

Abstract
Axonal degeneration is a hallmark of many neurological disorders. Studies in animal models of neurodegenerative diseases indicate that axonal degeneration is an early event in the disease process, and delaying this process can lead to decreased progression of the disease and survival extension. Overexpression of the Wallerian degeneration slow (Wlds) protein can delay axonal degeneration initiated via axotomy, chemotherapeutic agents, or genetic mutations. The Wlds protein consists of the N-terminal portion of the ubiquitination factor Ube4b fused to the nicotinamide adenine dinucleotide (NAD+) biosynthetic enzyme nicotinamide mononucleotide adenylyl transferase 1 (Nmnat1). We previously showed that the Nmnat1 portion of this fusion protein was the critical moiety for Wlds-mediated axonal protection. Here, we describe the development of an automated quantitative assay for assessing axonal degeneration. This method successfully showed that Nmnat1 enzymatic activity is important for axonal protection as mutants with reduced enzymatic activity lacked axon protective activity. We also found that Nmnat enzymes with diverse sequences and structures from various species, including Drosophila melanogaster, Saccharomyces cerevisiae, and archaebacterium Methanocaldococcus jannaschii, which encodes a protein with no homology to eukaryotic Nmnat enzymes, all mediate robust axonal protection after axotomy. Besides the importance of Nmnat enzymatic activity, we did not observe changes in the steady-state NAD+ level, and we found that inhibition of nicotinamide phosphoribosyltransferase (Nampt), which synthesizes substrate for Nmnat in mammalian cells, did not affect the protective activity of Nmnat1. These results provide the possibility of a role for new Nmnat enzymatic activity in axonal protection in addition to NAD+ synthesis.
Introduction
Axonal degeneration occurs in many neurological disorders and often precedes neuronal cell death. It appears to be a self-destructive process that is linked to, but independent from, apoptosis (Raff et al., 2002; Coleman, 2005). Insight into the process of axonal degeneration has been gained through studies of Wallerian degeneration slow (wlds) mice (Lunn et al., 1989; Perry et al., 1991). The wlds mutation results in the overexpression of a fusion protein (Wlds) composed of the N-terminal 70 aa of Ube4b linked to full-length nicotinamide mononucleotide adenylyl transferase 1 (Nmnat1) (Coleman et al., 1998). This Wlds fusion protein protects axons from degeneration initiated by a variety of insults both in vitro and in vivo (Mack et al., 2001; Conforti et al., 2007).
Initial insights into the molecular mechanism of axonal protection by Wlds were made using an in vitro DRG culture system in which overexpression of only the nicotinamide mononucleotide adenylyl transferase 1 (Nmnat1) portion of Wlds was sufficient to prevent axonal degeneration initiated by axotomy or vincristine (Araki et al., 2004). The Nmnat enzymes catalyze the last step in nicotinamide adenine dinucleotide (NAD) biosynthesis, which is synthesized via three major pathways in both prokaryotes and eukaryotes: the de novo, salvage, and Preiss-Handler independent. In the de novo pathway, NAD+ is synthesized from tryptophan, whereas it is generated from nicotinic acid and nicotinamide in the salvage pathway, and from nicotinamide riboside (NmR) in a recently discovered third route (Bieganowski and Brenner, 2004). Most NAD+ is produced via the salvage pathway in mammals by the actions of nicotinamide phosphoribosyltransferase (Nampt), which converts nicotinamide to nicotinamide mononucleotide (NMN), followed by the conversion of NMN to NAD+ via Nmnat (Berger et al., 2005). Interestingly, the overexpression of Nampt, as well as NAD+ itself and intermediates of the NAD+ synthetic pathway, including nicotinamide, NMN, nicotinic acid mononucleotide, or NmR, can also delay axonal degeneration (Wang et al., 2005; Kaneko et al., 2006; MacDonald et al., 2006; Sasaki et al., 2006). However, the axonal protection provided by these treatments is less potent than that provided by Nmnat overexpression. These studies indicate that the NAD+ biosynthetic pathways are protective against axonal degeneration; however, the mechanism of axonal protection may differ in each case.
Several studies of neuronal injury in Drosophila demonstrated that the mechanisms of axonal degeneration as well as Nmnat-mediated protection are evolutionarily conserved, as overexpression of Wlds or Drosophila Nmnat (dNmnat) were found to protect transected axons of mushroom body neurons (MacDonald et al., 2006). Others have demonstrated that dNmnat is essential for maintaining neuronal integrity in Drosophila (Zhai et al., 2006). Interestingly, Nmnat enzymatic activity was not required for prevention of neuronal cell death in this model; instead, a chaperone-like activity of dNmnat appeared to be important for neuronal survival. The contradictory nature of the reports thus far prompted us to undertake additional studies to examine the role of Nmnat enzymatic activity in axonal protection. We found that Nmnat enzymatic activity is essential for axonal protection, as mutations in Nmnat1 that severely reduced its ability to synthesize NAD+ compromise the ability to prevent degeneration (Araki et al., 2004; Jia et al., 2007). While Nmnat1 enzymatic activity is crucial, increased NAD+ levels alone do not provide axonal protection. In addition, Nmnat-mediated protection is not abrogated in neurons with severely reduced levels of NAD+. Finally, we found that Nmnat enzymes from multiple species, including the archaebacterium Methanocaldococcus jannaschii, promote axonal protection, highlighting the evolutionary conservation of pathways involved in axonal degeneration and stability.
Materials and Methods
Reagents
FK866 was obtained from National Institute of Mental Health Chemical Synthesis and Drug Supply Program (MH number F-901). All other reagents were from Sigma-Aldrich unless otherwise noted.
Animals
CD38-deficient (Cd38−/−) mice (Cockayne et al., 1998) and poly-ADP ribose polymerase-1 (PARP1)-deficient (Parp1−/−) mice (de Murcia et al., 1997) were each homozygous null (−/−), backcrossed >10 generations to C57BL/6J and then intercrossed to generate mice that were deficient in both CD38 and PARP1 (Cd38−/−Parp1−/− mice). Wlds mice were obtained from Harlan Bioproducts. All animals were cared for and used in accordance with institutional guidelines.
Plasmids
All Nmnat enzymes were epitope tagged with the 6xHis sequence (CATCACCATCACCATCAC) at the C terminus and sub-cloned into the lentivirus FUIV (ubiquitin promoter-gene of interest-IRES-enhanced YFP) vector (Araki et al., 2004). Plasmids encoding mouse Nmnat1 and Nmnat1 (W170A) were described previously (Araki et al., 2004; Sasaki et al., 2006). Other Nmnat1 mutants are as follows: Nmnat1 (H24A) was produced using PCR-based mutagenesis (Cormack, 1997). cytNmnat1 was described previously (Sasaki et al., 2006). mCherry fluorescent protein cDNA was obtained from the laboratory of Roger Tsien (University of California, San Diego, San Diego, CA) (Shaner et al., 2005). mCherry fusion protein of Nmnat1 or cytNmnat1 was cloned into lentivector plasmid FUW (Lois et al., 2002). Drosophila melanogaster NmnatB partial cDNA (dNmnat, GenBank NM_170185) was obtained from the Drosophila Genomics Resource Center (AT23490), and the missing 52 N-terminal nucleotides were restored by PCR. Saccharomyces cerevisiae Nmnat2 cDNA (yNmnat, GenBank NP_011524) was a gift from M. Johnston (Washington University, St. Louis, MO). A mammalian codon-optimized version of Methanocaldococcus jannaschii Nmnat (mjNmnat, GenBank NP_247520) was generated using overlapping oligonucleotides and PCR (Prodromou and Pearl, 1992).
