

Abstract
Drug resistance is a costly consequence of pathogen evolution and a major concern in public health. In this review, we show how population genetics can be used to study the evolution of drug resistance and also how drug resistance evolution is informative as an evolutionary model system. We highlight five examples from diverse organisms with particular focus on: (i) identifying drug resistance loci in the malaria parasite Plasmodium falciparum using the genomic signatures of selective sweeps, (ii) determining the role of epistasis in drug resistance evolution in influenza, (iii) quantifying the role of standing genetic variation in the evolution of drug resistance in HIV, (iv) using drug resistance mutations to study clonal interference dynamics in tuberculosis and (v) analysing the population structure of the core and accessory genome of Staphylococcus aureus to understand the spread of methicillin resistance. Throughout this review, we discuss the uses of sequence data and population genetic theory in studying the evolution of drug resistance.
Keywords: adaptation, clonal interference, epistasis, horizontal gene transfer, selective sweep, standing genetic variation
Introduction
Pathogen evolution is a major public health concern with enormous societal consequences around the world. Pathogens evolve quickly, allowing them to jump from multiple host species to humans (e.g. SARS coronavirus or Ebola virus; Woolhouse et al. 2005), to become more virulent and evade immune pressure within humans (e.g. influenza virus; Grenfell et al. 2004) and to become resistant to drugs used to combat them (WHO 2014d). In particular, drug resistance results in numerous deaths, increased hospitalizations and prolonged treatments (WHO 2014d). In addition to the costs borne by human health, the Centers for Disease Control and Prevention estimate the economic costs of drug resistance to be on the order of tens of billions of dollars a year in the United States alone (CDC 2011). While pharmaceutical intervention has played a critical role in our efforts to control epidemic pathogens, there is an urgent need to understand the evolution and spread of drug resistance.
Pathogens share common biological features that allow them to adapt rapidly under extreme selective pressures (imposed, for example, by drugs) on observable timescales. Pathogen populations with large sizes, high mutation rates and short generation times are likely to generate drug resistance mutations in a short amount of time. Pathogens also encompass a wide variety of organisms that include eukaryotes, prokaryotes and viruses. This means that our efforts to understand how pathogens evolve drug resistance need to account for factors such as: variable genome sizes [ranging from a few kilobases (Kb) in RNA viruses to many megabases (Mb) in Plasmodium falciparum], variable mutation rates (from 10−9 per base pair per generation in eukaryotes to 10−5 in some RNA viruses), alternative methods of genetic exchange (including viral re-assortment and horizontal gene transfer in bacteria) and clonal vs. recombinant forms of reproduction (see Box 1 for more on diverse modes of genetic exchange and recombination). In addition, the scale at which drug resistance evolves varies widely. For example, in HIV drug resistance mostly evolves within patients and is rarely transmitted (Wheeler et al. 2010), whereas in other pathogens (e.g. malaria), drug resistance is often transmitted between hosts, making certain drugs ineffective for a large fraction of new patients (Hyde 2005).
Box 1. Genetic exchange, re-assortment and recombination in Plasmodium falciparum, influenza, HIV, Mycobacterium tuberculosis and Staphylococcus aureus.
Genetic material can be moved between cells during asexual or sexual reproduction (i.e. vertical transmission), or it can be exchanged between cells within the same generation via horizontal gene transfer (HGT, see Box 3). These processes can allow the re-assortment of individual molecules of genetic material (e.g. chromosomal or viral reassortment), which generates new allelic combinations. Perhaps equally important to generating diversity is recombination, which occurs in all three domains of life as well as in viral organisms [homologous recombination appears to be the most conserved form of recombination across the tree of life, particularly as a means of repairing DNA double-strand breaks (Cromie et al. 2001)]. Recombination is the process by which individual molecules of genetic material are broken and then recombined to produce new combinations of alleles. Genetic exchange, chromosomal and viral re-assortment and recombination are critical to the generation and maintenance of genetic variation in natural populations. To understand adaptation in the genomes of the pathogens discussed in this review, we must have an understanding of these basic processes in each of the pathogens.
Plasmodium falciparum
This eukaryotic organism has 14 linear chromosomes (~23 Mb total) and a complex life cycle in which reproduction is asexual inside of human cells and sexual within the gut of the mosquito. Recombination occurs during meiosis/sexual reproduction and is quite high (see Box 2). Additionally, recombination is also used during the asexual stage to rearrange segments of the haploid genome, a potentially important factor in immune evasion during infection of the host (Jiang et al. 2011; Claessens et al. 2014).
Influenza virus A
Each viral particle contains eight RNA molecules (~14 kb total). When multiple virions inject their RNA into a single host cell, these RNA molecules can be shuffled before being re-encapsulated into a new virion (‘viral re-assortment’). While individual RNA molecules tend to evolve clonally and in the absence of recombination (Boni et al. 2008; Strelkowa & Lässig 2012), re-assortment of molecules between different virions plays an epidemiologically important role in influenza evolution (Holmes et al. 2005; Rambaut et al. 2008; Simon-Loriere & Holmes 2011).
Human Immunodeficiency Virus
This retrovirus’ genome is contained on one RNA molecule (~10 kb); however, each viral particle has two copies of it (i.e. diploidy). Upon injection of the virus’ genome into a host cell, it is reverse transcribed and then integrated into the host cell’s nuclear DNA. If multiple viral particles inject their genomes into the same host cell, then RNA molecules can be re-assorted to create heterozygous viral particles. Recombination between the two RNA molecules occurs, although note that instead of homologous recombination, retroviruses typically do copy-choice recombination in which the reverse transcriptase jumps between two different RNA molecules and thus creates a new allelic combination (Negroni & Buc 2000; Simon-Loriere & Holmes 2011).
Mycobacterium tuberculosis
This asexually reproducing bacterium has a circular chromosome (~4 Mb). Although there is some evidence for infrequent horizontal gene transfer and recombination, this species is thought to be primarily clonal and generally lack an accessory genome (described below and defined in Box 3) (Cole et al. 1998; Veyrier et al. 2009; Namouchi et al. 2012).
Staphylococcus aureus
This asexually reproducing bacterium has a circular chromosome of ~2.8 Mb in size, of which ~75% is thought to make up the core genome (i.e. shared by all members of the S. aureus species, defined in Box 3). The accessory genome includes the remaining ~25% of this circular chromosome as well as extrachromosomal elements. The mobile genetic elements making up the accessory genome can be present within bacteriophages, chromosomal casettes (e.g. SCCmec), plasmids, transposons and others. These elements primarily move in S. aureus via transduction (defined in Box 3) and can recombine onto the circular chromosome (e.g. SCCmec) or with each other (Lindsay & Holden 2004; Malachowa & Deleo 2010).
To prevent or at least manage drug resistance in pathogens, it is imperative to understand the evolutionary aspects of the problem. However, evolutionary biologists have traditionally left most of the study of drug resistance to the medicine and epidemiology communities (Read & Huijben 2009). This has changed in recent years with interesting population genetic work being done on natural populations of pathogens, some of which is featured in this review. This change is, at least in part, driven by the availability of genetic data for a variety of pathogens. A new cross-disciplinary field is emerging that uses large amounts of genetic data and new theoretical advances from the world of population genetics. Some of this work is directly aimed at finding ways to prevent drug resistance evolution or spread, whereas other work is more basic in nature and mostly uses the pathogen as a model system to learn about evolution in general. In the best cases, we learn lessons on how to prevent resistance and on general evolutionary principles simultaneously.
The aim of this review was to introduce researchers from different fields to new and exciting work on drug resistance evolution happening at the intersection of population genetics and medical fields such as virology and infectious disease. Perhaps more importantly this review is intended as an advertisement of the strengths of using natural pathogen populations to study evolutionary principles as well as the strengths of using evolutionary theory to better understand the dynamics of infectious diseases. To this end, we present five case studies at the intersection of evolutionary theory and pathogen drug resistance. While the five examples we have chosen are not meant to be exhaustive in any way, each example showcases an evolutionary question in the context of a different pathogen, each with a unique biology and differences in genome size, methods of genetic exchange, reproduction and recombination (see Box 1).
We will describe the following five examples:
Selective sweep mapping in the malaria parasite P. falciparum resistant to the antimalarial drug artemisinin. This is a great example of how population genetic methods originally developed for other sexually recombining eukaryotic organisms, such as humans, could be applied to P. falciparum, which experiences a very high rate of sexual recombination. The mapping of the drug resistance mutations in P. falciparum has allowed researchers to track the prevalence of artemisinin resistance worldwide.
The role of epistasis in oseltamivir (Tamiflu) resistance in influenza. The evolution and spread of oseltamivir resistance in influenza in 2007–2008 was unexpected. Even though the causal mutation for oseltamivir resistance was known, it was thought to be too costly to viral fitness to spread widely. Phylogenetic methods allowed researchers to discover mutations that interacted with the resistance mutation to ameliorate its detrimental effects, allowing the resistance mutation to spread. Influenza has become a model that helps researchers understand the importance of genetic interactions between sites during the course of evolution and the potential for predicting future evolution.
The importance of standing genetic variation (SGV) (also referred to as minority variants) to treatment failure for human immunodeficiency virus (HIV). Drug resistance mutations are well known in HIV and the evolution of drug resistance happens independently in different patients. This makes it possible to determine the importance of SGV for drug resistance in HIV. Several studies found that both SGV and de novo mutations contribute to drug resistance in HIV.
Clonal interference dynamics in Mycobacterium tuberculosis. Drug resistance mutations are also well known in M. tuberculosis. Through tracking these mutations, recent studies have revealed that clonal interference plays a large role in the evolutionary dynamics within patients. An examination of these dynamics offers insights into how multiply-resistant strains emerge and also provides a rare look into a natural population evolving without recombination.
Population structure analysis of the core and accessory genome of methicillin-resistant Staphylococcus aureus (MRSA). Staphylococcus aureus reproduces clonally, yet drug resistance is often acquired via the spread of mobile genetic elements (or horizontal gene transfer), such as the gene cassette SCCmec that confers methicillin resistance. While the genealogy of the core genome of MRSA reveals that relatively few lineages have spread across the globe, phylogenetic analysis of SCCmec reveals that methicillin resistance arises on a more local scale.
These five examples will be used to illustrate that the growing availability of sequence data facilitates the application of population genetic methods to important questions in the evolution of drug resistance.
Linkage disequilibrium statistics for the detection of selective sweeps in the eukaryotic pathogen Plasmodium falciparum
Malaria results in over 500 000 deaths every year (WHO 2014c) and is mainly caused by P. falciparum. Over the past 60 years, several antimalarial drugs have been deployed, but resistance to these drugs has developed repeatedly. The current front-line treatment, artemisinin-based combination therapies (ACTs), was introduced in the 1990s at a time when all other drugs were failing. While ACTs were initially very effective, by 2003, the first signs of resistance to artemisinin emerged in South-East Asia, and now resistance is widespread (Noedl et al. 2008; Dondorp et al. 2009, 2010; Kyaw et al. 2013; Ashley et al. 2014).
To better track the global spread of resistance to antimalarial drugs and understand the mechanism by which resistance develops, it is essential to know the locations in the P. falciparum genome conferring resistance. Here, we review the population genetic methods used to identify the kelch gene on chromosome 13 conferring resistance to artemisinin—a locus that until recently had gone relatively unnoticed and whose biological function is still unknown—and the experimental methods used to confirm the causal mutations. We highlight P. falciparum in this review because it is a pathogen that experiences strong selective pressures and, unlike most human pathogens, it is a eukaryotic organism that experiences high rates of recombination (see Box 2). As we show, these properties of P. falciparum make it relatively straightforward to apply standard population genetics methods to malaria in order to detect selected sites with great success, even though these methods were often designed for other eukaryotic model organisms.
Box 2. Common methods to detect selective sweeps in recombining eukaryotes, including Plasmodium falciparum.
In a selective sweep, an adaptive mutation rises in frequency in the population, dragging with it the linked genetic material on the same haplotype background to high frequency via hitchhiking (Maynard Smith & Haigh 1974; Kaplan et al. 1989; Kim & Stephan 2002). This can result in a pattern of low genetic variation, high linkage disequilibrium (LD) and high haplotype homozygosity in the vicinity of the adaptive mutation. Plasmodium falciparum experiences a high rate of recombination [58 cM/Mb (Su et al. 1999; Jiang et al. 2011) in contrast to 0.6, 0.56, 1.26, and 2.32 cM/Mb in rats, mice, humans and Drosophila melanogaster respectively (Jensen-Seaman et al. 2004; Comeron et al. 2012)] and strong selective pressures from the presence of antimalarial drugs, thereby allowing LD-based statistics to identify regions in its 23 Mb genome under selection with high resolution.
