

Abstract
Angiogenesis, the formation of new blood vessels from pre-existing vessels, in the central nervous system (CNS) is seen both as a normal physiological response as well as a pathological step in disease progression. Formation of the blood–brain barrier (BBB) is an essential step in physiological CNS angiogenesis. The BBB is regulated by a neurovascular unit (NVU) consisting of endothelial and perivascular cells as well as vascular astrocytes. The NVU plays a critical role in preventing entry of neurotoxic substances and regulation of blood flow in the CNS. In recent years, research on numerous acquired and hereditary disorders of the CNS has increasingly emphasized the role of angiogenesis in disease pathophysiology. Here, we discuss molecular mechanisms of CNS angiogenesis during embryogenesis as well as various pathological states including brain tumor formation, ischemic stroke, arteriovenous malformations, and neurodegenerative diseases.
Keywords: Central nervous system, Angiogenesis, Brain tumors, Embryogenesis, Ischemic stroke
Introduction
The central nervous system (CNS), consisting of the brain and spinal cord, possesses a highly specialized vasculature to meet the demands of this metabolically highly active tissue as well as to protect sensitive neurons from toxic metabolites and xenobiotics. The tissue-specific functions of CNS blood vessels are regulated by the neurovascular unit (NVU), which is characterized by cross-talk between neural and vascular cells. In the NVU, endothelial cells, perivascular cells (i.e., pericytes or vascular smooth muscle cells), and vascular astrocytes are closely opposed to each other and interact to form the blood–brain barrier (BBB) and provide the brain with a sufficient supply of oxygen and nutrients (Fig. 1). Endothelial cells form the inner lining of all blood vessels. The CNS endothelium differs from non-CNS endothelia in the presence of intercellular tight junctions and BBB-specific enzymes and transport proteins. Pericytes critically regulate BBB formation and CNS capillaries have a higher coverage of pericytes than capillaries of other tissues [1-3]. Astrocyte end-feet almost completely cover CNS blood vessels and regulate BBB function as well [4]. In addition, vascular astrocytes deliver nutrients from the blood to adjacent neurons [5, 6]. Furthermore, vascular astrocytes mediate neuronal regulation of blood flow in blood vessels of the brain [7].
Fig. 1.
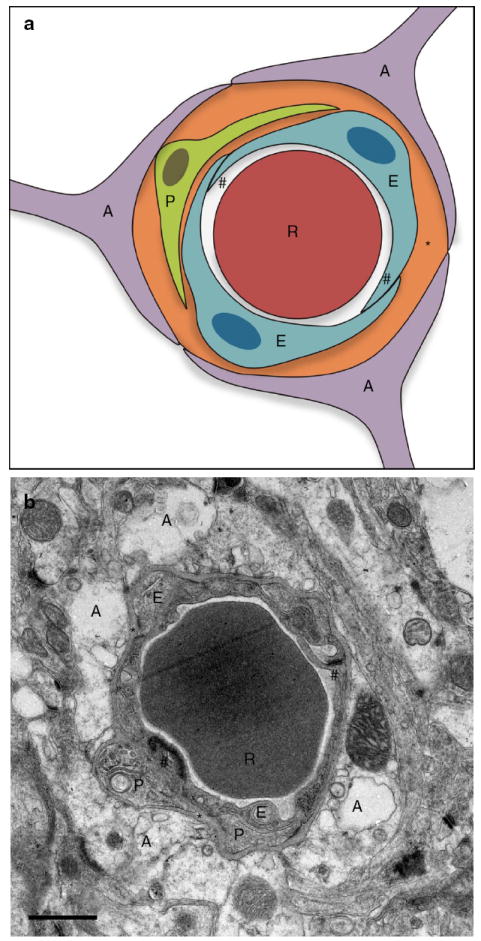
The neurovascular unit. Schematic representation (a) and electron microscopic image (b) of the neurovascular unit in a brain capillary of an adult mouse. A astrocyte end-foot, E endothelial cell, P pericyte, R red blood cell, asterisk basement membrane, hash symbol tight junction. Scale bar: 1 μm
Initial vascularization of embryonic tissues mainly occurs through vasculogenesis, the differentiation of mesoderm-derived angioblasts into blood vessels, whereas at later stages of embryogenesis and in the adult neovascularization mainly occurs through angiogenesis, the sprouting of new blood vessels from pre-existing vessels [8]. The developing CNS is exceptional as its vessels are exclusively formed by angiogenic sprouting of vessels from the perineural vascular plexus [8].
Angiogenesis is regulated by pro- and antiangiogenic factors [9, 10]. These molecules can be released by normal, tumor, endothelial, and stromal cells as well as from leukocytes and the extracellular matrix [11-15]. Proangiogenic factors include vascular endothelial growth factor A (VEGF), fibroblast growth factors, placental growth factor (PlGF), and interleukins, whereas angiostatin, endostatin, and thrombospondins 1 and 2 are putative antiangiogenic factors [13, 15-17]. In addition, certain metalloproteinases degrade extracellular matrix proteins, which can result in both induction and suppression of angiogenesis [12, 13, 15]. As long as expression of pro- and antiangiogenic factors is balanced, the “angiogenic switch” is off. If expression of proangiogenic factors is increased or expression of antiangiogenic factors is decreased, angiogenesis is induced. Induction of CNS angiogenesis contributes to pathological conditions such as brain tumor growth, neurodegenerative diseases, and arteriovenous malformations (AVMs). In contrast, CNS angiogenesis is required for embryonic development as well as recovery from ischemic stroke and brain injury.
This review summarizes our current knowledge of the vasculature of the CNS and the mechanisms underlying CNS angiogenesis during embryonic development and pathological conditions.
The blood–brain barrier
While the NVU is required to induce BBB properties in CNS blood vessels, the physical barrier of the BBB is confined to the endothelial compartment. All endothelia contain intercellular adherens junctions formed by transmembrane proteins of the cadherin, and intracellular proteins of the catenin, families. In addition to adherens junctions, CNS endothelial cells are interconnected by tight junctions similar to those found in most epithelia. Tight junctions, also known as occluding junctions or zonula occludens, are composed of a branching network of sealing strands. These strands are formed by transmembrane proteins of the claudin family and occludin as well as the intracellular ZO proteins (Fig. 2a, b). Tight junctions in the brain endothelium prevent passive diffusion of substances between adjacent cells (paracellular permeability). Only small non-polar molecules <400 Da (e.g., O2, CO2, steroid hormones) can passively diffuse through brain vessels. In addition to tight junctions, brain endothelial cells express specific transporters on the cell surface to transport small molecules such as glucose, amino acids, vitamins, and nucleosides across a concentration gradient from the blood into the brain parenchyma (transcellular permeability, Fig. 2c). Furthermore, CNS endothelial cells express transport proteins to actively transport large molecules such as transferrin, insulin, leptin, and LDL from the blood into the brain. On the other hand, they express transporters such as the multidrug resistance protein P-glycoprotein to actively export cell permeable xenobiotics back into the blood [18]. Brain endothelial cells also constitute a metabolic barrier eliminating chemicals that would otherwise move from the blood into the brain. Cytochrome P450-related enzymes oxidize unwanted substances within the cytoplasm of brain endothelial cells [19]. Likewise, monoamine oxidase (MAO) also contributes to the metabolic barrier to protect the brain from circulating neurotoxins and biogenic amines. In some cases, enzymatic activity within the BBB does not remove unwanted molecules but instead facilitates the transport of essential substances from blood to brain. Through its transpeptidase activity, gamma-glutamyl transpeptidase (GGT) assists in the transfer of amino acids across the BBB [20]. By modulating pyroglutamate levels, GGT indirectly stimulates a wide array of sodium-dependent amino acid transporters at the plasma membrane.
Fig. 2.
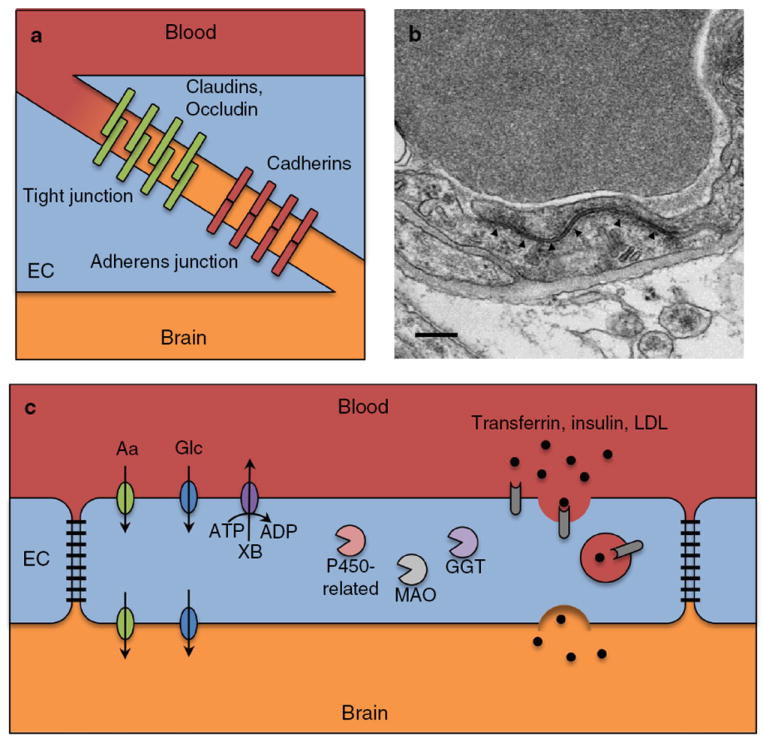
The blood–brain barrier. Schematic representation (a) and electron microscopic image (b) of tight and adherens junctions found in brain endothelium. b High-magnification electron microscopic image showing a tight junction (arrowheads) in a brain capillary of an adult mouse. Scale bar 200 nm. c Schematic representation of the cellular and molecular mechanisms regulating transcellular permeability of the blood–brain barrier. EC endothelial cell, Aa amino acids, Glc glucose, XB xenobiotics, MAO monoamine oxidase, GGT gamma-glutamyl transpeptidase, LDL low-density lipoprotein
CNS blood vessels have a higher coverage of pericytes than blood vessels of other organs, suggesting that they play an important role in BBB formation and maintenance [1]. Indeed, it has been shown that brain pericytes critically regulate endothelial tight junction assembly and BBB formation [2, 3]. Furthermore, vascular astrocytes have been reported to regulate BBB function [4]. Expression of BBB-specific genes is not intrinsic to brain endothelial cells as brain endothelial cell lines gradually lose BBB properties in culture [21]. Stewart and Wiley [22] demonstrated that avascular embryonic quail brain transplanted into chick gut is vascularized by chick peripheral vessels. These vessels acquire BBB features including tight junctions and the ability to restrict molecular diffusion into the brain tissue.
While the BBB protects the CNS from neurotoxic substances, it also prevents entry of most drugs into the brain. The BBB constitutes a major obstacle in the development of novel neurotherapeutics [23]. Several approaches have been developed to efficiently deliver drugs across the BBB, the most promising being drug conjugates that co-opt the transcytosis machinery of the BBB [24].
CNS angiogenesis during embryonic development
Vascularization of the developing CNS is preceded by the formation of the perineural vascular plexus by vasculogenesis. In mice, this process starts at the ventral region of the neural tube between E7.5 and E8.5 [25]. Subsequently, around E9.5, capillary sprouts invade the remaining neuroepithelium by angiogenesis. This is followed by extensive branching and arborization as capillary sprouts migrate from the pial surface to periventricular areas where angiogenic growth factors such VEGF are highly expressed and secreted by cells in the subventricular zone [26, 27]. This process continues through the remainder of embryonic development in all CNS tissues. While VEGF is a major growth factor inducing vasculogenesis and angiogenesis in CNS and non-CNS tissues, numerous other endothelial cell-autonomous and non-cell-autonomous genes regulating CNS angiogenesis during development have been identified (summarized in Fig. 3; Table 1). Endothelium-specific deletions in mice revealed that loci such as Vegfr2, Nrp1/2, Tgfbr1/2, β-catenin (Ctnnb1), and Gpr124 (discussed below) are required to elicit proper CNS angiogenesis. However, genetic studies revealed that the CNS parenchyma also plays a crucial role in CNS angiogenesis as deletion of genes such as Vegfa, Wnt7a/7b, Id1/3, and integrins αv or β8 (Itgav, Itgb8) in the neuroepithelium but not the endothelium results in angiogenic defects and CNS hemorrhage [28-30]. Interestingly, a common phenotype exhibited by numerous loci controlling developmental CNS angiogenesis is the formation of glomeruloid vascular malformations at the interface between the perineural vascular plexus and the neuroepithelium representing impaired angiogenic sprouting [28-36].
Fig. 3.

Endothelial cell-autonomous and non-cell-autonomous genes regulating developmental CNS angiogenesis. Immunofluorescence staining of an E10.5 mouse neural tube showing the two major compartments, neuroepithelium and endothelium, regulating developmental CNS angiogenesis. Neuroepithelial (green) and endothelial (red) cells were visualized by staining for nestin and CD31, respectively
Table 1.
