

Abstract
Activity-dependent changes in presynaptic function represent a critical mechanism by which synaptic strength is controlled. However, how changes in synaptic activity couple to presynaptic components to control synaptic vesicle release and recycling are poorly understood. Sphingosine kinase (SphK) is a sphingolipid metabolic enzyme whose activity-dependent recruitment to membrane regions within presynaptic terminals promotes neurotransmitter release. Here, we show that synaptic recruitment of SPHK-1, the SphK ortholog in C. elegans, is mediated by presynaptic calcium influx. Quantitative fluorescence imaging of live presynaptic terminals reveals that blocking presynaptic calcium influx reduces synaptic SPHK-1 abundance whereas increasing calcium influx increases SPHK-1 synaptic abundance. CALM-1, the calcium and integrin binding protein (CIB) ortholog, co-localizes with SPHK-1 at release sites and regulates muscarinic-mediated synaptic SPHK-1 recruitment. We identify two additional sphingolipid metabolic enzymes that are concentrated at presynaptic terminals, and mutants lacking one of these, HYL-1/ceramide synthase, have defects in synaptic transmission and in synaptic vesicle cycling. Finally, we show that SPHK-1 activity is required for the recruitment of the priming protein UNC-13/Munc13 to presynaptic terminals following activation by muscarinic signaling. These findings suggest that calcium-dependent regulation of local S1P metabolism at synapses may be an important mechanism by which synaptic vesicle priming factors are recruited to release sites to promote synaptic transmission.
Introduction
Regulation of neurotransmitter release allows synapses to alter their strength in response to changes in activity and environmental conditions. Presynaptic calcium influx plays a critical role in mediating these responses by regulating synaptic vesicle release and recycling, and by activating second messengers involved in synaptic plasticity (Catterall and Few, 2008). However, the molecular mechanisms by which calcium regulates presynaptic signaling pathways to modulate neurotransmitter release are poorly understood.
Sphingosine kinase (SphK) and its metabolites, the bioactive lipids sphingosine (SPH) and sphingosine-1-phosphate (S1P), regulate diverse cellular functions in the nervous system including cell survival, differentiation and neurotransmitter secretion (Okada et al., 2008). In particular, S1P facilitates neuronal firing and neurotransmitter release from neurons and neuroendocrine cells and SPH drives synaptic vesicle exocytosis by facilitating SNARE protein assembly (Okada et al., 2008; Darios et al., 2009). Genetic ablation or knockdown of SphK activity results in reductions in electrically evoked neurotransmitter release, defects in long-term potentiation, and accumulation of synaptic vesicles at presynaptic terminals (Kajimoto et al., 2007; Kanno et al., 2010; Chan et al., 2012). SphK rapidly translocates to presynaptic terminals in response to neuronal activity and to presynaptic heterotrimeric G protein signaling. Depolarization or treatment with muscarinic agonists results in rapid increase in synaptic translocation of SphK in hippocampal cultures or C. elegans motor neurons, respectively. On the other hand, hyperpolarization or reductions in muscarinic signaling decrease in SphK synaptic abundance (Kajimoto et al., 2007; Chan et al., 2012). Because studies in diverse cell types show that a critical step in regulating SphK activity is to promote its translocation to cellular membranes from cytosolic pools, identifying mechanisms underlying its translocation to synapses will be important for understanding presynaptic plasticity.
Calcium has been implicated in regulating SphK translocation to cellular membranes. In neuroblastoma cells, calmodulin inhibitors block translocation of SphK to plasma membranes (Young et al., 2003). In HEK cells, calcium mediates the interaction between SphK and calcium and integrin binding protein, CIB, and CIB knockdown impedes agonist-dependent SphK membrane translocation and S1P production (Jarman et al., 2010; Pitson, 2011). However, whether calcium mobilization mediates SphK translocation to synaptic membranes, how neuronal activity and heterotrimeric G protein signaling converge at synapses to regulate SphK, and how synaptic SphK, in turn, promotes neurotransmitter release are not known.
Here, we show that presynaptic calcium influx regulates the recruitment of SPHK-1 to presynaptic membranes in C. elegans motor neurons. In addition, we show that muscarinic-mediated recruitment of SPHK-1 to synapses requires presynaptic calcium influx as well as the interaction of SPHK-1 with the C. elegans CIB ortholog, CALM-1. Furthermore, we show that HYL-1/ceramide synthase, which also localizes to presynaptic terminals, antagonizes the effects of sphk-1 on synaptic transmission. Finally, we show that sphk-1 activity mediates the effects of muscarinic signaling by recruiting the synaptic vesicle priming protein UNC-13/Munc13 to synapses. Together, these findings suggest that local sphingolipid metabolism at synapses may be a previously unrecognized mechanism by which presynaptic calcium regulates presynaptic function.
Materials and Methods
C. elegans strains
All experiments were performed on young adult hermaphrodites. Strains sphk-1(ok1097) and hyl-1(gk203) were provided by the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources (NCRR). Strain calm-1(tm1353) was provided by National BioResource Project (Japan). The wild type reference strain was N2 Bristol. sphk-1(ok1097) was outcrossed 10 times and used for all experiments. All other strains used were outcrossed greater than 4 times. In addition, the following strains were used: egl-19(ad1006lf), egl-19(n2368gf), egl-30(js126gf) [strain information o n www.wormbase.org], unc-2(Ij1), unc-13(s69), unc-68(r1162), unc-104(e1265), nuIs46[myo-2-GFP, Punc-129-unc13S-GFP], nuIs152[ttx-3-mRFP, Punc-129-GFP-snb-1], nuIs197[myo-2-GFP, Punc-129-sphk-1-GFP]. All integrated transgenes were outcrossed 10 times.
Molecular biology
The promoter fragment of sphk-1 was amplified by PCR from mixed stage genomic DNA. sphk-1 mutations were made by standard molecular cloning techniques or by Quikchange PCR (Stratagene) targeting the following deletions: ΔCaM, (F217A, I272Q). The following primers were used for molecular cloning:
| gene | primer |
|
| |
| calm-1/CIB | 5’ CCCCCCGCTAGCAAAAATGGGAAACAATGCAAGTTC |
| 3’ CCCCCCGGTACCTTAAATTCTGATATGAAAAG | |
| calm-1 (promoter) | 5’ CCCCCCGCATGCGAAAATAACTTGTGGTATGATG |
| 3’ CCCCCCGGATCCCTGGTCACCTGATCTGAAATG | |
| hyl-1/CER synthase | 5’ ccccccgctagcaaaaATGTGGCGGATGTCATATTTCTGG |
| 3’ CCCCCCCCCGGGCTCCTTGCGAGGAGCTCTGC | |
| sphk-1 (promoter) | 5’ CCCCCCGCGATCGCTCTGTAAAAAATTGAAGAATTCG |
| 3’ CCCCCCGCGGCCGCGTGATGTTCGGTGCAAGTGC | |
| sphk-1 (gene) | 5’ CCCCCCGCTAGCAAAAATGTTCATAGTAGTGGTAAC |
| 3’ CCCCCCGGTACCCTAGGCAGTTGATGAGAAAACGG | |
| sphk-1 (NT1) | 5’ CCCCGCTAGCAAAAATGGAACAATGTCGTGGAAATC |
| 5’ CCCCCCACCGGTTCCATTCGTTTTTACTTTTG | |
| sphk-1 (NT2) | 5’ CCCCCCGCTAGCAAAAATGTTCATAGTAGTGGTAAC |
| 5’ CCCCCCACCGGTTCCATTCGTTTTTACTTTTG | |
| sphk-1 (NT3) | 5’ CCCCCCGCTAGCAAAAATGTTCATAGTAGTGGTAAC |
| CCCCCCaccggtAACTGAATACAAAGCTACGCTTTCCG | |
| sphk-1 (CT1) | 5’ CCCCCCGCTAGCAAAAATGGTGAATATCTATGCAGTTAC |
| 3’ CCCCCCACCGGTGGCAGTTGATGAGAAAACGG | |
| sphk-1(ΔCaM)* (FI-AQ) | 5’ CACAGATTCACTGTTATGGGAGCTCAACGCAGCTGCAATCTCCGATC |
| 3’ GATCGGAGATTGCAGCTGCGTTGAGCTCCCATAACAGTGAATCTGTG | |
| tag-38/S1P lyase | 5’ CCCCCCGCTAGCAAAAATGGATTTTGCACTGGAGC |
| CCCCCCACCGGTCTCTGTTAAAGCATAAACAGC | |
The cDNA was used to make the following plasmids by standard molecular biology techniques (plasmid name[promoter-gene-marker]): pDS221[Punc-129-sphk-1-gfp], pDS287[Punc-129-hyl-1-gfp], pDS297[Punc-129-sphk-1(NT1)-gfp], pDS300[Psphk-1-gfp-nls], pDS301[Punc-129-sphk-1-mCherry], pDS302[Pmyo-3-sphk-1-gfp], pDS303[Psphk-1-sphk-1-gfp], pDS306[Punc-129--sphk-1(ΔCaM)-gfp], pDS329[Pmyo-3-OMM(inverted)-mCherry], pDS338[Punc-129-tag-38-gfp], pDS386[Punc-129-sphk-1(NT3)-gfp], pDS387[Punc-129-sphk-1(NT2)-gfp], pDS388[Punc-129-sphk-1(CT1)- gfp], pDS389[Punc-129-calm-1-mCherry], pDS391[Punc-129-snn-1-mCherry], pDS394[Pcalm-1-gfp].
Transgenic lines
Transgenic strains were generated by injecting either N2 or nuIs197 animals with expression constructs (10 25 ng/μl) and the co-injection marker [KP#708 (Pttx-3-rfp, 40 ng/μl) or KP#1106 (Pmyo-2-gfp 10 ng/μl)]. Microinjection was performed using standard techniques as previously described (Mello et al., 1991). At least three lines for each transgene were examined and a single, representative transgene was used for the experiments. The following transgenic lines were made (name[plasmid]): vjEx38[pDS390], vjEx39[pDS300], vjEx84[pDS329], vjEx98[pDS391], vjEx133[pDS302], vjEx337[pDS306], vjEx651[pDS297], vjEx652[pDS386], vjEx653[pDS387], vjEx654[pDS388], vjEx655[pDS389], vjEx656[pDS287], vjEx657[pDS338], vjEx711[pDS394].
