

Abstract
The sporadic nature of Alzheimer's diease (AD) argues for an environmental link that may drive AD pathogenesis; however, the triggering factors and the period of its action are unknown. Recent studies in rodents have shown that exposure to lead (Pb) during brain development pre-determined the expression and regulation of the amyloid precursor protein (APP) and its amyloidogenic beta-amyloid (Aβ) product in old age. Here we report that the expression of AD-related genes (APP, BACE1) as well as their transcriptional regulator (Sp1) were elevated in aged (23-year old) monkeys exposed to Pb as infants. Furthermore, developmental exposure to Pb altered the levels, characteristics and intracellular distribution of Aβ staining and amyloid plaques in the frontal association cortex. These latent effects were accompanied by a decrease in DNA methyltransferase activity and higher levels of oxidative damage to DNA indicating that epigenetic imprinting in early life influenced the expression of AD-related genes and promoted DNA damage and pathogenesis. These data suggest that AD pathogenesis is influenced by early life exposures and argues for both an environmental trigger and a developmental origin of AD.
Keywords: Amyloidogenesis, development, environmental exposure, Pb, epigenetic regulation, transcription factor
Introduction
Alzheimer's disease (AD) is a progressive and irreversible neurodegenerative disorder which results in dementia and death. AD pathology is characterized by senile plaques and neurofibrillary tangles (NFTs), combined with massive neuronal loss, mainly in the hippocampus and association regions of the neocortex (Ball and Lo, 1977). The major constituents of senile plaques are 39−43 amino acid peptides (Aβ), snipped from a larger protein called Aβ precursor protein (APP) (Glenner and Wong, 1984; Masters et al., 1985; Goldgaber et al., 1987). Recent studies indicate that APP is processed by a group of secretases. The α-secretase generates a soluble product, while β-secretase and γ-secretase generate Aβ from APP.
The sporadic nature of most AD cases strongly argues for an environmental link that may drive AD pathogenesis; however, it is not clear when this may occur. Reconstructions of neonatal and medical histories of birth cohorts have led to the origin of ‘the Barker hypothesis’ (Barker et al., 1989; Osmond and Barker, 2000), which links early life experiences and adult diseases. These observations resulted in a new concept regarding certain adult diseases that emphasizes the role of environmental factors operating during the pre-conceptual, fetal and infantile phases of life (Gluckman and Hanson, 2004).
The pathological manifestations in AD patients are presumed to result from defects of old age; however, it is unlikely that the disease process begins late in life. Therefore, amyloidogenesis associated with AD can also be viewed as a pathological outcome that is evident during aging; however, the preceding initiating event may have occurred during early stages of brain development (Zawia and Basha, 2005). Specifically, this event would have been a latent early-life associated regulation (LEARn) alteration (Lahiri et al., 2007) affecting the expression of genes associated with a later-manifest condition.
Previous work from our laboratories showed that developmental exposure of rats to the metal Pb from birth to PND20 showed a delayed over-expression of APP and elevation of its amyloidogenic Aβ product in old age (Basha et al., 2005). We also observed elevations in the oxidative DNA marker 8-hydroxy-2’-deoxyguanosine (8-oxo-dG) in older rats that had been developmentally exposed to Pb (Bolin et al., 2006). These findings suggested that environmental influences occurring during brain development pre-determined the expression, regulation, and processing of APP later in life, potentially influencing the course of amyloidogenesis and oxidative damage.
In order to link these molecular and oxidative perturbations observed in rats to pathological consequences associated with AD, we have examined the brains of aged Cynomolgus monkeys who were similarly exposed to Pb as infants. Primates are among a few animal models that express amyloid plaques and other pathological features that are absent in wild-type/non-transgenic rodents. This study was undertaken to determine whether non-human primates which exhibit similar AD-like pathology in old age (Price and Sisodia, 1994) would be influenced by developmental perturbations, and to explore the potential mechanisms that could mediate such latent effects.
Materials and Methods
Animal Exposure
In 1980−81, a cohort of female monkeys (Macaca fascicularis) was randomly assigned at birth to one of two exposure groups: one received 1.5 mg/kg/day of lead acetate (Pb) from birth until 400 days of age via infant formula and vehicle after weaning, while the other group served as a control group and received formula or vehicle only. No overt signs of toxicity or health related problems were evident in the animals as a result of Pb exposure (Rice, 1990, 1992). They were then transferred to NIH facility until termination in 2003 at approximately 23 years of age and all animal procedures were conducted under the supervision of a licensed veterinarian according to a NIEHS/NIH approved animal protocol. As previously reported, the blood lead levels of these animals at 400 days of age averaged 19−26 μg/dl in Pb exposed monkeys as compared to 3−6 μg/dl in the controls (Rice, 1990, 1992). The monkeys were terminated in 2003 (23 years later) and multiple organ tissues, including the brain were collected, cut in 1cm sections, and immediately processed in 10% formalin for histopathology or frozen on dry ice and stored at −80°C. At this time, the animals were in good health and there were no indications of adverse health effects as a result of early Pb exposure.
Total RNA isolation, synthesis of cDNA, and Real Time PCR
RNA from various control and exposed monkey cortical tissues was isolated according to the TRIzol method (Invitrogen, CA). The RNA was reverse transcribed to obtain cDNAs, catalyzed by SuperScript III Reverse Transcriptase (RT). The RNA/primer mixture containing 500 ng of total RNA, 1μL of 10mM dNTP mix and 1μL Oligo(dT) was incubated at 65°C for 5 min. A reaction mixture containing 2 μL of 10× RT buffer [200 mM Tris-HCl (pH 8.4), 500 mM KCl], 4 μL of 25 mM MgCl2, 2 μL of 0.1 M DTT and 1 μL of RNaseOUT recombinant RNase inhibitor (40 U/μL) was added. One microliter of SuperScript III RT (200 U/μL) was then added and incubated at 50°C for 50 min. The reaction was terminated at 85°C for 5 min. One microliter of RNase H was added and the reaction was incubated for 20 min at 37°C. The resulting cDNA was stored at −20°C and used in the real time PCR step. The primer pairs used for APP, Sp1, BACE1 and GAPDH were as follows. Sp1 sense: 5’-CAA GCC CAA ACA ATC ACC TT-3’; antisense: 5’-CAA TGG GTG TGA GAG TGG TG-3’. BACE1 sense: 5’-TTT GTG GAG ATG GTG GAC AA-3’; antisense: 5’-CAG CAC CCA CTG CAA AGT TA-3’. APP sense: 5’-GCT GGC TGA ACC CCA GAT-3’; antisense: 5’-CCC ACT TCC CAT TCT GGA CAT-3’. GAPDH sense: 5’-TGA AGC AGG CGT CGG AGG G-3’; antisense: 5’-CGA AGG TGG AAG AGT GGG TG-3’. Each real time PCR reaction mix contained 1 uL of cDNA, 1 uL of primer mix (final concentration 200 nM), 10.5 uL of nuclease free water and 12.5 uL SYBR® GREEN PCR Master Mix (Applied Biosystems, CA). Each sample had triplicates. Real Time PCR was conducted for all of the above genes with respective primer pairs in a 7500 Real-Time PCR System following standard protocol. That was, 50 °C 2 min followed by 95 °C 10 min, then 40 cycles of 95 °C 15 sec and 60 °C 1min. Real time PCR products were checked with agarose gel to confirm that no non-specific products formed. Results were analyzed with 7500 system software with relative quantification method using GAPDH as endogenous control. Other housekeeping genes such as beta-actin were also utilized.
