

Abstract
Poliovirus 3AB protein is the first picornavirus protein demonstrated to have nucleic acid chaperone activity. Further characterization of 3AB demonstrates that the C-terminal 22 amino acids (3B region (also referred to as VPg), amino acid 88–109) of the protein is required for chaperone activity, as mutations in this region abrogate nucleic acid binding and chaperone function. Protein 3B alone has no chaperone activity as determined by established assays that include the ability to stimulate nucleic acid hybridization in a primer-template annealing assay, helix-destabilization in a nucleic acid unwinding assay or aggregation of nucleic acids. In contrast, the putative 3AB C-terminal cytoplasmic domain (C terminal amino acids 81–109, 3B + the last 7 C-terminal amino acids of 3A, termed 3B+7 in this report) possesses strong activity in these assays, albeit at much higher concentrations than 3AB. The characteristics of several mutations in 3B+7 are described here, as well as a model proposing that 3B+7 is the site of the “intrinsic” chaperone activity of 3AB while the 3A N-terminal region (amino acids 1–58) and/or membrane anchor domain (amino acids 59–80) serve to increase the effective concentration of the 3B+7 region leading to the potent chaperone activity of 3AB.
Keywords: nucleic acid chaperone, 3AB, poliovirus, virus replication, picornavirus
Viral Chaperone Proteins and the Role of 3AB in Poliovirus Replication
Nucleic acid chaperones are proteins that facilitate RNA rearrangements leading to thermodynamically more stable and biologically active conformations. They possess helix destabilizing (unwinding) and aggregation activity.1–4 The list of chaperone proteins among viruses has been growing rapidly and includes several retroviral proteins (nucleocapsid protein (NC),3 Viral infectivity factor (Vif),5 Transactivator of transcription (Tat)6 and Gag7), hepatitis C virus core protein,8,9 coronavirus nucleocapsid protein,10 hantavirus N protein,11 hepatitis delta virus D antigen12 and finally, poliovirus 3AB,13 which is the subject of this report. Exactly how chaperones function to enhance virus replication in vivo remains unclear. Development of in vivo/ in situ assays is complicated by the ubiquitous nature of viral chaperones, all of which appear to have multiple functions at various steps in the virus life-cycle. Still, several functions consistent with activities observed in vitro have been postulated and include among others, facilitating the folding of genomic RNA to biologically functional conformations, promoting the transition between genome replication, mRNA synthesis and packaging through a structural switch and enhancing recombination by accelerating binding of complementary nucleic acids (see below).
Protein 3AB is an integral part of the viral replication machinery. It anchors replication complexes to internal cellular membranes, interacts with several proteins required for genome replication and stimulates the viral polymerase (3Dpol) in vitro (see below). Details of the mechanism by which 3AB accomplishes these functions remain unclear. Protein 3AB is derived from the P3 genome region of poliovirus, which also codes for the viral polymerase (3D, referred to as 3Dpol) and viral proteases (3C, referred to as 3Cpro).14 The 84 kDa P3 precursor is first cleaved to produce two relatively stable intermediates, 3AB and 3CD (denoted 3CDpro), the latter possessing protease activity. Ultimately four proteins, 3A, 3B, 3Cpro and 3Dpol can be produced from the precursor. These individual proteins, as well as 3AB and 3CDpro play important roles in the synthesis of viral nucleic acids.
The ~12 kDa and 109 aa 3AB protein consists of two domains (see Fig. 2A). The N-terminal domain (3A) is ~10 kDa and the C-terminal domain (3B/VPg) is ~2 kDa. Evidence suggests that 3A, which contains a 22 amino acid membrane anchor domain, attaches 3AB to vesicles produced during viral infection that are the site of viral RNA synthesis.15 Protein 3AB also associates with itself, 3CDpro, 3Dpol and proteins from the P2 genome region.16–22 Although 3AB normally associates non-specifically with RNA,22,23 binding to 3CD produces a complex that associates strongly with the 5′ cloverleaf structure of the viral genome (Fig. 1) in vitro and may be involved with anchoring the genome to the vesicles.23–25
Figure 2.

(A–D) Depiction of 3AB sequence and results from FRET unwinding assay. (A) Amino acid sequence of poliovirus 3AB. Regions: 3A, 1–87: 3B (VPg), 88–109 (boxed); C-terminal cytoplasmic domain, 81–109 (referred to as 3B+7 in this proposal); membrane anchor domain, amino acids 59–80 (in bold italics). (B) Substrates for FRET unwinding assay. Complementary stem-loop forming oligos with 5′ fluorescing (FAM) or quenching DABCYL groups are shown. Hybridization leads to quenching of FAM fluorescence. Structures and ΔG values of −7.2 kcal/mol are as predicted from DNA fold.71 (C) FRET unwinding time course assay with wild type 3AB, 3AB-R104E, 3AB-Y90A, 3AB-R104E/Y90A, VPg (3B) and HIV NC, all at 2 μM. (D) FRET unwinding time course assay with wild type 3AB (2 μM) or 2, 8 and 100 μM 3B+7. Representative results are shown for all experiments and assays were repeated at least once and typically several times.
Figure 1.

Possible roles for chaperone activity in poliovirus/picornavirus replication. A schematic representation of the poliovirus genome along with important secondary structures in the non-translated regions (NTRs) is shown (from De Jesus NH, Virol J 2007; 4:70). Possible roles of 3AB or other chaperones are noted in boxed areas. See text for details.
Protein 3B is covalently attached to all newly synthesized viral plus and minus strands.26–29 This protein serves as the “primer” for RNA synthesis by 3Dpol. Uridylylation of 3B is required for the protein to prime RNA synthesis. Two uridine residues are covalently attached to 3B by 3Dpol. The template for the addition is a cis-acting replication element (CRE) located near the center of the genome.30,31 Association of 3B (or precursors containing 3B) and 3Dpol in this region results in template-directed addition of U residues to a tyrosine residue on 3B.32
Mutational analysis using purified proteins and yeast two-hybrid systems indicates that the 3AB-3Dpol complex is tethered mostly by interactions with amino acids in 3B as several mutations in this domain decrease binding.16,20,21,33 Both yeast two-hybrid and in vitro binding assays demonstrate that 3AB binds tightly to itself forming strongly associated dimers and more complex multimers.17,21,34 Dimerization maps to the 3A portion of the protein, notably, the hydrophobic membrane insertion domain (amino acids 59–80). However, this domain may not be required for dimerization as others have shown that when expressed by itself, the 1–58 region of 3A forms dimers.34 The ability of the 3AB to dimerize/multimerize could be pivotal to chaperone activity, especially in aggregation of nucleic acids.
In vitro, 3AB strongly stimulates the synthetic activity of 3Dpol, especially when low concentrations of the polymerase are used.17,23,35–37 The mechanism of stimulation has not been fully clarified but several possibilities have been put forth, including: stabilizing 3Dpol binding to primer-template and decreasing the dissociation rate constant (koff);37 stabilizing the enzymatic activity of 3Dpol; and enhancing primer binding to the template rather than direct 3Dpol stimulation.36 Whether the chaperone activity of 3AB plays a role in stimulation remains to be determined.
