

Abstract
The pathogenic mechanism(s) underlying neurodegenerative diseases associated with protein misfolding is unclear. Several studies have implicated ER stress pathways in neurodegenerative conditions, including prion disease, amyotrophic lateral sclerosis, Alzheimer’s disease and many others. The ER stress response and upregulation of ER stress-responsive chaperones is observed in the brains of patients affected with Creutzfeldt-Jacob disease and in mouse models of prion diseases. In particular, the processing of caspase-12, an ER-localized caspase, correlates with neuronal cell death in prion disease. However, the contribution of caspase-12 to neurodegeneration has not been directly addressed in vivo. We confirm that ER stress is induced and that caspase-12 is proteolytically processed in a murine model of infectious prion disease. To address the causality of caspase-12 in mediating infectious prion pathogenesis, we inoculated mice deficient in caspase-12 with prions. The survival, behavior, pathology and accumulation of proteinase K-resistant PrP are indistinguishable between caspase-12 knockout and control mice, suggesting that caspase-12 is not necessary for mediating the neurotoxic effects of prion protein misfolding.
INTRODUCTION
Prion diseases are neurodegenerative diseases of sporadic, inherited or acquired origin.1 While it is firmly established that the key pathogenic event in prion disease is the misfolding of the prion protein (PrP), the mechanism by which misfolded PrP causes neuronal dysfunction is poorly understood.2-4 The common neuropathological hallmarks of prion disease include widespread vacuolation, glial proliferation, synapse loss, and deposition of proteinase K-resistant aggregates of PrP.5 Neuronal cell death and necrosis have been observed in diverse organisms with prion disease.6,7 Understanding how protein misfolding causes neuronal dysfunction and death is vital for combating neurodegenerative diseases and other protein conformational disorders.
Different stress pathways have been implicated in prion toxicity, including oxidative stress, mitochondrial dysfunction, calcium imbalance and endoplasmic reticulum (ER) stress.6 Growing evidence suggests that the ER is potentially important not only for maturation of PrP, but also for the formation of misfolded and cytotoxic, disease-associated forms of the protein.8-11 Alteration of ER homeostasis correlates with synapse loss and neuronal death observed in infectious forms of prion disease.12-14 Furthermore, stress inducible ER chaperones, such as Grp58 and Grp78/BiP, are upregulated in the brains of human patients affected with Creutzfeldt-Jacob disease.12, 13, 15
Accumulating evidence suggests that a cellular overload of misfolded proteins in the ER can trigger caspase-dependent cell death.16 Caspases are evolutionarily conserved cysteine proteases that are critical for the cellular commitment to undergo apoptosis.17 This pathway includes the activation of the ER-localized Caspase-12 (C12) in rodents and its putative human ortholog, caspase-4, in addition to the executioner caspases, caspase-3 and-7.18-20 Furthermore, stable expression of C12 sensitizes cells to ER stress, which is associated with increased caspase-3 cleavage.21
Interestingly, it has been observed that C12 is proteolytically processed in the brains of prion infected mice.12 Given the prior observations of ER stress and C12 activation in prion disease, we hypothesized that intense ER stress associated with prion toxicity leads to C12 activation and neuronal cell death. To directly address this possibility, we inoculated mice deficient for C12 with RML prions and assessed their survival, behavior, pathology and accumulation of proteinase K-resistant PrP. We did not observe any alteration of these disease parameters in C12 knockouts (KO) compared to controls, suggesting that C12 is not required for prion pathogenesis.