Beta amyloid (Aß) 1−40 and 1−42 assay
The levels of Aß were measured using human Aß (1−40 and 1−42) assay kits (Immuno-Biological Laboratories, Gunma Japan). These kits were designed as solid phase sandwich ELISA with two kinds of highly specific antibodies. The assay conditions were followed according to the method described by Morishima-Kawashima (2000) et al., with slight modifications. Brain tissue was homogenized in Tris-Saline (TS) [50 mM Tris-HCl buffer, pH 7.4; 150 mM NaCl; 1 μg/mL TLCK (N-Alpha-p-tosyl-L-Lysine chloromethyl ketone); 1 μg/mL antipain; 0.5 mM DIFP (Diisopropyl fluorophosphates); 0.5 mM PMSF; 0.1% Protease Inhibitor cocktail] and centrifuged at 100,000 g for 20 min at 4°C. The pellet was resuspended in 4 volumes of TS and centrifuged at 70,000 g for 20 min at 4°C. The resultant pellet was dissolved in 500 μL of 6 M guanidine-HCl (in 50 mM Tris buffer, pH 7.6), incubated at room temperature (RT) for 30 min and centrifuged at 70,000 g for 20 min at 4°C. The resultant supernatant was collected and diluted by EIA buffer (supplied with the kit) to 12× to reduce sample Guanidine-HCl concentration, and aliquots (200 μg of protein in 100 μL EIA buffer) and assay standards were added to a 96 well plate [pre-coated with anti-human Aß (35−40) (1A10) Mouse IgG MoAb] and incubated over night at 4°C. The wells were washed 7× with EIA buffer. Then 100 μL of labeled antibody was added to each well containing sample or standard and incubated at 4°C for 1 hour. The wells were washed 9× with EIA buffer followed by the addition of 100 μL of TMB buffer, and incubated in the dark for 30 min at RT. The reaction was stopped by adding 100 μL of 1N H2SO4 and the colormetric absorption was performed at 450 nm. The levels of Aß in the test samples were calculated relative to the standard curve generated on each plate.
Immunohistochemistry
The cellular distribution of APP, SP1 and Aβ was examined in paraffin embedded brain tissue of Control (Con) and developmentally, Pb-exposed (Pb-E), 23-year old Cynomolgus monkeys. The sections were subjected to brief washes in 1× phosphate buffered saline (PBS) and 3% hydrogen peroxide. After rinsing, the sections were incubated in PBS containing 2% Bovine Serum Albumin (BSA) and 1% Triton X-100 blocking solution for 30 min, then incubated in the presence of primary antibody for APP (1:200; Sigma-Aldrich, St. Louis, MO), Aβ (1:50; Sigma-Aldrich), or SP1 (1:100; Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4°C. Sections were washed with PBS and incubated along with the species-specific biotinylated secondary antibody mouse/rabbit (1:200; Vector Labs, Burlingame, CA) for 30 min. The sections were incubated with Streptavidin (Vector Labs) for 30 min, rinsed briefly with PBS, and immunoreactivity was detected with the substrate, 3−3′ Diaminobenzidine-tetrahydrochloride (DAB) (Vector Labs). Coverslips were mounted with Permanent Mounting Medium (Vector Labs).
In all cases, negative controls were run on slides from each animal by omitting the primary antibody incubation. No signal was evident after incubation of tissues in secondary antibody alone.
Nuclear protein extraction
Nuclear proteins were extracted from the frontal association cortical tissue of control and Pb-exposed animals according to the method described by Dignam (1983) et al., with slight modifications. Tissue samples were homogenized with 1 ml PBS (phosphate buffered saline, pH 7.4) and centrifuged at 2500 g for 10 min at room temperature. The pellets obtained were suspended in 5 volumes of Buffer A (10 mM HEPES at pH 7.9, 1.5 mM MgCl2, 0.5 mM DTT, 0.5 mM EDTA, and 0.2 mM PMSF) and centrifuged at 6,000 g for 2 min at 4°C. The pellets were resuspended in 3 volumes of Buffer A and centrifuged at 6,000 g for 2 min at 4°C. The resulting pellets were then resuspended in 5 volumes of buffer C (20 mM HEPES at pH 7.9, 1.5 mM MgCl2, 0.5 mM DTT, 0.5 mM EDTA, 420 mM NaCl, 20% glycerol, 0.2 mM PMSF, 0.002 mg/ml aprotinin, and 0.0005 mg/ml leupeptin) and homogenized. The final suspensions were centrifuged at 12,000 g for 10 min. The supernatants were transferred to 1.5 ml tubes, snap frozen in an ethanol dry-ice bath, and stored at −80° C (Basha et al., 2005).
Primary mice cortical neuronal cell culture
Since the animals we studied were 23 years old and we did not have early time points to measure the progression of the molecular or epigenetic changes, we used a cell culture model to address some of the mechanistic questions. We used the C57BL/6 mouse as a source of primary cortical neurons based upon the wide use of this genetic background strain for various transgenic mice AD models. The C57BL/6 mice (Charles River Laboratories, Wilmington, MA) fetuses were used to generate primary cortical neuronal cultures. Animal usage was approved (Approval #AN00−01−007) and monitored by the Institutional Animal Care and Use Committee (IACUC) of the University of Rhode Island. Brains were excised and cortices dissected from day 15 mouse pups from the same dam. Meninges were removed and cortical tissue incubated for 15 min at 37°C in 5 ml Hanks' Balanced Salt Solution (HBSS) containing papain (2 mg/ml). Tissue was centrifuged at 125 g for 5 min and the resulting pellet was resuspended in HBSS and cells dissociated with trituration through a fire polished Pasteur pipette and repeated 3 times. Following centrifugation, cells were plated at a constant density of 6.5 × 104/well, in 24-well poly-D-Lysine pre-coated plates. The plating medium contained 2% B27, 0.5 mM L-Glutamine and 25 μM glutamic acid in NEUROBASAL medium (Invitrogen, CA) and maintained at 37°C in a humidified atmosphere of 5 % CO2. After 4 days in culture, half of the medium was replaced with fresh medium devoid of glutamic acid and the cultures were maintained. To determine methyltransferase activity, cells in the 24-well plates were treated with 0.1 μM Pb at the time of medium change for 24 h. Pb was removed and cells were aged for 7 days before nuclear extract was harvested and subjected to methyltransferase activity assay.
8-oxo-dG determination
8-oxo-dG levels were determined by HPLC as described previously (Bolin et al., 2006). Briefly, sample tissue was homogenized in nuclease free homogenization buffer followed by digestion with proteinase K to remove proteins. The DNA was precipitated by 3 consecutive organic extractions, precipitation by two volumes of ethanol (with respect to the aqueous volume), and incubated overnight at −20°C. The purified DNA was prepared for HPLC analysis by nuclease digestion into deoxynucleoside components. The amount of 8-oxo-dG and 2’-deoxyguanosine (2’-dG) was calculated by comparing the peak areas of 8-oxo-dG and 2’-dG obtained from the enzymatic hydrolysate of the DNA sample to a calibration curve for both compounds. Levels of 8-oxo-dG in the samples were expressed relative to the content of 2’-dG, e.g., the molar ratio of 8-oxo-dG/2’-dG (fmol 8-oxo-dG/nmol of 2’-dG). The HPLC method used for analysis of the samples was as follows: The mobile phase consisted of 100 mM sodium acetate, pH 5.2, with 5% methanol. Flow rate was kept at 1 mL/min using a Model 582 Solvent Delivery Module (ESA, Chelmsford, MA). DNA was analyzed using a reverse phase YMC basic HPLC column (4.6×150 mm) with a 3 μm particle size (YMC Inc. Wilmington, NC). 8-oxo-dG and 2’-dG were detected by a Model 5600A CoulArray Detector (ESA) with three model 6210 four-channel electrochemical cells; potentials were set at 175, 200, and 250 V for 8-oxo-dG and at 785, 850, and 890 V for 2’-dG. Data were recorded, stored, and analyzed with CoulArray for Windows32 Software (ESA). Data were expressed as femtomoles of 8-oxo-dG per nanomole of 2’-dG.
