

Abstract
Foodborne pathogens encounter rapidly changing environmental conditions during transmission, including exposure to temperatures below 37°C. The goal of this study was to develop a better understanding of the effects of growth temperatures and temperature shifts on regulation of invasion phenotypes and invasion-associated genes in Listeria monocytogenes. We specifically characterized the effects of L. monocytogenes growth at different temperatures (30°C vs. 37°C) on (i) the contributions to Caco-2 invasion of different regulators (including σB, PrfA, and 14 response regulators [RRs]) and invasion proteins (i.e., InlA and FlaA), and on (ii) gadA, plcA, inlA, and flaA transcript levels and their regulation. Overall, Caco-2 invasion efficiency was higher for L. monocytogenes grown at 30°C than for bacteria grown at 37°C ( p = 0.0051 for the effect of temperature on invasion efficiency; analysis of variance); the increased invasion efficiency of the parent strain 10403S (serotype 1/2a) observed after growth at 30°C persisted for 2.5 h exposure to 37°C. For L. monocytogenes grown at 30°C, the motility RRs DegU and CheY and σB, but not PrfA, significantly contributed to Caco-2 invasion efficiency. For L. monocytogenes grown at 37°C, none of the 14 RRs tested significantly contributed to Caco-2 invasion, whereas σB and PrfA contributed synergistically to invasion efficiency. At both growth temperatures there was significant synergism between the contributions to invasion of FlaA and InlA; this synergism was more pronounced after growth at 30°C than at 37°C. Our data show that growth temperature affects invasion efficiency and regulation of virulence-associated genes in L. monocytogenes. These data support increasing evidence that a number of environmental conditions can modulate virulence-associated phenotypes of foodborne bacterial pathogens, including L. monocytogenes.
Introduction
Listeria monocytogenes is a Gram-positive, non-spore-forming rod that is capable of causing disease in humans and animals. L. monocytogenes can enter intestinal epithelia via an internalization process initiated by the interaction of InlA, expressed on the surface of invading L. monocytogenes, and E-cadherin, expressed on the epithelial cell surface. Although the organism is widespread in nature, 99% of L. monocytogenes infections are foodborne (Mead et al., 1999). L. monocytogenes therefore encounter a variety of different environments and associated stress conditions during transmission from the environment through foods to humans, including a wide range of temperature, pH, and osmotic stress conditions.
The alternative sigma factor, σB, the pleiotropic transcriptional regulator PrfA, and two-component regulatory systems (TCS) have all been shown to regulate key processes important for L. monocytogenes stress response and virulence (Kallipolitis and Ingmer, 2001; Kazmierczak et al., 2003; Williams et al., 2005a; Scortti et al., 2007). In addition to a large stress response regulon, σB specifically regulates the transcription of genes involved in responses to stresses encountered during passage through the gastrointestinal system such as osmotic stress and acid stress. σB also regulates transcription of genes encoding different internalins that are involved in entry into host cells (Raffelsbauer et al., 1998; Kazmierczak et al., 2003; Kim et al., 2005; McGann et al., 2007b). PrfA regulates the L. monocytogenes core virulence genes plcA, plcB, hly, mpl, and actA, which are important for escape from the vacuole and cell-to-cell spread (Scortti et al., 2007) and also co-regulates, with σB, other virulence genes, including inlA (McGann et al., 2008). Among the 16 putative or confirmed TCS in L. monocytogenes, a number of them have been shown to regulate bacterial adaptation (Autret et al., 2003; Brondsted et al., 2003; Kallipolitis et al., 2003; Dons et al., 2004; Mandin et al., 2005; Sleator and Hill, 2005; Williams et al., 2005b; Larsen et al., 2006), including VirR, DegU and CheY, which have been implicated in regulating mechanisms contributing to host cell invasion (Dons et al., 2004; Knudsen et al., 2004; Mandin et al., 2005).
Growth temperature has been shown to have a profound effect on activities of key L. monocytogenes regulators (Leimeister-Wachter et al., 1992; Liu et al., 2002; Dons et al., 2004; Chan et al., 2007b; McGann et al., 2007a; van der Veen et al., 2007). For example, expression levels of the genes comprising the PrfA-regulated Listeria pathogenicity island I, which are important in escape from the host cell vacuole and cell-to-cell spread, are maximal at 37°C due to a prfA mRNA thermosensor that represses PrfA translation at lower temperatures (Johansson et al., 2002). Also, certain σB-dependent genes have been shown to be involved in adaptation to cold temperatures (Becker et al., 2000; Chan et al., 2007a), and σB-dependent internalin genes, including inlC2 and inlD, have been shown to be expressed at higher levels at temperatures ≤30°C than at 37°C (McGann et al., 2007a). Finally, L. monocytogenes motility, which has been shown to contribute to host cell invasion (Dons et al., 2004) and increased virulence in a mouse model (O’Neil and Marquis, 2006), is temperature dependent. At 37°C, the transcription of flaA (the gene encoding the flagellin structural protein) and other motility-associated genes is repressed by MogR (Grundling et al., 2004; Shen and Higgins, 2006), though the stringency of this repression may vary among different strains of L. monocytogenes. For example, repression of motility genes and associated motility phenotypes has been shown to be less stringent in L. monocytogenes 10403S than in other strains (Grundling et al., 2004; Way et al., 2004).
Although L. monocytogenes will have likely been adapted to a temperature below 37°C before ingestion, many studies on L. monocytogenes host cell invasion have been done only on L. monocytogenes cells grown at 37°C and have only determined the effects of single gene mutations on invasion (Cotter et al., 1999; Kallipolitis and Ingmer, 2001; Autret et al., 2003; Williams et al., 2005a). Therefore, a more comprehensive evaluation of the effect of growth temperature on the contributions of response regulators (RRs), transcriptional regulators, and motility genes as well as functional synergisms between select genes to host cell invasion is necessary to better understand how adaptation to environments outside a mammalian host affects L. monocytogenes virulence. The objective of this study was, thus, to characterize the effects of growth temperature (30°C vs. 37°C) on (i) the contributions of σB and PrfA, 14 RRs, and invasion proteins InlA and FlaA to Caco-2 invasion, and on (ii) transcript levels of inlA, flaA, gadA, and plcA. Growth temperatures of 30°C and 37°C were chosen because L. monocytogenes grown at 30°C and 37°C show very similar lag phase durations and growth rates, whereas L. monocytogenes grown at lower temperatures (e.g. 22.5°C; [Pal et al., 2008]) show considerably longer lag phase durations and slower growth rates. As Caco-2 invasion efficiency has been shown to vary with bacterial growth phase (Garner et al., 2006a), it is critical to use bacteria grown with similar growth parameters and synchronized to similar growth phases to ensure that differences in invasion efficiency represent a temperature effect rather than differences in growth phase. In addition, L. monocytogenes grown at 30°C have previously been shown to display differential expression of key virulence-associated characteristics, including (i) downregulation of PrfA activity (Johansson et al., 2002) and (ii) upregulation of motility (O’Neil and Marquis, 2006). Consistent with our choice of 30°C as a growth temperature that allows for appropriate comparison between L. monocytogenes grown at typical mammalian body temperature and bacteria grown under environmental conditions, others (Toledo-Arana et al., 2009) have recently used 30°C as a growth temperature representing the saprophytic stage of the life of L. monocytogenes. Despite these key reasons for using growth temperatures of 30°C and 37°C to study temperature-dependent phenotypes of L. monocytogenes, we appreciate that these temperatures represent an experimental model that does not necessarily reflect natural transmission where foodborne bacteria are more likely exposed to refrigeration or room temperatures or heat shock conditions before ingestion.
Materials and Methods
Bacterial strains and growth conditions
Bacterial strains used in this study are listed in Table 1. For invasion assays, L. monocytogenes 10403S (Bishop and Hinrichs, 1987) and associated mutant strains were grown to early stationary phase as previously described (McGann et al., 2007a). Briefly, a 12 to 18 h culture grown at 37°C with aeration (i.e., shaking at 220 rpm) in brain heart infusion broth (BHI) was diluted 1:100 into 5 mL of fresh BHI and grown at 37°C with aeration to OD600 = 0.4. This culture was diluted 1:100 into another 5 mL of fresh BHI and grown at 30°C or 37°C with aeration to early stationary phase (defined as growth to OD600 = 1.0, followed by an additional 3 h incubation). For temperature shift experiments, a 1 mL aliquot of the early stationary phase culture was centrifuged at 12,000 rpm for 10 min, resuspended in phosphate-buffered saline (PBS; 0.14 M NaCl, 2.7 mM KCl, 10 mM Na2PO4, and 1.8 mM KH2PO4; pH 7.2), and incubated statically at 30°C or 37°C for 0, 2.5, or 5 h.
Table 1.