Nampt small interfering RNA knock down
Oligonucleotides were designed to target Nampt using parameters set by Dharmacon on its small interfering RNA (siRNA) design website (Dharmacon) (Reynolds et al., 2004). Fifty-nine base pair oligonucleotides were designed containing both forward and reverse sequences for the 19 bp siRNA target sequences linked together with a hairpin loop, and ligated into AgeI–BamHI cut lentivirus transfer vector pFUIV downstream of the U6 promoter (Ryu et al., 2008). The Nampt target sequences used were GGAAAGACCATGAGAAAGA (siRNA #1) and GCAAGAGACTGCTGGCATA (siRNA #2).
DRG culture and in vitro axotomy
DRG drop cultures were performed based on a previously described method (Chen et al., 2008). DRGs were collected from embryonic day 12.5 mouse embryos. The DRGs from five embryos (~200 total DRGs) were collected in 500 μl of DMEM (Sigma D5671), centrifuged at 2000 × g for 1 min, and incubated in 500 μl of a solution containing 0.05% trypsin and 0.02% EDTA at 37°C for 15 min. An equal volume of DMEM containing 10% FBS was then added, the DRGs were triturated using a 1000 μl pipette, and the cells were centrifuged (2000 × g, 1 min), washed in DMEM containing 10% FBS, and resuspended in 40 μl of Neurobasal media (Invitrogen) containing 2% B27 (Invitrogen) and 50 ng/ml NGF (2.5S; Harlan Bioproducts) per one dissected embryo. The suspended DRG cells were cultured on plates coated with poly-D-lysine and laminin. The wells were initially coated with 250 μl of poly-D-lysine (0.1 mg/ml) (Sigma) solution overnight. The wells were washed twice with water and dried for 15 min, and then 250 μl of mouse laminin (2–5 μg/ml) (Invitrogen) was added for 1 h. Before the cells were seeded, the laminin was removed from the wells, and the plates were dried. The suspended DRG neurons (2 μl) were placed as a drop in the center of each well of a 24-well plate and incubated at 37°C with 5% CO2 for 15 min. After cell attachment, 500 μl of Neurobasal media containing 0.02% B27, 50 ng/ml NGF, 1 μM 5-fluoro-2′-deoxyuridine (Sigma), and 1 μM uridine (Sigma) was added to each well. After 5 d, 50% of the culture medium was replaced with media lacking 5-fluoro-2′-deoxyuridine and uridine. Under these conditions, the DRG cell bodies cluster in an ~3 mm diameter circle with axons extending in a radial pattern. Axotomy was performed at 14–28 DIV (days in vitro) by transecting axons away from the cell body cluster with a microscalpel.
Lentivirus infection
Lentiviruses were generated as described previously (Araki et al., 2004). DRG neurons were infected with 104–105 colony forming units (CFU) and transgene expression was allowed to accumulate for 5 d for overexpression or 8 d for siRNA experiments. Viral infection was monitored using fluorescent microscopy to visualize the GFP or Cherry reporter.
Automated quantification of axonal degeneration
For each well, phase-contrast images of four random fields of DRG axons were obtained using a 20× objective lens and an inverted microscope (Eclipse TE 300; Nikon). The images were captured using a 20 ms exposure on a CCD camera (Cool SNAP ES; Nikon) with MetaMorph image analysis software (Molecular Devices). For computer-based image analysis, it is important that the axon tracts be well separated; we therefore randomly selected fields of axons that were 1.5–3 mm away from the edge of the neuronal cell body cluster. To obtain images with an uniform background intensity, the gray level of the images was adjusted using the auto level function of Adobe Photoshop.
To quantify the axonal degeneration, we measured the area occupied by axons (total axon area) and by degenerated axons at various times after injury. The phase-contrast images were binarized such that pixel intensity of regions corresponding to axons were converted to black and all other regions were converted to white. The total number of black pixels was then defined as the total axon area. Healthy intact axons show a continuous tract, whereas degenerated axons have a particulate structure due to axonal fragmentation or beading. To detect degenerated axons, we used the particle analyzer module of ImageJ and counted the area of the small fragments or particles (size = 20–10,000 pixels) derived from the degenerated axons. A degeneration index (DI) was calculated as the ratio of fragmented axon area over total axon area. The production of the binary images and the function of the particle analysis as well as the accuracy of the DI in detecting axonal degeneration were optimized using multiple images of intact versus degenerating axons obtained from a large number of experiments using our DRG in vitro axotomy system.
NAD+ measurement
Cultured DRG neurons (15 DIV) were washed with PBS and lysed by addition of 1 M perchloric acid. Samples from six wells were pooled (total of 200 μl of 1 M perchloric acid extract). Extracts were centrifuged and the supernatant was collected and neutralized with 3 M K2CO3. Equal volumes of the neutralized extract were mixed with potassium phosphate buffer and assayed by HPLC using LC-18T HPLC column (Supelco) at flow rate of 1 ml/min and the absorbance at 254 nm was recorded. Each elution peak was compared with standards to identify and quantify NAD+. Protein concentrations were determined using the Bradford Protein Assay (Bio-Rad), and NAD+ levels were normalized to the protein amounts.
Nmnat enzymatic activity assay
To produce Nmnat proteins in bacteria, Nmnat1, Nmnat1 (H24A), and Nmnat1 (W170A) were cloned into pET3a plasmid (Invitrogen). The bacterially produced enzymes were purified using His-select affinity gel (Sigma). Protein concentrations were determined using the Bradford Protein Assay (Bio-Rad). To assess enzymatic activity, purified protein [0.2 μg for Nmnat1 and 2 μg for Nmnat1 (H24A) and Nmnat1 (W170A)] was diluted in ice-cold reaction buffer containing 20 mM HEPES-KOH, pH 7.4, 20 mM MgCl2, 1.5% EtOH, 0.5 U/μl alcohol dehydrogenase (Sigma), 0.5 mg/ml BSA, and 1 mM NMN (when ATP concentration dependency was measured) or 1 mM ATP (when NMN concentration dependency was measured). The reactions were performed at 37°C and initiated by the addition of NMN (final concentration = 0.02–10 mM) or ATP (final concentration = 0.05–5 mM). The reactions were monitored by measuring the absorbance at 340 nm (to measure NADH) at the indicated times. The enzymatic activity was calculated using the NADH molar extinction coefficient (1.0 OD at 340 nm = 6.22 M−1 · cm−1).