DNA methyltransferase assay
DNA methyltransferase activity was determined in nuclear extracts derived from the control and Pb-exposed frontal associated cortical tissue. Nuclear extracts were derived as described above and the DNA methyltransferase activity was assayed following the method as described by Takiguchi (2003) et al. Nuclear extracts containing 60 μg of protein (source of methyltransferase) were incubated with 50 ng of deoxyinosine-deoxycytidine (poly[dI.dC].poly[dI.dC]) double stranded DNA template (Sigma) as a substrate, 1 μM 3H-labeled S-adenosyl methionine (SAM) (79 Ci/mmol), 100 mM Tris-HCl (pH 8.2) in a final volume of 30 μL. The reaction was initiated by the addition of SAM for 1 hour at 37° C and terminated by chilling on ice. A 15 μL aliquot was spotted on DE81 filter paper and washed with sodium phosphate buffer (pH 7.0) twice, once with 70% ethanol, and once with 100% ethanol. The filter was dried and counted in a scintillation counter.
Statistical treatment
Data were analyzed using two-tailed Student's t-test and the values marked with an “*” were significantly different from the control group (p<0.05).
RESULTS
Pb levels
The exposure of these animals to low level of inorganic Pb from birth to 400 days resulted in blood Pb levels of 19−26 μg/dL when being dosed with Pb after weaning from infant formula. This level is slightly above the 10 μg/dL considered safe for humans by the CDC (1991), and is similar to the exposure scenario we had previously used in lifetime studies with rats (Basha et al., 2005). Sampling during young adulthood demonstrated clearance of Pb from the blood and levels were similar for both control and Pb exposed monkeys (Rice, 1992). Thus, any significant exposure was limited to the developing and adolescent period. In the aged animals, Pb levels in brain tissue were analyzed using Elemental Analyzer ICP-MS and we found that Pb levels for both exposed and control animals remained below the detection levels (<0.1 ng/g wet wt. of tissue).
Latent expression of APP, Aβ, BACE1 and Sp1
The amino acid sequence of APP of Cynomolgus monkeys is homologous (96%) to that of humans making it a good model for studies of the pathological effects of Aβ in the primate brain (Podlisny et al., 1991). The enzyme BACE1 plays a major role in the cleavage of APP (Singer et al., 2005) and transcription factor Sp1 (Lahiri and Robakis, 1991; Christensen et al., 2004) is a known regulator of both genes. Therefore, we compared APP, BACE1, and Sp1 mRNA levels in the control and developmentally Pb-exposed aged primates. The mRNA levels of all three genes (Fig. 1) were elevated in primates exposed to Pb as infants; however, the elevation in BACE1 was not statistically significant (p=0.063). Quantification of results indicated that APP mRNA levels increased 50% (p=0.045); Aβ1−40 levels increased 50% (p=0.011); and Aβ1−42 levels increased 100% (p=0.029), in a manner similar to that previously seen in rodents (Fig. 2A, 2B). While rodents have non plaque-forming Aβ with 3 amino acids different from that of primates, APP of Cynomolgus monkey shares 96% sequence homology with human, and both species have same plaque-forming Aβ peptides, which is more clinically relevant. Furthermore, the ratio of Aβ1−42 to Aβ1−40 increased due to developmental exposure to Pb (Fig. 2B). Exposure of cultured mouse primary neurons to these peptides confirmed that both of these peptides were toxic to the cells even in the soluble form and that Aβ1−42 was more cytotoxic than Aβ1−40 (Supplementary data: Figure-1).
Figure 1. Changes in mRNA expression of APP, BACE1 and Sp1 in the frontal association cortex of aged monkeys following developmental exposure to Pb.
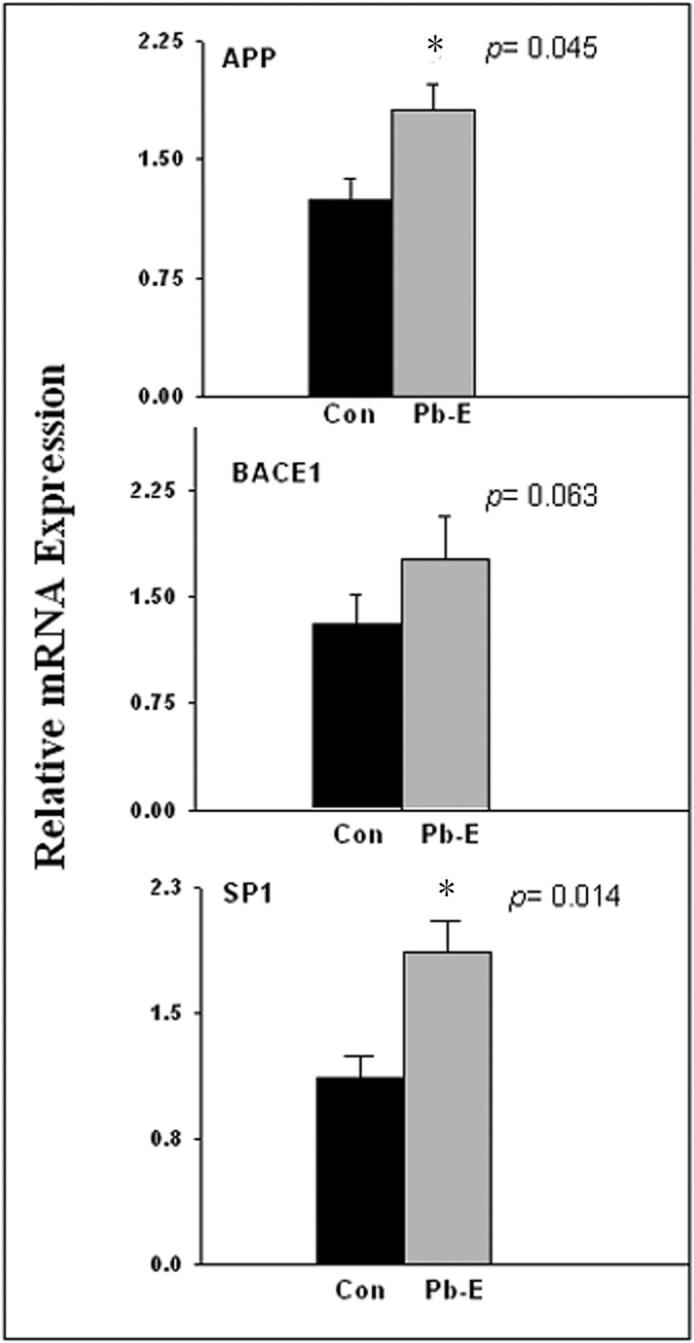
The frontal association cortical tissue of 23-year old Control (Con) and Pb-exposed (Pb-E) monkeys were analyzed for mRNA expression of APP, BACE1, and Sp1. The mRNA expression of APP, BACE1, and Sp1 were validated by real time PCR. Data shown represent the mean ± S.E.M. (4 animals in each group). Values marked with an “*” are significantly different from their corresponding controls (p<0.05) as determined by student's t-test.
