

Abstract
The CFTR chloride channel is activated by phosphorylation of serine residues in the regulatory (R) domain and then gated by ATP binding and hydrolysis at the nucleotide binding domains (NBDs). Studies of the ATP-dependent gating process in excised inside-out patches are very often hampered by channel rundown partly caused by membrane-associated phosphatases. Since the severed ΔR-CFTR, whose R domain is completely removed, can bypass the phosphorylation-dependent regulation, this mutant channel might be a useful tool to explore the gating mechanisms of CFTR. To this end, we investigated the regulation and gating of the ΔR-CFTR expressed in Chinese hamster ovary cells. In the cell-attached mode, basal ΔR-CFTR currents were always obtained in the absence of cAMP agonists. Application of cAMP agonists or PMA, a PKC activator, failed to affect the activity, indicating that the activity of ΔR-CFTR channels is indeed phosphorylation independent. Consistent with this conclusion, in excised inside-out patches, application of the catalytic subunit of PKA did not affect ATP-induced currents. Similarities of ATP-dependent gating between wild type and ΔR-CFTR make this phosphorylation-independent mutant a useful system to explore more extensively the gating mechanisms of CFTR. Using the ΔR-CFTR construct, we studied the inhibitory effect of ADP on CFTR gating. The Ki for ADP increases as the [ATP] is increased, suggesting a competitive mechanism of inhibition. Single channel kinetic analysis reveals a new closed state in the presence of ADP, consistent with a kinetic mechanism by which ADP binds at the same site as ATP for channel opening. Moreover, we found that the open time of the channel is shortened by as much as 54% in the presence of ADP. This unexpected result suggests another ADP binding site that modulates channel closing.
Keywords: chloride channel, single-channel kinetics, ABC transporter, gating mode, phosphorylation
Abbreviations used in this paper: ABC, ATP-binding cassette; BIM, bisindolylmaleimide; CHO, Chinese hamster ovary; CFTR, cystic fibrosis transmembrane conductance regulator; CPT-cAMP, 8-(4-chlorophenylthio)-cAMP; NBD, nucleotide binding domain; NMDG-Cl, N-methyl-d-glucamine chloride; PKI, PKA inhibitor; R, regulatory; WT, wild type
INTRODUCTION
The cystic fibrosis transmembrane conductance regulator (CFTR) is a member of the ATP-binding cassette transporter family, and the only ATP-binding cassette (ABC) transporter so far established as an ion channel. Mutations in this channel cause the genetic disease cystic fibrosis (Riordan et al., 1989). CFTR is composed of five domains: two membrane-spanning domains, each with six putative transmembrane segments, contribute to the formation of the chloride channel pore; two nucleotide-binding domains (NBD1 and NBD2), defined by the conserved Walker A and B motifs, play critical roles in CFTR gating; a regulatory domain (R domain), not found in other ABC transporters, contains several phosphorylation sites for cAMP-dependent protein kinase (PKA) and PKC. PKA activates CFTR by phosphorylating multiple serine residues in the R domain, while the opening and closing of the channel is coupled to ATP binding and hydrolysis at the nucleotide binding domains (NBDs) (for review see Gadsby and Nairn, 1999).
Several different kinetic models have been proposed to explain ATP-dependent gating of CFTR (e.g., Zeltwanger et al., 1999; Ikuma and Welsh, 2000; Vergani et al., 2003), but it remains unclear how each NBD utilizes ATP to open and close the channel. Compared with voltage-gated cation channels, CFTR gating is slow, with each opening and closing events lasting for hundreds of milliseconds to seconds. Our understanding of CFTR gating relies heavily on single-channel kinetic analysis that demands recordings that last for tens of minutes in order to collect sufficient gating events. However, stationary recordings for this length of time are technically challenging because PKA-dependent phosphorylation is absolutely required for WT-CFTR function and CFTR is often dephosphorylated by membrane-associated protein phosphatases in inside-out patches. The channel rundown by dephosphorylation could affect not only single-channel kinetic parameters, but also “steady-state” dose–response relationships (e.g., Szellas and Nagel, 2003). The severity of this technical problem varies among different expression systems. In some cases, dephosphorylation-induced channel rundown in excised patches takes place within seconds upon removal of PKA (e.g., Weinreich et al., 1999).
Another technical difficulty that hampers kinetic studies is to obtain membrane patches that contain only one channel. Although analytical methods have been developed to quantify gating kinetics from multichannel data (e.g., Fenwick et al., 1982; Csanády, 2000), these methods still cannot replace classical single-channel dwell-time analysis because of the necessity of using a preconceived gating model. The discovery of phosphorylation-independent CFTR constructs provides a potential solution for the problems described above. Recently, Csanády et al. (2000) constructed a CFTR whose R domain (amino acids 634–836) is completely deleted. This construct, when expressed in Xenopus oocytes, exhibits robust basal activity. However, addition of PKA causes a further increase in the ΔR-CFTR currents (~30%), suggesting the presence of phosphorylation site(s) outside the R domain. This conclusion contradicts an early report by Rich et al. (1993) that shows that all the functionally important phosphorylation sites are located in the R domain (see discussion for details).
One potential reason for this discrepancy is the difference in the expression system (mammalian cell line vs. Xenopus oocytes). We therefore examined if the ΔR-CFTR construct made by Csanády et al. (2000) is sensitive to modulation by PKA-dependent phosphorylation when it is expressed in a mammalian expression system. Our results show that the constitutive activity of ΔR-CFTR channels is not modified by application of cAMP agonists in cell-attached recordings or exogenous PKA in excised inside-out patches, indicating that these ΔR-CFTR mutant channels are completely phosphorylation independent, at least when expressed in Chinese hamster ovary (CHO) cells. Because the expression level is low, patches containing only a single channel can be frequently obtained. We found that gating parameters of ΔR-CFTR are very similar, if not identical, to those of WT-CFTR. These include single-channel Po, ATP dose–response relationship, and opening and closing rates. It is therefore concluded that, contrary to Csanády’s report, this ΔR-CFTR construct provides a valuable model system to study CFTR gating at a single-channel level when expressed in CHO cells. Indeed, our single-channel kinetic analysis in the presence of ADP reveals, for the first time, a new closed state, consistent with the idea that ADP inhibits CFTR opening by competing with ATP for a binding site. Surprisingly, ADP also reduces the open time of the channel, suggesting that binding of nucleotides affects the stability of the open state. This unexpected finding leads to further studies elaborated in the accompanying paper.
MATERIALS AND METHODS
Cell Culture
CHO cells were grown at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS. For whole-cell and cell-attached experiments, cell suspensions were prepared with trypsinization. For excised inside-out experiments, cells were plated on sterile glass chips in 35-mm tissue culture dishes 1–2 d before use.
Construction of the CFTR Mutant
The DNA construct (pBudCE4.1 split ΔR-CFTR) for expressing the ΔR-CFTR channel has been described in detail previously (Ai et al., 2004). In brief, the cDNAs encoding CFTR residues 1–633 and residues 837–1480 were subcloned into the expression vector pBudCE4.1 (Invitrogen) under the control of the CMV and EF1-α promoters, respectively. The plasmids pGEMHE-1-633 and pGEMHE-837-1480 were a gift from D. Gadsby (Rockefeller University, New York, NY).
Transient Expression of CFTR
To transiently express CFTR, CHO cells were grown in 35-mm tissue culture dishes one day before transfection. The plasmid pBudCE4.1 split ΔR-CFTR or pcDNA3.1 containing wild-type (WT)–CFTR were cotransfected with pEGFP-C3 (CLONTECH Laboratories, Inc.) encoding green fluorescent protein using SuperFect transfection reagent (QIAGEN) according to manufacturer’s protocols. The cells were incubated at 27°C for 1–2 d before use.
Whole-cell Experiments
Pipette electrodes were made from Corning 7056 glass capillaries (Warner Instrument). The pipette resistance was ~3 MΩ in the bath solution. The membrane potential was held with an EPC9 patch-clamp amplifier (HEKA) at 0 mV, voltage ramps (±100 mV, 2 s in duration, every 6 s) or voltage steps were generated with Pulse software (HEKA) to create the I–V relationships. Current traces were filtered at 1 kHz with a built-in four-pole Bessel filter and then digitized at 2 kHz into the computer. The currents were recorded at room temperature (~23°C). The pipette solution contained (in mM) 85 aspartic acid, 5 pyruvic acid, 10 EGTA, 20 tetraethylammonium-chloride, 5 tris creatine phosphate, 10 MgATP, 2 MgCl2, 5.5 glucose, and 10 HEPES (pH 7.4 with CsOH). Aspartate was replaced with Cl− for some experiments under symmetrical [Cl−] condition. The bath solution contained (in mM) 150 NaCl, 2 MgCl2, 1 CaCl2, 5 glucose, and 5 HEPES (pH 7.4 with NaOH). 20 mM sucrose was added to the bath solution to prevent activation of swelling-induced currents.
Single-channel Experiments
Patch-clamp experiments in excised inside-out mode were described in detail previously (e.g., Zeltwanger et al., 1999). In brief, single-channel CFTR currents were recorded at room temperature (~23°C) with an EPC9 or EPC10 amplifiers (HEKA). Data were filtered at 100 Hz with an eight-pole Bessel filter (Warner Instrument) and captured onto a hard disk at a sampling rate of 500 Hz. For cell-attached patches, the pipette potential is held at 50 mV. For excised inside-out patches, the membrane potential is held at −50 mV. The pipette solution contained (in mM) 140 N-methyl-d-glucamine chloride (NMDG-Cl), 2 MgCl2, 5 CaCl2, and HEPES (pH 7.4 with NMDG). The super-fusion solution for inside-out patches contained (in mM) 150 N-methyl-d-glucamine chloride (NMDG-Cl), 10 EGTA, 10 HEPES, 8 TRIS, and 2 MgCl2 (pH 7.4 with NMDG).