MATERIALS AND METHODS
Western blotting
Brain homogenates were prepared in glass dounce homogenizers as 10% homogenates (weight/volume) of whole brain in PBS from samples frozen at indicated timepoints. After sonication, large debris were pelleted by low speed centrifugation. Further dilutions were made into RIPA buffer (20 mM Tris pH 8.0, 150 mM NaCl, 0.1% SDS, 0.5% DOC, 0.5% triton X-100) containing a protease inhibitor cocktail (Roche) or lysis buffer (PBS, 150 mM NaCl, 1% Triton X-100 and 1% Tween 20) without protease inhibitor for proteinase K digests. The equivalent of 30–50 ug of total protein was loaded onto 4–12% SDS-PAGE (Novex NuPage, Invitrogen), transferred to nitrocellulose membranes, and analysed by Western blotting. The following antibodies and dilutions were used: 6H4 anti-PrP 1:10,000 (Prionics), SAF83 anti-PrP 1:3000 (Cayman), anti-Caspase-12, 1:5,000 (Exalpha); anti-Grp58, 1:5,000 (StressGene), 1:1,000 anti-phospho JNK and anti-JNK (Cell Signaling Technology). For proteinase K treatment, 10% brain homogenates from terminally ill WT or Caspase-12 KOs were diluted to 1% in lysis buffer and treated with 50 μg/ml proteinase K (Invitrogen) for one hour at 37°C.
Caspase-12 knockout mice
All animal experiments were approved by the MIT committee on animal care. Mice were genotyped from isopropanol extracted DNA isolated from tail clips. C12 KO mice were previously described (ref. 18) and subsequently backcrossed to C57Bl/6 for > 10 generations. The primers for genotyping were “TN1” CCAATGAGCACTCTAAAATGATGAACT and “TN2” TTACAAAACTGATCACGTGGACAAAG for amplification of the WT allele and “PGKP-1” TTGGCGCTACCGGTGGATGTGGA ATGTG and TN2 amplify the knockout allele. We noted that C12 knockout females often died while giving birth, so we generated the mice used in this study by intercrossing mice heterozygous for the C12 deletion allele.
Prion inoculations
For prion inoculations, brain homogenate was prepared as a 1% weight/volume suspension from the brains of terminally ill Rocky Mountain Laboratory Chandler (“RML”) strain infected mouse brain in PBS + 2% FBS. Prions were inoculated directly into the brain using a 26 gauge needle. The titer of prions used was approximately 6.5logLD50/30ul. The samples shown in (Fig. 1A) were from RML inoculated CD1 mice (Taconic Farms).
Figure 1.
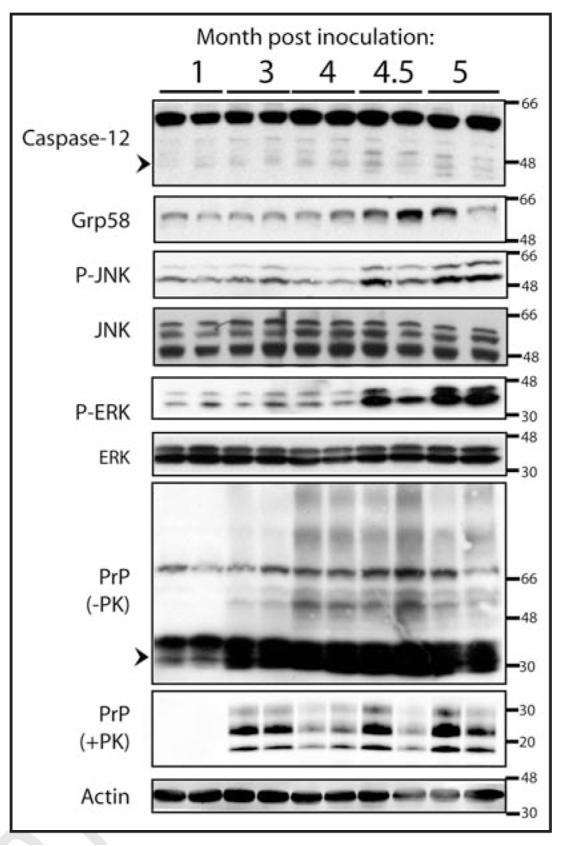
ER stress is activated in prion disease. Upregulation of ER stress markers was determined in prion infected CD1 mice (n = 2 per timepoint) after inoculation with 6.5logLD50 RML prions. Western blot analysis was performed to analyze the levels of C12, Grp58, JNK phosphorylation, ERK phosphorylation, total PrP and proteinase K-resistant PrP. The total level of JNK, ERK, and actin were measured as loading controls. Faint processing of C12 is visible at 4 months post inoculation (mpi) but more clearly at 4.5 and 5 mpi (active fragments of C12 are indicated by an arrow head). Phosphorylation of JNK (P-JNK) as well as induction of Grp58 was observed at 4.5 and 5 mpi. Phosphorylation of ERK was observed at 4.5 and 5 mpi. Higher order SDS-resistant PrP species were first visible at 3 mpi and thereafter (an arrow head marks the migration of monomeric PrP) along with proteinase K resistant PrP.