There are several statistics that utilize LD and haplotype homozygosity information to identify selective sweeps, including extended haplotype homozygosity (EHH), cross population extended haplotype homozygosity (XP-EHH), as well as iHS, nSL and H12 (Sabeti et al. 2002, 2007; Voight et al. 2006; Ferrer-Admetlla et al. 2014; Garud et al. 2015). EHH and XP-EHH have been particularly popular and successful in the detection of sites under selection in P. falciparum. EHH measures the decay in tracts of homozygosity radiating away from a SNP at a partial frequency in a single population (Sabeti et al. 2002). However, EHH is not powerful in detecting a selective sweep that has gone to fixation (Sabeti et al. 2002). In contrast, XP-EHH utilizes information from two populations, one where a sweep has presumably occurred, and another where a sweep has not (Sabeti et al. 2007). XP-EHH is thus powerful in detecting partial and complete sweeps that have occurred in one of the two populations considered. In addition to LD-based statistics, there are several site frequency spectrum (SFS) statistics which treat individual polymorphic sites in a sample independently from one another. Statistics such as Tajima’s D (Tajima 1989), Fay and Wu’s H (Fay & Wu 2000; Fay et al. 2002), and Sweepfinder (Nielsen et al. 2005) are sensitive to dips in neutral diversity and excesses in low and high frequency polymorphisms in the SFS (Braverman et al. 1995; Vitti et al. 2013). Another widely used SFS statistic is FST (Lewontin & Krakauer 1973), which compares genomic patterns between two populations. More specifically, FST measures the difference in average pairwise diversity measured in randomly sampled individuals across two different populations vs. within a single population, and can thus be an indicator of positive selection if one population diverges extremely in diversity from another. As we explain in this review, FST has been successfully used in conjunction with EHH and XP-EHH to detect selected sites in P. falciparum across multiple populations.
Association tests are a LD-based approach often used to identify drug resistance loci in multiple study systems. Association tests correlate a phenotype with the allelic states of polymorphisms in a sample from a single population (Anderson et al. 2011). In the case of P. falciparum, artemisinin resistance is characterized by slow parasite clearance rates, measured as the rate of decay in parasite density in the blood over time (Noedl et al. 2008; Dondorp et al. 2009). Therefore, this phenotype is used in studies to identify the underlying mutations conferring resistance to artemisinin. Association studies can result in many false positives because of a number of confounding factors such as population substructure and relatedness between individuals in a sample. Furthermore, association studies can be computationally slow in large sample sizes with large numbers of polymorphic sites. Efficient mixed models association (EMMA) is an association technique that aims to address these factors (Kang et al. 2008).
Using a variety of methods, several loci that confer resistance to older antimalarial drugs such as mefloquine, chloroquine, sulfadoxine–pyrimethamine and atovaquone (e.g. genes encoding the chloroquine resistance transporter, multidrug resistance protein, GTP-cyclohydrolase I, dihydropteroate synthetase, dihydrofolate reductase and cytochrome B) have been identified (Wootton et al. 2002; Nair et al. 2003; Ferdig et al. 2004; Sidhu et al. 2005; Musset et al. 2007; Vinayak et al. 2010; Anderson et al. 2011; Mita et al. 2011). Close examination of the genomic signatures immediately surrounding these loci reveals patterns of reduced diversity, elevated linkage disequilibrium (LD), and the presence of long haplotypes at high frequency (Wootton et al. 2002; Nair et al. 2003; Roper et al. 2004; Vinayak et al. 2010), all of which are associated with signatures of selective sweeps (Maynard Smith & Haigh 1974; Kaplan et al. 1989; Kim & Stephan 2002). The distinct genomic signatures resulting from drug resistance in P. falciparum suggest that novel loci conferring resistance to artemisinin can be discovered by searching for patterns of elevated LD and haplotype homozygosity. In Box 2, we discuss some of the popular population genomic methods used to identify selective sweeps and resistant loci in P. falciparum, including LD statistics such as XP-EHH (Sabeti et al. 2007), SFS statistics used to corroborate findings made with LD statistics, such as FST (Lewontin & Krakauer 1973) and genomewide association studies.
Population genomic methods identify a region on chromosome 13 associated with artemisinin resistance
Three recent studies by Cheeseman et al. (2012), Takala-Harrison et al. (2013), and Miotto et al. (2013) examined patterns of LD and haplotype homozygosity in the P. falciparum genome and identified a region on chromosome 13 associated with artemisinin resistance. This locus was later characterized as the kelch13 locus (Ariey et al. 2014). These three studies benefited greatly from deep samples of P. falciparum from multiple populations, some of which displayed the artemisinin drug resistance phenotype of slow parasite clearance rate while others did not (Noedl et al. 2008; Dondorp et al. 2009). This data allowed the authors to contrast genomic patterns in strains from geographical locations displaying the slow parasite clearance rate phenotype vs. those that did not.
In the first major study, Cheeseman et al. (2012) examined genomic data from samples from three populations: Laos, Thailand and Cambodia, where resistance is observed only in Thailand and Cambodia (Cheeseman et al. 2012). This data set offered the opportunity to contrast the Thai and Cambodia samples with Laos to discover putative loci conferring resistance to artemisinin. Cheeseman et al. (2012) utilized a two-step approach to identify genomic regions underlying resistance to artemisinin. First, the authors used the XP-EHH (Sabeti et al. 2007) and FST (Lewontin & Krakauer 1973) statistics and identified 33 regions as significant candidates under selection in at least one population. These statistics were particularly appropriate for their multipopulation data set because they are designed to contrast genomic signatures in two populations (Box 2). Cheeseman et al. (2012) found that 10 of the 33 regions discovered were associated with positive selection in studies examining different drugs, validating their approach. Second, Cheeseman et al. (2012) examined the associations of these 33 regions with the slow parasite clearance rate phenotype and identified two adjacent single-nucleotide polymorphisms (SNPs) 14 kb apart in their sample on chromosome 13 showing significant associations. Cheeseman et al. (2012) then performed fine mapping in this region and applied the EHH statistic (Box 2) to visually confirm the extended haplotype homozygosity pattern in a 35-kb region on chromosome 13 characteristic of a selective sweep.
Similar to the Cheeseman et al. (2012) study, Takala-Harrison et al. (2013) examined multiple populations from Cambodia, Thailand and Bangladesh for signatures of selection and association with the slow parasite clearance phenotype, with only Cambodian and Thai samples showing resistance phenotypes. The authors used efficient mixed-model association (EMMA, see Box 2) (Kang et al. 2008) to test the association of each SNP in their data set and the parasite clearance phenotype, thereby identifying four SNPs significantly associated, including two on chromosome 13. In conjunction with the association test, Takala-Harrison et al. (2013) applied XP-EHH (Sabeti et al. 2007) and FST (Lewontin & Krakauer 1973) to the Cambodia population, using the Thai and Bangladeshi populations as comparison populations. The authors found that only the polymorphisms on chromosome 13 previously identified with the EMMA test also had significant XP-EHH and FST values. Takala-Harrison et al. (2013) used HAPLOVIEW (Barrett et al. 2005) to visualize the extended haplotype homozygosity in the region.
Miotto et al. (2013) examined 10 locations in West Africa and South-East Asia and found that there was an exceptionally high amount of population substructure within Cambodia. Upon closer examination, the authors found that three of four clusters of P. falciparum found in western Cambodia showed slow parasite clearance rates, slow decay in LD, and loss of haplotype diversity, while the fourth cluster prevalent in northeastern Cambodia did not show any of these characteristics. This leads the authors to conclude that western Cambodia harbours at least three distinct populations of artemisinin-resistant P. falciparum. In particular, one of the subpopulations with resistant strains of P. falciparum showed a single haplotype extending across half of chromosome 13, corroborating the evidence from Cheeseman et al. (2012) and Takala-Harrison et al. (2013) that this locus is implicated in resistance to artemisinin.
Experimental confirmation of the kelch gene as a likely target of adaptation
While Cheeseman et al. (2012), Takala-Harrison et al. (2013) and Miotto et al. (2013) were all able to localize a putative locus on chromosome 13 strongly associated with the slow parasite clearance phenotype, it was only recently that Ariey et al. (2014) were able to identify causative mutations with high confidence. Ariey et al. (2014) used an in vitro drug selection technique to subject a parasite line to high doses of artemisinin for 5 years. Comparing the sequenced data from the selected line with that of a clonal population not experiencing any selection, the authors identified eight mutations in seven genes that were present in the artemisinin treatment group but absent from the control group. Ariey et al. (2014) narrowed their list of candidates and concluded that only the mutation appearing in the kelch gene on chromosome 13 appeared at the same time as when artemisinin resistance developed in their treatment group. To determine whether there was concordance between the presence of this mutation in the kelch gene and artemisinin-resistant parasites from Cambodia, Ariey et al. (2014) sequenced the locus at which these mutations were present in parasite samples from patients showing the drug-resistant phenotype and from patients who did not in different geographical locations in Cambodia. They found that mutations in the kelch gene were strongly associated with the slow clearance phenotype observed in the locations where malaria is prevalent. In this extended analysis, Ariey et al. (2014) identified 17 mutations in the kelch gene, and all were significantly associated with artemisinin resistance.
Several follow-up studies confirmed the findings of Ariey et al. (2014). Ashley et al. (2014) examined the geographical extent of resistance by tracking the prevalence of mutations in the kelch gene (see Fig. 1). The authors found several single point mutations in kelch significantly associated with slow parasite clearance rates. Recent work (Cheeseman et al. 2015; Miotto et al. 2015; Takala-Harrison et al. 2015; Tun et al. 2015) has also precisely mapped the origins and extent of mutations associated with artemisinin resistance (see Fig. 1). Ghorbal et al. (2014) used the CRISPR-Cas9 system to introduce a mutation (C580Y) implicated in artemisinin resistance (Ariey et al. 2014) into kelch, which produced the slow parasite clearance phenotype and demonstrated the first direct link between a mutant kelch and the characteristic phenotype. This work was expanded upon by Straimer et al. (2015) to confirm the role of multiple mutations and additional genetic factors in conferring artemisinin resistance.
Fig. 1.
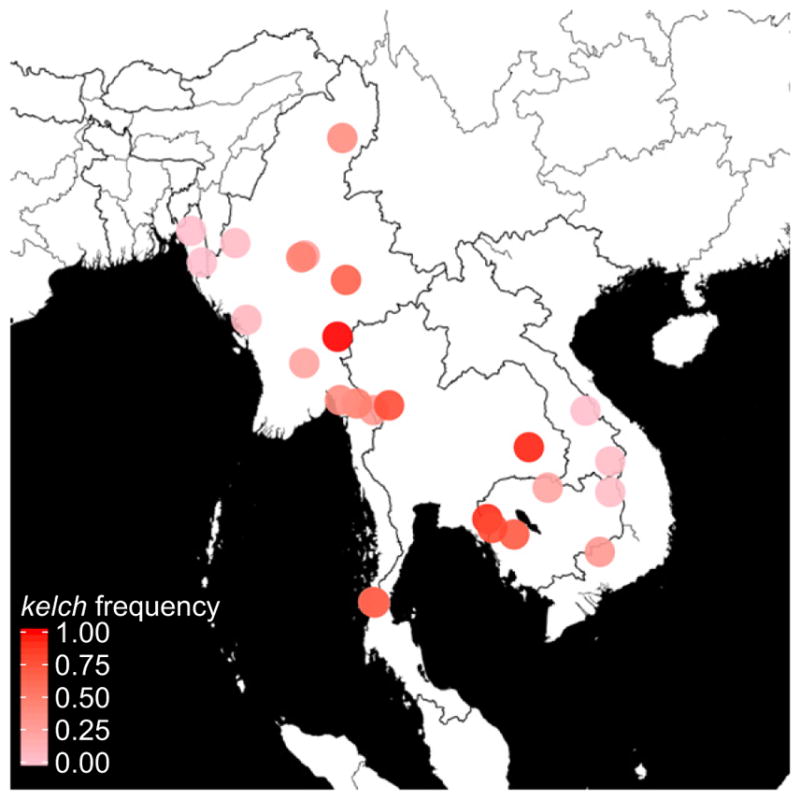
Distribution of the frequency of resistance mutations in kelch in Southeast Asia. Here, a resistance mutation is defined as any nonsynonymous mutation in highly conserved regions of kelch. The data presented are a compilation from Miotto et al. (2015), Tun et al. (2015), and Ariey et al. (2014). Map tiles by Stamen Design, under CC BY 3.0. Data by OpenStreetMap, under ODbL. Map generated using the GGMAP package (Kahle & Wickham 2013).
Of the five examples reviewed in this study, this example of using classic statistical methods to find the association between the kelch gene and artimisinin resistance in P. falciparum is perhaps the most familiar to population geneticists. As a eukaryotic pathogen with a high rate of recombination, sequence analysis of the P. falciparum genome was amenable to traditional genome scan approaches. Clever stratification of the samples gave additional power to the genomic scans, and in combination with novel experimental methods, researchers were able to find a locus under recent strong selection that was responsible for the evolution of resistance to artimisinin. The identification of the kelch gene by population genetics methods offers a clear example where standard methodologies to identify selective sweeps are powerful in identifying a causal locus. Given that P. falciparum can be manipulated experimentally to confirm computational predictions, this organism is an attractive choice for future studies of drug resistance, especially since malaria continues to be a costly disease and new drug-resistant loci may be unknown.
