

Abstract
Histamine dehydrogenase (HADH) from Nocardioides simplex catalyzes the oxidative deamination of histamine to produce imidazole acetaldehyde and ammonium. HADH is functionally related to trimethylamine dehydrogenase (TMADH) but HADH has strict substrate specificity towards histamine. HADH is a homodimer with each 76 kDa subunit containing two redox cofactors, a [4Fe-4S] cluster and an unusual covalently bound flavin mononucleotide, 6-S-cysteinyl-FMN. In order to understand the substrate specificity of HADH, we seek to determine its structure by X-ray crystallography. This enzyme has been expressed recombinantly in Escherichia coli and successfully crystallized in two forms. Diffraction data were collected to 2.7 Å resolution at the SSRL synchrotron with 99.7% completeness. The crystals belong to the orthorhombic space group P212121 with unit-cell parameters a = 101.14 Å, b = 107.03 Å, and c = 153.35 Å.
1. Introduction
Histamine is an essential biogenic amine that is present in prokaryotes and the tissues of animals and plants. In humans, histamine acts as a neurotransmitter, mediates allergic reactions, plays a role in cell proliferation, and is important in signaling the release of gastric acid into the stomach (Thurmond et al., 2008). Histamine receptors are the targets of drugs that treat allergies and stomach acidity, but there is very little structural information on the histamine binding sites of these proteins. Thus, model systems may prove useful for the understanding of histamine binding (Konkimalla & Chandra, 2003).
Histamine dehydrogenase (HADH) from Nocardioides simplex (N.simplex) catalyzes the oxidative deamination of histamine to form imidazole acetaldehyde and ammonium ion (Figure 1). HADH (Fujieda et al., 2004, Limburg et al., 2005) is related to trimethylamine dehydrogenase (TMADH, EC 1.5.99.7)(McIntire, 1990, Steenkamp, Kenney et al., 1978, Steenkamp, Mcintire et al., 1978), which contains a 6-S-cysteinyl FMN and a [4Fe-4S] cluster. HADH differs from TMADH in its substrate specificity. TMADH can act on short secondary and tertiary amines. In contrast, HADH is selective for the primary amine substrates, histamine (Km = 31 μM, kcat/Km = 2.1 × 105 M-1s-1), agmatine or decarboxylated arginine (Km = 37 μM, kcat/Km = 6.0 × 104 M-1s-1), and putrescine or butane-1,4-diamine (Km = 1280 μM, kcat/Km = 1500 M-1s-1) (Limburg et al., 2005). The selectivity for histamine over other biogenic amines suggests that HADH could be used in an amperometric biosensor. Current methods for histamine detection rely on either radioenzymatic or HLPC-based derivatization. The structural determination of HADH, with its unique selectivity for histamine, could prove useful in design of a histamine biosensor and provide important insights into histamine recognition by proteins. Furthermore, comparison between the structures of HADH and TMADH may provide insights into the mechanism of this class of enzymes, particularly in regards to the substrate binding site and the electronic interactions between the two cofactors.
Figure 1.

Histamine deamination reaction catalyzed by HADH.
2. Materials and Methods
2.1 Cloning and expression
The hadh gene was cloned from genomic DNA of N. simplex (ATCC 6946). PCR primers were designed to amplify the 2.1 Kbp fragment and incorporate EcoRI and XbaI sites for subsequent insertion into pUC19. The resulting hadh gene was sequenced in the forward and reverse directions and then subcloned between the NdeI and EcoRI sites of pET21b (Novagen) in order to express the recombinant protein without the C-terminal T7-tag or the N-terminal His-tag. The plasmid was transformed into E. coli Rosetta 2 (DE3) cells (Novagen). The cells were grown in 1 L of Terrific Broth media with 100 mg L-1 ampicillin at 37 °C and shaken at 225 rpm to an OD600 of 0.7. Protein expression was induced with 0.1 mM isopropyl β-d-1-thiogalactopyranoside. The addition of 250 mg L-1 iron sulfate and 50 mg L-1 riboflavin was also performed at an OD600 of 0.7 to ensure full incorporation of the iron-sulfur cluster and flavin mononucleotide. The temperature was reduced to 20 °C and the cultures were allowed to shake at 225 rpm overnight. The cells were harvested by centrifugation at 4°C. Selenomethionine-substituted protein was produced using the methionine-auxotrophic E. coli strain B834 (DE3) (Novagen) (Doublie, 1997).