Genes regulating developmental CNS angiogenesis
| Knocked-out gene/s | Compartment | Angiogenesis defects
|
CNS hemorrhage | CNS glomeruloid vascular malformations | References | |
|---|---|---|---|---|---|---|
| Non-CNS | CNS | |||||
| Vegf | Neuroepithelium | − | +++ | − | − | [40] |
| Id1 + Id3 | Global | Xenograft tumorsa, heart | ++ | ++ | ++ | [31, 32] |
| Id1 + Id3 | Endothelium | Heart | − | − | − | [47] |
| Wnt7a + Wnt7b | Embryo proper, neuroepithelium | − | ++ | ++ | ++ | [30] |
| β-catenin | Endothelium | − | +++ | +++ | − | [30] |
| Tgfβ1b+ Tgfβ3 | Global | Yolk sac | ++ | ++ | ++ | [33] |
| Tgfbr1 | Endothelium | Heart | ? | ++ | ? | [61] |
| Tgfbr2 | Endothelium | Heart | ? | ++ | ? | [62] |
| Integrin αv | Neuroepitheliumc | − | + | + | + | [28] |
| Integrin β8 | Neuroepitheliumc | − | + | + | + | [29] |
| Gpr124 | Global, endothelium | − | ++ | ++ | ++ | [34-36] |
Since Id1−/−; Id3−/− mice are embryonic lethal, experiments were performed using Id1+/−; Id3−/− mice, which are viable
The TGFβ1-RGE mutant, which phenocopies the TGFβ1 null allele was used
The conditional knockout in endothelial cells was also investigated but had no phenotype
VEGF signaling
Major triggers of VEGF expression during CNS development are hypoxia and glucose deprival [26, 37]. Cells respond to hypoxia by stabilizing transcription factors of the hypoxia inducible factor (HIF) family [26]. Hypoxia and glucose deprival also result in endoplasmic reticulum stress and activation of the unfolded protein response (UPR) [26, 38]. HIFs and the UPR have been shown to independently induce expression of VEGF [26, 37]. The VEGF family consists of several members including VEGF-A, -B, -C, -D, and PlGF, which bind to the VEGF receptors (VEGFR1-3) and the Neuropilin (Nrp-1 and -2) co-receptors [39]. VEGF signaling through VEGFR2/Nrp-1 is thought to be the main player in CNS angiogenesis [40, 41]. VEGF can be alternatively spliced into several isoforms, with the larger isoforms having large basic domains binding to heparin and proteoglycans found in the extracellular matrix [39]. Previous reports have implicated heparin-binding VEGFs in mediating migration of endothelial cells into avascular regions of the CNS during development [42]. Activation of VEGFR2 in endothelial cells activates several signaling pathways such as the MAP kinase and PI3 kinase/Akt pathways mediating migration, proliferation, and survival [43]. Genetic evidence showing an essential role for VEGF during vascularization of the CNS in mice originates from genetic studies in which Nestin-Cre was crossed into a floxed Vegfa background. In these studies, loss of Vegfa expression in the neuroepithelium leads to failure of CNS angiogenesis due to migratory arrest of endothelial cells at the periphery of the neuroepithelium [40]. This process of VEGF-mediated endothelial migration into the CNS has been further shown to be dependent on the levels of VEGF being produced by the neuroepithelium and is endothelial cell autonomous [41]. The importance of VEGF binding to heparin during CNS angiogenesis has been demonstrated by reduced vascular branching complexity, increased microvessel caliber, and decreased endothelial filopodia in mice lacking the VEGF heparin-binding isoforms VEGF164 and VEGF188, and solely producing VEGF120 [44].
Inhibitor of DNA-binding/differentiation proteins
Inhibitor of DNA-binding/differentiation proteins (Id1-4) comprise a family of proteins that heterodimerize with basic helix-loop-helix (bHLH) transcription factors to inhibit DNA binding of bHLH proteins [45]. Id proteins are key regulators of development where they function to prevent premature differentiation of stem cells [46]. Id1/Id3 double knockout mice are embryonic lethal and exhibit premature neuronal differentiation, defective CNS angiogenesis, forebrain hemorrhage, and impaired cardiac development [31, 32]. Id1 or Id3 single knockout mice do not have a phenotype, indicating functional redundancy of these genes. Interestingly, mice with an endothelial-specific double deletion of Id1/Id3 do not display CNS angiogenesis defects [47], suggesting a non-cell autonomous mechanism.
Wnt signaling
The canonical Wnt signaling pathway has been identified as an essential regulator of developmental CNS angiogenesis and BBB formation [30, 48, 49]. This pathway involves Wnt ligand-driven activation of Frizzled and LRP cell surface co-receptors, with Dishevelled-dependent stabilization of cytoplasmic β-catenin. Subsequent β-catenin translocation into the nucleus and binding to Lef-1/TCF transcription factors then induces transcription of Wnt-responsive genes [50]. In situ hybridization experiments showed that the Wnt ligands Wnt7a and Wnt7b are expressed in neural progenitor cells during CNS angiogenesis [49]. Functionally, Wnt7a/7b are required for developmental angiogenesis in the CNS. Invading vessels utilize paracrine Wnt signaling derived from the neuroepithelium to coordinate proper BBB development because loss-of-function of either Wnt7a/7b in neuroepithelium or of β-catenin/Ctnnb1 in endothelial cells leads to angiogenic defects and subsequent vascular hemorrhage [30, 49]. These mice exhibit angiogenic defects only in the CNS and not in the periphery, consistent with the observation that Wnt signaling is not activated in non-CNS endothelium [28, 45]. Wnt signaling appears to induce not only CNS angiogenesis but also expression of BBB components Glut-1 and Claudin-3 [30, 48, 49]. Either Wnt7a/7b or Cnttb1 deletion results in a lack of Glut-1 and Claudin-3 expression in the BBB. On the other hand, ectopic Wnt7a/7b expression induced Glut-1 expression outside the CNS, whereas addition of Wnt ligands to cultured brain endothelial cells induced both Glut-1 and Claudin-3 protein expression [30, 48, 49]. These findings emphasize the dual functional nature of Wnt-mediated vascular development in the CNS, whereby both angiogenesis and BBB formation are tightly coupled. Whether Wnts also plays a role in the final maturation of the NVU, including driving astrocytic associations with CNS blood vessels, is unclear. While the ligands required for Wnt-driven BBB development have been identified as Wnt7a/7b, the corresponding receptor(s) in CNS endothelial cells have not.
Integrin αvβ8
Mice with a null mutation in the integrin β8 gene (Itgb8) lack functional integrin αvβ8 and die either from failure of normal vasculogenesis in the yolk sac, causing death around E10, or abnormal brain angiogenesis, beginning at E11.5 and characterized by dilated vessels, endothelial cell hyperplasia and hemorrhage [51]. Mice with a null mutation in the integrin αv gene (Itgav) lack integrins αvβ1, αvβ3, αvβ5, αvβ6, and αvβ8 and die either from early defects in the placenta or from the same brain vascular pathology seen in Itgb8-null embryos [52]. Interestingly, similar brain vascular abnormalities occur when integrin β8 or αv is conditionally deleted in neuroepithelial cells but not in endothelial cells [28, 29] indicating a non-cell autonomous vascular defect.
TGFβ signaling
The three isoforms of transforming growth factor β (TGFβ1, -2, and -3) are multifunctional cytokines involved in development, cell differentiation, immune function, and cell cycle control. While they activate the same TGFβ receptor system, the phenotypes of the corresponding knockout mice do not overlap. Tgfb1-null mice die either during embryogenesis or from a lymphocyte-mediated inflammatory disease a few weeks after birth [53, 54]. Tgfb2-null mice die perinatally with defects in several organs, including the heart, skeleton, and genitourinary tract [55] and Tgfb3-null mice display a cleft palate and delayed lung development without embryonic lethality [56, 57]. TGFβ signaling plays a critical role in vascular development and function by regulating endothelial cell proliferation and migration, fibronectin synthesis, and mural cell differentiation [58, 59]. This is supported by the fact that embryonic lethality in some Tgfb1-null mouse embryos is caused by impaired yolk sac vasculogenesis [60]. In addition, conditional deletion of the TGFβ receptors 1 (Alk5) and 2 in endothelial cells results in embryonic lethality and CNS hemorrhage likely caused by defective CNS angiogenesis [61, 62]. Two integrins, αvβ6 and αvβ8, affect TGFβ activity directly by activating latent forms of TGFβ1 and TGFβ3 [63-66]. TGFβ latency occurs because the cytokine remains non-covalently associated with its propeptide, latency-associated peptide (LAP), after proteolytic processing of the pro-cytokine [67]. TGFβ1-LAP and TGFβ3-LAP contain the integrin-binding motif RGD (arginine–glycine–aspartic acid). Integrins αvβ6 and αvβ8 mediate the release of TGFβ1 or -3 from their respective LAPs by interacting with the RGD sequence, which either induces a conformational change in LAP, in the case of αvβ6 [68], or leads to proteolytic cleavage of LAP, in the case of αvβ8 [65]. Mice with a knock-in mutation in Tgfb1 that inactivates the integrin-binding site in LAP (RGD → RGE) have the same developmental defects as TGFβ1-null mice [69]. Thus, RGD-binding integrins play an important role in activating TGFβ1 during development of the yolk sac vasculature. Interestingly, Tgfb1RGE/RGE; Tgfb3−/− double-mutant mice display abnormal CNS angiogenesis accompanied by severe hemorrhage and formation of glomeruloid vascular malformations not seen in single-mutant mice, indicating that Tgfb1 and Tgfb3 have redundant functions in developmental CNS angiogenesis. The similarity between the phenotypes of the Tgfb1/Tgfb3 double-mutant and integrin αv/β8 knockout mice suggests that integrin αvβ8 could act upstream of TGFβ1 and -3 mediating their activation. TGFβ1 and -3 are both expressed in glial cells of the developing CNS [70-72]. Whether they are also expressed in endothelial cells during CNS development is unclear. Integrin αvβ8 is expressed in neuroepithelial but not endothelial cells [28], indicating that the molecular cross-talk in the NVU also regulates developmental CNS angiogenesis. Indeed, integrin αvβ8 expressed on astrocytes surrounding blood vessels has been proposed to act as an “angiogenic switch” in the CNS by activating TGFβ [73].
Gpr124
The orphan adhesion G protein-coupled receptor Gpr124/tumor endothelial marker 5 (TEM5) is mainly expressed in endothelial cells and pericytes of most vascular beds during embryogenesis and in CNS endothelial cells as well as pericytes of multiple organs in the adult [34]. Gpr124-null mice are embryonic lethal because of impaired angiogenic sprouting from the perineural vascular plexus into the forebrain and ventral neural tube accompanied by severe hemorrhage [34-36]. In these embryos, the telencephalon and ventral neural tube are essentially avascular. Only the typical glomeruloid vascular malformations are found at the periphery of the neuroepithelium. Furthermore, the defective Gpr124-null vessels fail to express the BBB marker Glut-1. On the other hand, compensatory upregulation of Glut-1 in the neuroepithelium occurs [34, 36]. Conversely, transgenic mice overexpressing Gpr124 under the endothelial-specific Tie2 promoter display increased vascular density and hyperplasia in the CNS [34]. Endothelial-specific knockout of Gpr124 completely phenocopies the global knockout indicating that Gpr124 functions endothelial cell-autonomously [34, 36]. In vitro migration assays showed enhanced migration of Gpr124-overexpressing brain endothelial cell lines towards primary forebrain cell conditioned medium or fetal bovine serum, indicating that Gpr124 regulates migration of brain endothelial cells [34, 36].
The striking similarity between the phenotypes of Gpr124 single-knockout and Wnt7a/7b double-knockout mice suggests an intersection between Gpr124 and Wnt signaling. However, using β-catenin reporter mice (TOP-Gal) we have shown that β-catenin activation, i.e., canonical Wnt signaling, is not impaired in Gpr124-null mice [34]. In addition, Gpr124 expression is not altered in β-catenin-null mice [34]. Whether Gpr124 regulates CNS angiogenesis by activation of G proteins remains to be determined. Anderson et al. performed microarray analysis of laser-microdissected forebrain blood vessels from wild-type and Gpr124-knockout embryos and found that 12 out of 27 TGFβ target genes were upregulated in the knockout embryos [35]. Based on these results, the authors conclude that Gpr124 signaling negatively regulates TGFβ signaling.
Brain tumor angiogenesis
Angiogenesis is required for growth and metastasis of all solid tumors. While primary brain tumors are rare (<2 % of all tumors diagnosed each year), their location and other physiological characteristics lead to a very poor prognosis [74]. In contrast to primary brain tumors, secondary brain tumors metastasize from other cancers, such as breast, lung, and colon carcinoma as well as melanoma. Glioblastoma multiforme (GBM) is the most common brain tumor accounting for about 40 % of all primary brain tumors and 70 % of all malignant gliomas [75, 76]. GBM is among the most vascularized and deadly tumors with a 5-year survival rate of 5 % [77]. When primary brain tumors or metastases grow beyond 1–2 mm in diameter within the brain parenchyma, angiogenesis is induced and the BBB becomes compromised structurally and functionally [78-85]. Typically, blood vessels within brain tumors are tortuous, disorganized, highly permeable, and characterized by abnormalities in their endothelial wall, pericyte coverage, and basement membrane [79-81, 86-89]. The vascular permeability of different xenograft tumors growing intracranially in mice is generally elevated [81, 90]. Some brain tumor vessels have pores in their walls as large as 550 nm in diameter [80]. Interestingly, some features of the BBB are retained in brain tumors. For example, when a tumor grows in the brain, it exhibits significantly reduced vascular permeability compared with the same tumor grown subcutaneously [79, 80]. Furthermore, although brain metastases from breast carcinomas are considerably more angiogenic than their orthotopic counterparts, the resulting tumor vessels are less permeable in the brain [91].