Evolution of oseltamivir resistance in influenza virus
Annual influenza epidemics are estimated to result in 3–5 million cases of severe illness and 250 000–500 000 deaths worldwide (WHO 2014b). Subtypes of the influenza type A virus are named according to the two surface proteins that allow influenza virus to bind and release from host cells, hemagglutinin (HA) and neuraminidase (NA). For example, H1N1 designates the particular subtype of influenza that was responsible for the 2009 swine flu epidemic. These two surface proteins, hemagglutinin and neuraminidase, are primary antigenic targets for the immune system and are also potential targets for pharmaceutical intervention. One common influenza drug is oseltamivir, which is marketed under the trade name Tamiflu. Oseltamivir works by inhibiting the influenza neuraminidase surface protein, the protein that cleaves sialic acid from the receptor of the host cell and allows replicated virus to spread to other uninfected cells (Moscona 2005). Resistance to oseltamivir in N1-containing influenza is conferred by a single histidine-to-tyrosine amino acid change at the 275th amino acid position in the neuraminidase protein. Note that this amino acid is often referred to as amino acid 274, which is its position in N2, a convention which we will follow here. The H274Y mutation was known from laboratory studies, but researchers predicted that fitness costs would prevent H274Y from spreading widely. However, oseltamivir resistance due to the H274Y mutation has been documented extensively following the 2007–2008 flu season when global surveillance indicated the increasing prevalence of H1N1 influenza viruses with acquired resistance (Moscona 2009). A combination of phylogenetics and elegant laboratory studies led to the discovery of two permissive mutations that mitigated the cost of the H274Y mutation, thus showing the importance of epistasis for influenza evolution. The study of the evolution of oseltamivir resistance has relied heavily on phylogenetic methods and taught us about the importance of epistasis, which, in turn, is of crucial importance for making predictions on influenza evolution.
Epistasis as a determining factor in the evolution of oseltamivir resistance
The interactions among sites in the genome can give rise to a phenomenon called epistasis where the effects of one mutation at a site are dependent on the presence or absence of mutations at other sites. Epistatic interactions, when they exist, ensure that the fitness effects of a drug resistance mutation are dependent on the genetic background on which it emerges. Epistasis in drug resistance evolution has been characterized by several in vitro studies, including the case of the five interacting mutations involved in cefotaxime resistance in Escherichia coli that produce predictable mutation orders (Weinreich et al. 2006) and the hundreds of interacting mutations that determine viral fitness in antiretroviral resistance in HIV (Hinkley et al. 2011).
In the case of oseltamivir resistance, the H274Y mutation was shown to have detrimental effects on viral fitness after it was first identified in the laboratory (Ives et al. 2002; Abed et al. 2004; Herlocher et al. 2004). This led researchers to believe that viruses carrying H274Y were ‘unlikely to be of clinical consequence’ (Ives et al. 2002). Why then did the resistant H1N1 strains subsequently reach a global frequency of nearly 95% (WHO 2009) during the 2008–2009 flu season? Bloom and others showed that the H274Y mutation reduces the amount of folded neuramindase that reaches the host cell surface and thereby reduces the virus’ fitness. They hypothesized that secondary mutations at other sites in the influenza genome may have acted to ameliorate deleterious effects of the H274Y mutation, permitting it to reach high global frequency (Bloom et al. 2010).
Bloom and colleagues took sequences from before and after 2007 and created a phylogenetic tree. They found five nonsynonymous mutations that separated the 2008 strains in which H274Y was common from the 1999 strains in which H274Y was found to have a strongly negative fitness effect. They then added the mutations one by one to the 1999 strain and found that two of the five mutations (R222Q, V234M) had a strong effect on the amount of neuraminidase at the cell surface. In viruses that had these two mutations, H274Y no longer reduced the fitness of the virus. In the absence of oseltamivir, the created triple-mutant virus (with H274Y, R222Q, V234M mutations) had a fitness comparable to wild type. In the presence of oseltamivir, the triple-mutant virus had much higher fitness than wild type (Bloom et al. 2010). A phylogeny with the status of these three mutations (H274Y, R222Q and V234M) is shown in Fig. 2.
Fig. 2.

Phylogenetic tree of neuraminidase (NA) subtype N1 illustrating the role of permissive mutations. Tree made with PhyML (Guindon et al. 2010) based on data from Bloom et al. (2010) and plotted using ggtree (Yu & Lam 2015). The phylogeny suggests an epistatic interaction between mutations R222Q, V234M and H274Y. Based on a maximum-likelihood reconstruction, branches with the H274Y resistance mutation are coloured red, whereas those without are coloured grey. Nonresistant branches with only the V234M mutation are coloured in light blue while nonresistant branches with both V234M and R222Q mutations are coloured in dark blue. The clade in which mutations, R222Q and V234M, are nearly fixed is shaded in blue. The phylogeny is consistent with the idea that the substitutions at site 222 and 234 act as ‘permissive’ mutations, ameliorating the fitness costs of H274Y on the previous background and allowing it to spread. The branch length scale is given in units of substitutions per site.
Subsequent studies also identified potential epistatic interactions between mutations in hemagglutinin and neuraminidase that impacted viral fitness and may have influenced the spread of oseltamivir resistance (Hensley et al. 2011; Ginting et al. 2012; Behera et al. 2015). Taken together these results suggest that epistasis had a profound impact on the evolution of resistance to oseltamivir and allowed the H274Y resistance mutation to spread through the global population. This work also provides cautionary evidence for the clinical assessment of drug resistance mutations. Secondary mutations that interact epistatically with drug resistance mutations are important factors that need to be incorporated when making predictions about the epidemiological consequences of drug resistance mutations. Knowing that even deleterious drug resistance mutations can spread when they emerge on permissive backgrounds tells us that we should pay close attention to identifying and monitoring potential epistatic interactions during virological surveillance.
Epistasis and the predictability of adaptive evolution
To what extent is epistasis a general feature of adaptive evolution? Both theoretical and empirical work predict that adaptation itself may enrich for epistatic interactions (Draghi et al. 2011; Draghi & Plotkin 2013; Gong et al. 2013; Rajon & Masel 2013; Szendro et al. 2013; Gong & Bloom 2014). This means that epistasis is likely to play a more general role in adaptive evolution and is not limited to specific case studies. It also means that researchers interested in adaptation need model systems in which to test theory regarding epistasis. Influenza resistance evolution has been a formative model for testing population genetic predictions regarding the role of epistasis in adaptive evolution.
Oseltamivir resistance evolution provided some of the first evaluations for methodologies that can potentially predict the sites within proteins that interact with one another based on sequence data and phylogenetic trees. Bloom and colleagues initially predicted the R194G mutation as a potential candidate for a positive epistatic interaction with H274Y using a phylogenetic method (Bloom & Glassman 2009) to infer stabilizing effects of mutations. They showed that R194G did indeed restore H274Y-mutant surface expression to wild type levels in the absence of oseltamivir (Bloom et al. 2010). Meanwhile Kryazhimskiy et al. (2011) examined hemagglutinin and neuraminidase sequences of multiple influenza strains and successfully predicted a known epistatic interaction (Collins et al. 2009) between H274Y and another mutation (D344N, see Fig. 3) in addition to the V234M and R222Q mutations identified by Bloom et al. (2010). Their methodology was based on a statistical ranking of the co-occurrence of specific amino acid substitutions within a phylogeny.
Fig. 3.

Phylogenetic tree of neuraminidase (NA) subtype N1 illustrating a putatively epistatic interaction. The same tree from Fig. 2 is used to illustrate the methodology presented in Kryazhimskiy et al. (2011). Nonresistant branches with the D344N mutation are coloured in blue. Branches with the H274Y resistance mutation are coloured in red. The phylogeny suggests an epistatic interaction between sites 344 and 275 where the H274Y substitution at site 275 (site 274 in N2 numbering scheme) appears multiple times immediately following the D344N substitution at site 344 (shaded in blue). This co-occurrence is consistent with the idea that the substitution at site 344 acts as a ‘permissive’ mutation and that these mutations interact in a positively epistatic manner.
Predicting the evolution of influenza is an intriguing prospect for evolutionary geneticists, which we will briefly discuss in the Discussion. These studies have shown that phylogenetic methods can not only affirm theoretical predictions regarding the patterns of epistasis but can also predict epistatic interactions that may be of clinical importance. They also establish a connection between protein phylogenies and fitness that may prove to be useful for future work in both population genetics and infectious disease.
The role of SGV for drug resistance evolution in HIV
HIV drug resistance has been a challenge since the introduction of HIV treatment because HIV adapts rapidly due to its high mutation rate, fast generation time and large population size (Coffin 1995). Although drug resistance in HIV has become less prevalent as treatments have improved (Lee et al. 2014; Feder et al. 2015), a fundamental question both for both clinicians and population geneticists remains: when exactly do drug-resistant mutations (DRMs) originate? Some patients may have pre-existing drug resistance mutations prior to receiving any drug therapy. Although these pre-existing mutations, also known as SGV, may offer no selective advantage (and could in fact be deleterious) before the onset of treatment when therapy begins, they quickly rise to high frequency in the HIV population and cause treatment failure (Pennings 2012). However, HIV can also acquire drug resistance mutations de novo after the onset of therapy.
Clinically, the extent to which drug resistance mutations arise from SGV or de novo mutation should inform treatment approaches. If SGV is critically important in the establishment of drug resistance, efforts should be focused on identifying any potential drug resistance mutations in the pretreatment HIV population to determine an appropriate regimen. However, if resistance occurs de novo after the start of treatment, patient adherence, drug penetrance and dosage should be the clinical focus. To this end, it is important to understand whether drug resistance mutations present before treatment onset can be identified in patient samples, and to assess the ultimate contribution of SGV to treatment failure.
From a population genetics perspective, drug resistance evolution in HIV allows us to understand the role of SGV in adaptation more generally. First, every infected person is an independent evolutionary replicate, and transmitted drug resistance is relatively rare. Around 2.8–11.5% of untreated patients have transmitted drug resistance mutations depending on the region of the world (Rhee et al. 2015), although these numbers are lower when considering drug resistance to any specific treatment. Therefore, most drug resistance emerges independently within a patient, and it is sufficient to follow a single patient over time to chart the evolution of an HIV population, in contrast to monitoring the acquisition of drug resistance mutations across multiple patients in a transmission chain. Second, the mutations conferring drug resistance are well catalogued (Bennett et al. 2009; Wensing et al. 2014), allowing us to quantify their frequency prior to treatment using allele-specific PCR (asPCR). Finally, adaptation can be rapid, allowing us to observe the evolution of drug resistance as it happens.
SGV contributes to treatment failure in HIV
Before an HIV-infected person starts treatment, a blood sample is taken to sequence the virus (see Schutten 2006 for an overview of genotyping assays). Sanger sequencing of HIV before the start of treatment is standard in clinical practice, but next-generation sequencing is not (Simen et al. 2009; Codoñer et al. 2011). A sequence of the protease, reverse transcriptase, and integrase genes are used to determine whether any drug resistance mutations are present at high frequencies, and if so, these results help the clinician and patient choose a combination of drugs with which to start treatment (Hirsch et al. 2008). If the majority of the viral population in a patient carries a resistance mutation, then this information will be used to choose a drug regimen that will work for the specific virus. However, drug resistance mutations at low population frequencies (at 20% or less) are not detected by standard sequencing protocols (Simen et al. 2009). An important question therefore is whether this low-frequency SGV is present in most patients, and if so, whether it allows the viral population to adapt and evolve drug resistance.
An important study by Paredes et al. (2010) used asPCR to determine whether drug resistance mutations were already present as minority variants in the viral population of 183 patients. These patients originally took part in a clinical trial, so blood samples from before the start of treatment and at treatment failure (if applicable) were stored for them. Treatment failure is defined as having virus detectable in the blood at levels higher than should be expected given that the patient is on treatment. The authors focused on two important resistance mutations in the reverse transcriptase gene, K103N and Y181C, because the patients were treated with a combination of reverse transcriptase inhibitors. In 73 of the 183 patients, they found that either K103N or Y181C was already present as a minority variant (mostly around 1% frequency), but they could not detect the mutation in the other 110 patients. Of the patients with the minority variant, treatment failed in 26 but was successful in 47. Out of the patients without the minority variants, treatment failed in only 16 but was successful in 94. Altogether, treatment failed in 23% of the 183 patients. When only considering the patients without detectable SGV, treatment failed in 15% of the patients. This means that in 8% of the patients, failure can be attributed to the presence of a minority variant (see Fig. 4).