Figure 2. Elevation of Aβ levels in the frontal association cortex of 23-year old Cynomolgus monkeys following developmental exposure to Pb.
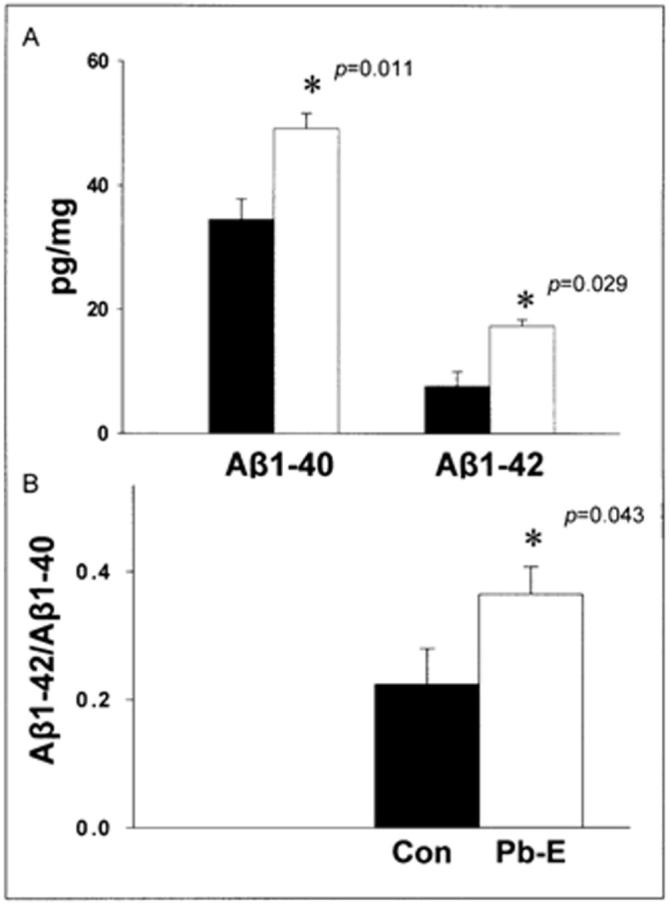
Brain tissue of Control (Con) and Pb-exposed (Pb-E) 23-year old Cynomolgus monkeys were used for the analysis of Aβ levels (A-B). The levels of Aβ were measured in the frontal association cortical tissue using ELISA as described in the methods section. Data shown represent the mean ± S.E.M. for 4 animals in each group. Values marked with an “*” are significantly different from their corresponding controls (p<0.05) as determined by a student's t- test.
Alteration of AD-pathology
Immunohistochemical analysis of the frontal association cortex was undertaken to determine if the observed molecular changes in APP expression and Aβ levels were accompanied by changes in the pathological features of the brains of these animals. The brains of the aged monkeys developmentally-exposed to Pb revealed an increase in the intracellular staining of total Aβ and dense-core plaques as compared to age-matched controls (Fig. 3). Higher magnification of intracellular Aβ staining revealed the accumulation of immunoreactive Aβ aggregates inside neuronal cells, the budding of some of these Aβ species from the membrane as well as their deposition in the extracellular space (Fig. 3). We also observed diffuse and cored plaques and NFTs morphologically similar to those observed in human brain. The Aβ plaques were found to be rich in Aβ1−42, the more amyloidogenic species of Aβ, and were detectable using Congo red staining.
Figure 3. Photomicrographs showing AD-like pathology in the frontal association cortex of 23-year old Cynomolgus monkeys following developmental exposure to Pb.
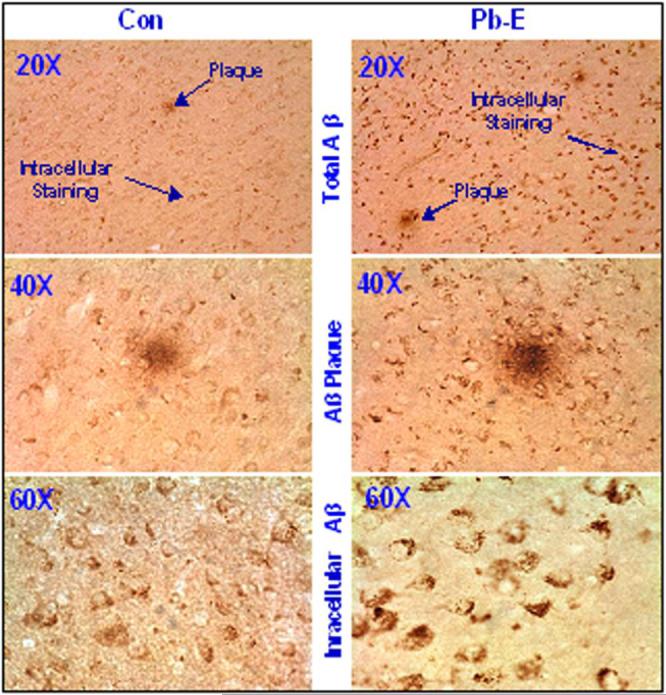
Brain tissue of Control (Con) and Pb-exposed (Pb-E) 23-year old Cynomolgus monkeys were used for the analysis of immunohistochemical analysis of AD-like pathology. Sections were prepared and stained with the Aβ-specific antibody which recognizes both Aβ1−40 and Aβ1−42 as discussed in the methods section. Arrows point to Aβ-containing plaques as well as granular and intracellular staining. The staining characteristics are similar to those reported in the literature.
DNA methylation
The latent expression of genes observed in these animals may be mediated through epigenetic pathways that are regulated via DNA methylation. To determine whether developmental exposure to Pb interfered with DNA-methylation patterns, we examined the activity of DNA methyltransferase 1 (DNMT1) in the 23-year old primate brain tissues. The activity of this methylating enzyme is selective for cytosine in a CpG dinucleotide, which is base-paired to a methylated CpG sequence on the complementary strand of DNA and is directly proportional to the abundance of methyl groups on CpG dinucleotides in the DNA (Poirier and Vlasova, 2002; Takiguchi et al., 2003). We found the activity of DNMT1 to be reduced by about 20% in brain tissue derived from developmentally Pb-exposed primates (Fig. 4B). Exposure of mouse primary cells from the cortex to low levels of Pb (0.1 μM) for a transient 24 hour period followed by aging of the cells for a week produced a similar trend in DNMT1 activity (Fig. 4A).
Figure 4. DNA methyltransferase activity in cortical neuronal cells of mice and in monkey brains.