VEGF is a major permeability and proangiogenic factor that is highly expressed in brain tumors and is partly responsible for the loss of the BBB during tumor growth (Fig. 4). Hypoxia and acidosis have been shown to independently regulate VEGF transcription in brain tumors [82]. VEGF expression can also be regulated by multiple oncogenes and tumor-suppressor genes (such as Ras, Src, and p53), hormones, cytokines, the UPR, and various signaling molecules (including nitric oxide and mitogen-activated protein kinases) [14, 37, 38, 92-94]. Finally, VEGF can also be released from other cells and the extracellular matrix [11, 14]. A direct consequence of VEGF-induced vascular permeability is increased interstitial fluid pressure (IFP). This is critical in brain tumors, since increased edema and fluid pressure can cause severe complications. In brain tumors in both rodents and patients, the IFP increases with tumor size and in mice it is higher than the cerebrospinal fluid (CSF) pressure [95]. Because of the raised IFP, interstitial fluid leaks from the tumors into the surrounding tissue, raising the CSF pressure until, ultimately, it becomes equal to the tumor IFP. In addition to the co-morbidity associated with edema, high IFP forms a barrier to drug delivery (Fig. 4) [96].
Fig. 4.
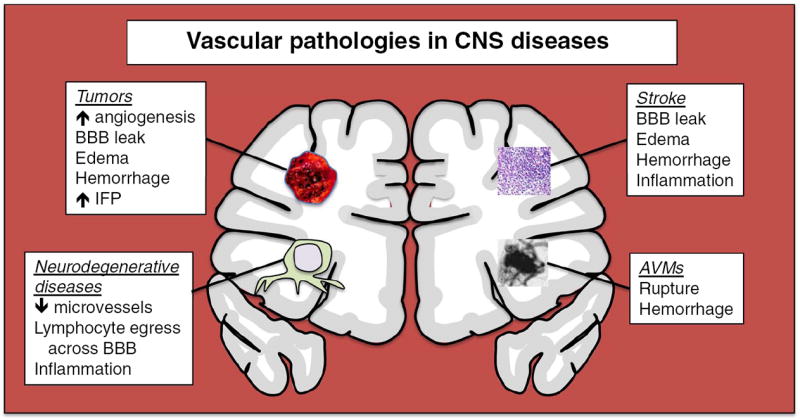
Vascular pathologies in CNS diseases. Tumors, neurodegenerative diseases, stroke, and arterio-venous malformations are major classes of CNS disorders that significantly alter the brain vasculature, with accompanying pathophysiologic consequences
Interleukin-8 is a chemokine with proangiogenic activity. High expression levels of interleukin-8 and hepatocyte growth factor have been detected in primary and recurrent glial tumors [97, 98]. Furthermore, expression of the chemokine CXCL12 and its receptor is induced in brain tumors and promotes angiogenesis [99]. Transcription profiles of gliomas from patients have shown expression of many proangiogenic factors including insulin-like growth factor 1 [100]. Stem cell factor and its receptor c-Kit have also been shown to play important roles in brain tumor angiogenesis [101]. Wnt signaling plays a crucial role in developmental CNS angiogenesis [30]. In contrast, Wnt has been shown to inhibit angiogenesis in brain tumors in the adult by inducing PDGFB expression and vessel normalization [102].
Inhibition of brain tumor angiogenesis represents a promising approach in the treatment of brain tumors and metastases. In 2009 the US Food and Drug Administration approved the VEGF-neutralizing antibody bevacizumab for the treatment of recurring GBM. A major mechanism by which bevacizumab improves the outcome of GBM is by reducing vascular permeability and edema, which are the major causes of morbidity and mortality associated with GBM [103]. The Notch signaling pathway is activated downstream of VEGF and mediates negative feedback and fine-tuning of VEGF signaling [104, 105]. Accordingly, it has been reported that inhibition of the Notch ligand Dll4 leads to non-productive angiogenesis and suppression of tumor growth in a glioma model [106]. However, prolonged treatment of mice with a Dll4-blocking antibody results in formation of vascular/endothelial cell-based tumors resembling hemangioblastoma [107].
CNS angiogenesis after brain injury: stroke and TBI
The CNS vascular system in adults is extremely stable under physiological conditions. However, this system can be disrupted both structurally and functionally under acute pathological conditions such as ischemic stroke and traumatic brain injury (TBI). Numerous studies have demonstrated that angiogenesis actively occurs after ischemic stroke and TBI and contributes to functional outcome during recovery. The underlying mechanisms of this improved functional recovery by angiogenesis include not only increased oxygen and nutrient supply to injured tissue, but also involve neurogenesis and synaptogenesis [108]. Cerebral vessels are important for neuroblast migration to the ischemic boundary zone [109], where angiogenesis is actively induced, and provide neurotrophic support to newly generated neurons [110, 111]. Upon injury, activated endothelial cells secrete stromal cell-derived factor 1α (SDF-1α), which attracts CXCR4-positive neuroblasts to migrate along with cerebral vessels to the injured region [112, 113].
A variety of cellular and molecular responses in brain take place after ischemic stroke and TBI, including excitotoxicity, reactive free radical generation, inflammation, ischemia/reperfusion injury, and result in neuronal death (Fig. 4). Since ischemia plays an important role in both stroke and TBI, this review will focus on cerebral ischemiainduced vascular injury and angiogenesis.
Acute phase: vascular injury and BBB breakdown
An ischemic cascade comprised of a series of biochemical events is rapidly initiated within minutes after occlusion of a cerebral vessel, leading to excitotoxicity and oxidative stress [114]. A variety of pro-inflammatory cytokines and proteolytic enzymes that are responsible for remodeling the extracellular matrix are subsequently upregulated, including TNF-α, IL-1β, IL-6, and matrix metalloproteinases (MMPs). These pro-inflammatory cytokines and MMPs all contribute to microvascular injury and BBB dysfunction [115].
Endothelial activation and expression of adhesion molecules is important for adhesion and infiltration of leukocytes [116], which then amplify the inflammatory cascade in the infarcted region and increase brain damage [117, 118]. Intercellular adhesions molecule-1 (ICAM-1) expression increases within hours after ischemia onset in humans [119] and inhibition of ICAM-1 reduces ischemic damage in rat [120]. E- and P-selectins are upregulated and mediate leukocyte invasion into brain during the early stages of ischemia [121, 122]. Moreover, soluble ICAM-1 levels are higher in patients with acute ischemic stroke compared to controls and are associated with poor short-term prognosis [123].
Numerous animal studies have shown that ischemic injury induces a molecular cascade in the NVU, leading to disruption of the BBB and vasogenic edema/hemorrhage [124]. Degradation of tight junction proteins, junctional adhesion molecules in the endothelial cell clefts, and basal lamina occurs a few hours after the onset of ischemia. Furthermore, there is a delayed secondary opening of the BBB occurring after 24–72 h due to a neuroinflammatory response. Experimental studies have demonstrated that expression and activation of MMPs accounts for the opening of BBB [124, 125]. MMPs in normal adult brain are either in a latent form (MMP-2) or at very low to undetectable expression levels (MMP-3 and -9). However, these MMPs can be rapidly activated or upregulated in cells of the NVU or leukocytes and released to act on multiple substrates in the extracellular space [126, 127]. In rodent stroke models, MMP-9 expression is upregulated a few hours after ischemia. Deletion of Mmp9 preserves tight junction protein levels and reduces infarct volume [128]. In humans, plasma MMP-9 but not MMP-2 level is increased and correlated to infarct volume and final functional outcome in cardioembolic stroke patients [129]. Concomitantly, Mmp2 knockout does not confer any protective effects against acute neural damage in mice, confounding the role of MMP-2 in BBB breakdown after stroke [130]. Cyclooxygenase-2 (Cox-2) is also involved in BBB opening but mainly in the second phase as part of a secondary inflammatory response [131].
Chronic phase: neovascularization by angiogenesis and vasculogenesis
Formation of new blood vessels plays an important role in the restoration of oxygen/nutrients and recovery phase of damaged neuronal tissue after ischemia, especially in the much larger volume of brain tissue surrounding the ischemic core, or penumbra. Intense and active neovascularization has been found in this area and is coupled with neurogenesis.
In rodent stroke models, endothelial cells in the peri-infarct brain tissue start to proliferate as early as 12–24 h following ischemic stroke and can continue proliferating for at least 21 days [132-134]. Accordingly, vessel density significantly increases in the peri-infarct region 3 days after ischemia [133, 134]. This angiogenic process occurs robustly and in a long-term way because vessel numbers in the ischemic area continue increasing even 21 days after ischemic injury [134, 135]. Importantly, studies on postmortem brain samples from patients with stroke demonstrated that cerebral ischemia induces active angiogenesis in the penumbra, which may contribute to longer survival of stroke patients [136].
New blood vessel formation is an intricately regulated and step-wise process. Important steps include degradation of surrounding matrix, proliferation and migration of endothelial cells, recruitment of pericytes, and stabilization and cessation of newly formed vessels [137]. As a result, an enormous variety of molecules including pro-angiogenic factors and molecules that are involved in other steps of angiogenesis are promptly induced after the onset of cerebral ischemia. A study by Hayashi et al. [134] analyzed the temporal expression profile of 96 angiogenesis-related genes in a mouse transient cerebral ischemia model. They found that 42, 29, and 13 angiogenesis-related genes are increased at 1 h, 1 day, and 21 days after ischemia, respectively. Moreover, some genes may demonstrate early or delayed induction or biphasic expression pattern, depending on their specific roles in angiogenesis. For example, VEGF, the most important mitogen for angiogenesis [39], and its receptor Flk1/VEGFR2 are rapidly upregulated at 1 h and reach the peak levels at 3 h after ischemia. Angiopoietin-1, which is involved in blood vessel maturation and stabilization [138], shows a delayed upregulation starting at 1 day and culminating at 7 days after stroke. On the other hand, vessel-stabilizing thrombospondins are rapidly induced at 1 h after ischemia but reduced at 1 day, and then increase again 3 days after stroke, indicating a shift from vascular protection to vascular remodeling. There have been many studies investigating the expression profile of different angiogenesis-implicated genes and their specific roles in post-ischemia angiogenesis (summarized in Table 2).
Table 2.
Molecules involved in angiogenesis post-brain injury
| Name | Role in angiogenesis | Expression level and induction time after ischemia | Cellular localization | References |
|---|---|---|---|---|
| VEGF | EC proliferation/migration; vascular permeability | Up; 1 h to 7 days | ECs; astrocytes; neurons; microglia/macrophages | [133, 134, 192-200] |
| VEGFR-1 (Flt-1) | Receptor of VEGF | Up; 48 h to 14 days | ECs | [133, 192, 197] |
| VEGFR-2 (Flk-1) | Receptor of VEGF | Up; 48 h to 7 days | ECs; neurons; microglia | [133, 195, 198] |
| PLGF | Ligand of VEGFR1; potentiating VEGF | Up; 3 days | Vessels | [201] |
| Neuropilins | Co-receptor of VEGF/PLGF | Up; 24 h to 28 days | ECs | [201, 202] |
| Angiopoietin-1/2 | Ligands of Tie2; vascular maturation and stabilization | Up; 1 h to 28 days | ECs; neurons | [132, 134, 135, 203, 204] |
| Tie1/2 | Vascular maturation and stabilization | Up 0 to 1 h→down→Up 3–14 days | ECs | [203, 205, 206] |
| PDGF-B | Pericyte recruitment; vascular stabilization | Up; hours to 7 days | ECs; neurons; macrophages | [207-209] |
| PDGFRβ | Receptor of PDGF-B | Up; 48 h to 14 days | Pericytes; neurons | [207-209] |
| Erythropoietin | EC proliferation | Up; 1–7 days | ECs; astrocytes; microglia | [210] |
| eNOS | EC proliferation/migration; vascular tone | Up; 1–7 days | ECs | [211] |
| TGFβ | Cellular migration; extracellular deposition | Up 12 h→down up 7 days | Vessels; astrocytes; | [212-215] |
| FGFs | EC growth | Up; 1–14 days | ECs; neurons; astrocytes; macrophages | [216-219] |
| Thrombospondins | Vessel stabilization | Up 1 h→down 1 day→up 3 days | ECs; neurons | [134] |
In addition to angiogenesis, the outgrowth of pre-existing vessels, formation of vessels by circulating bone marrow-derived endothelial progenitor cells (EPCs), or vasculogenesis [139-141] may also contribute to neovascularization after cerebral ischemia. Animal studies demonstrated that these circulating EPCs could home to infarcted brain tissues, differentiate into ECs in situ, and incorporate into the vasculature [142-144].
Angiogenesis in neurodegenerative diseases
Capillary loss and endothelial activation
Previous studies have shown that cerebral microvascular pathology precedes and accompanies age-related cognitive dysfunction and neurodegeneration. Capillary density decline has been found in aging, Alzheimer’s disease (AD), and leukoaraiosis (LA) [145]. Cerebral hypoperfusion triggers cognitive and degenerative changes in the brain and contributes to the pathologic process of AD [146]. A study on atherosclerosis revealed that severe circle of Willis atherosclerosis is associated with sporadic AD [147]. Mechanistically, impaired induction of HIF-1 and VEGF by hypoxia is proposed to be a major mechanism underlying capillary loss during aging [148-152]. Moreover, HIF-1 reduction is associated with neuronal loss [153]. In AD, amyloid β (Aβ) also plays an important role in the reduced capillary density by exerting both direct and indirect effects. Firstly, Aβ accumulates on capillaries and inhibits angiogenesis via anti-angiogenic activity [154, 155]. Secondly, it can bind to VEGF and co-deposit in plaques, resulting in decreased availability of VEGF [156], which is even upregulated in AD brains presumably due to hypoxia [157].
