

Abstract
Transforming Growth Factor-beta (TGF-β) has roles in embryonic development, the prevention of inappropriate inflammation and tumor suppression. However, TGF-β signaling also regulates pathological epithelial to-mesenchymal transition (EMT), inducing or progressing a number of diseases ranging from inflammatory disorders, to fibrosis and cancer. However, TGF-β signaling does not proceed linearly but rather induces a complex network of cascades that mutually influence each other and cross-talk with other pathways to successfully induce EMT. Particularly, there is substantial evidence for cross-talk between αV integrins and TGF-β during EMT, and anti-integrin therapeutics are under development as treatments for TGF-β-related disorders. However, TGF-β’s complex signaling network makes the development of therapeutics to block TGF-β mediated pathology challenging. Moreover, despite our current understanding of integrins and TGF-β function during EMT, the precise mechanism of their role during physiological versus pathological EMT is not fully understood. This review focuses on the circle of regulation between αV integrin and TGF-β signaling during TGF-β induced EMT, which pose as a significant driver to many known TGF-β-mediated disorders.
EMT and pathogenesis: the good, the bad and the ugly
Epithelial to-mesenchymal transition (EMT) contributes to diverse developmental events including blastocyst implantation, gastrulation, generation of the neural crest, closure of the palate and normal wound healing [1–3]. Paradoxically, EMT also plays a role in pathological wound healing, tissue fibrosis, and aspects of the EMT-like transitions that occur in many types of cancer [4,5]. Morphologically, EMT is defined as the loss of epithelial characters such as apical-basolateral polarity, tight and adherens junctions and the ability to synthesize basement membranes. Concomitantly, these cells develop a fibroblastic morphology by rearranging their actin cytoskeleton, become migratory by forming filopodia and lamellopodia, interact with stromal extracellular matrices (ECM) due to changes in cell surface matrix receptors such as integrins, begin direct synthesis of stromal ECM and, in some cases, particularly in scar formation, become contractile myofibroblasts [3,6,7]. However, due to its functional diversity and complexity, scientists have long disagreed on a clear definition of EMT. A recent international meeting on EMT proposed to solve this dispute by classifying EMT into three different types: Type1, Type2 and Type3 (see figure 1) [8,9]. Type1 EMT occurs during the earliest stages of development, for example, during implantation and embryogenesis. Type2 EMT is defined as that which occurs in more mature epithelial tissues. In contrast to Type1 EMT, Type2 EMT can be triggered by inflammation or wound-healing responses and may lead to fibrosis. Finally, Type 3 EMT is the loss of epithelial and the gain of mesenchymal characters associated with cancer progression and metastasis.
Figure 1.
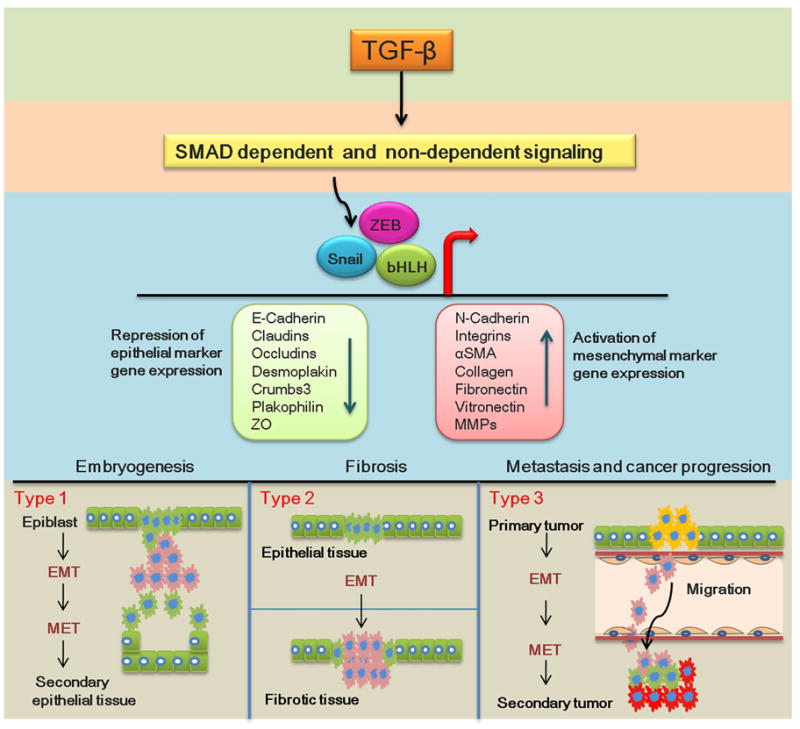
Epithelial mesenchymal transition (EMT) occurs when epithelial cells lose their epithelial cell characteristics and become mesenchymal. Mesenchymal cells can return to an epithelial phenotype, a process called mesenchymal-epithelial transition (MET). Type1 EMT: During embryogenesis, the primitive epithelium (the epiblast) undergoes EMT forming primary mesenchyme that can migrate and undergo MET to form secondary epithelia that differentiate into new epithelial tissues. Type2 EMT: In mature or adult tissues, epithelial cells can also undergo EMT following local cellular disorganization caused by various stressors, inflammation or wounding but fail to undergo MET leading to fibroblast production and finally fibrosis. Type3 EMT: Epithelial cancer cells can undergo EMT to acquire a more migratory mesenchymal phenotype that allows them to invade secondary epithelia and proliferate as secondary tumors. During the process, migrating mesenchymal cells will have to intravasate, migrate through vasculature and extravasate to invade secondary tissue, undergo MET and proliferate forming secondary tumors. Green (epithelial cells), Pink (mesenchymal), Yellow (primary tumor, Red (secondary tumor)
Transforming Growth Factor-β-a central Player in EMT
While several growth factors participate in EMT [10], Transforming Growth Factor-beta (TGF-β) is the most studied, with evidence suggesting that it can drive all three classes of EMT [11,12]. Further, TGF-β also regulates a wide array of other cellular processes including cell division, differentiation, motility, apoptosis and tumor suppression [13,14]. There are three known isoforms of TGF-β in mammals, TGF-β1, TGF-β2 and TGF-β3 [15]. The expression and function of all three isoforms varies dramatically among tissues and can even vary from species to species [16,17]. Therefore, the mechanism by which TGF-β function is regulated and its signals transmitted within cells is very complex. TGF-β is synthesized within the secretory pathway as a precursor, which is cleaved to form the functional 25kDa TGF-β homodimer and the latency-associated peptide (LAP). LAP and TGF-β homodimer remain associated as a non-covalently bound complex known as the small latent complex (SLC). The SLC remains in the secretory pathway until it is bound by Latent TGF-β-BindingProtein (LTBP) to form the Large Latent Complex (LLC). The LLC is secreted and bound either covalently or non-covalently to the ECM [18,19]. In most cases, the LLC will remain in the ECM until it is further processed to release active TGF-β [20]. Upon its release, the active TGF-β cytokine ignites TGF-β signaling by first binding to the type II TGF-β receptor, inducing a conformational change in its serine/threonine kinase domain and recruiting the type I TGF-β receptor to form the active receptor complex. Once the receptor complex is formed, the type II receptor kinase phosphorylates multiple serine and threonine residues in the TTSGSGSG sequence of the cytoplasmic GS region of the type I receptor, leading to the activation of TGF-β induced signaling [21].