Strains Used in This Study
| Strain designationa | Genotype | Description of gene product | Referenceb |
|---|---|---|---|
| X1-001 | Parent strain | Strain 10403S; serotype 1/2a | Bishop and Hinrichs (1987) |
| K4-006 | ΔinlA | Internalin A, required for internalization into selected host cells | Bakardjiev et al. (2004) |
| H6-199 | ΔflaA | Flagellum structural protein | O’Neil and Marquis (2006) |
| B4-007 | ΔlisRKc | LisRK, contributes to log-phase acid resistance | This study |
| B2-078 | ΔagrA | Response regulator (RR), contributes to protein secretion | This study |
| B2-080 | ΔresD | RR, contributes to virulence gene repression in presence of select carbohydrates | This study |
| B2-086 | Δlmo1022 | Putative RR, no known role | This study |
| B2-096 | Δlmo1060 | Putative RR, no known role | This study |
| C5-017 | Δlmo2010 | Putative RR, no known role | This study |
| C5-019 | Δlmo2583 | Putative RR, no known role | This study |
| B2-100 | ΔphoP | RR, similar to Bacillus subtilis PhoP | This study |
| C5-041 | ΔvirR | Novel RR involved in invasion of Caco-2 cells | This study |
| C5-036 | Δlmo1507 | Putative RR, no known role | This study |
| C5-033 | ΔdegU | RR, regulates expression of motility genes | This study |
| B2-104 | ΔkdpE | RR, contributes to growth at high osmoloarity and low temperature | This study |
| B2-105 | ΔcheY | RR, contributes to chemotaxis | This study |
| B2-102 | ΔcesR | RR, contributes to ethanol and β-lactam tolerance | This study |
| A1-254 | ΔsigB | Alternative sigma factor σB, regulates general stress response | Wiedmann et al. (1998) |
| B2-046 | ΔprfA | PrfA, regulation of virulence genes | Cheng and Portnoy (2003) |
| B2-068 | ΔsigBΔprfA | McGann et al. (2007b) | |
| I1-001 | ΔinlAΔflaA | Marquis Lab |
All strain designations carry the prefix FSL.
References where a given mutant was previously described.
This mutant includes an internal deletion of the lisR gene, which also removed a portion of the ribosome binding site of lisK sensory kinase gene.
RR, response regulator.
Mutant construction
Internal, in-frame, nonpolar deletion mutant alleles in 14 RR genes (Table 1) were created using splicing overlap extension polymerase chain reaction (PCR) (see Supplemental Table S1, available online at www.liebertonline.com for primers; all supplemental material is located at http://author.cals.cornell.edu/cals/foodsci/research/labs/wiedmann/links/ivy2010-1.cfm) and cloned into the temperature-sensitive suicide shuttle vector pKSV7 (Smith and Youngman, 1992) as previously detailed by our group (Wiedmann et al., 1998). Mutant alleles were introduced into L. monocytogenes 10403S using standard allelic exchange mutagenesis procedures (Camilli et al., 1993). Mutant strains were confirmed with sequencing. lmo0287 is likely to be essential, as null mutants in this gene could not be constructed here, consistent with similar studies by others (Kallipolitis and Ingmer, 2001; Williams et al., 2005a).
Caco-2 invasion assays
The Caco-2 cell-line (ATCC HTB-37) was maintained at 37°C (4%–6% CO2 and 85% humidity) in a Caco-2 medium, which was Dulbecco’s minimal essential medium with Earle’s salts, 1% sodium pyruvate, 20% fetal bovine serum, 1.0% nonessential amino acids, 1.5 g/L sodium bicarbonate, and, when appropriate, penicillin G and streptomycin (each at 100 μg/mL) (all reagents were obtained from Gibco, a subsidiary of Invitrogen). All invasion assays were performed at 37°C as previously described (Garner et al., 2006b) with minor modifications. Briefly, 48 h before the assay, Caco-2 cells were seeded (from a culture passaged no >60 times) into 24-well tissue culture plates (Corning Inc.) at a density of 5×104 cells/well in the Caco-2 medium without antibiotics. For infection, ~2×107 CFU L. monocytogenes were added to each well (representing a multiplicity of infection of ~200). All inocula were enumerated on BHI agar plates. Thirty minutes postinfection, the Caco-2 monolayers were washed three times with PBS to remove any unassociated L. monocytogenes, and the medium was replaced with a fresh Caco-2 medium. Forty-five minutes after infection, the medium was replaced with the Caco-2 medium plus 150 μg/mL gentamycin to kill any extracellular L. monocytogenes. At 90 min postinfection, Caco-2 cells were washed three times with PBS and lysed with ice-cold distilled water. Intracellular L. monocytogenes were enumerated by plating the appropriate dilutions of the Caco-2 lysate on BHI agar, using a spiral plater (Spiral Biotech). At least three independent trials of the invasion assays were performed with duplicate wells tested for each treatment in each replicate.
Quantitative reverse transcriptase-PCR (qRT-PCR)
Transcript levels of inlA, flaA, plcA, gadA, rpoB, sigB, and prfA were quantified for select strains grown to early stationary phase at 30°C or 37°C using TaqMan probes and primers and the ABI Prism 7000 Sequence Detection System as previously described (Sue et al., 2003; Chaturongakul and Boor, 2006; McGann et al., 2007a) with one exception: copy numbers for each gene were normalized to rpoB levels. Primers and probes for inlA, plcA, gadA, sigB, rpoB, and prfA have been previously described (Sue et al., 2004; Kim et al., 2005; Kazmierczak et al., 2006). flaA Taqman primers ( flaA-F: 5′-TCGTAAAAATAACGAAGGCATGAC-3′; flaA-R: AGA ACTGTTAATACGTTTACCAGATGCT-3′) and the flaA MGB probe (FAM-5′-CAAGCGCAAGAAC-3′NFQ) were designed using Primer Express 1.0 (Applied Biosystems).
Statistical analyses
All statistical analyses were performed in JMP 7.0 (SAS Institute Inc.). Invasion efficiencies were initially analyzed using a one-way analysis of variance (ANOVA; α = 0.05). Strain and, where appropriate, date of experiment were included as variables in the model. For each ANOVA, data were log-transformed to ensure that the data set satisfied ANOVA assumptions of normality of residuals and equality of variances. To determine whether a particular mutant strain differed from the parent strain (e.g., in invasion efficiency), a post hoc Dunnett’s many to one test was used (Shun et al., 2003). To compare invasion efficiencies for a given strain exposed to multiple conditions (i.e., as shown in Table 2) or among strains (in cases where a strain with a double mutation was included in the comparison), a post hoc Tukey honestly significantly different (HSD) test was used. For qRT-PCR, normalized log copy numbers for a given gene were compared among strains by using one-way ANOVA, followed by Tukey HSD. To measure whether two gene deletions showed an effect on invasion efficiency or transcript level that is more than additive (which indicates synergism), the parent strain, single mutants, and double mutants were assigned unique allelic states by coding dummy variables (e.g., “gene1” and “gene2”) for each allelic state with 1 meaning the gene is present and 0 meaning the gene is absent. A two-way ANOVA was performed to determine the effect of each allelic state on invasion efficiency or transcript level in these analyses. “Date of experiment,” “gene1,” “gene2,” and “gene1*gene2” were included as effects in the two-way ANOVA model. A significant effect of gene1*gene2 in the model (i.e., p < 0.05) indicates a more than additive effect of the double mutation (i.e., deletion of both gene1 and gene2 in the same genetic background has a greater effect than the sum of the effects of deleting gene1 alone and gene2 alone).
Table 2.
Caco-2 Invasion Efficiencies of Listeria monocytogenes 10403s and Select Mutants After Exposure to Varying Temperature Treatments
| Growth temperature | Temperature of PBS hold (2.5 h) | Caco-2 invasion efficiency (calculated as [CFU recovered/CFU infected] × 100) (SD) fora |
||||
|---|---|---|---|---|---|---|
| 10403S | ΔcheY | ΔdegU | ΔflaA | ΔinlA | ||
| 30°C | None | 0.67 (0.17)A | 0.018 (0.007)B,* | 0.015 (0.008)B,* | 0.014 (0.010)B,* | 0.024 (0.010)A,* |
| 30°C | 30°C | 0.64 (0.19)A | 0.013 (0.003)B,* | 0.010 (0.005)B,* | 0.010 (0.006)B,* | 0.023 (0.007)A,* |
| 30°C | 37°C | 0.36 (0.10)A | 0.006 (0.002)B,* | 0.007 (0.002)B,* | 0.007 (0.004)B,* | 0.014 (0.005)A,* |
| 37°C | None | 0.14 (0.03)B | 0.14 (0.06)A | 0.20 (0.05)A | 0.14 (0.06)A | 0.0008 (0.0003)B,* |
| 37°C | 37°C | 0.11 (0.04)B | 0.12 (0.05)A | 0.14 (0.10)A | 0.09 (0.06)A | 0.0007 (0.0007)B,* |
| 37°C | 30°C | 0.11 (0.03)B | 0.13 (0.05)A | 0.17 (0.08)A | 0.14 (0.07)A | 0.0007 (0.0005)B,* |
Within a given column values with identical letters (A or B) are not significantly different (p ≥ 0.05; post hoc Tukey HSD test). Within a given row, invasion efficiencies that are lower for a given mutant than the parent strain exposed to the same condition (p ≤ 0.05; post hoc Dunnett’s test) are marked with an asterisk (*). Data represent the mean and standard deviation of three biological replicates.
PBS, phosphate-buffered saline; SD, standard deviation; ANOVA, analysis of variance; HSD, honestly significantly different.
Results
Contributions of RRs to Caco-2 invasion
Overall ANOVA analysis showed a highly significant effect of temperature on invasion efficiency ( p = 0.0051), and all RR mutants (except ΔdegU and ΔcheY) and 10403S had numerically higher mean Caco-2 invasion efficiencies after growth to early stationary phase in BHI at 30°C than growth at 37°C. For several strains, including 10403S, ΔresD, Δlmo1022, ΔphoP, ΔcesR, and ΔkdpE, this difference was statistically significant ( p < 0.05; t-test; Fig. 1).
FIG. 1.