Decreased vascularity in the brain leads to hypoxia, which in turn stimulates the upregulation of proangiogenic factors and endothelial activation [158]. In a transgenic mouse model of AD, endothelial cell activation occurs in an age-dependent manner and is associated with Aβ deposition [159]. Activated endothelial cells elaborate a number of proteases, inflammatory factors, and other products with biologic activity that may promote neuronal death (Fig. 4) [158].
BBB disruption
Disruption of the BBB exists in many neurological disorders, including AD, multiple sclerosis (MS), and amyotrophic lateral sclerosis (ALS) [160]. In a mouse AD model, compromised BBB occurs long before other disease pathology occurs, such as consolidated amyloid plaques [161]. BBB disruption is also found in AD patients manifested by magnetic resonance imaging (MRI) [162]. The exact cause of BBB dysfunction in AD remains unclear but accumulation of Aβ and neuroinflammation have been suggested to play a role [163-165]. Interestingly, Aβ immunization restores the disrupted BBB in a mouse AD model, demonstrating that Aβ may directly affect the BBB [166]. Similar to AD, disruption of BBB is observed in mouse ALS models before motor neuron degeneration and the neurovas-cular inflammatory response occurs [167]. MS is an autoimmune disease seen in young adults where the immune system erroneously attacks oligodendrocytes forming the insulating myelin sheath of axons. Disruption of BBB is an early event in MS, evidenced by in vivo gadolinium uptake on MRI and post-mortem evidence of focal microvascular leakage. While the exact etiology of MS remains unknown, it is generally believed that leukocyte activation and transendothelial migration play an important role in initiation of CNS inflammation in MS [168]. Treatment of MS with the humanized monoclonal antibody natalizumab targeting the α4-chain of α4β1 integrin, also known as very late antigen-4 (VLA-4), expressed on circulating immune cells, inhibits leukocyte adhesion and extravasation into the CNS, effectively reducing CNS inflammation and secondary BBB breakdown [169]. The presence and mechanisms of BBB disruption in other neurological diseases, such as epilepsy and neuromyelitis optica, have been recently reviewed by Obermeier and colleagues [115].
Arteriovenous malformations in the brain
AVMs are a collection of abnormally clustered blood vessels resulting from shunting from arterial to venous circulation. AVMs in the brain have a high risk of rupture with subsequent intracerebral and subarachnoid hemorrhage due to the fragility of these improperly formed vessels in the absence of an intervening capillary bed (Fig. 4). Most AVMs are sporadic, but the genetic component of AVM pathogenesis has been studied in hereditary hemorrhagic telangiectasia (HHT, Osler-Weber-Rendu disease), an autosomal dominant disorder presenting with high frequency of AVMs with hemorrhage from superficial AVMs in the nose and GI tract, and larger AVMs in major organs including liver, brain, and lung [170]. Cerebral AVMs and pulmonary AVMs occur in 15–20 % of HHT cases [170]. HHT is the most frequent cause of hemorrhagic stroke in young adults [171, 172].
The TGFβ superfamily plays a critical role in vascular development [173]. TGFβ is necessary for formation and remodeling of the primary vascular plexus through regulation of endothelial cell proliferation, migration, and differentiation. It also plays a role in recruiting pericytes and vascular smooth muscle cells to newly formed blood vessels. HHT is frequently associated with mutations in the SMAD4, endoglin, and ALK1 (ACVRL1) genes of the TGFβ pathway [174-180]. Heterozygous mutations in endoglin lead to HHT type 1 [170]. Endoglin encodes a homodimeric integral membrane glycoprotein that exhibits high expression on vascular endothelial cells [181, 182]. Endoglin has been demonstrated to interact with the ligand-binding receptors of multiple members of the TGFβ superfamily, including activin and bone morphogenic proteins (BMPs) [183]. Heterozygous mutations in ALK1, an activin-like kinase receptor type I, lead to HHT type 2, which has a later disease onset and reduced penetrance in comparison to HHT1 [184, 185]. Patients with mutations in the SMAD4 gene and juvenile polyposis may also develop HHT [186]. Environmental causes may also play a role in AVM development in HHT patients, which is supported by a study showing that excisional skin wounding induces AVMs in Alk1-deficient mice [187].
Targeted deletion of TGFβ signaling pathway components Alk1, Tgfrb1 (Alk5), Tgfbr2, and endoglin in mice results in AVMs similar to HHT [173]. The Alk1 knockout phenotype has been elucidated in both the zebrafish and the mouse model and both demonstrate dilation of cranial vessels with an abnormal direct connection to veins [188, 189]. Additionally, Alk1 deletion decreases expression of the arterial marker ephrin-B2 [189]. Mice with endoglin deletion develop AVMs and fail to confine intraembryonic hematopoiesis to arteries, without demonstrating a phenotype of vessel dilation or downregulation of arterial marker ephrin-B2, suggesting a role for endoglin as an accessory coreceptor modulating Alk1 signaling [189]. Endothelial cell-specific inducible deletion of endoglin results in an abnormal increase in endothelial cell proliferation observed in veins of adult skin and all vessels of neonatal retina [190]. Expression of molecular regulators of arterial and venous identity, including Jagged-1, ephrin-B2, apelin receptor (Aplnr), and EphB4, is preserved in the vasculature of the retina in endoglin mutants, arguing against loss of arterial and venous molecular identity as the primary cause of AVM formation in HHT pathogenesis [190]. However, endoglin deletion in mouse embryos increases arterial expression of chicken ovalbumin upstream promoter (COUP) transcription factor II, a venous-specific marker [191].
Conclusions
CNS capillaries differ from the vasculature of other organs through the presence of the BBB, which is critically regulated by a NVU containing endothelial cells, pericytes, and astrocytes. The balance of pro- and antian-giogenic factors maintaining vascular homeostasis in the CNS may be disrupted in pathological conditions and acute vascular injury. Insult to the BBB can occur as a result of ischemia, leading to the upregulation of proinflammatory cytokines, metalloproteinases, and other proteolytic enzymes that remodel the extracellular matrix. The importance of proper regulation of angiogenesis and BBB integrity is attested to by both the pathophysiology of disorders as diverse as brain AVMs, CNS neoplasms, stroke and MS, as well as the therapeutic efficacy of VEGF inhibitors such as bevacizumab for brain tumors and leukocyte adhesion inhibitors such as natalizumab for MS. Although numerous mouse and human genetic models have identified crucial regulatory molecules governing CNS angiogenesis, the further inquiry into the regulation of vascularization and cerebrovascular integrity during physiological and pathological states is critical to the future development of pharmacologic therapies in a broad variety of neurological disorders.
Acknowledgments
We thank Dr. Jiyong Zhang of Brown University for his advice and help in preparing the figures. H. Z. was supported by a HHMI Medical Student Research Fellowship. This review was supported by funding from the NIH (1R01NS064517) and American Heart Association (12PILT12850014) to C. J. K.
Contributor Information
Mario Vallon, Email: mvallon@stanford.edu, Division of Hematology, Stanford University School of Medicine, 269 Campus Drive, CCSR 3100, Stanford, CA 94305, USA.
Junlei Chang, Division of Hematology, Stanford University School of Medicine, 269 Campus Drive, CCSR 3100, Stanford, CA 94305, USA.
Haijing Zhang, Division of Hematology, Stanford University School of Medicine, 269 Campus Drive, CCSR 3100, Stanford, CA 94305, USA.
Calvin J. Kuo, Email: cjkuo@stanford.edu, Division of Hematology, Stanford University School of Medicine, 269 Campus Drive, CCSR 3100, Stanford, CA 94305, USA.
References
- 1.Shepro D, Morel NM. Pericyte physiology. FASEB J. 1993;7:1031–1038. doi: 10.1096/fasebj.7.11.8370472. [DOI] [PubMed] [Google Scholar]
- 2.Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, Johansson BR, Betsholtz C. Pericytes regulate the blood–brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- 3.Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature. 2010;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood–brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 5.Figley CR, Stroman PW. The role(s) of astrocytes and astrocyte activity in neurometabolism, neurovascular coupling, and the production of functional neuroimaging signals. Eur J Neurosci. 2011;33:577–588. doi: 10.1111/j.1460-9568.2010.07584.x. [DOI] [PubMed] [Google Scholar]
- 6.Figley CR. Lactate transport and metabolism in the human brain: implications for the astrocyte-neuron lactate shuttle hypothesis. J Neurosci. 2011;31:4768–4770. doi: 10.1523/JNEUROSCI.6612-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, Carmignoto G. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003;6:43–50. doi: 10.1038/nn980. [DOI] [PubMed] [Google Scholar]
- 8.Flamme I, Frolich T, Risau W. Molecular mechanisms of vasculogenesis and embryonic angiogenesis. J Cell Physiol. 1997;173:206–210. doi: 10.1002/(SICI)1097-4652(199711)173:2<206:AID-JCP22>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 9.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 10.Folkman J. Anti-angiogenesis: new concept for therapy of solid tumors. Ann Surg. 1972;175:409–416. doi: 10.1097/00000658-197203000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fukumura D, Xavier R, Sugiura T, Chen Y, Park EC, Lu N, Selig M, Nielsen G, Taksir T, Jain RK, Seed B. Tumor induction of VEGF promoter activity in stromal cells. Cell. 1998;94:715–725. doi: 10.1016/s0092-8674(00)81731-6. [DOI] [PubMed] [Google Scholar]
- 12.Fang J, Shing Y, Wiederschain D, Yan L, Butterfield C, Jackson G, Harper J, Tamvakopoulos G, Moses MA. Matrix metalloproteinase-2 is required for the switch to the angiogenic phenotype in a tumor model. Proc Natl Acad Sci USA. 2000;97:3884–3889. doi: 10.1073/pnas.97.8.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Folkman J. Antiangiogenesis in cancer therapy—endostatin and its mechanisms of action. Exp Cell Res. 2006;312:594–607. doi: 10.1016/j.yexcr.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 14.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 15.O’Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, Moses M, Lane WS, Cao Y, Sage EH, Folkman J. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. 1994;79:315–328. doi: 10.1016/0092-8674(94)90200-3. [DOI] [PubMed] [Google Scholar]
- 16.Dameron KM, Volpert OV, Tainsky MA, Bouck N. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science. 1994;265:1582–1584. doi: 10.1126/science.7521539. [DOI] [PubMed] [Google Scholar]
- 17.Good DJ, Polverini PJ, Rastinejad F, Le Beau MM, Lemons RS, Frazier WA, Bouck NP. A tumor suppressor-dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc Natl Acad Sci USA. 1990;87:6624–6628. doi: 10.1073/pnas.87.17.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alavijeh MS, Chishty M, Qaiser MZ, Palmer AM. Drug metabolism and pharmacokinetics, the blood–brain barrier, and central nervous system drug discovery. NeuroRx. 2005;2:554–571. doi: 10.1602/neurorx.2.4.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hawkins RA, O’Kane RL, Simpson IA, Vina JR. Structure of the blood–brain barrier and its role in the transport of amino acids. J Nutr. 2006;136:218S–226S. doi: 10.1093/jn/136.1.218S. [DOI] [PubMed] [Google Scholar]
- 21.Schrade A, Sade H, Couraud PO, Romero IA, Weksler BB, Niewoehner J. Expression and localization of claudins-3 and -12 in transformed human brain endothelium. Fluids Barriers CNS. 2012;9:6. doi: 10.1186/2045-8118-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stewart PA, Wiley MJ. Developing nervous tissue induces formation of blood–brain barrier characteristics in invading endothelial cells: a study using quail–chick transplantation chimeras. Dev Biol. 1981;84:183–192. doi: 10.1016/0012-1606(81)90382-1. [DOI] [PubMed] [Google Scholar]