The best studied pathway mediating TGF-β function is the SMAD dependent (canonical) pathway which initiates when receptor-regulated SMAD 2 and/or 3 (r-SMAD2/3) is recruited to the activated TGF-β receptor by the SMAD anchor for receptor activation (SARA) [22]. The type I TGF-β receptor then phosphorylates r-SMAD2/3 inducing a conformational changes in the MH2 domain of r-SMAD2/3 and its subsequent dissociation from the receptor complex. Once phosphorylated and released, the phosphorylated r-SMAD2/3 attains a high affinity towards the co-SMAD (SMAD4) and binds to it [23]. The complex formed by r-SMAD2/3 and co-SMAD4 translocates to the nucleus where, in the case of EMT, it represses epithelial gene transcription while transcriptionally activating the expression of mesenchymal genes as well as other transcription factors capable of regulating EMT, such as members of Snail, ZEB, and bHLH families [16,24]. This canonical pathway can also induces EMT via cross talk with other signaling pathways. For instance, SMAD-mediated TGF-β signaling can activate integrin linked kinase (ILK) which allows ILK to phosphorylate GSK-3β and Akt, leading to β-catenin nuclear translocation and activation of other transcription factors, resulting in EMT of renal tubular epithelial cells [25,26]. TGF-β can also mediate EMT through its non-canonical (SMAD independent) pathways [27]. For instance, TGF-β induced activation of Erk/MAP kinase, Rho GTPase and the PI3 kinase/Akt pathways can result in all three EMT types [28,29]. This is an area of active investigation; more non-canonical pathways and additional levels of regulation are still being discovered [30].
Recently it was shown that TGF-β signaling can regulate the expression of microRNAs which play a crucial role in regulating EMT [31]. MicroRNAs belonging to the miR-200 and miR-205family can prevent EMT by downregulating the EMT associated transcription factors ZEB1 andSIP1 [32]. Since miR-200 and miR-205 expression is reduced upon TGF-β stimulation, this suggests that TGF-β is a key regulator of the expression of microRNAs that block EMT [33]. It is likely that other microRNAs either synergize with or antagonize TGF-β signaling during TGF-β induced EMT, thus, investigating how TGF-β signaling regulates microRNA expression during EMT is important for the eventual development of therapeutics that target TGF-β function.
Changes in cell-cell/cell-ECM adhesion in EMT- The role of integrins during EMT
Interactions between cells and ECM convey micro-environmental cues that influence cell behavior and function [34,35]. Integrins are heterodimeric transmembrane proteins consisting of one α- and one β-subunit. Integrins facilitate interactions between cells and the ECM and play major roles in cell proliferation, differentiation, adhesion and migration [36–38]. At least 24 different integrin heterodimers exist which recognize diverse components of the ECM including laminin, collagen, heparan sulfate proteoglycans, vitronectin, fibronectin, osteopontin, bone sialoprotein, thrombospondin, fibrinogen, and tenascin [39,40]. Further, changes in ECM composition, such as the increased expression of fibronectin, vitronectin and Type I collagen seen during EMT, can switch integrins from an inactive low affinity to an active high affinity ligand binding state [41,42]. This can result in outside–in integrin signaling mediated via adaptor proteins that bind to the cytoplasmic tail of integrins such as ILK, paxillin, FAK and PINCH. Likewise, intracellular cues can also activate integrin associated proteins affecting integrin affinity for extracellular ligands, a process known as inside-out signaling [43].
Thus, in the case of EMT, perturbations of the ECM microenviroment or intracellular cues can persuade integrins to dictate adhesion changes between cells and the ECM (and in some cases cells to cells), subsequently inducing disassembly of tight and adherens junctions, dissolution of desmosomes, actin reorganization and loss of epithelial apical-basal polarity which are all associated with EMT. In extremis, these changes initiate focal adhesion complex formation leading to cell migration and invasion [44]. For example, β5 integrin regulates cell-cell/cell-ECM adhesion changes upon TGF-β signaling and β5 integrin depletion reduces the invasiveness of breast carcinoma cells by impairing the dissociation of tight junctions and/or reducing cell-ECM adhesion [45]. Further, β1 integrin engagement with collagen type I results in loss of E-cadherin and indirect upregulation of N-cadherin, suggesting that integrin activation can directly result in EMT [46,47]. Integrins can also facilitate changes in cell-ECM contact during EMT by binding to membrane type 1-matrix metalloproteinase (MT1-MMP) to control their function [48].
TGF-β as a regulator of integrin expression during EMT
Many αV integrins, especially αVβ3, αVβ5 and αVβ6 are expressed at low levels in healthy epithelial tissues but upregulate during both type2 and type3 EMT [49–51]. For example, αVβ6 integrin is highly upregulated during pulmonary inflammation, fibrosis, and cervical squamous cell carcinoma, where its increased expression correlates with poor prognosis [52,53]. This implies that the TGF-β signaling that induces EMT also plays an essential role in the upregulation of integrin expression. A recent study showed that TGF-β1 signaling during renal fibrosis enhances SMAD3 binding to the β1 integrin promoter, triggering an upregulation of β1 integrin gene expression. Interestingly, knockdown or functional blockade of either β1 integrin or SMAD3, slowed the progression of renal fibrosis [54]. Another study showed that TGF-β1 induced signaling increased both β3 integrin subunit mRNA and protein levels as well as surface expression of αVβ3 in human lung fibroblasts via a β3 integrin, c-Src-, and p38 MAPK-dependent pathway [55].
TGF-β mediated integrin upregulation can also be indirect. TGF-β is a key regulator of ECM production, deposition and remodeling [56,57],while integrin expression is partially regulated by the cellular microenvironment [42]. Thus, TGF-β mediated upregulation of ECM protein expression can indirectly stimulate integrin expression. For example, TGF-β induced fibronectin expression in MRC-5 human myofibroblasts subsequently induces the expression of αVβ3, αVβ1, and α1β1 integrins [58]. TGF-β induced signaling can also regulate integrin activation states by mediating the expression of integrin associated adaptor proteins such as PINCH-1, which binds to integrin-linked kinase (ILK) and plays an essential role in integrin signaling [59–61]. Further, TGF-β signaling can directly activate integrins by phosphorylating the cytoplasmic tail of the β-integrin subunit. A recent study reports that TGF-β1 promotes vascular invasion by phosphorylating β1 integrin’s threonine 788–789 via the TGF-β receptor, inducing inside out signaling that ultimately drives hepatocellular carcinoma invasion [62]. Thus, by regulating integrin expression and function, TGF-β gains the ability to regulate cell biology in a tissue and cell specific fashion. However, in spite of the emerging understanding of how TGF-β signaling can regulate integrins during EMT progression, it is much less clear why these processes are tissue and cell specific.
αV integrins activate TGF-β
TGF-β is secreted in a latent form which must be activated extracellularly to efficiently trigger receptor-mediated TGF-β signaling. Diverse activation mechanisms have been demonstrated which allow for regulation of TGF-β function in different cellular/tissue contexts [63]. Among the three TGF-β isoforms, only the LAPs of TGF-β1 and TGF-β3 contain an integrin binding motif, arginine–glycine–aspartic acid (RGD). Notably, many αV integrins including αVβ1, αVβ3, αVβ5, αVβ6 and αVβ8 can interact with this RGD sequence resulting in activation of TGF-β1 and TGF-β3 [52,64–67]. Not surprisingly, mice with a nonfunctional variant of the RGD sequence in their TGF-β1 LAP express normal levels of latent TGF-β1, but display features similar to that of TGF-β1 knockouts [52,68]. Moreover, mice lacking αVβ6 and αVβ8, phenocopy TGF-β1 and TGF-β3 knockouts confirming the importance of αV integrins in TGF-β1 and TGF-β3 activation [69]. Furthermore, antibody-mediated blockade of αV integrin function, particularly αVβ6, downregulates the TGF-β induced EMT and inflammation known to causes fibrosis, metastasis and cancer [70–73]. Two different models of how αV integrins can activate TGF-β are proposed, both of which have significant experimental support. It appears that the choice of TGF-β activation mechanism is cell type specific and is influenced by whether the appropriate integrin is expressed and the specific cell physiological context. However, it is not known whether the use of a particular TGF-β activating mechanism influences the resulting TGF-β signaling.