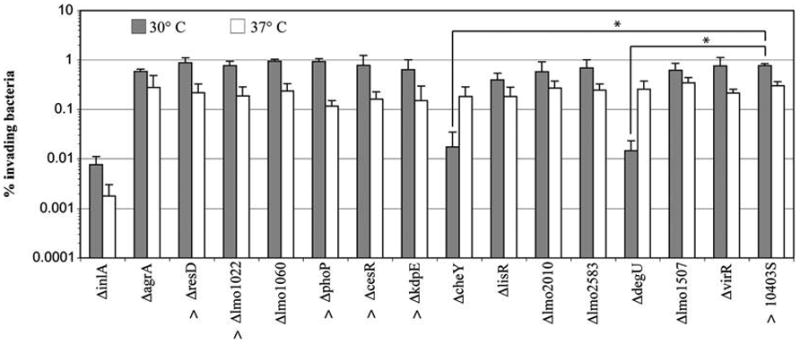
Caco-2 invasion efficiencies of Listeria monocytogenes 10403S and 14 mutant strains with deletions in genes encoding RRs. Bacteria were grown to early stationary phase (growth to OD600 = 1.0, followed by 3 h of incubation) with aeration (i.e., shaking at 220 rpm) at 30°C or 37°C. Invasion assays were performed at 37°C. Data represent the mean of at least three biological replicates. Error bars represent SD. A ΔinlA strain was included as a control. Overall ANOVA analyses of the 14 RR mutants and the parent strain showed a significant effect of temperature ( p = 0.0051); all strains (except ΔcheY and ΔdegU) showed numerically higher invasion efficiencies when grown at 30°C. Strains showing statistically higher invasion after growth at 30°C than at 37°C ( p < 0.05; t-test) are denoted by ^. For 10403S and RR mutants, ANOVA showed a significant effect of strain on invasion efficiency for bacteria grown at 30°C ( p < 0.0001). For bacteria grown at 37°C this effect was not significant ( p = 0.3900). Among RR mutants grown at 30°C, ΔcheY and ΔdegU showed significantly lower invasion efficiencies than the parent strain ( p < 0.0001; post hoc Dunnett’s; denoted by *). RR, response regulator; ANOVA, analysis of variance; SD, standard deviation.
For the L. monocytogenes parent strain and the 14 RR mutants grown at 30°C, ANOVA showed a significant effect of strain on Caco-2 invasion efficiency ( p < 0.0001). Specifically, ΔdegU and ΔcheY both had ~50-fold lower mean invasion efficiencies than the parent strain ( p < 0.0001 for each; Dunett’s test) when grown at 30°C (Fig. 1). These results confirm the findings of other studies that have shown a role for CheY in host cell invasion (Dons et al., 2004). Unlike all other strains tested, the ΔcheY and ΔdegU strains showed numerically higher invasion efficiency after growth at 37°C than at 30°C (Fig. 1).
For L. monocytogenes parent strain 10403S and the 14 RR mutants grown to early stationary phase at 37°C, the effect of the factor “strain” on Caco-2 invasion efficiencies was not statistically significant ( p = 0.3900; ANOVA). Therefore, the appropriate conservative statistical test (i.e., ANOVA) found no statistically significant reduction in Caco-2 invasion for any of the 14 RR mutants grown at 37°C.
As L. monocytogenes motility has been reported in other L. monocytogenes strains to be regulated by DegU and CheY at temperatures ≤30°C (Dons et al., 2004; Knudsen et al., 2004; Williams et al., 2005a; Mauder et al., 2008), we conducted swarming assays to evaluate motility of the L. monocytogenes 10403S, ΔdegU, ΔcheY, and ΔflaA strains. While 10403S was clearly motile when grown at 30°C, ΔdegU, ΔcheY, and ΔflaA showed reduced swarming (6%, 6%, and 1%, respectively, of the swarming area for 10403S grown at 30°C; see Supplemental Fig. S1, available online at www.liebertonline.com). When grown at 37°C, the parent strain showed limited and considerably reduced swarming compared to 30°C (Supplemental Fig. S1), and the three mutant strains showed no detectable swarming at this temperature. The finding that 10403S exhibited some swarming at 37°C is consistent with previous reports that L. monocytogenes 10403S (the parent strain used here) shows limited flaA expression even at 37°C (Grundling et al., 2004; Way et al., 2004).
Effects of temperature shifts and holds on growth temperature-dependent phenotypes
On the basis of the data reported above, we evaluated whether the increased invasion efficiency of 10403S at 30°C is maintained after a shift to 37°C. While L. monocytogenes 10403S grown at 30°C and held (in PBS) at 37°C for 2.5 h (30°C–37°C) showed numerically (about twofold) reduced invasion efficiency compared to bacteria either before the shift or bacteria shifted to 30°C for 2.5 h (Table 2), the invasion efficiency of the “30°C–37°C” treatment group was not significantly different from the control groups grown and held at 30°C (30°C–30°C in Table 2) ( p > 0.05; Tukey HSD) but was significantly higher than control groups grown and held at 37°C (37°C–37°C) ( p < 0.05; Tukey HSD). These results suggest that increased invasion efficiency of L. monocytogenes grown at 30°C is largely maintained during a 2.5 h hold at 37°C in PBS. When L. monocytogenes 10403S was grown at 30°C, and shifted to and held at 37°C for 5 h (in PBS), the invasion efficiency was reduced (0.15% invasion efficiency) and virtually identical to that of L. monocytogenes grown at 37°C (0.14% invasion efficiency) with no significant difference ( p > 0.05; Tukey HSD). This indicates that prolonged exposure of L. monocytogenes grown at 30°C to 37°C (in PBS) reduces invasion efficiencies to values typical for bacteria grown at 37°C. Growth and de novo protein synthesis (which are unlikely to occur in PBS) thus do not seem to be required for reduced invasiveness observed at 37°C.
When the L. monocytogenes parent strain grown at 37°C was switched to 30°C and held at this temperature for 2.5 h, bacteria showed no changes in invasion efficiency and maintained lower invasion efficiency typical for bacteria grown at 37°C ( p < 0.05; Tukey HSD; Table 2), suggesting that the increased invasion phenotype cannot be induced by simply switching the organism to 30°C but requires growth at 30°C. Controls included in these experiments showed that exposure to PBS for 2.5 h had no effect on invasion efficiency; bacteria grown at 30°C and exposed to PBS at 30°C for 2.5 h as well as bacteria grown at 37°C and exposed to PBS at 37°C for 2.5 h did not differ in their invasion efficiencies from bacteria grown at 30°C or 37°C, respectively, without subsequent exposure to PBS ( p > 0.05; Tukey HSD; Table 2).
As L. monocytogenes ΔdegU, ΔcheY, and ΔflaA showed lower invasion than the parent strain after growth to early stationary phase at 30°C, we also tested whether this invasion defect was maintained after a shift from 30°C to 37°C, followed by a 2.5 h hold at this temperature (in PBS). All three strains (ΔdegU, ΔcheY, and ΔflaA) maintained lower invasion efficiencies than the parent strain even after bacteria grown at 30°C were held for 2.5 h at 37°C (Table 2). Invasion efficiencies for these strains grown at 30°C and shifted to 37°C for 2.5 h were not significantly different ( p > 0.05; Tukey HSD; Table 2) from invasion efficiencies for (i) strains grown at 30°C (before shift) or (ii) strains grown at 30°C and subsequently held at 30°C in PBS for 2.5 h (Table 2). The invasion efficiencies for ΔdegU, ΔcheY, and ΔflaA grown at 30°C and held for 2.5 h at 37°C also continued to be significantly lower than the invasion efficiencies for the same strains grown at 37°C ( p < 0.05; Tukey HSD); invasion efficiencies for these three strains in the “30°C–37°C” treatment group were ~20 to 30-fold lower than the same strains grown at 37°C (Table 2). These three mutant strains thus maintained their invasion deficient phenotype even after 2.5 h exposure to 37°C. To test the effect of prolonged exposure to 37°C on the reduced invasion efficiency of motility-deficient mutant strains, invasion efficiency of the ΔflaA strain was also evaluated for bacteria grown at 30°C and shifted to 37°C with a hold in PBS at 37°C for 5 h. Even after a hold at 37°C for 5 h, the ΔflaA strain maintained reduced invasion efficiency (0.006%) compared to the invasion efficiency of either the parent strain grown at 37°C (0.14%) or ΔflaA grown at 37°C (0.14%; both comparisons had p < 0.05; Tukey HSD), suggesting that increased invasion efficiency at 37°C, of a ΔflaA mutant, requires growth at 37°C and probably de novo protein synthesis (which is unlikely to occur in PBS). When the ΔdegU, ΔcheY, and ΔflaA strains were grown at 37°C and shifted to 30°C with a hold at 30°C for 2.5 h, all three strains maintained the higher invasion efficiency as they displayed when grown at 37°C (Table 2).
Temperature-dependent contributions of InlA and FlaA to Caco-2 invasion
In initial experiments, the L. monocytogenes ΔinlA strain showed 4.4-fold higher invasion efficiency when grown at 30°C than at 37°C (Fig. 1). In subsequent experiments, when grown at 37°C, the ΔinlA strain showed, on average, greater than 100-fold lower invasion efficiency than either the parent strain or the ΔdegU, ΔcheY, or ΔflaA strains grown at 37°C (Table 2). However, when grown at 30°C, the ΔinlA strain showed only a 26-fold lower average invasion efficiency than the parent strain (Table 2). These experiments also confirmed a significantly higher invasion efficiency for the ΔinlA grown at 30°C than at 37°C ( p < 0.05; Tukey HSD; Table 2). Temperature shifts (i.e., 30°C–37°C, 2.5 h or 37°C–30°C, 2.5 h) did not significantly affect these statistical differences (Table 2). These results indicate an InlA-independent increase in Caco-2 invasion for L. monocytogenes grown at 30°C (as compared to 37°C), which is maintained after a shift to 37°C.