Behavioral analysis
Behavioral analysis was performed essentially as previously described (ref. 22). Briefly, C12 WT and KO mice (n = 6–9 per group) were singly housed and video recorded for a single 24 hour light-dark cycle (using dim red lighting during the dark cycle) beginning at one month post inoculation (mpi), then at 2, 3, 3.5, 4, 4.5 and 5 mpi video data was analyzed using HomecageScan 2.0 software (Clever Systems, Inc.). Data in (Fig. 1C—E) represents the mean and standard error of the mean pooling across each genotype at each time point. Data is shown beginning at 3 mpi which is prior to the onset of symptoms in RML infected C57BL/6 mice.
Neuropathological analysis
Brains were immersion fixed in formalin, paraffin embedded, and sectioned coronally. Five micron sections were stained with hematoxylin and eosin. For GFAP immunostaining, sections were deparaffinized and then incubated in 98.6 degree waterbath at pH 6.0 for 40 minutes, after which the sections were stained using an automated stainer, using a rabbit anti-cow GFAP antibody at 1:400 dilution (Dako). A blinded analysis of vacuolation and gliosis in the hippocampus, thalamus, striatum, cortex and cerebellum was conducted.
Statistical analysis
Statistical analysis was performed using GraphPad Prism. A Wilcoxon rank sum test was used for analysis of behavioral data and a log rank test was used for analysis of survival data. No statistically significant (p < 0.05) differences were detected.
RESULTS
First, we confirmed that Rocky Mountain Laboratory (RML) prion strain infection of CD mice leads to the activation of C12. Indeed, caspase activation was evident by its proteolytic processing at the later and end stage of disease, 4.5 and 5 months post inoculation (mpi.) as previously reported in Figure 1. In addition, a progressive upregulation of the ER chaperone Grp58 was observed after prion infection (Fig. 1). This response was consistent with, but less robust, than previous reports using a different prion strain, 139A.12, 13 Phosphorylation of Jun N-terminal kinase (JNK), which is activated by ER stress23 was induced in prion inoculated mice at 4.5 mpi (Fig. 1, P-JNK). Furthermore phosphorylation of extracellular signal-regulated kinase (ERK), another marker of ER stress24 was activated at later stages of prion disease (4.5 and 5 mpi) (Fig. 1). We also tested activation of the p38 MAPK but did not detect an induction at any stage of prion disease (data not shown). We noted an increase in aggregated PrP during the course of infection as indicated by a smear of SDS-resistant higher order oligomeric structures of PrP using the 6H4 antibody and appearance of proteinase K-resistant PrP fragments appearing at 3 mpi (Fig. 1).
To determine whether C12 promotes the pathogenesis of infectious prion disease in vivo, we infected mice deficient for C1218 with prions. This C12 knockout line has no known phenotypes other than resistance to ER-stress induced apoptosis as previously described (ref. 18). We inoculated C12KO (n = 13) and control mice (n = 26) (which were C12+/+ and C12+/-) intracranially with RML strain of murine prions (6.5 log LD50/dose) and assessed survival. Surprisingly, we observed no significant differences in the duration of time required for the C12 KO and WT groups to succumb to disease. Median survival was 171 days post inoculation for C12 KO and 169 days post inoculation for C12 WT (p = 0.13, log rank test) (Fig. 2).
Figure 2.