Conformational change activation mechanism
Upon binding to the LAP of TGF-β1 and TGF-β3, αV integrins can exert adhesion-mediated cell forces inducing conformational changes of the LAP, which results in structural deformation of the latent complex and liberation of active TGF-β [52]. Drugs which block myofibroblast contraction such as ML-7, blebbistatin, cytochalasin D and the α-SMA contraction inhibitor SMA-FP, also inhibited activation of TGF-β1 by contraction-inducing drugs [74,75]. Analogously, studies that inhibited αVβ5 integrin function by forming αvβ5 -Thy1 complexes also inhibit contraction-induced latent TGF-β1 activation [76]. Since TGF-β signaling induces a contractile cytoskeleton along with α-SMA expression during EMT, this can result in further TGF-β activation [77]. While this direct activation does not require proteolysis of either the LAP or LTBP, it does require a mechanically stiff ECM and contractile cytoskeleton which provides sufficient forces to liberate TGF-β from its latent complex [20,78].
Conformational change activation mechanism with proteolysis
Not all integrin interactions with the LAP directly activate TGF-β, particularly in the absence of a stiff ECM. Moreover, active TGF-β must be physically proximal to a type II TGF-β receptor for activation of TGF-β signaling. Notably, several MMPs such MMP-9 and MMP-2 can proteolytically cleave the LAP and/or LTBP to liberate TGF-β from its ECM bound stores [79,80]. Further, αV integrins can interact with MMPs, such as MMP2 and MMP9, to tether them to the cell surface [81,82]. Thus, in certain physiological contexts, αV integrins can simultaneously promote proximity of MMPs to the LAP and sequester the LLC close to the type II TGF-β receptor. Depending on the cell/tissue type, this mechanism can function without significant cell traction [78,83], although a contractile cytoskeleton and a mechanically resistant ECM can also participate [75,78].
Overall, αV integrins can activate TGF-β, which induces EMT, which then in turn creates an ideal microenvironment for further unregulated TGF-β activation resulting in pathological EMT. Recently, the known complexity of this mechanism has been increased by a report showing that αV integrins also interact with the TGF-β co-receptor, endoglin, and this interaction can enhance αV integrin mediated TGF-β activation[84]. However, the extent to which cell type, magnitude of the ECM’s mechanical resistance, the relative proximity of latent TGF-β to proteinases and other known TGF-β activators such as pH, thrombospondin-1 and reactive oxygen species [20] play a role, remains unclear.
TGF-β and integrin signaling cross talk during EMT
Inactive integrins are not usually bound to the ECM or tightly linked to the actin cytoskeleton, instead, they are scattered diffusely over the cell surface. However, upon their binding to ECM, a purposeful association with the actin cytoskeleton is induced, leading to integrin clustering into focal adhesion complexes, which, depending on the properties of the ECM, will instigate integrin signaling [85,86]. This signal is propagated to the intracellular integrin associated adaptor proteins such as ILK, Src, PTKs and FAK; which subsequently activate other downstream players such as MAPK, Ras/Rho, small GTPases, PI3K and AKT [87]. On the other hand, canonical TGF-β signaling initiates upon receptor activation and SMAD phosphorylation but can indirectly activate MAPK and PI3K pathways as well [88]. In addition, non-canonical TGF-β signaling can directly activate MAPK, PI3, Ras/Rho and small GTPase pathways [30]. Therefore, cross-talk between TGF-β and integrin signaling can occur downstream of initial receptor activation and regulate various cellular processes [89] which may override the tumor suppressing function of TGF-β [90–92] or lead to other TGF-β associated disorders [93]. TGF-β signaling can also result in either direct or indirect phosphorylation of integrin adaptor proteins, while integrins can associate with receptor tyrosine kinases (RTKs) to activate TGF-β signaling resulting in tumor invasion and metastasis [51]. Further, TGF-β1 signaling can induce tyrosine phosphorylation of FAK including its autophosphorylation site, Tyr-397, leading to inside-out integrin signaling that subsequently induces myofibroblast differentiation in fibrotic diseases [87]. TGF-β1 induced signaling can also phosphorylate other proteins such as the p85 subunit of PI3K, and serine 473 of Akt resulting in the upregulation of αV integrin expression that is necessary for the migration of human chondrosarcoma cells [88].
On the other hand, integrin association with RTKs may activate the integrin–receptor-tyrosine-kinase signaling that is necessary for development, tumor invasion and metastasis. For example, TGF-β signaling can lead to the transcriptional upregulation of αVβ3 integrin expression which then enhances the functional interaction between the type II TGF-β receptor and αVβ3 integrins; a situation that potentiates the proliferative effects of TGF-β induced EMT in human lung fibroblasts [94]. Type II TGF-β receptor and αVβ3 integrin interactions can also auto-stimulate type II TGF-β receptor phosphorylation on its tyrosine residues via Src; initiating a TGF-β signaling induced stimulation of MAPKs that leads to EMT and invasion in epithelial cells [95]. Overall, more potential points of cross talk between integrin and TGF-β signaling are being discovered, yet even now, the mechanistic differences that allow TGF-β to suppress tumorigenic processes in healthy tissue and promote tumor formation by cancer cells still remain unclear.
Disease perspectives
Active TGF-β has a high affinity for its receptors, so, in most cases, a released/active TGF-β ligand will initiate a TGF-β signaling cascade as long as TGF-β receptors are within reach [96]. Since different cellular functions require distinct levels of TGF-β signaling [78], tight regulation of latent TGF-β activation is necessary to prevent diverse diseases including inflammation, autoimmune disorders, fibrosis, cancer and cataract [97–101]. Conversely, inadequate levels of active TGF-β due to mutation of either the TGF-β genes or those for TGF-β activators can lead to pathology. For instance, TGF-β1 deficient mice exhibit a multifocal, mixed inflammatory cell response and tissue necrosis, leading to organ failure and death 20 days after birth [102,103]. In humans, inadequate TGF-β signaling can result in various disorders including brain hemorrhage and immune system-associated disorders [104,105]. Notably, restoring normal TGF-β signaling and/or inhibiting its inappropriate expression in experimental animals reverses some TGF-β associated pathologies and stands as promising therapeutic approach (See table 1). TGF-β induced signaling is also known to destabilize E-cadherin mediated cell-cell adhesion during EMT [106]. Fascinatingly, a study in renal fibrosis revealed that bone morphogenic protein (BMP)-7 can reverse TGF-β1 induced EMT by restoring E-cadherin expression hence halting EMT progression. These results suggest that cross talk between BMP-7 and TGF-β is essential to regulate pathological EMT [107]. αV integrins can activate TGF-β by binding to its LAP and evidence of such activation has been linked in EMT progression. Therefore, blocking the undesirable activities of αV integrins without interfering with their beneficial functions could impede EMT progression during cancer, wound healing and fibrosis.