Fig. 4.

Determining the amount of drug resistance from standing genetic variation (SGV) in HIV-1 using two studies. In two different studies, the percentage of patients acquiring drug resistance is partitioned into those whose HIV populations acquired drug resistance from SGV (light blue) and those whose populations acquired drug resistance via de novo mutation (dark blue). The total rate of failure is higher in the Paredes study than in the Margot study, but the percentage that is attributable to SGV is similar in the two studies. The calculation of the percentage of patients failing from SGV from the Paredes et al. (2010) study is shown in the inset. Patients are partitioned into those starting without drug-resistant SGV (left) and those with drug-resistant SGV (right). Eleven patients are expected to have failed due to de novo mutations based on the sample size (15% of 73 patients), and the remaining 15 failures are attributed to failure due to drug resistance from SGV.
The estimate from Paredes et al. (2010) that 8% of treatment failures come from SGV may be an underestimate because the authors only looked at the presence of two drug resistance mutations, and they may not have detected the variants in all cases. However, the two mutations are the most important for the treatment they looked at, and the result is fairly similar to a different estimate which we will describe below.
A less direct method to study the role of SGV for the evolution of HIV drug resistance was developed by one of us (Pennings 2012). We re-analysed data from a previous study (Margot et al. 2006) and looked for excess adaptation early during treatment. We found that in the study of interest, resistance evolution happened at a constant rate in the second and third year of treatment. In both of those years, the virus acquired resistance in 3.5% of the patients who previously had a virus without drug resistance. Such a constant rate of evolution of drug resistance was also observed in several other trials (e.g. UK Collaborative Group on HIV Drug Resistance and UK CHIC Study Group and Others 2010). In years 2 and 3 of treatment, SGV that was present before treatment probably plays no role, so the 3.5% likely reflects evolution of resistance from de novo mutations. In the first year of treatment, it is expected that both new mutations and SGV contribute to the evolution of resistance, so if resistance evolved in more than 3.5% of the patients’ viral populations, this suggests that SGV played a role.
In Margot et al. (2006), there were 600 patients in total in the first year of treatment. The expected number of people with resistance after 1 year was 21 (this is 3.5%, the rate that is seen in year 2 and year 3). In reality, there were 57 people with resistance, so 36 more than expected. Thirty-six of 600 is 6% (see Fig. 4), so it was estimated that SGV leads to resistance in 6% of the patients.
One may note that the total number of failing patients is much higher in Paredes et al. (2010). This may be because they have a stricter criterion for treatment success [<200 copies for Paredes et al. (2010) vs. <400 copies for Margot et al. (2006)]. Also, the patients were on similar but not the same treatments (3TC/AZT/EFV or ABC/3TC/AZT/EFV in the Paredes study and 3TC/ TDF/EFV or 3TC/d4T/EFV in the Margot study).
Quantifying the importance of SGV in drug resistance evolution
In the light of different overall rates of evolution in the two studies, it is perhaps surprising to see that the estimated rate of evolution from SGV is similar (8% and 6%). When Pennings used a third data set to estimate the rate of evolution of resistance due to SGV—based on a trial of long treatment interruptions with 435 patients (Danel et al. 2009)—her estimate was again 6%, which suggests that these estimates are fairly robust (Pennings 2012).
Adaptation from SGV has been discussed extensively in the literature; however, there are relatively few studies that have attempted to quantify its relative importance as a mode of adaptation. Altogether, the results from studying HIV suggest that SGV plays an important and quantitatively predictable role in treatment failures for this particular disease. SGV may also play a large role in treatment outcomes for other diseases, both those caused by pathogens and others such as cancer (Bozic et al. 2013). Next-generation sequencing can be used to determine whether drug-resistant SGV exists in patients prior to treatment, thus informing clinical decisions for individual patients. Apart from its clinical relevance, the particular example of HIV drug resistance evolution is also well-suited for validating evolutionary theory regarding adaptation from SGV (Orr & Betancourt 2001; Hermisson & Pennings 2005; Barrett & Schluter 2008).
Clonal interference among nonrecombining populations of Mycobacterium tuberculosis
Mycobacterium tuberculosis, the causative agent of tuberculosis (TB), is a nonrecombining bacterium that is believed to reproduce entirely clonally (although see Namouchi et al. 2012). Mycobacterium tuberculosis latently infects approximately one-third of the world’s population, and tuberculosis caused ~1.5 million deaths in 2014. The vast majority of these deaths occurred in low- or middle-wealth countries (WHO 2014a). Although tuberculosis is often effectively treated with combination therapies of antibiotics, drug resistance is common to nearly all known drugs. Multiply drug-resistant and extensively drug-resistant strains can be particularly difficult to treat, requiring longer courses of more expensive medications. Therefore, much work has been done in categorizing the genomic locations and probable mechanisms of these drug resistance mutations.
Many mutations have been identified that confer a large degree of drug resistance within patients. These drug resistance mutations have been found by collecting M. tuberculosis samples, assaying their growth in media containing different antibiotics and identifying associations between mutations and resistance profiles. A comprehensive list of drug resistance mutations can be found in the Tuberculosis Drug Resistance Database (Sandgren et al. 2009) where 1178 drug resistance mutations of various effect sizes to nine drugs are recorded within more than 30 different genes.
Despite the apparent adaptability of M. tuberculosis, diversity in intrapatient populations has historically been considered to be very low (Sreevatsan et al. 1997; Musser et al. 2000). It is only recently that studies have found that considerable diversity exists within M. tuberculosis populations (Al-Hajoj et al. 2010; Navarro et al. 2011; Sun et al. 2012; Eldholm et al. 2014). By combining new sequencing data with the knowledge of well-documented drug resistance mutations in M. tuberculosis, we can test our theoretical understanding of how clonal evolution proceeds and arrive at a more complete picture of how M. tuberculosis drug resistance emerges within a patient.
The dynamics of clonal interference in TB
Recombination is thought to be evolutionarily adaptive because it allows different adaptive mutations that reside on different genetic backgrounds in a population to appear together (Fisher 1930; Muller 1932; Felsenstein 1974). In addition, it can unlink positively selected traits from deleterious passenger mutations and mitigate the effects of linked selection (Hill & Robertson 1966; Birky & Walsh 1988; Good & Desai 2014). In populations with no recombination, each mutation remains on the genetic background on which it originally occurred. Therefore, if multiple positively selective traits enter the population simultaneously on different backgrounds, these traits cannot be recombined to augment each other. Instead, they must compete against each other as they both rise in frequency in the population (Muller 1932). This process is known as clonal interference (Gerrish & Lenski 1998).
While clonal interference has been extensively modelled (Rouzine et al. 2003; Desai & Fisher 2007; Park & Krug 2007; Martens & Hallatschek 2011; Neher 2013) and investigated in laboratory experiments (Miralles et al. 1999; Pepin & Wichman 2008; Kvitek & Sherlock 2013; Lang et al. 2013), few systems exist that allow us to test these predictions in natural populations. Globally, influenza seems to evolve clonally across years (Strelkowa & Lässig 2012), but intrahost populations of nonrecombining bacteria allow for many semi-independent evolutionary trajectories to be compared over much shorter time scales. Although there is evidence that HIV experiences clonal interference within patients, recombination complicates its study (Pandit & de Boer 2014). There is also a strong interest in clonal evolution in cancer, but sampling at different time points is often much harder for cancer than for M. tuberculosis or HIV, which makes cancer as a study system more difficult (Ding et al. 2012; Walter et al. 2012).
Intrahost evolution of M. tuberculosis provides a fairly straightforward way to examine clonal interference in vivo and to understand the extent to which it can slow the evolutionary process. Because there is no recombination, allele frequencies from pooled resequencing can, in some cases, be reconstructed into haplotypes (see below). Its large genome size (~4.5 Mb), well-documented drug resistance mutations, and long time course of infection mean that patterns of diversity can be tracked over time. Although previous studies have shown the presence of competing clonal lineages in M. tuberculosis populations in a patient using neutral markers (Al-Hajoj et al. 2010; Navarro et al. 2011), we can further use our understanding of drug resistance mutations to better understand the evolutionary dynamics of within-host populations. Among patients who are infected with drug-sensitive strains of M. tuberculosis, drug resistance can evolve within a patient during treatment. By tracking the frequencies of drug resistance mutations over time in a single patient, we can observe clonal interference between lineages that each carry beneficial mutations in real time.
Sun et al. (2012) conducted the first analysis of this kind by tracking drug resistance allele frequencies in three patients across at least two time points. The first patient was entirely free of drug resistance mutations at the onset of treatment, but by the second time point of sampling had four segregating drug resistance mutations. At the final time point, 94% of the sample had a single drug resistance mutation, and 6% was divided among other drug-resistant strains, including those containing drug resistance mutations not present at the second time point. This suggests that while M. tuberculosis rapidly acquires resistance mutations, different drug-resistant strains compete against each other due to lack of recombination. The second patient entered the study with an M. tuberculosis population fixed for a certain drug resistance mutation (rpoB L533P) but remained sensitive to the antibiotic rifampicin. Eighteen months later, this patient had a population whose genetic composition was dominated by a different drug resistance mutation (rpoB H526Y) and was now resistant to rifampicin, suggesting that successive sweeps of alternative drug resistance mutations can lead to multidrug-resistant strains.
Eldholm et al. (2014) performed a similar study, but with much greater sequencing depth, in a single patient who was followed over 42 months as extensive drug resistance was acquired by the pathogen. The patient, who was started on a standard antibiotic regimen (pyrazinamide, rifampicin and isoniazid), was given increasingly uncommon drugs as their tuberculosis population acquired more drug resistance mutations. The dynamics of clonal interference are shown in Fig. 5, based on data from Eldholm et al. (2014). Allele frequencies were measured at several time points, so alleles with similar frequency trajectories can be assumed to be on the same background or in the same clone. Therefore, the clonal frequency changes can be inferred across time.
Fig. 5.

Clonal interference in Mycobacterium tuberculosis. Dynamics of clonal interference among drug resistance mutations (stars) and mutations not known to be drug resistance mutations (dots) arising within a patient with tuberculosis. Each expanding colour block represents an M. tuberculosis lineage starting from one or more mutations and spreading through the population. Lineages are coloured depending on whether the mutations they possess ultimately fix in the population (green) or are lost (blue). Lineages that begin with mutations not known to be drug resistance mutations (non-DRMs) are coloured in brown. The background on which each new lineage emerges is overlapped at the starting point of each colour block. Trajectories are inferred from allele frequencies at the point of sampling (black vertical lines). Mutations for which the inferred trajectories do not closely match clonal dynamics are marked in red. Data from Eldholm et al. (2014).
Eldholm et al. (2014) identified 12 drug resistance mutations reaching frequency >25%, of which only seven ultimately fixed (shown in green), suggesting that clonal interference may purge over 40% of strongly selected drug resistance mutations even after reaching high frequencies (shown in blue).
The knowledge of these positively selected drug resistance mutations can also allow us to understand the dynamics of co-occurring hitchhiking mutations that were already on the background on which the drug resistance mutation arose. Eldholm et al. (2014) found that 23 mutations not directly associated with drug resistance also fixed in the population over the 42 weeks of sampling. While these mutations may have been neutral, compensatory, or positively selected through adaptation unrelated to drugs, it is also possible that some or all of them are slightly deleterious but rose in frequency due to their linkage to beneficial mutations.
From Eldholm et al. (2014) it may be noted that, in most cases, the drug resistance mutations were added to haplotypes one by one, which suggests that each individual mutation leads to an increase in fitness. This is troubling, because part of the rationale behind using multiple drugs at the same time is that it should make it much harder for the pathogen to evolve resistance, as it needs to acquire multiple mutations at the same time. It has been suggested that imperfect drug penetration may explain the evolution of resistance in tuberculosis despite multidrug therapy (Lipsitch & Levin 1998; Moreno-Gamez et al. 2015).
The study of clonal interference, thus far largely investigated within experimental and theoretical models, has a unique application in the study of clonal dynamics of natural populations of the human pathogen M. tuberculosis. Using the presence of drug resistance mutations as a positive control for strong directional selection, we are able to better understand the evolutionary dynamics of clonally evolving pathogen populations such as M. tuberculosis. Although we currently only have detailed data for a few well-studied patients that include deep coverage of the entire genome across multiple time points, it appears to be the case that clonal interference in M. tuberculosis populations occurs in a way that is similar to what is seen in laboratory experiments. With the knowledge of hundreds of drug resistance mutations and the availability of affordable whole-genome sequencing, we expect to see many more studies of within-host dynamics of tuberculosis in the near future. We especially look forward to studies where population’s genetics can be used to understand genetic patterns associated with diverse treatment outcomes.