To measure the enzymatic activities of dNmnat, yNmnat, mjNmnat, and mouse Nmnat1, each respective lentivirus transfer vector was transfected into HEK 293T using Fugene6. Three days after transfection, the cells were homogenized in buffer containing 20 mM HEPES-KOH, pH 7.4, 200 mM NaCl, and protease inhibitor cocktail (Invitrogen). The lysates were centrifuged at 10,000 × g for 10 min, and total protein concentration in the cleared lysates was determined. Each lysate (5 μg) was incubated in reaction buffer (25 μl) containing 20 mM HEPES-KOH, pH 7.4, 20 mM MgCl2, 1.5% EtOH, 0.5 U/μl alcohol dehydrogenase, 1 mM NMN, and 1 mM ATP at 37°C for 1 h. The amount of NADH produced was measured by absorbance at 340 nm, and the enzymatic activity was calculated as above.
Surgical procedures
Four-month-old wild-type, Wlds mutant, or CD38−/−;PARP1−/− mice were anesthetized by intraperitoneal injection of 2,2,2-tribromoethanol at a dose of 500 mg/kg. The sciatic nerve was exposed in the hip and cut, and the distal stump was deflected onto an adjacent muscle to prevent regeneration. Nerve lesions were produced on the right side, and left contralateral intact nerves were used as controls. Animals were killed 7 d after surgery. Both the right and left sciatic nerves were collected and fixed with osmium tetroxide. Sections of sciatic nerve were stained with toluidine blue to assess myelinated axon profiles.
Results
Quantitative analysis of axonal degeneration
In transgenic mice overexpressing Wlds protein, the extent of axonal protection is directly correlated with transgene expression level, with complete loss of protection occurring at a level <20% of that present in wlds mutant mice (Mack et al., 2001). Thus, the varying results obtained with respect to Nmnat- versus Wlds-mediated axonal protection (Araki et al., 2004; Wang et al., 2005; MacDonald et al., 2006; Conforti et al., 2007) may be due to different levels of Nmnat1 and Wlds expression. Alternatively, they may result from different threshold amounts required for protection, different culture conditions, or the nonquantitative nature of the axonal degeneration assays.
To address the genesis of these discrepant results, we first developed a quantitative, automated axonal degeneration assay using ImageJ software (NIH). This algorithm is used to calculate a DI that is based on the ratio of the areas of fragmented axons versus total axonal area. A key factor in this system is the usage of DRG drop cultures grown in defined media (Neurobasal with B27) (see Materials and Methods) that provide a minimum variance of neuronal number and area of the axonal outgrowth halo size. To assess the axonal state, phase-contrast images of axons were taken before and after injury, and the axon-containing fields were binarized. Degenerated axon fragments were detected using the particle analyzer algorithm of ImageJ, and the total fragmented axon area versus total axonal area was used to generate a degeneration index or “DI” (Fig. 1A). The reproducibility of this method was demonstrated by analyzing multiple (n = 48) pictures of axons randomly taken from 12 different wells of DRG neurons [nontransected control (DI = 0.092 ± 0.039) vs 72 h after axotomy (DI = 0.52 ± 0.13)]. To test the correlation of the degeneration index with the visual rating system used by Araki et al. (2004), pictures from the same fields of axons were taken every hour for 24 h after axotomy and analyzed by both methods. The resulting plot shows a time-dependent increase in the calculated DI after axotomy (see Materials and Methods) that is well correlated with qualitative visual assessment (Fig. 1B,C). Through these comparative experiments, a DI >0.2 was established as the level at which axonal degeneration was clearly evident. This value is used throughout these studies.
Figure 1.

Quantitative axonal degeneration assay. A, Representative pictures of original images, binarized images, and fragmented axons from intact and degenerated DRG axons. B, DRG neurons were cultured for 14 d, and the axons were then transected. Representative images of the same fields of axons at the indicated times after axotomy are shown. The time after axotomy is shown above, and the degeneration index is shown below, each image. C, Images from the same field after axotomy were analyzed by two different methods. The bar graph shows the percentage of remaining axons ±SD calculated with the visual rating system used by Araki et al. (2004). The degeneration index calculated from these images was plotted against time after axotomy and displayed as a line graph. The extent of axonal degeneration detected by these two methods is well correlated.
Enzymatic activity is important for Nmnat1-mediated axonal protection
We previously reported that enzymatically inactive Nmnat1 (W170A) and the corresponding Wlds (W258A) mutant do not promote axonal protection (Araki et al., 2004). However, studies of Drosophila Nmnat demonstrated that neurodegeneration could be prevented with mutants that have greatly reduced enzymatic activity (Zhai et al., 2006). To reexamine this issue, we produced a number of additional mutants and performed in vitro assays to accurately measure their enzymatic and axonal protective activities. The Nmnat1 (W170A) mutant is predicted to disrupt binding of NMN based on its molecular structure (Zhou et al., 2002). The His24 residue of human Nmnat1 as well as the homologous His residue in Nmnat2 are thought to be crucial for ATP binding, transition state stabilization, and enzymatic activity (Garavaglia et al., 2002; Yalowitz et al., 2004). We generated mouse Nmnat1 (H24A) and cloned it, along with Nmnat1 (W170A) and wild-type Nmnat1, into the pET3a bacterial expression vector. Recombinant Nmnat proteins were produced in Escherichia coli and purified by affinity chromatography. The enzymatic activities of wild-type and mutant Nmnat1 proteins were measured at a range of ATP and NMN concentrations as described in Materials and Methods. Previous analysis using human Nmnat1 showed that its Km for NMN was 23–59 μM with a kcat of 14–54 s−1 depending on the experimental conditions (Raffaelli et al., 2002; Berger et al., 2005; Sorci et al., 2007). Our analysis of mouse Nmnat1 revealed similar values, with a Km = 27.6 μM and kcat = 5.5 s−1 for NMN and a Km = 50.4 μM and kcat = 4.5 s−1 for ATP. Under these experimental conditions, the enzymatic activity of the Nmnat1 (H24A) and Nmnat1 (W170A) mutants was significantly lower than that of wild-type Nmnat1 (Table 1). For example, the kcat/Km value for wild-type Nmnat1 is 2.0 × 105 M−1 · s−1 (NMN as a substrate) and 8.9 × 104 M−1 · s−1 (ATP as a substrate), whereas the Nmnat1 (W170A) and Nmnat1 (H24A) mutants had kcat/Km values of 1.5 M−1 · s−1 (NMN as a substrate) and 2.7 × 102 M −1 · s−1 (ATP as a substrate), respectively. These experiments demonstrated that the NAD+ synthesizing activity of these mutants was greatly reduced [<10−5 for Nmnat1 (W170A) and <10−3 for Nmnat1 (H24A)].
Table 1.
Kinetic parameters of purified recombinant Nmnat1 proteins
| Enzyme | Substrate | Km (μM) | Vmax (μmol · min−1 · μg−1) | kcat (s−1) |
|---|---|---|---|---|
| Nmnat1 | ATP | 50.4 | 8.1 | 4.5 |
| NMN | 27.6 | 9.9 | 5.5 | |
| Nmnat1 (W170A) | NMN | 1.4 × 103 | 0.0037 | 0.0021 |
| Nmnat1 (H24A) | ATP | 47.3 | 0.024 | 0.013 |
To examine the axonal protective activity of these Nmnat mutants, we performed axonal degeneration assays using DRG neurons infected with lentiviruses expressing wild-type or mutant Nmnat1 or EGFP (control). While axonal protection mediated by wild-type Nmnat1 was robust, the DI of neurons expressing Nmnat1 (W170A) or Nmnat1 (H24A) was similar to that of EGFP-expressing neurons (Fig. 2A,B). These experiments demonstrated that mutants with greatly reduced enzymatic activity were also defective in promoting axonal protection.