Behavioral analysis of locomotion activity and sensitivity to aldicarb
For analysis of sensitivity to inhibitors of acetylcholinesterase, paralysis of adult worms was scored every 10 minutes, starting at 40 minutes using 1.0 mM aldicarb (Chem Services, West Chester, PA). For each experiment, three plates of twenty worms per genotype were placed on NGM plates supplemented with aldicarb, and the number of worms paralyzed on each plate was counted to extract the percent paralyzed at each time-point per genotype. The percentages were averaged at each time point per genotype and plotted graphically. For each experiment, the genotype was blind to the scorer and the analysis was repeated at least two times. For experiments using arecoline, worms were pretreated with plates containing either control M9 or arecoline (15mM in M9) for 2 hours, which was determined to be the minimum concentration that produced maximal aldicarb hypersensitivity, in order to minimize non-specific effects of the drug. For experiments using EGTA (ethylene glycol tetraacetic acid), worms were pretreated in drops of either EGTA (50mM, pH 7.2) or water (pH 7.2) for 2 hours at room temperature. Concentrations were determined by experiments previously successful in other studies (Xu et al., 2001; O’Halloran et al., 2009; Earls et al., 2010). Statistical differences were determined at each time point by Student t-tests.
Microscopy and analysis
Fluorescence microscopy experiments were performed as previously described (Ch’ng et al., 2008). Briefly, adult worms were paralyzed using 2,3-Butanedione monoxime (BDM, 30 μg/μL, Sigma) and mounted on 2% agarose pads for imaging. Images of synapses were captured from dorsal axons of DA neurons near the posterior gonadal bend of the worm. Quantification spanned >100 synapses for all experiments and specific n values are shown in Table 1. For all fluorescence microscopy experiments, images were captured with a Nikon eclipse 90i microscope equipped with a Nikon PlanApo 100× objective (NA = 1.4) and a Photometrics Coolsnap ES2 camera. Metamorph 7.0 software (Universal Imaging/Molecular Devices) was used to capture serial image stacks, and the maximum intensity projection was used for analysis of the dorsal cords. Linescans of the maximum intensity projection image were also recorded using Metamorph. The fluorescence intensity values were then quantified using Puncta 6.0 software written with Igor Pro (Wavemetrics), as previously described (Dittman and Kaplan, 2006; Ch’ng et al., 2008). For all experiments, fluorescence values were normalized to the values of 0.5 μm FluoSphere beads (Invitrogen) captured during each imaging session. This was performed to provide a standard for comparing absolute fluorescence levels between animals from different sessions.
Table 1. Quantification of fluorescence analysis of synaptic protein distribution.
GFP-tagged transgenes were analyzed by fluorescence microscopy. Data is presented as comparisons of fluorescence values of mutants compared to WT.
| Transgene | Mutant | arecolinea | Punctal Fluorescence | Cord Fluorescenceb | Width | Density | n |
|---|---|---|---|---|---|---|---|
| SPHK-1 (FL) | wild type | - | 100% | n/a | 100% | 100% | |
| SPHK-1 (FL) | wild type | + | 122.64±11.28%* | n/a | 105.72±9.33% | 101.97±6.64% | 20 |
| SPHK-1 (FL)d | wild type | + | 127.50±24.38%* | n/a | 108.94±18.03% | 101.17±11.02% | 8 |
| SPHK-1 (FL) | unc-2/VGCCα | - | 78.20±9.34%* | n/a | 96.81±7.06% | 91.81±5.26% | 30 |
| SPHK-1 (FL) | unc-2/VGCCα | + | 87.28±10.49% | n/a | 90.42±6.48% | 98.23±5.53% | 30 |
| SPHK-1 (FL) | unc-68/RyR | - | 65.25±12.42%** | n/a | 106.41±10.05% | 115.98±19.82% | 49 |
| SPHK-1 (FL) | egl-19/L-VGCCα | - | 82.42±10.52%* | n/a | 112.20±7.10%* | 108.42±7.15% | 34 |
| SPHK-1 (FL) | egl-19gf/L-VGCCα | - | 135.42±17.00%* | n/a | 101.47±5.61% | 112.73±6.20%* | 37 |
| SPHK-1 (FL) | egl-30gf/Gαqc | - | 117.37±13.95%* | n/a | 113.09±7.97% | 92.68±4.59% | 36 |
| SPHK-1 (FL) | egl-30gf;unc-2 | - | 68.22±8.02%*** | n/a | 90.37±6.90% | 87.025.15%*** | 30 |
| SPHK-1(NT1)d | wild type | - | 39.55±16.89%* | 396.25±201.70%* | 120.98±13.04%* | 134.71±14.45%* | 8 |
| SPHK-1(NT1)d | wild type | + | 56.22±37.38% | 46.45±44.25% | 81.23±10.92% | 110.29±17.24% | 8 |
| SPHK-1(NT2)d | wild type | - | 97.62±14.37% | 79.48±38.40% | 150.92±13.56%** | 99.58±8.71% | 8 |
| SPHK-1(NT2)d | wild type | + | 90.29±10.65% | 81.71±46.12% | 86.46±9.57% | 109.74±9.89% | 12 |
| SPHK-1 (FL)d | calm-1/CIB | - | 79.05±9.69%* | n/a | 96.43±6.69% | 93.67±5.19% | 35 |
| SPHK-1 (FL)d | calm-1/CIB | + | 95.35±10.20% | n/a | 101.66±5.14% | 100.96±5.23% | 32 |
| SPHK-1(ΔCaM)d | wild type | + | 107.10±21.83% | 93.29±54.85% | 100.91±12.27% | 94.57±10.75% | 12 |
| SNB-1/synaptobrevi n | hyl-1/CER synthase | - | 123.79±13.21%* | 124.54±21.03% | 106.41±6.64% | 92.14±3.84%* | 29 |
| TAG-38/S1P lyased | wild type | + | 116.58±19.94% | 147.35±59.88% | 104.68±12.30% | 101.96±9.00% | 13 |
| UNC-13S/Munc13 | egl-30(gf) | - | 114.51±6.52%** | 121.60±22.43% | 111.76±7.97% | 95.91±4.10% | 33 |
| UNC-13S/Munc13 | sphk-1 | - | 102.34±6.64% | 83.96±18.13% | 104.97±8.33% | 95.22±4.50% | 26 |
| UNC-13S/Munc13 | egl-30(gf);sphk-1 | - | 98.97±6.62% | 101.19±16.15% | 123.63±10.73% | 87.70±4.90% | 33 |
|
| |||||||
| Transgene | Mutant | EGTAa | Punctal Fluorescence | Cord Fluorescenceb | Width | Density | n |
|
| |||||||
| SPHK-1 (FL) | egl-19(gf) | - | 100% | n/a | 100% | 100% | 25 |
| SPHK-1 (FL) | egl-19(gf) | + | 78.11±8.90%* | n/a | 89.91±6.85% | 104.40±5.87% | 23 |
Values for drug-treatment experiments are expressed as fluorescence values of drug-treated / control treated animals. Arecoline and EGTA treatment was for two hours and control treatment was M9 and water, respectively.
SPHK-1-GFP cord fluorescence was omitted because it was not reliably detected above background, except for SPHK-1 truncation variants.
Data was also used in (Chan et al. 2012).
Transgene is an extrachromasomal array.
Abbreviations: n/a, not applicable; EGTA, ethylene glycol tetraacetic acid; FL, full-length; NT, N-terminal deletion; CaM, calcium/calmodulin binding domain; RyR, ryanodine receptor, gf, gain-of-function mutation; CER, ceramide; CIB, calcium and integrin binding protein.
p<0.05,
p<0.005,
p<0.0005.
We found that the conditions used for imaging showed that SPHK-1-GFP were predominantly localized to synaptic puncta, as determined previously (Chan et al., 2012). There was little or no difference between their interpunctal fluorescence (in axons) and the auto-fluorescence observed at C. elegans axons. Therefore, we excluded the interpunctal fluorescence in our analysis for these markers (except for Figure 4c and Table 1, in which mutations in SPHK-1-GFP caused increased interpunctal fluorescence).
Figure 4. Structural determinants that localize SPHK-1 at synapses and cell bodies.
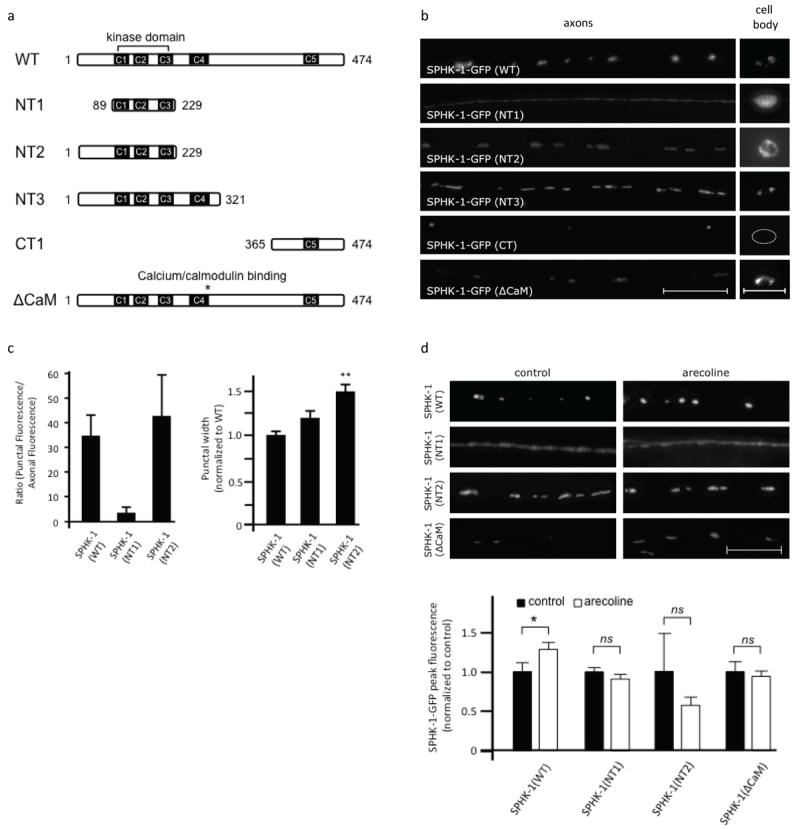
(a) Diagram of wild type SPHK-1-GFP and its variants used. (b) Representative images of the localization of SPHK-1-GFP and indicated variants in DA/DB motor neuron axons (left) and cell body (right). (c) Quantification of the ratio of punctal fluorescence to interpunctal fluorescence (left) and punctal width (right) of SPHK-1(WT)-GFP, SPHK-1(NT1)-GFP, and SPHK-1(NT3)-GFP in DA/DB motor neurons. (d) Representative images (left) and quantification (right) of SPHK-1-GFP (WT and the indicated variants) in DA/DB motor neurons with pretreatment of control M9 (black bars) or arecoline (15mM in M9; white bars). For all, *p<0.05 and **p<0.005, Student’s t-Tests, and scale bars are 10μm.