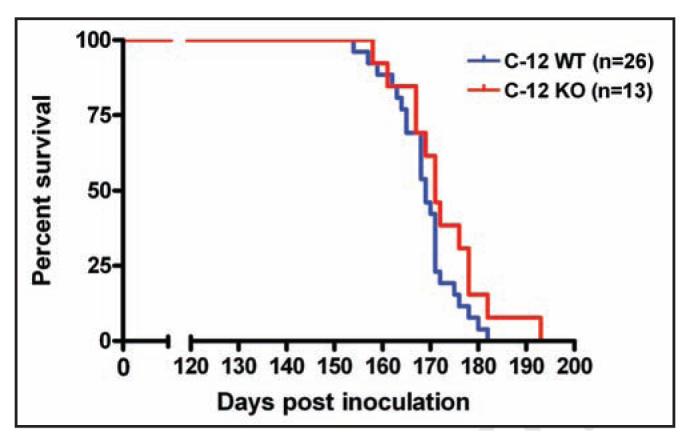
The survival after prion inoculation of C12 WT (either +/+ or +/- for C12) and KO. The median survival for C12 WT was 169 days and median survival for C12 KO was 171 days (P = 0.13, log rank test).
We also examined the onset of behavioral symptoms using high resolution automated video-based behavior analysis of home cage activities, a recently developed technique that robustly distinguishes between diseased mice inoculated with prions versus mock inoculated controls.22 As previously reported, the symptoms of RML prion disease in C57BL/6 mice (the strain background used in this study) include decreased grooming and hanging, as well as hyperactivity as measured by the percent of total time spent walking. We video recorded C12 WT and KO mice (n = 6–9 per group) following prion inoculation. Their behaviors were indistinguishable, grooming and hanging showed a similar decline (Fig. 3A and B) while walking showed a similar increase (Fig. 3C), suggesting that deletion of C12 during prion disease does not alter the brain circuits that are responsible for the behaviors examined.
Figure 3.

Behavioral analysis of prion inoculated C12 WT and KO mice (n = 6–9 per group). Mice were single housed and video recorded for 24 hours and their behaviors were quantitated by Homecagescan 2.0 software. A similar decrease in the amount of time spent grooming (A) and hanging (B) was observed and a similar dramatic increase in activity as measured by the percent of total time spent walking (C) was observed in C12 WT and KO mice inoculated with prions. Data is shown as mean values plus standard error of the mean. No statistically significant differences (at a threshold of p < 0.05) were observed for grooming, hanging and walking (Wilcoxon rank sum test).
In addition, we conducted detailed neuropathological analysis of C12 KO and control brains at 4.5 and 5 mpi. Their disease state was first assessed at the light microscopic level by hematoxylin and eosin staining. Histological analysis of brain sections revealed extensive but very similar cell damage and vacuolation in C12 KO and WT prion infected brains compared to uninoculated control brains (Fig. 4A, i–iii). In addition to promoting ER stress-induced cell death, C12 may function in the inflammatory response.25,26 To this end, we determined GFAP immunoreactivity, a measure of inflammation in the brain expressed as GFAP synthesis in reactive astrocytes. GFAP staining increased during disease progression but was indistinguishable between C12 WT and KOs (Figs. 4A, iv–vi).
Figure 4.

Analysis of spongiform changes in C12 WT and KO brain sections (A, i and iii) show a similar amount of vacuolation, indicated by arrows, in the hippocampus of prion inoculated mice but no vacuolation in uninoculated mice [C12 WT is shown in (A, i)]. The amount of gliosis was examined by staining for GFAP, which did not show staining in uninoculated samples [C12 WT is shown in (A, iv)] but showed abundant staining in prion inoculated samples from C12 WT and C12 KO (v and vi). For all of these parameters, blinded analysis did not reveal any differences between prion inoculated C12 KO and control brains. (B) The amount of proteinase K resistant PrP was assayed in whole brain homogenates taken from prion inoculated C12 WT (n = 5) and C12 KO (n = 5) mice (treated for 50ug/ml PK for one hour at 37C), which all showed ample PK resistant PrP by immunoblotting with SAF83. Total PrP inputs are shown to ensure equivalent loading.