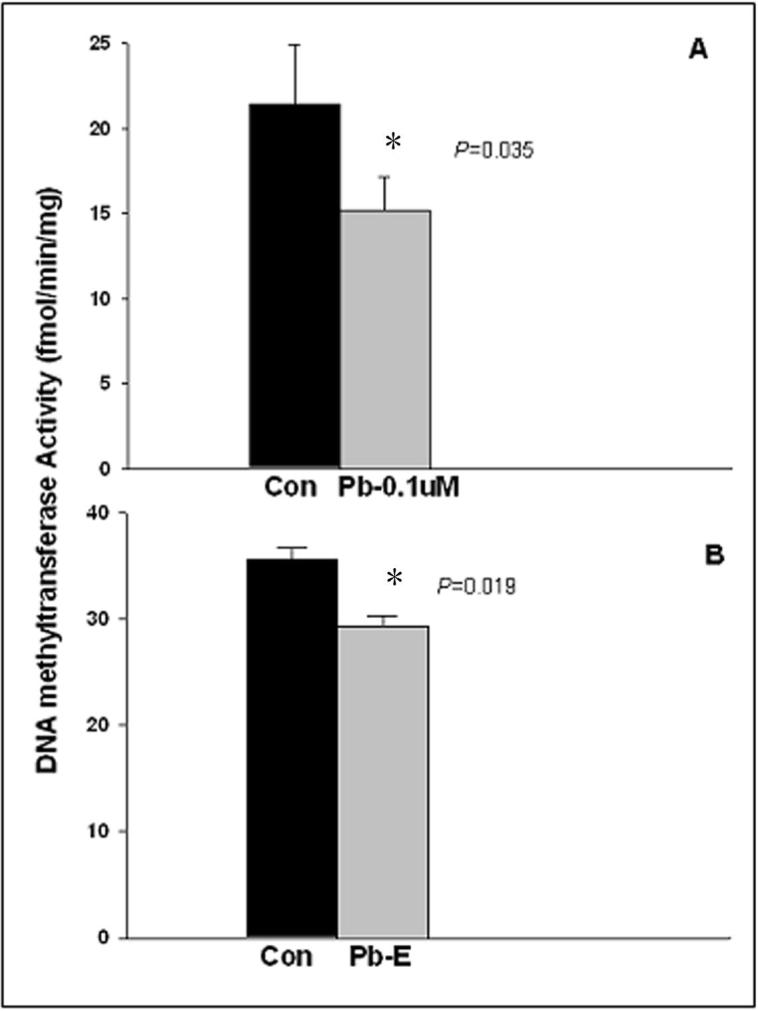
(A) Control and Pb-exposed mouse cortical neuronal cells, and (B) Frontal association cortical tissue of 23-year old control (Con) and Pb-exposed (Pb-E) monkeys were used to estimate the DNA methyltransferase activity as described in the methods section. Data shown represent the mean ± S.E.M. for 4 independent determinations (4 animals in each group for monkey data). Values marked with an “*” are significantly different from their corresponding controls (p<0.05) as determined by a student's t-test.
DNA oxidation
In addition to the alteration in APP and Aβ deposition, age-related accumulation of oxidative damage is suspected to play a role in the pathogenesis of AD. Aβ levels are well known to induce functional disturbance in vivo through their pro-oxidant and neurotoxic properties (Castellani et al., 2006). Along with the increase seen in Aβ levels in the cortex of developmentally Pb-exposed animals, higher levels of the biomarker of oxidative DNA damage, 8-oxo-dG were noted (Fig. 5).
Figure 5. Oxidative DNA damage in control and infantile-exposed aged monkey brains.
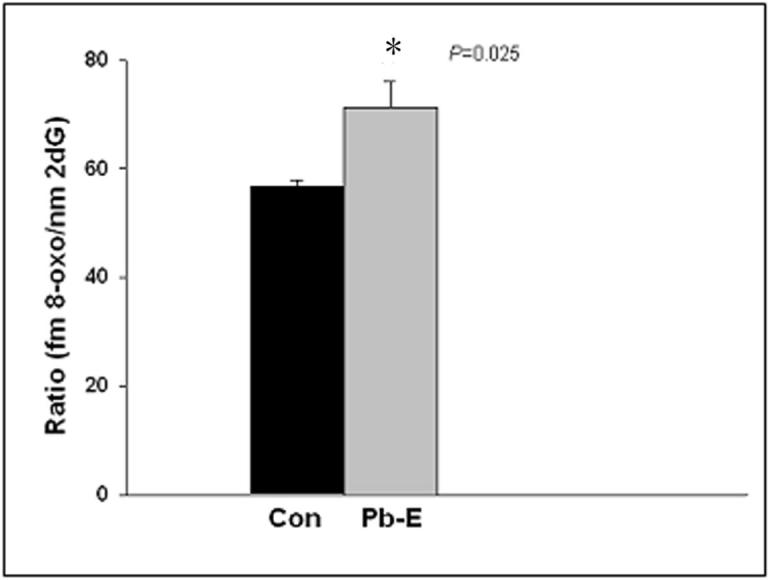
Frontal association cortical tissues were obtained from 23-year old control (Con) and Pb-exposed (Pb-E) monkeys and used to measure the marker of oxidative DNA damage as described in the methods section. Data shown represent the mean ± S.E.M. for 4 animals in each group. Values marked with an “*” are significantly different from their corresponding controls (p<0.05) as determined by a student's t- test.
Discussion
The data presented in this paper show that monkeys exposed to Pb at birth to 400 days up-regulate the expression of APP, BACE1, and Sp1 in old age. The up-regulation of both APP and BACE1 gene expression is mediated by Sp1, and the essentiality of Sp1 as a mediator of these delayed transcriptional up-regulations has been previously shown by us (Basha et al., 2005). Moreover, both APP and BACE1 are rich in CpG dinucleotides and GC box elements (Pollwein et al., 1992) making them subjected to epigenetic reprogramming and transcriptional regulation via DNA methylation pathways (Lahiri et al., 2007). This is further reinforced by preliminary microarray screens which showed that more than 95% of the differentially expressed genes screened in control versus Pb-exposed monkeys were also CpG-rich (supplementary data: Table-1). This transcriptional reprogramming at the gene level is also translated into biological consequences. Levels of amyloidogenic products of APP were also increased in the aged animals that were exposed to Pb as infants (Fig. 2A). This is consistent with earlier findings in rodents (Basha et al., 2005) and argues for a transcriptional process that promotes neurodegeneration in old age.
These molecular and biochemical changes observed in 23-year old animals are accompanied by altered features of AD-like pathology in the exposed monkeys. Our intracellular staining closely resembles what is seen in humans and other animal models (Mochizuki et al., 1996; Schmitz et al., 2004). It reveals granular, oval, and crescent shaped Aβ localization in pyramidal cells and globular shaped neurons in layers II-IV of the cortex. The occurrence of these molecular, biochemical and pathological changes in primates that develop plaques and tangles in old age, in response to developmental exposure to Pb, suggests that developmental exposure can influence latent pathogenesis, hence bearing a direct relevance to humans. The possibility that developmental exposure to Pb could result in the formation of AD pathology in humans is further supported by findings in a patient who survived from severe Pb toxicity at 2 years of age, but died of severe mental deterioration at the age of 42 (Niklowitz and Mandybur, 1975). The brain of this patient revealed many senile plaques and NFTs (Niklowitz and Mandybur, 1975). Although this is a single case, the plausibility that early exposure to Pb could be a risk factor for AD warrants further study.
One way to achieve permanent changes or long-term alteration in gene expression is to alter the structural make-up of the DNA bases that determine the sequence-specific DNA-binding of a transcription factor. Our findings with changed DNA methyltransferase activity argue that exposure to Pb early in life interferes with gene imprinting and could thus leave a permanent molecular scar on the DNA. Consistent with such a potential mechanism is the decreased activity of the DNA methylating enzyme in the aged animals exposed to Pb as infants, as well as the delayed decrease in the activity of this enzyme in cells that had prior exposure to Pb (Fig. 4).