To further investigate the contributions of InlA and flagellin to Caco-2 invasion after growth at 30°C and 37°C and to specifically determine whether there is synergism between InlA and flagellin in facilitating invasion, 10403S, ΔflaA, ΔinlA, and ΔinlAΔflaA were grown at 30°C or 37°C and tested for their Caco-2 invasion efficiencies (Fig. 2). After growth at 30°C, ΔflaA showed ~30-fold lower invasion efficiency than the parent strain (Fig. 2). At both 30°C and 37°C, the invasion efficiency of the ΔinlAΔflaA strain was lower ( p < 0.05; Tukey HSD) than that of the parent strain (0.007% vs. 0.680% for 37°C and 0.003% vs. 2.064% for 30°C, respectively). Further, two-way ANOVA analysis showed a significant “flaA*inlA” interaction effect on Caco-2 invasion for L. monocytogenes grown at both 30°C ( p = 0.0015) and 37°C ( p = 0.0269). Interestingly, the F-ratio of the interaction effect for bacteria grown at 30°C (F = 20.15) was higher than that of bacteria grown at 37°C (F = 6.97), suggesting a greater contribution of the interaction effect to the observed variance at 30°C than at 37°C. These statistical findings indicate that the effects of deleting inlA and flaA on Caco-2 invasion are more than additive, indicating a synergism between InlA and FlaA in facilitating Caco-2 invasion for bacteria grown at either 30°C or 37°C.
FIG. 2.
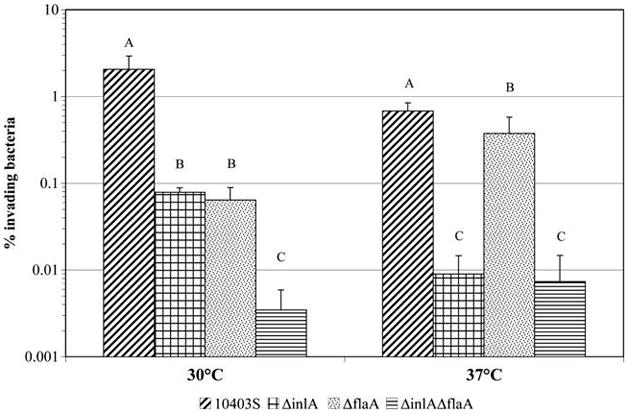
Caco-2 invasion efficiencies of L. monocytogenes 10403S, ΔinlA, ΔflaA, and ΔinlAΔflaA strains grown to early stationary phase with aeration (i.e., shaking at 220 rpm) at either 30°C or 37°C. Data represent the mean of four biological replicates. Error bars represent SDs. Strains with different letters had significantly different invasion efficiencies at a given growth temperature ( p < 0.05; post hoc Tukey HSD); lower invasion for the ΔflaA strain than the parent strain (for growth at 37°C) was only borderline significant ( p = 0.0279; Tukey HSD). Two-way ANOVA was performed within each temperature to determine effects of single and double mutations on invasion efficiency (see Table 3 for p-values). HSD, honestly significantly different.
Temperature-dependent contributions of transcriptional regulators σB and PrfA to Caco-2 invasion
To determine growth temperature effects on the contributions of σB and PrfA to Caco-2 invasion, ΔprfA, ΔsigB, and ΔsigBΔprfA strains grown to early stationary phase at 30°C or 37°C were used for Caco-2 invasion assays (performed at 37°C). After growth at 30°C, the invasion efficiency of ΔprfA was 2.737%, which was not significantly different ( p > 0.05; Tukey HSD) than the parent strain (2.064% invasion efficiency); the ΔsigB (0.376% invasion) and ΔsigBΔprfA (0.231% invasion) strains were significantly less invasive ( p < 0.05; Tukey HSD) than the parent strain (Fig. 3). These results indicate that only σB, not PrfA, is involved in invasion when L. monocytogenes is grown at 30°C. When the bacteria were grown at 37°C, ΔprfA (0.322%), ΔsigB (0.060%), and ΔsigBΔprfA (0.018%) had significantly lower invasion efficiencies ( p < 0.05; Tukey HSD) than the parent strain (0.680%) (Fig. 3); the ΔsigBΔprfA strain also showed lower invasion efficiency than the parent strain and the ΔprfA strain ( p < 0.05; Tukey HSD). A two-way ANOVA also showed a significant “sigB*prfA” interaction effect, indicating a synergism between σB and PrfA in regulating Caco-2 invasion for L. monocytogenes grown at 37°C (Table 3).
FIG. 3.
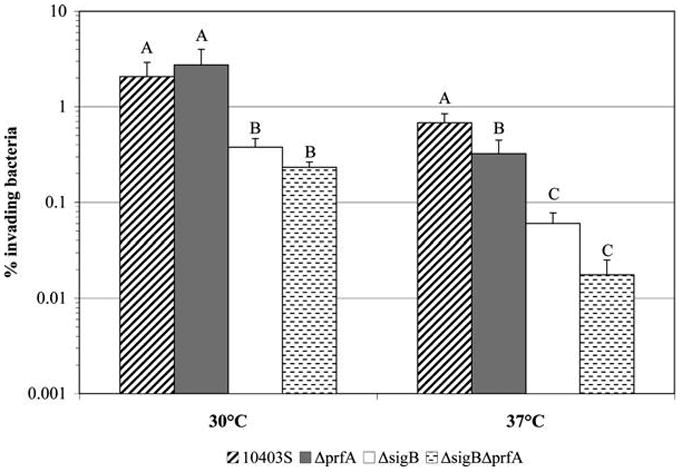
Caco-2 invasion efficiencies of L. monocytogenes 10403S, ΔprfA, ΔsigB, and ΔsigBΔprfA strains grown to early stationary phase with aeration (i.e., shaking at 220 rpm) at either 30°C or 37°C. Data for the parent strain (10403S) are the same as those shown in Figure 2. Data represent the mean of four biological replicates. Error bars represent SD. Strains with different letters have significantly different invasion efficiencies at a given growth temperature ( p < 0.05; post hoc Tukey HSD). Two-way ANOVA was performed within each temperature to determine effects of single and double mutations on invasion efficiency (see Table 3 for p-values).
Table 3.
Effects of Various Gene Deletions on Listeria monocytogenes Caco-2 Invasion Efficiency
| Deletion variablea |
p-Values from two-way ANOVA for L. monocytogenes grown at |
|
|---|---|---|
| 30°C | 37°C | |
| sigB | 0.0001b | < 0.0001b |
| prfA | 0.4401 | 0.0014c |
| sigB*prfA | 0.226 | 0.0057c |
| inlA | 0.001b | < 0.0001b |
| flaA | 0.0009b | 0.0256d |
| inlA*flaA | 0.0015c | 0.0269d |
The variables listed in this column represent either single-gene deletions (e.g., “sigB”) or interactions between two gene deletions (e.g., “sigB*prfA”). The p-values for the single-gene deletions measure the individual effect of deleting each respective gene. The “gene*gene” variable measures synergistic deletion effects by comparing the effect of deleting both genes to the effect of deleting either one gene or the other; significant values are marked with
p ≤ 0.001,
p ≤ 0.01, or
Temperature-dependent regulation of genes involved in Caco-2 invasion
To determine the effects of growth temperature on σB and PrfA-dependent regulation of L. monocytogenes genes involved in invasion, we determined transcript levels for inlA, flaA, plcA, gadA, sigB, prfA, and rpoB in 10403S, and ΔsigB, ΔprfA, and ΔsigBΔprfA grown to early stationary phase at 30°C or 37°C. Neither prfA nor sigB transcript levels differed between the parent strain grown at 30°C and 37°C ( p > 0.05, t-test). The transcript levels for the σB-dependent gadA and the PrfA-dependent plcA (measured as indicators of σB and PrfA activity, respectively) (Fig. 4) also did not differ significantly between bacteria grown at 30°C and 37°C ( p > 0.05; t-test), suggesting no differences in PrfA and σB activity between L. monocytogenes 10403S grown to early stationary phase in BHI at these two temperatures.
FIG. 4.
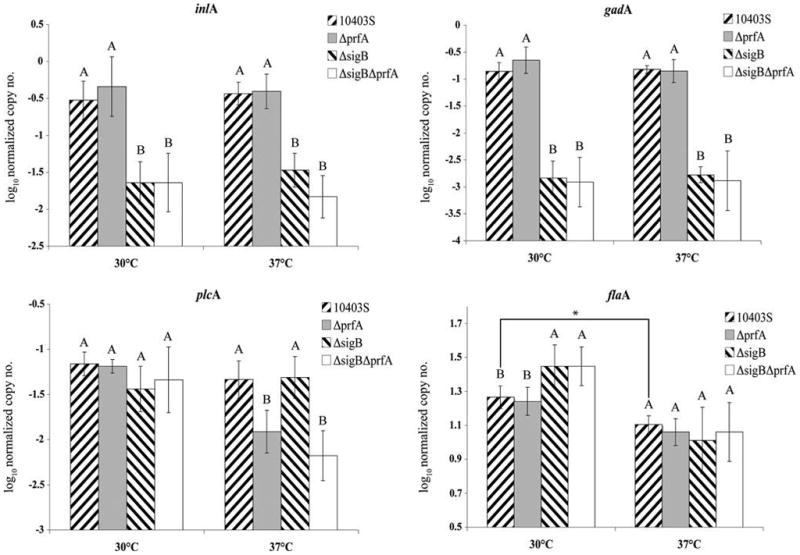
Normalized transcript levels of four genes in L. monocytogenes 10403S, ΔsigB, ΔprfA, and ΔsigBΔprfA grown to early stationary phase with aeration (i.e., shaking at 220 rpm) at 30°C or 37°C. Data presented as log10 (target gene mRNA level/rpoB mRNA level). Bars represent the average of four biological replicates and error bars represent SD. Strains with different letters (e.g., A, B) had significantly different transcript levels ( p < 0.05; post hoc Tukey HSD). Two-way ANOVA was performed for each gene at each temperature to determine effects of single and double mutations on transcript levels (see Supplemental Table S2 for p-value summary). For each gene, a t-test was used to compare transcript levels in 10403S between 30°C and 37°C. Only flaA levels were significantly different between temperatures ( p = 0.0014; t-test; indicated by *).