Finally, we assessed the amount of prion replication in C12 KOs by western blotting as measured by the accumulation in the brain of proteinase K resistant PrP, the classic surrogate marker for prion infection. In terminally ill C12 KO and control brains (n = 5 of each group), we observed high levels of proteinase K resistant PrP in whole brain homogenates but no notable differences between C12 KOs and controls (Fig. 4B).
DISCUSSION
A common feature of many neurodegenerative diseases is the accumulation of misfolded proteins, which is thought to be linked to neuronal dysfunction and death.27-29 The pathways leading to neuro-degeneration have yet to be elucidated for many neurodegenerative disorders. Interestingly, disease progression and neuronal loss correlate with C12 processing in many different models of neurological disorders associated with protein misfolding, including Parkinson’s, Alzheimer’s and Huntington’s disease, amyotrophic lateral sclerosis, and spinocerebellar ataxias.18,19,21,30-34 The processing of C12 has been observed in prion-diseased brains from diverse species.12 To our knowledge, our study is the first to address the necessity of C12 in mediating neurodegeneration. Since C12 knockouts inoculated with RML prions do not show alterations in survival, behavior, or pathology, C12 is not required for prion pathogenesis. However, it is possible that inoculation with different prion strains or a lower dose of prions than that used in our study could have revealed a subtle protective role for C12 deletion. Our results do not rule out ER stress as a pathogenic mechanism in prion disease, in that there may be compensatory mechanisms for induction of cellular death or damage in C12 knockout mice. A very recent study of prion disease in neuronal knockouts of the transcription factor XBP-1, which is a key mediator of the unfolded protein response and activated in prion infection, showed no alterations in prion pathogenesis disease using the 139A murine prion strain.35
Many in vitro and in vivo models have been developed to understand the molecular basis of prion neurotoxicity. Yet at present, there is conflicting evidence-possibly due to differences in the etiology of the disease in various model systems-on the role of apoptotic cell death in prion diseases. Studies by Chiesa et al.36 utilizing a transgenic mouse expressing a familial mutant PrP support a role for Bax in promoting the execution of apoptotic cell death. In a different model of prion toxicity, where a truncated PrP mutant (PrPΔ32-34) shows cerebellar toxicity only in a PrP null background,37 apoptotic pathways are also implicated. Two groups report a delay in disease onset and neuronal death in PrPΔ32-134 transgenic mice deleted for Bax38 or overexpressing Bcl-2.39 Moreover, Heitz, et al. provide evidence that Purkinje cell loss is mediated by Bax/Bcl-2 cell death pathways in a related model in which the PrP homolog, doppel, is ectopically expressed in the brains of PrP deficient mice40,41 whereas Dong et al. have opposing results.42
In infectious forms of prion disease, the apoptotic pathways mediated by Bax are unlikely to play a major role. Inoculation of Bax KO mice with a BSE derived prion strain did not alter survival or pathology.43 Also, Bax deletion or transgenic over expression of Bcl-2 specifically in neurons does not protect and may even subtly enhance several aspects of RML prion-mediated neurodegeneration.44 It is likely that both apoptotic and nonapoptotic forms of cell death are activated in prion disease in a context dependent manner. Teasing apart the causal pathways of prion neurotoxicity, and that of other misfolded proteins, remains an enormous challenge in neurodegenerative disease research.
ACKNOWLEDGEMENTS
We are grateful to Artur Topolszki (WIBR) for providing expert technical assistance with all aspects of mouse work and to Marie Hardwick (Johns Hopkins) for providing the Caspase-12 knockouts. Funding for this work was provided by the US Department of Defense (DAMD17-00-1-0296), Ellison Medical Research Foundation, and the Howard Hughes Medical Institute (to S.L.) and FONDAP (15010006), FONDECYT (1070444) and the HighQ foundation (to C.H.).