- 23.Pardridge WM. The blood–brain barrier: bottleneck in brain drug development. NeuroRx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gabathuler R. Approaches to transport therapeutic drugs across the blood–brain barrier to treat brain diseases. Neurobiol Dis. 2010;37:48–57. doi: 10.1016/j.nbd.2009.07.028. [DOI] [PubMed] [Google Scholar]
- 25.Hogan KA, Ambler CA, Chapman DL, Bautch VL. The neural tube patterns vessels developmentally using the VEGF signaling pathway. Development. 2004;131:1503–1513. doi: 10.1242/dev.01039. [DOI] [PubMed] [Google Scholar]
- 26.Breier G, Risau W. The role of vascular endothelial growth factor in blood vessel formation. Trends Cell Biol. 1996;6:454–456. doi: 10.1016/0962-8924(96)84935-x. [DOI] [PubMed] [Google Scholar]
- 27.Breier G, Albrecht U, Sterrer S, Risau W. Expression of vascular endothelial growth factor during embryonic angiogenesis and endothelial cell differentiation. Development. 1992;114:521–532. doi: 10.1242/dev.114.2.521. [DOI] [PubMed] [Google Scholar]
- 28.McCarty JH, Lacy-Hulbert A, Charest A, Bronson RT, Crowley D, Housman D, Savill J, Roes J, Hynes RO. Selective ablation of alphav integrins in the central nervous system leads to cerebral hemorrhage, seizures, axonal degeneration and premature death. Development. 2005;132:165–176. doi: 10.1242/dev.01551. [DOI] [PubMed] [Google Scholar]
- 29.Proctor JM, Zang K, Wang D, Wang R, Reichardt LF. Vascular development of the brain requires beta8 integrin expression in the neuroepithelium. J Neurosci. 2005;25:9940–9948. doi: 10.1523/JNEUROSCI.3467-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stenman JM, Rajagopal J, Carroll TJ, Ishibashi M, McMahon J, McMahon AP. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science. 2008;322:1247–1250. doi: 10.1126/science.1164594. [DOI] [PubMed] [Google Scholar]
- 31.Lyden D, Young AZ, Zagzag D, Yan W, Gerald W, O’Reilly R, Bader BL, Hynes RO, Zhuang Y, Manova K, Benezra R. Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature. 1999;401:670–677. doi: 10.1038/44334. [DOI] [PubMed] [Google Scholar]
- 32.Fraidenraich D, Stillwell E, Romero E, Wilkes D, Manova K, Basson CT, Benezra R. Rescue of cardiac defects in id knockout embryos by injection of embryonic stem cells. Science. 2004;306:247–252. doi: 10.1126/science.1102612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mu Z, Yang Z, Yu D, Zhao Z, Munger JS. TGFbeta1 and TGFbeta3 are partially redundant effectors in brain vascular morphogenesis. Mech Dev. 2008;125:508–516. doi: 10.1016/j.mod.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 34.Kuhnert F, Mancuso MR, Shamloo A, Wang HT, Choksi V, Florek M, Su H, Fruttiger M, Young WL, Heilshorn SC, Kuo CJ. Essential regulation of CNS angiogenesis by the orphan G protein-coupled receptor GPR124. Science. 2010;330:985–989. doi: 10.1126/science.1196554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson KD, Pan L, Yang XM, Hughes VC, Walls JR, Dominguez MG, Simmons MV, Burfeind P, Xue Y, Wei Y, Macdonald LE, Thurston G, Daly C, Lin HC, Economides AN, Valenzuela DM, Murphy AJ, Yancopoulos GD, Gale NW. Angiogenic sprouting into neural tissue requires Gpr124, an orphan G protein-coupled receptor. Proc Natl Acad Sci USA. 2011;108:2807–2812. doi: 10.1073/pnas.1019761108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cullen M, Elzarrad MK, Seaman S, Zudaire E, Stevens J, Yang MY, Li X, Chaudhary A, Xu L, Hilton MB, Logsdon D, Hsiao E, Stein EV, Cuttitta F, Haines DC, Nagashima K, Tessarollo L, St Croix B. GPR124, an orphan G protein-coupled receptor, is required for CNS-specific vascularization and establishment of the blood–brain barrier. Proc Natl Acad Sci USA. 2011;108:5759–5764. doi: 10.1073/pnas.1017192108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghosh R, Lipson KL, Sargent KE, Mercurio AM, Hunt JS, Ron D, Urano F. Transcriptional regulation of VEGF-A by the unfolded protein response pathway. PLoS One. 2010;5:e9575. doi: 10.1371/journal.pone.0009575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Alam GN, Ning Y, Visioli F, Dong Z, Nor JE, Polverini PJ. The unfolded protein response induces the angiogenic switch in human tumor cells through the PERK/ATF4 pathway. Cancer Res. 2012;72:5396–5406. doi: 10.1158/0008-5472.CAN-12-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 40.Raab S, Beck H, Gaumann A, Yuce A, Gerber HP, Plate K, Hammes HP, Ferrara N, Breier G. Impaired brain angiogenesis and neuronal apoptosis induced by conditional homozygous inactivation of vascular endothelial growth factor. Thromb Haemost. 2004;91:595–605. doi: 10.1160/TH03-09-0582. [DOI] [PubMed] [Google Scholar]
- 41.Haigh JJ, Morelli PI, Gerhardt H, Haigh K, Tsien J, Damert A, Miquerol L, Muhlner U, Klein R, Ferrara N, Wagner EF, Betsholtz C, Nagy A. Cortical and retinal defects caused by dosage-dependent reductions in VEGF-A paracrine signaling. Dev Biol. 2003;262:225–241. doi: 10.1016/s0012-1606(03)00356-7. [DOI] [PubMed] [Google Scholar]
- 42.Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C, Alitalo K, Shima D, Betsholtz C. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. 2003;161:1163–1177. doi: 10.1083/jcb.200302047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling—in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 44.Ruhrberg C, Gerhardt H, Golding M, Watson R, Ioannidou S, Fujisawa H, Betsholtz C, Shima DT. Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev. 2002;16:2684–2698. doi: 10.1101/gad.242002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perk J, Iavarone A, Benezra R. Id family of helix-loop-helix proteins in cancer. Nat Rev Cancer. 2005;5:603–614. doi: 10.1038/nrc1673. [DOI] [PubMed] [Google Scholar]
- 46.Yokota Y. Id and development. Oncogene. 2001;20:8290–8298. doi: 10.1038/sj.onc.1205090. [DOI] [PubMed] [Google Scholar]
- 47.Zhao Q, Beck AJ, Vitale JM, Schneider JS, Gao S, Chang C, Elson G, Leibovich SJ, Park JY, Tian B, Nam HS, Fraidenraich D. Developmental ablation of Id1 and Id3 genes in the vasculature leads to postnatal cardiac phenotypes. Dev Biol. 2011;349:53–64. doi: 10.1016/j.ydbio.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liebner S, Corada M, Bangsow T, Babbage J, Taddei A, Czupalla CJ, Reis M, Felici A, Wolburg H, Fruttiger M, Taketo MM, von Melchner H, Plate KH, Gerhardt H, Dejana E. Wnt/beta-catenin signaling controls development of the blood–brain barrier. J Cell Biol. 2008;183:409–417. doi: 10.1083/jcb.200806024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci USA. 2009;106:641–646. doi: 10.1073/pnas.0805165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Amerongen R, Nusse R. Towards an integrated view of Wnt signaling in development. Development. 2009;136:3205–3214. doi: 10.1242/dev.033910. [DOI] [PubMed] [Google Scholar]
- 51.Zhu J, Motejlek K, Wang D, Zang K, Schmidt A, Reichardt LF. Beta8 integrins are required for vascular morphogenesis in mouse embryos. Development. 2002;129:2891–2903. doi: 10.1242/dev.129.12.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bader BL, Rayburn H, Crowley D, Hynes RO. Extensive vasculogenesis, angiogenesis, and organogenesis precede lethality in mice lacking all alpha v integrins. Cell. 1998;95:507–519. doi: 10.1016/s0092-8674(00)81618-9. [DOI] [PubMed] [Google Scholar]
- 53.Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci USA. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet. 1995;11:415–421. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- 57.Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, Ding J, Ferguson MW, Doetschman T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet. 1995;11:409–414. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bohnsack BL, Hirschi KK. Red light, green light: signals that control endothelial cell proliferation during embryonic vascular development. Cell Cycle. 2004;3:1506–1511. doi: 10.4161/cc.3.12.1334. [DOI] [PubMed] [Google Scholar]
- 59.Perrella MA, Jain MK, Lee ME. Role of TGF-beta in vascular development and vascular reactivity. Miner Electrolyte Metab. 1998;24:136–143. doi: 10.1159/000057361. [DOI] [PubMed] [Google Scholar]
- 60.Dickson MC, Martin JS, Cousins FM, Kulkarni AB, Karlsson S, Akhurst RJ. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knockout mice. Development. 1995;121:1845–1854. doi: 10.1242/dev.121.6.1845. [DOI] [PubMed] [Google Scholar]
- 61.Sridurongrit S, Larsson J, Schwartz R, Ruiz-Lozano P, Kaartinen V. Signaling via the Tgf-beta type I receptor Alk5 in heart development. Dev Biol. 2008;322:208–218. doi: 10.1016/j.ydbio.2008.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robson A, Allinson KR, Anderson RH, Henderson DJ, Arthur HM. The TGFbeta type II receptor plays a critical role in the endothelial cells during cardiac development. Dev Dyn. 2010;239:2435–2442. doi: 10.1002/dvdy.22376. [DOI] [PubMed] [Google Scholar]
- 63.Annes JP, Rifkin DB, Munger JS. The integrin alphaV-beta6 binds and activates latent TGFbeta3. FEBS Lett. 2002;511:65–68. doi: 10.1016/s0014-5793(01)03280-x. [DOI] [PubMed] [Google Scholar]
- 64.Araya J, Cambier S, Morris A, Finkbeiner W, Nishimura SL. Integrin-mediated transforming growth factor-beta activation regulates homeostasis of the pulmonary epithelial-mesenchymal trophic unit. Am J Pathol. 2006;169:405–415. doi: 10.2353/ajpath.2006.060049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, Nishimura SL. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J Cell Biol. 2002;157:493–507. doi: 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 67.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116:217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 68.Annes JP, Chen Y, Munger JS, Rifkin DB. Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1. J Cell Biol. 2004;165:723–734. doi: 10.1083/jcb.200312172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang Z, Mu Z, Dabovic B, Jurukovski V, Yu D, Sung J, Xiong X, Munger JS. Absence of integrin-mediated TGFbeta1 activation in vivo recapitulates the phenotype of TGFbeta1-null mice. J Cell Biol. 2007;176:787–793. doi: 10.1083/jcb.200611044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Krieglstein K, Strelau J, Schober A, Sullivan A, Unsicker K. TGF-beta and the regulation of neuron survival and death. J Physiol Paris. 2002;96:25–30. doi: 10.1016/s0928-4257(01)00077-8. [DOI] [PubMed] [Google Scholar]
- 71.Pelton RW, Dickinson ME, Moses HL, Hogan BL. In situ hybridization analysis of TGF beta 3 RNA expression during mouse development: comparative studies with TGF beta 1 and beta 2. Development. 1990;110:609–620. doi: 10.1242/dev.110.2.609. [DOI] [PubMed] [Google Scholar]
- 72.Pelton RW, Saxena B, Jones M, Moses HL, Gold LI. Immunohistochemical localization of TGF beta 1, TGF beta 2, and TGF beta 3 in the mouse embryo: expression patterns suggest multiple roles during embryonic development. J Cell Biol. 1991;115:1091–1105. doi: 10.1083/jcb.115.4.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cambier S, Gline S, Mu D, Collins R, Araya J, Dolganov G, Einheber S, Boudreau N, Nishimura SL. Integrin alpha(v)beta8-mediated activation of transforming growth factor-beta by perivascular astrocytes: an angiogenic control switch. Am J Pathol. 2005;166:1883–1894. doi: 10.1016/s0002-9440(10)62497-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor A, Feuer EJ, Thun MJ. Cancer statistics, 2005. CA Cancer J Clin. 2005;55:10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 75.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, Chin L, DePinho RA, Cavenee WK. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 76.Meyer MA. Malignant gliomas in adults. N Engl J Med. 2008;359:1850. doi: 10.1056/NEJMc086380. [DOI] [PubMed] [Google Scholar]
- 77.Behin A, Hoang-Xuan K, Carpentier AF, Delattre JY. Primary brain tumours in adults. Lancet. 2003;361:323–331. doi: 10.1016/S0140-6736(03)12328-8. [DOI] [PubMed] [Google Scholar]