Potential Role of 3AB’s Chaperone Activity in Replication
As described above, 3AB and its cleavage product 3B are putatively involved in several steps related to replication. The mechanistic details for many of these processes remain to be determined. The chaperone activity of 3AB may be important to several of these functions (see Fig. 1). For example, helix destabilizing activity could help unfold the genome to allow access for 3Dpol and other host proteins and a general chaperone activity could aid proper folding of the highly structured genome. Complex RNA structures at the 5′, 3′ and central regions of the genome are critical for genome replication (most notably the 5′ cloverleaf structure,25,38–40 and CRE in the 2C central region of the genome (see above)) and translation (the internal ribosome entry site (IRES) in the 5′ leader region41,42). Chaperones could also be involved in promoting “structural switches” that lead to plus strands being shuffled into different pathways like translation, minus sense RNA synthesis or genome packaging.
In addition, 3AB may also be involved in recombination which occurs with high frequency in polio and other plus sense RNA viruses.43–55 Copy-choice type recombination occurs by a mechanism that involves transfer (often referred to as “strand transfer” or “template-switching”) of an RNA being synthesized on one viral genome (generally during synthesis of the minus strand in polio50) to another where synthesis continues. Chaperone proteins like HIV-NC can stimulate copy-choice recombination in reconstituted in vitro systems through accelerating binding of complementary nucleic acids.56–65
Discovery of 3AB as a Chaperone Protein and Summary of Recent Advances
In an earlier report, we showed that bacterially expressed purified 3AB protein possessed helix destabilizing activity and was able to promote the hybridization of complementary nucleic acids in the absence of nucleotides (as an energy source).13 Enhancement of annealing occurred for both RNA and DNA and even highly structured nucleic acids. Hybrid formation was concentration-dependent and required enough 3AB to completely or partially coat the nucleic acid strands. Conditions for optimal activity were established and included low divalent cation (1 mM Mg2+) and salt concentrations (20 mM KCl). In the current report we show that the 3B region of 3AB is necessary but not sufficient for chaperone activity as specific mutations in this domain abolish chaperone activity while 3B alone has no chaperone activity even at very high concentrations. Interestingly, the putative C-terminal cytoplasmic domain of 3AB, consisting of 3B and the last 7 amino acids at the C-terminal end of 3A (termed 3B+7 in this report), does have chaperone activity at high concentrations. These results form the basis of the working model of 3AB chaperone function presented in this report.
Results
Assays used to examine 3AB chaperone activity
The chaperone activity of 3AB was evaluated using three different assays that measure annealing of complementary nucleic acids and aggregation. The primer-template annealing assay indirectly measures helix destabilization and aggregation activities by testing the ability of a chaperone to stimulate annealing of a primer to a larger template and causing it to gel-shift.13 Chaperone proteins presumably accelerate annealing in the assay through aggregation and partial unwinding of the large template. The aggregation assay tests the chaperone’s ability to induce aggregate formation, a common, though not exclusive property of chaperones. Incubation of chaperones with nucleic acid can result in the formation of large aggregates consisting of the chaperone and nucleic acid (which is generally labeled with radioactivity in the assay). Aggregated material can be isolated by slow speed centrifugation in a microfuge.65 The fluorescence resonance energy transfer (FRET) unwinding assay measures unwinding and annealing stimulation. The assay illustrated in Figures 2 was developed in the DeStefano lab for examining nucleic acid chaperones.13,64,66 It is rapid and produces immediate results which typically correlate well with the primer-template annealing assay. Complementary nucleic acid strands that have little structure or form stem-loops are used in the assay. One complement has a fluorescing group (FAM) at the 5′ end and the second has a quenching group (DABCYL) at the 3′ end. The thermodynamic stability of the strands can be varied by changing the G-C content of the stem.66 When the individual complements form stem-loops, they hybridize poorly at low temperature (30–37°C is typically used). In the presence of a chaperone, partial unwinding of the stem structures enhances annealing by exposing complementary regions. Annealing is measured by the quenching of the fluorescence signal that occurs when the FAM and DABCYL groups become juxtaposed upon hybrid formation. In assays using complements with highly stable stem regions (as in the current report), the major parameter tested is the ability of the chaperone to unwind the stem. Gel-shift assays were used to evaluate the affinity of the mutated and wild type proteins for nucleic acids.
Mutations in the 3B region of 3AB inhibit chaperone activity while 3B by itself shows no chaperone activity
Amino acids in the 3B region of 3AB are known to be pivotal for 3AB binding to nucleic acids and complex formation with 3Dpol. Previous results showed that the R104E (arginine to glutamate) mutation severely disrupts binding to nucleic acid.22 The effect of this mutation on chaperone activity was tested using the FRET unwinding assay and, as expected, no significant chaperone activity was detected (Fig. 2C and Table 1). A second mutation, Y90A (tyrosine to alanine), was also tested. Tyrosine 90 is the amino acid that is uridylylated by 3Dpol during replication (see Introduction). In addition, aromatic amino acids are pivotal to the chaperone activity of HIV nucleocapsid protein (NC) and are presumed important in the disruption of secondary strucutures. The Y90A mutation also decreased chaperone activity although a low level of activity was detected. A double mutant with R104E and Y90A (Y90A/ R104E) was also tested and found to be similar to R104E.
Table 1.
Results for 3AB, 3B+7, 3B, and various mutants in chaperone assays
| a Protein and concentration | b FRET assay | b,c Agg | b,d P-T Ann. | e FRET rate constant (k) |
|---|---|---|---|---|
| 3AB (2 μM) | +++ | +++ | +++ | 0.79 ± 0.21 |
| 3AB-Y90 > A (2 μM) | + | − | − | ND |
| 3AB-R104 > E (2 μM) | − | ND | − | ND |
| 3AB-Y90A/R104E (2 μM) | − | − | − | ND |
| 3B (100 μM) | − | − | − | 0.030 ± 0.001 |
| f3B+7 (100 μM) | +++ | ++ | ++ | 0.59 ± 0.07 |
| 3B+7-K81A (100 μM) | + | ND | ND | 0.13 ± 0.02 |
| 3B+7-F83A (100 μM) | + | ND | ND | 0.095 ± 0.049 |
| 3B+7-H86A (100 μM) | ++ | + | + | 0.21 ± 0.06 |
| 3B+7-Y90 > A (100 μM) | + | − | − | 0.15 ± 0.02 |
| 3B+7-R104A (100 μM) | − | ND | ND | ND |
| 3B+7-R104 > E (100 μM) | − | − | − | 0.031 ± 0.016 |
3B and 3B+7 proteins were made by chemical synthesis. All others were expressed and purified from E. coli.
+++, ~75–100%; ++, ~25–74%; +, ~5–24%; −, <5%; ND, Not Determined. All relative to 3AB wild type. See Figure 2 for FRET unwinding assay.
Aggregation, determined by formation of a nucleic acid pellet using slow speed centrifugation (see Methods).
Rate constant (k) for FRET assays was determined as described in Methods. Results are an average of two or more experiments +/− standard deviation.
In FRET assays, 2 μM 3B+7 showed little stimulation while 8 μM showed some stimulation (+).
It was clear from the above results that the 3B region of 3AB was pivotal to chaperone acitivity. To test its role further, the 22 amino acid 3B protein was chemically synthesized and tested for chaperone activity. No activity was observed, even at very high concentrations that were ~50 times greater than the concentration where 3AB showed maximal activity (Table 1). Based on these results, we concluded that the 3B region of 3AB was necessary but not sufficient for chaperone activity.