Table 1.
Attempts to target αV integrin function as a therapeutic strategy to treat TGF-β associated disorders
| Integrin | Disorder | Experimental findings |
|---|---|---|
| αVβ3 αVβ5 |
|
|
| αVβ6 |
|
|
| αVβ8 |
|
|
Integrin antagonists show clinical promise for the treatment of TGF-β induced EMT associated disorders such as inflammation, fibrosis and cancer [108]. Most of the therapeutic approaches currently under investigation target integrin function using anti-integrin agents including both naturally occurring and engineered peptides that can mimic their RGD ligand, or antibodies that can act as integrin antagonists [109–111]. For example, clinical administration of a peptide antagonist of the αVβ3 receptor successfully inhibits pathological angiogenesis seen in cancer, proliferative retinopathy, rheumatoid arthritis, and psoriasis [112,113]. Likewise, TGF-β-mediated enhancement of glioma cell migration via the upregulation of αVβ3 integrin expression is abrogated by echistatin, a Arg-Gly-Asp (RGD) containing snake venom which is a potent antagonist of αVβ3integrin [114].
Several integrin targeted therapies are in clinical development for the treatment of cancer [119]. For instance, Cilengitide or EMD12197 (Merck KGaA, Darmstadt, Germany), is a small cyclic RGD designed peptide that selectively and competitively antagonizes ligand binding to αVβ3 and αVβ5 [120] which is being evaluated in a phase III clinical study for treatment of glioblastoma [121]. A number of monoclonal antibodies are also in clinical development. CNTO 95 is a fully humanized monoclonal antibody targeting αV integrin which shows anti-tumor and anti-metastatic activity in animal models and is in a Phase I clinical trial for the treatment of solid tumors [122,123]. Likewise, Vitaxin, also known as MEDI-522 or Abegrin, is also a humanized monoclonal antibody that can block the interaction of αVβ3 with various ligands such as osteopontin, latent TGF-β and vitronectin [116]. Vitaxin is currently in clinical trials for the treatment of stage IV metastatic melanoma and androgen-independent prostate cancer [124]. Notably, MedImmune Inc. ended advanced human testing of Vitaxin to treat rheumatoid arthritis and psoriasis in 2004 because it failed to show clinical benefits in initial studies [125]. More recently, it was shown that pre-treament of osteoclasts with macrophage colony stimulating factor (M-CSF), which is known to activate αVβ3, enhanced Vitaxin’s inhibitory effect. Furthermore, the PI3-kinase inhibitor wortmannin abolished M-CSF’s effects on the action of Vitaxin suggesting that Vitaxin’s inhibitory effects require an activated form of αVβ3 integrin and that PI3-kinase signaling is involved in the process [126]. On the other hand, numerous studies have shown that PI3K-Akt signaling is involved in TGF-β induced EMT and cell migration [127,128]. This exemplifies how understanding the cross-talk between αV integrins and TGF-β signaling can enhance the therapeutic potential of not only Vitaxin, but other integrin antagonists as well, to make better and more successful therapeutics.
Completing the circle
Integrins and their signaling can regulate EMT by both perturbing cell adhesion and stimulating EMT associated gene expression. In addition, integrins containing the αV subunit can activate latent TGF-β to result in TGF-β induced EMT. Concomitantly, TGF-β signaling can activate integrins and also upregulate integrin expression. TGF-β signaling can also induce a contractile cytoskeleton and a stiff cellular microenvironment to further facilitate latent TGF-β activation by αV integrins. This altogether creates a feed-forward circle of cross regulation between αV integrins and TGF-β (Figure 2) that can drive the EMT responsible for TGF-β associated disorders ranging from inflammation, fibrosis and cancer. Since experimental approaches that interfere with αV integrin function block TGF-β mediated disease pathogenesis in experimental models; elucidation of the molecular participants in both physiological and pathological EMT as well as differences in their regulation, may lead to selective therapies for TGF-β associated disorders.
Figure 2.

αV integrins recognize a RGD motif present in the LAP of TGF-β. This binding induces either adhesion-mediated cell forces and/or brings latent TGF-β into the proximity of MMPs which consequently lead to the liberation/activation of the TGF-β homodimer from its latent complex. Upon activation, the TGF-β homodimer will bind to the Type II TGF-β receptor initiating TGF-β-Smad signaling which upregulates the expression of αV integrins in addition to that of other EMT markers. These newly formed integrins can liberate more TGF-β from its latent complex, sustaining and reinforcing TGF-β induced EMT progression. This cooperative feed forward loop between αV integrins and TGF-β can lead to the unregulated TGF-β signaling responsible for a number of TGF-β-associated disorders.
Acknowledgments
This work was supported by National Eye Institute Grant # EY015279 and its training supplement # EY015279 S1. Special thanks to all members of Dr. Melinda Duncan’s laboratory group for critical reading of the manuscript.
Glossary
- AKT
Akt family serine/threonine protein kinase
- α-SMA
α-smooth muscle actin
- ECM
Extracellular matrix
- EMT
Epithelial-to-mesenchymal transition
- FAK
Focal adhesion kinase
- ILK
Integrin-linked kinase
- LAP
Latency-associated protein
- LLC
Large latent complex
- LTBP
Latent TGF-β binding protein
- MAPK
Mitogen-activated protein kinase
- MET
Mesenchymal to-epithelial transition
- MMP
Matrix metalloproteinase
- PI(3)K
Phosphatidylinositol-3-kinase
- PINCH
Five LIM domains containing focal adhesion protein
- PTKs
Protein tyrosine Kinases
- RAS
RAS protein subfamily of small GTPases
- RGD
Arginine–glycine–aspartate
- ROCK
Rho-associated kinase
- RTKs
Receptor tyrosine Kinases
- SIP1
SMAD interacting protein 1
- SLC
Small latent complex
- SMAD
SMA-Mothers against decapentaplegic family member
- r-SMAD
Receptor-regulated SMAD
- Src
Src family of non-receptor tyrosine kinases
- TGF-β
Transforming growth factor-beta
Footnotes
Conflicts of interest:
The authors confirm that there are no conflicts of interest.