Reagents
Forskolin, purchased from Alexis, was stored as 20 mM stock in DMSO at 4°C. Genistein were purchased from Sigma-Aldrich and stored as 100 mM stock in DMSO at −20°C. CPT-cAMP (8-(4-chlorophenylthio)-cAMP) and Mg-ATP, purchased from Sigma-Aldrich, were stored in water at −20°C. PKA was purchased from Promega. PKI was purchased from Alexis. Bisindolylmaleimide (BIM) and PMA were purchased from Sigma-Aldrich. ADP was purchased from Calbiochem.
Data Analysis
The steady-state mean current and single channel amplitudes were calculated with Igor software (Wavemetrics). Curve fitting of the dose–response relationships were also performed with Igor. Single channel Po, mean burst duration, and mean interburst duration were obtained from patches with few channels (<4) with a program developed by Dr. Csanády (Csanády, 2000). To determine the true ATP-dependent parameters, the ATP-independent brief closures (flickers) observed within a burst had to be removed. To do that, we employed the three-state model shown below:
(SCHEME 1).
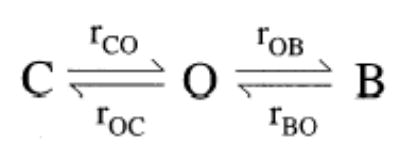
where O and C are open and closed states, respectively. B is a blocked state induced by an intrinsic blocker (Zhou et al., 2001). Rate constants rCO, rOC, rOB, and rBO are extracted by a simultaneous fit to the dwell-time histograms of all conductance levels. Mean interburst, burst durations, and channel open probability (Po), were calculated as τib = 1/rCO, τb = (1/rOC)(1 + rOB/rBO), Po = 1/(1 + rOC/rCO + rOB/rBO), respectively. Then, the burst time (τb) corresponds to the ATP-dependent open time of the channel (τo), and the interburst time (τib) corresponds to the ATP-dependent closed time of the channel (τc). To avoid confusions, we will use throughout this manuscript the terms open and closed time to refer to these ATP-dependent events.
For single-channel dwell-time analysis (Fig. 8), we pooled the current recordings obtained from several patches containing only a single ΔR-CFTR channel. The digitized current records were further filtered digitally at 50 Hz and analyzed with a program developed by Y. Sohma in our lab. The program automatically detects events using a 50% threshold-crossing method. The pooled open and closed interval durations were log-binned at a resolution of 10 bins per log unit and plotted as the square root of the number of intervals per bin with the constant bin width on a logarithmic time axis (Sigworth and Sine, 1987). To exclude the short-lived, ATP-independent flickery events (Zhou et al., 2001), we reconstructed dwell-time histograms by setting up a cutoff of 50 ms (e.g., Carson et al., 1995; Li et al., 1996; Zeltwanger et al., 1999). Least square estimation with sums of exponential components was performed to obtain open and closed time constants. The intervals shorter than dead times were excluded from the fitting.
Figure 8.
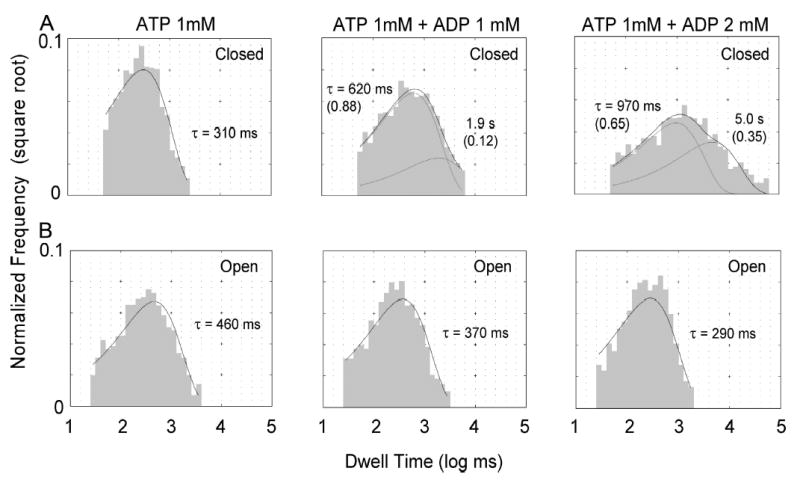
Single-channel dwell time histograms. Events from several single-channel recordings were pulled together to obtain these dwell time histograms to determine the open and closed times distributions in the presence of 1 mM ATP, 1 mM ATP + 1 mM ADP, and 1 mM ATP + 2 mM ADP. (A) The closed time distributions can be fitted with a double exponential function in the presence of ADP, indicating the presence of a new, longer closed time constant. The data could be fitted with a single exponential (τ = ~2 s), but in the case of 2 mM ADP, the fitted curve fails to capture nearly all the closed events >10 s (not depicted). (B) The open time dwell time histograms show that the mean open time decreases as the concentration of ADP increases.
All values are presented as mean ± SEM. Student’s t test was performed with Sigmaplot (SPSS Science), P < 0.05 was considered significant.
Online Supplemental Material
The supplemental material for this paper consists of two figures (available at http://www.jgp.org/cgi/content/full/jgp.200409227/DC1). Fig. S1 shows the ATP dose–response for ΔR-CFTR mutant channels. Fig. S2 shows the change from slow gating mode to fast gating mode as we switch from on-cell mode to excised inside-out mode in WT-CFTR channels.
RESULTS
Phosphorylation-independent Regulation of ΔR-CFTR
It was reported that ΔR-CFTR, expressed in Xenopus oocytes, is constitutively active and that the whole-cell ΔR-CFTR channel currents can be increased by cAMP stimulation (Csanády et al., 2000). Surprisingly, when expressed in CHO cells, constitutively active ΔR-CFTR currents do not respond to forskolin or CPT-cAMP stimulation. Fig. 1 A shows a representative cell-attached recording of ΔR-CFTR. Addition of 10 μM forskolin plus 100 μM CPT-cAMP, both maximally effective concentrations for CFTR activation (Hwang et al., 1997; Al-Nakkash and Hwang, 1999), failed to increase the basal activity, but 20 μM genistein, a known CFTR potentiator (e.g., Yang et al., 1997), increased the current by approximately twofold (n = 4).
Figure 1.
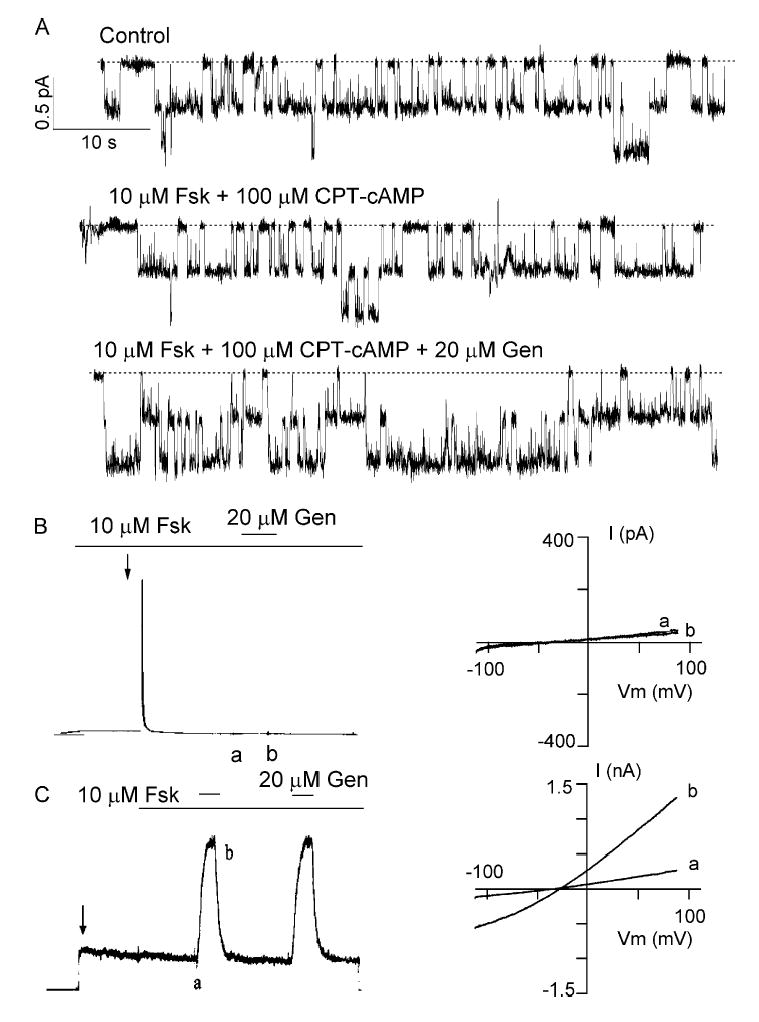
Effects of PKA activators and inhibitors on ΔR-CFTR. (A) A cell-attached recording showing that addition of 10 μM forskolin (Fsk) + 100 μM CPT-cAMP failed to increase the basal current of ΔR-CFTR. However, addition of 20 μM genistein (Gen) could potentiate the current (n = 4). (B and C) Effects of PKI, a peptide inhibitor of PKA, on whole-cell currents from either WT- or ΔR-CFTR. For WT-CFTR, in the presence of forskolin, a very brief outward current was obtained immediately after the whole-cell configuration was formed (arrow). Then, the currents decayed rapidly. Subsequent application of genistein did not potentiate the currents. For ΔR-CFTR, basal currents were seen without any cAMP stimulant after the whole-cell configurations are formed (arrow). Subsequent application of forskolin did not alter the currents. But, genistein could potentiate the channel activity. Similar results were obtained from seven cells. Ramp I–V curves (right) were taken as marked in the raw current traces.