Further attempts to determine which region(s) of 3AB is required for chaperone activity using deletion mutagenesis have thus far been unsuccessful due to severe aggregate formation during protein expression. Protein 3AB is notoriously difficult to work with because of this tendency. Our lab is currently testing different expression systems to try and increase the solubility of 3AB and 3AB mutations during purification. In the meantime we have pursued an interesting finding which showed that a protein consisting of 3B plus the last 7 amino acids at the C-termiuns of 3A (termed 3B+7 in this report) possess chaperone activivity (see below).
The putative C-terminal cytoplasmic domain of 3AB possess chaperone activity at hgh concentrations
Since 3AB is a membrane protein, it is likely that the intrinsic chaperone activity resides in a portion of the protein that is cyctoplasmic allowing direct intereaction with nucleic acids. Models for 3AB interactions with membranes suggest that the both the first 58 amino acid in the N-terminus and the last 29 amino acids in the C-terminus are cytoplasmic (see Fig. 6). Since interactions with nucleic acids map mostly to the C-terminus, this was clearly the region most likely to contain intrinsic chaperone activity, although none was detected in 3B alone (see above). The last 7 amino acids of 3A are proposed to lie just outside the membrane and are connected to the membrane insertion domain. These 7 amino acids and the 22 of 3B compose the C-terminal cytoplasmic domain. Chemically synthesized 3B+7 was tested in the FRET unwinding assay at various concentrations. Although very low chaperone activity was detected at concentrations where 3AB showed high activity (2 μM), activity increased at higher concentrations (8 μM) and high activity was observed with 100 μM 3B+7 (Fig. 2D).
Figure 6.
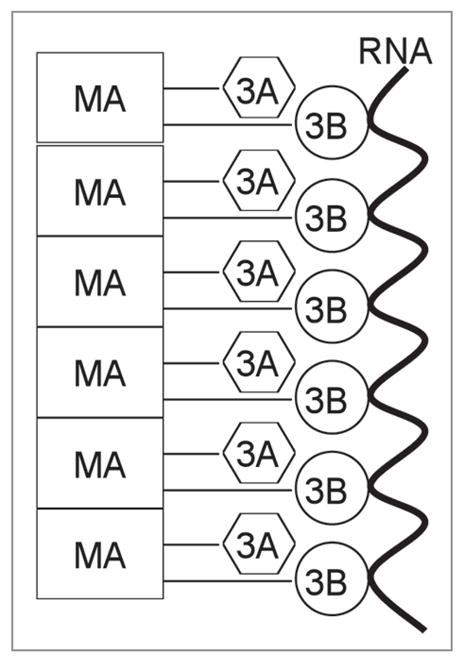
Model for in vitro 3AB chaperone activity. See text for description. MA, membrane anchor domain.
Several 3B+7 proteins containing mutations were chemically synthesized and tested in the FRET unwinding assay (Table 1). Proteins containing the same mutation as those tested in 3AB were tested first (3B+7 R104E and Y90A, as well as R104A) to see if they had a similar affect on the activity of 3B+7. Similar to what was observed with 3AB, R104E dramatically reduced the chaperone activity of 3B+7 while Y90A was inhibitory but to a lesser extent. An R104A mutation was also strongly inhibitory. Additional mutations including F83A (phenylalanine to alanine) and K81A (lysine to alanine) showed results similar to Y90A while H86A (histidine to alanine) showed greater chaperone activity than those mutants but slighlty less than wild type. From the limited number of mutations that were tested, 3B+7 appeared to mimic wild type 3AB’s behavior, albeit at the much higher concentration than was required to observe activity.
Wild type 3AB is much more potent than 3B+7 in the primer-template annealing assay
Proteins were next compared in the primer-template annealing assay using a fixed time point protein titration. Wild type 3AB stimulated annealing strongly as the concentration of the protein was increased (Fig. 3). Peak activity occurred at ~1 μM under the conditions used, similar to what was previously observed.13 In contrast, the Y90A and Y90A/R104E mutants showed no annealing activity in this assay (Table 1). Protein 3B+7 showed some annealing activity in the assay though reduced compared to wild type protein, even when very high concentrations were used (Fig. 4). The H86A mutation in 3B+7 resulted in annealing that was detectable but lower than non-mutated 3B+7, consistent with the modestly lower level of activity in the FRET unwinding assay for these proteins (Table 1). Other tested mutations in 3B+7 including Y90A and R104E and Y90A/R104E, showed no activity in the assay as did 3B (Table 1).
Figure 3.

Primer-template annealing assay with wild type 3AB. A 25 nucleotide 5′ end-labeled RNA primer that was complementary to a 230 nucleotide RNA template was used in the assay. The primer and template were incubated with decreasing amounts of wild type 3AB (l-r: 2, 1, 0.5, 0.25, 0.013, 0.0063 or 0.0031 μM). Samples were processed and run on a 6% native polyacrylamide gel as described under Methods. The positions of the primer and annealed primer-template hybrid are indicated. Three controls (A–C) are as described below the panel. Representative results are shown and the assay was repeated several times.
Figure 4.

Primer-template annealing assay with wild type 3B+7. The amount of 3B+7 protein used was: l-r, 200, 100, 50, 25, 12.5, 6.3 or 3.1 μM. See Figure 3 for details.
Wild type 3AB and 3B+7 induce aggregate formation while mutated versions of these proteins with low unwinding and primer-template annealing activity do not
Large aggregates that can form in the presence of chaperone proteins and nucleic acids can help to induce the formation of hybrids.65,67 An aggregation assay was performed with wild type 3AB, Y90A, Y90A/R104E, 3B+7 (and various mutations) and 3B. Wild type 3AB induced aggregate formation in a concentration-dependent manner (data not shown), similar to what was observed in the primer-template annealing assay. Full-length proteins were tested at 2 μM and 3B or 3B+7 at 100 μM. Of those proteins tested wild type 3AB showed the highest level of aggregation (Table 1) and was essentially equal to HIV NC, a protein known to aggregate efficiently (NC data not shown). In addtion, 3B+7 showed aggregation activity that was slighlty lower than 3AB while 3B+7-H86A showed some activity. All other tested proteins did not aggregate the RNA template.
Affinity of 3AB and mutated proteins for nucleic acid
Gel-shift assays can be used to estimate the binding affinity of 3AB for nucleic acids. Although this method does not yield an accurate equilibrium binding constant, it is useful for comparing various 3AB mutations.22 A simpler nitrocellulose filter binding assay proved unreliable in our hands as the amount of nucleic acid retained on the filters in the presence of 3AB was low and variable. Wild type 3AB was able to gel shift nucleic acid in this assay consistent with previous reports (Fig. 5, note: the major band of shifted nucleic acid runs slightly below the wells in the assay). In contrast, mutations Y90A, R104E and Y90A/R104E did not shift any nucleic acid indicating that they have lower affinity for nucleic acid (Fig. 5). The result is completely expected for R104E and Y90A/R104E as the R104E mutation was previously shown to abrogate nucleic acid binding.22 Since the Y90A mutation showed low but measurable activity in the unwinding assay it would be expected that nucleic acid binding would occur, however, it was not detected by gel shift, perhaps because binding was too weak (see Discussion). Protein 3B+7 was unable to shift nucleic acids in the gel shift assay or reliably retain nucleic acid on nitrocellulose filters (data not shown).
Figure 5.