2.2 Purification
Cells (10 g) were resuspended in 20 mM potassium phosphate buffer (pH = 7.4) containing 0.1 M KCl. Cells were disrupted by ultrasonication, and centrifuged at 40,000 g for 30 min. The supernatant was loaded onto a 100 mL Toyopearl-DEAE column pre-equilibrated with 20 mM potassium phosphate (pH = 7.4) containing 0.1 M KCl, and proteins were eluted with a 300 mL linear gradient increasing from 0.1 to 0.3 M KCl (20 mM potassium phosphate buffer, pH 7.4). Fractions exhibiting an Abs444/Abs382 ratio of 1.0 or higher were pooled and (NH4)2SO4 was added to a final concentration of 0.8 M. The protein solution was then loaded on to a 100 mL Toyopearl Butyl-650 column (Tosoh Bioscience) pre-equilibrated with 50 mM potassium phosphate buffer (pH = 7.4) containing 0.8 M (NH4)2SO4. Bound proteins were eluted with a 300 mL gradient decreasing from 0.8 to 0 M (NH4)2SO4 (50 mM potassium phosphate buffer, pH = 7.4). Fractions exhibiting an Abs444/Abs382 ratio of 1.2 or higher were pooled and concentrated to 1 mL using an Amicon Ultra-30 centrifugal unit (Millipore). This protein was loaded on to a HiLoad Superdex 200 sizing column (GE Biosciences) and eluted with 50 mM Tris-HCl (pH 7.4) containing 0.15 M KCl. Fractions with an Abs444/Abs382 ratio of 1.38 or higher were concentrated to >20 mg/mL. The purity of HADH was confirmed with SDS-PAGE and protein concentration was determined by a BCA assay (Thermo Fisher Scientific). The kcat and Km for histamine were determined to be 9.8 s-1 and 24 μM respectively using a standard assay (Siddiqui et al., 2000). Selenomethionine-substituted protein was purified as described above and the incorporation of selenomethionine determined by MALDI-TOF mass spectrometry at KU Analytical Proteomics Lab.
2.3 Crystallization
Prior to crystallization, protein was concentrated to 20 mg/mL in 50 mM Tris-HCl buffer (pH 7.4) containing 0.15 M KCl. Screening to identify crystallization conditions was performed by the hanging-drop vapor-diffusion method using commercially available sparse matrix screening kits (Hampton Research and Emerald Biosystems). Equal volumes of protein and reservoir solution (1 μL + 1 μL) were mixed and equilibrated against 750 μL reservoir solution at 293 K.
2.4 Data collection and processing
Initial unsubstituted HADH crystals were screened for X-ray diffraction in-house on an R-AXIS IV++ detector with Cu Kα X-rays generated by a Rigaku RU-H3RHB rotating-anode generator and focused using an Osmic confocal optical system (Rigaku, Japan) at KU Protein Structure Laboratory. A full data set was collected, but initial attempts at molecular replacement using the TMADH structure (PDB 1DJN) (Trickey et al., 2000) as a search model were not successful. Subsequently, SeMet-HADH crystals were screened and a complete MAD data set collected at beamline BL9-2 at the Stanford Synchrotron Radiation Laboratory (SSRL) using the Stanford Automated Mounting (SAM) system (Cohen, 2002). Data collection was performed at 100 K using an oscillation angle of 1° per frame over a total of 270° to yield a redundant data set. The diffraction data was processed with MOSFLM (Leslie, 2006) and scaled with SCALA from the CCP program suite (Collaborative Computational Project, 1994).