- 78.Fidler IJ, Yano S, Zhang RD, Fujimaki T, Bucana CD. The seed and soil hypothesis: vascularisation and brain metastases. Lancet Oncol. 2002;3:53–57. doi: 10.1016/s1470-2045(01)00622-2. [DOI] [PubMed] [Google Scholar]
- 79.Yuan F, Salehi HA, Boucher Y, Vasthare US, Tuma RF, Jain RK. Vascular permeability and microcirculation of gliomas and mammary carcinomas transplanted in rat and mouse cranial windows. Cancer Res. 1994;54:4564–4568. [PubMed] [Google Scholar]
- 80.Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, Jain RK. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci USA. 1998;95:4607–4612. doi: 10.1073/pnas.95.8.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Monsky WL, Fukumura D, Gohongi T, Ancukiewcz M, Weich HA, Torchilin VP, Yuan F, Jain RK. Augmentation of transvascular transport of macromolecules and nanoparticles in tumors using vascular endothelial growth factor. Cancer Res. 1999;59:4129–4135. [PubMed] [Google Scholar]
- 82.Fukumura D, Xu L, Chen Y, Gohongi T, Seed B, Jain RK. Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res. 2001;61:6020–6024. [PubMed] [Google Scholar]
- 83.Izumi Y, Xu L, di Tomaso E, Fukumura D, Jain RK. Tumour biology: herceptin acts as an anti-angiogenic cocktail. Nature. 2002;416:279–280. doi: 10.1038/416279b. [DOI] [PubMed] [Google Scholar]
- 84.Bullitt E, Zeng D, Gerig G, Aylward S, Joshi S, Smith JK, Lin W, Ewend MG. Vessel tortuosity and brain tumor malignancy: a blinded study. Acad Radiol. 2005;12:1232–1240. doi: 10.1016/j.acra.2005.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Deeken JF, Loscher W. The blood–brain barrier and cancer: transporters, treatment, and Trojan horses. Clin Cancer Res. 2007;13:1663–1674. doi: 10.1158/1078-0432.CCR-06-2854. [DOI] [PubMed] [Google Scholar]
- 86.Winkler F, Kozin SV, Tong RT, Chae SS, Booth MF, Garkavtsev I, Xu L, Hicklin DJ, Fukumura D, di Tomaso E, Munn LL, Jain RK. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. 2004;6:553–563. doi: 10.1016/j.ccr.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 87.Plate KH, Mennel HD. Vascular morphology and angiogenesis in glial tumors. Exp Toxicol Pathol. 1995;47:89–94. doi: 10.1016/S0940-2993(11)80292-7. [DOI] [PubMed] [Google Scholar]
- 88.Guo P, Hu B, Gu W, Xu L, Wang D, Huang HJ, Cavenee WK, Cheng SY. Platelet-derived growth factor-B enhances glioma angiogenesis by stimulating vascular endothelial growth factor expression in tumor endothelia and by promoting peri-cyte recruitment. Am J Pathol. 2003;162:1083–1093. doi: 10.1016/S0002-9440(10)63905-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zagzag D, Hooper A, Friedlander DR, Chan W, Holash J, Wiegand SJ, Yancopoulos GD, Grumet M. In situ expression of angiopoietins in astrocytomas identifies angiopoietin-2 as an early marker of tumor angiogenesis. Exp Neurol. 1999;159:391–400. doi: 10.1006/exnr.1999.7162. [DOI] [PubMed] [Google Scholar]
- 90.Jain RK. The next frontier of molecular medicine: delivery of therapeutics. Nat Med. 1998;4:655–657. doi: 10.1038/nm0698-655. [DOI] [PubMed] [Google Scholar]
- 91.Monsky WL, Mouta Carreira C, Tsuzuki Y, Gohongi T, Fukumura D, Jain RK. Role of host microenvironment in angiogenesis and microvascular functions in human breast cancer xenografts: mammary fat pad versus cranial tumors. Clin Cancer Res. 2002;8:1008–1013. [PubMed] [Google Scholar]
- 92.Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol. 2002;20:4368–4380. doi: 10.1200/JCO.2002.10.088. [DOI] [PubMed] [Google Scholar]
- 93.Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25:581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- 94.Fukumura D, Kashiwagi S, Jain RK. The role of nitric oxide in tumour progression. Nat Rev Cancer. 2006;6:521–534. doi: 10.1038/nrc1910. [DOI] [PubMed] [Google Scholar]
- 95.Boucher Y, Salehi H, Witwer B, Harsh GRT, Jain RK. Interstitial fluid pressure in intracranial tumours in patients and in rodents. Br J Cancer. 1997;75:829–836. doi: 10.1038/bjc.1997.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jain RK. Barriers to drug delivery in solid tumors. Sci Am. 1994;271:58–65. doi: 10.1038/scientificamerican0794-58. [DOI] [PubMed] [Google Scholar]
- 97.Salmaggi A, Eoli M, Frigerio S, Silvani A, Gelati M, Corsini E, Broggi G, Boiardi A. Intracavitary VEGF, bFGF, IL-8, IL-12 levels in primary and recurrent malignant glioma. J Neurooncol. 2003;62:297–303. doi: 10.1023/a:1023367223575. [DOI] [PubMed] [Google Scholar]
- 98.Schmidt NO, Westphal M, Hagel C, Ergun S, Stavrou D, Rosen EM, Lamszus K. Levels of vascular endothelial growth factor, hepatocyte growth factor/scatter factor and basic fibro-blast growth factor in human gliomas and their relation to angiogenesis. Int J Cancer. 1999;84:10–18. doi: 10.1002/(sici)1097-0215(19990219)84:1<10::aid-ijc3>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 99.Li M, Ransohoff RM. The roles of chemokine CXCL12 in embryonic and brain tumor angiogenesis. Semin Cancer Biol. 2009;19:111–115. doi: 10.1016/j.semcancer.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 100.Tso CL, Freije WA, Day A, Chen Z, Merriman B, Perlina A, Lee Y, Dia EQ, Yoshimoto K, Mischel PS, Liau LM, Cloughesy TF, Nelson SF. Distinct transcription profiles of primary and secondary glioblastoma subgroups. Cancer Res. 2006;66:159–167. doi: 10.1158/0008-5472.CAN-05-0077. [DOI] [PubMed] [Google Scholar]
- 101.Sun L, Hui AM, Su Q, Vortmeyer A, Kotliarov Y, Pastorino S, Passaniti A, Menon J, Walling J, Bailey R, Rosenblum M, Mikkelsen T, Fine HA. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell. 2006;9:287–300. doi: 10.1016/j.ccr.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 102.Reis M, Czupalla CJ, Ziegler N, Devraj K, Zinke J, Seidel S, Heck R, Thom S, Macas J, Bockamp E, Fruttiger M, Taketo MM, Dimmeler S, Plate KH, Liebner S. Endothelial Wnt/beta-catenin signaling inhibits glioma angiogenesis and normalizes tumor blood vessels by inducing PDGF-B expression. J Exp Med. 2012;209:1611–1627. doi: 10.1084/jem.20111580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Vredenburgh JJ, Desjardins A, Herndon JE, 2nd, Dowell JM, Reardon DA, Quinn JA, Rich JN, Sathornsumetee S, Gururangan S, Wagner M, Bigner DD, Friedman AH, Friedman HS. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13:1253–1259. doi: 10.1158/1078-0432.CCR-06-2309. [DOI] [PubMed] [Google Scholar]
- 104.Hellstrom M, Phng LK, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, Alva J, Nilsson AK, Karlsson L, Gaiano N, Yoon K, Rossant J, Iruela-Arispe ML, Kalen M, Gerhardt H, Betsholtz C. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature. 2007;445:776–780. doi: 10.1038/nature05571. [DOI] [PubMed] [Google Scholar]
- 105.Phng LK, Gerhardt H. Angiogenesis: a team effort coordinated by notch. Dev Cell. 2009;16:196–208. doi: 10.1016/j.devcel.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 106.Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, Lin HC, Yancopoulos GD, Thurston G. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature. 2006;444:1032–1037. doi: 10.1038/nature05355. [DOI] [PubMed] [Google Scholar]
- 107.Yan M, Callahan CA, Beyer JC, Allamneni KP, Zhang G, Ridgway JB, Niessen K, Plowman GD. Chronic DLL4 blockade induces vascular neoplasms. Nature. 2010;463:E6–E7. doi: 10.1038/nature08751. [DOI] [PubMed] [Google Scholar]
- 108.Xiong Y, Mahmood A, Chopp M. Angiogenesis, neurogenesis and brain recovery of function following injury. Curr Opin Investig Drugs. 2010;11:298–308. [PMC free article] [PubMed] [Google Scholar]
- 109.Yamashita T, Ninomiya M, Hernandez Acosta P, Garcia-Verdugo JM, Sunabori T, Sakaguchi M, Adachi K, Kojima T, Hirota Y, Kawase T, Araki N, Abe K, Okano H, Sawamoto K. Subventricular zone-derived neuroblasts migrate and differentiate into mature neurons in the post-stroke adult striatum. J Neurosci. 2006;26:6627–6636. doi: 10.1523/JNEUROSCI.0149-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Leventhal C, Rafii S, Rafii D, Shahar A, Goldman SA. Endothelial trophic support of neuronal production and recruitment from the adult mammalian subependyma. Mol Cell Neurosci. 1999;13:450–464. doi: 10.1006/mcne.1999.0762. [DOI] [PubMed] [Google Scholar]
- 111.Palmer TD, Willhoite AR, Gage FH. Vascular niche for adult hippocampal neurogenesis. J Comp Neurol. 2000;425:479–494. doi: 10.1002/1096-9861(20001002)425:4<479::aid-cne2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 112.Hill WD, Hess DC, Martin-Studdard A, Carothers JJ, Zheng J, Hale D, Maeda M, Fagan SC, Carroll JE, Conway SJ. SDF-1 (CXCL12) is upregulated in the ischemic penumbra following stroke: association with bone marrow cell homing to injury. J Neuropathol Exp Neurol. 2004;63:84–96. doi: 10.1093/jnen/63.1.84. [DOI] [PubMed] [Google Scholar]
- 113.Imitola J, Raddassi K, Park KI, Mueller FJ, Nieto M, Teng YD, Frenkel D, Li J, Sidman RL, Walsh CA, Snyder EY, Khoury SJ. Directed migration of neural stem cells to sites of CNS injury by the stromal cell-derived factor 1alpha/CXC chemokine receptor 4 pathway. Proc Natl Acad Sci USA. 2004;101:18117–18122. doi: 10.1073/pnas.0408258102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lakhan SE, Kirchgessner A, Hofer M. Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J Transl Med. 2009;7:97. doi: 10.1186/1479-5876-7-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood–brain barrier. Nat Med. 2013;19:1584–1596. doi: 10.1038/nm.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yilmaz G, Granger DN. Cell adhesion molecules and ischemic stroke. Neurol Res. 2008;30:783–793. doi: 10.1179/174313208X341085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, Iwaki T, Okada Y, Iida M, Cua DJ, Iwakura Y, Yoshimura A. Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat Med. 2009;15:946–950. doi: 10.1038/nm.1999. [DOI] [PubMed] [Google Scholar]
- 118.Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, Giese T, Veltkamp R. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009;15:192–199. doi: 10.1038/nm.1927. [DOI] [PubMed] [Google Scholar]
- 119.Lindsberg PJ, Carpen O, Paetau A, Karjalainen-Lindsberg ML, Kaste M. Endothelial ICAM-1 expression associated with inflammatory cell response in human ischemic stroke. Circulation. 1996;94:939–945. doi: 10.1161/01.cir.94.5.939. [DOI] [PubMed] [Google Scholar]
- 120.Zhang RL, Chopp M, Li Y, Zaloga C, Jiang N, Jones ML, Miyasaka M, Ward PA. Anti-ICAM-1 antibody reduces ischemic cell damage after transient middle cerebral artery occlusion in the rat. Neurology. 1994;44:1747–1751. doi: 10.1212/wnl.44.9.1747. [DOI] [PubMed] [Google Scholar]
- 121.Goussev AV, Zhang Z, Anderson DC, Chopp M. P-selectin antibody reduces hemorrhage and infarct volume resulting from MCA occlusion in the rat. J Neurol Sci. 1998;161:16–22. doi: 10.1016/s0022-510x(98)00262-7. [DOI] [PubMed] [Google Scholar]
- 122.Zhang R, Chopp M, Zhang Z, Jiang N, Powers C. The expression of P- and E-selectins in three models of middle cerebral artery occlusion. Brain Res. 1998;785:207–214. doi: 10.1016/s0006-8993(97)01343-7. [DOI] [PubMed] [Google Scholar]
- 123.Rallidis LS, Zolindaki MG, Vikelis M, Kaliva K, Papadopoulos C, Kremastinos DT. Elevated soluble intercellular adhesion molecule-1 levels are associated with poor short-term prognosis in middle-aged patients with acute ischaemic stroke. Int J Cardiol. 2009;132:216–220. doi: 10.1016/j.ijcard.2007.11.031. [DOI] [PubMed] [Google Scholar]
- 124.Yang Y, Rosenberg GA. Blood–brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke. 2011;42:3323–3328. doi: 10.1161/STROKEAHA.110.608257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yang Y, Thompson JF, Taheri S, Salayandia VM, McAvoy TA, Hill JW, Yang Y, Estrada EY, Rosenberg GA. Early inhibition of MMP activity in ischemic rat brain promotes expression of tight junction proteins and angiogenesis during recovery. J Cereb Blood Flow Metab. 2013;33:1104–1114. doi: 10.1038/jcbfm.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Park KP, Rosell A, Foerch C, Xing C, Kim WJ, Lee S, Opdenakker G, Furie KL, Lo EH. Plasma and brain matrix metalloproteinase-9 after acute focal cerebral ischemia in rats. Stroke. 2009;40:2836–2842. doi: 10.1161/STROKEAHA.109.554824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Morancho A, Rosell A, Garcia-Bonilla L, Montaner J. Metalloproteinase and stroke infarct size: role for anti-inflammatory treatment? Ann N Y Acad Sci. 2010;1207:123–133. doi: 10.1111/j.1749-6632.2010.05734.x. [DOI] [PubMed] [Google Scholar]