To further test if ΔR-CFTR currents are indeed phosphorylation independent, we used PKI, a PKA inhibitor, in the whole-cell configuration, which allows introducing PKI directly into the cell. Previously, Hwang et al. (1992) showed that introducing 1 mM PKI into the cell through the patch pipette can abolish cAMP-stimulated CFTR currents in cardiac myocytes. We first tested effects of PKI on WT-CFTR. In cells expressing WT-CFTR, an outward current was observed upon breaking into the whole-cell configuration in the presence of 10 μM forskolin (Fig. 1 B). However, this current was completely abolished within 1 min due to a rapid diffusion of the pipette PKI into the cell. Subsequent application of 20 μM genistein did not potentiate the current (Fig. 1 B), consistent with the notion that genistein only potentiates CFTR that is already activated by PKA-dependent phosphorylation (Yang et al., 1997; cf. Illek et al., 1995). In contrast, ΔR-CFTR basal currents were obtained without any cAMP stimulant after the whole-cell configuration was formed. The pipette PKI has little effect on this current. Although a slow decay of the ΔR-CFTR current over a course of tens of minutes is observed, this is likely due to phosphorylation-independent rundown since a similar irreversible decrease of the cAMP-dependent current is very often seen with the WT-CFTR (unpublished data). Application of 10 μM forskolin did not alter the PKI-resistant current, but 20 μM genistein further increased the current (n = 7) (Fig. 1 C).
Although the cAMP–PKA pathway constitutes the major regulatory mechanism for CFTR, PKC may also be involved in the phosphorylation of CFTR (Tabcharani et al., 1991; Button et al., 2001; Chappe et al., 2003; Chen et al., 2004). We therefore tested whether ΔR-CFTR is sensitive to PKC modulation by using both a PKC activator, PMA, and an inhibitor, BIM. Fig. 2 A shows a continuous current trace of whole-cell ΔR-CFTR currents. A ramp voltage >100 mV was applied every 6 s to monitor any conductance change over the time course of the experiment. The basal ΔR-CFTR current is insensitive to 1 μM BIM (n = 7). In cell-attached patches (Fig. 2 B), the PKC activator PMA (500 nM) did not change ΔR-CFTR channel activity (n = 6). In both cases, however, genistein, serving as a positive control, increased ΔR-CFTR currents.
Figure 2.
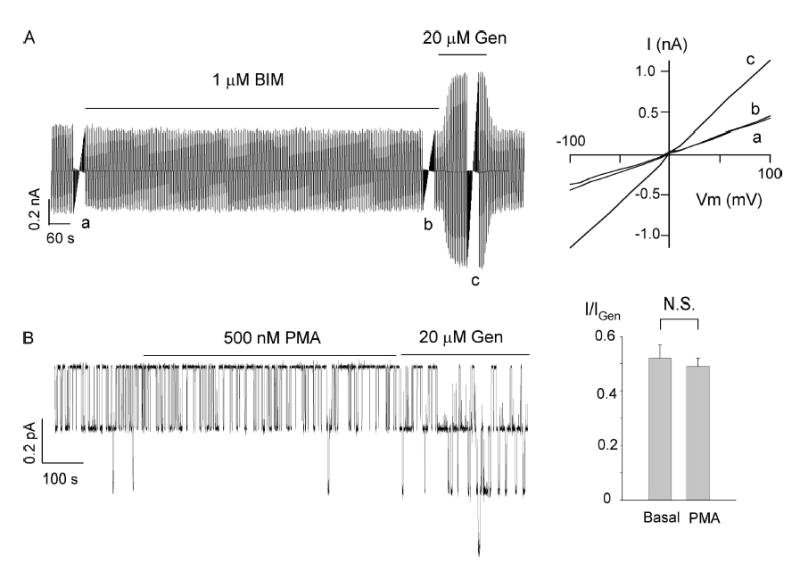
Effect of PKC on ΔR-CFTR. (A) A representative whole-cell ΔR-CFTR current trace showing lack of effects of BIM, a PKC inhibitor, on the basal current. The inset shows the I–V relationships at different conditions as marked. Similar results were obtained from seven cells. (B) A continuous ΔR-CFTR current trace in a cell-attached mode. PMA, a PKC activator, did not alter the ΔR-CFTR currents. Summary of the mean currents in the presence or absence of PMA. Values were normalized by mean currents with genistein (n = 6).
Sensitivity of the ΔR-CFTR channel to PKA-dependent phosphorylation was further tested in excised inside-out patches. Fig. 3 A shows the current trace of a single ΔR-CFTR channel with 2.75 mM ATP. Consistent with the results shown above, subsequent application of PKA (25 U/ml) did not change the open probability. Fig. 3 B shows expanded traces in the presence or absence of PKA. In six similar experiments, the mean single-channel Po in the presence of 2.75 mM ATP alone is not significantly different from that in the presence of PKA plus 2.75 mM ATP (Fig. 3 B). We thus conclude that ΔR-CFTR channels, when expressed in CHO cells, are constitutively active in the presence of ATP and are phosphorylation independent.
Figure 3.
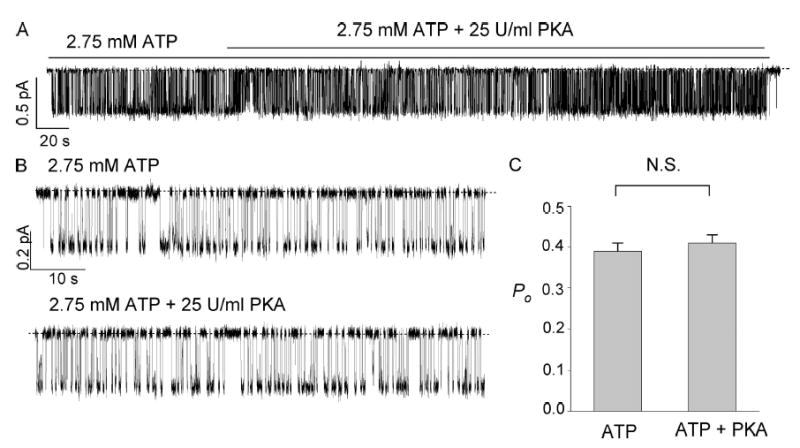
PKA did not alter the ATP-induced ΔR-CFTR chloride channel currents. (A) Single-channel currents were induced by ATP application 2 min after patch excision. Subsequent addition of PKA did not alter the channel activity. (B) Expanded traces of the recording in A. (C) Summary of the measured Po with 2.75 mM ATP alone and with both 2.75 mM ATP and PKA (25 U/ml) (n = 6).
ATP-dependent Gating of ΔR-CFTR
To further explore the possibility of using ΔR-CFTR channels to study the ATP-dependent gating process, we performed detailed experiments to examine gating behavior of ΔR-CFTR in response to ATP in excised inside-out patches and compared the gating parameters to those of WT-CFTR.
The ΔR-CFTR channel activity in the presence of different [ATP] was monitored to obtain a dose–response relationship. The single channel open probability (Po) was plotted against [ATP], and a fit with the Michaelis-Menten equation yielded a K1/2 value of 89 ± 25 μM, which, within the error range, is quite similar to that of WT-CFTR (137 ± 28 μM; Zeltwanger et al., 1999). Despite some scattered differences, the Po values for ΔR-CFTR are also very close to those of WT-CFTR (see Fig. S1, available at http://www.jgp.org/cgi/content/full/jgp.200409227/DC1).
Fig. 4 A shows representative single-channel traces of ΔR-CFTR in the presence of different [ATP]. Similar to WT-CFTR, ΔR-CFTR exhibits two types of closings, an ATP-sensitive long closing and flickery closings that appear to be ATP-independent. For single-channel kinetic analysis, recordings from patches containing up to three channels were analyzed using the program developed by Csanády (2000). Fig. 4 B shows relationships between [ATP] and mean open time (τo), or mean closed time (τc). Both gating parameters are remarkably similar to those of WT-CFTR (Zeltwanger et al., 1999). Thus, the function of NBDs, the gating machinery for CFTR, appears to be intact despite a complete removal of the R domain. We therefore conclude that ΔR-CFTR, being phosphorylation independent and behaving similarly to WT channels, is an ideal construct to explore more extensively the gating mechanisms of CFTR.
Figure 4.
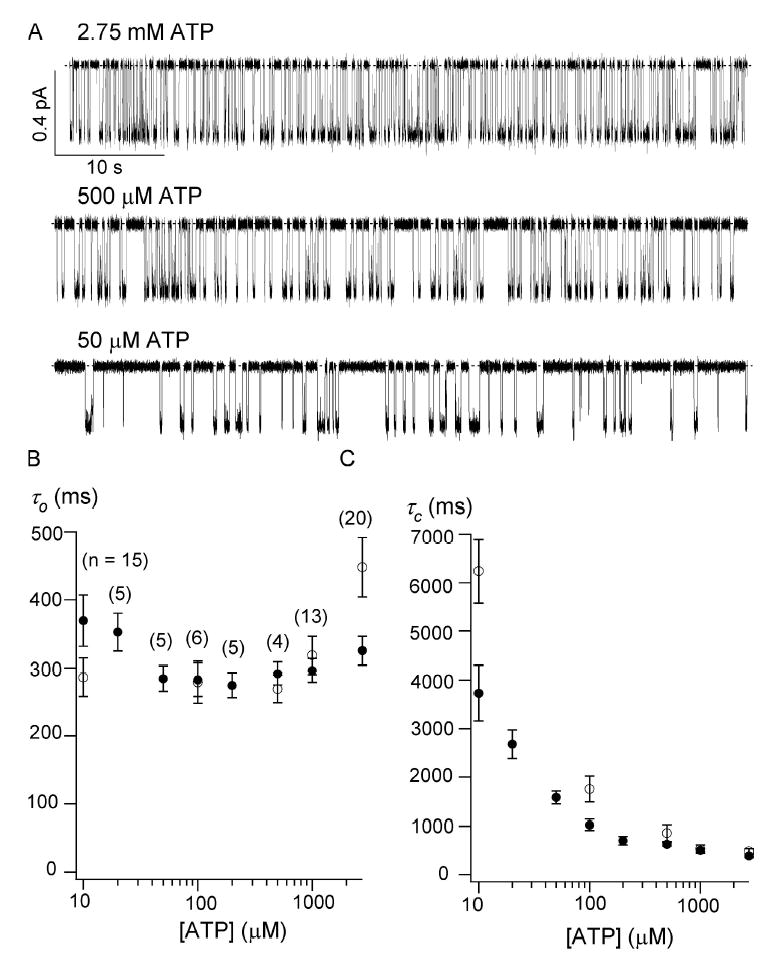
Effect of different concentrations of ATP on single-channel kinetics. (A) Representative single-channel ΔR-CFTR traces with different concentrations of ATP in excised inside-out patches. Each trace represents a 60-s recording. ATP concentration dependence of the mean open time (B) and the mean closed time (C). All values are represented by mean ± SEM. Overlaid open circles (○) represent the corresponding values measured for WT-CFTR channels (from Zeltwanger et al., 1999).