Figure 2.

Nmnat enzymatic activity is critical for axonal protection. A, Representative images of axons of DRG neurons infected with lentivirus expressing EGFP, Nmnat1, Nmnat1 (H24A), or Nmnat1 (W170A) are shown. Axons from Nmnat1-expressing DRG neurons were protected after axotomy, whereas axons from Nmnat1 (H24A)- or Nmnat1 (W170A)-expressing neurons degenerated similarly to those expressing EGFP. B, The quantification of axonal degeneration in DRG neurons expressing EGFP, Nmnat1, Nmnat1 (H24A), or Nmnat1 (W170A). Twelve fields were evaluated for each condition, and each experiment was repeated three times. The degeneration index value ± SD at 24, 48, and 72 h is displayed. Only Nmnat1 shows significant axonal protection ( p < 0.001, n = 3).
Nmnat-mediated axonal protection is evolutionarily conserved
Nmnat1 mutants with decreased enzymatic activity have reduced protective capacity, whereas other enzymatically active Nmnat isoforms like the mitochondrial Nmnat3 are protective (Sasaki et al., 2006; Press and Milbrandt, 2008), supporting the idea that Nmnat enzymatic activity is important for axonal protection. However, although the sequences of Nmnat3 and Nmnat1 diverge considerably outside of the active site, the possibility remains that regions conserved between these two proteins could support additional functions or interactions that are also crucial for axonal protection (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). In this regard, it should be noted that the mutations used to reduce the NAD+ synthetic activity of Nmnat could also inhibit other hypothetical functions. To circumvent this issue, we tested enzymes with NAD+ synthetic activity from other species for their ability to protect mammalian axons. NAD+ synthetic activity is conserved from human to archaebacterium, although the molecular structures of the cognate enzymes vary greatly (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). For instance, mouse Nmnat1 is 36% identical to dNmnat and 22% identical to yNmnat (Emanuelli et al., 2003), with each of these homologs sharing one Nmnat conserved domain (CD02165 in NCBI). On the other hand, the archaebacterium Methanocaldococcus jannaschii Nmnat (mjNmnat) is a very small protein with no detectable homology to mammalian Nmnat isoforms. In addition, structural and biochemical studies indicate that mjNmnat has different substrate specificities and binding properties from mammalian Nmnat enzymes (D’Angelo et al., 2000; Zhou et al., 2002). These studies are consistent with the finding that mjNmnat contains a conserved domain CD02166 that is highly divergent from the conserved domain CD02165 present in all eukaryotic Nmnat enzymes (Garavaglia et al., 2002; Marchler-Bauer et al., 2002).
To examine the enzymatic and axonal protective activities of these NAD+ synthetic enzymes, we generated lentiviruses expressing C-terminal His-tagged dNmnat, yNmnat, or mjNmnat. These Nmnat enzymes as well as mouse Nmnat1 were expressed in HEK293 cells (supplemental Fig. S2A,B, available at www.jneurosci.org as supplemental material), and enzymatic assays demonstrated that each of them possessed robust NAD+ synthetic activity (Fig. 3A). Immunocytochemistry with an anti-His antibody demonstrated that these enzymes were all localized predominantly in the cytoplasm, in a pattern comparable to cyt-Nmnat1 (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material). To assess their ability to protect axons from degeneration, the Nmnat proteins were expressed in DRG neurons via lentivirus infection, and the degree of axonal degeneration after axotomy was measured. Interestingly, we found that Nmnat enzymes from all species, including the Archaea protein from Methanocaldococcus jannaschii, provided robust axonal protection (Fig. 3B; supplemental Fig. S2C, available at www.jneurosci.org as supplemental material). Thus, although these Nmnat enzymes have little, if any, primary sequence homology, as well as different crystal structures, they all possess both NAD+ synthetic and axonal protective activities, strongly suggesting that these two Nmnat-mediated functions are linked.
Figure 3.

Nmnat axonal protective activity is conserved throughout evolution. A, The Nmnat enzymatic activities from HEK293T cell lysates expressing EGFP, Nmnat1, dNmnat, yNmnat, or mjNmnat demonstrated that each enzyme had similar NAD+ synthetic activity. Error bars represent SD. B, The rate of axonal degeneration after injury was quantified in DRG neurons expressing EGFP, mouse Nmnat1, dNmnat, yNmnat, or mjNmnat. Twelve fields were evaluated for each condition, and each experiment was repeated three times. The degeneration index value ± SD at 24, 48, and 72 h is displayed. All Nmnat enzymes, from mammalian to Archaea, delayed axonal degeneration of injured DRG neurons in a comparable manner. Nmnat1-, dNmnat-, yNmnat-, or mjNmnat- versus EGFP-expressing neurons, p <0.001 (n = 3).
Elevated neuronal NAD+ levels do not provide axonal protection
Axonal protection by Nmnat overexpression is strongly dependent on its enzymatic activity; however, no increases in NAD+ levels are observed in these neurons. In addition, exogenous application of NAD+ as well as NAD+ intermediates also provides axonal protection (Araki et al., 2004; Sasaki et al., 2006). To reexamine the role of NAD+ in axonal protection, we sought to increase NAD+ levels by reducing NAD+ consumption. The first of two important enzymes in NAD+ breakdown is CD38, a glycohydrolase found on plasma membrane and intracellular membranes that converts NAD+ to ADP-ribose, cyclic ADP-ribose, and nicotinamide (Chini et al., 2002; Lund, 2006). The second enzyme is PARP1, which modifies various nuclear proteins by poly(ADP-ribosyl)ation via converting NAD+ to poly-ADP-ribose and nicotinamide (Schreiber et al., 2006).
To determine whether decreased CD38 and/or PARP1 activity altered NAD+ levels, we prepared brain lysates from mice deficient in CD38 (Cockayne et al., 1998) alone or CD38 and PARP1. We measured NAD+ levels by HPLC and found that they were elevated in Cd38−/− (276 ± 8 nmol of NAD+/g protein) brain compared with wild-type brain (196 ± 4 nmol of NAD+/g protein), similar to previous results (Young et al., 2006). Interestingly, NAD levels were further elevated in the brains of Cd38−/−Parp1−/− mice (420 ± 23 nmol of NAD+/g protein) compared with normal controls. To confirm that NAD+ levels were elevated in neurons specifically, we performed similar experiments on cultured DRG neurons from these mutant animals. We found that Cd38−/−Parp1−/− DRG neurons had twofold higher levels of NAD+ than wild type (Cd38−/−Parp1−/−: 23 ± 2 pmol of NAD+/μg protein vs wild type: 12 ± 3 pmol of NAD+/μg protein) and that Cd38−/− neurons had intermediate levels (15 ± 3 pmol of NAD+/μg protein). We then infected these DRG neurons with cytNmnat1 lentivirus and, after 7 d, measured NAD+ levels. As expected, we found that Nmnat overexpression did not alter steady-state NAD+ levels in wild-type, Cd38−/−, or Cd38−/−Parp1−/− neurons (data not shown).