For bacteria grown to early stationary phase at 30°C, inlA transcript levels were 10 times lower in the ΔsigB strain than the parent strain ( p < 0.05; Tukey HSD; Fig. 4), whereas there was no significant effect of the prfA deletion on inlA transcript levels ( p = 0.1282; two-way ANOVA) (Supplemental Table S2, available online at www.liebertonline.com). These data indicate that inlA transcription is σB dependent, but PrfA independent under these conditions. For L. monocytogenes grown at 37°C, inlA transcript levels were significantly lower in ΔsigB than in the parent strain ( p < 0.05; ANOVA; Tukey HSD; Fig. 4), whereas inlA transcript levels were not significantly different in ΔprfA compared to the parent strain ( p > 0.05; Tukey HSD; Fig. 4). However, the sigB*prfA interaction effect on inlA transcript levels was borderline significant ( p = 0.0799; two-way ANOVA; Supplemental Table S2), suggesting contributions of both σB and PrfA to regulation of inlA transcription in L. monocytogenes grown at 37°C.
In the L. monocytogenes parent strain, flaA transcript levels were higher after growth at 30°C (1.26; standard deviation = 0.07) than at 37°C (1.104; standard deviation = 0.05) ( p = 0.0014, t-test; data shown in Fig. 4). Although the difference is small, these results indicate at least some temperature-dependent regulation of flaA transcription in 10403S. flaA transcript levels were not significantly different between ΔprfA and the parent strain for either growth temperature ( p > 0.05; Tukey HSD; Fig. 4). In L. monocytogenes grown at 37°C, flaA transcript levels were not different between the ΔsigB strain and the parent strain ( p > 0.05; Tukey HSD; Fig. 4). However, at 30°C flaA transcription was significantly higher in ΔsigB and ΔsigBΔprfA than in the parent strain ( p < 0.05; ANOVA, Tukey HSD; Fig. 4), and two-way ANOVA showed a significant “sigB effect” on flaA transcript levels ( p = 0.0003; Supplemental Table S2). These results are consistent with other studies that suggest a role of σB in down-regulating chemotaxis genes (Raengpradub et al., 2008; Toledo-Arana et al., 2009).
Discussion
L. monocytogenes grown at 30 °C shows a higher Caco-2 invasion efficiency than bacteria grown at 37 °C
Out data showed that, with the exception of mutants with deletions of motility-related genes, all L. monocytogenes strains showed higher Caco-2 invasion efficiency when grown at 30°C than at 37°C. This finding supports previous work (Dons et al., 2004) reporting that a different L. monocytogenes strain (12067) showed about 10-fold increased association with Caco-2 cells and twofold increased invasion of Caco-2 cells for bacteria grown at 24°C (where 12067 was motile) compared to 37°C (where this strain was not motile); this previous study did not include a statistical evaluation of these differences in invasion efficiencies (Dons et al., 2004). As most L. monocytogenes, including strain 10403S, show increased flagellar motility when grown at 30°C, a trait that contributes to Caco-2 cell invasion (Dons et al., 2004) and intestinal colonization in mice (O’Neil and Marquis, 2006), expression of motility genes at ≤30°C may explain the increased Caco-2 invasion of L. monocytogenes grown at ≤30°C compared to bacteria grown at 37°C.
Interestingly, other bacterial pathogens have also been shown to differ in their invasiveness and virulence depending on growth temperature (Konkel and Tilly, 2000). While in a number of bacterial pathogens virulence genes have been found to be more highly expressed at 37°C than at lower temperatures (Maurelli, 1989), some pathogens have been shown to express invasion factors at higher levels when grown at temperatures below 37°C. For example, Yersinia pseudotuberculosis invA, which encodes an invasin required for host cell invasion, appears to be expressed at higher levels in bacteria grown at 28°C than at 37°C (based on visual examination of Western blot data) (Isberg et al., 1988). Yersinia enterocolitica has also been shown to express a motility phenotype when grown below 37°C, whereas motility, which may play a role in initiation of host cell invasion, is down-regulated in bacteria grown at 37°C (Young et al., 2000). Therefore, adaptation to environments outside the host may increase the virulence potential of L. monocytogenes and other bacterial pathogens.
Enteric pathogens transmitted to humans from food and environmental sources often experience a sudden change in environmental temperature when they are ingested. Our results show that invasion phenotypes of the parent strain and the motility mutants grown at 30°C persisted for 2.5 h after the bacteria were switched to 37°C. As 70% to 90% of human stomach contents are emptied after 2 h (Bennink et al., 1999), our findings, in combination with other studies (Dons et al., 2004; O’Neil and Marquis, 2006), suggest that L. monocytogenes grown at temperatures that permit motility [i.e., 12°C–30°C (Di Bonaventura et al., 2008)] may have increased invasion potential that could be maintained during gastric passage. As regulation of motility appears to differ considerably between L. monocytogenes strains (Grundling et al., 2004; Way et al., 2004), further studies will need to use different strains to validate our findings on growth temperature dependence of Caco-2 invasion efficiency.
In addition to growth temperature, other studies (Garner et al., 2006a; Andersen et al., 2007) have shown that exposure to other environmental conditions (e.g., organic acids, anaerobic conditions) appear to affect the virulence potential of L. monocytogenes. For example L. monocytogenes grown in an oxygen-limited environment displayed 100-fold increased invasion efficiency in Caco-2 cells as well as increased virulence in a guinea pig model, including 10 to 100-fold higher L. monocytogenes fecal shedding levels, compared to numbers for bacteria grown under aerobic conditions (Andersen et al., 2007). Further, L. monocytogenes grown in the presence of sodium lactate or NaCl showed an about 10-fold higher invasion efficiency than bacteria grown without these compounds (Garner et al., 2006a). Overall, growth temperatures as well as other preinvasion environmental conditions (e.g., anaerobiosis) thus appear to affect the regulation of genes with roles in host attachment and invasion across different environmentally transmitted pathogens, including L. monocytogenes.
CheY and DegU significantly contribute to invasion in L. monocytogenes grown ≤30°C
Our data, in conjunction with other reports (e.g., Dons et al., 2004), clearly show that DegU and CheY play a temperature-dependent role in enhancing invasion of host cells across different L. monocytogenes strain backgrounds. For example, ΔcheA, ΔcheY, and ΔcheYA mutants in an L. monocytogenes 12067 background all showed about 100-fold reduced Caco-2 cell invasion as well as reduced motility as compared to their parent strain after growth at 24°C (Dons et al., 2004), whereas Williams et al. (2005a) found that L. monocytogenes EGD ΔdegU and ΔcheY strains, grown at 37°C, did not show reduced invasion efficiency of Cos-1 fibroblast cells. Previous studies have shown increased swarming and flagellar motility of L. monocytogenes grown at 24°C compared to L. monocytogenes grown at 37°C (Dons et al., 2004; Knudsen et al., 2004; Shen and Higgins, 2006), due to repression of L. monocytogenes motility genes at 37°C (Grundling et al., 2004; Shen and Higgins, 2006). Therefore, contributions of motility genes to virulence phenotypes are generally apparent only when L. monocytogenes is grown at temperatures where the organism is typically motile (i.e., ≤30°) (Dons et al., 2004; Knudsen et al., 2004; Williams et al., 2005b; O’Neil and Marquis, 2006). While DegU has also been shown to contribute to motility in L. monocytogenes grown at 24°C and to virulence in a mouse model (Williams et al., 2005b), we are not aware of any previous studies reporting a specific effect of a degU null mutation on invasion of human intestinal epithelial cells.
While we found no contributions of other RRs, besides DegU and CheY, to invasion of Caco-2 cells, some studies have shown contributions of other RRs to virulence phenotypes in L. monocytogenes strains other than 10403S and in other invasion models. For example, Williams et al. (2005a) reported that RRs Lmo1507 and LisR are involved in invasion of Cos-1 cells. A deletion of lisK (encoding the LisR-associated sensory kinase) in L. monocytogenes LO28 was also found to affect virulence in mice (Cotter et al., 1999), further supporting contributions of LisRK to L. monocytogenes virulence in some models. Similarly, L. monocytogenes EGD with a deletion of the RR VirR, grown at 37°C, has also been reported to be deficient in Caco-2 cell invasion (Mandin et al., 2005), even though the L. monocytogenes 10403S ΔvirR strain used in our current study did not show evidence for reduced invasion, again possibly reflecting strain differences. Overall, different TCS thus appears to contribute to L. monocytogenes virulence and virulence-associated characteristics, even though contributions appear to differ based on growth conditions, strain backgrounds, and assays used.
