery A, Palmer SE, Seidel J, Gatti RA, Varon R, Oettinger MA, Neitzel H, Jeggo PA, Concannon P. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol Cell. 2001;8:1175–1185. doi: 10.1016/s1097-2765(01)00408-7. [DOI] [PubMed] [Google Scholar]
- 94.Andreae J, Varon R, Sperling K, Seeger K. Polymorphisms in the DNA ligase IV gene might influence the risk of acute lymphoblastic leukemia in children. Leukemia. 2007;21:2226–2227. doi: 10.1038/sj.leu.2404783. [DOI] [PubMed] [Google Scholar]
- 95.Chen X, Zhong S, Zhu X, Dziegielewska B, Ellenberger T, Wilson GM, MacKerell AD, Jr, Tomkinson AE. Rational design of human DNA ligase inhibitors that target cellular DNA replication and repair. Cancer Res. 2008;68:3169–3177. doi: 10.1158/0008-5472.CAN-07-6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brady N, Gaymes TJ, Cheung M, Mufti GJ, Rassool FV. Increased error-prone NHEJ activity in myeloid leukemias is associated with DNA damage at sites that recruit key nonhomologous end-joining proteins. Cancer Res. 2003;63:1798–1805. [PubMed] [Google Scholar]
- 97.Nowicki M, F R, Koptyra M, Slupianek A, Stoklosa T, Gloc E, Nieborowska-Skorska M, Blasiak J, Skorski T. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double strand breaks. Blood. 2004;104:3746–3753. doi: 10.1182/blood-2004-05-1941. [DOI] [PubMed] [Google Scholar]
- 98.Gaymes TJ, Mufti GJ, Rassool FV. Myeloid leukemias have increased activity of the nonhomologous end-joining pathway and concomitant DNA misrepair that is dependent on the Ku70/86 heterodimer. Cancer Res. 2002;62:2791–2797. [PubMed] [Google Scholar]
- 99.Friedberg EC, Walker EH, Siede W. DNA repair and mutagenesis. ASM press; Washington, DC: 1995. [Google Scholar]
- 100.Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, Abraham RT. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999;59:4375–4382. [PubMed] [Google Scholar]
- 101.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9159. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 102.Kennedy RD, Chen CC, Stuckert P, Archila EM, De la Vega MA, Moreau LA, Shimamura A, D'Andrea AD. Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. J Clin Invest. 2007;117:1440–1449. doi: 10.1172/JCI31245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hine CM, Seluanov A, Gorbunova V. Use of the Rad51 promoter for targeted anti-cancer therapy. Proc Natl Acad Sci USA. 2008;105:20810–20815. doi: 10.1073/pnas.0807990106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ohnishi T, Taki T, Hiraga S, Arita N, Morita T. In vitro and in vivo potentiation of radiosensitivity of malignant gliomas by antisense inhibition of the RAD51 gene. Biochem Biophys Res Commun. 1998;245:319–324. doi: 10.1006/bbrc.1998.8440. [DOI] [PubMed] [Google Scholar]
- 105.Husain A, He G, Venkatraman ES, Spriggs DR. BRCA1 up-regulation is associated with repair-mediated resistance to cis-diamminedichloroplatinum(II) Cancer Res. 1998;58:1120–1123. [PubMed] [Google Scholar]
- 106.Dupre A, Boyer-Chatenet L, Sattler RM, Modi AP, Lee JH, Nicolette ML, Kopelovich L, Jasin M, Baer R, Paull TT, Gautier J. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat Chem Biol. 2008;4:119–125. doi: 10.1038/nchembio.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Powell SN, Kachnic LA. Therapeutic exploitation of tumor cell defects in homologous recombination. Anticancer Agents Med Chem. 2008;8:448–460. doi: 10.2174/187152008784220267. [DOI] [PubMed] [Google Scholar]
- 108.Markowitz J, Chen I, Gitti R, Baldisseri DM, Pan Y, Udan R, Carrier F, MacKerell AD, Jr, Weber DJ. Identification and characterization of small molecule inhibitors of the calcium-dependent S100B–p53 tumor suppressor interaction. J Med Chem. 2004;47:5085–5093. doi: 10.1021/jm0497038. [DOI] [PubMed] [Google Scholar]
- 109.Ame JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004;26:882–893. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 110.Okano S, Lan L, Caldecott KW, Mori T, Yasui A. Spatial and temporal cellular responses to single-strand breaks in human cells. Mol Cell Biol. 2003;23:3974–3981. doi: 10.1128/MCB.23.11.3974-3981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Menissier de Murcia J, Ricoul M, Tartier L, Niedergang C, Huber A, Dantzer F, Schreiber V, Ame JC, Dierich A, LeMeur M, Sabatier L, Chambon P, de Murcia G. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J. 2003;22:2255–2263. doi: 10.1093/emboj/cdg206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 113.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 114.Tentori L, Leonetti C, Scarsella M, d'Amati G, Portarena I, Zupi G, Bonmassar E, Graziani G. Combined treatment with temozolomide and poly(ADP-ribose) polymerase inhibitor enhances survival of mice bearing hematologic malignancy at the central nervous system site. Blood. 2002;99:2241–2244. doi: 10.1182/blood.v99.6.2241. [DOI] [PubMed] [Google Scholar]