Gel-shift assay to test protein-nucleic acid binding. Proteins (type and amount as indicated) were incubated with a 40 nucleotide 5′ end-labeled RNA and run on a 6% native polyacrylamide as described in Methods. The positions of the unshifted 40-mer and shifted material are indicated. Two controls are as described below the part. Representative results are shown and the assays were repeated twice with similar results.
Discussion
This report presents the most recent advances in the understanding of how poliovirus 3AB functions as a nucleic acid chaperone. This work has been hampered by difficulties encountered while making and expressing mutated 3AB proteins, especially truncations and deletions. Despite this, the importance of the 3B region of the protein in chaperone activity has been clearly demonstrated as mutations in this region severely weaken or abolish chaperone activity (Table 1). Thus far all of the 3B region mutations tested in the context of the full-length protein (Y90A, R104E and Y90A/R104E) showed highly reduced nucleic acid binding which explains the low chaperone acitivity. The 3B region of 3AB is known to be pivotal for nucleic acid binding so the results are not surprising.22 For single point mutations to have such a dramatic effect on nucleic acid binding it is likely that they disrupt the structure of the 3B region. Both Y90A and R104E make additional contacts with other amino acids in the 3B (VPg) solution structure and the mutations tested would likely disrupt some or all of the interactions.68,69 In fact, R104 interacts with nearly every amino acid residue in the C-terminal end of 3B in the proposed solution structure. Both R104 and Y90 lie on the same face of 3B along with several other amino residues that are conserved among picornavirues (G88, G92, K96, K97, P101 and Q109). We are currently evaluating the affect of mutations in these other conserved residues on chaperone activity.
The most interesting result from this work was the finding that the putative C-terminal cytoplasmic domain of 3AB (termed 3B+7 in this work), but not 3B by itself, possessed chaperone activity. In the FRET unwinding assay, which measures mostly the ability of a chaperone to unwind nucleic acids, 3B+7 showed activity that was approximately 50-fold lower than 3AB on a per mole basis. This was based on saturation of the rate of annealing in the FRET unwinding assay requiring ~2 μM 3AB and ~100 μM 3B+7. Still, each protein was able to stimulate unwinding to about the same maximal rate. Mutations in 3B+7 behaved similar to the same mutations in the wild type protein for the few that were examined including R104E, Y90A and Y90A/R104E, with the R104E and the double mutant showing essentially no activity in the FRET unwinding assay and Y90A, a low level of activity. This suggests that 3B+7 may be a reliable though incomplete (see below) model for examining the chaperone activity of 3AB. A R104A mutation in 3B+7 also showed very little activity indicating that the additional negative charge of R104E was not responsible for the loss of activity.
Since 3B+7 showed chaperone activity while 3B did not, it was clear that the 7 additional amino acids from the C-terminal end of 3A were pivotal. Three mutations were made in this region and tested in the context of 3B+7. Two of them, K81A and F83A, showed reduced activity similar to the Y90A mutation in 3B+7. In contrast, H86A showed more activity but still less than wild type 3B+7. The results reiterates the importance of postiviely charged and aromatic amino acid in chaperone acitivity. We are currently in the process of placing the K81A, F83A and H86A mutations in the full-length protein to determine if they have the same effect on chaperone activity.
It is also interesting that the 29 amino acids of 3B+7 comprise the entire putative C-terminal cytoplasmic domain of 3AB. This means that intrisic chaperone activity in vitro requires only that part of the C-terminal region of the protein that would be available for nucleic acid binding in vivo. The membrane anchor domain (amino acid 59–80) would presumably be embeded in membranes in vivo and this would prevent direct access to nucleic acid. The N-terminal 58 amino acids of 3A are also predicted to be cytoplasmic and could therefore be involved with direct contacts to nucleic acids. However, judging from the limited affect of mutation in this region on nucleic acid binding,22 a direct role in chaperone activity seems less likely, although an indirect role that enhances activity is possible for this region and the membrane anchor domain (see below).
At this point, it is difficult to speculate precisely what roles the 3B and +7 regions play in chaperone acitivity or how these region interact. One possible explanation is that 3B+7 simply binds nucleic acid better than 3B. While this remains possible, we were unable to detect binding in gel-shift or filter binding assays with either protein and 3B did not aggregate nucleic acids. Still, it is clear that 3B+7 does interact with nucleic acids as it would not show activity in the FRET unwinding and primer-template annealing assays if it did not. Possible explanations for the lack of binding in gel-shift assays are that 3B+7 is too small to produce a reliable shift, the binding is too weak to survive this assay or the protein simply does not work in this type of assay, as is the case for many other nucleic acid binding proteins. As for the filter binding assay, even full-length 3AB did not work well in this assay, so it was not surprising that 3B+7 did not. We are currently working on assays that use fluorescently labled nucleic acid to test binding. These assays should help in analyzing differences in binding between various mutants and wild type protein.
Another possible explanation of why both 3B and the additional 7 amino acids are required for activity is that they form a folded complex together that has intrisic chaperone activity. Although possible, this explanation is weakened by the fact that 3B by itself forms a defined structure in solution that has been solved using NMR.68,69 In addition, chemically synthesized 3B is active in uridylylation assays in which two UMP residues are added to tyrosine 90 by 3Dpol in the presence of poly(rA) or the CRE genome region, the natural template for uridylylation (see Introduction). These findings suggest 3B forms an active complex, at least for uridylylation, in the absence of the 7 amino acid from 3A and that chemically synthesized 3B folds correctly.
It was interesting that the various assays used to test chaperone activity gave different results with certain 3AB mutations. Wild type 3AB showed high activity in the FRET unwinding assay while 3AB-Y90A showed low but measurable activity. The same senario was observed with 3B+7 and 3B+7-Y90A, albeit at higher protein concentrations. In contrast, only wild type 3AB gel-shifted nucleic acid and only 3AB, 3B+7 and 3B+7-H86A aggregated RNA template. In the primer-template annealing assay wild type 3AB was most active and 3B+7 also showed modest activity and 3B+7-H86A low activity. None of the other mutated proteins, even those that showed some activity in the FRET unwinding assay, were able to anneal primer-template. This may be due to the primer-template annealing assay requiring both unwinding and aggregation activity while the FRET unwinding assay requires mostly the former. Both wild type 3AB and 3B+7 unwind and aggregate well based on the FRET unwinding and aggregation assays, respectively. This leads to greater annealing in the primer-template assay which is dependent on both activities. The only other mutated protein to show some activity in the primer-template annealing assay was 3B+7-H86A. This protein had low but measurable aggregation activity and reasonably high activity in the FRET-unwinding assay. This may have resulted in the low level of primer-template annealing that was observed. These results also point out the importance of measuring chaperone activity in more than one type of assay and using assays that focus on particular aspects of that activity (i.e., unwinding, aggregation or both).