Statistical Analysis
A Student’s t-test was used to determine significance when comparing the fluorescence of SPHK-1-GFP in different conditions (significance was set at p<0.05). Propagation of standard error of the mean (S.E.M.) was performed for all comparisons. Statistical analyses for all experimental comparisons are presented in Table 1. Quantification of synaptic marker abundance at puncta (punctal fluorescence), synaptic marker abundance in-between puncta (axonal fluorescence), the size of puncta (width fluorescence), and the distance between puncta (interpunctal interval) were performed as previously described (Ch’ng et al., 2008) and are useful measures of the structural and functional properties of presynaptic terminals.
Results
Sphingosine kinase expression and localization in C. elegans
Mammalian sphingosine kinases are broadly expressed in many tissues including the brain, and they associate with diverse cellular membranes, including the nuclear envelope, plasma membrane, ER, and mitochondria (Siow et al., 2011). We previously showed that C. elegans sphingosine kinase, sphk-1, is expressed in motor neurons of the ventral nerve cord, where it localizes to presynaptic membranes near synaptic vesicle release sites (Chan et al., 2012). Here, we determined the expression and localization pattern of SPHK-1 in other neuronal compartments and in other tissues. Transgenic animals expressing GFP under the control of a fully rescuing sphk-1 promoter fragment displayed fluorescence in multiple tissues, including the hypodermis (skin), muscles, the intestine, excretory canal cells, and some neurons in the head and tail (Figure 1a).
Figure 1. Expression and localization of sphingosine kinase in C. elegans.
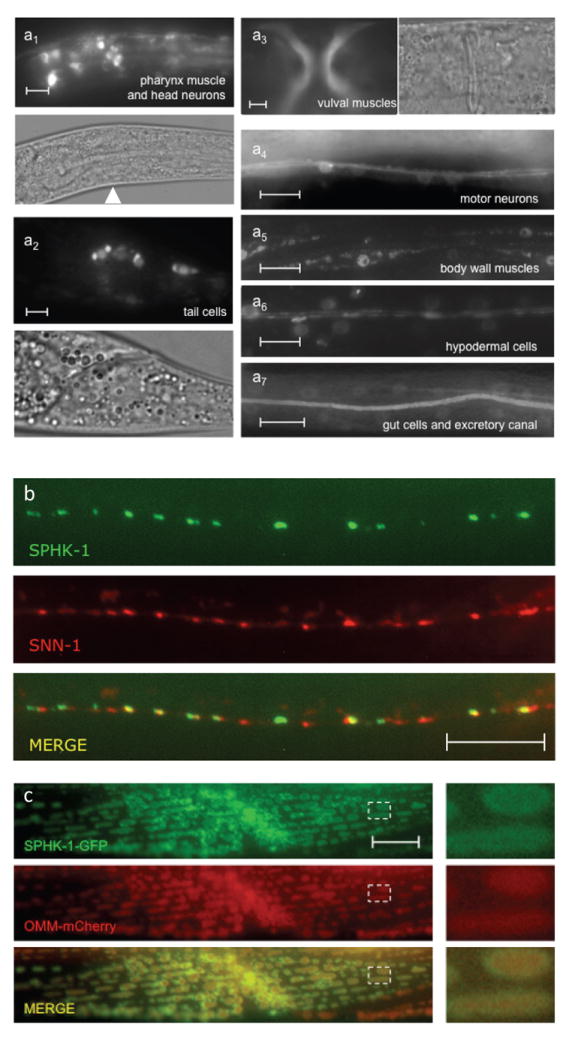
(a) Expression of a sphk-1 promoter fragment driving either GFP (a3, a4, and a7) or nuclear localized GFP (a1, a2, a5, and a6) in adults. Analogous DIC images are also shown for a1-a3. Arrowhead indicates neurons in the nerve ring. (b) Representative images showing the localization of SPHK-1-GFP with SNN-1/synapsin (SNN-1-mCherry). (c) Representative images showing the localization of SPHK-1-GFP and an outer mitochondrial membrane marker (OMM-mCherry) in body wall muscles (expressed under the myo-3 promoter). The dotted box was magnified 4x and shown to the right of the images. All scale bars are 10μm.
In DA/DB class cholinergic motor neurons, functional GFP-tagged SPHK-1 fusion proteins (SPHK-1-GFP) were detected in extra-nuclear regions in cell bodies (Figure 4b), likely corresponding to Golgi (Sumakovic et al., 2009), occasionally in puncta in dendrites (data not shown), and in puncta at en passant presynaptic terminals (Chan et al., 2012). Co-localization studies revealed that SPHK-1-GFP was closely apposed to but not colocalized with the SV and actin-associated protein SNN-1/synapsin (Figure 1b). In body wall muscles, SPHK-1-GFP was primarily observed on the outer surface of mitochondria as demonstrated by its colocalization with a RFP-tagged outer membrane mitochondrial marker (Figure 1c). Thus, SPHK-1 appears to localize to different cellular membranes depending on cell type.
Synaptic recruitment of SPHK-1 is regulated by presynaptic calcium
Previous studies have found that neuronal depolarization or hyperpolarization results in corresponding decreases or increases in sphingosine kinase synaptic abundance (Kajimoto et al., 2007; Chan et al., 2012). To explore whether calcium influx into motor neurons mediates the effects of neuronal activity on SPHK-1 synaptic recruitment, we examined SPHK-1-GFP in animals with defects in presynaptic calcium entry. To detect changes in synaptic SPHK-1-GFP fluorescence, we averaged the fluorescence intensity and distribution of several hundred SPHK-1-GFP puncta from DA/DB class motor axons (using the unc-129 promoter) from about thirty live animals. Among the parameters examined were: punctal fluorescence, which measures synaptic abundance, punctal width, which measures distribution within a synapse, and punctal density, which measures the number of puncta per unit length of an axon.
Calcium enters synaptic terminals from two main sources: the extracellular space or internal stores. We first tested the effects of mutations that alter calcium entry through voltage gated calcium channels. unc-2 encodes the C. elegans α1 subunit of the non-L type voltage-gated calcium channel (VGCCα1). UNC-2/VGCCα1 localizes to presynaptic active zones, and unc-2/VGCCα1 mutations reduce calcium influx in certain sensory neurons (Richmond and Jorgensen, 1999; Mathews et al., 2003; Saheki and Bargmann, 2009). We found that loss-of-function mutations in unc-2/VGCCα1 reduced SPHK-1 average punctal fluorescence by about 20% (Figure 2a and Table 1). The decrease in punctal fluorescence was due to a reduction in the fraction of puncta with high fluorescence intensities and a corresponding increase in the fraction of puncta with low intensities, as revealed by cumulative probability plots (Figure 2b). In contrast, unc-2/VGCCα1 mutations did not significantly alter either SPHK-1-GFP punctal width or punctal density, indicating that unc-2/VGCCα1 specifically affects SPHK-1 abundance, possibly by regulating its translocation to synaptic terminals. In addition, unc-2/VGCCα1 does not impact overall synaptic structure since the punctal fluorescence of active and periactive zone proteins (eg. UNC-10/RIM1, SYD-2/Lyprin-α, SNN-1/Synapsin, and ITSN-1/Intersectin) are not significantly altered in unc-2/VGCCα1 mutants (Ch’ng et al., 2008). unc-2/VGCCα1 mutations severely reduce acetylcholine release from motor neurons and dramatically alter synaptic vesicle cycling (Richmond et al., 2001; Ch’ng et al., 2008). Thus, it is possible that the decreases observed in SPHK-1 synaptic abundance are a secondary effect of alterations in synaptic vesicle cycling or release in these mutants. This is likely not to be the case because synaptic SPHK-1-GFP abundance is not detectably altered in unc-13/Munc13 or unc-18/Munc18 mutants (Chan et al., 2012), in which synaptic vesicle release and cycling are blocked but calcium entry remains intact (Richmond et al., 2001; Arellano-Carbajal et al., 2011). Thus calcium entry through UNC-2/VGCCα1 positively regulates SPHK-1 synaptic abundance independently of its effects on synaptic vesicle release.
Figure 2. Calcium mediates the basal and muscarinic-mediated recruitment of SPHK-1 at synapses.
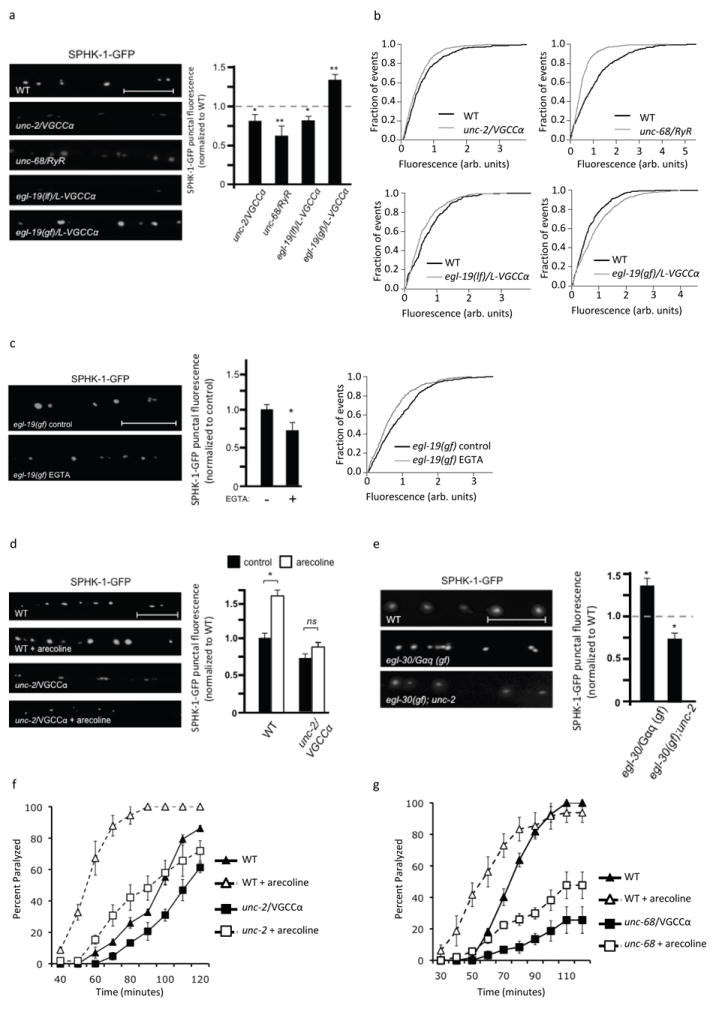
(a) Representative images (left) and quantification (right) of SPHK-1-GFP punctal fluorescence in the indicated mutants. (b) Cumulative probability plots showing the distribution of SPHK-1-GFP intensities per punctum. (c) Representative images (left), quantification (middle), and cumulative probability plot (right) of SPHK-1-GFP punctal fluorescence in egl-19/L-VGCCα1 gain-of-function mutants, treated with water (pH 7.2) or EGTA (50mM, pH 7.2). (d and e) Representative images (left) and quantification (right) of SPHK-1-GFP punctal fluorescence in the indicated mutants. For d, the indicated strains were analyzed following a 2 hour treatment with M9 (salt solution-black bars) or arecoline (15mM in M9; white bars) and the data are normalized to WT (control) fluorescence. (f and g) Rates of worm paralysis of the unc-2/VGCCα1 (e) or unc-68/RyR (f) mutants upon exposure of the acetylcholine esterase inhibitor aldicarb (1.0mM) following a 2 hour pretreatment with control M9 (solid lines) or arecoline (15mM in M9; dotted lines). For all, *p<0.05 and **p<0.005, Student’s t-Tests and scale bars are 10μm.