Evolution of drug resistance within populations of Staphylococcus aureus bacteria via horizontal gene transfer
There is a growing need to address drug resistance within bacterial species that exhibit horizontal gene transfer (HGT, see Box 3), which is the sharing of genetic material between cells. Resistance to antibiotics has been increasingly reported for many bacterial species with HGT, including S. aureus, Helicobacter pylori, Salmonella enterica, E. coli and many others. Most bacterial species primarily reproduce clonally, passing their current genomes vertically to the next generation. In species with HGT, there are also genetic elements that can be passed between individuals within the current generation and recombined into their genomes. HGT can move genes, sets of genes or even chromosomes between cells of even distantly related species (Nelson et al. 1999; Keeling & Palmer 2008; Syvanen 2012; Meric et al. 2015). This makes defining a bacterial species difficult, let alone tracking the spread of drug resistance.
Box 3. Horizontal gene transfer and the bacterial core and accessory genomes.
Bacteria have a circular chromosome (typically 1–10 Mb in size and in 1–2 copies per cell) that generally contains the ‘core’ or ‘essential’ genome and typically codes for fundamental functions like DNA repair and replication, cell structures and shapes, and metabolic functions. Many species of bacteria have an additional ‘accessory’ or ‘satellite’ genome that is typically 0.1–10% of the size of the core genome and can be present on the main circular chromosome or on extrachromosomal elements, such as plasmids. The core genome is usually defined as the set of genetic regions that are present within all members of the species, and the accessory genome is that which can vary between individual strains or cells. The accessory genome, whether on the main chromosome or not, can include chromosomal cassettes, bacteriophages, pathogenicity islands, genomic islands and transposons (Lindsay & Holden 2004). The accessory genome is sometimes called ‘dispensable’ because it is not required for survival in an optimal environment; however, not only can the accessory genome encode information essential to a bacterium’s survival in some environments, but bacteria have evolved elaborate and fascinating methods of sharing accessory genes between cells (Amabile-Cuevas 1993; Stokes & Gillings 2011).
The sharing of genetic information between cells is called horizontal gene transfer (HGT). While bacteria are asexual and typically defined as ‘clonal’, both HGT (distinct from sex because it alters the genome of the recipient) and new mutations can generate novel genetic variation within a population of bacteria. HGT can occur both within and across species, and there are even cases of transfer across kingdoms (Keeling & Palmer 2008). The actual methods of transferring DNA through the cell membrane include transformation, transduction, and conjugation. Transformation is the uptake of naked DNA into the cell, generally mediated by membrane proteins with some DNA specificity. It is a method restricted to species that have evolved to uptake DNA. Transduction is transfer of DNA that is mediated by phages (or bacteriophages, viruses that infect and replicate within a bacterium), thus it is restricted by which hosts a phage is capable of infecting. There are two types: generalized transduction, where random bacterial DNA is incorporated into phage DNA, and specialized transduction, where the incorporation is limited to a specific set of bacterial genes. Perhaps the most common method of HGT is conjugation, which requires cell-to-cell contact. One famous method of conjugation is the E. coli F conjugative plasmids, which code for cellular structures that bring the cells into contact (called ‘pili’) as well as for machinery to inject the DNA into the recipient cell. The genetic material transferred via HGT can be recombined onto the main bacterial chromosome or onto extrachromosomal elements. Often ‘cassettes’ of genes evolve together and can spread through a bacterial population, including in many cases of drug resistance (Amabile-Cuevas 1993; Stokes & Gillings 2011).
One useful framework for understanding the spread of drug resistance in species with HGT is the concept of a selective sweep. In general, in a selective sweep, a new adaptive mutation arises and spreads through a population, causing a corresponding reduction in genetic diversity around the adaptive site (Maynard Smith & Haigh 1974). In clonal populations (e.g. populations of M. tuberculosis), a selective sweep can lead to the removal of entire lineages and a strong genomewide reduction in diversity. By contrast, in recombining populations (e.g. populations of P. falciparum) the extent of the reduction in diversity is inversely proportional to the ratio of recombination and selection, r/s [such that the greatest reduction in diversity occurs when recombination (r) is weak and selection (s) is strong] (Kaplan et al. 1989). Selective sweeps in organisms that exhibit HGT can be best understood by distinguishing between gene-specific and genomewide selective sweeps (Shapiro & Polz 2014). Gene-specific selective sweeps occur when an adaptive mutation or gene can be transmitted across a population faster via HGT than by clonal expansion, which means that the adaptive mutation spreads independent of the rest of the genome. Perhaps more familiarly, a genomewide selective sweep occurs when an adaptive mutation spreads via clonal expansion of the genome that first acquired the adaptive mutation. Another way to frame gene-specific and genomewide selective sweeps can be to think again of the ratio of recombination (here mediated via HGT) to selection, r/s, and how it impacts genetic variation in the genome. When r/s ≫ 1 gene-specific selective sweeps are more likely to occur, and they will have little impact on genomewide diversity. When r/s ≪ 1 genomewide selective sweeps are more likely, and genomic diversity can be strongly reduced. Thus far, it appears that many adaptive events in microbial species with HGT may fall into the r/s ≫ 1 regime which leads to gene-specific sweeps (Shapiro & Polz 2014), although more data are needed. For example, what looks to be a high r/s ratio may be partially be explained by the slowing of genomewide sweeps due to clonal interference and perhaps selection for low-frequency genotypes (Shapiro & Polz 2014). Nevertheless, it is useful to frame the evolution of bacterial species with HGT in the context of both genomewide and gene-specific sweeps.
A more traditional framework for studying the evolution of bacterial genomes with HGT has been to study evolution within the ‘core’ genome, defined as the gene regions that are shared by all isolates within a species, vs. the ‘accessory’ genome, which is the genetic material that is not shared by all isolates of a species (see Box 3). We will use the example of S aureus and the accessory element SCCmec to explore the evolution of drug resistance within a bacterial species capable of horizontal gene transfer (note that SCCmec recombines onto the circular chromosome as a gene cassette, see Boxes 1 and 3). We have chosen to consider MRSA not only because it is a major emerging health threat but also because it is an instructive example in how the phylogenetic relationships between global isolates reveal different evolutionary histories depending on whether the core genome or an accessory genetic element is considered.
Clinical background and historical analysis of the Staphylococcus aureus genome
Staphylococcus aureus is a human commensal that is known for causing dangerous skin, blood stream and other hospital-associated infections as well as having the ability to become resistant to the majority of antibiotics (Lowy 2003; Stryjewski & Corey 2014). In addition to methicillin resistance, which first emerged in the 1960s, there is also a growing number of cases of resistance to other antibiotics, including vancomycin, quinolones, aminoglycosides, streptogramins, oxazolidinones and rifamycins (Lowy 2003; Stryjewski & Corey 2014). MRSA can be particularly deadly. For example, mortality rates for patients with MRSA bloodstream infections has been reported to be ~30% (De Kraker et al. 2011). In the USA ~94 000 infections and ~19 000 deaths per year are caused by MRSA (Stockman 2009). Drug resistance can arise via new mutations within the genome of a single clone (Strahilevitz & Hooper 2005; Howden et al. 2008), via horizontal gene transfer between clones (Coombs et al. 2011) and even via horizontal gene transfer from another bacterial species (Hanssen & Ericson Sollid 2006; Bloemendaal et al. 2010; Malachowa & Deleo 2010; Smyth et al. 2012). Staphylococcus aureus strains acquire methicillin resistance via the mobile genetic element SCCmec [most likely via transduction (defined in Box 3) (Maslanova et al. 2013)], which integrates as a cassette of genes into the bacterium’s chromosome.
The genome of S. aureus is ~2.8 Mbp total, where ~2.3 Mbp compose the core genome and ~0.5 Mbp compose the accessory genome. The most common method for analysing the genomes of S. aureus isolates has been multilocus sequence typing (MLST), in which a standardized set of housekeeping genes from the core genome are sequenced and categorized into allelic types, allowing placement of each isolate into a defined ‘clonal complex’ (Enright & Day 2000; Enright et al. 2002; Feil et al. 2004; Maiden et al. 2014). MLST has historically been applied to the core genome, revealing that S. aureus is a highly clonal species with relatively few recombination events in the core genome (Robinson & Enright 2004). When MLST is applied to international collections of isolates, it appears that a small number of clonal lineages are responsible for most infections. For example, Oliveira et al. (2002) applied MLST to over 3000 MRSA isolates from hospitals across Europe, South America, and the USA, and found that just five clones caused 70% of the infections (Oliveira et al. 2002). As findings such as these emerged, it was thought that methicillin resistance was likely to have spread across the population of S. aureus when relatively few clones acquired SCCmec and then clonally expanded (i.e. genomewide sweeps) (Kreiswirth et al. 1993; Crisóstomo et al. 2001; Feil & Enright 2004). However, closer analysis of the population structure of the S. aureus accessory genome reveals a different story.
The mobile element SCCmec as a clinical indicator of hospital-and community-associated infections
In the first few decades of MRSA outbreaks, it was found that the strains causing hospital-associated (HA-MRSA) vs. community-associated (CA-MRSA) infections displayed distinct clinical and genetic differences (these differences may be eroding, as will be addressed later). HA-MRSA tends to be resistant to a wider array of antibiotics and often causes blood stream and other hospital-related infections in individuals with additional medical conditions. CA-MRSA tends to be more virulent (caused by virulence genes also transmitted via HGT) and often causes skin infections in otherwise healthy individuals (David & Daum 2010; Chua et al. 2014). These clinical differences are partly mediated by the mobile genetic element called SCCmec, a chromosomal gene cassette that confers resistance to methicillin and can also confer resistance to other antibiotics (Katayama et al. 2000; Hanssen & Ericson Sollid 2006). Interestingly, genetic changes within this gene cassette track successive epidemic waves of MRSA (Katayama et al. 2000; Chambers & DeLeo 2009), and extensive work has been done to investigate the source and spread of this mobile element, both as a phylogenetically informative sequence and as an adaptive factor in its own right. SCCmec is currently classified into 11 types (with more subtypes) using mutations and the orientation of segments within the cassette (Milheiriço et al. 2007; IWG-SCC 2015).
There have been a number of studies showing that the population structure of S. aureus is different between HA-MRSA and CA-MRSA strains (David & Daum 2010; Mediavilla et al. 2012; Stryjewski & Corey 2014). Both HA-MRSA and CA-MRSA strains are thought to arise when methicillin-sensitive clones newly acquire a SCCmec cassette from a source population, most likely the staphylococcus species S. epidermidis (Wu et al. 1996; Hanssen et al. 2004, 2005; Meric et al. 2015). It has been found that HA-MRSA strains tend to carry the longer SCCmec types I, II, and III elements in just a few clonal backgrounds, whereas CA-MRSA strains tends to carry the shorter SCCmec types IV and V elements in diverse clonal backgrounds (Enright et al. 2002; Robinson & Enright 2003; David & Daum 2010; Coombs et al. 2011). For example, the early HA-MRSA pandemic could be relatively neatly classified into just five clonal complexes (Musser & Kapur 1992; Fitzgerald et al. 2001; Enright et al. 2002; Robinson & Enright 2003). In contrast, as researchers began analysing the strains responsible for the emerging CA-MRSA health threat, they found these strains to display more diversity and a stronger association between clonal type and geographic location (Chua et al. 2010; David & Daum 2010; Coombs et al. 2011). As of 2006 the SCCmec type IV element alone was found to have entered at least nine different clonal complexes of S. aureus (Lina et al. 2006).
The clinical and genetic differences between hospital-associated (HA-) and community-associated (CA-) MRSA are eroding in the modern pandemic, mainly due to the emergence of CA-MRSA strains that seed an increasing number of hospital outbreaks (Chambers & DeLeo 2009; David & Daum 2010; Mediavilla et al. 2012; Hsu et al. 2015). A study of MRSA infections in 2004–2005 in the city of San Francisco, California, found that ~90% of MRSA infections were acquired in the community (Liu et al. 2008). In the United States, there is currently a predominant CA-MRSA clone, called USA300 (typically containing SCCmec Type IV), that is responsible for the vast majority of community-associated infections in addition to causing hospital outbreaks (Seybold et al. 2006; Chambers & DeLeo 2009). Mathematical models predict that these CA-MRSA strains will eventually replace the strains traditionally categorized as HA-MRSA, rendering the community-associated and hospital-associated categories far less useful (D’Agata et al. 2009). The success of these CA-MRSA strains is potentially mediated by the arrival of the SCCmec Type IV cassette, which may be particularly suited to recurrent horizontal transfer because it confers faster growth rates at little or no fitness cost (Okuma et al. 2002; Diep et al. 2008; Chambers & DeLeo 2009; Mediavilla et al. 2012).