The current results and previous results from others have allowed us to formulate a working model to explain the in vitro chaperone activity of 3AB (Fig. 6). We hypothesize that the intrinsic chaperone activity of 3AB resides in the 3B+7 region based on the chaperone activity of this protein at high concentrations (Table 1). The membrane anchor domain of the 3A region (amino acid 59–80) serves to “concentrate” the 3B+7 region by stacking on itself. This leads to an increase in the effective concentration of the 3B+7 region such that a lower concentration of the full-length protein, in comparison to 3B+7 alone, is required to obtain high chaperone activity. In Figure 6, the membrane anchor domain is shown as the major stacking agent, although the N-terminal cytoplasmic domain (amino acids 1–58, labeled in Fig. 3A) could also be involved. This region has been shown by NMR to dimerize when expressed in the absence of the rest of the protein so interactions leading to multimer formation are plausible.34 In addition to being consistent with the current results, the proposed model is also consistent with 3AB’s ability to form homomultimeric complexes.17,21 Note that no membranes are required for the proposed stacking as the assays were conducted in the absence of phospholipids. Still, cellular membranes may help catalyze stacking and this will be examined in the future using membranes in in vitro reactions. We are currently testing this model by making point mutations in the membrane anchor and N-terminal region of 3AB. Truncations and deletions will also be tested permitting that these proteins are able to be produced.
Ultimately we hope to find point mutations in 3AB that abrogate or weaken chaperone activity but have little affect on nucleic acid binding or uridylylation. These mutations can be tested in live virus to determine what steps in the replication cycle they effect. This would help to establish a clear role for chaperone activity at particular steps in viral replication, something that has been very difficult to do for most chaperones.
Materials and Methods
Materials
T3 RNA polymerase, calf intestinal phosphatase (CIP), DNase I (RNase free) and ribonucleotides, were obtained from Roche Applied Science. RNasin was obtained from Promega, restriction enzymes and T4 polynucleotide kinase from New England Biolabs and proteinase K from Stratagene. All synthetic oligonucleotides were custom-ordered from Integrated DNA technologies Inc. Chemically synthesized proteins including 3B, 3B+7 and all 3B+7 proteins with mutations were from GenScript Inc., and were of 90% purity or greater. Sephadex G-25 spin columns were from Harvard Apparatus. RNeasy mini kits were from Qiagen Inc., Radiolabeled compounds were from Perkin Elmer. All other chemicals were obtained from Sigma Chemical Co., Fisher Scientific or VWR scientific.
Methods
Preparation of recombinant proteins poliovirus 3AB and HIV-1 NC by expression in Escherichia coli
Protein 3AB of poliovirus type 1 (Mahoney strain) was expressed in E. coli using plasmid pGEX-3AB, kindly provided by Dr. Stephen Plotch (formerly of Wyeth-Ayerst Research). Expression and purification were performed as described previously.17 Purified 3AB was stored at −70°C in buffer containing 50 mM Tris-HCl (pH = 8), 1 mM DTT, 0.05% Triton X-100 and 10% glycerol (3AB buffer). HIV-1 NC protein was produced using a vector kindly provided by Dr. Charles McHenry (Univ. of Colorado). The protein was expressed in E. coli and purified as previously described.70
5′ end-labeling of oligonucleotides
Reactions for primer labeling were done in a 50 μl volume containing 70 mM Tris-HCl, pH = 7.6, 10 mM MgCl2, 5 mM DTT, 10 μl of γ-32P ATP (3,000 Ci/mmol, 10 μCi/μl) and 2 μl (20 units) of T4 poly-nucleotide kinase. The reaction mixture was incubated for 30 minutes at 37°C and then the T4 polynucleotide kinase was heat inactivated for 10 minutes at 70°C according to the manufacturer’s recommendation. The material was then passed through a Sephadex G-25 spin column to eliminate free nucleotides and exchange buffer.
Preparation of RNA templates
Run-off transcripts with T3 RNA polymerase were synthesized using the manufacturer’s protocol. Plasmid pBSM13+ was cleaved with Bgl I and T3 polymerase was used to prepare run-off transcripts ~230 nucleotides in length. After transcription for 2 hours, 15 units of DNase I (RNase-free) was added and incubation was continued for 20 min. Reactions were then processed with a RNeasy mini kit according to the manufactures’ protocol. The length and purity of the RNA was evaluated by gel electrophoresis to assure that it was full-length (data not shown). The RNA was then quantified by spectrophotometric analysis. The equation used to calculate the molecular weight was: ([A × 382.2] + [G × 344.2] + [C × 304.2] + [U × 305]). The molecular weight was used to calculate the molar concentration of RNA using the standard conversion of 1 OD260 ≈ 40 μg/ml for single stranded RNA.
Fluorescence resonance energy transfer (FRET) unwinding assay
Two complementary 42 nucleotide DNAs, one with a 5′ fluorescein-6-carboxamidohexyl (FAM) (FAM-CAT TAT CGG ATA GTG GAA CCT AGC TTC GAC TAT CGG ATA ATC-3′) group and the second with a 3′4-[[(4-dimethylamino)phenyl]-azo] benzenesulfonicamino (DABCYL) group (5′-GAT TAT CCG ATA GTC GAA GCT AGG TTC CAC TAT CCG ATA ATG-3′-DABCYL) were used in the assays. Using mfold71 and conditions of 20 mM KCl, 1 mM MgCl2and 30°C, each strand was predicted to form a stem-loop structure with a ΔG value of −7.2 kcal/mol (Fig. 2B). Annealing assays were completed at 30°C using a Cary Eclipse fluorescent spectrophotometer (Varian). FAM and DABCYL DNAs (10 and 20 nM, resp.) were separately incubated for 5 min at 30°C in the presence or absence of 3AB or other proteins (concentration as indicated) in 35 μl of buffer containing 50 mM HEPES (pH = 7), 20 mM KCl, 5 mM DTT, 1 mM MgCl2, 6 mM Tris-HCl (pH = 8), 0.014% Triton X-100 and 2.9% glycerol. The reactions were started by mixing the FAM and DABCYL samples in a quartz cuvette (final concentrations 5 and 10 nM, for FAM and DABCYL DNAs, respectively). The excitation wavelength was 494 nm with a bandwidth of 5 nm. The emission bandwidth was 10 nm and the spectrum was observed at 520 nm. The emission spectrum was taken every 15 seconds for up to 16 minutes. An intensity ratio (Ir) was determined by dividing the peak intensity at a given time (It) by the peak intensity at time zero (I0) (Ir = It/I0). This value was plotted versus time for the different concentrations of protein used. For some proteins rate constant values (k) describing the rate of annealing for the complementary strands were calculated. These values were calculated from the t1/2values by dividing 0.693 by t1/2 as described.59
Primer-template annealing assay
The 230 nucleotide RNA transcript (10 nM final concentration) (described above) and a 25 nucleotide complementary 5′ 32P-labeled RNA oligonucleotide (10 nM final concentration, 5′-CCU CUU CGC UAU UAC GCC AG-3′) that hybridized to based 155–179 from the 5′ end of the transcript were incubated in 50 mM HEPES (pH = 7), 5 mM DTT, 1.4 mM MgCl2 and 28.5 mM KCl. This material was pre-incubated for 3 mins at 30°C in 7 μl total volume. Three μl of 3AB or other derivatives (concentration as indicated) or 3AB buffer (see above) was added after the pre-incubation to start the reaction which was continued for 15 minutes. One μl of proteinase K (final concentration 1 μg/μl) in 10 mM Tris-HCl pH = 8, 50 mM EDTA pH = 8 was then added to each sample and incubation was continued for 10 minutes at 37°C. After this, 5 μl of stop mix (20% glycerol, 20 mM EDTA pH = 8, 0.2% SDS, 0.4 μg/μl tRNA, 0.1% bromophenol blue) was added. There were also two controls. One control contained the pre-incubated reaction and 3AB buffer and was heated at 65°C for 5 mins and then slow cooled. This control was used to show where the hybrid migrated on the gel. A second control contained BSA (final concentration 0.1 μg/μl) in 3AB buffer instead of 3AB. All controls were processed as above. Products were analyzed on a 6% native polyacrylamide gel as describe below. Dried gels were exposed to an imaging screen and visualized and quantified using a Fujifilm FLA-7000.