To determine whether high NAD+ levels themselves were sufficient to promote axonal protection, we performed in vitro axonal degeneration assays and compared the responses of wild-type, Cd38−/−, and Cd38−/−Parp1−/− DRG neurons. Surprisingly, we found that despite much higher levels of NAD+, axons of Cd38−/−Parp1−/− DRG neurons degenerated similarly to those of wild-type neurons (Fig. 4A). However, if we expressed cytNmnat1 in these neurons, then minimal axonal degeneration was observed at 72 h regardless of genotype (Fig. 4A). These results demonstrated that Nmnat can promote axonal protection in Cd38−/−Parp1−/− neurons, in which NAD+ levels are high, indicating that loss of these proteins has no detrimental effect on the Nmnat axonal protective pathway.
Figure 4.

Axonal degeneration proceeds normally in Cd38−/−Parp1−/− mice despite high NAD+ levels. A, In vitro axonal degeneration assays were performed using DRG neurons from mice with the indicated genotypes infected with lentivirus expressing cytNmnat1 or EGFP. The DI was calculated as outlined in Materials and Methods. Sixteen fields were evaluated for each condition, and each experiment was repeated three times. The degeneration index value ± SD at 0, 24, 48, and 72 h is displayed. *Significant difference ( p < 0.001) between neurons expressing cytNmnat1 and those expressing EGFP of the indicated genotypes. B, Sciatic nerves in mice of the indicated genotypes were transected, and the distal segments were harvested 7 d later. Transverse sections of the distal segments stained with toluidine blue are displayed. While axons from wlds mice are preserved, degenerating axon profiles are abundant in nerves from Cd38−/−Parp1−/− and wild-type (wt) mice.
To investigate whether high neuronal NAD+ levels influenced axonal degeneration in vivo, we performed sciatic nerve transection experiments using 4-month-old wild-type, Cd38−/−Parp1−/−, and Wlds (positive control) mice. Seven days after axotomy, the distal segment of the transected nerves as well as the uninjured contralateral nerves were collected and analyzed by toluidine blue staining. We found that the transected nerves from Cd38−/−Parp1−/− appeared very similar to those of wild-type animals, with severe axonal degeneration and loss of myelin profiles, whereas nerves from Wlds mice were well preserved and looked similar to the uninjured nerves (Fig. 4B). Together, these experiments demonstrate that high levels of NAD+ alone are insufficient to protect axons from degenerating after injury, both in vitro and in vivo.
Inhibition of Nampt does not interfere with Nmnat-mediated axonal protection
Our previous experiments showed that increased levels of NAD+ do not promote axonal protection, whereas Nmnat enzymatic activity is clearly required. If Nmnat-mediated NAD+ production is essential for axonal protection, we reasoned that reducing intracellular NAD+ levels would affect its protective capability. While there are no compounds available to inhibit any of the three mammalian Nmnat proteins, FK866 is a specific inhibitor of Nampt, the rate-limiting enzyme in NAD+ biosynthesis that converts nicotinamide to NMN (Hasmann and Schemainda, 2003). We treated DRG neurons with FK866 (1 μM) for up to 96 h and found that NAD+ levels were decreased dramatically within 24 h (Fig. 5A). Nmnat overexpression did not prevent the FK866 reduction in NAD+ levels. To test whether cytNmnat1 can prevent axonal degeneration in neurons with severely reduced NAD+ levels, we performed axonal degeneration assays. Neurons expressing either GFP or cytNmnat1 were grown in media containing FK866 or DMSO (control) for 24 h, and then axons were transected and monitored for axonal degeneration. While there was no evidence of axonal degeneration in cultures treated with FK866 for up to 96 h, axotomy produced complete axonal degeneration within 72 h. Interestingly, at 24 h FK866 treatment provided modest axonal protection, reminiscent of the protection provided by other molecules in the NAD salvage pathway (Sasaki et al., 2006). When cytNmnat1 was expressed in FK866-treated neurons, which have very low levels of NAD+, axonal protection was robust and long lasting, similar to that observed in control neurons (Fig. 5B). These results indicate that Nmnat-mediated axonal protection is not correlated with intracellular NAD+ levels.
Figure 5.
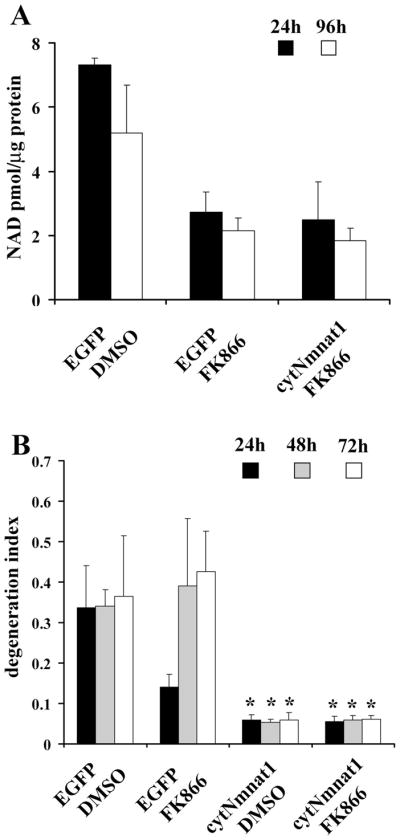
The Nampt inhibitor FK866 reduces neuronal NAD+ levels but does not affect Nmnat-mediated axonal protection. A, FK866 (1 μM) treatment of DRG neurons expressing either EGFP or cytNmnat1 reduced NAD+ levels by ~65% within 24 h. Error bars represent SD. B, Axonal degeneration assays were performed using neurons expressing EGFP or cytNmnat1 that were treated with FK866 or DMSO (control). The degeneration indices were calculated. Twelve fields were evaluated for each condition, and each experiment was repeated three times. The degeneration index value ± SD at 24, 48, and 72 h is displayed. *Significant difference (p < 0.001) between neurons expressing cytNmnat1 and those expressing EGFP of the indicated genotypes.
To confirm this finding, we generated multiple lentiviruses expressing Nampt siRNAs to knock down Nampt expression. DRG neurons were infected with these lentiviruses, and two siRNAs (siNampt1 and siNampt2) were identified that reduced Nampt mRNA levels by 75% and 90%, respectively. Neurons expressing these siRNAs were harvested after 8 d, and NMN levels were reduced by 50–70%, while NAD+ levels were reduced by 70–90% (Fig. 6A and data not shown). Surprisingly, we found no evidence of axonal degeneration in these cultures even after 10 d of growth, despite the reduced neuronal NAD+ levels (Fig. 6A). We next tested the ability of cytNmnat1 as well as Nmnat1 to promote axonal protection in neurons expressing siNampt2, in which neuronal NAD+ levels were reduced by ~80%. Axonal degeneration assays demonstrated that both nuclear and cytosolic forms of Nmnat1 provided robust axonal protection in neurons expressing siNampt2 (Fig. 6B and data not shown), again demonstrating that severely reduced NAD+ (and NMN) levels do not affect Nmnat-mediated axonal protection.