High-resolution population structure analysis of Staphylococcus aureus using the mobile element SCCmec
As the clinical and genetic differences between CA- and HA-MRSA strains have eroded in the modern pandemic, so too has the emphasis on their differences within recent literature. Instead, attention is moving towards leveraging new sequencing technologies (Maiden et al. 2014) to achieve higher resolution into the global patterns of methicillin resistance acquisition. Overall, analyses of the core genome of MRSA strains suggest that relatively few methicillin-resistant clones have dispersed widely across the globe (Robinson & Enright 2003), while analyses of the accessory genome of MRSA suggest that new strains emerge locally via frequent de novo transfers of the SCCmec element. In other words, it appears that there has been clonal expansion of relatively few core genomes, while the accessory genome frequently goes through gene-specific sweeps. To illustrate this, Lina et al. (2006) applied MLST to both the core genome and the SCCmec element and found no association between clonal complex background and SCCmec element sequence type. The authors therefore concluded that SCCmec appears to transfer both repeatedly across distinct clonal complexes (particularly in the case of SCCmec IV) and repeatedly within a clonal complex, which suggests that SCCmec spreads through gene-specific sweeps. Chua et al. showed in another study that MRSA from geographically and genetically distant isolates (i.e. distantly related core genomes) can nevertheless have relatively conserved accessory elements, including SCCmec type IV (Chua et al. 2011), which again suggests that transfer happens often and sweeps are gene-specific. Another example is a study by Nübel et al. (2008) where the authors used an improved sequence typing method of the core and accessory genome to investigate the genetic relationships within a single clonal lineage (ST5, belonging to clonal complex CC5) using 135 isolates from across 22 countries and six continents. In contrast to lower resolution studies that came before, their analyses of the core genome sequences found geographically-associated phylogenetic clades within the ST5 clonal lineage. Additionally, their analyses of the SCCmec sequence revealed that at least 23 independent transfers of SCCmec occurred into this clonal group alone, with acquisitions appearing to occur locally (rather than a single acquisition then disseminating globally; Fig. 6). We could refer to this case of multiple origins of the adaptive SCCmec element as a soft, gene-specific sweep (Pennings & Hermisson 2006). The authors estimate that previous calculations for the rate of SCCmec acquisitions were at least an order of magnitude too low.
Fig. 6.

Minimum-spanning tree constructed from core genomic sequences of 135 isolates of the ST5 Staphylococcus aureus clone collected across the globe. Figure reproduced from fig. 1A,B of Nübel et al. (2008), Copyright (2008) National Academy of Sciences, USA. The sizes of circles are proportional to the haplotype frequency, and the line length is proportional to the number of mutational steps between haplotypes. In panel (A) haplotypes are painted with colours in proportion to the country of origin that isolates with this haplotype were found. Fourteen lineages (labelled ‘A’ through ‘N’) tend to cluster with geographical origin emerge, suggesting that specific haplotypes of the core genome of the S. aureus ST5 clone are largely endemic to a geographical area. In panel (B) the same minimum-spanning tree is used, but now haplotypes are painted with colours in proportion to the SCCmec element type co-occurring in the isolate. Some SCCmec elements are nontypeable (‘nt’), likely due to being novel variants. The most parsimonious distribution of acquisition events of SCCmec is indicated with roman numerals placed along the branches of the tree (where the roman numeral corresponds to the SCCmec type), although the actual number of SCCmec acquisitions may be higher. Note that while the different SCCmec types are present across the globe, the acquisition events tend to occur at the tips of the tree, suggesting that methicillin resistance tends to evolve within a local S. aureus strain as opposed to occurring once and then disseminating globally.
Taken together, we now know that genetically diverse core genomes can nevertheless contain closely related resistance alleles and that frequent transfers can occur even within a clonal lineage. This makes understanding the evolutionary history of these alleles complicated, because alleles can be transmitted independently of the clonal expansion of core genome lineages. Nevertheless, these studies of drug resistance in S. aureus illustrate that researchers can achieve fine resolution into the demographic and adaptive history of S. aureus using improved sequencing technologies, in this case facilitated by the exploration of both the core and accessory genomes.
As researchers begin to use whole-genome sequencing in S. aureus, an even finer resolution into the evolutionary history of MRSA outbreaks is possible. For example, Holden et al. (2013) were able to pinpoint both the population size changes and geographic origin of a specific clone (EMRSA-15/ST22), and Harris et al. (2013) were able to construct the transmission pathway of a hospital outbreak between staff and patients. Thus, the growing availability of sequence data and whole-genome sequencing technologies has provided the ability to determine that resistance may generally be unconstrained by global transmission dynamics and instead can be tracked on local spatial scales. This presents researchers with the opportunity to use sequencing as a tool to monitor transmission networks within hospitals to limit the numbers of hospital-acquired drug-resistant infections. Overall, the transmission and repeated evolution of drug resistance in bacterial populations capable of HGT presents important and interesting problems for reconstructing population structure and spatial distributions of resistance.
Discussion
The costs that drug-resistant pathogens impose on human health are enormously high, making any tool that can lead to increased understanding of the evolution and spread of drug resistance of great potential value. It is therefore important that tools from the field of evolutionary genetics, including population genetic and phylogenetic ones, are applied to pathogen populations where these tools could provide meaningful insights. In addition to the possibility of helping to prevent the evolution and spread of drug resistance, valuable lessons can be learned to deepen our understanding of evolutionary biology, making pathogens attractive study organisms. In this review we highlighted five evolutionary genetic studies in pathogens that we hope give a sampling of the range of biological lessons that can be learned from the study of drug resistance in pathogens. First, we showed how classic population genetic methods for finding selective sweeps were used to successfully identify the region involved with resistance to artemisinin in the malaria parasite Plasmodium falciparum. Next, we described how phylogenetic methods helped uncover the role of epistasis in the evolution of influenza surface proteins in general and in the evolution of oseltamivir resistance in particular. In our third example, we highlighted the importance of SGV in the evolution of resistance to antiretrovirals in patients with HIV. Fourth, we showed that drug resistance mutations can be used to explore the dynamics of clonal interference within a patient infected with Mycobacterium tuberculosis. Finally, we discussed the complications involved in studying bacterial populations and how horizontal gene transfer of genetic elements contributes to the spread of antibiotic resistance in Staphylococcus aureus. These five examples are not meant to be exhaustive, but rather to show the wealth of possible study organisms and evolutionary questions that can be addressed. We hope that these five examples convince the reader that the evolutionary genetics of drug resistance is an appealing field.
In each of the cases we highlighted, the study of drug resistance has been useful in expanding our understanding of basic evolutionary processes. The insights gained from these examples are not limited to the organisms mentioned in this review, but the discoveries that can be made do, in some ways, depend on the organism, its biology, and the availability of data. For example, the work on epistasis in influenza was only possible because of the extensive sequence collection that is continuously happening around the world. The work on mapping a resistance allele in the malaria parasite P. falciparum also depended on the availability of data but additionally was possible because P. falciparum has a high recombination rate that facilitates linkage-based analyses. The described studies on SGV in HIV were possible because the relevant mutations were known and because evolution of resistance can happen independently in every patient, giving researchers the ability to work with replicate evolutionary histories. The work in M. tuberculosis was possible, once again, because the resistance mutations are well known and because haplotypes could be reconstructed from allele frequencies due to a lack of recombination. Finally, the example of drug resistance evolution in MRSA shows how horizontal gene transfer can be studied when extensive multilocus genotyping data are available from isolates collected across the globe.
Similar analyses can and are being performed in other pathogens. For example, we highlighted work on selective sweeps in P. falciparum, but selective sweeps and hitchhiking are also being studied in HIV (Zanini & Neher 2013; Pennings et al. 2014; Feder et al. 2015), in hepatitis C virus (Bull et al. 2011), and in influenza (Koelle et al. 2006; Strelkowa & Lässig 2012). We used MRSA as an example for the importance of horizontal gene transfer, but there are many other such examples. For example, a recent study on pneumococcus describes horizontal gene transfer and soft selective sweeps related to drug resistance and vaccine escape (Croucher et al. 2014). Another example of a group of pathogens in which the evolution of drug resistance can be studied are the herpes viruses. Cytomegalovirus (CMV) is the herpes virus for which drug resistance is best studied and for which many resistance mutations are known (Hakki & Chou 2011), and population geneticists have recently started to analyse CMV sequences (Renzette et al. 2015). Drug resistance also evolves in other herpes viruses, such as varicella-zoster virus and herpes simplex virus (Piret & Boivin 2014; Brunnemann et al. 2015), but these viruses have not been extensively analysed using population genetic or evolutionary genetic methods.
In all these cases, the role of evolutionary genetics is to connect information contained in genomic sequences to the processes underlying pathogen evolution, and this will require careful attention to the diverse biological properties of pathogens as we have discussed in this review (see Box 1). Pathogen evolution represents an enormous opportunity for population geneticists to study major questions in evolutionary biology and also contributes to our understanding of some of the most important issues in public health. By combining innovative population genetics approaches with appropriate data collection, we hope that future studies will make beneficial contributions to the fields of evolutionary biology and public health.
The advantages of pathogens as evolutionary model systems
The field of population genetics traditionally leans heavily on the use of fruit flies, yeast, Escherichia coli, and humans as model organisms. However, it is becoming clear that many pathogens can also be excellent model systems. Pathogen evolution as a model evolutionary system has many advantages. Pathogens receive a lot of attention from biomedical researchers, and as a consequence, they are well studied and extensively sampled. Some pathogens already function as model organisms in the lab [e.g., influenza (Foll et al. 2014), HIV (van Opijnen & Berkhout 2005), polio (Acevedo et al. 2014) and E. coli (Toprak et al. 2012)], which allows researchers to assess characteristics of population genetic and epidemiological relevance from laboratory populations, for example the cell surface expression assays that were performed in the previously mentioned study of oseltamivir resistance in influenza. In some pathogen systems, researchers can also apply known selective pressures with precision in a laboratory environment, thus allowing high-resolution measurements of an organism’s response to selection. Additionally, because some pathogens evolve drug resistance quickly and repeatedly within patients, there are cases where many replicate evolutionary histories can be studied (HIV, HCV, TB, etc.).
Pathogens are also among the organisms for which the most genetic sequence data are available for population genetics analysis: In 2014, over 270 000 S. aureus sequences, 50 000 influenza sequences, 40 000 HIV sequences, and 5000 malaria sequences were released in GenBank alone (Benson et al. 2013; accessed 26 May 2015). Moreover, decreased costs of next-generation sequencing will make the amount of new data for pathogens increase exponentially. Throughout this review, we highlight uses of sequence data and population genetics theory in studying pathogens as evolutionary model systems. We believe that pathogen evolution will continue to be a fruitful area of research for evolutionary biologists where fundamental insights can lead to beneficial public health consequences.
Frontiers in the study of pathogen evolution and drug resistance
As more sequencing data emerge in the foreseeable future, there are some specific questions that we would like to see addressed at the intersection of population genetics and drug resistance in pathogens. First, we are expecting more work that bridges within- vs. between-host dynamics (e.g. Lythgoe & Fraser 2012). This is important because considerable genetic variation is created and selected for within the host; however, the within-host processes of short-lived infections have not yet received sufficient attention. We are expecting more within-host work, including time series and deep samples for pathogens that are traditionally studied at the between-host level. The M. tuberculosis work described in this review is a good example, but new within-patient work on influenza is also being done (Rogers et al. 2015). Second, with deeper samples and more time series, it should become increasingly possible to estimate selection pressures on pathogens and to estimate epistatic interactions between mutations. For some of this work, methods that are currently used for identifying mutations and evaluating selection in viruses grown in cell culture (Lou et al. 2013; Foll et al. 2014) will hopefully find use in clinical samples taken from infected patients. Extending these methods to patient data can potentially give researchers the ability to study the evolutionary dynamics of pathogens within patients over the entire course of an infection.
Another important goal for these evolutionary studies of pathogens was to increase our predictive power of pathogen evolution. Until very recently predicting evolution seemed like science fiction, but recent work is changing our perceptions of what is possible. In addition to the predictive phylogenetic methodologies mentioned in our discussion of epistasis in influenza, a method that uses the shapes of protein phylogenies to predict evolutionary trajectories was evaluated using influenza data (Neher et al. 2014). The work shows that predictability is low when evolution occurs by big steps, but much higher when evolution proceeds by small steps (Neher et al. 2014). Although we tend to fix our attention on large adaptive events, such as drug resistance or immune escape, evolution by small steps may be more common than previously expected (Bhatt et al. 2011; Strelkowa & Lässig 2012; Gong et al. 2013). In an organism like influenza where new vaccines are manufactured every year, the application of these and other new predictive methodologies that use population genetics theory (Luksza & Lässig 2014) are potentially groundbreaking.
We hope that the works on drug resistance reviewed here offer a glimpse into a growing field at the intersection of studies in pathogen evolution and evolutionary genetics. We also hope to have illustrated how these fields of study can benefit greatly from each other and that this review contributes to the exciting future of this field.