To test whether calcium plays a permissive or instructive role in SPHK-1 recruitment, we examined egl-19, which encodes the α1 subunit of the C. elegans L-type voltage-gated calcium channel (L-VGGCα1). egl-19/L-VGGCα1 is expressed in motor neurons of the ventral nerve cord (Hunt-Newbury et al., 2007), and egl-19/L-VGGCα1 loss-of-function mutants have reduced calcium influx, whereas egl-19/L-VGGCα1 gain-of-function mutants have increased calcium entry (Kerr et al., 2000; Frokjaer-Jensen et al., 2006). We observed a 20% decrease and 34% increase in SPHK-1 abundance in egl-19/L-VGGCα1 loss-of-function and gain-of-function mutants, respectively, compared to wild type controls (Figure 2a, b and Table 1). The increase in SPHK-1-GFP fluorescence in egl-19/L-VGGCα1 gain-of-function mutants was blocked by acute treatment with the calcium chelator EGTA (Figure 2c). Thus, egl-19/L-VGGCα1 activation elicits a calcium-dependent increase in synaptic SPHK-1-GFP due to acute changes in calcium influx. Together, these results indicate that calcium influx is both necessary and sufficient for SPHK-1 recruitment, and suggest that calcium is instructive in this process.
We next examined the contribution of calcium entry through calcium stores to SPHK-1 recruitment. unc-68 encodes a ryanodine receptors (RyR), which mediates calcium efflux from intracellular calcium stores. unc-68/RyR mutants have reductions in neurotransmitter release from motor neurons (Liu et al., 2005). We found that mutations that reduce the activity of unc-68/RyR significantly reduced SPHK-1-GFP peak fluorescence compared to wild type controls (Figure 2a, b and Table 1), without affecting either punctual width or density (Table 1). Thus, it appears that calcium from both intracellular and extracellular sources is needed for SPHK-1 recruitment.
Calcium entry is required for muscarinic-dependent SPHK-1 recruitment
Activating a presynaptic muscarinic pathway either by acute treatment of C. elegans with the muscarinic agonist arecoline or by constitutively activating the heterotrimeric G protein Gαq (egl-30/Gαq(gf)) significantly increases SPHK-1-GFP punctal fluorescence (Figure 2d, 2e, Table 1 and (Chan et al., 2012)), suggesting that muscarinic signaling regulates SPHK-1 translocation to release sites. We examined whether muscarinic-regulated translocation of SPHK-1 is dependent on calcium influx. The increase in SPHK-1 punctal fluorescence caused by either arecoline or by a constitutively active egl-30/Gαq mutant was completely blocked by unc-2/VGCCα1 mutations (Figure 2d, 2e and Table 1). These results suggest that presynaptic calcium is required for muscarinic-mediated recruitment of SPHK-1 to synapses.
Arecoline treatment causes animals to become hypersensitive to the paralytic effects of the acetylcholine esterase inhibitor aldicarb (Lackner et al., 1999). Aldicarb prevents removal of acetylcholine from the synaptic cleft, causing muscle hypercontraction and paralysis. Hypersensitivity to aldicarb is observed under conditions of increased neuromuscular function, including enhanced neurotransmitter release (Mahoney et al., 2006; Vashlishan et al., 2008). sphk-1 is required for the effects of arecoline treatment on aldicarb hypersensitivity (Chan et al., 2012), suggesting that sphk-1 mediates the effects of arecoline on synaptic transmission. If the recruitment of SPHK-1 by presynaptic calcium were required for the effects of muscarinic signaling on synaptic transmission, then we would expect calcium mobilization mutants to block the aldicarb hypersensitivity caused by arecoline treatment. Consistent with this idea, we found that unc-2/VGCCα1 or unc-68/RyR mutants had a severely blunted response to arecoline treatment compared to wild type animals, indicating that the effects of muscarinic stimulation on aldicarb responsiveness are partially dependent on calcium entry. (Figure 2f, g). Together these results suggest that presynaptic calcium promotes both basal and muscarinic-potentiated synaptic transmission by recruiting SPHK-1 to presynaptic terminals.
CALM-1/CIB is presynaptic and mediates SPHK-1 recruitment
We next examined the molecular mechanism by which SPHK-1 might be recruited to synapses by calcium. SphKs do not contain sequence motifs that are known to bind calcium directly. However, SphKs contain a conserved calcium/calmodulin binding motif that has been shown to interact with calcium/calmodulin as well as calcium and integrin binding protein (CIB) (Young et al., 2003; Jarman et al., 2010). CIB is a family of calcium responsive proteins with diverse biological functions, including cell survival, growth, migration and synaptic plasticity (Kauselmann et al., 1999; Leisner et al., 2005; Yoon et al., 2009; Frost and Olson, 2010). CIB1 recently has been implicated in the calcium-dependent recruitment of human SphK1 to the plasma membrane in HEK cells (Jarman et al., 2010). C. elegans encodes an ortholog to the CIB family, calm-1, which shares 57% similarity to mammalian CIB1 and 48% similarity to mammalian CIB2. calm-1 mRNA is expressed in motor neurons (Fox et al., 2005). We found that a calm-1 transcriptional reporter, in which 1852bp of the calm-1 promoter drove the expression of GFP, was expressed in body wall muscle and in cholinergic motor neurons of the ventral cord (Figure 3a). We examined the subcellular localization of a CALM-1/CIB fusion protein (CALM-1-mCherry) in motor neuron axons and found that it adopted a punctate pattern of fluorescence, reminiscent of SPHK-1-GFP. CALM-1-mCherry puncta largely colocalized with SPHK-1-GFP puncta at presynaptic terminals (Figure 3b).
Figure 3. Muscarinic-mediated recruitment of SPHK-1 at synapses depends on CALM-1/CIB.
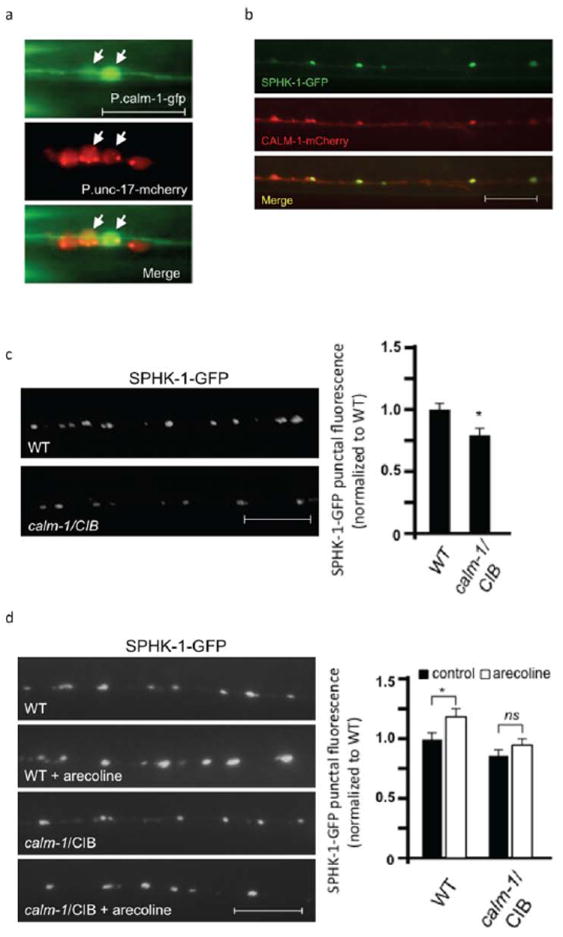
(a) Fluorescence images showing GFP expressed under the calm-1 transcriptional reporter (P.calm-1-gfp) in neurons of the ventral cord, which colocalize with mCherry expressed under a cholingeric neuron reporter (P.unc-17-mCherry). (b) Representative images showing the localization of SPHK-1-GFP and CALM-1/CIB-1 (CALM-1-mCherry) in DA/DB motor neurons (both using the unc-129 promoter). Arrows indicate regions of colocalization and arrowheads indicate regions where CALM-1-mCherry was observed without SPHK-1-GFP. (c) Representative images (left) and quantification (right) of SPHK-1-GFP punctal fluorescence in wild type or calm-1/CIB mutants. (d) Representative images (left) and quantification (right) of SPHK-1-GFP punctal fluorescence in wild type or calm-1/CIB mutants following pretreatment with control M9 (salt solution-black bars) or arecoline (15mM in M9; white bars). For a, the scale bar is 5μm, and for b-d, the scale bars are 10μm.
Next, we examined the requirement of calm-1/CIB in regulating SPHK-1 synaptic recruitment. calm-1(tm1353) mutants, which have a 618 nucleotide deletion that removes part of the calm-1 promoter and the first exon, showed a significant decrease in SPHK-1-GFP synaptic abundance (Figure 3c and Table 1), without altering SPHK-1 abundance in neuronal cell bodies (336±45 arbitrary fluorescence units in WT vs. 342±38 units in calm-1/CIB mutants, p>0.05). We also found that arecoline-mediated recruitment of SPHK-1-GFP to synapses was blocked in calm-1/CIB mutants (Figure 3d and Table 1). Together these results suggest that presynaptic CALM-1 promotes the recruitment of SPHK-1 to release sites.
The calcium/calmodulin-binding domain of SPHK-1 is required for muscarinic-mediated SPHK-1 recruitment
To investigate how calcium and muscarinic signaling might converge to regulate SPHK-1 abundance, we next sought to identify the SPHK-1 sequence determinants that are responsible for the synaptic translocation of SPHK-1 in response to calcium or muscarinic signaling. SPHK-1 contains five conserved domains, termed C1-C5, which share greater than 50% amino acid similarity with human SphK1 and SphK2 (Figure 4a). We examined the synaptic abundance of GFP-tagged SPHK-1 variants in which various conserved domains had been deleted (Figure 4a). A SPHK-1-GFP variant that contains only the kinase domain (SPHK-1(NT1)) adopted a diffuse pattern of fluorescence in axons and in cell bodies (Figure 4a, b), implying a redistribution of the protein to the cytosol (Hengst et al., 2010; Siow et al., 2011). Quantification of the localization pattern of the kinase domain of SPHK-1 in axons revealed a reduction in punctal fluorescence and a corresponding increase in inter-punctal fluorescence compared to wild type SPHK-1-GFP (Figure 4c).