3. Results and Discussion
3.1 Expression, flavin content, SeMet incorporation, and purity
The recombinant unsubstituted HADH was over-expressed in E. coli BL21DE3 Rosetta strain. The Rosetta strain was used as hadh has a high GC content (> 85 %) whereby there are a number of rare codons in the sequence. Expression using this system yielded ∼25 mg purified protein L-1 of E. coli culture. Expression in the methionine-auxotrophic E. coli strain B834 (DE3) reduced yield to ∼1/10 of the Rosetta expression. In order to ensure that the resulting enzyme had a full content of 6-S-cysteinyl-FMN and [4Fe-4S], the media was supplemented with the precursors for both cofactors. The percentage of flavination can be determined from the visible spectrum where a ratio of Abs444/Abs382 of 1.4 is consistent with a full complement of 6-S-cysteinyl-FMN (Fujieda et al., 2004). Both unsubstituted and SeMet-HADH protein had ratios above 1.37. MALDI-TOF mass spectrometry suggested that 10 of the 13 methionines were substituted. The recombinant HADH has the same activity towards histamine (both kcat and kcat/Km) as the native enzyme. As estimated by SDS-PAGE, recombinant HADH has the expected molecular weight of 76 kDa and was purified to > 99% homogeniety.
3.2. Crystallization
Initial yellow crystals grew as both square rods and square “pizza box” crystals in 0.1 HEPES buffer (pH 7.4) containing 2.0 M (NH4)2SO4 and 2% 400-PEG (v/v) in four to seven days (Figure 2). The growth of these crystals was then optimized using the same solution, except the protein concentration was varied from 5 - 20 mg/mL. These crystals rapidly dissolved upon opening of the sealed wells, but crystallization in the same buffer with the addition of 2% - 4% of glycerol (v/v) obviated this problem and allowed harvesting of diffraction quality crystals. The crystals used for data collection were square rods that grew to approximately 0.05 × 0.05 × 0.8 mm in size. We determined that a solution consisting of the mother liquor with the addition of 25% glycerol was sufficient for cryoprotection of these crystals.
Figure 2.
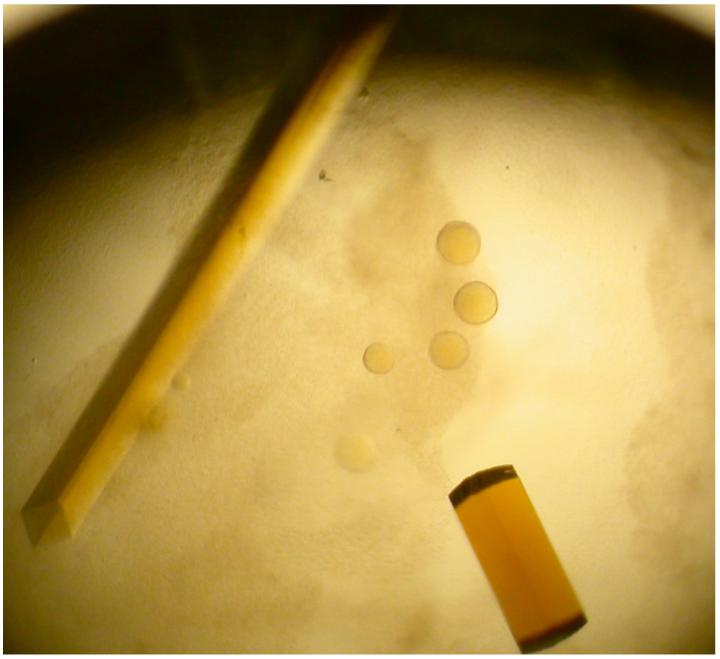
Crystals of unsubstituted recombinant HADH grown in 0.1 M HEPES buffer (pH 7.4) containing 2.0 M (NH4)2SO4, 2% 400-PEG (v/v), and 4% glycerol (v/v). Crystal to the left is a square rod like those used to collect data, while the crystal on the rod is one of the square “pizza box” crystals viewed edge-on.
3.3 Data collection and preliminary X-ray diffraction analysis
Although we collected a complete 3-wavelength MAD data set, structure determination using SOLVE/RESOLVE was not immediately successful. However, using only the single 0.98 Å data we were able to determine the structure by molecular replacement using the polypeptide of TMADH from Methylophilus methylotrophus (PDB 1DJN) (Trickey et al., 2000) as a search model and the program Phaser (LLG>1012) (Mccoy et al., 2007). The X-ray diffraction data used to solve the HADH structure were collected to a resolution of 2.7 Å (Figure 3) with 99.7 completeness and an Rsym of 16% (Table 1). The crystals belong to the orthorhombic space group P212121 with unit-cell parameters a = 101.14 Å, b = 107.03 Å, and c = 153.35 Å. Each asymmetric unit contained two molecules of HADH with a calculated Matthews coefficient Vm of 2.79 Å3 Da-1 giving an estimated solvent content of 55.96% (Matthews, 1968). Refinement and model building are currently ongoing using Refmac (Murshudov et al., 1997) and Coot (Emsley & Cowtan, 2004).