While InlA and FlaA show significant synergism in their contributions to Caco-2 invasion, synergism is less pronounced after growth at 37 °C
While flagellin has been identified as a critical part of the motility machinery involved in Caco-2 invasion in L. monocytogenes grown at temperatures that allow motility gene expression (i.e., ≤30°C) (Dons et al., 2004; O’Neil and Marquis, 2006), we found flagellin and InlA contribute synergistically to Caco-2 cell invasion in L. monocytogenes 10403S grown at 30°C and 37°C. While studies in other L. monocytogenes strains (e.g., 12067, EDGe) have shown that contributions of motility factors to host cell invasion typically are only apparent in L. monocytogenes grown at 30°C or less (Dons et al., 2004; Shen and Higgins, 2006), some studies (Grundling et al., 2004; Way et al., 2004) have shown increased flagellar motility at 37°C for strain 10403S as compared to other strains. For example, flaA repression by MogR was shown to be less stringent in 10403S than in EGDe (Grundling et al., 2004), and 10403S activated a flagellum-dependent innate immune response even after growth at 37°C (Way et al., 2004). Critical contributions of InlA to invasion of human intestinal epithelial cells (Dramsi et al., 1993; Lingnau et al., 1995) and virulence after oral infection (Lecuit et al., 1999; Garner et al., 2006b) have been well established for host species carrying the E-cadherin allotype that allows for InlA binding (e.g., humans and guinea pigs). Although we observed a synergism between the contributions of FlaA and InlA to Caco-2 invasion in L. monocytogenes grown at both 30°C and 37°C, we also found that the ΔinlA strain shows increased invasion efficiency when grown at 30°C compared to 37°C, suggesting InlA-independent contributions of flagellar motility to Caco-2 cell invasion. This observation further supports the importance of flagellar motility in L. monocytogenes virulence.
σB, but not PrfA, contributes to invasion of bacteria grown at 30 °C, whereas σB and PrfA show synergistic contributions to invasion if bacteria are grown at 37 °C
Our data showed that (i) σB contributes to Caco-2 invasion for L. monocytogenes grown at both 30°C and 37°C and (ii) σB positively regulates inlA transcription at both of these temperatures. The observation that σB contributes to Caco-2 invasion in bacteria grown at both temperatures is consistent with a number of studies that have shown that σB is critical for invasion of intestinal cells in vitro (Kim et al., 2004; Garner et al., 2006b) and in a guinea pig model of listeriosis (Garner et al., 2006b). While McGann et al. (2007a) found higher transcript levels in L. monocytogenes grown at 30°C than bacteria grown at 37°C, for certain σB-dependent internalin genes (i.e., inlC2, inlD, lmo331, and lmo0610) and opuCA, which has been shown to be regulated by both σA and σB (Cetin et al., 2004; Chan et al., 2007a), the same study also found that inlA transcript levels were similar in L. monocytogenes grown at 30°C and 37°C (McGann et al., 2007a). Another study also reported that genes found to be σB dependent in the intestine were generally not differentially expressed at 30°C and 37°C (Toledo-Arana et al., 2009). Overall, these data suggest that σB activity and σB-dependent regulation of invasion is similar in L. monocytogenes grown at 30°C and 37°C, even though some σB-dependent genes (e.g., opuCA and inlC2D) may be regulated by additional temperature-dependent mechanisms (McGann et al., 2007a; Chan et al., 2007a). We did find though that σB-dependent negative regulation of flaA transcript levels, which was previously described by two studies (Raengpradub et al., 2008; Toledo-Arana et al., 2009), was only apparent here in bacteria grown at 30°C, even though this regulation has previously been reported in bacteria exposed to salt stress at 37°C (Raengpradub et al., 2008). Temperature effects on σB-dependent regulation of some genes may thus be dependent on other environmental conditions (e.g., osmotic stress).
Contributions of PrfA to Caco-2 cell invasion in L. monocytogenes grown at 37°C, but not in bacteria grown at 30°C, are consistent with previous findings of low PrfA activity in L. monocytogenes grown at temperatures < 37°C (Leimeister-Wachter et al., 1992) as well as data showing that PrfA-dependent inlA expression is significantly higher at 37°C than at 25°C (Dramsi et al., 1993). Interestingly, in our study here, prfA transcript levels and PrfA activity (as measured by plcA transcription levels) were not significantly different in the parent strain between 30°C and 37°C growth conditions; this observation may reflect low baseline PrfA activity in L. monocytogenes grown at 37°C in BHI (i.e., the conditions used here) as low levels of easily catabolized sugar and/or presence of other compounds (e.g., charcoal) seem to be required to induce PrfA activity at 37°C (Ripio et al., 1996; Milenbachs et al., 1997; Gilbreth et al., 2004). While PrfA activity and PrfA-dependent phenotypes are thus clearly temperature dependent, with maximum PrfA activity in bacteria grown under certain conditions at 37°C, it is increasingly clear that transcriptional patterns and phenotypic characteristics of L. monocytogenes are governed by complex, environmental-condition-dependent interactions between multiple regulators.
Synergisms between PrfA and σB were confirmed here through formal statistical analyses that showed (i) a statistically significant interaction effect between sigB and prfA deletions on Caco-2 invasion in bacteria grown at 37°C, but not in bacteria grown at 30°C and (ii) a borderline significant interaction effect between sigB and prfA deletions on inlA transcript levels in bacteria grown at 37°C, but not at 30°C. While this type of temperature-dependent synergism has not previously been described, contributions of both σB and PrfA to Caco-2 cell invasion and co-regulation of L. monocytogenes virulence genes, including inlA, have been reported previously (Lingnau et al., 1995; Kazmierczak et al., 2003; Sue et al., 2004; Kim et al., 2005; McGann et al., 2007b). The observation that flaA also appears to be downregulated by σB, possibly with an antisense RNA type mechanism (Toledo-Arana et al., 2009), further supports a temperature-dependent regulatory network involving PrfA and σB that affects multiple effector proteins contributing to L. monocytogenes invasion and virulence, even though possible (temperature-dependent) contributions of PrfA itself to transcription of flaA and other motility regulated genes (Michel et al., 1998; Milohanic et al., 2003) will require further confirmation. For example, while others (Ripio et al., 1997) previously reported (based on visual examination of an RNA slot blot) negative regulation of flaA transcription by PrfA* (i.e., a PrfA protein that is constitutively active) in L. monocytogenes grown at 20°C, we did not find any effect of the prfA deletion on flaA transcript levels.
Conclusions
Overall, our data show that L. monocytogenes use a number of regulatory mechanisms to modulate virulence gene expression, particularly expression of genes important for invasion, under different temperatures. Specifically, modulation of gene expression in L. monocytogenes grown at temperatures < 37°C (mimicking environmental conditions before host infection) appears to increase the invasiveness of this pathogen, priming it for subsequent infection of a mammalian host. Importantly, our data also suggest an initial specific model for regulation of key invasion-associated genes at transition of L. monocytogenes from environment to host. During growth at temperatures below mammalian body temperatures, CheY and DegU activity induce a motility phenotype, and although PrfA-dependent virulence gene expression is minimal, InlA expression is assured by σB-dependent inlA transcription. After introduction into the host environment, passage through the gastrointestinal system can further activate σB (e.g., through acid and osmotic stress), priming the cell for intestinal cell invasion through increased inlA transcription (Sue et al., 2004). Once L. monocytogenes enter the intracellular environments, PrfA-dependent gene expression becomes critical for intracellular survival and spread (Freitag et al., 1993) with σB taking on a modulating role by downregulating expression of genes encoding cytolysins, which may cause excessive host cell damage (Ollinger et al., 2008). Concurrently, motility appears to be downregulated in L. monocytogenes grown at 37°C, including through σB-dependent mechanisms (Raengpradub et al., 2008; Toledo-Arana et al., 2009), possibly facilitating evasion of Toll-like-receptor-mediated host responses (Hayashi et al., 2001; Torres et al., 2004).
Supplementary Material
Acknowledgments
We would like to acknowledge Dr. Hélène Marquis for donation of the ΔflaA and ΔinlAΔflaA strains, and thank Sara Milillo, Courtney Lucas Stelling, Karlyn Beer, and Wan-Lin Su for help with plasmid construction, as well as the members of the Wiedmann and Boor laboratories at Cornell University for their support and suggestions. This work was funded by USDA Special Research Grants 2003-34459-12999 and 2004-34459-14296 (to M.W.) and NIH-NIAID (R01 AI052151 to K.J.B.). Reid A. Ivy was supported by the USDA-CSREES Food and Agricultural Sciences National Needs Graduate Fellowship Grants Program (2005-38420-15776).
Footnotes
Disclosure Statement
No competing financial interests exist.