Addition of the N-terminus of SPHK-1 to the kinase domain (SPHK-1(NT2)) restored both synaptic localization and presumed Golgi localization (Figure 4a, b). However, SPHK-1(NT2)-GFP synaptic puncta were significantly dimmer and wider than wild type controls, and fluorescence was also found on the plasma membrane in cell bodies in three out of three lines we examined (Figure 4b, 4c and Table 1). Adding the conserved domain C4 to this variant (SPHK-1(NT3)) restored wild type levels of SPHK-1 at synapses and restored Golgi localization in cell bodies (Figure 4a, b). However, synaptic puncta remained significantly wider than wild type controls (Figure 4b and Table 1). A 110 amino acid fragment (SPHK-1(CT1)) formed puncta in axons that were similar in size to SPHK-1(WT)-GFP puncta. However, these puncta were much less frequent compared to SPHK-1(WT)-GFP, and SPHK-1(CT1) was not detectable in cell bodies (Figure 4a, b). Finally, a full length SPHK-1 variant containing a mutation in the C4 domain that corresponds to the calcium/calmodulin (CaM) binding domain (SPHK-1(ΔCaM)-GFP) adopted a pattern that was similar to SPHK-1(NT2): wider and dimmer synaptic puncta and mislocalization in cell bodies (Figure 4a, b and (Chan et al., 2012)). Together, these results suggest that the region N-terminal of the kinase domain can direct SPHK-1 to synapses (and to Golgi), but does not restrict SPHK-1 within synapses. Similarly, the C4 domain does not appear to restrict SPHK-1 to small regions within synapses; however, it is required for normal SPHK-1 levels at synapses. On the other hand, the similarity of the localization patterns SPHK-1(NT2) and the SPHK-1(ΔCaM) variants, both of which lack the CaM domain in C4, suggests that the CaM domain is essential for recruiting and restricting SPHK-1 within synapses. However the C4 domain is likely to not be sufficient for synaptic clustering of SPHK-1 since SPHK-1(NT3), which has the C4 domain, fails to cluster. Thus, SPHK-1 may require both the C4 and the C5 domains to restrict SPHK-1 within synapses.
To identify the sequence determinants required for muscarinic-mediated SPHK-1 synaptic recruitment, we next tested the effects of acute arecoline treatment (2 hours) on the abundance of the SPHK-1 variants at synapses. Although arecoline treatment significantly increased the punctal fluorescence of SPHK-1-GFP transgenes, arecoline treatment had no effect on the distribution or abundance of the SPHK-1(NT1)-GFP, SPHK-1(NT3)-GFP, or SPHK-1(CaM)-GFP variants (Figure 4d and Table 1). These results suggest that sequence determinants that lie C-terminal to the C4 domain as well as the CaM-binding site are important for arecoline responsiveness. Our observation that either calm-1/CIB mutants or deletion of the CaM-binding site block muscarinic-mediated SPHK-1 translocation suggests that CALM-1/CIB acting on the CaM-binding site of SPHK-1 may be critical for the ability of muscarinic signaling to recruit SPHK-1 to synapses.
HYL-1/ceramide synthase and TAG-38/S1P lyase are concentrated at presynaptic terminals
Multiple enzymes catalyze the generation and degradation of sphingolipids, and they are thought to function coordinately to fine-tune levels of each sphingolipid metabolite. If the regulation of synaptic levels of SPH and S1P by SPHK-1 is important for synaptic function, then we expect that additional enzymes involved in SPH or S1P metabolism might also function at synapses. To test this idea, we examined two enzymes that are predicted to directly affect levels of SPH and S1P: ceramide (CER) synthase, which converts SPH to CER, and S1P lyase, which breaks down S1P into phosphoethanolamine and hexadecanal (Figure 5a). C. elegans encodes three predicted CER synthases. One of these, hyl-1, is expressed in motor neurons (Fox et al., 2005), and mutants lacking hyl-1 display hyperactive locomotion (Chan et al., 2012). We generated a GFP-tagged HYL-1 fusion protein (HYL-1-GFP) and found that it adopted a punctate pattern of fluorescence in motor neuron axons (Figure 5b). HYL-1-GFP largely colocalized with the SV marker SNB-1-mCherry (Figure 5d), but was mostly apposed to SPHK-1-mCherry and SNN-1-mCherry puncta (Figure 5b, d). HYL-1-GFP puncta were eliminated in mutants lacking unc-104/kinesin, which is a motor protein that transports SVs and other organelles to presynaptic terminals (Figure 5c). These results suggest that HYL-1 associates with synaptic vesicle pools (or other presynaptic organelles) at presynaptic terminals.
Figure 5. Localization of HYL-1/CER synthase and TAG-38/S1P lyase at synapses.
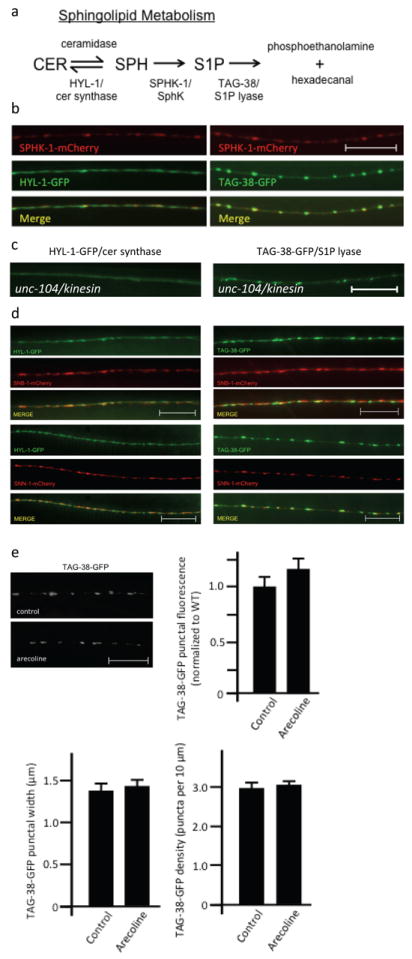
(a) Diagram of the sphingolipid metabolism pathway and its enzymes that have orthologs in C. elegans. CER, ceramide; SPH, sphingosine; S1P, sphingosine-1-phosphate. (b) Representative images showing the localization of SPHK-1-mCherry with HYL-1-GFP (left) or TAG-38-GFP (right) in DA/DB motor neurons. (c) Representative image of HYL-1-GFP (left) or TAG-38-GFP (right) at synapses of DA/DB motor neurons in mutants lacking the synaptic vesicle trafficking protein unc-104/Kinesin KIF1A. (d) Representative images showing the localization of HYL-1-GFP (left) or TAG-38-GFP (right) with SNB-1/synaptobrevin (mCherry-SNB-1; top) and SNN-1/synapsin (SNN-1-mCherry; bottom) in DA/DB motor neurons. (e) Representative images (top) and quantification (bottom) of TAG-38-GFP in DA/DB motor neurons following pretreatment of control M9 (salt solution-black bars) or arecoline (15mM in M9; white bars). For all, scale bars are 10μm.
C. elegans encodes two predicted S1P lyases, spl-1 and tag-38. Whereas spl-1 expression has not been detected in the nervous system [wormbase.org], tag-38 cDNA is expressed in motor neurons (Fox et al., 2005). Therefore, we examined a GFP-tagged TAG-38/S1P lyase fusion protein (TAG-38-GFP) and found that it adopted a highly punctate pattern of fluorescence in axons that largely overlapped with SPHK-1-mCherry or SNN-1-mCherry puncta (Figure 5b, d). TAG-38-GFP localized less well with the SV markers SNB-1 and remained highly punctate in unc-104/kinesin mutants (Figure 5c). Together these results suggest that like SPHK-1, TAG-38/S1P lyase associates with presynaptic membranes at release sites, but not synaptic vesicles. Given the striking similarity between the localization of TAG-38/S1P lyase and SPHK-1, we wondered whether TAG-38/S1P lyase synaptic abundance might also be regulated by muscarinic signaling. However, we found that arecoline treatment did not significantly change TAG-38-GFP axonal punctal fluorescence, width, or density (Figure 5e and Table 1). Therefore, muscarinic signaling appears to specifically regulate SPHK-1, but not TAG-38/S1P lyase, at release sites.
hyl-1/ceramide synthase antagonizes sphk-1
We previously showed that hyl-1/CER synthase mutants have hyperactive locomotion rates compared to wild type controls (Chan et al., 2012). To examine the role of hyl-1/CER synthase in synaptic transmission more directly, we examined aldicarb responses and SV cycling in hyl-1/CER synthase mutants. We found that hyl-1/CER synthase mutants where hypersensitive to aldicarb (Figure 6a), consistent with increased ACh release at NMJs. hyl-1/CER synthase mutants also displayed increases in punctual fluorescence of the SV marker SNB-1-GFP (Figure 6b and Table 1). Increases in punctal fluorescence are indicative of larger SV pools at synaptic terminals, and may arise from alterations in SV trafficking, biogenesis or release. The increase in locomotion rates, the aldicarb hypersensitivity, and the increase in SV abundance of hyl-1/CER synthase mutants are similar to those observed in constitutively active egl-30/Gαq(gf) mutants (Ch’ng et al., 2008). Interestingly, both egl-30/Gαq(gf) mutants and hyl-1/CER synthase mutants are predicted to have elevated S1P levels: egl-30/Gαq(gf) mutants have more SPHK-1 at synapses (Chan et al., 2012), and hyl-1/CER synthase mutants have five-fold higher levels of the S1P precursor SPH (Menuz et al., 2009). Thus, elevated S1P levels may account for the similar phenotypes observed in these mutants.
Figure 6. HYL-1/ceramide synthase antagonizes SPHK-1.
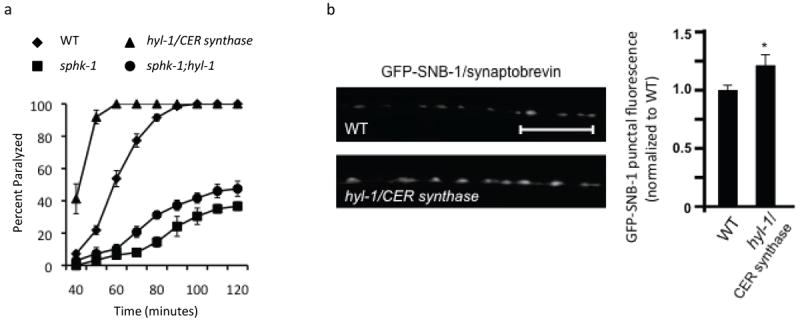
(a) Rates of worm paralysis of WT controls and hyl-1/CER synthase mutants upon exposure to the acetylcholine esterase inhibitor aldicarb (1.0mM). (b) Representative images (left) and quantification (right) of GFP-tagged synaptobrevin (GFP-SNB-1) in axons of DA/DB motor neurons analyzed in the WT and hyl-1/CER synthase mutants. *p<0.05, Student’s t-Tests and the scale bar is 10μm.