This is further supported by studies that show that regions of the human APP promoter upstream of −500bp displayed tissue and brain region-specific profiles of methylation, which roughly reflect APP expression patterns (Rogaev et al., 1994) and age-related reduction in methylcytosine (−224 to −101) that occur on the human APP promoter (Tohgi et al., 1999). To link the association of an epigenetic phenomenon in current model, we hypothesize that genes that are regulated by methylation can be reprogrammed in adulthood due to infantile exposure to inorganic Pb. This hypothesis was supported by our microarray screening of 588 neurobiology-related genes. We found that most of the genes (20 out of 22) that were altered due to infantile exposure to Pb were rich (>60%) in CpG dinucleotides (Supplementary data: Table-1). However, our own work herein and elsewhere (Basha et al., 2005) has shown that while SP1 and APP are up-regulated in a latent fashion after early exposure to Pb in both Cynomolgus monkeys and rats, other genes regulated by SP1, such as BACE1 (Christensen et al., 2004) do not respond in rodents (Bolin et al., 2006) and show a modest trend in primates.
In addition, SP1 regulates a wide variety of other genes that have not been shown to respond in this latent fashion to early Pb exposure. This does not exclude SP1 from an active role in latent Pb-induced pathogenesis. The specific position of an affected SP1 site may explain the difference. For example, active SP1 sites in the APP gene appear in its 5’-UTR (Villa et al., 2004) while the active SP1 site in the BACE1 promoter is approximately 1kb upstream of the transcription start (Christensen et al., 2004). Alternatively, general density of CpG dinucleotides in combination with presence of transcription factor sites (such as SP1) may explain the differential effects between APP and BACE1. Comparison of the cumulative potential methyl CpG dinucleotides 2 kilobases (kb) upstream of the transcription start sites indicated that the proximal promoter regions surveyed by Bolin et al suggested a difference in methylation site density centered around −100 (Bolin et al., 2006).
Ample evidence has accumulated that oxidative damage to macromolecules such as DNA, protein, and lipids (Cecchi et al., 2002; Esposito et al., 2006), as well as, a down-regulation in antioxidant enzymes are associated with AD (Smith and Perry, 1995). We have previously found elevations in the oxidative DNA marker 8-oxo-dG in older rats that had been developmentally exposed to Pb (Bolin et al., 2006). Here we also find a similar accumulation of 8-oxo-dG (Fig. 5). This latent accumulation of oxidized DNA could possibly come from two sources. The latent increase in Aβ could promote the formation of reactive oxygen species, thus damaging the DNA; and/or epigenetic modulation in the methylation pattern of cytosines could interfere with the repair of oxidized guanines or render them more susceptible to oxidative damage (Evans and Cooke, 2004).
Few studies address both DNA methylation and DNA oxidative damage as an epigenetic phenomenon. Researchers using synthetic DNA oligonucleotides with both methylation and oxidative damage in a single CpG site or oxidized dG at a complementary strand have found oxidation of guanine in a CpG dinucleotide reduced the MBD (methyl group binding domain) binding to that site (Valinluck et al., 2004). Even the oxidation on the guanine in the opposite strand diminished the MBD binding but not so much as the same strand G-oxidation. When 5-methylcytosine was oxidized to 5-hydroxymethylcytosine, its affinity to MBD is greatly reduced to the same level as unmethylated cytosine. We thus hypothesize that epigenetic mechanisms such as DNA methylation can influence oxidative damage and make organisms more susceptible to the pathogenesis of AD and that this process can be modulated by environmental exposure.
We, therefore, propose a model (Fig. 6) of interaction between methylation and oxidative damage in genes reprogramming. Using the APP gene proximal promoter/5’-UTR sequence as an example, in undamaged genes, MECP2 (methyl CpG binding protein 2) would compete against transcription factors such as SP1 for regulation. The resulting competition would down-regulate a given gene. If target DNA is altered via oxidation to 8-oxo-dG or 5-hydroxymethylcytosine, MECP2 binding may be partially or completely blocked, but insufficiently to permit sufficient competing SP1 interaction with the sequence to change expression (Fig. 6B). Normal changes of development, maturity, and aging result in triggering of latent SP1 regulatory alterations, which significantly increase levels of the transcription factor (Fig. 6C). The greater levels of SP1, when combined with partial blockage of MECP2 binding, permit sufficient SP1 interaction with the sequence to result in greater target gene expression. It is likely that other transcription factors may function as competitive targets for MECP2 in addition to SP1.
Figure 6. Model of effects of oxidative damage on SP1-mediated expression of APP via interference with MECP2 binding of methylated CpG dinucleotides.

The APP promoter and 5'-UTR from −1000 base pairs to the “ATG” start codon at +148 are shown. CpG dinucleotides are indicated on the sequence, as is the +1 transcription start site. (A) Undamaged DNA, showing the binding of one or more MECP2 to methylated CpG, competing against SP1. APP expression is at normal levels. (B) Conversion of G residues to 8-oxo-dG or mC to 5-hydroxymethylcytosine blocks binding of some MECP2 to methylated CpG, potentially permitting additional binding of SP1 but with insufficient levels to significantly alter target gene expression levels. (C) Normal changes of development, maturity and aging result in triggering of latent SP1 regulatory alterations, which significantly increase levels of the transcription factor, permitting SP1 to out-compete remaining MeCP2 binding, increasing expression of target genes.
Adult diseases such as schizophrenia have also been linked to infection, fetal malnutrition or hypoxia in early life (Dalman et al., 1999; Van Erp et al., 2002; Boksa and El-Khodor, 2003). A study by Bilbo et al. (2005) showed that perinatal exposure to an infectious agent affected how the nervous system responded to an immune challenge and memory consolidation later in adulthood. The authors also found that neonatal pathogen exposure decreased the number of adult hippocampal astrocytes, increased their reactivity, and decreased brain IL-1β levels following adult lipopolysaccharide (LPS) exposure. Work in autism spectrum disorders has demonstrated at least some contribution of differential methylation (Jiang et al., 2004). Hypomethylation of the DRD2 and HTR2A genes have been implicated in both schizophrenia and bipolar disorder (Abdolmaleky et al., 2004). On the other hand, hypermethylation of the RELN gene has been shown to associate with schizophrenia (Abdolmaleky et al., 2005), indicating that latent pathogenic effects of epigenetic perturbations may work on more than one mechanism, while our work specifically addressed latent effects of Pb, other metals may have a similar effect (Poirier and Vlasova, 2002 ;Takiguchi et al., 2003).
In conclusion, this paper presents novel findings in primates that implicate an environmental agent (Pb) in the pathogenesis of AD and demonstrate that development is an important period of vulnerability which could increase future susceptibility to neurodegeneration and AD pathology.
Acknowledgements
This research was supported by grants (ES013022 and AG027246) from the National Institutes of Health (NIH) awarded to NHZ and URI core facility grant (P20RR016457) funded by the National Center for Research Resources (NCRR), a component of NIH. Work at FCP's lab was supported by the division of intramural research of NIEHS/NIH, NIA grant1R15AG023604-01; and work at DKL's lab was supported by NIH P20 RR15583-07 and NIH P20RRP20RR017670-04, Alzheimer's association and NIH grant AG18379 and AG18884.