- 128.Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood–brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001;21:7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Montaner J, Alvarez-Sabin J, Molina C, Angles A, Abilleira S, Arenillas J, Gonzalez MA, Monasterio J. Matrix metalloproteinase expression after human cardioembolic stroke: temporal profile and relation to neurological impairment. Stroke. 2001;32:1759–1766. doi: 10.1161/01.str.32.8.1759. [DOI] [PubMed] [Google Scholar]
- 130.Asahi M, Sumii T, Fini ME, Itohara S, Lo EH. Matrix metalloproteinase 2 gene knockout has no effect on acute brain injury after focal ischemia. NeuroReport. 2001;12:3003–3007. doi: 10.1097/00001756-200109170-00050. [DOI] [PubMed] [Google Scholar]
- 131.Candelario-Jalil E, Gonzalez-Falcon A, Garcia-Cabrera M, Leon OS, Fiebich BL. Post-ischaemic treatment with the cyclooxygenase-2 inhibitor nimesulide reduces blood–brain barrier disruption and leukocyte infiltration following transient focal cerebral ischaemia in rats. J Neurochem. 2007;100:1108–1120. doi: 10.1111/j.1471-4159.2006.04280.x. [DOI] [PubMed] [Google Scholar]
- 132.Beck H, Acker T, Wiessner C, Allegrini PR, Plate KH. Expression of angiopoietin-1, angiopoietin-2, and tie receptors after middle cerebral artery occlusion in the rat. Am J Pathol. 2000;157:1473–1483. doi: 10.1016/S0002-9440(10)64786-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Marti HJ, Bernaudin M, Bellail A, Schoch H, Euler M, Petit E, Risau W. Hypoxia-induced vascular endothelial growth factor expression precedes neovascularization after cerebral ischemia. Am J Pathol. 2000;156:965–976. doi: 10.1016/S0002-9440(10)64964-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hayashi T, Noshita N, Sugawara T, Chan PH. Temporal profile of angiogenesis and expression of related genes in the brain after ischemia. J Cereb Blood Flow Metab. 2003;23:166–180. doi: 10.1097/01.WCB.0000041283.53351.CB. [DOI] [PubMed] [Google Scholar]
- 135.Zhang ZG, Zhang L, Tsang W, Soltanian-Zadeh H, Morris D, Zhang R, Goussev A, Powers C, Yeich T, Chopp M. Correlation of VEGF and angiopoietin expression with disruption of blood–brain barrier and angiogenesis after focal cerebral ischemia. J Cereb Blood Flow Metab. 2002;22:379–392. doi: 10.1097/00004647-200204000-00002. [DOI] [PubMed] [Google Scholar]
- 136.Krupinski J, Kaluza J, Kumar P, Kumar S, Wang JM. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke. 1994;25:1794–1798. doi: 10.1161/01.str.25.9.1794. [DOI] [PubMed] [Google Scholar]
- 137.Conway EM, Collen D, Carmeliet P. Molecular mechanisms of blood vessel growth. Cardiovasc Res. 2001;49:507–521. doi: 10.1016/s0008-6363(00)00281-9. [DOI] [PubMed] [Google Scholar]
- 138.Fagiani E, Christofori G. Angiopoietins in angiogenesis. Cancer Lett. 2013;328:18–26. doi: 10.1016/j.canlet.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 139.Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 140.Chang J, Li Y, Huang Y, Lam KS, Hoo RL, Wong WT, Cheng KK, Wang Y, Vanhoutte PM, Xu A. Adiponectin prevents diabetic premature senescence of endothelial progenitor cells and promotes endothelial repair by suppressing the p38 MAP kinase/p16INK4A signaling pathway. Diabetes. 2010;59:2949–2959. doi: 10.2337/db10-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chang J, Atochin DN, Li Q, Lam KS, Xu A, Huang PL. Bone marrow-derived circulating endothelial progenitor cells contribute to eNOS-regulated endothelial repair and vasodilation after arterial injury in vivo. J Cardiol Vasc Med. 2013;1:1–8. [Google Scholar]
- 142.Hess DC, Hill WD, Martin-Studdard A, Carroll J, Brailer J, Carothers J. Bone marrow as a source of endothelial cells and NeuN-expressing cells after stroke. Stroke. 2002;33:1362–1368. doi: 10.1161/01.str.0000014925.09415.c3. [DOI] [PubMed] [Google Scholar]
- 143.Zhang ZG, Zhang L, Jiang Q, Chopp M. Bone marrow-derived endothelial progenitor cells participate in cerebral neovascularization after focal cerebral ischemia in the adult mouse. Circ Res. 2002;90:284–288. doi: 10.1161/hh0302.104460. [DOI] [PubMed] [Google Scholar]
- 144.Beck H, Voswinckel R, Wagner S, Ziegelhoeffer T, Heil M, Helisch A, Schaper W, Acker T, Hatzopoulos AK, Plate KH. Participation of bone marrow-derived cells in long-term repair processes after experimental stroke. J Cereb Blood Flow Metab. 2003;23:709–717. doi: 10.1097/01.WCB.0000065940.18332.8D. [DOI] [PubMed] [Google Scholar]
- 145.Brown WR, Thore CR. Review: cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol Appl Neurobiol. 2011;37:56–74. doi: 10.1111/j.1365-2990.2010.01139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.de la Torre JC, Stefano GB. Evidence that Alzheimer’s disease is a microvascular disorder: the role of constitutive nitric oxide. Brain Res Brain Res Rev. 2000;34:119–136. doi: 10.1016/s0165-0173(00)00043-6. [DOI] [PubMed] [Google Scholar]
- 147.Roher AE, Esh C, Kokjohn TA, Kalback W, Luehrs DC, Seward JD, Sue LI, Beach TG. Circle of Willis atherosclerosis is a risk factor for sporadic Alzheimer’s disease. Arterioscler Thromb Vasc Biol. 2003;23:2055–2062. doi: 10.1161/01.ATV.0000095973.42032.44. [DOI] [PubMed] [Google Scholar]
- 148.Black JE, Polinsky M, Greenough WT. Progressive failure of cerebral angiogenesis supporting neural plasticity in aging rats. Neurobiol Aging. 1989;10:353–358. doi: 10.1016/0197-4580(89)90048-1. [DOI] [PubMed] [Google Scholar]
- 149.Frenkel-Denkberg G, Gershon D, Levy AP. The function of hypoxia-inducible factor 1 (HIF-1) is impaired in senescent mice. FEBS Lett. 1999;462:341–344. doi: 10.1016/s0014-5793(99)01552-5. [DOI] [PubMed] [Google Scholar]
- 150.Rivard A, Fabre JE, Silver M, Chen D, Murohara T, Kearney M, Magner M, Asahara T, Isner JM. Age-dependent impairment of angiogenesis. Circulation. 1999;99:111–120. doi: 10.1161/01.cir.99.1.111. [DOI] [PubMed] [Google Scholar]
- 151.Rivard A, Berthou-Soulie L, Principe N, Kearney M, Curry C, Branellec D, Semenza GL, Isner JM. Age-dependent defect in vascular endothelial growth factor expression is associated with reduced hypoxia-inducible factor 1 activity. J Biol Chem. 2000;275:29643–29647. doi: 10.1074/jbc.M001029200. [DOI] [PubMed] [Google Scholar]
- 152.Chavez JC, LaManna JC. Hypoxia-inducible factor-1alpha accumulation in the rat brain in response to hypoxia and ischemia is attenuated during aging. Adv Exp Med Biol. 2003;510:337–341. doi: 10.1007/978-1-4615-0205-0_55. [DOI] [PubMed] [Google Scholar]
- 153.Rapino C, Bianchi G, Di Giulio C, Centurione L, Cacchio M, Antonucci A, Cataldi A. HIF-1alpha cytoplasmic accumulation is associated with cell death in old rat cerebral cortex exposed to intermittent hypoxia. Aging Cell. 2005;4:177–185. doi: 10.1111/j.1474-9726.2005.00161.x. [DOI] [PubMed] [Google Scholar]
- 154.Paris D, Patel N, DelleDonne A, Quadros A, Smeed R, Mullan M. Impaired angiogenesis in a transgenic mouse model of cerebral amyloidosis. Neurosci Lett. 2004;366:80–85. doi: 10.1016/j.neulet.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 155.Paris D, Townsend K, Quadros A, Humphrey J, Sun J, Brem S, Wotoczek-Obadia M, DelleDonne A, Patel N, Obregon DF, Crescentini R, Abdullah L, Coppola D, Rojiani AM, Crawford F, Sebti SM, Mullan M. Inhibition of angiogenesis by Abeta peptides. Angiogenesis. 2004;7:75–85. doi: 10.1023/B:AGEN.0000037335.17717.bf. [DOI] [PubMed] [Google Scholar]
- 156.Yang SP, Bae DG, Kang HJ, Gwag BJ, Gho YS, Chae CB. Co-accumulation of vascular endothelial growth factor with beta-amyloid in the brain of patients with Alzheimer’s disease. Neurobiol Aging. 2004;25:283–290. doi: 10.1016/S0197-4580(03)00111-8. [DOI] [PubMed] [Google Scholar]
- 157.Kalaria RN, Cohen DL, Premkumar DR, Nag S, LaManna JC, Lust WD. Vascular endothelial growth factor in Alzheimer’s disease and experimental cerebral ischemia. Brain Res Mol Brain Res. 1998;62:101–105. doi: 10.1016/s0169-328x(98)00190-9. [DOI] [PubMed] [Google Scholar]
- 158.Grammas P, Sanchez A, Tripathy D, Luo E, Martinez J. Vascular signaling abnormalities in Alzheimer disease. Cleve Clin J Med. 2011;78(Suppl 1):S50–S53. doi: 10.3949/ccjm.78.s1.09. [DOI] [PubMed] [Google Scholar]
- 159.Schultheiss C, Blechert B, Gaertner FC, Drecoll E, Mueller J, Weber GF, Drzezga A, Essler M. In vivo characterization of endothelial cell activation in a transgenic mouse model of Alzheimer’s disease. Angiogenesis. 2006;9:59–65. doi: 10.1007/s10456-006-9030-4. [DOI] [PubMed] [Google Scholar]
- 160.Rosenberg GA. Neurological diseases in relation to the blood–brain barrier. J Cereb Blood Flow Metab. 2012;32:1139–1151. doi: 10.1038/jcbfm.2011.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ujiie M, Dickstein DL, Carlow DA, Jefferies WA. Blood–brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation. 2003;10:463–470. doi: 10.1038/sj.mn.7800212. [DOI] [PubMed] [Google Scholar]
- 162.Farrall AJ, Wardlaw JM. Blood–brain barrier: ageing and microvascular disease—systematic review and meta-analysis. Neurobiol Aging. 2009;30:337–352. doi: 10.1016/j.neurobiolaging.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 163.Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B. RAGE mediates amyloid-beta peptide transport across the blood–brain barrier and accumulation in brain. Nat Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- 164.Kumar-Singh S, Pirici D, McGowan E, Serneels S, Ceuterick C, Hardy J, Duff K, Dickson D, Van Broeckhoven C. Dense-core plaques in Tg2576 and PSAPP mouse models of Alzheimer’s disease are centered on vessel walls. Am J Pathol. 2005;167:527–543. doi: 10.1016/S0002-9440(10)62995-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Paul J, Strickland S, Melchor JP. Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer’s disease. J Exp Med. 2007;204:1999–2008. doi: 10.1084/jem.20070304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Dickstein DL, Biron KE, Ujiie M, Pfeifer CG, Jeffries AR, Jefferies WA. Abeta peptide immunization restores blood–brain barrier integrity in Alzheimer disease. FASEB J. 2006;20:426–433. doi: 10.1096/fj.05-3956com. [DOI] [PubMed] [Google Scholar]
- 167.Zhong Z, Deane R, Ali Z, Parisi M, Shapovalov Y, O’Banion MK, Stojanovic K, Sagare A, Boillee S, Cleveland DW, Zlokovic BV. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci. 2008;11:420–422. doi: 10.1038/nn2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Larochelle C, Alvarez JI, Prat A. How do immune cells overcome the blood–brain barrier in multiple sclerosis? FEBS Lett. 2011;585:3770–3780. doi: 10.1016/j.febslet.2011.04.066. [DOI] [PubMed] [Google Scholar]
- 169.McCormack PL. Natalizumab: a review of its use in the management of relapsing-remitting multiple sclerosis. Drugs. 2013;73:1463–1481. doi: 10.1007/s40265-013-0102-7. [DOI] [PubMed] [Google Scholar]
- 170.Guttmacher AE, Marchuk DA, White RI., Jr Hereditary hemorrhagic telangiectasia. N Engl J Med. 1995;333:918–924. doi: 10.1056/NEJM199510053331407. [DOI] [PubMed] [Google Scholar]
- 171.da Costa L, Wallace MC, Ter Brugge KG, O’Kelly C, Willinsky RA, Tymianski M. The natural history and predictive features of hemorrhage from brain arteriovenous malformations. Stroke. 2009;40:100–105. doi: 10.1161/STROKEAHA.108.524678. [DOI] [PubMed] [Google Scholar]
- 172.Kim H, Marchuk DA, Pawlikowska L, Chen Y, Su H, Yang GY, Young WL. Genetic considerations relevant to intracranial hemorrhage and brain arteriovenous malformations. Acta Neurochir Suppl. 2008;105:199–206. doi: 10.1007/978-3-211-09469-3_38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.ten Dijke P, Arthur HM. Extracellular control of TGFbeta signalling in vascular development and disease. Nat Rev Mol Cell Biol. 2007;8:857–869. doi: 10.1038/nrm2262. [DOI] [PubMed] [Google Scholar]
- 174.Bayrak-Toydemir P, McDonald J, Markewitz B, Lewin S, Miller F, Chou LS, Gedge F, Tang W, Coon H, Mao R. Genotype-phenotype correlation in hereditary hemorrhagic telangiectasia: mutations and manifestations. Am J Med Genet A. 2006;140:463–470. doi: 10.1002/ajmg.a.31101. [DOI] [PubMed] [Google Scholar]