Different Gating Modes
Stable recordings of ΔR-CFTR provide a unique opportunity to observe gating behavior over a long duration. In addition, bypassing the PKA-dependent activation step allows examination of the gating pattern immediately following patch excision. We observed three different gating modes: slow gating mode, fast gating mode, and a high Po mode. The slow gating mode is characterized by long openings as well as long closings, and is often observed immediately after excision of the membrane. Within a few minutes, it switches spontaneously to the fast gating mode. Fig. 5 A presents a 3-min single-channel trace showing such a switch. This switch is not phosphorylation dependent since the addition of PKA did not switch the channel back to the original slow gating mode. It is interesting, however, to note that the Po values for slow gating mode and fast gating mode are similar, 0.45 ± 0.06 (n = 6) and 0.42 ± 0.06 (n = 8), respectively, in the presence of 2.75 mM ATP.
Figure 5.
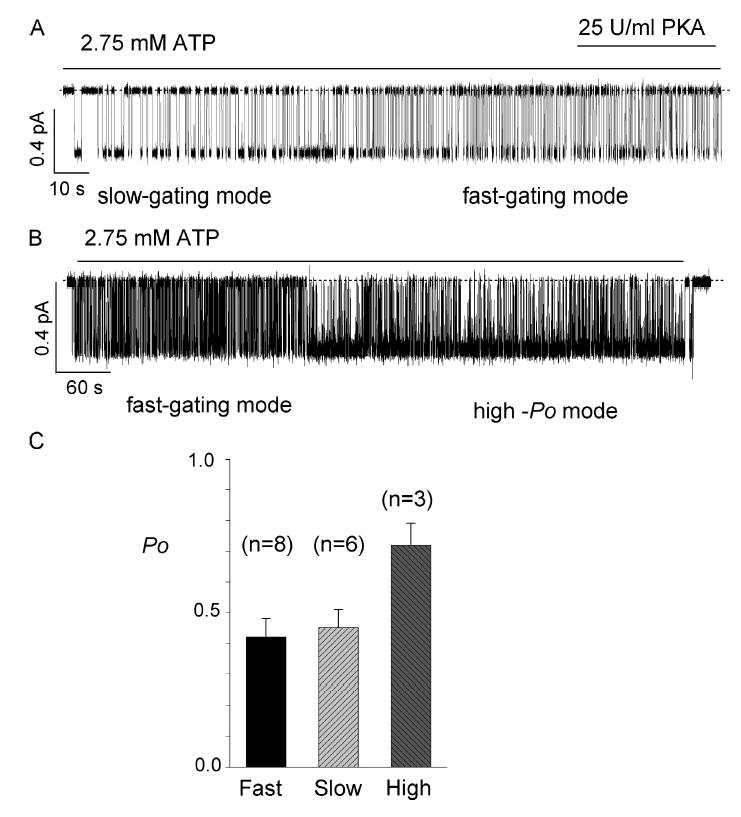
Mode shifts of ΔR-CFTR. (A) A single-channel recording of the ΔR-CFTR channel in an excised inside-out patch. The slow gating mode, i.e., long openings and closings of the channel, is often observed immediately after excision of the membrane patch. The slow gating mode usually switches to the fast gating mode spontaneously within a few minutes after patch excision. (B) A sample trace showing a spontaneous mode switch from the fast gating mode to the high Po mode. The mode switch occurred 4 min after the excision of membrane, and lasts for >6 min. (C) Summary of Po of ΔR-CFTR in different modes. *, P < 0.01.
In addition to the gating modes described above, we also observed a rarely occurring state whose Po is significantly higher than that of the fast or slow gating modes. Fig. 5 B shows a spontaneous switch of the gating mode from a fast one to the high Po mode. This high-Po mode is characterized by a Po of 0.72 ± 0.07 (n = 3) in the presence of 2.75 mM ATP. The higher Po is likely due to a prolonged open time and a shortened closed time. Unlike the switch between fast and slow gating modes, which usually occurs in the first few minutes of recordings, the switch to the high Po mode can take place at any time after excision. The mechanism underlying this mode shift is unknown but may explain partly the discrepancy in the literature for the different Po values reported for WT-CFTR (see discussion for details).
Effect of ADP in Fast Gating Mode
It has been reported that ADP inhibits the ATP-dependent WT-CFTR activity (Anderson and Welsh, 1992; Gunderson and Kopito, 1994; Winter et al., 1994; Schultz et al., 1995). Although it is proposed that ADP and ATP compete for a binding site that opens the channel (Anderson and Welsh, 1992), a recent report by Randak and Welsh (2003), however, suggest a potential alternative mechanism for nucleotides’ action on CFTR gating. Because of the structural similarity between ATP and ADP, the competitive mechanism seems appealing, but definite single-channel kinetic evidence is lacking. We first show that ADP inhibits ATP-induced ΔR-CFTR currents in a dose-dependent manner (Fig. 6). As predicted for a competitive mechanism, the magnitude of ADP-dependent inhibition is a function of [ATP]. As the [ATP] is increased, a higher [ADP] is needed to achieve a same magnitude of inhibition. The apparent Ki values for ADP inhibition are as follows: 26.3 ± 5.1 μM with 75 μM ATP, 51.2 ± 5.1 μM with 200 μM ATP, 180.1 ± 38.6 μM with 500 μM ATP, and 205.1 ± 43.7 μM with 1 mM ATP (Fig. 6 B). These results strongly suggest that ADP inhibits the CFTR channel by competitively binding to an ATP binding site.
Figure 6.
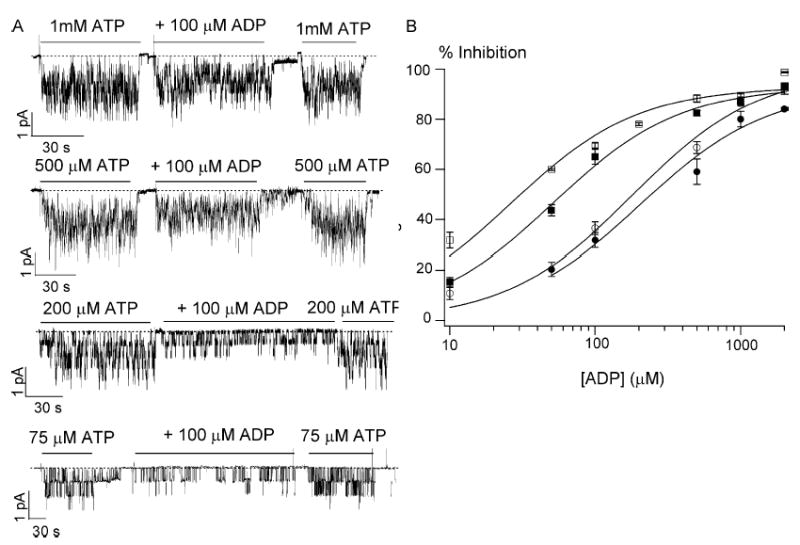
Inhibition of ATP-induced ΔR-CFTR currents by ADP. (A) Inhibitory effects of ADP on the ΔR-CFTR currents induced with different ATP concentrations. (B) The dose–response relationship between [ADP] and the magnitude of inhibition in the presence of four different ATP concentrations: (□) 75 μM ATP, (▪) 200 μM ATP, (○) 500 μM ATP, and (•) 1 mM ATP. All data points are presented as mean ± SEM of at least four values obtained from different patches. Data are fitted with the Michaelis-Menten equation.
Fig. 7 shows effects of ADP at the single-channel level. Even by eye inspection, one can discern the increase of the closed time upon addition of ADP. Using the analysis program for multichannel recordings (see materials and methods), we were able to demonstrate that the Po decreases in the presence of ADP is mostly due to an increase of the mean closed time (τc = 437 ± 36 ms for 1 mM ATP to τc = 1288 ± 197 ms for 1 mM ATP + 1mM ADP, n = 11). Although the mean open time remains almost unchanged (τo = 333 ± 18 ms for 1 mM ATP and 318 ± 26 ms for 1 mM ATP + 1 mM ADP), one should note that a simple three-state scheme was assumed for data analysis so that the τo values presented here can only be considered as a rough estimate (Fig. 7).
Figure 7.
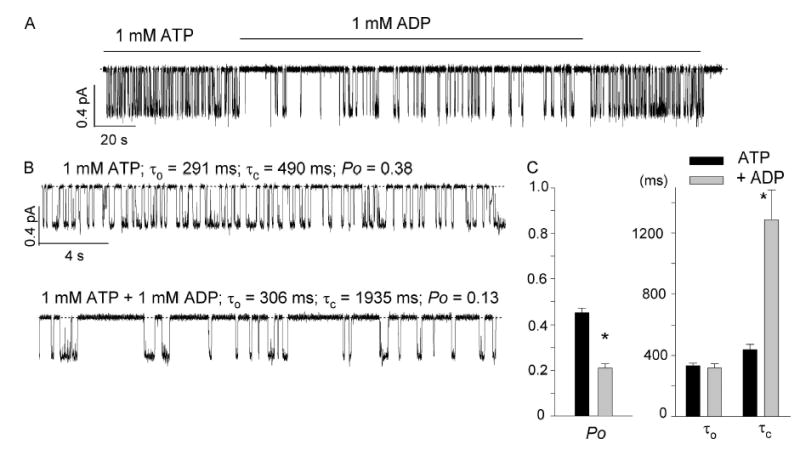
Effect of ADP on ΔR-CFTR single-channel current. (A) A representative single-channel current trace in an excised inside-out patch in the presence or absence of ADP. (B) Expanded traces with ATP alone (top) and with both ATP and ADP (bottom). (C) Summary of single-channel kinetic parameters (n = 11). *, P < 0.05.