References
- Andersen JB, Roldgaard B, Christensen B, Licht T. Oxygen restriction increases the infective potential of Listeria monocytogenes in vitro in Caco-2 cells and in vivo in guinea pigs. BMC Microbiol. 2007;7:55. doi: 10.1186/1471-2180-7-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autret N, Raynaud C, Dubail I, Berche P, Charbit A. Identification of the agr locus of Listeria monocytogenes: role in bacterial virulence. Infect Immun. 2003;71:4463–4471. doi: 10.1128/IAI.71.8.4463-4471.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakardjiev AI, Stacy BA, Fisher SJ, Portnoy DA. Listeriosis in the pregnant guinea pig: a model of vertical transmission. Infect Immun. 2004;72:489–497. doi: 10.1128/IAI.72.1.489-497.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker LA, Evans SN, Hutkins RW, Benson AK. Role of Sigma B in adaptation of Listeria monocytogenes to growth at low temperature. J Bacteriol. 2000;182:7083–7087. doi: 10.1128/jb.182.24.7083-7087.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennink R, Peeters M, Van den Maegdenbergh V, Geypens B, Rutgeerts P, De Roo M, Mortelmans L. Evaluation of small-bowel transit for solid and liquid test meal in healthy men and women. Eur J Nucl Med Mol Imaging. 1999;26:1560–1566. doi: 10.1007/s002590050495. [DOI] [PubMed] [Google Scholar]
- Bishop DK, Hinrichs DJ. Adoptive transfer of immunity to Listeria monocytogenes. The influence of in vitro stimulation on lymphocyte subset requirements. J Immunol. 1987;139:2005–2009. [PubMed] [Google Scholar]
- Brondsted L, Kallipolitis BH, Ingmer H, Knochel S. kdpE and a putative RsbQ homologue contribute to growth of Listeria monocytogenes at high osmolarity and low temperature. FEMS Microbiol Lett. 2003;219:233–239. doi: 10.1016/S0378-1097(03)00052-1. [DOI] [PubMed] [Google Scholar]
- Camilli A, Tilney LG, Portnoy DA. Dual roles of plcA in Listeria monocytogenes pathogenesis. Mol Microbiol. 1993;8:143–157. doi: 10.1111/j.1365-2958.1993.tb01211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cetin MS, Zhang C, Hutkins RW, Benson AK. Regulation of transcription of compatible solute transporters by the general stress sigma factor, Sigma B, in Listeria monocytogenes. J Bacteriol. 2004;186:794–802. doi: 10.1128/JB.186.3.794-802.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YC, Boor KJ, Wiedmann M. Sigma B-dependent and -independent mechanisms contribute to transcription of Listeria monocytogenes cold stress genes during cold shock and cold growth. Appl Environ Microbiol. 2007a;73:6019–6029. doi: 10.1128/AEM.00714-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YC, Raengpradub S, Boor KJ, Wiedmann M. Microarray-based characterization of the Listeria monocytogenes cold regulon in log- and stationary-phase cells. Appl Environ Microbiol. 2007b;73:6484–6498. doi: 10.1128/AEM.00897-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturongakul S, Boor KJ. Sigma B activation under environmental and energy stress conditions in Listeria monocytogenes. Appl Environ Microbiol. 2006;72:5197–5203. doi: 10.1128/AEM.03058-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng LW, Portnoy DA. Drosophila S2 cells: an alternative infection model for Listeria monocytogenes. Cell Microbiol. 2003;5:875–885. doi: 10.1046/j.1462-5822.2003.00327.x. [DOI] [PubMed] [Google Scholar]
- Cotter PD, Emerson N, Gahan CGM, Hill C. Identification and disruption of lisRK, a genetic locus encoding a two-component signal transduction system involved in stress tolerance and virulence in Listeria monocytogenes. J Bacteriol. 1999;181:6840–6843. doi: 10.1128/jb.181.21.6840-6843.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Bonaventura G, Piccolomini R, Paludi D, D’Orio V, Vergara A, Conter M, Ianieri A. Influence of temperature on biofilm formation by Listeria monocytogenes on various food-contact surfaces: relationship with motility and cell surface hydrophobicity. J Appl Microbiol. 2008;104:1552–1561. doi: 10.1111/j.1365-2672.2007.03688.x. [DOI] [PubMed] [Google Scholar]
- Dons L, Eriksson E, Jin Y, Rottenberg ME, Kristensson K, Larsen CN, Bresciani J, Olsen JE. Role of flagellin and the two-component CheA/CheY system of Listeria monocytogenes in host cell invasion and virulence. Infect Immun. 2004;72:3237–3244. doi: 10.1128/IAI.72.6.3237-3244.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dramsi S, Kocks C, Forestier C, Cossart P. Internalin-mediated invasion of epithelial cells by Listeria monocytogenes is regulated by the bacterial growth state, temperature and the pleiotropic activator PrfA. Mol Microbiol. 1993;9:931–941. doi: 10.1111/j.1365-2958.1993.tb01223.x. [DOI] [PubMed] [Google Scholar]
- Freitag NE, Rong L, Portnoy DA. Regulation of the PrfA transcriptional activator of Listeria monocytogenes: multiple promoter elements contribute to intracellular growth and cell-to-cell spread. Infect Immun. 1993;61:2537–2544. doi: 10.1128/iai.61.6.2537-2544.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner MR, James KE, Callahan MC, Wiedmann M, Boor KJ. Exposure to salt and organic acids increases the ability of Listeria monocytogenes to invade Caco-2 cells but decreases its ability to survive gastric stress. Appl Environ Microbiol. 2006a;72:5384–5395. doi: 10.1128/AEM.00764-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner MR, Njaa BL, Wiedmann M, Boor KJ. Sigma B contributes to Listeria monocytogenes gastrointestinal infection but not to systemic spread in the guinea pig infection model. Infect Immun. 2006b;74:876–886. doi: 10.1128/IAI.74.2.876-886.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbreth SE, Benson AK, Hutkins RW. Catabolite repression and virulence gene expression in Listeria monocytogenes. Curr Microbiol. 2004;49:95–98. doi: 10.1007/s00284-004-4204-z. [DOI] [PubMed] [Google Scholar]
- Grundling A, Burrack LS, Bouwer HGA, Higgins DE. From the cover: Listeria monocytogenes regulates flagellar motility gene expression through MogR, a transcriptional repressor required for virulence. PNAS. 2004;101:12318–12323. doi: 10.1073/pnas.0404924101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410:1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- Isberg RR, Swain A, Falkow S. Analysis of expression and thermoregulation of the Yersinia pseudotuberculosis inv gene with hybrid proteins. Infect Immun. 1988;56:2133–2138. doi: 10.1128/iai.56.8.2133-2138.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson J, Mandin P, Renzoni A, Chiaruttini C, Springer M, Cossart P. An RNA thermosensor controls expression of virulence genes in Listeria monocytogenes. Cell. 2002;110:551–561. doi: 10.1016/s0092-8674(02)00905-4. [DOI] [PubMed] [Google Scholar]
- Kallipolitis BH, Ingmer H. Listeria monocytogenes response regulators important for stress tolerance and pathogenesis. FEMS Microbiol Lett. 2001;204:111–115. doi: 10.1111/j.1574-6968.2001.tb10872.x. [DOI] [PubMed] [Google Scholar]
- Kallipolitis BH, Ingmer H, Gahan CG, Hill C, Sogaard-Andersen L. CesRK, a two-component signal transduction system in Listeria monocytogenes, responds to the presence of cell wall-acting antibiotics and affects {beta}-lactam resistance. Antimicrob Agents Chemother. 2003;47:3421–3429. doi: 10.1128/AAC.47.11.3421-3429.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmierczak MJ, Mithoe SC, Boor KJ, Wiedmann M. Listeria monocytogenes Sigma B regulates stress response and virulence functions. J Bacteriol. 2003;185:5722–5734. doi: 10.1128/JB.185.19.5722-5734.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmierczak MJ, Wiedmann M, Boor KJ. Contributions of Listeria monocytogenes Sigma B and PrfA to expression of virulence and stress response genes during extra- and intracellular growth. Microbiology. 2006;152:1827–1838. doi: 10.1099/mic.0.28758-0. [DOI] [PubMed] [Google Scholar]
- Kim H, Boor KJ, Marquis H. Listeria monocytogenes Sigma B contributes to invasion of human intestinal epithelial cells. Infect Immun. 2004;72:7374–7378. doi: 10.1128/IAI.72.12.7374-7378.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Marquis H, Boor KJ. Sigma B contributes to Listeria monocytogenes invasion by controlling expression of inlA and inlB. Microbiology. 2005;151:3215–3222. doi: 10.1099/mic.0.28070-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen GM, Olsen JE, Dons L. Characterization of DegU, a response regulator in Listeria monocytogenes, involved in regulation of motility and contributes to virulence. FEMS Microbiol Lett. 2004;240:171–179. doi: 10.1016/j.femsle.2004.09.039. [DOI] [PubMed] [Google Scholar]
- Konkel ME, Tilly K. Temperature-regulated expression of bacterial virulence genes. Microb Infect. 2000;2:157–166. doi: 10.1016/s1286-4579(00)00272-0. [DOI] [PubMed] [Google Scholar]
- Larsen MH, Kallipolitis BH, Christiansen JK, Olsen JE, Ingmer H. The response regulator ResD modulates virulence gene expression in response to carbohydrates in Listeria monocytogenes. Mol Microbiol. 2006;61:1622–1635. doi: 10.1111/j.1365-2958.2006.05328.x. [DOI] [PubMed] [Google Scholar]