- 175.Lesca G, Genin E, Blachier C, Olivieri C, Coulet F, Brunet G, Dupuis-Girod S, Buscarini E, Soubrier F, Calender A, Danesino C, Giraud S, Plauchu H. Hereditary hemorrhagic telangiectasia: evidence for regional founder effects of ACVRL1 mutations in French and Italian patients. Eur J Hum Genet. 2008;16:742–749. doi: 10.1038/ejhg.2008.3. [DOI] [PubMed] [Google Scholar]
- 176.Letteboer TG, Zewald RA, Kamping EJ, de Haas G, Mager JJ, Snijder RJ, Lindhout D, Hennekam FA, Westermann CJ, van Ploos Amstel JK. Hereditary hemorrhagic telangiectasia: ENG and ALK-1 mutations in Dutch patients. Hum Genet. 2005;116:8–16. doi: 10.1007/s00439-004-1196-5. [DOI] [PubMed] [Google Scholar]
- 177.McDonald J, Bayrak-Toydemir P, Pyeritz RE. Hereditary hemorrhagic telangiectasia: an overview of diagnosis, management, and pathogenesis. Genet Med. 2011;13:607–616. doi: 10.1097/GIM.0b013e3182136d32. [DOI] [PubMed] [Google Scholar]
- 178.Saba HI, Morelli GA, Logrono LA. Brief report: treatment of bleeding in hereditary hemorrhagic telangiectasia with aminocaproic acid. N Engl J Med. 1994;330:1789–1790. doi: 10.1056/NEJM199406233302504. [DOI] [PubMed] [Google Scholar]
- 179.McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;8:345–351. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- 180.Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, Stenzel TT, Speer M, Pericak-Vance MA, Diamond A, Guttmacher AE, Jackson CE, Attisano L, Kucherlapati R, Porteous ME, Marchuk DA. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13:189–195. doi: 10.1038/ng0696-189. [DOI] [PubMed] [Google Scholar]
- 181.Gougos A, Letarte M. Primary structure of endoglin, an RGD-containing glycoprotein of human endothelial cells. J Biol Chem. 1990;265:8361–8364. [PubMed] [Google Scholar]
- 182.ten Dijke P, Goumans MJ, Pardali E. Endoglin in angiogenesis and vascular diseases. Angiogenesis. 2008;11:79–89. doi: 10.1007/s10456-008-9101-9. [DOI] [PubMed] [Google Scholar]
- 183.Barbara NP, Wrana JL, Letarte M. Endoglin is an accessory protein that interacts with the signaling receptor complex of multiple members of the transforming growth factor-beta superfamily. J Biol Chem. 1999;274:584–594. doi: 10.1074/jbc.274.2.584. [DOI] [PubMed] [Google Scholar]
- 184.Vincent P, Plauchu H, Hazan J, Faure S, Weissenbach J, Godet J. A third locus for hereditary haemorrhagic telangiectasia maps to chromosome 12q. Hum Mol Genet. 1995;4:945–949. doi: 10.1093/hmg/4.5.945. [DOI] [PubMed] [Google Scholar]
- 185.Johnson DW, Berg JN, Gallione CJ, McAllister KA, Warner JP, Helmbold EA, Markel DS, Jackson CE, Porteous ME, Marchuk DA. A second locus for hereditary hemorrhagic telangiectasia maps to chromosome 12. Genome Res. 1995;5:21–28. doi: 10.1101/gr.5.1.21. [DOI] [PubMed] [Google Scholar]
- 186.Gallione CJ, Repetto GM, Legius E, Rustgi AK, Schelley SL, Tejpar S, Mitchell G, Drouin E, Westermann CJ, Marchuk DA. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4) Lancet. 2004;363:852–859. doi: 10.1016/S0140-6736(04)15732-2. [DOI] [PubMed] [Google Scholar]
- 187.Park SO, Wankhede M, Lee YJ, Choi EJ, Fliess N, Choe SW, Oh SH, Walter G, Raizada MK, Sorg BS, Oh SP. Real-time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J Clin Invest. 2009;119:3487–3496. doi: 10.1172/JCI39482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Roman BL, Pham VN, Lawson ND, Kulik M, Childs S, Lekven AC, Garrity DM, Moon RT, Fishman MC, Lechleider RJ, Weinstein BM. Disruption of acvrl1 increases endothelial cell number in zebrafish cranial vessels. Development. 2002;129:3009–3019. doi: 10.1242/dev.129.12.3009. [DOI] [PubMed] [Google Scholar]
- 189.Sorensen LK, Brooke BS, Li DY, Urness LD. Loss of distinct arterial and venous boundaries in mice lacking endoglin, a vascular-specific TGFbeta coreceptor. Dev Biol. 2003;261:235–250. doi: 10.1016/s0012-1606(03)00158-1. [DOI] [PubMed] [Google Scholar]
- 190.Mahmoud M, Allinson KR, Zhai Z, Oakenfull R, Ghandi P, Adams RH, Fruttiger M, Arthur HM. Pathogenesis of arteriovenous malformations in the absence of endoglin. Circ Res. 2010;106:1425–1433. doi: 10.1161/CIRCRESAHA.109.211037. [DOI] [PubMed] [Google Scholar]
- 191.Mancini ML, Terzic A, Conley BA, Oxburgh LH, Nicola T, Vary CP. Endoglin plays distinct roles in vascular smooth muscle cell recruitment and regulation of arteriovenous identity during angiogenesis. Dev Dyn. 2009;238:2479–2493. doi: 10.1002/dvdy.22066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Kovacs Z, Ikezaki K, Samoto K, Inamura T, Fukui M. VEGF and flt. Expression time kinetics in rat brain infarct. Stroke. 1996;27:1865–1872. doi: 10.1161/01.str.27.10.1865. Discussion 1872–1863. [DOI] [PubMed] [Google Scholar]
- 193.Hayashi T, Abe K, Suzuki H, Itoyama Y. Rapid induction of vascular endothelial growth factor gene expression after transient middle cerebral artery occlusion in rats. Stroke. 1997;28:2039–2044. doi: 10.1161/01.str.28.10.2039. [DOI] [PubMed] [Google Scholar]
- 194.Cobbs CS, Chen J, Greenberg DA, Graham SH. Vascular endothelial growth factor expression in transient focal cerebral ischemia in the rat. Neurosci Lett. 1998;249:79–82. doi: 10.1016/s0304-3940(98)00377-2. [DOI] [PubMed] [Google Scholar]
- 195.Lennmyr F, Ata KA, Funa K, Olsson Y, Terent A. Expression of vascular endothelial growth factor (VEGF) and its receptors (Flt-1 and Flk-1) following permanent and transient occlusion of the middle cerebral artery in the rat. J Neuropathol Exp Neurol. 1998;57:874–882. doi: 10.1097/00005072-199809000-00009. [DOI] [PubMed] [Google Scholar]
- 196.Lee MY, Ju WK, Cha JH, Son BC, Chun MH, Kang JK, Park CK. Expression of vascular endothelial growth factor mRNA following transient forebrain ischemia in rats. Neurosci Lett. 1999;265:107–110. doi: 10.1016/s0304-3940(99)00219-0. [DOI] [PubMed] [Google Scholar]
- 197.Plate KH, Beck H, Danner S, Allegrini PR, Wiessner C. Cell type specific upregulation of vascular endothelial growth factor in an MCA-occlusion model of cerebral infarct. J Neuropathol Exp Neurol. 1999;58:654–666. doi: 10.1097/00005072-199906000-00010. [DOI] [PubMed] [Google Scholar]
- 198.Issa R, Krupinski J, Bujny T, Kumar S, Kaluza J, Kumar P. Vascular endothelial growth factor and its receptor, KDR, in human brain tissue after ischemic stroke. Lab Invest. 1999;79:417–425. [PubMed] [Google Scholar]
- 199.Slevin M, Krupinski J, Slowik A, Kumar P, Szczudlik A, Gaffney J. Serial measurement of vascular endothelial growth factor and transforming growth factor-beta1 in serum of patients with acute ischemic stroke. Stroke. 2000;31:1863–1870. doi: 10.1161/01.str.31.8.1863. [DOI] [PubMed] [Google Scholar]
- 200.Gunsilius E, Petzer AL, Stockhammer G, Kahler CM, Gastl G. Serial measurement of vascular endothelial growth factor and transforming growth factor-beta1 in serum of patients with acute ischemic stroke. Stroke. 2001;32:275–278. doi: 10.1161/01.str.32.1.275-b. [DOI] [PubMed] [Google Scholar]
- 201.Beck H, Acker T, Puschel AW, Fujisawa H, Carmeliet P, Plate KH. Cell type-specific expression of neuropilins in an MCA-occlusion model in mice suggests a potential role in post-ischemic brain remodeling. J Neuropathol Exp Neurol. 2002;61:339–350. doi: 10.1093/jnen/61.4.339. [DOI] [PubMed] [Google Scholar]
- 202.Zhang ZG, Tsang W, Zhang L, Powers C, Chopp M. Up-regulation of neuropilin-1 in neovasculature after focal cerebral ischemia in the adult rat. J Cereb Blood Flow Metab. 2001;21:541–549. doi: 10.1097/00004647-200105000-00008. [DOI] [PubMed] [Google Scholar]
- 203.Lin TN, Wang CK, Cheung WM, Hsu CY. Induction of angiopoietin and Tie receptor mRNA expression after cerebral ischemia-reperfusion. J Cereb Blood Flow Metab. 2000;20:387–395. doi: 10.1097/00004647-200002000-00021. [DOI] [PubMed] [Google Scholar]
- 204.Zhang Z, Chopp M. Vascular endothelial growth factor and angiopoietins in focal cerebral ischemia. Trends Cardiovasc Med. 2002;12:62–66. doi: 10.1016/s1050-1738(01)00149-9. [DOI] [PubMed] [Google Scholar]
- 205.Zhang ZG, Chopp M, Lu D, Wayne T, Zhang RL, Morris D. Receptor tyrosine kinase tie 1 mRNA is upregulated on cerebral microvessels after embolic middle cerebral artery occlusion in rat. Brain Res. 1999;847:338–342. doi: 10.1016/s0006-8993(99)02013-2. [DOI] [PubMed] [Google Scholar]
- 206.Lin TN, Nian GM, Chen SF, Cheung WM, Chang C, Lin WC, Hsu CY. Induction of Tie-1 and Tie-2 receptor protein expression after cerebral ischemia-reperfusion. J Cereb Blood Flow Metab. 2001;21:690–701. doi: 10.1097/00004647-200106000-00007. [DOI] [PubMed] [Google Scholar]
- 207.Iihara K, Sasahara M, Hashimoto N, Uemura Y, Kikuchi H, Hazama F. Ischemia induces the expression of the platelet-derived growth factor-B chain in neurons and brain macrophages in vivo. J Cereb Blood Flow Metab. 1994;14:818–824. doi: 10.1038/jcbfm.1994.102. [DOI] [PubMed] [Google Scholar]
- 208.Krupinski J, Issa R, Bujny T, Slevin M, Kumar P, Kumar S, Kaluza J. A putative role for platelet-derived growth factor in angiogenesis and neuroprotection after ischemic stroke in humans. Stroke. 1997;28:564–573. doi: 10.1161/01.str.28.3.564. [DOI] [PubMed] [Google Scholar]
- 209.Renner O, Tsimpas A, Kostin S, Valable S, Petit E, Schaper W, Marti HH. Time- and cell type-specific induction of platelet-derived growth factor receptor-beta during cerebral ischemia. Brain Res Mol Brain Res. 2003;113:44–51. doi: 10.1016/s0169-328x(03)00085-8. [DOI] [PubMed] [Google Scholar]
- 210.Bernaudin M, Marti HH, Roussel S, Divoux D, Nouvelot A, MacKenzie ET, Petit E. A potential role for erythropoietin in focal permanent cerebral ischemia in mice. J Cereb Blood Flow Metab. 1999;19:643–651. doi: 10.1097/00004647-199906000-00007. [DOI] [PubMed] [Google Scholar]
- 211.Veltkamp R, Rajapakse N, Robins G, Puskar M, Shimizu K, Busija D. Transient focal ischemia increases endothelial nitric oxide synthase in cerebral blood vessels. Stroke. 2002;33:2704–2710. doi: 10.1161/01.str.0000033132.85123.6a. [DOI] [PubMed] [Google Scholar]
- 212.Wiessner C, Gehrmann J, Lindholm D, Topper R, Kreutzberg GW, Hossmann KA. Expression of transforming growth factor-beta 1 and interleukin-1 beta mRNA in rat brain following transient forebrain ischemia. Acta Neuropathol. 1993;86:439–446. doi: 10.1007/BF00228578. [DOI] [PubMed] [Google Scholar]
- 213.Krupinski J, Kumar P, Kumar S, Kaluza J. Increased expression of TGF-beta 1 in brain tissue after ischemic stroke in humans. Stroke. 1996;27:852–857. doi: 10.1161/01.str.27.5.852. [DOI] [PubMed] [Google Scholar]
- 214.Yamashita K, Gerken U, Vogel P, Hossmann K, Wiessner C. Biphasic expression of TGF-beta1 mRNA in the rat brain following permanent occlusion of the middle cerebral artery. Brain Res. 1999;836:139–145. doi: 10.1016/s0006-8993(99)01626-1. [DOI] [PubMed] [Google Scholar]
- 215.Haqqani AS, Nesic M, Preston E, Baumann E, Kelly J, Stanimirovic D. Characterization of vascular protein expression patterns in cerebral ischemia/reperfusion using laser capture microdissection and ICAT-nanoLC-MS/MS. FASEB J. 2005;19:1809–1821. doi: 10.1096/fj.05-3793com. [DOI] [PubMed] [Google Scholar]
- 216.Chen HH, Chien CH, Liu HM. Correlation between angiogenesis and basic fibroblast growth factor expression in experimental brain infarct. Stroke. 1994;25:1651–1657. doi: 10.1161/01.str.25.8.1651. [DOI] [PubMed] [Google Scholar]
- 217.Hara Y, Tooyama I, Yasuhara O, Akiyama H, McGeer PL, Handa J, Kimura H. Acidic fibroblast growth factor-like immunoreactivity in rat brain following cerebral infarction. Brain Res. 1994;664:101–107. doi: 10.1016/0006-8993(94)91959-3. [DOI] [PubMed] [Google Scholar]
- 218.Lin TN, Te J, Lee M, Sun GY, Hsu CY. Induction of basic fibroblast growth factor (bFGF) expression following focal cerebral ischemia. Brain Res Mol Brain Res. 1997;49:255–265. doi: 10.1016/s0169-328x(97)00152-6. [DOI] [PubMed] [Google Scholar]
- 219.Issa R, AlQteishat A, Mitsios N, Saka M, Krupinski J, Tarkowski E, Gaffney J, Slevin M, Kumar S, Kumar P. Expression of basic fibroblast growth factor mRNA and protein in the human brain following ischaemic stroke. Angiogenesis. 2005;8:53–62. doi: 10.1007/s10456-005-5613-8. [DOI] [PubMed] [Google Scholar]