Figure 6.

Neurons expressing Nampt siRNA have low levels of NAD+, but Nmnat-mediated axonal protection is intact. A, DRG neurons were infected with lentiviruses expressing EGFP or cytNmnat1 as well as siRNAs targeting Nampt or luciferase. After 8 d, NAD+ levels were measured using HPLC. Both Nampt siRNAs effectively reduced NAD+ levels. Overexpression of cytNmnat1 had no effect on NAD+ levels. Values are from three independent experiments; *p < 0.001 versus luciferase siRNA-expressing neurons. Error bars represent SD. B, Axonal degeneration assays were performed using neurons infected with lentiviruses expressing the indicated siRNA and either cytNmnat1 or EGFP, and degeneration indices were calculated. Sixteen fields were evaluated for each condition, and each experiment was repeated three times. The degeneration index value ± SD at 0, 24, 48, and 72 h is displayed. *Significant difference ( p < 0.001) between cytNmnat1- and EGFP-expressing neurons expressing either luciferase or Nampt siRNA. Note that Nampt siRNA had no effect on Nmnat-mediated axonal protection.
Discussion
Axonopathy is an early and common component of many neurological disorders and warrants investigation as a therapeutic target. The concept that axonal degeneration is an active process has been largely derived from studies of wlds mutant mice. In deciphering the molecular mechanism of axonal protection by the Wlds protein, we discovered that Nmnat1 alone could protect against axonal damage (Araki et al., 2004). While this finding has been duplicated in a number of laboratories (Wang et al., 2005; MacDonald et al., 2006; Press et al., 2008), others have reported that only wlds, and not Nmnat alone, can provide axonal protection (Conforti et al., 2007). Several groups studying axonal degeneration in Drosophila have found that Wlds protein as well as dNmnat can inhibit axonal degeneration after injury; however, neither protein could prevent developmental axonal pruning (Hoopfer et al., 2006; MacDonald et al., 2006). These results were the first evidence that Nmnat could protect axons in vivo. In contrast, Nmnat1 transgenic mice were reported to show no delay in axonal degeneration after sciatic nerve transection or in injury experiments with cultured transgenic DRGs (Conforti et al., 2007). However, another group recently showed that Nmnat3 transgenic mice displayed significant axonal protection in the sciatic nerve transection model, whereas Nmnat1 transgenic mice did not (Yahata and Araki, 2007). Thus, it appears that the Nmnat is the component of the Wlds protein that is most important for its axonal protective activity.
In keeping with the evolutionarily conserved nature of Nmnat axonal protection and the conservation of enzymes capable of synthesizing NAD+, we found that Nmnat enzymes from a variety of species can promote axonal protection. Interestingly, Nmnat from the archaebacterium Methanocaldococcus jannaschii shares no detectable conservation with mammalian enzymes, yet has robust NAD+ synthetic and axonal protective activities. Indeed, the conserved domain (NCBI CD02165) that corresponds to the active site in eukaryotic Nmnat enzymes is replaced in bacteria and archaebacteria with the highly divergent domain (NCBI CD02166) (Garavaglia et al., 2002; Marchler-Bauer et al., 2002). The robust protection against axonal degeneration promoted by these widely divergent Nmnat enzymes strongly supports the importance of Nmnat enzymatic activity in this protective function.
In further examining the importance of Nmnat enzymatic function in preventing axonal degeneration, we found that mutations in Nmnat located in either the ATP or NMN binding pockets severely reduced NAD+ production and axonal protection. These studies are consistent with other studies in mammalian neurons demonstrating that Nmnat1 or WldS mutants with decreased NAD+ synthetic activity failed to prevent axonal degeneration (Araki et al., 2004; Jia et al., 2007). However, they are at odds with studies in Drosophila that found that neuronal losses (as opposed to axonal degeneration) in Drosophila lacking dNmnat could be complemented with either wild-type or enzymatically deficient Nmnat enzymes (Zhai et al., 2006). These results indicated that other functions of dNmnat were critical for preventing neuronal degeneration, and they subsequently showed that, in addition to NAD+ synthesis, dNmnat is a stress-response protein that acts as a chaperone for neuronal maintenance and protection (Zhai et al., 2008). It is presently unclear whether chaperone activity is also necessary for axonal protection in mammals. If this is the case, then this function, and therefore interactions between Nmnat and its partners, must be conserved in Nmnat proteins from many species, even those with minimal homology to animal Nmnat enzymes. Furthermore, we tested the axonal protective capability of two chemical chaperones, trimethylamine N-oxide dihydrate and 4-phenyl butyric acid, widely studied for their ability to prevent apoptosis (Ozcan et al., 2006; Wei et al., 2008); however, no changes in axotomy-induced axonal degeneration were observed (data not shown). Overall, it appears that Nmnat-mediated protection of axons in mammals requires functions different from those required for prevention of neurodegeneration in Drosophila.
Nmnat enzymes convert NMN via the transfer of an adenylate group to NAD+. The association between Nmnat axonal protection and its enzymatic activity indicated that NAD+ is a crucial moiety in axonal protection (Araki et al., 2004). Further experimentation demonstrated that NAD+ itself, along with NMN and nicotinamide riboside, could also provide some measure of axonal protection (Sasaki et al., 2006). This protection was less impressive and shorter lived than that provided by Nmnat expression, but led to the conclusion that NAD was the active moiety preventing axonal degeneration. Furthermore, others have shown that Nmnat or Wlds overexpression helps maintain NAD+ levels after axon transection, leading them to postulate that steady-state levels of NAD+ are important for maintenance of axonal integrity (Wang et al., 2005). Together, this body of evidence strongly supported the idea that increased NAD+ production was the mechanism by which Nmnat promoted axonal protection.
On the other hand, Nmnat enzymatic activity is approximately fourfold higher in wlds mutant mice, yet NAD+ levels are similar to those of wild-type brain (Mack et al., 2001). Similarly, we and others have observed no increases in NAD+ levels in DRG neurons or non-neuronal cells over-expressing Nmnat1 [Revollo et al. (2004) and data not shown]. This failure to demonstrate increased levels of NAD+ in tissues of Wlds mice or in cells engineered to overexpress Nmnat is inconsistent with the idea that increased NAD+ levels are responsible for Nmnat-mediated axonal protection. In this study, we examined neurons with increased levels of NAD+ due to deficiencies in CD38 and PARP1 (Cockayne et al., 1998), enzymes that are major consumers of NAD+. Despite the elevated levels of NAD+ under steady-state conditions, injured axons from these neurons degenerated with normal kinetics, indicating that increased intracellular NAD+ levels were not protective. We did find that exogenous addition of NAD+ to Cd38−/−Parp1−/− DRG neurons provided axonal protection similar to that observed with NAD+-treated wild-type neurons (data not shown). Furthermore, we found that FK866, which structurally mimics molecules in the NAD salvage pathway, could provide short-term axonal protection even as it lowered NMN levels. This result suggested that perhaps Nmnat protection is achieved by reducing the levels of its substrate NMN (i.e., removing a potential inhibitor or axonal stability) rather than increasing the concentration of its product NAD+. However, genetic inhibition of Nampt greatly reduced neuronal NMN and NAD+ levels, yet did not lead to axonal protection (or degeneration). Most importantly, axonal protection by Nmnat overexpression was unaffected by these severely reduced NAD+ and NMN levels, implying that Nmnat-mediated neuronal protection does not operate through altering neuronal NAD+ or NMN levels and that the short-lived axonal protection provided by exogenous NAD+ occurs via a different mechanism than does the more robust Nmnat-mediated protection.