Acknowledgments
We would like to thank Ian Cheeseman for helpful discussions regarding the evolution of artemisinin resistance, Caitlin Pepperell for helpful discussions regarding tuberculosis, Vegard Eldholm for the help of interpreting the clonal interference data, Jesse Bloom for providing influenza sequences to verify substitution identities and Joshua Plotkin for clarification on the method discussed in this review to predict epistatic interactions. B.W., N.G., A.F. and Z.A. are members of the laboratory of Dmitri Petrov at Stanford University.
Footnotes
All authors designed the study and contributed to the writing and editing of the manuscript. B.A.W. wrote the chapter on Influenza, N.R.G. wrote the chapter on Malaria, Z.J.A. wrote the chapter on MRSA, A.F.F. wrote the chapter on TB and P.S.P. wrote the chapter on HIV.
References
- Abed Y, Goyette N, Boivin G. A reverse genetics study of resistance to neuraminidase inhibitors in an influenza A/H1N1 virus. Antiviral Therapy. 2004;9:577–582. [PubMed] [Google Scholar]
- Acevedo A, Brodsky L, Andino R. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature. 2014;505:686–690. doi: 10.1038/nature12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hajoj SAM, Akkerman O, Parwati I, et al. Microevolution of Mycobacterium tuberculosis in a tuberculosis patient. Journal of Clinical Microbiology. 2010;48:3813–3816. doi: 10.1128/JCM.00556-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amabile-Cuevas CF. Molecular Biology Intelligence Unit. R. G. Landes; Boca Raton, Florida: 1993. Origin, Evolution and Spread of Antibiotic Resistance Genes. [Google Scholar]
- Anderson T, Nkhoma S, Ecker A, Fidock D. How can we identify parasite genes that underlie antimalarial drug resistance? Pharmacogenomics. 2011;12:59–85. doi: 10.2217/pgs.10.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariey F, Witkowski B, Amaratunga C, et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 2014;505:50–55. doi: 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley EA, Dhorda M, Fairhurst RM, et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. The New England Journal of Medicine. 2014;371:411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett RDH, Schluter D. Adaptation from standing genetic variation. Trends in Ecology & Evolution. 2008;23:38–44. doi: 10.1016/j.tree.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Behera AK, Basu S, Cherian SS. Molecular mechanism of the enhanced viral fitness contributed by secondary mutations in the hemagglutinin protein of oseltamivir resistant H1N1 influenza viruses: modeling studies of antibody and receptor binding. Gene. 2015;557:19–27. doi: 10.1016/j.gene.2014.12.003. [DOI] [PubMed] [Google Scholar]
- Bennett DE, Camacho RJ, Otelea D, et al. Drug resistance mutations for surveillance of transmitted HIV-1 drug-resistance: 2009 update. PLoS ONE. 2009;4:e4724. doi: 10.1371/journal.pone.0004724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Cavanaugh M, Clark K, et al. Genbank. Nucleic Acids Research. 2013;41:D36–D42. doi: 10.1093/nar/gks1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt S, Holmes EC, Pybus OG. The genomic rate of molecular adaptation of the human influenza A virus. Molecular Biology and Evolution. 2011;28:2443–2451. doi: 10.1093/molbev/msr044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birky CW, Walsh JB. Effects of linkage on rates of molecular evolution. Proceedings of the National Academy of Sciences of the USA. 1988;85:6414–6418. doi: 10.1073/pnas.85.17.6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloemendaal A, Brouwer EC, Fluit AC. Methicillin resistance transfer from Staphylocccus epidermidis to methicillin-susceptible Staphylococcus aureus in a patient during antibiotic therapy. PLoS ONE. 2010;5:e11841. doi: 10.1371/journal.pone.0011841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom JD, Glassman MJ. Inferring stabilizing mutations from protein phylogenies: application to influenza hemagglutinin. PLoS Computational Biology. 2009;5:e1000349. doi: 10.1371/journal.pcbi.1000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom JD, Gong LI, Baltimore D. Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. Science. 2010;328:1272–1275. doi: 10.1126/science.1187816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boni MF, Zhou Y, Taubenberger JK, Holmes EC. Homologous recombination is very rare or absent in human influenza A virus. Journal of Virology. 2008;82:4807–4811. doi: 10.1128/JVI.02683-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozic I, Reiter JG, Allen B, et al. Evolutionary dynamics of cancer in response to targeted combination therapy. eLife. 2013;2:e00747. doi: 10.7554/eLife.00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braverman JM, Hudson RR, Kaplan NL, Langley CH, Stephan W. The hitchhiking effect on the site frequency spectrum of DNA polymorphisms. Genetics. 1995;140:783–796. doi: 10.1093/genetics/140.2.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunnemann AK, Bohn-Wippert K, Zell R, et al. Drug resistance of clinical varicella-zoster virus strains confirmed by recombinant thymidine kinase expression and by targeted resistance mutagenesis of a cloned wild-type isolate. Antimicrobial Agents and Chemotherapy. 2015;59:2726–2734. doi: 10.1128/AAC.05115-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull RA, Luciani F, McElroy K, et al. Sequential bottlenecks drive viral evolution in early acute hepatitis C virus infection. PLoS Pathogens. 2011;7:e1002243. doi: 10.1371/journal.ppat.1002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC. [accessed 26 May 2015];Antimicrobial resistance posing growing health threat. 2011 Available from http://www.cdc.gov/media/releases/2011/p0407_antimicrobialresistance.html.
- Chambers HF, DeLeo FR. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nature Reviews Microbiology. 2009;7:629–641. doi: 10.1038/nrmicro2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman IH, Miller BA, Nair S, et al. A major genome region underlying artemisinin resistance in malaria. Science. 2012;336:79–82. doi: 10.1126/science.1215966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman IH, McDew-White M, Phyo AP, Sriprawat K, Nosten F, Anderson TJ. Pooled sequencing and rare variant association tests for identifying the determinants of emerging drug resistance in malaria parasites. Molecular Biology and Evolution. 2015;32:1080–1090. doi: 10.1093/molbev/msu397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua K, Seemann T, Harrison PF, et al. Complete genome sequence of Staphylococcus aureus strain JKD6159, a unique Australian clone of ST93-IV community methicillin-resistant Staphylococcus aureus. Journal of Bacteriology. 2010;192:5556–5557. doi: 10.1128/JB.00878-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua KYL, Seemann T, Harrison PF, et al. The dominant Australian community-acquired methicillin-resistant Staphylococcus aureus clone ST93-IV [2B] is highly virulent and genetically distinct. PLoS ONE. 2011;6:e25887. doi: 10.1371/journal.pone.0025887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua KYL, Howden BP, Jiang JH, Stinear T, Peleg AY. Population genetics and the evolution of virulence in Staphylococcus aureus. Infection, Genetics and Evolution. 2014;21:554–562. doi: 10.1016/j.meegid.2013.04.026. [DOI] [PubMed] [Google Scholar]
- Claessens A, Hamilton WL, Kekre M, et al. Generation of antigenic diversity in Plasmodium falciparum by structured rearrangement of var genes during mitosis. PLoS Genetics. 2014;10:e1004812. doi: 10.1371/journal.pgen.1004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codoñer FM, Pou C, Thielen A, et al. Added value of deep sequencing relative to population sequencing in heavily pre-treated HIV-1-infected subjects. PLoS ONE. 2011;6:e19461. doi: 10.1371/journal.pone.0019461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffin JM. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science. 1995;267:483–489. doi: 10.1126/science.7824947. [DOI] [PubMed] [Google Scholar]
- Cole ST, Brosch R, Parkhill J, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- Collins P, Haire L, Lin Y, et al. Structural basis for oseltamivir resistance of influenza viruses. Vaccine. 2009;27:6317–6323. doi: 10.1016/j.vaccine.2009.07.017. [DOI] [PubMed] [Google Scholar]
- Comeron JM, Ratnappan R, Bailin S. The many landscapes of recombination in Drosophila melanogaster. PLoS Genetics. 2012;8:e1002905. doi: 10.1371/journal.pgen.1002905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombs GW, Monecke S, Pearson JC, et al. Evolution and diversity of community-associated methicillin-resistant Staphylococcus aureus in a geographical region. BMC Microbiology. 2011;11:215. doi: 10.1186/1471-2180-11-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisóstomo MI, Westh H, Tomasz A, Chung M, Oliveira DC, de Lencastre H. The evolution of methicillin resistance in Staphylococcus aureus: similarity of genetic backgrounds in historically early methicillin-susceptible and - resistant isolates and contemporary epidemic clones. Proceedings of the National Academy of Sciences of the USA. 2001;98:9865–9870. doi: 10.1073/pnas.161272898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromie GA, Connelly JC, Leach DRF. Recombination at double-strand breaks and DNA ends: conserved mechanisms from phage to humans. Molecular Cell. 2001;8:1163–1174. doi: 10.1016/s1097-2765(01)00419-1. [DOI] [PubMed] [Google Scholar]
- Croucher NJ, Chewapreecha C, Hanage WP, et al. Evidence for soft selective sweeps in the evolution of pneumococcal multidrug resistance and vaccine escape. Genome Biology and Evolution. 2014;6:1589–1602. doi: 10.1093/gbe/evu120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Agata EMC, Webb GF, Horn MA, Moellering RC, Ruan S. Modeling the invasion of community acquired methicillin-resistant Staphylococcus aureus into hospitals. Clinical Infectious Diseases. 2009;48:274–284. doi: 10.1086/595844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danel C, Moh R, Chaix ML, et al. Two-months-off, four-months-on antiretroviral regimen increases the risk of resistance, compared with continuous therapy: a randomized trial involving West African adults. The Journal of Infectious Diseases. 2009;199:66–76. doi: 10.1086/595298. [DOI] [PubMed] [Google Scholar]
- David MZ, Daum RS. Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clinical Microbiology Reviews. 2010;23:616–687. doi: 10.1128/CMR.00081-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Kraker MEA, Wolkewitz M, Davey PG, Grundmann H. Clinical impact of antimicrobial resistance in European hospitals: excess mortality and length of hospital stay related to methicillin-resistant Staphylococcus aureus bloodstream infections. Antimicrobial Agents and Chemotherapy. 2011;55:1598–1605. doi: 10.1128/AAC.01157-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai MM, Fisher DS. Beneficial mutation selection balance and the effect of linkage on positive selection. Genetics. 2007;176:1759–1798. doi: 10.1534/genetics.106.067678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diep BA, Stone GG, Basuino L, et al. The arginine catabolic mobile element and staphylococcal chromosomal cassette mec linkage: convergence of virulence and resistance in the USA300 clone of methicillin-resistant Staphylococcus aureus. The Journal of Infectious Diseases. 2008;197:1523–1530. doi: 10.1086/587907. [DOI] [PubMed] [Google Scholar]
- Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondorp AM, Nosten F, Yi P, et al. Artemisinin resistance in Plasmodium falciparum malaria. The New England Journal of Medicine. 2009;361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondorp AM, Yeung S, White L, et al. Artemisinin resistance: current status and scenarios for containment. Nature Reviews Microbiology. 2010;8:272–280. doi: 10.1038/nrmicro2331. [DOI] [PubMed] [Google Scholar]
- Draghi JA, Plotkin JB. Selection biases the prevalence and type of epistasis along adaptive trajectories. Evolution. 2013;67:3120–3131. doi: 10.1111/evo.12192. [DOI] [PubMed] [Google Scholar]
- Draghi JA, Parsons TL, Plotkin JB. Epistasis increases the rate of conditionally neutral substitution in an adapting population. Genetics. 2011;187:1139–1152. doi: 10.1534/genetics.110.125997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldholm V, Norheim G, von der Lippe B, et al. Evolution of extensively drug-resistant Mycobacterium tuberculosis from a susceptible ancestor in a single patient. Genome Biology. 2014;15:490. doi: 10.1186/s13059-014-0490-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright M, Day N. Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. Journal of Clinical Microbiology. 2000;38:1008–1015. doi: 10.1128/jcm.38.3.1008-1015.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright MC, Robinson DA, Randle G, Feil EJ, Grundmann H, Spratt BG. The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA) Proceedings of the National Academy of Sciences of the USA. 2002;99:7687–7692. doi: 10.1073/pnas.122108599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Wu CI. Hitchhiking under positive Darwinian selection. Genetics. 2000;155:1405–1413. doi: 10.1093/genetics/155.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Wyckoff GJ, Wu CI. Testing the neutral theory of molecular evolution with genomic data from Drosophila. Nature. 2002;415:1024–1026. doi: 10.1038/4151024a. [DOI] [PubMed] [Google Scholar]