References
- 1.Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95:13363–83. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aguzzi A, Heikenwalder M. Pathogenesis of prion diseases: Current status and future outlook. Nat Rev Microbiol. 2006;4:765–75. doi: 10.1038/nrmicro1492. [DOI] [PubMed] [Google Scholar]
- 3.Caughey B, Baron GS. Prions and their partners in crime. Nature. 2006;443:803–10. doi: 10.1038/nature05294. [DOI] [PubMed] [Google Scholar]
- 4.Steele AD, Lindquist S, Aguzzi A. The prion protein knockout mouse: A phenotype under challenge. Prion. 2007;1:83–93. doi: 10.4161/pri.1.2.4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jeffrey M, Gonzalez L. Pathology and pathogenesis of bovine spongiform encephalopathy and scrapie. Curr Top Microbiol Immunol. 2004;284:65–97. doi: 10.1007/978-3-662-08441-0_3. [DOI] [PubMed] [Google Scholar]
- 6.Hetz CA, Soto C. Stressing out the ER: A role of the unfolded protein response in prion-related disorders. Curr Mol Med. 2006;6:37–43. doi: 10.2174/156652406775574578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liberski PP, Sikorska B, Bratosiewicz-Wasik J, Gajdusek DC, Brown P. Neuronal cell death in transmissible spongiform encephalopathies (prion diseases) revisited: From apoptosis to autophagy. Int J Biochem Cell Biol. 2004;36:2473–90. doi: 10.1016/j.biocel.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 8.Ma J, Lindquist S. Wild-type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation. Proc Natl Acad Sci USA. 2001;98:14955–60. doi: 10.1073/pnas.011578098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ivanova L, Barmada S, Kummer T, Harris DA. Mutant prion proteins are partially retained in the endoplasmic reticulum. J Biol Chem. 2001;276:42409–21. doi: 10.1074/jbc.M106928200. [DOI] [PubMed] [Google Scholar]
- 10.Drisaldi B, Stewart RS, Adles C, Stewart LR, Quaglio E, Biasini E, Fioriti L, Chiesa R, Harris DA. Mutant PrP is delayed in its exit from the endoplasmic reticulum, but neither wild-type nor mutant PrP undergoes retrotranslocation prior to proteasomal degradation. J Biol Chem. 2003;278:21732–43. doi: 10.1074/jbc.M213247200. [DOI] [PubMed] [Google Scholar]
- 11.Hetz C, Castilla J, Soto C. Perturbation of endoplasmic reticulum homeostasis facilitates prion replication. J Biol Chem. 2007;282:12725–33. doi: 10.1074/jbc.M611909200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hetz C, Russelakis-Carneiro M, Maundrell K, Castilla J, Soto C. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. Embo J. 2003;22:5435–45. doi: 10.1093/emboj/cdg537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hetz C, Russelakis-Carneiro M, Walchli S, Carboni S, Vial-Knecht E, Maundrell K, Castilla J, Soto C. The disulfide isomerase Grp58 is a protective factor against prion neurotoxicity. J Neurosci. 2005;25:2793–802. doi: 10.1523/JNEUROSCI.4090-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown AR, Rebus S, McKimmie CS, Robertson K, Williams A, Fazakerley JK. Gene expression profiling of the preclinical scrapie-infected hippocampus. Biochem Biophys Res Commun. 2005;334:86–95. doi: 10.1016/j.bbrc.2005.06.060. [DOI] [PubMed] [Google Scholar]
- 15.Yoo BC, Krapfenbauer K, Cairns N, Belay G, Bajo M, Lubec G. Overexpressed protein disulfide isomerase in brains of patients with sporadic Creutzfeldt-Jakob disease. Neurosci Lett. 2002;334:196–200. doi: 10.1016/s0304-3940(02)01071-6. [DOI] [PubMed] [Google Scholar]