To study in detail the closed time distribution we pooled data from eight single-channel patches and analyzed the dwell time histograms without the need of assuming a particular kinetic scheme. We observed that the closed time distribution with 1 mM ATP + 1 mM ADP was broader than the distribution with 1 mM ATP alone, suggesting the presence of a second closed time constant (Fig. 8 A). However, the difference between these two mean closed times is not large enough to allow a clear separation of the two kinetic components. Based on the hypothesis that ADP and ATP compete for the binding site that opens the channel, one would predict that a higher [ADP] will further increase the ADP-induced closed time. To test this idea, we increased the ADP concentration to 2 mM. Indeed, the closed dwell time histogram becomes even broader and the data can now be better fitted with a double exponential function. As far as we know, this result provides the first piece of single-channel kinetic evidence for competitive inhibition of channel opening by ADP. Surprisingly, when the open time histograms were fitted with a single exponential function, a shortening of the open time by ADP was seen. The shortening of the open time is modest in the presence of 1 mM ADP (only 20%), but becomes more evident when the ADP concentration is increased to 2 mM (37%) (Fig. 8 B).
Effect of ADP in Slow Gating Mode
While single-channel dwell time analysis shows an effect of ADP on the mean open time (Fig. 8), we were surprised that analysis of multichannel recording failed to reveal this effect (Fig. 7). One possibility, already mentioned above, is that the assumed gating scheme for multichannel kinetic analysis is oversimplified. Thus, a 20–40% change of the open time constant by ADP was not detected. One should also note that since the fast gating mode represents the most commonly occurring state of the channel, the results described above reflect mostly the effects of ADP on ΔR-CFTR gating in the fast gating mode. If the lifetime of the ADP-induced open state is not drastically different from that with ATP alone, it will be difficult to detect this state. We therefore examined effects of ADP on gating kinetics while the channel is in the slow gating mode when the mean open time is somewhat longer. The experiments were performed in the first 3–5 min following patch excision.
The patch membranes were exposed to 1 mM ATP for 1–2 min, then to 1 mM ADP plus 1 mM ATP for 1–3 min, and back to 1 mM ATP again. This bracketing was very important as a control because of the spontaneous switch of the gating mode. Fig. 9 A shows a continuous current trace obtained in a patch with a single channel. As expected, the closed time is dramatically prolonged in the presence of ADP. In addition, the current trace clearly shows a reversible shortening of the open time in the presence of ADP. Due to the short duration of the slow gating mode, it was very difficult to obtain a large number of patches that could be used for kinetic analysis since most of the time the channel switched to the fast gating mode before the appropriate bracketing was performed. Out of >30 patches, only 6 of them could be analyzed. The results showed that the open time was reduced by an average of 54 ± 6%. The mean open time was reduced from 828 ± 53 ms with 1 mM ATP alone, to 384 ± 41 ms with 1 mM ATP plus 1 mM ADP; the mean closed time increased from 614 ± 101 ms with 1 mM ATP to 1993 ± 373 ms for 1 mM ATP plus 1 mM ADP. Detailed studies of this novel effect of ADP are described in the accompanying paper.
Figure 9.
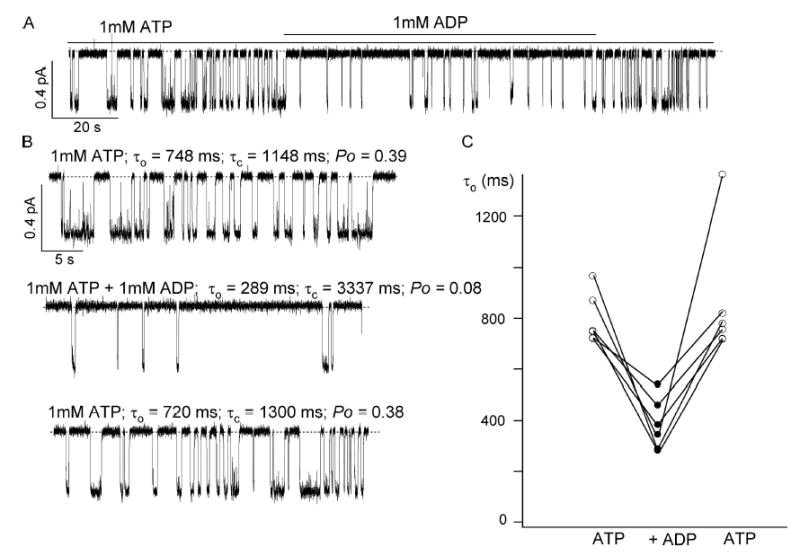
Effect of ADP on the mean open time in the slow gating mode. (A) A representative single-channel ΔR-CFTR current trace in slow gating mode. (B) Expanded traces with ATP alone and with both ATP and ADP from the recording shown in A. (C) Reversible shortening of the mean open time by ADP from six patches where ΔR-CFTR channels are in slow gating mode.
DISCUSSION
Characterization of the ΔR-CFTR Construct
Being a member of the ABC transporter family, CFTR utilizes the free energy of ATP binding/hydrolysis to perform its function (Gadsby and Nairn, 1999). While other members of the family harvest this energy to actively transport substrates across the cell membrane (Kuchler and Thorner, 1992), CFTR, being a bona fide ion channel, uses ATP to control the opening and closing of its gate. The exquisite sensitivity of electrophysiological techniques makes it possible to study detailed kinetic events at a single-molecule level, a goal not easily attainable for other members of this superfamily. However, studying the ATP-dependent gating of CFTR has proven to be a difficult task mainly because of the technical hurdles associated with obtaining long stable single-channel recordings needed for rigorous kinetic analysis (Powe et al., 2002). Channel “rundown” in excised inside-out patches often presents an insurmountable problem (see Powe et al., 2002 for detailed discussion). This technical difficulty of channel rundown may at least partially account for the enormous variation of reported kinetic parameters for WT-CFTR (see below).
One known mechanism for this rundown is the dephosphorylation of the CFTR by membrane-associated protein phosphatases. Although including phosphatase inhibitors such as okadaic acid may be of some value to inhibit phosphatases 1 and 2A (e.g., Berger et al., 1993; Hwang et al., 1993), no known specific inhibitors have been reported for phosphatase 2C, a major class of phosphatases that dephosphorylate CFTR (Travis et al., 1997; Luo et al., 1998). To complicate matters further, a recent study suggests that PP2C through its close association with CFTR is a strong candidate for the endogenous membrane-bound phosphatase responsible for dephosphorylating the channel inside an intact cell as well as in the membrane after patch excision (Zhu et al., 1999). It is therefore questionable whether using phosphatase inhibitors can amend this problem.
In theory, one can also include PKA throughout the experiment to promote CFTR phosphorylation. The antagonism between added kinases and membrane-associated phosphatases allows for stable, steady-state conditions during recording. However, it is inevitable that the kinase/phosphatase-driven transitions between partially phosphorylated and dephosphorylated states become nested within transitions solely due to ATP-dependent gating, adding further variability to the extracted kinetic parameters.
Rich et al. (1991) first reported that CFTR channels become constitutively active after removal of part of the R domain (amino acids 708–835). Since several of the PKA consensus sequences remain in this partial ΔR construct, the channel remains responsive to cAMP stimulation and therefore is not immune to dephosphorylation-dependent rundown in excised patches (compare Ma et al., 1997). Later Rich et al. (1993) reported a phosphorylation-independent channel, obtained after the mutation of serine 660 in their partial ΔR construct. More recently, Csanády et al. (2000) obtained a functional CFTR channel with the R domain completely deleted (amino acids 634–836). However, they reported that the basal activity of this ΔR-CFTR, expressed in Xenopus oocytes, could be increased ~30% by the application of PKA. Although a suggestion of the presence of PKA phosphorylation site(s) outside of the R domain was made, so far there is no evidence that any serine residue outside of the R domain can be phosphorylated in intact CFTR in vivo or in vitro (Cheng et al., 1991; Picciotto et al., 1992; Neville et al., 1997). In fact, using mass spectroscopic methods, the most sensitive biochemical technique for identification of phosphorylated peptides, Csanády et al. (2005) failed to detect phosphorylation of extra R domain serine. Indeed, after removal of 15 PKA consensus sites (major as well as minor), 14 of which are located in the R domain, CFTR can no longer be activated by PKA (Seibert et al., 1999).
We expressed the ΔR-CFTR construct reported by Csanády et al. (2000) in CHO cells and characterized in detail its regulation and gating mechanisms. Contrary to the results reported by Csanády et al. (2000), we found that the basal ΔR-CFTR currents cannot be enhanced by the application of cAMP agonists in cell-attached and whole-cell experiments, nor can the single-channel Po of ΔR-CFTR be increased by the presence of exogenous PKA in excised inside-out patches. The reason for this discrepancy is unknown. It should be noted, however, that two different expression systems were used: mammalian cells versus Xenopus oocytes. Nevertheless, the fact that ΔR-CFTR, expressed in CHO cells, is resistant to dephosphorylation makes this construct an ideal candidate for studying ATP-dependent gating, at least in CHO cells.