Gel-shift assay to test nucleic acid binding
A 40 nucleotide 5′ 32P-labeled RNA (2 nM final concentration, 5′-GAG UGC ACC AUA UGC CAU UCA GGC UAC GCA ACU GUU GGG A-3′) was mixed with different amounts of 3AB or other proteins (see Fig. 5) in 10 μl of a buffer containing 10 mM HEPES pH = 7, 5 mM KCl and 5 mM DTT. Two μl of 6X loading buffer (40% sucrose, 0.25% (w/v) bromophenol blue, 0.25% (w/v) xylene cyanol) was added and the samples were loaded onto a 6% native gel prepared in 0.5x Tris-borate-EDTA buffer.72 Samples were electrophoresed at 15 mA for approximately 1.5 hours and gels were processed as described above.
Polyacrylamide gel electrophoresis
Six % (w/v) native polyacrylamide (29:1 acrylamide:bisacrylamide) gels were prepared and electrophoresis was performed as described.72
Acknowledgments
We would like to thank Dr. Stephen Plotch (formerly of Wyeth-Ayerst Research) for the plasmids for 3AB, Dr. Charles McHenry (Univ. of Colarado) for the expression plasmid for HIV NC protein and Dr. Gauri Nair for critical reading of the manuscript. This work was supported by National Institutes of General Medicine grant number GM051140 and undergraduate fellowships from the Howard Hughes Medical Institute awarded to E. Eden and M. Shah.
References
- 1.Zuniga S, Sola I, Cruz JL, Enjuanes L. Role of RNA chaperones in virus replication. Virus Res. 2009;139:253–66. doi: 10.1016/j.virusres.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herschlag D. RNA chaperones and the RNA folding problem. J Biol Chem. 1995;270:20871–4. doi: 10.1074/jbc.270.36.20871. [DOI] [PubMed] [Google Scholar]
- 3.Levin JG, Guo J, Ioulia R, Musier-Forsyth K. Nucleic acid chaperone activity of HIV-1 nucleocapsid protein: critical role in reverse transcription and molecular mechanism. Prog Nucleic Acids Res Mol Biol. 2005;80:217–86. doi: 10.1016/S0079-6603(05)80006-6. [DOI] [PubMed] [Google Scholar]
- 4.Rajkowitsch L, Chen D, Stampfl S, Semrad K, Waldsich C, Mayer O, et al. RNA chaperones, RNA annealers and RNA helicases. RNA Biol. 2007;4:118–30. doi: 10.4161/rna.4.3.5445. [DOI] [PubMed] [Google Scholar]
- 5.Henriet S, Sinck L, Bec G, Gorelick RJ, Marquet R, Paillart JC. Vif is a RNA chaperone that could temporally regulate RNA dimerization and the early steps of HIV-1 reverse transcription. Nucleic Acids Res. 2007;35:5141–53. doi: 10.1093/nar/gkm542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuciak M, Gabus C, Ivanyi-Nagy R, Semrad K, Storchak R, Chaloin O, et al. The HIV-1 transcriptional activator Tat has potent nucleic acid chaperoning activities in vitro. Nucleic Acids Res. 2008;36:3389–400. doi: 10.1093/nar/gkn177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rowley GL, Ma Q-F, Bathurst IC, Barr PJ, Kenyon GL. Stabilization and Activiation of Recombinant human Immunodeficiency Virus-1 Reverse Transcriptase-P66. Biochem Biophys Res Commun. 1990;167:673–9. doi: 10.1016/0006-291x(90)92078-e. [DOI] [PubMed] [Google Scholar]
- 8.Cristofari G, Ivanyi-Nagy R, Gabus C, Boulant S, Lavergne JP, Penin F, et al. The hepatitis C virus Core protein is a potent nucleic acid chaperone that directs dimerization of the viral (+) strand RNA in vitro. Nucleic Acids Res. 2004;32:2623–31. doi: 10.1093/nar/gkh579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ivanyi-Nagy R, Lavergne JP, Gabus C, Ficheux D, Darlix JL. RNA chaperoning and intrinsic disorder in the core proteins of Flaviviridae. Nucleic Acids Res. 2008;36:712–25. doi: 10.1093/nar/gkm1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zuniga S, Sola I, Moreno JL, Sabella P, Plana-Duran J, Enjuanes L. Coronavirus nucleocapsid protein is an RNA chaperone. Virology. 2007;357:215–27. doi: 10.1016/j.virol.2006.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mir MA, Panganiban AT. Characterization of the RNA chaperone activity of hantavirus nucleocapsid protein. J Virol. 2006;80:6276–85. doi: 10.1128/JVI.00147-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang ZS, Su WH, Wang JL, Wu HN. Selective strand annealing and selective strand exchange promoted by the N-terminal domain of hepatitis delta antigen. J Biol Chem. 2003;278:5685–93. doi: 10.1074/jbc.M207938200. [DOI] [PubMed] [Google Scholar]
- 13.Destefano JJ, Titilope O. Poliovirus Protein 3AB Displays Nucleic Acid Chaperone and Helix-Destabilizing Activities. J Virol. 2006;80:1662–71. doi: 10.1128/JVI.80.4.1662-1671.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Houghton M. In Fields BN, Knipe DM, Howley PM, (Eds. ) Philidelphia: Lippincott-Raven. 1996;1:1035–58. [Google Scholar]
- 15.Towner JS, Ho TV, Semler BL. Determinants of membrane association for poliovirus protein 3AB. J Biol Chem. 1996;271:26810–8. doi: 10.1074/jbc.271.43.26810. [DOI] [PubMed] [Google Scholar]
- 16.Hope DA, Diamond SE, Kirkegaard K. Genetic dissection of interaction between poliovirus 3D polymerase and viral protein 3AB. Journal of Virology. 1997;71:9490–8. doi: 10.1128/jvi.71.12.9490-9498.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plotch SJ, Palant O. Poliovirus protein 3AB form a complex with and stimulates the activity of the viral RNA polymerase, 3Dpol. J Virol. 1995;69:7169–79. doi: 10.1128/jvi.69.11.7169-7179.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li N, Cui ZQ, Wen JK, Zhang ZP, Wei HP, Zhou YF, et al. Live cell imaging of protein interactions in poliovirus RNA replication complex using fluorescence resonance energy transfer (FRET) Biochem Biophys Res Commun. 2008;368:489–94. doi: 10.1016/j.bbrc.2008.01.094. [DOI] [PubMed] [Google Scholar]
- 19.Yin J, Liu Y, Wimmer E, Paul AV. Complete protein linkage map between the P2 and P3 non-structural proteins of poliovirus. J Gen Virol. 2007;88:2259–67. doi: 10.1099/vir.0.82795-0. [DOI] [PubMed] [Google Scholar]
- 20.Strauss DM, Wuttke DS. Characterization of protein-protein interactions critical for poliovirus replication: analysis of 3AB and VPg binding to the RNA-dependent RNA polymerase. J Virol. 2007;81:6369–78. doi: 10.1128/JVI.02252-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiang W, Cuconati A, Hope D, Kirkegaard K, Wimmer E. Complete protein linkage map of poliovirus P3 proteins: interaction of polymerase 3Dpol with VPg and with genetic variants of 3AB. J Virol. 1998;72:6732–41. doi: 10.1128/jvi.72.8.6732-6741.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiang W, Cuconati A, Paul AV, Cao X, Wimmer E. Molecular dissection of the multifunctional poliovirus RNA-binding protein 3AB. Rna. 1995;1:892–904. [PMC free article] [PubMed] [Google Scholar]