- Lecuit M, Dramsi S, Gottardi C, Fedor-Chaiken M, Gumbiner B, Cossart P. A single amino acid in E-cadherin responsible for host specificity towards the human pathogen Listeria monocytogenes. EMBO J. 1999;18:3956–3963. doi: 10.1093/emboj/18.14.3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leimeister-Wachter M, Domann E, Chakraborty T. The expression of virulence genes in Listeria monocytogenes is thermoregulated. J Bacteriol. 1992;174:947–952. doi: 10.1128/jb.174.3.947-952.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingnau A, Domann E, Hudel M, Bock M, Nichterlein T, Wehland J, Chakraborty T. Expression of the Listeria monocytogenes EGD inlA and inlB genes, whose products mediate bacterial entry into tissue culture cell lines, by PrfA-dependent and -independent mechanisms. Infect Immun. 1995;63:3896–3903. doi: 10.1128/iai.63.10.3896-3903.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Graham JE, Bigelow L, Morse PD, II, Wilkinson BJ. Identification of Listeria monocytogenes genes expressed in response to growth at low temperature. Appl Environ Microbiol. 2002;68:1697–1705. doi: 10.1128/AEM.68.4.1697-1705.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandin P, Fsihi H, Dussurget O, Vergassola M, Milohanic E, Toledo-Arana A, Lasa I, Johansson J, Cossart P. VirR, a response regulator critical for Listeria monocytogenes virulence. Mol Microbiol. 2005;57:1367–1380. doi: 10.1111/j.1365-2958.2005.04776.x. [DOI] [PubMed] [Google Scholar]
- Mauder N, Williams T, Fritsch F, Kuhn M, Beier D. Response regulator DegU of Listeria monocytogenes controls temperature-responsive flagellar gene expression in its un-phosphorylated state. J Bacteriol. 2008;190:4777–4781. doi: 10.1128/JB.00258-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurelli AT. Temperature regulation of virulence genes in pathogenic bacteria: a general strategy for human pathogens? Microb Pathog. 1989;7:1–10. doi: 10.1016/0882-4010(89)90106-x. [DOI] [PubMed] [Google Scholar]
- McGann P, Ivanek R, Wiedmann M, Boor KJ. Temperature-dependent expression of Listeria monocytogenes internalin and internalin-like genes suggests functional diversity of these proteins among the Listeriae. Appl Environ Microbiol. 2007a;73:2806–2814. doi: 10.1128/AEM.02923-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGann P, Raengpradub S, Ivanek R, Wiedmann M, Boor KJ. Differential regulation of Listeria monocytogenes internalin and internalin-like genes by Sigma B and PrfA as revealed by subgenomic microarray analyses. Foodborne Pathog Dis. 2008;5:417–435. doi: 10.1089/fpd.2008.0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGann P, Wiedmann M, Boor KJ. The alternative sigma factor Sigma B and the virulence gene regulator PrfA both regulate transcription of Listeria monocytogenes internalins. Appl Environ Microbiol. 2007b;73:2919–2930. doi: 10.1128/AEM.02664-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead PS, Slutsker L, Dietz V, McCaig LF, Bresee JS, Shapiro C, Griffin PM, Tauxe RV. Food-related illness and death in the United States. Emerg Infect Dis. 1999;5:607–625. doi: 10.3201/eid0505.990502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel E, Mengaud J, Galsworthy S, Cossart P. Characterization of a large motility gene cluster containing the cheR, motAB genes of Listeria monocytogenes and evidence that PrfA downregulates motility genes. FEMS Microbiol Lett. 1998;169:341–347. doi: 10.1111/j.1574-6968.1998.tb13338.x. [DOI] [PubMed] [Google Scholar]
- Milenbachs AA, Brown DP, Moors M, Youngman P. Carbon-source regulation of virulence gene expression in Listeria monocytogenes. Mol Microbiol. 1997;23:1075–1085. doi: 10.1046/j.1365-2958.1997.2711634.x. [DOI] [PubMed] [Google Scholar]
- Milohanic E, Glaser P, Coppee J-Y, Frangeul L, Vega Y, Vazquez-Boland JA, Kunst F, Cossart P, Buchrieser C. Transcriptome analysis of Listeria monocytogenes identifies three groups of genes differently regulated by PrfA. Mol Microbiol. 2003;47:1613–1625. doi: 10.1046/j.1365-2958.2003.03413.x. [DOI] [PubMed] [Google Scholar]
- Ollinger J, Wiedmann M, Boor KJ. SigB- and PrfA-dependent transcription of genes previously classified as putative constituents of the Listeria monocytogenes PrfA Regulon. Foodborne Pathog Dis. 2008;5:281–293. doi: 10.1089/fpd.2008.0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neil HS, Marquis H. Listeria monocytogenes flagella are used for motility, not as adhesins, to increase host cell invasion. Infect Immun. 2006;74:6675–6681. doi: 10.1128/IAI.00886-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal A, Labuza TP, Diez-Gonzalez F. Comparison of primary predictive models to study the growth of Listeria monocytogenes at low temperatures in liquid cultures and selection of fastest growing ribotypes in meat and turkey product slurries. Food Microbiol. 2008;25:460–470. doi: 10.1016/j.fm.2008.01.009. [DOI] [PubMed] [Google Scholar]
- Raengpradub S, Wiedmann M, Boor KJ. Comparative analysis of the Sigma B-dependent stress responses in Listeria monocytogenes and Listeria innocua strains exposed to selected stress conditions. Appl Environ Microbiol. 2008;74:158–171. doi: 10.1128/AEM.00951-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffelsbauer D, Bubert A, Engelbrecht F, Scheinpflug J, Simm A, Hess J, Kaufmann SH, Goebel W. The gene cluster in-lC2DE of Listeria monocytogenes contains additional new internalin genes and is important for virulence in mice. Mol Gen Genet. 1998;260:144–158. doi: 10.1007/s004380050880. [DOI] [PubMed] [Google Scholar]
- Ripio MT, Brehm K, Lara M, Suarez M, Vazquez-Boland JA. Glucose-1-phosphate utilization by Listeria monocytogenes is PrfA dependent and coordinately expressed with virulence factors. J Bacteriol. 1997;179:7174–7180. doi: 10.1128/jb.179.22.7174-7180.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripio MT, DomÌnguez-Bernal G, Suárez M, Brehm K, Berche P, Vázquez-Boland JA. Transcriptional activation of virulence genes in wild-type strains of Listeria monocytogenes in response to a change in the extracellular medium composition. Res Microbiol. 1996;147:371–384. doi: 10.1016/0923-2508(96)84712-7. [DOI] [PubMed] [Google Scholar]
- Scortti M, Monzo HJ, Lacharme-Lora L, Lewis DA, Vazquez-Boland JA. The PrfA virulence regulon. Microb Infect. 2007;9:1196–1207. doi: 10.1016/j.micinf.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Shen A, Higgins DE. The MogR transcriptional repressor regulates nonhierarchal expression of flagellar motility genes and virulence in Listeria monocytogenes. PLoS Pathog. 2006;2:0283–0295. doi: 10.1371/journal.ppat.0020030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shun Z, Silverberg A, Chang C, Ouyang P. Dunnett’s many-to-one test and least square means. J Biopharm Stat. 2003;13:17–28. doi: 10.1081/BIP-120017723. [DOI] [PubMed] [Google Scholar]
- Sleator RD, Hill C. A novel role for the LisRK two-component regulatory system in listerial osmotolerance. Clin Microbiol Infect. 2005;11:599–601. doi: 10.1111/j.1469-0691.2005.01176.x. [DOI] [PubMed] [Google Scholar]
- Smith K, Youngman P. Use of a new integrational vector to investigate compartment-specific expression of the Bacillus subtilis spollM gene. Biochimie. 1992;74:705–711. doi: 10.1016/0300-9084(92)90143-3. [DOI] [PubMed] [Google Scholar]
- Sue D, Boor KJ, Wiedmann M. Sigma B-dependent expression patterns of compatible solute transporter genes opuCA and lmo1421 and the conjugated bile salt hydrolase gene bsh in Listeria monocytogenes. Microbiology. 2003;149:3247–3256. doi: 10.1099/mic.0.26526-0. [DOI] [PubMed] [Google Scholar]
- Sue D, Fink D, Wiedmann M, Boor KJ. Sigma B-dependent gene induction and expression in Listeria monocytogenes during osmotic and acid stress conditions simulating the intestinal environment. Microbiology. 2004;150:3843–3855. doi: 10.1099/mic.0.27257-0. [DOI] [PubMed] [Google Scholar]
- Toledo-Arana A, Dussurget O, Nikitas G, Sesto N, Guet-Revillet H, Balestrino D, Loh E, Gripenland J, Tiensuu T, Vaitkevicius K, Barthelemy M, Vergassola M, Nahori M-A, Soubigou G, Regnault B, Coppee J-Y, Lecuit M, Johansson J, Cossart P. The Listeria transcriptional landscape from saprophytism to virulence. Nature. 2009;459:950–956. doi: 10.1038/nature08080. [DOI] [PubMed] [Google Scholar]
- Torres D, Barrier M, Bihl F, Quesniaux VJF, Maillet I, Akira S, Ryffel B, Erard F. Toll-like Receptor 2 is required for optimal control of Listeria monocytogenes infection. Infect Immun. 2004;72:2131–2139. doi: 10.1128/IAI.72.4.2131-2139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veen S, Hain T, Wouters JA, Hossain H, de Vos WM, Abee T, Chakraborty T, Wells-Bennik MHJ. The heat-shock response of Listeria monocytogenes comprises genes involved in heat shock, cell division, cell wall synthesis, and the SOS response. Microbiology. 2007;153:3593–3607. doi: 10.1099/mic.0.2007/006361-0. [DOI] [PubMed] [Google Scholar]
- Way SS, Thompson LJ, Lopes JE, Hajjar AM, Kollmann TR, Freitag NE, Wilson CB. Characterization of flagellin expression and its role in Listeria monocytogenes infection and immunity. Cell Microbiol. 2004;6:235–242. doi: 10.1046/j.1462-5822.2004.00360.x. [DOI] [PubMed] [Google Scholar]
- Wiedmann M, Arvik TJ, Hurley RJ, Boor KJ. General stress transcription factor Sigma B and its role in acid tolerance and virulence of Listeria monocytogenes. J Bacteriol. 1998;180:3650–3656. doi: 10.1128/jb.180.14.3650-3656.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams T, Bauer S, Beier D, Kuhn M. Construction and characterization of Listeria monocytogenes mutants with in-frame deletions in the response regulator genes identified in the genome sequence. Infect Immun. 2005a;73:3152–3159. doi: 10.1128/IAI.73.5.3152-3159.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams T, Joseph B, Beier D, Goebel W, Kuhn M. Response regulator DegU of Listeria monocytogenes regulates the expression of flagella-specific genes. FEMS Microbiol Lett. 2005b;252:287–298. doi: 10.1016/j.femsle.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Young GM, Badger JL, Miller VL. Motility is required to initiate host cell invasion by Yersinia enterocolitica. Infect Immun. 2000;68:4323–4326. doi: 10.1128/iai.68.7.4323-4326.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.