The current report shows that ATP-dependent gating of ΔR-CFTR is very similar to WT-CFTR gating. The single-channel Po, the ATP dose response, and the open and closed times agree very well with our previous studies of WT-CFTR channels (Zeltwanger et al., 1999). However the kinetic parameters in the current manuscript do not agree with those reported by Csanády et al. (2000). At maximal ATP concentration (2 mM), their Po is much lower than the one reported in this paper (0.19 ± 0.01 vs. 0.39 ± 0.01). The open time of the channel is about the same (297 ± 35 ms vs. 325 ± 21 ms), but their closed time is significantly longer (1297 ± 181 ms vs. 388 ± 34 ms). We do not know what accounts for these differences. Different expression systems may be partly responsible. Factors other than phosphorylation/dephosphorylation (e.g., PIP2, PDZ binding proteins) should also be considered (Wang et al., 2000; Raghuram et al., 2001; Himmel and Nagel, 2004).
Perhaps because of its inefficient assembly, ΔR-CFTR expression in the cell membrane is low. We were able to obtain a significant number of patches that contain only a single ΔR-CFTR channel. This distinct advantage, plus the fact that ΔR-CFTR is resistant to dephosphorylation-induced rundown, makes this ΔR-CFTR construct a useful tool to explore the mechanisms of CFTR gating. It may also provide a useful system to test the effect of pharmacological reagents since the phosphorylation step is bypassed (e.g., Ai et al., 2004).
Different Gating Modes
Overcoming the dephosphorylation-induced rundown also allows us to obtain long-lasting steady recordings of ΔR-CFTR channels, which reveal shifts of gating modes. Mode shifts are commonly observed in ion channel studies, reported for numerous other channels such as the Ca-activated maxi K channels (McManus and Mangleby, 1988), fast Cl channels from rat skeletal muscle (Blatz and Mangleby, 1986), and Na channels from mouse myocardial cells (Bohle and Benndorf, 1995). Although in most cases the exact mechanism for mode shifts is yet to be identified, some are known to be phosphorylation dependent (e.g., Yue et al., 1990). In the current study, we do not understand the mechanism for the mode shifts of CFTR, but it appears that CFTR’s gating modes are not related to PKA-dependent phosphorylation.
Does WT-CFTR exhibit mode shifts? Although the current study does not provide direct evidence that these mode shifts occur in WT-CFTR, we speculate that they do exist for the following reasons. First, the reported Po values for WT-CFTR vary in the literature (see Table I). Some of these differences may be due to different levels of phosphorylation (e.g., a lower Po value reported by Mathews et al., 1998). However, there seems to be a pattern of observations regarding the Po of WT-CFTR. For example, most studies showed a Po of ~0.4 for WT-CFTR, a number very similar to that of ΔR-CFTR in fast and slow gating modes, but much higher Po values have been reported (Gunderson and Kopito, 1994; Ramjeesingh et al., 1999). Interestingly, the Po value of ~0.7 in these studies is similar to that of ΔR-CFTR in the high Po mode. Second, it is known for years that CFTR gating is much slower when studied in the cell-attached configuration than that in excised inside-out patches (compare Fig. 1 and Fig. 3; also Hwang et al., 1997 vs. Zeltwanger et al., 1999). This change of gating behavior of WT-CFTR is consistent with our observation that ΔR-CFTR usually exhibits slow gating right after patch excision. Lastly, in a few patches with WT-CFTR activated with cAMP agonists in the cell-attached configuration before patch excision, we did observe a change from slow to fast gating mode after patch excision (Fig. S2).
TABLE I.
Summary of Single-channel Po for WT-CFTR
| Manuscript | Cell type | Configuration | Stimulant | Po |
|---|---|---|---|---|
| Gunderson and Kopito, 1994 | HEK293 | bilayer | 1 mM ATP | 0.69–0.76 |
| Ma et al., 1997 | HEK293 | bilayer | 2 mM ATP | 0.364 ± 0.042 |
| Ramjeesingh et al., 1999 | sf9 | bilayer | 1 mM ATP | 0.68 ± 0.05 |
| Aleksandrov et al., 2000 | CHO | bilayer | 0.3 mM ATP | 0.16–0.38 |
| Csanády et al., 2000 | Xenopus oocytes | excised | 2 mM ATP + PKA | 0.36 ± 0.03 |
| Tabcharani et al., 1991 | CHO | excised | 0.5 mM ATP + PKA | 0.407 ± 0.023 |
| Chang et al., 1993 | CHO | excised | 1 mM ATP + PKA | 0.46 ± 0.8 |
| Mathews et al., 1998 | CHO | excised | 1 mM ATP + PKA | 0.24 ± 0.04 |
| Rich et al., 1993 | HeLa | excised | 0.88 mM ATP | 0.35 ± 0.01 |
| Sheppard et al., 1993 | HeLa | excised | 1 mM ATP + PKA | 0.34 ± 0.09 |
| Ikuma and Welsh, 2000 | HeLa | excised | 1 mM ATP | 0.33 ± 0.02 |
| Anderson and Welsh, 1992 | HeLa | excised | 2.53 mM ATP | 0.44 ± 0.04 |
| Winter et al., 1994 | HeLa/NIH3T3 | excised | 3 mM ATP | 0.44 ± 0.06 |
| Denning et al., 1992 | NIH3T3 | excised | 1 mM ATP | 0.34 ± 0.02 |
| Zeltwanger et al., 1999 | NIH3T3 | excised | 2.75 mM ATP | 0.41 ± 0.03 |
| Venglarik et al., 1994 | L | excised | 1 mM ATP | 0.42 ± 0.03 |
| Cai et al., 2003 | C127 | excised | 1 mM ATP + PKA | 0.57 ± 0.04 |
| Raghuram et al., 2003 | Calu-3 | excised | 2 mM ATP + PKA | 0.43 ± 0.06 |
| Fischer and Machen, 1994 | NIH3T3 | cell attached | 1 μM Fsk | 0.5 ± 0.02 |
| Hwang et al., 1997 | NIH3T3 | cell attached | 10 μM Fsk | 0.27 ± 0.03 |
| Ai et al., 2004 | NIH3T3 | cell attached | 10 μM Fsk | 0.2 ± 0.02 |
| Haws et al., 1992 | 16HBE | cell attached | 10 μM Fsk | 0.33 ± 0.3 |
One may ask why mode shifts have never been reported for WT-CFTR. First, a time-dependent rundown of WT-CFTR makes it difficult to interpret any time-dependent changes of gating behavior. Second, in order for WT-CFTR to function in excised inside-out patches, PKA and ATP have to be applied once the membrane patch is excised. Phosphorylation by exogenous PKA usually takes a few minutes to reach a steady state. Therefore, it is likely that mode shifts, if occur, may be buried in the recording that precedes the steady state. In addition, even if mode shifts are observed, presteady-state channel activity is seldom used for data analysis.
Effect of ADP on CFTR Gating
After establishing ΔR-CFTR channels as useful tools for studying CFTR gating, we first investigated the mechanism of ADP-induced inhibition of CFTR channel activity. Previous studies showed that ADP can inhibit CFTR channel activity presumably by competing with ATP for a common binding site since the magnitude of inhibition is diminished upon increasing [ATP] (Anderson and Welsh, 1992; Schultz et al., 1995). Subsequently, Gunderson and Kopito (1994) and Winter et al. (1994) showed that ADP increased the closed time with little effect on the open time. Using noise analysis of macroscopic CFTR currents, Schultz et al. (1995) confirmed that ADP decreases the Po of CFTR by decreasing the opening rate. However, since ATP is likely hydrolyzed during CFTR gating cycle, many potential kinetic steps can be affected to account for this inhibition. Without demonstrating an ADP-dependent closed state, a definite kinetic mechanism for this ADP inhibition of the opening rate remains to be determined.
In this paper we provide, for the first time, single-channel kinetic evidence that ADP competes with ATP by binding to the binding site that opens the channel. First, the ADP dose–response curves shift to the right as the ATP concentration increases, so that the Ki is higher with higher ATP concentration, supporting a competitive mechanism (also see Anderson and Welsh, 1992). More importantly, using single-channel dwell time analysis, we were able to observe a new closed state in the presence of ADP. This new closed state was suggested by Schultz et al. (1995), but their only single-channel recording was not long enough to collect enough events to adequately separate it from the ATP-dependent closed state. Indeed, the fact that both ATP-dependent gating and ADP-induced inhibition are slow events demands extremely long recordings for single-channel analysis. The steady channel activity of ΔR-CFTR allows us to use a higher ADP/ATP ratio that effectively separates two closed time constants. With this result, we conclude then that ADP competitively inhibits CFTR channel opening. It should be noted that our results do not indicate that an ADP-bound channel cannot open. It simply means that ADP-bound channels have a much lower opening rate compared with ATP-bound ones.
Which NBD does ADP bind to inhibit channel opening? Recent biochemical studies (Szabo et al., 1999; Aleksandrov et al., 2002; Basso et al., 2003) suggest that ATP is occluded in NBD1 (i.e., the off rate is extremely slow). Therefore, association and dissociation of ATP at NBD1 cannot account for the electrophysiologically observed, millisecond-to-second gating transitions. Vergani et al. (2003) found that mutations of either Walker A lysine, K464, or K1250 result in a rightward shift of the dose–response of ATP in CFTR activation. The same paper shows that the D1370N mutation at the Walker B motif in NBD2 also decreases the apparent affinity of ATP (compare Bompadre et al., 2005). Incorporating both electrophysiological and biochemical data, Vergani et al. (2003) proposes (Scheme 2) that binding of ATP to NBD1 is prerequisite for ATP binding at NBD2 to open the channel, but association and dissociation of ATP at NBD1 (first C↔C·ATP transitions in Scheme 2) are not involved in the observed gating transitions because of the slow off rate of ATP at NBD1. Based on this scheme, we propose that ADP competes with ATP at NBD2 for channel opening.
(SCHEME 2).