- 23.Lama J, Sanz MA, Rodriguez PL. Role of 3AB protein in poliovirus genome replication. J Biol Chem. 1995;270:14430–8. doi: 10.1074/jbc.270.24.14430. [DOI] [PubMed] [Google Scholar]
- 24.Harris KS, Xiang W, Alexander L, Lane WS, Paul AV, Wimmer E. Interaction of poliovirus polypeptide 3CDpro with the 5′ and 3′ termini of the poliovirus genome. Identification of viral and cellular cofactors needed for efficient binding. J Biol Chem. 1994;269:27004–14. [PubMed] [Google Scholar]
- 25.Xiang W, Harris KS, Alexander L, Wimmer E. Interaction between the 5′-terminal cloverleaf and 3AB/3CDpro of poliovirus is essential for RNA replication. J Virol. 1995;69:3658–67. doi: 10.1128/jvi.69.6.3658-3667.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baron MH, Baltimore D. Anti-VPg antibody inhibition of the poliovirus replicase reaction and production of covalent complexes of VPg-related proteins and RNA. Cell. 1982;30:745–52. doi: 10.1016/0092-8674(82)90279-3. [DOI] [PubMed] [Google Scholar]
- 27.Semler BL, Anderson CW, Hanecak R, Dorner LF, Wimmer E. A membrane-associated precursor to poliovirus VPg identified by immunoprecipitation with antibodies directed against a synthetic heptapeptide. Cell. 1982;28:405–12. doi: 10.1016/0092-8674(82)90358-0. [DOI] [PubMed] [Google Scholar]
- 28.Takegami T, Kuhn RJ, Anderson CW, Wimmer E. Membrane-dependent uridylylation of the genome-linl\kerd protein PPg of poliovirus. Proc Natl Acad Sci USA. 1983;80:7447–51. doi: 10.1073/pnas.80.24.7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takegami T, Semler BI, Anderson CW, Wimmer E. Membrane fractions active in poliovirus RNA replication contain VPg precursor polypeptides. Virology. 1983;128:33–47. doi: 10.1016/0042-6822(83)90316-1. [DOI] [PubMed] [Google Scholar]
- 30.Paul AV, Boom JH, Filippov D, Wimmer E. Protein-primed RNA synthesis by purified RNA polymerase. Nature. 1998;393:280–4. doi: 10.1038/30529. [DOI] [PubMed] [Google Scholar]
- 31.Paul AV, Rieder E, Kim DW, van Boom JH, Wimmer E. Identification of an RNA hairpin in poliovirus RNA that serves as the primary template in the In vitro uridylylation of VPg [In Process Citation] J Virol. 2000;74:10359–70. doi: 10.1128/jvi.74.22.10359-10370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fujita K, Krishnakumar SS, Franco D, Paul AV, London E, Wimmer E. Membrane topography of the hydrophobic anchor sequence of poliovirus 3A and 3AB proteins and the functional effect of 3A/3AB membrane association upon RNA replication. Biochemistry. 2007;46:5185–99. doi: 10.1021/bi6024758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lyle JM, Clewell A, Richmond K, Richards OC, Hope DA, Schultz SC, et al. Similar structural basis for membrane localization and protein priming by an RNA-dependent RNA polymerase. J Biol Chem. 2002;277:16324–31. doi: 10.1074/jbc.M112429200. [DOI] [PubMed] [Google Scholar]
- 34.Strauss DM, Glustrom LW, Wuttke DS. Towards an understanding of the poliovirus replication complex: the solution structure of the soluble domain of the poliovirus 3A protein. J Mol Biol. 2003;330:225–34. doi: 10.1016/s0022-2836(03)00577-1. [DOI] [PubMed] [Google Scholar]
- 35.Paul AV, Cao X, Harris KS, Lama J, Wimmer E. Studies with poliovirus polymerase 3Dpol. Stimulation of poly(U) synthesis in vitro by purified poliovirus protein 3AB. J Biol Chem. 1994;269:29173–81. [PubMed] [Google Scholar]
- 36.Richards OC, Ehrenfeld E. Effects of poliovirus 3AB protein on 3D polymerase-catalyzed reaction. J Biol Chem. 1998;273:12832–40. doi: 10.1074/jbc.273.21.12832. [DOI] [PubMed] [Google Scholar]
- 37.Rodriguez-Wells V, Plotch SJ, DeStefano JJ. Primer-dependent synthesis by poliovirus RNA-dependent RNA polymerase (3D(pol)) Nucleic Acids Res. 2001;29:2715–24. doi: 10.1093/nar/29.13.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andino R, Rieckhof GE, Achacoso PL, Baltimore D. Poliovirus RNA synthesis utilizes an RNP complex formed around the 5′-end of viral RNA. EMBO J. 1993;12:3587–98. doi: 10.1002/j.1460-2075.1993.tb06032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andino R, Rieckhof GE, Baltimore D. A functional ribonucleoprotein complex forms around the 5′ end of poliovirus RNA. Cell. 1990;63:369–80. doi: 10.1016/0092-8674(90)90170-j. [DOI] [PubMed] [Google Scholar]
- 40.Rohll JB, Percy N, Ley R, Evans DJ, Almond JW, Barclay WS. The 5′-untranslated regions of picornavirus RNAs contain independent functional domains essential for RNA replication and translation. J Virol. 1994;68:4384–91. doi: 10.1128/jvi.68.7.4384-4391.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jackson RJ, Howell MT, Kaminski A. The novel mechanism of initiation of picornavirus RNA translation. Trends Biochem Sci. 1990;15:477–83. doi: 10.1016/0968-0004(90)90302-r. [DOI] [PubMed] [Google Scholar]
- 42.Jang SK, Pestova TV, Hellen CU, Witherell GW, Wimmer E. Cap-independent translation of picornavirus RNAs: structure and function of the internal ribosomal entry site. Enzyme. 1990;44:292–309. doi: 10.1159/000468766. [DOI] [PubMed] [Google Scholar]
- 43.Agut H, Kean KM, Bellocq C, Fichot O, Girard M. Intratypic recombination of polioviruses: evidence for multiple crossing-over sites on the viral genome. J Virol. 1987;61:1722–5. doi: 10.1128/jvi.61.5.1722-1725.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arnold JJ, Cameron CE. Poliovirus RNA-dependent RNA polymerase (3Dpol) is sufficient for template switching in vitro. J Biol Chem. 1999;274:2706–16. doi: 10.1074/jbc.274.5.2706. [DOI] [PubMed] [Google Scholar]
- 45.Duggal R, Cuconati A, Gromeier M, Wimmer E. Genetic recombination of poliovirus in a cell-free system. Proc Natl Acad Sci USA. 1997;94:13786–91. doi: 10.1073/pnas.94.25.13786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duggal R, Wimmer E. Genetic recombination of poliovirus in vitro and in vivo: temperature-dependent alteration of crossover sites. Virology. 1999;258:30–41. doi: 10.1006/viro.1999.9703. [DOI] [PubMed] [Google Scholar]