Why then is Nmnat enzymatic activity required for axonal protection, yet intracellular levels of its known product NAD+ are seemingly unimportant for this function? One potential explanation is that Nmnat enzymatic activity is required for production of metabolites other than NAD+ that are important for axonal protection. For example, mjNmnat structural studies demonstrate that it has structural similarity to glutaminyl-tRNA synthetase, glycerol-3-phosphate cytidylyltransferase, and phosphopantetheine adenylyltransferase (D’Angelo et al., 2000). These proteins belong to a superfamily of nucleotidyltransferase α/β phosphodiesterases that transfer a nucleotide monophosphate moiety onto various substrates. The ability of mjNmnat to protect axons along with its functional similarity to nucleotidyltransferases could be important clues for understanding how Nmnat-mediated axonal protection is achieved. Our results showing that Nmnat-mediated axonal protection proceeds despite inhibition of Nampt and the consequent decreased production of NMN and NAD+ is consistent with this idea. Furthermore, these experiments also cast doubt on the possibility that NAD+ synthesized by Nmnat is rapidly converted to an “axonal protective” metabolite, such that Nmnat-mediated increases in NAD+ itself are never observed. However, it is possible that newly synthesized NAD+ is compartmentalized, such that Nmnat overexpression results in increased NAD+ levels in a critical compartment but that overall intracellular levels are not affected. In this scenario, NAD+ levels in the “critical compartment” are sensitive to Nmnat-mediated production but are unaffected by decreases or increases in total intracellular NAD+ levels. Indeed, there is precedent for this as mitochondrial NAD+ levels appear to be maintained independently of cytoplasmic NAD+ levels (Yang et al., 2007). NAD+ biosynthetic enzymes may exist as a multienzyme complex similar to the recently described complex of enzymes involved in purine biosynthesis (i.e., the purinosome) (An et al., 2008). The dynamic clustering of these enzymes, which allows for compartmentalization of purine intermediates, may have a counterpart in NAD+ synthesis. Nmnat overexpression could alter the dynamics of such a complex and provide increased NAD+ to a particular compartment and/or process important for axonal stability. However, such compartmentalization is perhaps an unlikely explanation, as Nmnat proteins located within the nucleus, cytosol, or mitochondria can all promote axonal protection (Sasaki et al., 2006).
In summary, axonal protection by Nmnat requires enzymatic activity and can be provided by evolutionarily diverse enzymes capable of converting NMN to NAD+. However, increased intracellular NAD+ levels or severely reduced NAD+ and NMN levels do not mediate or block Nmnat-mediated axonal protection. These results indicate that decreased levels of NAD+ in degenerating axons are not the cause, but instead are result of the degeneration process. Finally, these results suggest that Nmnat mediates axonal protection by acting on additional substrates to produce molecules other than NAD+.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Neuroscience Blueprint Center Core Grant P30 NS057105 to Washington University, the Hope Center for Neurological Disorders, National Institutes of Health Grants NS040745 (J.M.), AG13730 (J.M.), and AI063399 (F.E.L.), Muscular Dystrophy Association Grant 10040 (J.M.), and Ministry of Health, Labour and Welfare of JAPAN (SHA4431). We thank Kelli Simburger, Amy Strickland, Amber Neilson, and Tim Fahrner for experimental assistance and members of the Milbrandt laboratory and Robert Baloh for their comments on this manuscript and helpful discussions. We also thank Dr. Gilbert de Murcia for providing PARP1-deficient mice.
Y.S., B.P.S.V., F.E.L., J.M., and Washington University may derive benefit from a licensing agreement with Sirtris Pharmaceuticals, which did not provide any support for this work.
References
- An S, Kumar R, Sheets ED, Benkovic SJ. Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science. 2008;320:103–106. doi: 10.1126/science.1152241. [DOI] [PubMed] [Google Scholar]
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem. 2005;280:36334–36341. doi: 10.1074/jbc.M508660200. [DOI] [PubMed] [Google Scholar]
- Bieganowski P, Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell. 2004;117:495–502. doi: 10.1016/s0092-8674(04)00416-7. [DOI] [PubMed] [Google Scholar]
- Chen Y, Stevens B, Chang J, Milbrandt J, Barres BA, Hell JW. NS21: Re-defined and modified supplement B27 for neuronal cultures. J Neurosci Methods. 2008;171:239–247. doi: 10.1016/j.jneumeth.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chini EN, Chini CC, Kato I, Takasawa S, Okamoto H. CD38 is the major enzyme responsible for synthesis of nicotinic acid-adenine dinucleotide phosphate in mammalian tissues. Biochem J. 2002;362:125–130. doi: 10.1042/0264-6021:3620125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockayne DA, Muchamuel T, Grimaldi JC, Muller-Steffner H, Randall TD, Lund FE, Murray R, Schuber F, Howard MC. Mice deficient for the ecto-nicotinamide adenine dinucleotide glycohydrolase CD38 exhibit altered humoral immune responses. Blood. 1998;92:1324–1333. [PubMed] [Google Scholar]
- Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889–898. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- Coleman MP, Conforti L, Buckmaster EA, Tarlton A, Ewing RM, Brown MC, Lyon MF, Perry VH. An 85-kb tandem triplication in the slow Wallerian degeneration (Wlds) mouse. Proc Natl Acad Sci U S A. 1998;95:9985–9990. doi: 10.1073/pnas.95.17.9985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Fang G, Beirowski B, Wang MS, Sorci L, Asress S, Adalbert R, Silva A, Bridge K, Huang XP, Magni G, Glass JD, Coleman MP. NAD(+) and axon degeneration revisited: Nmnat1 cannot substitute for Wld(S) to delay Wallerian degeneration. Cell Death Differ. 2007;14:116–127. doi: 10.1038/sj.cdd.4401944. [DOI] [PubMed] [Google Scholar]
- Cormack B. Directed mutagenesis using the polymerase chain reaction. Curr Protoc Mol Biol. 1997:8.5.1–8.5.10. doi: 10.1002/0471142727.mb0805s37. [DOI] [PubMed] [Google Scholar]
- D’Angelo I, Raffaelli N, Dabusti V, Lorenzi T, Magni G, Rizzi M. Structure of nicotinamide mononucleotide adenylyltransferase: a key enzyme in NAD(+) biosynthesis. Structure. 2000;8:993–1004. doi: 10.1016/s0969-2126(00)00190-8. [DOI] [PubMed] [Google Scholar]
- de Murcia JM, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, Oliver FJ, Masson M, Dierich A, LeMeur M, Walztinger C, Chambon P, de Murcia G. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci U S A. 1997;94:7303–7307. doi: 10.1073/pnas.94.14.7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelli M, Amici A, Carnevali F, Pierella F, Raffaelli N, Magni G. Identification and characterization of a second NMN adenylyltransferase gene in Saccharomyces cerevisiae. Protein Expr Purif. 2003;27:357–364. doi: 10.1016/s1046-5928(02)00645-9. [DOI] [PubMed] [Google Scholar]