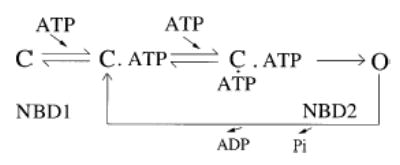
Surprisingly, we found that ADP not only induces a new closed state but also shortens the open time. The open time of the channel was reduced in the presence of ADP by as much as 54%. An effect of ADP on the open time has been suggested by Weinreich et al. (1999). While performing macroscopic relaxation analysis, they observed that ADP increases the relaxation rate of macroscopic currents (reflecting channel closing) upon nucleotide removal. Like what has been discussed above, the mechanism of this effect of ADP is unknown without detailed single-channel kinetic analysis. Here we provide the first single-channel kinetic evidence that ADP affects the open state stability of the CFTR channel. This novel effect of ADP on the open time constant cannot be easily explained by the gating model described in Scheme 2 since both nucleotide binding sites have to be occupied by ATP for channel opening. We hypothesize that ADP binds at a site that is different from the binding site for channel opening, as proposed by Weinreich et al. (1999). In theory, one would predict the presence of a discrete open time constant corresponding to the new open state of the channel when ADP is bound. While our single-channel dwell time analysis shows a shortening of the open time in the presence of ADP, we were not able to resolve two open time constants in WT-CFTR. We reason that if the difference between the time constant of ADP-bound open state and that of ATP-bound one is not extremely large, dissecting different states is a technical difficulty that is hard to overcome. Nevertheless, the use of the ΔR-CFTR construct gives us a unique opportunity to be able to collect enough single-channel events that reveal this phenomenon. Observing the slow gating mode allows us to verify the ADP effect on the open time. In the accompanying paper, we used various CFTR mutants to further elaborate the kinetic mechanism for this interesting observation.
Supplementary Material
Acknowledgments
We are grateful to Drs. Joseph Mindell, Kevin Gillis, and Xiaoqin Zou for their critical reading of the manuscript. We thank Shenghui Hu for technical assistance.
Footnotes
T. Ai’s present address is Texas Heart Institute, St. Luke’s Episcopal Hospital, Houston, TX 77030.
The online version of this article contains supplemental material.
This work is supported by the National Institutes of Health (DK55835 and HL53445 to T.C. Hwang). T. Ai is a recipient of postdoctoral fellowship from the American Heart Association. S.G. Bompadre is a recipient of NRSA (DK062565). Y. Sohma is supported by the Japan Society for the Promotion of Science (15590196).
Olaf S. Andersen served as editor.
References
- Ai T, Bompadre SG, Wang X, Hu S, Li M, Hwang TC. Capsaicin potentiates wild-type and mutant CFTR chloride channel currents. Mol Pharmacol. 2004;65:1415–1426. doi: 10.1124/mol.65.6.1415. [DOI] [PubMed] [Google Scholar]
- Aleksandrov AA, Chang XB, Aleksandrov L, Riordan JR. The non-hydrolytic pathway of the cystic fibrosis transmembrane conductance regulator ion channel gating. J Physiol. 2000;528:259–265. doi: 10.1111/j.1469-7793.2000.00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleksandrov L, Aleksandrov AA, Chang XB, Riordan JR. The first nucleotide binding domain of cystic fibrosis transmembrane conductance regulator is a site of stable nucleotide interaction, whereas the second is a site of rapid turnover. J Biol Chem. 2002;277:15419–15425. doi: 10.1074/jbc.M111713200. [DOI] [PubMed] [Google Scholar]
- Al-Nakkash L, Hwang TC. Activation of wild-type and ΔF508-CFTR by phosphodiesterase inhibitors. Pflugers Arch. 1999;437:553–561. doi: 10.1007/s004240050817. [DOI] [PubMed] [Google Scholar]
- Anderson MP, Welsh MJ. Regulation by ATP and ADP of CFTR chloride channels that contain mutant nucleotide-binding domains. Science. 1992;257:1701–1704. doi: 10.1126/science.1382316. [DOI] [PubMed] [Google Scholar]
- Basso C, Vergani P, Nairn AC, Gadsby DC. Prolonged nonhydrolytic interaction of nucleotide with CFTR’s NH2-terminal nucleotide binding domain and its role in channel gating. J Gen Physiol. 2003;122:333–348. doi: 10.1085/jgp.200308798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger HA, Travis SM, Welsh MJ. Regulation of the cystic fibrosis transmembrane conductance regulator Cl channels by specific kinases and protein phosphatases. J Biol Chem. 1993;268:2037–2047. [PubMed] [Google Scholar]
- Blatz AL, Mangleby KL. Quantitative description of three modes of activity of fast chloride channels from rat skeletal muscle. J Physiol. 1986;378:141–174. doi: 10.1113/jphysiol.1986.sp016212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohle T, Benndorf K. Multimodal action of single Na channels in myocardial mouse cells. Biophys J. 1995;68:121–130. doi: 10.1016/S0006-3495(95)80166-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bompadre SG, Cho JH, Wang X, Sohma Y, Zou X, Li M, Hwang TC. CFTR gating II: effects of nucleotide binding on the stability of open states. J Gen Physiol. 2005;125:377–394. doi: 10.1085/jgp.200409228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Button B, Reuss L, Altenberg GA. PKC-mediated stimulation of amphibian CFTR depends on a single phosphorylation consensus site. Insertion of this site confers PKC sensitivity to human CFTR. J Gen Physiol. 2001;117:457–467. doi: 10.1085/jgp.117.5.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Scott-Ward TS, Sheppard DN. Voltage-dependent gating of the cystic fibrosis transmembrane conductance regulator Cl− channel. J Gen Physiol. 2003;122:605–620. doi: 10.1085/jgp.200308921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson MR, Travis SM, Welsh MJ. The two nucleotide-binding domains of cystic fibrosis transmembrane conductance regulator (CFTR) have distinct functions in controlling channel activity. J Biol Chem. 1995;270:1711–1717. doi: 10.1074/jbc.270.4.1711. [DOI] [PubMed] [Google Scholar]
- Chang XB, Tabcharani JA, Hou YX, Jensen TJ, Kartner N, Alon NW, Hanrahan JW, Riordan JR. Protein kinase A (PKA) still activates CFTR chloride channel after mutagenesis of all 10 PKA consensus phosphorylation sites. J Biol Chem. 1993;268:11304–11311. [PubMed] [Google Scholar]
- Chappe V, Hinkson DA, Zhu T, Chang XB, Riordan JR, Hanrahan JW. Phosphorylation of protein kinase C sites in NBD1 and the R domain control CFTR channel activation by PKA. J Physiol. 2003;548:39–52. doi: 10.1113/jphysiol.2002.035790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Altenberg GA, Reuss L. Mechanism of activation of Xenopus CFTR by stimulation of PKC. Am J Physiol Cell Physiol. 2004;287:C1256–C1263. doi: 10.1152/ajpcell.00229.2004. [DOI] [PubMed] [Google Scholar]
- Cheng SH, Rich DP, Marshall J, Gregory RJ, Welsh MJ, Smith AE. Phosphorylation of the R domain by cAMP-dependent protein kinase regulates CFTR chloride channel. Cell. 1991;66:1027–1036. doi: 10.1016/0092-8674(91)90446-6. [DOI] [PubMed] [Google Scholar]
- Csanády L. Rapid kinetic analysis of multichannel records by a simultaneous fit to all dwell-time histograms. Biophys J. 2000;78:785–799. doi: 10.1016/S0006-3495(00)76636-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csanády L, Chan KW, Seto-Young D, Kopsco DC, Nairn AC, Gadsby DC. Severed channels probe regulation of gating of cystic fibrosis transmembrane conductance regulator by its cytoplasmic domains. J Gen Physiol. 2000;116:477–500. doi: 10.1085/jgp.116.3.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csanády L, Seto-Young D, Chan KW, Cenciarelli C, Angel BB, Qin J, McLachlin DT, Krutchinsky AN, Chait BT, Nairn AC, Gadsby DC. Preferential phosphorylation of serine 768 dampens activation of CFTR channels by PKA. J Gen Physiol. 2005;125:171–186. doi: 10.1085/jgp.200409076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- Fenwick EM, Marty A, Neher E. Sodium and calcium channels in bovine chromaffin cells. J Physiol. 1982;331:599–635. doi: 10.1113/jphysiol.1982.sp014394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer H, Machen TE. CFTR displays voltage dependence and two gating modes during stimulation. J Gen Physiol. 1994;104:541–566. doi: 10.1085/jgp.104.3.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadsby DC, Nairn AC. Control of CFTR channel gating by phosphorylation and nucleotide hydrolysis. Physiol Rev. 1999;79:S77–S107. doi: 10.1152/physrev.1999.79.1.S77. [DOI] [PubMed] [Google Scholar]
- Gunderson KL, Kopito RR. Effects of pyrophosphate and nucleotide analogs suggest a role for ATP hydrolysis in cystic fibrosis transmembrane regulator channel gating. J Biol Chem. 1994;269:19349–19353. [PubMed] [Google Scholar]
- Haws C, Krouse ME, Xia Y, Gruenert DC, Wine JJ. CFTR channels in immortalized human airway cells. Am J Physiol. 1992;263:L692–L707. doi: 10.1152/ajplung.1992.263.6.L692. [DOI] [PubMed] [Google Scholar]
- Himmel B, Nagel G. Protein kinase-independent activation of CFTR by phosphatidylinositol phosphates. EMBO Rep. 2004;5:85–90. doi: 10.1038/sj.embor.7400034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang TC, Horie M, Nairn AC, Gadsby DC. Role of GTP-binding proteins in the regulation of mammalian cardiac chloride conductance. J Gen Physiol. 1992;99(4):465–489. doi: 10.1085/jgp.99.4.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang TC, Horie M, Gadsby DC. Functionally distinct phospho-forms underlie incremental activation of protein kinase-regulated Cl− conductance in mammalian heart. J Gen Physiol. 1993;101:629–650. doi: 10.1085/jgp.101.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang TC, Wang F, Yang I, Reenstra WW. Genistein potentiates wild-type and ΔF508 CFTR channel. Am J Physiol. 1997;273:C988–C998. doi: 10.1152/ajpcell.1997.273.3.C988. [DOI] [PubMed] [Google Scholar]