Supplementary Material
Supplementary data: Figure-1 Cytotoxicity of Aβ1−40 and Aβ1−42 in mice primary neuronal cells: Mice primary cortical neuronal cells were exposed to various concentrations (0.0/0.1/1.0/10.0 μM) of soluble (non-aggregated) Aβ1−40 or Aβ1−42 obtained from commercial source (American peptide company, Sunnyvale, CA). After 48 hours of exposure, cell viability was tested by MTT assay using Promega (Madison, WI) kit. Results revealed that both the peptides were toxic to the cells even at lower concentrations and Aβ1−42 was more cytoxic than Aβ1−40. Data shown were Mean ± S.E. derived from 3−4 independent observations.
Supplementary data: Table 1 Microarray screening of neurobilogy related genes: Microarray array analysis of about 588 neurobiology-related human genes was conducted to identify the genes that are altered due to infantile exposure to Pb in the frontal association cortex of 23-year old cynomolgus monkeys. The results showed that the expression profile of only a few genes (22) was changed due to early life exposure to Pb. Most of the altered genes belonged to neurotransmitter, growth receptors and signal transduction pathways. We further conducted searches on various databases (Ensembl and NCBI nucleotides) to determine if the regulatory regions of these genes were rich in CpG dinucleotides (>60%). We found that most of these altered genes (with the exception of two) were abundant in CpG dinucleotides in their 5’untranslated regions (5’UTR). These findings provide the initial evidence for the association of epigenetic pathway to explain such latent effects; however, these studies require validation by high throughput methylation sequence profiling.
References
- Abdolmaleky HM, Smith CL, Faraone SV, Shafa R, Stone W, Glatt SJ, Tsuang MT. Methylomics in psychiatry: Modulation of gene-environment interactions may be through DNA methylation. Am J Med Genet B Neuropsychiatr Genet. 2004;127:51–59. doi: 10.1002/ajmg.b.20142. [DOI] [PubMed] [Google Scholar]
- Abdolmaleky HM, Cheng KH, Russo A, Smith CL, Faraone SV, Wilcox M, Shafa R, Glatt SJ, Nguyen G, Ponte JF, Thiagalingam S, Tsuang MT. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am J Med Genet B Neuropsychiatr Genet. 2005;134:60–66. doi: 10.1002/ajmg.b.30140. [DOI] [PubMed] [Google Scholar]
- Ball MJ, Lo P. Granulovacuolar degeneration in the ageing brain and in dementia. J Neuropathol Exp Neurol. 1977;36:474–487. doi: 10.1097/00005072-197705000-00006. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet. 1989;2:577–580. doi: 10.1016/s0140-6736(89)90710-1. [DOI] [PubMed] [Google Scholar]
- Basha MR, Wei W, Bakheet SA, Benitez N, Siddiqi HK, Ge YW, Lahiri DK, Zawia NH. The fetal basis of amyloidogenesis: exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain. J Neurosci. 2005;25:823–829. doi: 10.1523/JNEUROSCI.4335-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilbo SD, Levkoff LH, Mahoney JH, Watkins LR, Rudy JW, Maier SF. Neonatal infection induces memory impairments following an immune challenge in adulthood. Behav Neurosci. 2005;119:293–301. doi: 10.1037/0735-7044.119.1.293. [DOI] [PubMed] [Google Scholar]
- Boksa P, El-Khodor BF. Birth insult interacts with stress at adulthood to alter dopaminergic function in animal models: possible implications for schizophrenia and other disorders. Neurosci Biobehav Rev. 2003;27:91–101. doi: 10.1016/s0149-7634(03)00012-5. [DOI] [PubMed] [Google Scholar]
- Bolin CM, Basha R, Cox D, Zawia NH, Maloney B, Lahiri DK, Cardozo-Pelaez F. Exposure to lead and the developmental origin of oxidative DNA damage in the aging brain. Faseb J. 2006;20:788–790. doi: 10.1096/fj.05-5091fje. [DOI] [PubMed] [Google Scholar]
- Castellani RJ, Lee HG, Perry G, Smith MA. Antioxidant protection and neurodegenerative disease: the role of amyloid-beta and tau. Am J Alzheimers Dis Other Demen. 2006;21:126–130. doi: 10.1177/153331750602100213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecchi C, Fiorillo C, Sorbi S, Latorraca S, Nacmias B, Bagnoli S, Nassi P, Liguri G. Oxidative stress and reduced antioxidant defenses in peripheral cells from familial Alzheimer's patients. Free Radic Biol Med. 2002;33:1372–1379. doi: 10.1016/s0891-5849(02)01049-3. [DOI] [PubMed] [Google Scholar]
- Christensen MA, Zhou W, Qing H, Lehman A, Philipsen S, Song W. Transcriptional regulation of BACE1, the beta-amyloid precursor protein beta-secretase, by Sp1. Mol Cell Biol. 2004;24:865–874. doi: 10.1128/MCB.24.2.865-874.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalman C, Allebeck P, Cullberg J, Grunewald C, Koster M. Obstetric complications and the risk of schizophrenia: a longitudinal study of a national birth cohort. Arch Gen Psychiatry. 1999;56:234–240. doi: 10.1001/archpsyc.56.3.234. [DOI] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito L, Raber J, Kekonius L, Yan F, Yu GQ, Bien-Ly N, Puolivali J, Scearce-Levie K, Masliah E, Mucke L. Reduction in mitochondrial superoxide dismutase modulates Alzheimer's disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J Neurosci. 2006;26:5167–5179. doi: 10.1523/JNEUROSCI.0482-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MD, Cooke MS. Factors contributing to the outcome of oxidative damage to nucleic acids. Bioessays. 2004;26:533–542. doi: 10.1002/bies.20027. [DOI] [PubMed] [Google Scholar]
- Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA. Living with the past: evolution, development, and patterns of disease. Science. 2004;305:1733–1736. doi: 10.1126/science.1095292. [DOI] [PubMed] [Google Scholar]
- Goldgaber D, Lerman MI, McBride OW, Saffiotti U, Gajdusek DC. Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer's disease. Science. 1987;235:877–880. doi: 10.1126/science.3810169. [DOI] [PubMed] [Google Scholar]
- Jiang YH, Sahoo T, Michaelis RC, Bercovich D, Bressler J, Kashork CD, Liu Q, Shaffer LG, Schroer RJ, Stockton DW, Spielman RS, Stevenson RE, Beaudet AL. A mixed epigenetic/genetic model for oligogenic inheritance of autism with a limited role for UBE3A. Am J Med Genet A. 2004;131:1–10. doi: 10.1002/ajmg.a.30297. [DOI] [PubMed] [Google Scholar]
- Lahiri DK, Robakis NK. The promoter activity of the gene encoding Alzheimer beta-amyloid precursor protein (APP) is regulated by two blocks of upstream sequences. Brain Res Mol Brain Res. 1991;9:253–257. doi: 10.1016/0169-328x(91)90009-m. [DOI] [PubMed] [Google Scholar]
- Lahiri DK, Maloney B, Basha MR, Ge YW, Zawia NH. How and when environmental agents and dietary factors affect the course of Alzheimer's disease: the “LEARn” model (latent early-life associated regulation) may explain the triggering of AD. Curr Alzheimer Res. 2007;4:219–228. doi: 10.2174/156720507780362164. [DOI] [PubMed] [Google Scholar]
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki A, Peterson JW, Mufson EJ, Trapp BD. Amyloid load and neural elements in Alzheimer's disease and nondemented individuals with high amyloid plaque density. Exp Neurol. 1996;142:89–102. doi: 10.1006/exnr.1996.0181. [DOI] [PubMed] [Google Scholar]