References
- 1.Duband JL, Thiery JP. Appearance and distribution of fibronectin during chick embryo gastrulation and neurulation. Developmental Biology. 1982;94:337–50. doi: 10.1016/0012-1606(82)90352-9. [DOI] [PubMed] [Google Scholar]
- 2.Vićovac L, Aplin JD. Epithelial-mesenchymal transition during trophoblast differentiation. Acta Anat (Basel) 1996;156:202–16. doi: 10.1159/000147847. [DOI] [PubMed] [Google Scholar]
- 3.Hay ED. An overview of epithelio-mesenchymal transformations. Acta Anat. 1995;154:8–20. doi: 10.1159/000147748. [DOI] [PubMed] [Google Scholar]
- 4.Chaffer CL, Thompson EW, Williams ED. Mesenchymal to Epithelial Transition in Development and Disease. Cells Tissues Organs. 2007;185:7–19. doi: 10.1159/000101298. [DOI] [PubMed] [Google Scholar]
- 5.Jose Miguel L-N, Nieto MA. Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Molecular Medicine. 2009;1:303–14. doi: 10.1002/emmm.200900043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. The Journal of Clinical Investigation. 2003;112:1776–84. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thiery JP, Acloque H, Huang RY, et al. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 8.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. The Journal of Clinical Investigation. 2009;119:1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. The Journal of Clinical Investigation. 2009;119:1429–37. doi: 10.1172/JCI36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strutz F, Zeisberg M, Ziyadeh FN, et al. Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int. 2002;61:1714–28. doi: 10.1046/j.1523-1755.2002.00333.x. [DOI] [PubMed] [Google Scholar]
- 11.Nawshad A, LaGamba D, Polad A, et al. Transforming Growth Factor-L̂2 Signaling during Epithelial-Mesenchymal Transformation: Implications for Embryogenesis and Tumor Metastasis. Cells Tissues Organs. 2005;179:11–23. doi: 10.1159/000084505. [DOI] [PubMed] [Google Scholar]
- 12.Bhowmick NA, Ghiassi M, Bakin A, et al. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell. 2001;12:27–36. doi: 10.1091/mbc.12.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taipale J, Saharinen J, Keski-Oja J. Extracellular matrix-associated transforming growth factor-beta: role in cancer cell growth and invasion. Cancer Res Adv. 1998:75. doi: 10.1016/s0065-230x(08)60740-x. [DOI] [PubMed] [Google Scholar]
- 14.Yue J, Mulder KM. Transforming growth factor-[beta] signal transduction in epithelial cells. Pharmacology & Therapeutics. 2001;91:1–34. doi: 10.1016/s0163-7258(01)00143-7. [DOI] [PubMed] [Google Scholar]
- 15.Derynck R, Lindquist PB, Lee A, et al. A new type of transforming growth factor-beta, TGF-beta 3. EMBO J. 1988;7:3737–43. doi: 10.1002/j.1460-2075.1988.tb03257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Massagué J. TGF-β in Cancer. Cell. 2008;134:215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Massague J, Cheifetz S, Laiho M, et al. Transforming growth factor-beta. Cancer Surv. 1992;12:81–103. [PubMed] [Google Scholar]
- 18.Nunes I, Gleizes P-E, Metz CN, et al. Latent Transforming Growth Factor-β Binding Protein Domains Involved in Activation and Transglutaminase-dependent Cross-Linking of Latent Transforming Growth Factor-β. The Journal of Cell Biology. 1997;136:1151–63. doi: 10.1083/jcb.136.5.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Q, Sivakumar P, Barley C, et al. Potential Role for Heparan Sulfate Proteoglycans in Regulation of Transforming Growth Factor-β (TGF-β) by Modulating Assembly of Latent TGF-β-binding Protein-1. Journal of Biological Chemistry. 2007;282:26418–30. doi: 10.1074/jbc.M703341200. [DOI] [PubMed] [Google Scholar]
- 20.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGF{beta} activation. J Cell Sci. 2003;116:217–24. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 21.Shi Y, Massagué J. Mechanisms of TGF-[beta] Signaling from Cell Membrane to the Nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 22.Wu G, Chen Y-G, Ozdamar B, et al. Structural Basis of Smad2 Recognition by the Smad Anchor for Receptor Activation. Science. 2000;287:92–7. doi: 10.1126/science.287.5450.92. [DOI] [PubMed] [Google Scholar]
- 23.Souchelnytskyi S, Rönnstrand L, Heldin C-H, et al. Protein Kinase Protocols. Humana Press; 2001. Phosphorylation of Smad Signaling Proteins by Receptor Serine/Threonine Kinases; pp. 107–20. [DOI] [PubMed] [Google Scholar]
- 24.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–28. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 25.Li Y, Tan X, Dai C, et al. Inhibition of integrin-linked kinase attenuates renal interstitial fibrosis. J Am Soc Nephrol. 2009;20:1907–18. doi: 10.1681/ASN.2008090930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Delcommenne M, Tan C, Gray V, et al. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci U S A. 1998;95:11211–6. doi: 10.1073/pnas.95.19.11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-[beta] family signalling. Nature. 2003;425:577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 28.Zavadil J, Bitzer M, Liang D, et al. Genetic programs of epithelial cell plasticity directed by transforming growth factor-beta. Proc Natl Acad Sci USA. 2001;98:6686–91. doi: 10.1073/pnas.111614398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu J, Lamouille S, Derynck R. TGF-[beta]-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–72. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128–39. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zavadil J, Narasimhan M, Blumenberg M, et al. Transforming Growth Factor-β and microRNA:mRNA Regulatory Networks in Epithelial Plasticity. Cells Tissues Organs. 2007;185:157–61. doi: 10.1159/000101316. [DOI] [PubMed] [Google Scholar]
- 32.Gregory PA, Bert AG, Paterson EL, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 33.Pandit KV, Corcoran D, Yousef H, et al. Inhibition and Role of let-7d in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2010;182:220–9. doi: 10.1164/rccm.200911-1698OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slavkin HC. Combinatorial process for extracellular matrix influences on gene expression: a hypothesis. J Craniofac Genet Dev Biol. 1982;2:179–89. [PubMed] [Google Scholar]
- 35.Lukashev ME, Werb Z. ECM signalling: orchestrating cell behaviour and misbehaviour. Trends in Cell Biology. 1998;8:437–41. doi: 10.1016/s0962-8924(98)01362-2. [DOI] [PubMed] [Google Scholar]
- 36.Hynes RO. Integrins: Bidirectional, Allosteric Signaling Machines. Cells. 2002;110:673–87. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 37.Hynes RO. INTEGRINS - VERSATILITY, MODULATION, AND SIGNALING IN CELL-ADHESION. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 38.Hynes RO. Targeted mutations in cell adhesion genes: What have we learned from them? Developmental Biology. 1996;180:402–12. doi: 10.1006/dbio.1996.0314. [DOI] [PubMed] [Google Scholar]