Figure 3.

Diffraction pattern of HADH taken at beamline BL9-2, SSRL. The resolution of this data set was 2.7 Å.
Table 1.
X-ray data collection statistics for HADH. Values in parentheses are for the last resolution shell
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 101.14 b = 107.03 c = 153.35 |
| Resolution (Å) | 84.5 – 2.70 (2.77 – 2.70) |
| No. of measurements | 451,076 |
| No. of unique reflections | 46,309 |
| Redundancy | 9.7 (9.2) |
| Completeness (%) | 99.7 (98.8) |
| Rsym (%) | 16.0 (38.5) |
| Average I/σ(I) | 16.8 (6.3) |
Acknowledgements
Home source X-ray data was collected at the Protein Structure Laboratory at The University of Kansas. High-resolution data was collected at the Stanford Synchrotron Radiation Laboratory (SSRL), which provided excellent diffraction facilities and support. The SSRL is operated by the Department of Energy, Office of Basic Energy Sciences. The SSRL Biotechnology Program is supported by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and by the Department of Energy, Office of Biological and Environmental Research. This research was supported by NIH GM079446 (JL), NIH 5P20 RR17708 (COBRE Center in Protein Structure and Function), and funds from the University of Kansas Center for Research.
References
- Cohen AE, Ellis PJ, Miller MD, Deacon AM, Phizackerley RP. Journal of Applied Crystallography. 2002;35:720–726. doi: 10.1107/s0021889802016709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project Acta Crystallographica. 1994;D50:760–763. N. [Google Scholar]
- Doublie S. Methods in Enzymology. 1997;276:523–530. [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Acta Crystallographica Section D-Biological Crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Fujieda N, Satoh A, Tsuse N, Kano K, Ikeda T. Biochemistry. 2004;43:10800–10808. doi: 10.1021/bi049061q. [DOI] [PubMed] [Google Scholar]
- Konkimalla VB, Chandra N. Biochemical and Biophysical Research Communications. 2003;309:425–431. doi: 10.1016/j.bbrc.2003.08.023. [DOI] [PubMed] [Google Scholar]
- Leslie AGW. Acta Crystallographica Section D-Biological Crystallography. 2006;62:48–57. doi: 10.1107/S0907444905039107. [DOI] [PubMed] [Google Scholar]
- Limburg J, Mure M, Klinman JP. Archives of Biochemistry and Biophysics. 2005;436:8–22. doi: 10.1016/j.abb.2004.11.024. [DOI] [PubMed] [Google Scholar]
- Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Journal of Applied Crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcintire WS. Methods in Enzymology. 1990;188:250–260. doi: 10.1016/0076-6879(90)88042-9. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallographica Section D-Biological Crystallography. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Siddiqui JA, Shoeb SM, Takayama S, Shimizu E, Yorifuji T. FEMS Microbiology Letters. 2000;189:183–187. doi: 10.1111/j.1574-6968.2000.tb09227.x. [DOI] [PubMed] [Google Scholar]
- Steenkamp DJ, Kenney WC, Singer TP. Journal of Biological Chemistry. 1978;253:2812–2817. [PubMed] [Google Scholar]
- Steenkamp DJ, Mcintire W, Kenney WC. Journal of Biological Chemistry. 1978;253:2818–2824. [PubMed] [Google Scholar]
- Thurmond RL, Gelfand EW, Dunford PJ. Nature Review Drug Discovery. 2008;7:41–53. doi: 10.1038/nrd2465. [DOI] [PubMed] [Google Scholar]
- Trickey P, Basran J, Lian LY, Chen ZW, Barton JD, Sutcliffe MJ, Scrutton NS, Mathews FS. Biochemistry. 2000;39:7678–7688. doi: 10.1021/bi9927181. [DOI] [PubMed] [Google Scholar]