- Morishima-Kawashima M, Oshima N, Ogata H, Yamaguchi H, Yoshimura M, Sugihara S, Ihara Y. Effect of apolipoprotein E allele epsilon4 on the initial phase of amyloid beta-protein accumulation in the human brain. Am J Pathol. 2000;157:2093–2099. doi: 10.1016/s0002-9440(10)64847-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niklowitz WJ, Mandybur TI. Neurofibrillary changes following childhood lead encephalopathy. J Neuropathol Exp Neurol. 1975;34:445–455. doi: 10.1097/00005072-197509000-00006. [DOI] [PubMed] [Google Scholar]
- Osmond C, Barker DJ. Fetal, infant, and childhood growth are predictors of coronary heart disease, diabetes, and hypertension in adult men and women. Environ Health Perspect. 2000;3(108 Suppl):545–553. doi: 10.1289/ehp.00108s3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podlisny MB, Tolan DR, Selkoe DJ. Homology of the amyloid beta protein precursor in monkey and human supports a primate model for beta amyloidosis in Alzheimer's disease. Am J Pathol. 1991;138:1423–1435. [PMC free article] [PubMed] [Google Scholar]
- Poirier LA, Vlasova TI. The prospective role of abnormal methyl metabolism in cadmium toxicity. Environ Health Perspect. 2002;5(110 Suppl):793–795. doi: 10.1289/ehp.02110s5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollwein P, Masters CL, Beyreuther K. The expression of the amyloid precursor protein (APP) is regulated by two GC-elements in the promoter. Nucleic Acids Res. 1992;20:63–68. doi: 10.1093/nar/20.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DL, Sisodia SS. Cellular and molecular biology of Alzheimer's disease and animal models. Annu Rev Med. 1994;45:435–446. doi: 10.1146/annurev.med.45.1.435. [DOI] [PubMed] [Google Scholar]
- Rice DC. Lead-induced behavioral impairment on a spatial discrimination reversal task in monkeys exposed during different periods of development. Toxicol Appl Pharmacol. 1990;106:327–333. doi: 10.1016/0041-008x(90)90251-o. [DOI] [PubMed] [Google Scholar]
- Rice DC. Effect of lead during different developmental periods in the monkey on concurrent discrimination performance. Neurotoxicology. 1992;13:583–592. [PubMed] [Google Scholar]
- Rogaev EI, Lukiw WJ, Lavrushina O, Rogaeva EA, St George-Hyslop PH. The upstream promoter of the beta-amyloid precursor protein gene (APP) shows differential patterns of methylation in human brain. Genomics. 1994;22:340–347. doi: 10.1006/geno.1994.1393. [DOI] [PubMed] [Google Scholar]
- Schmitz C, Rutten BP, Pielen A, Schafer S, Wirths O, Tremp G, Czech C, Blanchard V, Multhaup G, Rezaie P, Korr H, Steinbusch HW, Pradier L, Bayer TA. Hippocampal neuron loss exceeds amyloid plaque load in a transgenic mouse model of Alzheimer's disease. Am J Pathol. 2004;164:1495–1502. doi: 10.1016/S0002-9440(10)63235-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer O, Marr RA, Rockenstein E, Crews L, Coufal NG, Gage FH, Verma IM, Masliah E. Targeting BACE1 with siRNAs ameliorates Alzheimer disease neuropathology in a transgenic model. Nat Neurosci. 2005;8:1343–1349. doi: 10.1038/nn1531. [DOI] [PubMed] [Google Scholar]
- Smith MA, Perry G. Free radical damage, iron, and Alzheimer's disease. J Neurol Sci. 1995;(134 Suppl):92–94. doi: 10.1016/0022-510x(95)00213-l. [DOI] [PubMed] [Google Scholar]
- Takiguchi M, Achanzar WE, Qu W, Li G, Waalkes MP. Effects of cadmium on DNA-(Cytosine-5) methyltransferase activity and DNA methylation status during cadmium-induced cellular transformation. Exp Cell Res. 2003;286:355–365. doi: 10.1016/s0014-4827(03)00062-4. [DOI] [PubMed] [Google Scholar]
- Tohgi H, Utsugisawa K, Nagane Y, Yoshimura M, Genda Y, Ukitsu M. Reduction with age in methylcytosine in the promoter region −224 approximately −101 of the amyloid precursor protein gene in autopsy human cortex. Brain Res Mol Brain Res. 1999;70:288–292. doi: 10.1016/s0169-328x(99)00163-1. [DOI] [PubMed] [Google Scholar]
- Valinluck V, Tsai HH, Rogstad DK, Burdzy A, Bird A, Sowers LC. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res. 2004;32:4100–4108. doi: 10.1093/nar/gkh739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Erp TG, Saleh PA, Rosso IM, Huttunen M, Lonnqvist J, Pirkola T, Salonen O, Valanne L, Poutanen VP, Standertskjold-Nordenstam CG, Cannon TD. Contributions of genetic risk and fetal hypoxia to hippocampal volume in patients with schizophrenia or schizoaffective disorder, their unaffected siblings, and healthy unrelated volunteers. Am J Psychiatry. 2002;159:1514–1520. doi: 10.1176/appi.ajp.159.9.1514. [DOI] [PubMed] [Google Scholar]
- Villa A, Santiago J, Belandia B, Pascual A. A response unit in the first exon of the beta-amyloid precursor protein gene containing thyroid hormone receptor and Sp1 binding sites mediates negative regulation by 3,5,3'-triiodothyronine. Mol Endocrinol. 2004;18:863–873. doi: 10.1210/me.2003-0260. [DOI] [PubMed] [Google Scholar]
- Zawia NH, Basha MR. Environmental risk factors and the developmental basis for Alzheimer's disease. Rev Neurosci. 2005;16:325–337. doi: 10.1515/revneuro.2005.16.4.325. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data: Figure-1 Cytotoxicity of Aβ1−40 and Aβ1−42 in mice primary neuronal cells: Mice primary cortical neuronal cells were exposed to various concentrations (0.0/0.1/1.0/10.0 μM) of soluble (non-aggregated) Aβ1−40 or Aβ1−42 obtained from commercial source (American peptide company, Sunnyvale, CA). After 48 hours of exposure, cell viability was tested by MTT assay using Promega (Madison, WI) kit. Results revealed that both the peptides were toxic to the cells even at lower concentrations and Aβ1−42 was more cytoxic than Aβ1−40. Data shown were Mean ± S.E. derived from 3−4 independent observations.
Supplementary data: Table 1 Microarray screening of neurobilogy related genes: Microarray array analysis of about 588 neurobiology-related human genes was conducted to identify the genes that are altered due to infantile exposure to Pb in the frontal association cortex of 23-year old cynomolgus monkeys. The results showed that the expression profile of only a few genes (22) was changed due to early life exposure to Pb. Most of the altered genes belonged to neurotransmitter, growth receptors and signal transduction pathways. We further conducted searches on various databases (Ensembl and NCBI nucleotides) to determine if the regulatory regions of these genes were rich in CpG dinucleotides (>60%). We found that most of these altered genes (with the exception of two) were abundant in CpG dinucleotides in their 5’untranslated regions (5’UTR). These findings provide the initial evidence for the association of epigenetic pathway to explain such latent effects; however, these studies require validation by high throughput methylation sequence profiling.