If elevated synaptic S1P promotes synaptic transmission, then we would predict that the aldicarb hypersensitivity of hyl-1 mutants should be dependent on S1P production by SPHK-1. Indeed, we found that sphk-1;hyl-1 double mutants were nearly as aldicarb resistant as sphk-1 single mutants (Figure 6a). Together, these data suggest that high SPH levels alone cannot account for the increased locomotion rates, aldicarb hypersensitivity, and accumulation of SVs in hyl-1/CER synthase mutants. Instead, these observations indicate that the conversion of SPH to S1P by SPHK-1 is important for neurotransmission.
sphk-1 is required for muscarinic-mediated recruitment of UNC-13S to synapses
How might SPHK-1 recruitment to presynaptic terminals couple to the SV release machinery to promote acetylcholine release? Munc13 is a presynaptic protein involved in priming of secretory vesicles through its interaction with the SNARE protein syntaxin (Brose et al., 2000; Madison et al., 2005). The association of Munc13 with release sites is regulated by diacylglycerol and is proposed to control vesicle priming and subsequent neurotransmitter release (Richmond et al., 1999; Madison et al., 2005). Like SPHK-1, C. elegans Munc13, UNC-13, is recruited to presynaptic terminals by EGL-30/Gαq-dependent muscarinic signaling (Lackner et al., 1999). We previously found that SPHK-1-GFP punctal fluorescence did not change in unc-13/Munc13 mutants (Chan et al., 2012), suggesting that UNC-13/Munc13 is not required for SPHK-1 synaptic recruitment.
We next examined whether the synaptic abundance of GFP-tagged UNC-13 (UNC-13S-GFP) was dependent on sphk-1 activity. We found no change in synaptic abundance of UNC-13S-GFP in sphk-1 mutants compared to wild type controls under normal conditions (Figure 7a and Table 1). However, sphk-1 activity was essential for muscarinic signaling-dependent UNC-13S-GFP recruitment. Activating muscarinic signaling (using gain-of-function egl-30/Gαq mutants) significantly increased UNC-13S-GFP punctal fluorescence (Figure 7a and Table 1). egl-30/Gαq activation appears to specifically recruit UNC-13S-GFP to synapses because egl-30/Gαq mutations did not significantly alter the punctal fluorescence of another active zone protein, UNC-10/RIM1 (Ch’ng et al., 2008). The increase in UNC-13S-GFP punctal fluorescence was completely blocked by sphk-1 mutations: punctal UNC-13S-GFP fluorescence was reduced to wild type levels in egl-30(gf);sphk-1 double mutants (Figure 7a and Table 1). Thus, muscarinic signaling may selectively recruit UNC-13S to synaptic membranes via a sphk-1-dependent mechanism (Figure 7b).
Figure 7. Muscarinic-mediated recruitment of UNC-13/Munc13 requires SPHK-1.
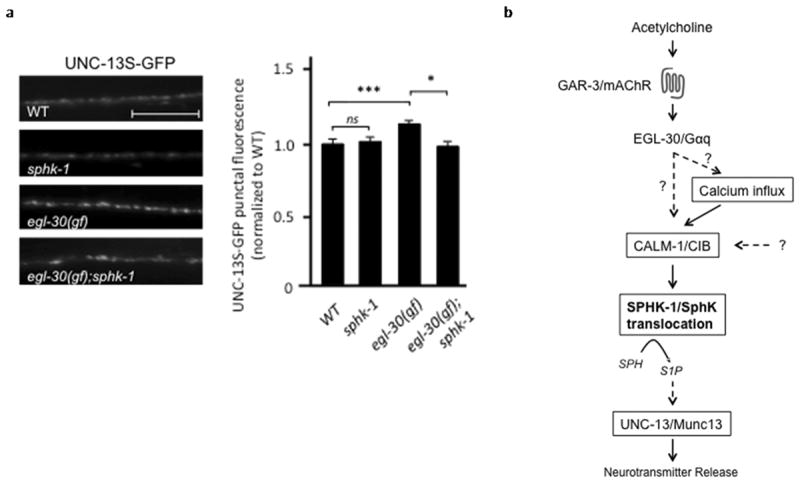
(a) Representative images (left) and quantification (right) of GFP-tagged UNC-13S (UNC-13S-GFP) in axons of DA/DB motor neurons analyzed in the indicated strains. *p<0.05 and ***p<0.0005, Student’s t-Tests and the scale bar is 10μm. (b) Model of the muscarinic- and calcium-mediated SPHK-1 translocation for the facilitating effects of muscarinic signaling pathways on neurotransmitter release in C. elegans cholinergic motor neurons. Dotted lines represent unknown pathways or pathways that are likely to be connected via multiple proteins.
Discussion
Changes in presynaptic structure and function represent important mechanisms by which modulatory pathways regulate synaptic strength. In this study, we found that muscarinic signaling regulates the abundance of SPHK-1 at synaptic terminals via a calcium dependent mechanism involving CIB. Synaptic SPHK-1 in turn promotes the recruitment of at least one priming factor, UNC-13/Munc13, in response to muscarinic signaling. Thus, local sphingolipid metabolism appears to be an important way by which synaptic vesicle priming is regulated.
Calcium mobilization regulates the abundance of SPHK-1 at synapses
We found that calcium mobilization regulates SPHK-1 synaptic abundance and is required for SPHK-1 translocation in response to muscarinic signaling. First, mutants with reduced extracellular calcium influx through non-L or L type voltage gated calcium channels (unc-2/VGCCα1 or egl-19/L-VGCCα1) or reduced intracellular calcium influx through ryanodine receptors (unc-68/RyR) decreased steady-state synaptic SPHK-1 abundance. Second, increasing calcium influx (in egl-19/L-VGCCα1 gain-of-function mutants) increased synaptic SPHK-1 abundance in a calcium-dependent manner. Interestingly, the changes in SPHK-1 synaptic abundance we observed in egl-19 loss- and gain-of function mutants (18% decrease and 34% increase) correlate with the changes in calcium influx observed in the respective mutants (19% decrease and 19% increase) (Kerr et al., 2000). Third, calcium influx through unc-2/VGCCα1 was necessary for muscarinic-mediated recruitment of SPHK-1 to synapses and for muscarinic-induced aldicarb hypersensitivity. Fourth, SPHK-1 variants lacking only the calcium/calmodulin-binding domain of SPHK-1 were mislocalized at synapses and were not responsive to synaptic recruitment by muscarinic signaling. We previously showed that SPHK-1 translocation can occur within 15 minutes in vivo (Chan et al., 2012), but in principal, SPHK-1 synaptic recruitment could occur over much more rapid timescales. A previous study has observed that agonist-dependent translocation of SphK in HEK cells occurs on the order of seconds (ter Braak et al., 2009). Thus, calcium dependent recruitment of SPHK-1 by muscarinic signaling may encode a presynaptic plasticity mechanism during cholinergic activity.
The observation that mutations in any one of three calcium channels reduce SPHK-1-GFP abundance suggests that these channels act non redundantly to promote SPHK-1 recruitment. Previous studies demonstrate that these channel types can promote calcium entry at presynaptic terminals. UNC-68/RyR is required for proper synaptic release from motor terminals and UNC-68 and mammalian RyRs have been shown to localize at presynaptic terminals (Bouchard et al., 2003; Liu et al., 2005; Nizami et al., 2010). Similarly, EGL-19 contributes to the generation of calcium waves in axons following axotomy (Ghosh-Roy et al., 2010). Finally, UNC-2/VGCCα1 is concentrated at presynaptic active zones and positively regulates release at neuromuscular junctions (Saheki and Bargmann, 2009). However, RyRs also function in cell bodies, where the endoplasmic reticulum is abundant, and EGL-19/L-VGCCα1 promotes calcium influx in neuron cell bodies (Frokjaer-Jensen et al., 2006). Thus we cannot rule out that these channels may function at a distance (in motor neuron somas) to regulate SPHK-1 recruitment to presynaptic terminals.
Activation of muscarinic receptors drives diverse signaling pathways downstream of Gαq, including those that alter calcium influx into the cytoplasm. In neurons, activation of the M3 subtype of muscarinic receptor classically leads phospholipase C (PLC)-dependent changes in potassium channel conductance, resulting in changes in neuronal activity (Wess et al., 2007; Brown, 2010). However, muscarinic activation of M3 muscarinic receptors has also been shown to induce calcium influx and to require voltage-sensitive calcium channels and TRP channels (Michel et al., 2005; Tai et al., 2006). In other cell types such as beta cells, M3 receptor activation leads to PLC-mediated changes in calcium release form internal stores, which drives insulin secretion (Gromada and Hughes, 2006). Thus, calcium influx is an important downstream event stimulated by muscarinic activation and can alter many cellular functions.
How do presynaptic muscarinic signaling and calcium influx converge on synaptic SPHK-1 recruitment? One possibility is that muscarinic signaling directly recruits SPHK-1 to synapses in a calcium-dependent manner. Alternatively, muscarinic signaling might promote calcium influx, which in turn could lead to SPHK-1 recruitment. Our results point to a model whereby muscarinic activation leads to CALM-1/CIB-dependent SPHK-1 recruitment and/or stabilization at synapses (Figure 7b). In support of this model, CALM-1/CIB and SPHK-1 co-localized at presynaptic membranes in neurons. In addition, calm-1/CIB was required for normal SPHK-1-GFP levels at synapses and for the muscarinic-dependent recruitment of SPHK-1-GFP to synapses. The calcium/calmodulin binding domain also was required for SPHK-1-GFP restriction to discrete membrane domains within presynaptic terminals. The function of CALM-1 in neurons may be analogous to its function in HEK cells, where calcium-dependent binding of CIB to the CaM binding domain of SphK is proposed to promote SphK membrane translocation and increased S1P production (Jarman et al., 2010). Interestingly, mammalian CIB1-positive cells are decreased in cortical regions of brains of Alzheimer’s disease patients and CIB1 is thought to bind presenilin 2, suggesting that CIB proteins may play a role in the neurodegeneration associated with Alzheimer’s disease (Stabler et al., 1999; Bernstein et al., 2005). Given that SphK and S1P levels are also lower in brains of Alzheimer’s disease patients (He et al., 2010; Piccinini et al., 2010), it is intriguing to speculate that CIB and SphK may be coordinately misregulated during disease states.