- Ikuma M, Welsh MJ. Regulation of CFTR Cl channel gating by ATP binding and hydrolysis. Proc Natl Acad Sci USA. 2000;97:8675–8680. doi: 10.1073/pnas.140220597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illek B, Fischer H, Santos GF, Widdicombe JH, Machen TE, Reenstra WW. cAMP-independent activation of CFTR Cl channels by the tyrosine kinase inhibitor genistein. Am J Physiol. 1995;268:C886–C893. doi: 10.1152/ajpcell.1995.268.4.C886. [DOI] [PubMed] [Google Scholar]
- Kuchler K, Thorner J. Secretion of peptides and proteins lacking hydrophobic signal sequences: the role of adenosine triphosphate-driven membrane translocators. Endocr Rev. 1992;13:499–514. doi: 10.1210/edrv-13-3-499. [DOI] [PubMed] [Google Scholar]
- Li C, Ramjeesingh M, Wang W, Garami E, Hewryk M, Lee D, Rommens JM, Galley K, Bear CE. ATPase activity of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1996;271:28463–28468. doi: 10.1074/jbc.271.45.28463. [DOI] [PubMed] [Google Scholar]
- Luo J, Pato MD, Riordan JR, Hanrahan JW. Differential regulation of single CFTR channels by PP2C, PP2A, and other phosphatases. Am J Physiol. 1998;274:C1397–C1410. doi: 10.1152/ajpcell.1998.274.5.C1397. [DOI] [PubMed] [Google Scholar]
- Ma J, Zhao J, Drumm ML, Xie J, Davis PB. Function of the R domain in the cystic fibrosis transmembrane conductance regulator chloride channel. J Biol Chem. 1997;272:28133–28141. doi: 10.1074/jbc.272.44.28133. [DOI] [PubMed] [Google Scholar]
- Mathews CJ, Tabcharani JA, Hanrahan JW. The CFTR chloride channel: nucleotide interactions and temperature-dependent gating. J Membr Biol. 1998;163(1):55–66. doi: 10.1007/s002329900370. [DOI] [PubMed] [Google Scholar]
- McManus OB, Mangleby KL. Kinetic states and modes of single large-conductance calcium-activated potassium channels in cultured rat skeletal muscle. J Physiol. 1988;402:79–120. doi: 10.1113/jphysiol.1988.sp017195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neville DCA, Rozanas CR, Price EM, Gruis DB, Verkman AS, Townsend RR. Evidence for phosphorylation of serine 753 in CFTR using a novel metal-ion affinity resin and matrix-assisted laser desorption mass spectroscopy. Protein Sci. 1997;6:2436–2445. doi: 10.1002/pro.5560061117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Cohn JA, Bertuzzi G, Greengard P, Nairn AC. Phosphorylation of the cystic fibrosis transmembrane regulator. J Biol Chem. 1992;267:12742–12752. [PubMed] [Google Scholar]
- Powe A, Zhou Z, Hwang TC, Nagel G. Quantitative analysis of ATP-dependent gating of CFTR. Methods Mol Med. 2002;70:67–98. doi: 10.1385/1-59259-187-6:67. [DOI] [PubMed] [Google Scholar]
- Raghuram V, Mak DD, Foskett JK. Regulation of cystic fibrosis transmembrane conductance regulator single-channel gating by bivalent PDZ-domain-mediated interaction. Proc Natl Acad Sci USA. 2001;98(3):1300–1305. doi: 10.1073/pnas.031538898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuram V, Hormuth H, Foskett JK. A kinase-regulated mechanism controls CFTR channel gating by disrupting bivalent PDZ domain interactions. Proc Natl Acad Sci USA. 2003;100:9620–9625. doi: 10.1073/pnas.1633250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramjeesingh M, Li C, Garemi E, Galley KA, Huan LJ, Wang Y, Bear CE. Walker mutations reveal loose relationship between catalytic and channel-gating activities of purified CFTR (cystic fibrosis transmembrane conductance regulator) Biochemistry. 1999;38:1463–1468. doi: 10.1021/bi982243y. [DOI] [PubMed] [Google Scholar]
- Randak C, Welsh MJ. An intrinsic adenylate kinase activity regulates gating of the ABC transporter CFTR. Cell. 2003;115:837–850. doi: 10.1016/s0092-8674(03)00983-8. [DOI] [PubMed] [Google Scholar]
- Rich DP, Gregory RJ, Anderson MP, Manavalan P, Smith AE, Welsh MJ. Effect of deleting the R domain on CFTR-generated chloride channels. Science. 1991;253:205–207. doi: 10.1126/science.1712985. [DOI] [PubMed] [Google Scholar]
- Rich DP, Berger HA, Cheng SH, Travis SM, Saxena M, Smith AE, Welsh MJ. Regulation of the cystic fibrosis transmembrane conductance regulator Cl channel by negative charge in the R domain. J Biol Chem. 1993;268:20259–20267. [PubMed] [Google Scholar]
- Riordan JR, Rommens JM, Kerem BS, Alon N, Rozmahel R, Grzelzak Z, Zielenski J, Lok S, Plavsic N, Chou JL, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1072. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Schultz BD, Venglarik CJ, Bridges RJ, Frizzell RA. Regulation of CFTR Cl− channel gating by ADP and ATP analogs. J Gen Physiol. 1995;105:329–361. doi: 10.1085/jgp.105.3.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibert FS, Chang XB, Aleksandrov AA, Clarke DM, Hanrahan JW, Riordan JR. Influence of phosphorylation by protein kinase A on CFTR at the cell surface and endoplasmic reticulum. Biochim Biophys Acta. 1999;1461:275–283. doi: 10.1016/s0005-2736(99)00163-7. [DOI] [PubMed] [Google Scholar]
- Sheppard DN, Rich DP, Ostedgaard LS, Gregory RJ, Smith AE, Welsh MJ. Mutations in CFTR associated with mild-disease-form Cl− channels with altered pore properties. Nature. 1993;362:160–164. doi: 10.1038/362160a0. [DOI] [PubMed] [Google Scholar]
- Sigworth FJ, Sine SM. Data transformations for improved display and fitting of single-channel dwell time histograms. Biophys J. 1987;52:1047–1054. doi: 10.1016/S0006-3495(87)83298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo K, Szakacs G, Hegeds T, Sarkadi B. Nucleotide occlusion in the human cystic fibrosis transmembrane conductance regulator. Different patterns in the two nucleotide binding domains. J Biol Chem. 1999;274:12209–12212. doi: 10.1074/jbc.274.18.12209. [DOI] [PubMed] [Google Scholar]
- Szellas T, Nagel G. Apparent affinity of CFTR for ATP is increased by continuous kinase activity. FEBS Lett. 2003;535:141–146. doi: 10.1016/s0014-5793(02)03892-9. [DOI] [PubMed] [Google Scholar]
- Tabcharani JA, Chang XB, Riordan JR, Hanrahan JW. Phosphorylation-regulated Cl− channel in CHO cells stably expressing the cystic fibrosis gene. Nature. 1991;352:628–631. doi: 10.1038/352628a0. [DOI] [PubMed] [Google Scholar]
- Travis SM, Berger HA, Welsh MJ. Protein phosphatase 2C dephosphorylates and inactivates cystic fibrosis transmembrane conductance regulator. Proc Natl Acad Sci USA. 1997;94:11055–11060. doi: 10.1073/pnas.94.20.11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venglarik CJ, Schultz BD, Frizzell RA, Bridges RJ. ATP alters current fluctuations of cystic fibrosis transmembrane conductance regulator: evidence for a three-state activation mechanism. J Gen Physiol. 1994;104:123–146. doi: 10.1085/jgp.104.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani P, Nairn AC, Gadsby DC. On the mechanism of MgATP-dependent gating of CFTR Cl− channels. J Gen Physiol. 2003;121:17–36. doi: 10.1085/jgp.20028673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Zeltwanger S, Hu S, Hwang TC. Deletion of phenylalanine 508 causes attenuated phosphorylation-dependent activation of the CFTR chloride channel. J Physiol. 2000;524:637–648. doi: 10.1111/j.1469-7793.2000.00637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreich F, Riordan JR, Nagel G. Dual effects of ADP and adenylylimidodiphosphate on CFTR channel kinetics show binding to two different nucleotide binding sites. J Gen Physiol. 1999;114:55–70. doi: 10.1085/jgp.114.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter MC, Sheppard DN, Carson MR, Welsh MJ. Effect of ATP concentration on CFTR Cl− channels: a kinetic analysis of channel regulation. Biophys J. 1994;66:1398–1403. doi: 10.1016/S0006-3495(94)80930-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang IC, Cheng TH, Wang F, Price EM, Hwang TC. Modulation of CFTR chloride channels by calyculin A and genistein. Am J Physiol. 1997;272:C142–C155. doi: 10.1152/ajpcell.1997.272.1.C142. [DOI] [PubMed] [Google Scholar]
- Yue DT, Herzig S, Marban E. β-Adrenergic stimulation of calcium channels occurs by potentiation of high-activity gating modes. Proc Natl Acad Sci USA. 1990;87:753–757. doi: 10.1073/pnas.87.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeltwanger S, Wang F, Wang GT, Gillis KD, Hwang TC. Gating of cystic fibrosis transmembrane conductance regulator chloride channels by adenosine triphosphate hydrolysis. Quantitative analysis of a cyclic gating scheme. J Gen Physiol. 1999;113:541–554. doi: 10.1085/jgp.113.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Hu S, Hwang TC. Voltage-dependent flickery block of an open cystic fibrosis transmembrane conductance regulator (CFTR) channel pore. J Physiol. 2001;532:435–448. doi: 10.1111/j.1469-7793.2001.0435f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu T, Dahan D, Evagelidis A, Zheng S, Luo J, Hanrahan JW. Association of cystic fibrosis transmembrane conductance regulator and protein phosphatase 2C. . J. Biol Chem. 1999;274:29102–29107. doi: 10.1074/jbc.274.41.29102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.