- 39.Ruoslahti E. RGD and other recognition sequences for integrins. Annual Review of Cell and Developmental Biology. 1996;12:697–715. doi: 10.1146/annurev.cellbio.12.1.697. [DOI] [PubMed] [Google Scholar]
- 40.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Imamichi Y, Menke A. Signaling Pathways Involved in Collagen-Induced Disruption of the E-Cadherin Complex during Epithelial-Mesenchymal Transition. Cells Tissues Organs. 2007;185:180–90. doi: 10.1159/000101319. [DOI] [PubMed] [Google Scholar]
- 42.Boudreau NJ, Jones PL. Extracellular matrix and integrin signalling: the shape of things to come. Biochem J. 1999;339 (Pt 3):481–8. [PMC free article] [PubMed] [Google Scholar]
- 43.Anthis NJ, Campbell ID. Trends in Biochemical Sciences. 2011. The tail of integrin activation. In Press, Corrected Proof. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer and Metastasis Reviews. 2009;28:15–33. doi: 10.1007/s10555-008-9169-0. [DOI] [PubMed] [Google Scholar]
- 45.Bianchi A, Gervasi ME, Bakin A. Role of β5-integrin in epithelial-mesenchymal transition in response to TGF-β. Cell Cycle. 2010;9:1647–59. doi: 10.4161/cc.9.8.11517. [DOI] [PubMed] [Google Scholar]
- 46.Koenig A, Mueller C, Hasel C, et al. Collagen Type I Induces Disruption of E-Cadherin–Mediated Cell-Cell Contacts and Promotes Proliferation of Pancreatic Carcinoma Cells. Cancer Research. 2006;66:4662–71. doi: 10.1158/0008-5472.CAN-05-2804. [DOI] [PubMed] [Google Scholar]
- 47.Shintani Y, Fukumoto Y, Chaika N, et al. Collagen I mediated up-regulation of N-cadherin requires cooperative signals from integrins and discoidin domain receptor 1. The Journal of Cell Biology. 2008;180:1277–89. doi: 10.1083/jcb.200708137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gonzalo P, Moreno V, Gálvez BG, et al. MT1-MMP and integrins: Hand-to-hand in cell communication. BioFactors. 2010;36:248–54. doi: 10.1002/biof.99. [DOI] [PubMed] [Google Scholar]
- 49.Breuss JM, Gallo J, DeLisser HM, et al. Expression of the beta 6 integrin subunit in development, neoplasia and tissue repair suggests a role in epithelial remodeling. J Cell Sci. 1995;108:2241–51. doi: 10.1242/jcs.108.6.2241. [DOI] [PubMed] [Google Scholar]
- 50.Breuss JM, Gillett N, Lu L, et al. Restricted distribution of integrin beta 6 mRNA in primate epithelial tissues. J Histochem Cytochem. 1993;41:1521–7. doi: 10.1177/41.10.8245410. [DOI] [PubMed] [Google Scholar]
- 51.Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5:816–26. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- 52.Munger JS, Huang X, Kawakatsu H, et al. A Mechanism for Regulating Pulmonary Inflammation and Fibrosis: The Integrin [alpha]v[beta]6 Binds and Activates Latent TGF [beta]1. Cell. 1999;96:319–28. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 53.Hazelbag S, Kenter GG, Gorter A, et al. Overexpression of the αvβ6 integrin in cervical squamous cell carcinoma is a prognostic factor for decreased survival. The Journal of Pathology. 2007;212:316–24. doi: 10.1002/path.2168. [DOI] [PubMed] [Google Scholar]
- 54.Yeh Y-C, Wei W-C, Wang Y-K, et al. Transforming Growth Factor-[beta]1 Induces Smad3-Dependent [beta]1 Integrin Gene Expression in Epithelial-to-Mesenchymal Transition during Chronic Tubulointerstitial Fibrosis. Am J Pathol. 2010;177:1743–54. doi: 10.2353/ajpath.2010.091183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pechkovsky DV, Scaffidi AK, Hackett TL, et al. Transforming Growth Factor beta 1Induces αVβ3 Integrin Expression in Human Lung Fibroblasts via a β3 Integrin-, c-Src-, and p38 MAPK-dependent Pathway. Journal of Biological Chemistry. 2008;283:12898–908. doi: 10.1074/jbc.M708226200. [DOI] [PubMed] [Google Scholar]
- 56.Schiller M, Javelaud D, Mauviel A. TGF-[beta]-induced SMAD signaling and gene regulation: consequences for extracellular matrix remodeling and wound healing. Journal of Dermatological Science. 2004;35:83–92. doi: 10.1016/j.jdermsci.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 57.Verrecchia F, Mauviel A. Transforming Growth Factor-[bgr] Signaling Through the Smad Pathway: Role in Extracellular Matrix Gene Expression and Regulation. 2002;118:211–5. doi: 10.1046/j.1523-1747.2002.01641.x. [DOI] [PubMed] [Google Scholar]
- 58.Honda E, Yoshida K, Munakata H. Transforming Growth Factor-beta; Upregulates the Expression of Integrin and Related Proteins in MRC-5 Human Myofibroblasts. The Tohoku Journal of Experimental Medicine. 2010;220:319–27. doi: 10.1620/tjem.220.319. [DOI] [PubMed] [Google Scholar]
- 59.Wu C. Integrin-linked kinase and PINCH: partners in regulation of cell-extracellular matrix interaction and signal transduction. Journal of cell science. 1999;112 (Pt 24):4485–9. doi: 10.1242/jcs.112.24.4485. [DOI] [PubMed] [Google Scholar]
- 60.Wu C. PINCH, N(i)ck and the ILK: network wiring at cell-matrix adhesions. Trends in Cell Biology. 2005;15:460–6. doi: 10.1016/j.tcb.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 61.Li Y, Dai C, Wu C, et al. PINCH-1 Promotes Tubular Epithelial-to-Mesenchymal Transition by Interacting with Integrin-Linked Kinase. Journal of the American Society of Nephrology. 2007;18:2534–43. doi: 10.1681/ASN.2007030315. [DOI] [PubMed] [Google Scholar]
- 62.Fransvea E, Mazzocca A, Antonaci S, et al. Targeting transforming growth factor (TGF)-βRI inhibits activation of β1 integrin and blocks vascular invasion in hepatocellular carcinoma. Hepatology. 2009;49:839–50. doi: 10.1002/hep.22731. [DOI] [PubMed] [Google Scholar]
- 63.Miyazono K, Heldin CH. Latent forms of TGF-beta: molecular structure and mechanisms of activation. Ciba Found Symp. 1991;157:81–9. doi: 10.1002/9780470514061.ch6. discussion 9–92. [DOI] [PubMed] [Google Scholar]
- 64.Mu D, Cambier S, Fjellbirkeland L, et al. The integrin αvβ8 mediates epithelial homeostasis through MT1-MMP–dependent activation of TGF-β1. The Journal of Cell Biology. 2002;157:493–507. doi: 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Annes JP, Rifkin DB, Munger JS. The integrin [alpha]V[beta]6 binds and activates latent TGF[beta]3. FEBS Letters. 2002;511:65–8. doi: 10.1016/s0014-5793(01)03280-x. [DOI] [PubMed] [Google Scholar]
- 66.Annes JP, Chen Y, Munger JS, et al. Integrin {alpha}V{beta}6-mediated activation of latent TGF-{beta} requires the latent TGF-{beta} binding protein-1. J Cell Biol. 2004;165:723–34. doi: 10.1083/jcb.200312172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ludbrook SB, Barry ST, Delves CJ, et al. The integrin alphavbeta3 is a receptor for the latency-associated peptides of transforming growth factors beta1 and beta3. Biochem J. 2003;369:311–8. doi: 10.1042/BJ20020809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang Z, Mu Z, Dabovic B, et al. Absence of integrin-mediated TGF-beta1 activation in vivo recapitulates the phenotype of TGF-beta1-null mice. The Journal of Cell Biology. 2007;176:787–93. doi: 10.1083/jcb.200611044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aluwihare P, Mu Z, Zhao Z, et al. Mice that lack activity of αvβ6- and αvβ8-integrins reproduce the abnormalities of Tgfb1- and Tgfb3-null mice. J Cell Sci. 2009;122:227–32. doi: 10.1242/jcs.035246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang B, Dolinski BM, Kikuchi N, et al. Role of αvβ6 integrin in acute biliary fibrosis. Hepatology. 2007;46:1404–12. doi: 10.1002/hep.21849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Horan GS, Wood S, Ona V, et al. Partial Inhibition of Integrin {alpha}v 6 Prevents Pulmonary Fibrosis without Exacerbating Inflammation. Am J Respir Crit Care Med. 2008;177:56–65. doi: 10.1164/rccm.200706-805OC. [DOI] [PubMed] [Google Scholar]