Sphingolipid enzymes and presynaptic function
The generation of S1P by SphK is the last step in an enzymatic cascade that converts sphingomyelin (SM) to S1P. Spatial and temporal regulation of the enzymes in this cascade is likely to be critical in precisely regulating the levels and cellular distribution of S1P. We found that at least two sphingolipid enzymes, in addition to SPHK-1, are localized to presynaptic terminals: HYL-1/CER synthase and TAG-38/S1P lyase. HYL-1/CER synthase appears to be associated with SV pools whereas SPHK-1 and TAG-38 appear to be associated together on the presynaptic plasma membrane near release sites. It is possible that HYL-1/CER synthase may associate with mitochondria at presynaptic terminals, since it localizes to mitochondria in other cell types (Yu et al., 2007; Deng et al., 2008). The mechanism by which synaptic TAG-38/S1P lyase is regulated appears to be distinct from that of SPHK-1 because SPHK-1-GFP, but not TAG-38-GFP, abundance is altered by muscarinic signaling. We speculate that the balance of sphingolipids at synapses is dynamically regulated by the convergence of multiple pathways that individually regulate different enzymatic components both spatially and temporally at presynaptic terminals.
HYL-1/CER synthase drives the sphingolipid cascade towards more SM and less S1P production, whereas SphK drives this cascade toward S1P production. Accordingly, hyl-1/CER synthase mutants have locomotion and aldicarb phenotypes that are opposite to those of sphk-1 mutants. The effects of hyl-1 mutants on NMJ function may be due to increased S1P levels, which would explain why hyl-1 aldicarb hypersensitivity is blocked by sphk-1 mutations. However, the presence of TAG-38/S1P lyase at synapses raises the possibility that S1P may be rapidly degraded, and that perhaps the degradation products of S1P may be driving synaptic transmission.
SPHK-1 is required for G protein dependent recruitment of UNC-13/Munc13 in neurons
One cellular mechanism by which G protein pathways regulate neuronal activity is through the recruitment of specific synaptic proteins to release sites. UNC-13/Munc13 is a critical priming protein that promotes synaptic vesicle maturation prior to exocytosis (Lackner et al., 1999; Richmond et al., 1999; Brose et al., 2000; Madison et al., 2005). We found that UNC-13S-GFP, but not the active zone protein UNC-10/RIM1-GFP, is recruited to release sites by constitutively activate egl-30/Gαq mutations (Ch’ng et al., 2008), and this recruitment was dependent on sphk-1. However, sphk-1 mutations did not change UNC-13S-GFP puncta fluorescence under normal conditions, suggesting that SPHK-1 activity may specifically recruit UNC-13S during situations where neuronal activity is high (e.g. muscarinic/G protein activity). This result is consistent with the observation that sphk-1 mutants do not have defects in spontaneous SV release but only exhibit defects in neurotransmitter release in response to evoked stimuli (Chan et al., 2012). How might SPHK-1 recruit UNC-13/Munc13 to synapses? UNC-13/Munc13 is recruited to release sites by DAG binding to its C1 domain (Lou et al., 2008). SphK has been reported to activate phospholipase D (PLD), which catalyzes the production of the DAG precursor phosphatidic acid (Pasquare et al., 2011). Thus, local regulation of DAG by a SphK-PLD pathway at synapses might control UNC-13 synaptic recruitment. However, SphK may regulate UNC-13S abundance by other mechanisms since UNC-13/Munc13 synaptic abundance is also regulated by calcium/calmodulin, the calcium binding protein Doc2, and by ubiquitination (Mochida et al., 1998; Aravamudan et al., 1999; Zikich et al., 2008).
We propose that SPHK-1 at synapses may promote neurotransmitter release by two mechanisms. First, when muscarinic signaling is not activated, SPHK-1 may promote release independently of UNC-13/Munc13. Second, under conditions of increased activity (e.g., during muscarinic stimulation) SPHK-1 activity recruits UNC-13/Munc13 to release sites, where it can prime synaptic vesicle for release. Identifying the synaptic targets of S1P will help to elucidate the molecular mechanisms by which SPHK-1 acts.
Acknowledgments
We thank the Caenorhabditis Genetic center and the National BioResource Project for strains used in this study. We thank Q. Cn’ng, T. Staab, H. Wang and M. Olahova for critical reading of this manuscript, S. Gorda, C. Gillian, J. Delcarson and T. Staab for technical assistance, and A. van der Bliek for the mitochondrial markers. This work was supported by grants from the American Heart Association and the National Institutes of Health (NIH) to D.S. (NS071085).
Footnotes
Conflict of interest: The authors declare no competing financial interests
References
- Aravamudan B, Fergestad T, Davis WS, Rodesch CK, Broadie K. Drosophila UNC-13 is essential for synaptic transmission. Nat Neurosci. 1999;2:965–971. doi: 10.1038/14764. [DOI] [PubMed] [Google Scholar]
- Arellano-Carbajal F, Briseno-Roa L, Couto A, Cheung BH, Labouesse M, de Bono M. Macoilin, a conserved nervous system-specific ER membrane protein that regulates neuronal excitability. PLoS Genet. 2011;7:e1001341. doi: 10.1371/journal.pgen.1001341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein HG, Blazejczyk M, Rudka T, Gundelfinger ED, Dobrowolny H, Bogerts B, Kreutz MR, Kuznicki J, Wojda U. The Alzheimer disease-related calcium-binding protein Calmyrin is present in human forebrain with an altered distribution in Alzheimer’s as compared to normal ageing brains. Neuropathol Appl Neurobiol. 2005;31:314–324. doi: 10.1111/j.1365-2990.2005.00646.x. [DOI] [PubMed] [Google Scholar]
- Bouchard R, Pattarini R, Geiger JD. Presence and functional significance of presynaptic ryanodine receptors. Progress in Neurobiology. 2003;69:391–418. doi: 10.1016/s0301-0082(03)00053-4. [DOI] [PubMed] [Google Scholar]
- Brose N, Rosenmund C, Rettig J. Regulation of transmitter release by Unc-13 and its homologues. Curr Opin Neurobiol. 2000;10:303–311. doi: 10.1016/s0959-4388(00)00105-7. [DOI] [PubMed] [Google Scholar]
- Brown DA. Muscarinic acetylcholine receptors (mAChRs) in the nervous system: some functions and mechanisms. J Mol Neurosci. 2010;41:340–346. doi: 10.1007/s12031-010-9377-2. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Few AP. Calcium channel regulation and presynaptic plasticity. Neuron. 2008;59:882–901. doi: 10.1016/j.neuron.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Ch’ng Q, Sieburth D, Kaplan JM. Profiling synaptic proteins identifies regulators of insulin secretion and lifespan. PLoS Genet. 2008;4:e1000283. doi: 10.1371/journal.pgen.1000283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JP, Hu Z, Sieburth D. Recruitment of sphingosine kinase to presynaptic terminals by a conserved muscarinic signaling pathway promotes neurotransmitter release. Genes Dev. 2012;26:1070–1085. doi: 10.1101/gad.188003.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darios F, Wasser C, Shakirzyanova A, Giniatullin A, Goodman K, Munoz-Bravo JL, Raingo J, Jorgacevski J, Kreft M, Zorec R, Rosa JM, Gandia L, Gutierrez LM, Binz T, Giniatullin R, Kavalali ET, Davletov B. Sphingosine facilitates SNARE complex assembly and activates synaptic vesicle exocytosis. Neuron. 2009;62:683–694. doi: 10.1016/j.neuron.2009.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Yin X, Allan R, Lu DD, Maurer CW, Haimovitz-Friedman A, Fuks Z, Shaham S, Kolesnick R. Ceramide biogenesis is required for radiation-induced apoptosis in the germ line of C. elegans. Science. 2008;322:110–115. doi: 10.1126/science.1158111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman JS, Kaplan JM. Factors regulating the abundance and localization of synaptobrevin in the plasma membrane. Proc Natl Acad Sci U S A. 2006;103:11399–11404. doi: 10.1073/pnas.0600784103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earls LR, Hacker ML, Watson JD, Miller DM., 3rd Coenzyme Q protects Caenorhabditis elegans GABA neurons from calcium-dependent degeneration. Proc Natl Acad Sci U S A. 2010;107:14460–14465. doi: 10.1073/pnas.0910630107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox RM, Von Stetina SE, Barlow SJ, Shaffer C, Olszewski KL, Moore JH, Dupuy D, Vidal M, Miller DM., 3rd A gene expression fingerprint of C. elegans embryonic motor neurons. BMC Genomics. 2005;6:42. doi: 10.1186/1471-2164-6-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frokjaer-Jensen C, Kindt KS, Kerr RA, Suzuki H, Melnik-Martinez K, Gerstbreih B, Driscol M, Schafer WR. Effects of voltage-gated calcium channel subunit genes on calcium influx in cultured C. elegans mechanosensory neurons. J Neurobiol. 2006;66:1125–1139. doi: 10.1002/neu.20261. [DOI] [PubMed] [Google Scholar]
- Frost RJ, Olson EN. Separating the good and evil of cardiac growth by CIB1 and calcineurin. Cell Metab. 2010;12:205–206. doi: 10.1016/j.cmet.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh-Roy A, Wu Z, Goncharov A, Jin Y, Chisholm AD. Calcium and cyclic AMP promote axonal regeneration in Caenorhabditis elegans and require DLK-1 kinase. J Neurosci. 2010;30:3175–3183. doi: 10.1523/JNEUROSCI.5464-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromada J, Hughes TE. Ringing the dinner bell for insulin: muscarinic M3 receptor activity in the control of pancreatic beta cell function. Cell Metab. 2006;3:390–392. doi: 10.1016/j.cmet.2006.05.004. [DOI] [PubMed] [Google Scholar]