- 16.Hetz C, Bernasconi P, Fisher J, Lee AH, Bassik MC, Antonsson B, Brandt GS, Iwakoshi NN, Schinzel A, Glimcher LH, Korsmeyer SJ. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1 alpha. Science. 2006;312:572–6. [Google Scholar]
- 17.Degterev A, Boyce M, Yuan J. A decade of caspases. Oncogene. 2003;22:8543–67. doi: 10.1038/sj.onc.1207107. [DOI] [PubMed] [Google Scholar]
- 18.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 19.Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K, Tsujimoto Y, Tohyama M. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J Cell Biol. 2004;165:347–56. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rao RV, Hermel E, Castro-Obregon S, del Rio G, Ellerby LM, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program: Mechanism of caspase activation. J Biol Chem. 2001;276:33869–74. doi: 10.1074/jbc.M102225200. [DOI] [PubMed] [Google Scholar]
- 21.Hitomi J, Katayama T, Taniguchi M, Honda A, Imaizumi K, Tohyama M. Apoptosis induced by endoplasmic reticulum stress depends on activation of caspase-3 via caspase-12. Neurosci Lett. 2004;357:127–30. doi: 10.1016/j.neulet.2003.12.080. [DOI] [PubMed] [Google Scholar]
- 22.Steele AD, Jackson WS, King OD, Lindquist S. The power of automated high-resolution behavior analysis revealed by its application to mouse models of Huntington‘s and prion diseases. Proc Natl Acad Sci USA. 2007;104:1983–8. doi: 10.1073/pnas.0610779104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–6. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 24.Arai K, Lee SR, van Leyen K, Kurose H, Lo EH. Involvement of ERK MAP kinase in endoplasmic reticulum stress in SH-SY5Y human neuroblastoma cells. J Neurochem. 2004;89:232–9. doi: 10.1111/j.1471-4159.2004.02317.x. [DOI] [PubMed] [Google Scholar]
- 25.Saleh M, Mathison JC, Wolinski MK, Bensinger SJ, Fitzgerald P, Droin N, Ulevitch RJ, Green DR, Nicholson DW. Enhanced bacterial clearance and sepsis resistance in caspase-12-deficient mice. Nature. 2006;440:1064–8. doi: 10.1038/nature04656. [DOI] [PubMed] [Google Scholar]
- 26.Obeng EA, Boise LH. Caspase-12 and caspase-4 are not required for caspase-dependent endoplasmic reticulum stress-induced apoptosis. J Biol Chem. 2005;280:29578–87. doi: 10.1074/jbc.M502685200. [DOI] [PubMed] [Google Scholar]
- 27.Gitler AD, Shorter J. Prime time for alpha-synuclein. J Neurosci. 2007;27:2433–4. doi: 10.1523/JNEUROSCI.0094-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steele AD, Yi CH. Neuromuscular denervation: Bax up against the wall in amyotrophic lateral sclerosis. J Neurosci. 2006;26:12849–51. doi: 10.1523/JNEUROSCI.4086-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aguzzi A, Haass C. Games played by rogue proteins in prion disorders and Alzheimer‘s disease. Science. 2003;302:814–8. doi: 10.1126/science.1087348. [DOI] [PubMed] [Google Scholar]
- 30.Aoyama K, Burns DM, Suh SW, Garnier P, Matsumori Y, Shiina H, Swanson RA. Acidosis causes endoplasmic reticulum stress and caspase-12-mediated astrocyte death. J Cereb Blood Flow Metab. 2005;25:358–70. doi: 10.1038/sj.jcbfm.9600043. [DOI] [PubMed] [Google Scholar]
- 31.Larner SF, Hayes RL, McKinsey DM, Pike BR, Wang KK. Increased expression and processing of caspase-12 after traumatic brain injury in rats. J Neurochem. 2004;88:78–90. doi: 10.1046/j.1471-4159.2003.02141.x. [DOI] [PubMed] [Google Scholar]
- 32.Wootz H, Hansson I, Korhonen L, Napankangas U, Lindholm D. Caspase-12 cleavage and increased oxidative stress during motoneuron degeneration in transgenic mouse model of ALS. Biochem Biophys Res Commun. 2004;322:281–6. doi: 10.1016/j.bbrc.2004.07.118. [DOI] [PubMed] [Google Scholar]