- 72.Koopman Van Aarsen LA, Leone DR, Ho S, et al. Antibody-Mediated Blockade of Integrin αvβ6 Inhibits Tumor Progression In vivo by a Transforming Growth Factor-β–Regulated Mechanism. Cancer Research. 2008;68:561–70. doi: 10.1158/0008-5472.CAN-07-2307. [DOI] [PubMed] [Google Scholar]
- 73.Bates RC, Bellovin DI, Brown C, et al. Transcriptional activation of integrin β6 during the epithelial-mesenchymal transition defines a novel prognostic indicator of aggressive colon carcinoma. The Journal of Clinical Investigation. 2005;115:339–47. doi: 10.1172/JCI23183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hinz B, Gabbiani G, Chaponnier C. The NH2-terminal peptide of alpha smooth muscle actin inhibits force generation by the myofibroblast in vitro and in vivo. The Journal of Cell Biology. 2002;157:657–63. doi: 10.1083/jcb.200201049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wipff P-J, Rifkin DB, Meister J-J, et al. Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. The Journal of Cell Biology. 2007;179:1311–23. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou Y, Hagood JS, Lu B, et al. Thy-1-Integrin alpha(v)beta(5) Interactions Inhibit Lung Fibroblast Contraction-induced Latent Transforming Growth Factor- beta Activation and Myofibroblast Differentiation. Journal of Biological Chemistry. 2010;285:22382–93. doi: 10.1074/jbc.M110.126227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kurosaka D, Kato K, Nagamoto T, et al. Growth factors influence contractility and alpha-smooth muscle actin expression in bovine lens epithelial cells. Investigative Ophthalmology & Visual Science. 1995;36:1701–8. [PubMed] [Google Scholar]
- 78.Wipff P-J, Hinz B. Integrins and the activation of latent transforming growth factor [beta]1 - An intimate relationship. European Journal of Cell Biology. 2008;87:601–15. doi: 10.1016/j.ejcb.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 79.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes & Development. 2000;14:163–76. [PMC free article] [PubMed] [Google Scholar]
- 80.Dallas SL, Rosser JL, Mundy GR, et al. Proteolysis of latent transforming growth factor-beta (TGF-beta)-binding protein-1 by osteoclasts. A cellular mechanism for release of TGF-beta from bone matrix. J Biol Chem. 2002;277:21352–60. doi: 10.1074/jbc.M111663200. [DOI] [PubMed] [Google Scholar]
- 81.Brooks PC, Stromblad S, Sanders LC, et al. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell. 1996;85:683–93. [Google Scholar]
- 82.Rolli M, Fransvea E, Pilch J, et al. Activated integrin αvβ3 cooperates with metalloproteinase MMP-9 in regulating migration of metastatic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:9482–7. doi: 10.1073/pnas.1633689100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-L̂2 and promotes tumor invasion and angiogenesis. Genes & Development. 2000;14:163–76. [PMC free article] [PubMed] [Google Scholar]
- 84.Rivera LB, Brekken RA. SPARC promotes pericyte recruitment via inhibition of endoglin-dependent TGF-β1 activity. The Journal of Cell Biology. 2011;193:1305–19. doi: 10.1083/jcb.201011143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schoenwaelder SM, Burridge K. Bidirectional signaling between the cytoskeleton and integrins. Current Opinion in Cell Biology. 1999;11:274–86. doi: 10.1016/s0955-0674(99)80037-4. [DOI] [PubMed] [Google Scholar]
- 86.Berrier AL, Yamada KM. Cell–matrix adhesion. J Cell Physiol. 2007;213:565–73. doi: 10.1002/jcp.21237. [DOI] [PubMed] [Google Scholar]
- 87.Dedhar S. Integrins and signal transduction. Curr Opin Hematol. 1999;6:37–43. doi: 10.1097/00062752-199901000-00007. [DOI] [PubMed] [Google Scholar]
- 88.Guo X, Wang X-F. Signaling cross-talk between TGF-[beta]/BMP and other pathways. Cell Res. 2009;19:71–88. doi: 10.1038/cr.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cary LA, Han DC, Guan JL. Integrin-mediated signal transduction pathways. Histol Histopathol. 1999;14:1001–9. doi: 10.14670/HH-14.1001. [DOI] [PubMed] [Google Scholar]
- 90.Wendt MK, Schiemann WP. Therapeutic targeting of the focal adhesion complex prevents oncogenic TGF-beta signaling and metastasis. Breast Cancer Res. 2009;11:R68. doi: 10.1186/bcr2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sieg DJ, Hauck CR, Ilic D, et al. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2:249–56. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- 92.Playford MP, Schaller MD. The interplay between Src and integrins in normal and tumor biology. Oncogene. 2004;23:7928–46. doi: 10.1038/sj.onc.1208080. [DOI] [PubMed] [Google Scholar]
- 93.Worthington JJ, Klementowicz JE, Travis MA. TGFbeta: a sleeping giant awoken by integrins. Trends in Biochemical Sciences. 2011;36:47–54. doi: 10.1016/j.tibs.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 94.Scaffidi AK, Petrovic N, Moodley YP, et al. alpha(v)beta(3) Integrin interacts with the transforming growth factor beta (TGFbeta) type II receptor to potentiate the proliferative effects of TGFbeta1 in living human lung fibroblasts. J Biol Chem. 2004;279:37726–33. doi: 10.1074/jbc.M403010200. [DOI] [PubMed] [Google Scholar]
- 95.Galliher A, Schiemann W. beta3 Integrin and Src facilitate transforming growth factor-beta mediated induction of epithelial-mesenchymal transition in mammary epithelial cells. Breast Cancer Research. 2006;8:R42. doi: 10.1186/bcr1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Massague J. Subunit structure of a high-affinity receptor for type beta-transforming growth factor. Evidence for a disulfide-linked glycosylated receptor complex. Journal of Biological Chemistry. 1985;260:7059–66. [PubMed] [Google Scholar]
- 97.Roberts AB, Wakefield LM. The two faces of transforming growth factor beta in carcinogenesis. Proc Natl Acad Sci U S A. 2003;100:8621–3. doi: 10.1073/pnas.1633291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Derynck R, Akhurst RJ, Balmain A. TGF-[beta] signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–29. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 99.Irina GL, Nevins WT, Natalia N, et al. Regulation of pulmonary inflammation and fibrosis through expression of integrins alphaVbeta3 and alphaVbeta5 on pulmonary T lymphocytes. Arthritis & Rheumatism. 2009;60:1530–9. doi: 10.1002/art.24435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ghannad F, Nica D, Garcia Fulle MI, et al. Absence of {alpha}v{beta}6 Integrin Is Linked to Initiation and Progression of Periodontal Disease. Am J Pathol. 2008;172:1271–86. doi: 10.2353/ajpath.2008.071068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ulrike S, Stefan P, Afschin S, et al. Upregulation of αVβ6 integrin, a potent TGF-β1 activator, and posterior capsule opacification. Journal of cataract and refractive surgery. 2005;31:595–606. doi: 10.1016/j.jcrs.2004.05.058. [DOI] [PubMed] [Google Scholar]