- He X, Huang Y, Li B, Gong CX, Schuchman EH. Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol Aging. 2010;31:398–408. doi: 10.1016/j.neurobiolaging.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengst JA, Guilford JM, Conroy EJ, Wang X, Yun JK. Enhancement of sphingosine kinase 1 catalytic activity by deletion of 21 amino acids from the COOH-terminus. Arch Biochem Biophys. 2010;494:23–31. doi: 10.1016/j.abb.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt-Newbury R, et al. High-throughput in vivo analysis of gene expression in Caenorhabditis elegans. PLoS Biol. 2007;5:e237. doi: 10.1371/journal.pbio.0050237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarman KE, Moretti PA, Zebol JR, Pitson SM. Translocation of sphingosine kinase 1 to the plasma membrane is mediated by calcium- and integrin-binding protein 1. J Biol Chem. 2010;285:483–492. doi: 10.1074/jbc.M109.068395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimoto T, Okada T, Yu H, Goparaju SK, Jahangeer S, Nakamura S. Involvement of sphingosine-1-phosphate in glutamate secretion in hippocampal neurons. Mol Cell Biol. 2007;27:3429–3440. doi: 10.1128/MCB.01465-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno T, Nishizaki T, Proia RL, Kajimoto T, Jahangeer S, Okada T, Nakamura S. Regulation of synaptic strength by sphingosine 1-phosphate in the hippocampus. Neuroscience. 2010;171:973–980. doi: 10.1016/j.neuroscience.2010.10.021. [DOI] [PubMed] [Google Scholar]
- Kauselmann G, Weiler M, Wulff P, Jessberger S, Konietzko U, Scafidi J, Staubli U, Bereiter-Hahn J, Strebhardt K, Kuhl D. The polo-like protein kinases Fnk and Snk associate with a Ca(2+)- and integrin-binding protein and are regulated dynamically with synaptic plasticity. EMBO J. 1999;18:5528–5539. doi: 10.1093/emboj/18.20.5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr R, Lev-Ram V, Baird G, Vincent P, Tsien RY, Schafer WR. Optical imaging of calcium transients in neurons and pharyngeal muscle of C. elegans. Neuron. 2000;26:583–594. doi: 10.1016/s0896-6273(00)81196-4. [DOI] [PubMed] [Google Scholar]
- Lackner MR, Nurrish SJ, Kaplan JM. Facilitation of synaptic transmission by EGL-30 Gqalpha and EGL-8 PLCbeta: DAG binding to UNC-13 is required to stimulate acetylcholine release. Neuron. 1999;24:335–346. doi: 10.1016/s0896-6273(00)80848-x. [DOI] [PubMed] [Google Scholar]
- Leisner TM, Liu M, Jaffer ZM, Chernoff J, Parise LV. Essential role of CIB1 in regulating PAK1 activation and cell migration. J Cell Biol. 2005;170:465–476. doi: 10.1083/jcb.200502090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Chen B, Yankova M, Morest DK, Maryon E, Hand AR, Nonet ML, Wang ZW. Presynaptic ryanodine receptors are required for normal quantal size at the Caenorhabditis elegans neuromuscular junction. J Neurosci. 2005;25:6745–6754. doi: 10.1523/JNEUROSCI.1730-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X, Korogod N, Brose N, Schneggenburger R. Phorbol esters modulate spontaneous and Ca2+-evoked transmitter release via acting on both Munc13 and protein kinase C. J Neurosci. 2008;28:8257–8267. doi: 10.1523/JNEUROSCI.0550-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison JM, Nurrish S, Kaplan JM. UNC-13 interaction with syntaxin is required for synaptic transmission. Curr Biol. 2005;15:2236–2242. doi: 10.1016/j.cub.2005.10.049. [DOI] [PubMed] [Google Scholar]
- Mahoney TR, Luo S, Nonet ML. Analysis of synaptic transmission in Caenorhabditis elegans using an aldicarb-sensitivity assay. Nat Protoc. 2006;1:1772–1777. doi: 10.1038/nprot.2006.281. [DOI] [PubMed] [Google Scholar]
- Mathews EA, Garcia E, Santi CM, Mullen GP, Thacker C, Moerman DG, Snutch TP. Critical residues of the Caenorhabditis elegans unc-2 voltage-gated calcium channel that affect behavioral and physiological properties. J Neurosci. 2003;23:6537–6545. doi: 10.1523/JNEUROSCI.23-16-06537.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menuz V, Howell KS, Gentina S, Epstein S, Riezman I, Fornallaz-Mulhauser M, Hengartner MO, Gomez M, Riezman H, Martinou JC. Protection of C. elegans from anoxia by HYL-2 ceramide synthase. Science. 2009;324:381–384. doi: 10.1126/science.1168532. [DOI] [PubMed] [Google Scholar]
- Michel FJ, Fortin GD, Martel P, Yeomans J, Trudeau LE. M3-like muscarinic receptors mediate Ca2+ influx in rat mesencephalic GABAergic neurones through a protein kinase C-dependent mechanism. Neuropharmacology. 2005;48:796–809. doi: 10.1016/j.neuropharm.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Mochida S, Orita S, Sakaguchi G, Sasaki T, Takai Y. Role of the Doc2 alpha-Munc13-1 interaction in the neurotransmitter release process. Proc Natl Acad Sci U S A. 1998;95:11418–11422. doi: 10.1073/pnas.95.19.11418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nizami S, Lee VW, Davies J, Long P, Jovanovic JN, Sihra TS. Presynaptic roles of intracellular Ca(2+) stores in signalling and exocytosis. Biochem Soc Trans. 2010;38:529–535. doi: 10.1042/BST0380529. [DOI] [PubMed] [Google Scholar]
- O’Halloran DM, Altshuler-Keylin S, Lee JI, L’Etoile ND. Regulators of AWC-mediated olfactory plasticity in Caenorhabditis elegans. PLoS Genet. 2009;5:e1000761. doi: 10.1371/journal.pgen.1000761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T, Kajimoto T, Jahangeer S, Nakamura SI. Sphingosine kinase/sphingosine 1-phosphate signalling in central nervous system. Cell Signal. 2008 doi: 10.1016/j.cellsig.2008.07.011. [DOI] [PubMed] [Google Scholar]
- Pasquare SJ, Gaveglio VL, Giusto NM. Regulation of phosphatidic Acid metabolism by sphingolipids in the central nervous system. J Lipids. 2011;2011:342576. doi: 10.1155/2011/342576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccinini M, Scandroglio F, Prioni S, Buccinna B, Loberto N, Aureli M, Chigorno V, Lupino E, DeMarco G, Lomartire A, Rinaudo MT, Sonnino S, Prinetti A. Deregulated sphingolipid metabolism and membrane organization in neurodegenerative disorders. Mol Neurobiol. 2010;41:314–340. doi: 10.1007/s12035-009-8096-6. [DOI] [PubMed] [Google Scholar]
- Pitson SM. Regulation of sphingosine kinase and sphingolipid signaling. Trends Biochem Sci. 2011;36:97–107. doi: 10.1016/j.tibs.2010.08.001. [DOI] [PubMed] [Google Scholar]
- Richmond JE, Jorgensen EM. One GABA and two acetylcholine receptors function at the C. elegans neuromuscular junction. Nat Neurosci. 1999;2:791–797. doi: 10.1038/12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Davis WS, Jorgensen EM. UNC-13 is required for synaptic vesicle fusion in C. elegans. Nat Neurosci. 1999;2:959–964. doi: 10.1038/14755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Weimer RM, Jorgensen EM. An open form of syntaxin bypasses the requirement for UNC-13 in vesicle priming. Nature. 2001;412:338–341. doi: 10.1038/35085583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saheki Y, Bargmann CI. Presynaptic CaV2 calcium channel traffic requires CALF-1 and the alpha(2)delta subunit UNC-36. Nat Neurosci. 2009;12:1257–1265. doi: 10.1038/nn.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siow DL, Anderson CD, Berdyshev EV, Skobeleva A, Natarajan V, Pitson SM, Wattenberg BW. Sphingosine kinase localization in the control of sphingolipid metabolism. Adv Enzyme Regul. 2011;51:229–244. doi: 10.1016/j.advenzreg.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stabler SM, Ostrowski LL, Janicki SM, Monteiro MJ. A myristoylated calcium-binding protein that preferentially interacts with the Alzheimer’s disease presenilin 2 protein. J Cell Biol. 1999;145:1277–1292. doi: 10.1083/jcb.145.6.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumakovic M, Hegermann J, Luo L, Husson SJ, Schwarze K, Olendrowitz C, Schoofs L, Richmond J, Eimer S. UNC-108/RAB-2 and its effector RIC-19 are involved in dense core vesicle maturation in Caenorhabditis elegans. J Cell Biol. 2009;186:897–914. doi: 10.1083/jcb.200902096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai C, Kuzmiski JB, MacVicar BA. Muscarinic enhancement of R-type calcium currents in hippocampal CA1 pyramidal neurons. J Neurosci. 2006;26:6249–6258. doi: 10.1523/JNEUROSCI.1009-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Braak M, Danneberg K, Lichte K, Liphardt K, Ktistakis NT, Pitson SM, Hla T, Jakobs KH, Meyer zu Heringdorf D. Galpha(q)-mediated plasma membrane translocation of sphingosine kinase-1 and cross-activation of S1P receptors. Biochim Biophys Acta. 2009;1791:357–370. doi: 10.1016/j.bbalip.2009.01.019. [DOI] [PubMed] [Google Scholar]
- Vashlishan AB, Madison JM, Dybbs M, Bai J, Sieburth D, Ch’ng Q, Tavazoie M, Kaplan JM. An RNAi screen identifies genes that regulate GABA synapses. Neuron. 2008;58:346–361. doi: 10.1016/j.neuron.2008.02.019. [DOI] [PubMed] [Google Scholar]
- Wess J, Eglen RM, Gautam D. Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nat Rev Drug Discov. 2007;6:721–733. doi: 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- Xu K, Tavernarakis N, Driscoll M. Necrotic cell death in C. elegans requires the function of calreticulin and regulators of Ca(2+) release from the endoplasmic reticulum. Neuron. 2001;31:957–971. doi: 10.1016/s0896-6273(01)00432-9. [DOI] [PubMed] [Google Scholar]
- Yoon KW, Cho JH, Lee JK, Kang YH, Chae JS, Kim YM, Kim J, Kim EK, Kim SE, Baik JH, Naik UP, Cho SG, Choi EJ. CIB1 functions as a Ca(2+)-sensitive modulator of stress-induced signaling by targeting ASK1. Proc Natl Acad Sci U S A. 2009;106:17389–17394. doi: 10.1073/pnas.0812259106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young KW, Willets JM, Parkinson MJ, Bartlett P, Spiegel S, Nahorski SR, Challiss RA. Ca2+/calmodulin-dependent translocation of sphingosine kinase: role in plasma membrane relocation but not activation. Cell Calcium. 2003;33:119–128. doi: 10.1016/s0143-4160(02)00205-1. [DOI] [PubMed] [Google Scholar]
- Yu J, Novgorodov SA, Chudakova D, Zhu H, Bielawska A, Bielawski J, Obeid LM, Kindy MS, Gudz TI. JNK3 signaling pathway activates ceramide synthase leading to mitochondrial dysfunction. J Biol Chem. 2007;282:25940–25949. doi: 10.1074/jbc.M701812200. [DOI] [PubMed] [Google Scholar]
- Zikich D, Mezer A, Varoqueaux F, Sheinin A, Junge HJ, Nachliel E, Melamed R, Brose N, Gutman M, Ashery U. Vesicle priming and recruitment by ubMunc13-2 are differentially regulated by calcium and calmodulin. J Neurosci. 2008;28:1949–1960. doi: 10.1523/JNEUROSCI.5096-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]