- 115.Liu SK, Coackley C, Krause M, Jalali F, Chan N, Bristow RG. A novel poly(ADP-ribose) polymerase inhibitor, ABT-888, radiosensitizes malignant human cell lines under hypoxia. Radiother Oncol. 2008;88:258–268. doi: 10.1016/j.radonc.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 116.Lord CJ, Ashworth A. Targeted therapy for cancer using PARP inhibitors. Curr Opin Pharmacol. 2008;8:363–369. doi: 10.1016/j.coph.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 117.Evers B, Drost R, Schut E, de Bruin M, van der Burg E, Derksen PW, Holstege H, Liu X, van Drunen E, Beverloo HB, Smith GC, Martin NM, Lau A, O'Connor MJ, Jonkers J. Selective inhibition of BRCA2-deficient mammary tumor cell growth by AZD2281 and cisplatin. Clin Cancer Res. 2008;14:3916–3925. doi: 10.1158/1078-0432.CCR-07-4953. [DOI] [PubMed] [Google Scholar]
- 118.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JH, de Bono JS. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 119.Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, De Greve J, Lubinski J, Shanley S, Messiou C, A'Hern R, Tutt A, Ashworth A, Stone J, Carmichael J, Schellens JH, de Bono JS, Kaye SB. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 28:2510–2519. doi: 10.1200/JCO.2009.26.9589. [DOI] [PubMed] [Google Scholar]
- 120.Wang H, Wang X, Zhang P, Wang Y. The Ku-dependent non-homologous end-joining but not other repair pathway is inhibited by high linear energy transfer ionizing radiation. DNA Repair (Amst) 2008;7:725–733. doi: 10.1016/j.dnarep.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 121.Eriksson A, Lewensohn R, Nilsson A. Expression and activity of DNA-dependent protein kinase in normal human leukocytes. Anticancer Res. 2000;20:3051–3058. [PubMed] [Google Scholar]
- 122.Grawunder U, Finnie N, Jackson SP, Riwar B, Jessberger R. Expression of DNA-dependent protein kinase holoen-zyme upon induction of lymphocyte differentiation and V(D)J recombination. Eur J Biochem. 1996;241:931–940. doi: 10.1111/j.1432-1033.1996.00931.x. [DOI] [PubMed] [Google Scholar]
- 123.Okaichi K, Suzuki K, Morita N, Ikeda M, Takahashi H, Matsuda N, Watanabe M, Okumura Y. Low dose of wortmannin reduces radiosensitivity of human glioblastoma cells through the p53 pathway. Oncol Rep. 2002;9:859–862. [PubMed] [Google Scholar]
- 124.Shinohara ET, Geng L, Tan J, Chen H, Shir Y, Edwards E, Halbrook J, Kesicki EA, Kashishian A, Hallahan DE. DNA-dependent protein kinase is a molecular target for the development of noncytotoxic radiation-sensitizing drugs. Cancer Res. 2005;65:4987–4992. doi: 10.1158/0008-5472.CAN-04-4250. [DOI] [PubMed] [Google Scholar]
- 125.Rosenzweig KE, Youmell MB, Palayoor ST, Price BD. Radiosensitization of human tumor cells by the phosphatidylinositol3-kinase inhibitors wortmannin and LY294002 correlates with inhibition of DNA-dependent protein kinase and prolonged G2-M delay. Clin Cancer Res. 1997;3:1149–1156. [PubMed] [Google Scholar]
- 126.Ellenberger T, Tomkinson AE. Eukaryotic DNA ligases: structural and functional insights. Annu Rev Biochem. 2008;77:313–338. doi: 10.1146/annurev.biochem.77.061306.123941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhong S, Chen X, Zhu X, Dziegielewska B, Bachman KE, Ellenberger T, Ballin JD, Wilson GM, Tomkinson AE, Mackerell AD., Jr Identification and validation of human DNA ligase inhibitors using computer-aided drug design. J Med Chem. 2008;51:4553–4562. doi: 10.1021/jm8001668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Vijayakumar S, Dziegielewska B, Levin DS, Song W, Yin J, Yang A, Matsumoto Y, Bermudez VP, Hurwitz J, Tomkinson AE. Phosphorylation of human DNA ligase I regulates its interaction with replication factor C and its participation in DNA replication and DNA repair. Mol Cell Biol. 2009;29:2042–2052. doi: 10.1128/MCB.01732-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lakshmipathy U, Campbell C. Antisense-mediated decrease in DNA ligase III expression results in reduced mitochondrial DNA integrity. Nucleic Acids Res. 2001;29:668–676. doi: 10.1093/nar/29.3.668. [DOI] [PMC free article] [PubMed] [Google Scholar]