- 102.Sterner-Kock A, Thorey IS, Koli K, et al. Disruption of the gene encoding the latent transforming growth factor-β binding protein 4 (LTBP-4) causes abnormal lung development, cardiomyopathy, and colorectal cancer. Genes & Development. 2002;16:2264–73. doi: 10.1101/gad.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shull MM, Ormsby I, Kier AB, et al. Targeted disruption of the mouse transforming growth factor-[beta]1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–9. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cambier S, Gline Stephanie, Mu Dezhi, et al. Integrin alpha (v)beta8-mediated activation of transforming growth factor-beta by perivascular astrocytes: an angiogenic control switch. Am J Pathol. 2005;166:1883–94. doi: 10.1016/s0002-9440(10)62497-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Travis MA, Reizis B, Melton AC, et al. Loss of integrin αVβ8 on dendritic cells causes autoimmunity and colitis in mice. Nature. 2007;449:361–5. doi: 10.1038/nature06110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vogelmann R, Nguyen-Tat MD, Giehl K, et al. TGFbeta-induced downregulation of E-cadherin-based cell-cell adhesion depends on PI3-kinase and PTEN. Journal of cell science. 2005;118:4901–12. doi: 10.1242/jcs.02594. [DOI] [PubMed] [Google Scholar]
- 107.Zeisberg M, Bottiglio C, Kumar N, et al. Bone morphogenic protein-7 inhibits progression of chronic renal fibrosis associated with two genetic mouse models. Am J Physiol Renal Physiol. 2003;285:F1060–7. doi: 10.1152/ajprenal.00191.2002. [DOI] [PubMed] [Google Scholar]
- 108.Wang Z, Chui W-K, Ho PC. Integrin targeted drug and gene delivery. Expert Opinion on Drug Delivery. 2010;7:159–71. doi: 10.1517/17425240903468696. [DOI] [PubMed] [Google Scholar]
- 109.Chernousov MA, Carey DJ. [alpha]V[beta]8 integrin is a Schwann cell receptor for fibrin. Exp Cell Res. 2003;291:514–24. doi: 10.1016/s0014-4827(03)00409-9. [DOI] [PubMed] [Google Scholar]
- 110.Palmade FS-CO, Regnouf de Vains JB, Coquelet C, Bonne C. Inhibition of cell adhesion to lens capsule by LCM 1910, an RGD-derived peptide. J Ocul Pharmacol. 1994;10:623–32. doi: 10.1089/jop.1994.10.623. [DOI] [PubMed] [Google Scholar]
- 111.Oharazawa HIN, Ohara K, Reddy VN. Inhibitory Effects of Arg-Gly-Asp (RGD) Peptide on Cell Attachment and Migration in a Human Lens Epithelial Cell Line. Ophthalmic Res. 2005;37:191–6. doi: 10.1159/000086595. [DOI] [PubMed] [Google Scholar]
- 112.Del Gatto A, Zaccaro L, Grieco P, et al. Novel and Selective αvβ3 Receptor Peptide Antagonist: Design, Synthesis, and Biological Behavior. Journal of Medicinal Chemistry. 2006;49:3416–20. doi: 10.1021/jm060233m. [DOI] [PubMed] [Google Scholar]
- 113.D’Andrea LD, Del Gatto A, Pedone C, et al. Peptide-based Molecules in Angiogenesis. Chemical Biology & Drug Design. 2006;67:115–26. doi: 10.1111/j.1747-0285.2006.00356.x. [DOI] [PubMed] [Google Scholar]
- 114.Platten M, Wick W, Wild-Bode C, et al. Transforming Growth Factors [beta]1 (TGF-[beta]1) and TGF-[beta]2 Promote Glioma Cell Migration via Up-Regulation of [alpha]V[beta]3 Integrin Expression. Biochemical and Biophysical Research Communications. 2000;268:607–11. doi: 10.1006/bbrc.2000.2176. [DOI] [PubMed] [Google Scholar]
- 115.Coleman KR, Braden GA, Willingham MC, et al. Vitaxin, a Humanized Monoclonal Antibody to the Vitronectin Receptor ({alpha}vβ3), Reduces Neointimal Hyperplasia and Total Vessel Area After Balloon Injury in Hypercholesterolemic Rabbits. Circ Res. 1999;84:1268–76. doi: 10.1161/01.res.84.11.1268. [DOI] [PubMed] [Google Scholar]
- 116.Wilder RL. Integrin alpha V beta 3 as a target for treatment of rheumatoid arthritis and related rheumatic diseases. Ann Rheum Dis. 2002;61:ii96–ii9. doi: 10.1136/ard.61.suppl_2.ii96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Asano Y, Ihn H, Yamane K, et al. Increased Expression of Integrin L̂±vL̂23 Contributes to the Establishment of Autocrine TGF-L̂2 Signaling in Scleroderma Fibroblasts. The Journal of Immunology. 2005;175:7708–18. doi: 10.4049/jimmunol.175.11.7708. [DOI] [PubMed] [Google Scholar]
- 118.Araya J, Cambier S, Markovics JA, et al. Squamous metaplasia amplifies pathologic epithelial-mesenchymal interactions in COPD patients. The Journal of Clinical Investigation. 2007;117:3551–62. doi: 10.1172/JCI32526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nemeth JA, Nakada MT, Trikha M, et al. Alpha-v Integrins as Therapeutic Targets in Oncology. Cancer Investigation. 2007;25:632–46. doi: 10.1080/07357900701522638. [DOI] [PubMed] [Google Scholar]
- 120.Nisato R, Tille J-C, Jonczyk A, et al. αvβ3 and αvβ5 integrin antagonists inhibit angiogenesis in vitro. Angiogenesis. 2003;6:105–19. doi: 10.1023/B:AGEN.0000011801.98187.f2. [DOI] [PubMed] [Google Scholar]
- 121.Tabatabai G, Weller M, Nabors B, et al. Targeting integrins in malignant glioma. Targeted Oncology. 2010;5:175–81. doi: 10.1007/s11523-010-0156-3. [DOI] [PubMed] [Google Scholar]
- 122.Chen Q, Manning C, Millar H, et al. CNTO 95, a fully human anti αv integrin antibody, inhibits cell signaling, migration, invasion, and spontaneous metastasis of human breast cancer cells. Clinical and Experimental Metastasis. 2008;25:139–48. doi: 10.1007/s10585-007-9132-4. [DOI] [PubMed] [Google Scholar]
- 123.Mullamitha SA, Ton NC, Parker GJM, et al. Phase I Evaluation of a Fully Human Anti–αv Integrin Monoclonal Antibody (CNTO 95) in Patients with Advanced Solid Tumors. Clinical Cancer Research. 2007;13:2128–35. doi: 10.1158/1078-0432.CCR-06-2779. [DOI] [PubMed] [Google Scholar]
- 124.Tucker GC. Integrins: molecular targets in cancer therapy. Curr Oncol Rep. 2006;8:96–103. doi: 10.1007/s11912-006-0043-3. [DOI] [PubMed] [Google Scholar]
- 125.Rosenwald MS. The Washington Post. 2004. MedImmune Ends Some Vitaxin Testing. [Google Scholar]
- 126.Gramoun A, Shorey S, Bashutski JD, et al. Effects of Vitaxin®, a novel therapeutic in trial for metastatic bone tumors, on osteoclast functions in vitro. Journal of Cellular Biochemistry. 2007;102:341–52. doi: 10.1002/jcb.21296. [DOI] [PubMed] [Google Scholar]
- 127.Bakin AV, Tomlinson AK, Bhowmick NA, et al. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. 2000;275:36803–10. doi: 10.1074/jbc.M005912200. [DOI] [PubMed] [Google Scholar]
- 128.Runyan CE, Schnaper HW, Poncelet AC. The phosphatidylinositol 3-kinase/Akt pathway enhances Smad3-stimulated mesangial cell collagen I expression in response to transforming growth factor-beta1. J Biol Chem. 2004;279:2632–9. doi: 10.1074/jbc.M310412200. [DOI] [PubMed] [Google Scholar]