- Garavaglia S, D’Angelo I, Emanuelli M, Carnevali F, Pierella F, Magni G, Rizzi M. Structure of human NMN adenylyltransferase. A key nuclear enzyme for NAD homeostasis. J Biol Chem. 2002;277:8524–8530. doi: 10.1074/jbc.M111589200. [DOI] [PubMed] [Google Scholar]
- Hasmann M, Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003;63:7436–7442. [PubMed] [Google Scholar]
- Hoopfer ED, McLaughlin T, Watts RJ, Schuldiner O, O’Leary DD, Luo L. Wlds protection distinguishes axon degeneration following injury from naturally occurring developmental pruning. Neuron. 2006;50:883–895. doi: 10.1016/j.neuron.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Jia H, Yan T, Feng Y, Zeng C, Shi X, Zhai Q. Identification of a critical site in Wld(s): essential for Nmnat enzyme activity and axon-protective function. Neurosci Lett. 2007;413:46–51. doi: 10.1016/j.neulet.2006.11.067. [DOI] [PubMed] [Google Scholar]
- Kaneko S, Wang J, Kaneko M, Yiu G, Hurrell JM, Chitnis T, Khoury SJ, He Z. Protecting axonal degeneration by increasing nicotinamide adenine dinucleotide levels in experimental autoimmune encephalomyelitis models. J Neurosci. 2006;26:9794–9804. doi: 10.1523/JNEUROSCI.2116-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- Lund FE. Signaling properties of CD38 in the mouse immune system: enzyme-dependent and -independent roles in immunity. Mol Med. 2006;12:328–333. doi: 10.2119/2006-00099.Lund. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur J Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- MacDonald JM, Beach MG, Porpiglia E, Sheehan AE, Watts RJ, Freeman MR. The Drosophila cell corpse engulfment receptor draper mediates glial clearance of severed axons. Neuron. 2006;50:869–881. doi: 10.1016/j.neuron.2006.04.028. [DOI] [PubMed] [Google Scholar]
- Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, Coleman MP. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A, Panchenko AR, Shoemaker BA, Thiessen PA, Geer LY, Bryant SH. CDD: a database of conserved domain alignments with links to domain three-dimensional structure. Nucleic Acids Res. 2002;30:281–283. doi: 10.1093/nar/30.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Görgün CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry VH, Brown MC, Lunn ER. Very slow retrograde and Wallerian degeneration in the CNS of C57BL/Ola mice. Eur J Neurosci. 1991;3:102–105. doi: 10.1111/j.1460-9568.1991.tb00815.x. [DOI] [PubMed] [Google Scholar]
- Press C, Milbrandt J. Nmnat delays axonal degeneration caused by mitochondrial and oxidative stress. J Neurosci. 2008;28:4861–4871. doi: 10.1523/JNEUROSCI.0525-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodromou C, Pearl LH. Recursive PCR: a novel technique for total gene synthesis. Protein Eng. 1992;5:827–829. doi: 10.1093/protein/5.8.827. [DOI] [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- Raffaelli N, Sorci L, Amici A, Emanuelli M, Mazzola F, Magni G. Identification of a novel human nicotinamide mononucleotide adenylyltransferase. Biochem Biophys Res Commun. 2002;297:835–840. doi: 10.1016/s0006-291x(02)02285-4. [DOI] [PubMed] [Google Scholar]
- Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279:50754–50763. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. Rational siRNA design for RNA interference. Nat Biotechnol. 2004;22:326–330. doi: 10.1038/nbt936. [DOI] [PubMed] [Google Scholar]
- Ryu EJ, Yang M, Gustin JA, Chang LW, Freimuth RR, Nagarajan R, Milbrandt J. Analysis of peripheral nerve expression profiles identifies a novel myelin glycoprotein, MP11. J Neurosci. 2008;28:7563–7573. doi: 10.1523/JNEUROSCI.1659-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Araki T, Milbrandt J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J Neurosci. 2006;26:8484–8491. doi: 10.1523/JNEUROSCI.2320-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- Sorci L, Cimadamore F, Scotti S, Petrelli R, Cappellacci L, Franchetti P, Orsomando G, Magni G. Initial-rate kinetics of human NMN-adenylyltransferases: substrate and metal ion specificity, inhibition by products and multisubstrate analogues, and isozyme contributions to NAD+ biosynthesis. Biochemistry. 2007;46:4912–4922. doi: 10.1021/bi6023379. [DOI] [PubMed] [Google Scholar]
- Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, He Z. A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol. 2005;170:349–355. doi: 10.1083/jcb.200504028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Kim SJ, Zhang Z, Tsai PC, Wisniewski KE, Mukherjee AB. ER and oxidative stresses are common mediators of apoptosis in both neurodegenerative and non-neurodegenerative lysosomal storage disorders and are alleviated by chemical chaperones. Hum Mol Genet. 2008;17:469–477. doi: 10.1093/hmg/ddm324. [DOI] [PubMed] [Google Scholar]
- Yahata N, Araki T. Differential axonal protection against Wallerian degeneration in vivo is observed in mice overexpressing family members of nicotinamide mononucleotide adenylyltransferase(Nmnat) Soc Neurosci Abstr. 2007;33:377.20. [Google Scholar]
- Yalowitz JA, Xiao S, Biju MP, Antony AC, Cummings OW, Deeg MA, Jayaram HN. Characterization of human brain nicotinamide 5′-mononucleotide adenylyltransferase-2 and expression in human pancreas. Biochem J. 2004;377:317–326. doi: 10.1042/BJ20030518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, de Cabo R, Sauve AA, Sinclair DA. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130:1095–1107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young GS, Choleris E, Lund FE, Kirkland JB. Decreased cADPR and increased NAD+ in the Cd38−/− mouse. Biochem Biophys Res Commun. 2006;346:188–192. doi: 10.1016/j.bbrc.2006.05.100. [DOI] [PubMed] [Google Scholar]
- Zhai RG, Cao Y, Hiesinger PR, Zhou Y, Mehta SQ, Schulze KL, Verstreken P, Bellen HJ. Drosophila NMNAT maintains neural integrity independent of its NAD synthesis activity. PLoS Biol. 2006;4:e416. doi: 10.1371/journal.pbio.0040416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai RG, Zhang F, Hiesinger PR, Cao Y, Haueter CM, Bellen HJ. NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature. 2008;452:887–891. doi: 10.1038/nature06721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Kurnasov O, Tomchick DR, Binns DD, Grishin NV, Marquez VE, Osterman AL, Zhang H. Structure of human nicotinamide/nicotinic acid mononucleotide adenylyltransferase. Basis for the dual substrate specificity and activation of the oncolytic agent tiazofurin. J Biol Chem. 2002;277:13148–13154. doi: 10.1074/jbc.M111469200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.