- 47.Gmyl AP, Belousov EV, Maslova SV, Khitrina EV, Chetverin AB, Agol VI. Nonreplicative RNA recombination in poliovirus. J Virol. 1999;73:8958–65. doi: 10.1128/jvi.73.11.8958-8965.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jarvis TC, Kirkegaard K. The polymerase in its labyrinth: mechanisms and implications of RNA recombination. Trends Genet. 1991;7:186–91. doi: 10.1016/0168-9525(91)90434-R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jarvis TC, Kirkegaard K. Poliovirus RNA recombination: mechanistic studies in the absence of selection. EMBO J. 1992;11:3135–45. doi: 10.1002/j.1460-2075.1992.tb05386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kirkegaard K, Baltimore D. The mechanism of RNA recombination in poliovirus. Cell. 1986;47:433–43. doi: 10.1016/0092-8674(86)90600-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nagy PD, Simon AE. New insights into the mechanisms of RNA recombination. Virology. 1997;235:1–9. doi: 10.1006/viro.1997.8681. [DOI] [PubMed] [Google Scholar]
- 52.Pierangeli A, Bucci M, Forzan M, Pagnotti P, Equestre M, Perez Bercoff R. ‘Primer alignment-and-extension’: a novel mechanism of viral RNA recombination responsible for the rescue of inactivated poliovirus cDNA clones. J Gen Virol. 1999;80:1889–97. doi: 10.1099/0022-1317-80-8-1889. [DOI] [PubMed] [Google Scholar]
- 53.Romanova LI, Blinov VM, Tolskaya EA, Viktorova EG, Kolesnikova MS, Guseva EA, et al. The primary structure of crossover regions of intertypic poliovirus recombinants: a model of recombination between RNA genomes. Virology. 1986;155:202–13. doi: 10.1016/0042-6822(86)90180-7. [DOI] [PubMed] [Google Scholar]
- 54.Simon AE. Replication, recombination and symptom-modulation properties of the satellite RNAs of turnip crinkle virus. Curr Top Microbiol Immunol. 1999;239:19–36. doi: 10.1007/978-3-662-09796-0_2. [DOI] [PubMed] [Google Scholar]
- 55.Tolskaya EA, Romanova LI, Blinov VM, Viktorova EG, Sinyakov AN, Kolesnikova MS, et al. Studies on the recombination between RNA genomes of poliovirus: the primary structure and nonrandom distribution of crossover regions in the genomes of intertypic poliovirus recombinants. Virology. 1987;161:54–61. doi: 10.1016/0042-6822(87)90170-x. [DOI] [PubMed] [Google Scholar]
- 56.Darlix JL, Vincent A, Gabus C, de Rocquigny H, Roques B. Transactivation of the 5′ to 3′ viral DNA strand transfer by nucleocapsid protein during reverse transcription of HIV1 RNA. C R Acad Sci III. 1993;316:763–71. [PubMed] [Google Scholar]
- 57.Allain B, Lapadat-Tapolsky M, Berlioz C, Darlix JL. Transactivation of the minus-strand DNA transfer by nucleocapsid protein during reverse transcription of the retroviral genome. EMBO J. 1994;13:973–81. doi: 10.1002/j.1460-2075.1994.tb06342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peliska JA, Balasubramanian S, Giedroc DP, Benkovic SJ. Recombinant HIV-1 nucleocapsid protein accelerates HIV-1 reverse transcriptase catalyzed DNA strand transfer reactions and modulates RNase H activity. Biochemistry. 1994;33:13817–23. doi: 10.1021/bi00250a036. [DOI] [PubMed] [Google Scholar]
- 59.You JC, McHenry CS. Human immunodeficiency virus nucleocapsid protein accelerates strand transfer of the terminally redundant sequences involved in reverse transcription. J Biol Chem. 1994;269:31491–5. [PubMed] [Google Scholar]
- 60.DeStefano JJ. Human immunodeficiency virus nucleocapsid protein stimulates strand transfer from internal regions of heteropolymeric RNA templates. Arch Virol. 1995;140:1775–89. doi: 10.1007/BF01384341. [DOI] [PubMed] [Google Scholar]
- 61.Rodriguez-Rodriguez L, Tsuchihashi Z, Fuentes GM, Bambara RA, Fay PJ. Influence of human immuno-deficiency virus nucleocapsid protein on synthesis and strand transfer by the reverse transcriptase in vitro. J Biol Chem. 1995;270:15005–11. doi: 10.1074/jbc.270.25.15005. [DOI] [PubMed] [Google Scholar]
- 62.Cameron CE, Ghosh M, Le Grice SF, Benkovic SJ. Mutations in HIV reverse transcriptase which alter RNase H activity and decrease strand transfer efficiency are suppressed by HIV nucleocapsid protein. Proc Natl Acad Sci USA. 1997;94:6700–5. doi: 10.1073/pnas.94.13.6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guo J, Henderson LE, Bess J, Kane B, Levin JG. Human immunodeficiency virus type 1 nucleocapsid protein promotes efficient strand transfer and specific viral DNA synthesis by inhibiting TAR-dependent self-priming from minus-strand strong-stop DNA. J Virol. 1997;71:5178–88. doi: 10.1128/jvi.71.7.5178-5188.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Heath MJ, Derebail SS, Gorelick RJ, DeStefano JJ. Differing roles of the N-terminal and C-terminal zinc fingers in HIV-1 nucleocapsid protein enhanced nucleic acid annealing. J Biol Chem. 2003;278:30755–63. doi: 10.1074/jbc.M303819200. [DOI] [PubMed] [Google Scholar]
- 65.Anthony RM, Destefano JJ. In vitro synthesis of long DNA products in reactions with HIV-RT and nucleocapsid protein. J Mol Biol. 2007;365:310–24. doi: 10.1016/j.jmb.2006.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Narayanan N, Gorelick RJ, DeStefano JJ. Structure/ function mapping of amino acids in the N-terminal zinc finger of the human immunodeficiency virus type 1 nucleocapsid protein: residues responsible for nucleic acid helix destabilizing activity. Biochemistry. 2006;45:12617–28. doi: 10.1021/bi060925c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tanchou V, Delaunay T, Bodeus M, Roques B, Darlix JL, Benarous R. Conformational changes between human immunodeficiency virus type 1 nucleocapsid protein NCp7 and its precursor NCp15 as detected by anti-NCp7 monoclonal antibodies. J Gen Virol. 1995;76:2457–66. doi: 10.1099/0022-1317-76-10-2457. [DOI] [PubMed] [Google Scholar]
- 68.Schein CH, Oezguen N, van der Heden van Noort GJ, Filippov DV, Paul A, Kumar E, et al. NMR solution structure of poliovirus uridylylated peptide linked to the genome (VPgpU) Peptides. 2008 doi: 10.1016/j.peptides.2010.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schein CH, Oezguen N, Volk DE, Garimella R, Paul A, Braun W. NMR structure of the viral peptide linked to the genome (VPg) of poliovirus. Peptides. 2006;27:1676–84. doi: 10.1016/j.peptides.2006.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.You JC, McHenry CS. HIV nucleocapsid protein. Expression in Escherichia coli, purification and characterization. J Biol Chem. 1993;268:16519–27. [PubMed] [Google Scholar]
- 71.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–15. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]