- 33.Shibata M, Hattori H, Sasaki T, Gotoh J, Hamada J, Fukuuchi Y. Activation of caspase-12 by endoplasmic reticulum stress induced by transient middle cerebral artery occlusion in mice. Neuroscience. 2003;118:491–9. doi: 10.1016/s0306-4522(02)00910-7. [DOI] [PubMed] [Google Scholar]
- 34.Kalai M, Lamkanfi M, Denecker G, Boogmans M, Lippens S, Meeus A, Declercq W, Vandenabeele P. Regulation of the expression and processing of caspase-12. J Cell Biol. 2003;162:457–67. doi: 10.1083/jcb.200303157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hetz C, Lee AH, Gonzalez-Romero D, Thielen P, Castilla J, Soto C, Glimcher LH. The Unfolded Protein Response transcription factor XBP-1 does not influence Prion replication or pathogenesis. Proc Natl Acad Sci USA. 2008;105:757–62. doi: 10.1073/pnas.0711094105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chiesa R, Piccardo P, Dossena S, Nowoslawski L, Roth KA, Ghetti B, Harris DA. Bax deletion prevents neuronal loss but not neurological symptoms in a transgenic model of inherited prion disease. Proc Natl Acad Sci USA. 2005;102:238–43. doi: 10.1073/pnas.0406173102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shmerling D, Hegyi I, Fischer M, Blattler T, Brandner S, Gotz J, Rulicke T, Flechsig E, Cozzio A, von Mering C, Hangartner C, Aguzzi A, Weissmann C. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell. 1998;93:203–14. doi: 10.1016/s0092-8674(00)81572-x. [DOI] [PubMed] [Google Scholar]
- 38.Li A, Barmada SJ, Roth KA, Harris DA. N-terminally deleted forms of the prion protein activate both Bax-dependent and Bax-independent neurotoxic pathways. J Neurosci. 2007;27:852–9. doi: 10.1523/JNEUROSCI.4244-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nicolas O, Gavin R, Braun N, Urena JM, Fontana X, Soriano E, Aguzzi A, Del Rio JA. Bcl-2 overexpression delays caspase-3 activation and rescues cerebellar degeneration in prion-deficient mice that overexpress amino-terminally truncated prion. Faseb J. 2007;21:3107–17. doi: 10.1096/fj.06-7827com. [DOI] [PubMed] [Google Scholar]
- 40.Heitz S, Lutz Y, Rodeau JL, Zanjani H, Gautheron V, Bombarde G, Richard F, Fuchs JP, Vogel MW, Mariani J, Bailly Y. BAX contributes to Doppel-induced apoptosis of prion-protein-deficient Purkinje cells. Dev Neurobiol. 2007;67:670–86. doi: 10.1002/dneu.20366. [DOI] [PubMed] [Google Scholar]
- 41.Heitz S, Gautheron V, Lutz Y, Rodeau JL, Zanjani HS, Sugihara I, Bombarde G, Richard F, Fuchs JP, Vogel MW, Mariani J, Bailly Y. BCL-2 counteracts Doppel-induced apoptosis of prion-protein-deficient Purkinje cells in the Ngsk Prnp(0/0) mouse. Dev Neurobiol. 2007;68:332–48. doi: 10.1002/dneu.20555. [DOI] [PubMed] [Google Scholar]
- 42.Dong J, Li A, Yamaguchi N, Sakaguchi S, Harris DA. Doppel induces degeneration of cerebellar purkinje cells independently of Bax. Am J Pathol. 2007;171:599–607. doi: 10.2353/ajpath.2007.070262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coulpier M, Messiaen S, Hamel R. Fernandez de Marco M, Lilin T, Eloit M. Bax deletion does not protect neurons from BSE-induced death. Neurobiol Dis. 2006;23:603–11. doi: 10.1016/j.nbd.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 44.Steele AD, King OD, Jackson WS, Hetz CA, Borkowski AW, Thielen P, Wollmann R, Lindquist S. Diminishing apoptosis by deletion of Bax or overexpression of Bcl-2 does not protect against infectious prion toxicity in vivo. J Neurosci. 2007;27:13022–7. doi: 10.1523/JNEUROSCI.3290-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]