- Feder AF, Rhee SY, Shafer RW, Petrov DA, Pennings PS. More efficacious drugs lead to harder selective sweeps in the evolution of drug resistance in HIV-1. 2015 doi: 10.7554/eLife.10670. bioRxiv 024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil EJ, Enright MC. Analyses of clonality and the evolution of bacterial pathogens. Current Opinion in Microbiology. 2004;7:308–313. doi: 10.1016/j.mib.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. Journal of Bacteriology. 2004;186:1518–1530. doi: 10.1128/JB.186.5.1518-1530.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. The evolutionary advantage of recombination. Genetics. 1974;78:737–756. doi: 10.1093/genetics/78.2.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdig MT, Cooper RA, Mu J, et al. Dissecting the loci of low-level quinine resistance in malaria parasites. Molecular Microbiology. 2004;52:985–997. doi: 10.1111/j.1365-2958.2004.04035.x. [DOI] [PubMed] [Google Scholar]
- Ferrer-Admetlla A, Liang M, Korneliussen T, Nielsen R. On detecting incomplete soft or hard selective sweeps using haplotype structure. Molecular Biology and Evolution. 2014;31:1275–1291. doi: 10.1093/molbev/msu077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RA. The Genetical Theory of Natural Selection: A Complete Variorum Edition. Oxford University Press; Oxford: 1930. [Google Scholar]
- Fitzgerald JR, Sturdevant DE, Mackie SM, Gill SR, Musser JM. Evolutionary genomics of Staphylococcus aureus: insights into the origin of methicillin-resistant strains and the toxic shock syndrome epidemic. Proceedings of the National Academy of Sciences of the USA. 2001;98:8821–8826. doi: 10.1073/pnas.161098098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foll M, Poh YP, Renzette N, et al. Influenza virus drug resistance: a time-sampled population genetics perspective. PLoS Genetics. 2014;10:e1004185. doi: 10.1371/journal.pgen.1004185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garud NR, Messer PW, Buzbas EO, Petrov DA. Recent selective sweeps in North American Drosophila melanogaster show signatures of soft sweeps. PLoS Genetics. 2015;11:e1005004. doi: 10.1371/journal.pgen.1005004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrish PJ, Lenski RE. The fate of competing beneficial mutations in an asexual population. Genetica. 1998;102:127–144. [PubMed] [Google Scholar]
- Ghorbal M, Gorman M, Macpherson CR, Martins RM, Scherf A, Lopez-Rubio JJ. Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPRCas9 system. Nature Biotechnology. 2014;32:819–821. doi: 10.1038/nbt.2925. [DOI] [PubMed] [Google Scholar]
- Ginting TE, Shinya K, Kyan Y, et al. Amino acid changes in hemagglutinin contribute to the replication of oseltamivir-resistant H1N1 influenza viruses. Journal of Virology. 2012;86:121–127. doi: 10.1128/JVI.06085-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong LI, Bloom JD. Epistatically interacting substitutions are enriched during adaptive protein evolution. PLoS Genetics. 2014;10:e1004328. doi: 10.1371/journal.pgen.1004328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong LI, Suchard MA, Bloom JD. Stability-mediated epistasis constrains the evolution of an influenza protein. eLife. 2013;2:e00631. doi: 10.7554/eLife.00631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good BH, Desai MM. Deleterious passengers in adapting populations. Genetics. 2014;198:1183–1208. doi: 10.1534/genetics.114.170233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenfell BT, Pybus OG, Gog JR, et al. Unifying the epidemiological and evolutionary dynamics of pathogens. Science. 2004;303:327–332. doi: 10.1126/science.1090727. [DOI] [PubMed] [Google Scholar]
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of phyml 3.0. Systematic Biology. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Hakki M, Chou S. The biology of cytomegalovirus drug resistance. Current Opinion in Infectious Diseases. 2011;24:605. doi: 10.1097/QCO.0b013e32834cfb58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanssen AM, Ericson Sollid JU. SCCmec in staphylococci: genes on the move. FEMS Immunology and Medical Microbiology. 2006;46:8–20. doi: 10.1111/j.1574-695X.2005.00009.x. [DOI] [PubMed] [Google Scholar]
- Hanssen AM, Kjeldsen G, Sollid JUE. Local variants of staphylococcal cassette chromosome mec in sporadic methicillin-resistant Staphylococcus aureus and methicillin-resistant coagulase-negative staphylococci: evidence of horizontal gene transfer? Antimicrobial Agents and Chemotherapy. 2004;48:285–296. doi: 10.1128/AAC.48.1.285-296.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanssen AM, Fossum A, Mikalsen J, Halvorsen DS, Bukholm G, Sollid JUE. Dissemination of community-acquired methicillin-resistant Staphylococcus aureus clones in northern Norway: sequence types 8 and 80 predominate. Journal of Clinical Microbiology. 2005;43:2118–2124. doi: 10.1128/JCM.43.5.2118-2124.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SR, Cartwright EJP, Török ME, et al. Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: a descriptive study. The Lancet Infectious Diseases. 2013;13:130–136. doi: 10.1016/S1473-3099(12)70268-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley SE, Das SR, Gibbs JS, et al. Influenza A virus hemagglutinin antibody escape promotes neuraminidase antigenic variation and drug resistance. PLoS ONE. 2011;6:e15190. doi: 10.1371/journal.pone.0015190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlocher ML, Truscon R, Elias S, et al. Influenza viruses resistant to the antiviral drug oseltamivir: transmission studies in ferrets. Journal of Infectious Diseases. 2004;190:1627–1630. doi: 10.1086/424572. [DOI] [PubMed] [Google Scholar]
- Hermisson J, Pennings PS. Soft sweeps: molecular population genetics of adaptation from standing genetic variation. Genetics. 2005;169:2335–2352. doi: 10.1534/genetics.104.036947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill WG, Robertson A. The effect of linkage on limits to artificial selection. Genetical Research. 1966;8:269. [PubMed] [Google Scholar]
- Hinkley T, Martins J, Chappey C, et al. A systems analysis of mutational effects in HIV-1 protease and reverse transcriptase. Nature Genetics. 2011;43:487–489. doi: 10.1038/ng.795. [DOI] [PubMed] [Google Scholar]
- Hirsch MS, Günthard HF, Schapiro JM, et al. Antiretroviral drug resistance testing in adult HIV-1 infection: 2008 recommendations of an international aids society-USA panel. Clinical Infectious Diseases. 2008;47:266–285. doi: 10.1086/589297. [DOI] [PubMed] [Google Scholar]
- Holden MTG, Hsu LY, Kurt K, et al. A genomic portrait of the emergence, evolution, and global spread of a methicillin-resistant Staphylococcus aureus pandemic. Genome Research. 2013;23:653–664. doi: 10.1101/gr.147710.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes EC, Ghedin E, Miller N, et al. Whole-genome analysis of human influenza a virus reveals multiple persistent lineages and reassortment among recent H3N2 viruses. PLoS Biology. 2005;3:1579. doi: 10.1371/journal.pbio.0030300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howden BP, Stinear TP, Allen DL, Johnson PDR, Ward PB, Davies JK. Genomic analysis reveals a point mutation in the two-component sensor gene graS that leads to intermediate vancomycin resistance in clinical Staphylococcus aureus. Antimicrobial Agents and Chemotherapy. 2008;52:3755–3762. doi: 10.1128/AAC.01613-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu LY, Harris SR, Chlebowicz MA, et al. Evolutionary dynamics of methicillin-resistant Staphylococcus aureus within a healthcare system. Genome Biology. 2015;16:1–13. doi: 10.1186/s13059-015-0643-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde JE. Drug-resistant malaria. Trends in Parasitology. 2005;21:494–498. doi: 10.1016/j.pt.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ives J, Carr J, Mendel D, et al. The H274Y mutation in the influenza A/H1N1 neuraminidase active site following oseltamivir phosphate treatment leave virus severely compromised both in vitro and in vivo. Antiviral Research. 2002;55:307–317. doi: 10.1016/s0166-3542(02)00053-0. [DOI] [PubMed] [Google Scholar]
- IWG-SCC. [accessed 26 May 2015];Scc classification. 2015 Available from http://www.sccmec.org/Pages/SCC_ClassificationEN.html.
- Jensen-Seaman MI, Furey TS, Payseur BA, et al. Comparative recombination rates in the rat, mouse, and human genomes. Genome Research. 2004;14:528–538. doi: 10.1101/gr.1970304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Li N, Gopalan V, et al. High recombination rates and hotspots in a Plasmodium falciparum genetic cross. Genome Biology. 2011;12:R33. doi: 10.1186/gb-2011-12-4-r33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle D, Wickham H. ggmap: Spatial visualization with ggplot2. The R Journal. 2013;5:144–161. [Google Scholar]
- Kang HM, Zaitlen NA, Wade CM, et al. Efficient control of population structure in model organism association mapping. Genetics. 2008;178:1709–1723. doi: 10.1534/genetics.107.080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan NL, Hudson RR, Langley CH. The “hitchhiking effect” revisited. Genetics. 1989;123:887–899. doi: 10.1093/genetics/123.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama Y, Ito T, Hiramatsu K. A new class of genetic element, Staphylococcus cassette chromosome mec, encodes methicillin resistance in Staphylococcus aureus. Antimicrobial Agents and Chemotherapy. 2000;44:1549–1555. doi: 10.1128/aac.44.6.1549-1555.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling PJ, Palmer JD. Horizontal gene transfer in eukaryotic evolution. Nature Reviews Genetics. 2008;9:605–618. doi: 10.1038/nrg2386. [DOI] [PubMed] [Google Scholar]
- Kim Y, Stephan W. Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics. 2002;160:765–777. doi: 10.1093/genetics/160.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koelle K, Cobey S, Grenfell B, Pascual M. Epochal evolution shapes the phylodynamics of interpandemic influenza A (H3N2) in humans. Science. 2006;314:1898–1903. doi: 10.1126/science.1132745. [DOI] [PubMed] [Google Scholar]
- Kreiswirth B, Kornblum J, Arbeit RD, et al. Evidence for a clonal origin of methicillin resistance in Staphylococcus aureus. Science. 1993;259:227–230. doi: 10.1126/science.8093647. [DOI] [PubMed] [Google Scholar]
- Kryazhimskiy S, Dushoff J, Bazykin GA, Plotkin JB. Prevalence of epistasis in the evolution of influenza A surface proteins. PLoS Genetics. 2011;7:e1001301. doi: 10.1371/journal.pgen.1001301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvitek DJ, Sherlock G. Whole genome, whole population sequencing reveals that loss of signaling networks is the major adaptive strategy in a constant environment. PLoS Genetics. 2013;9:e1003972. doi: 10.1371/journal.pgen.1003972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyaw MP, Nyunt MH, Chit K, et al. Reduced susceptibility of Plasmodium falciparum to artesunate in southern Myanmar. PLoS ONE. 2013;8:e57689. doi: 10.1371/journal.pone.0057689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GI, Rice DP, Hickman MJ, et al. Pervasive genetic hitchhiking and clonal interference in forty evolving yeast populations. Nature. 2013;500:571–574. doi: 10.1038/nature12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FJ, Amin J, Carr A. Efficacy of initial antiretroviral therapy for HIV-1 infection in adults: a systematic review and meta-analysis of 114 studies with up to 144 weeks’ follow-up. PLoS ONE. 2014;9:e97482. doi: 10.1371/journal.pone.0097482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewontin RC, Krakauer J. Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics. 1973;74:175–195. doi: 10.1093/genetics/74.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lina G, Durand G, Berchich C, et al. Staphylococcal chromosome cassette evolution in Staphylococcus aureus inferred from ccr gene complex sequence typing analysis. Clinical Microbiology and Infection. 2006;12:1175–1184. doi: 10.1111/j.1469-0691.2006.01548.x. [DOI] [PubMed] [Google Scholar]
- Lindsay JA, Holden MTG. Staphylococcus aureus: superbug, super genome? Trends in Microbiology. 2004;12:378–385. doi: 10.1016/j.tim.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Lipsitch M, Levin B. Population dynamics of tuberculosis treatment: mathematical models of the roles of non-compliance and bacterial heterogeneity in the evolution of drug resistance. The International Journal of Tuberculosis and Lung Disease. 1998;2:187–199. [PubMed] [Google Scholar]
- Liu C, Graber CJ, Karr M, et al. A population-based study of the incidence and molecular epidemiology of methicillin-resistant Staphylococcus aureus disease in San Francisco, 2004–2005. Clinical Infectious Diseases. 2008;46:1637–1646. doi: 10.1086/587893. [DOI] [PubMed] [Google Scholar]
- Lou DI, Hussmann JA, McBee RM, et al. High-throughput dna sequencing errors are reduced by orders of magnitude using circle sequencing. Proceedings of the National Academy of Sciences of the USA. 2013;110:19872–19877. doi: 10.1073/pnas.1319590110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowy F. Antimicrobial resistance: the example of Staphylococcus aureus. The Journal of Clinical Investigation. 2003;111:1265–1273. doi: 10.1172/JCI18535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luksza M, Lässig M. A predictive fitness model for influenza. Nature. 2014;507:57–61. doi: 10.1038/nature13087. [DOI] [PubMed] [Google Scholar]
- Lythgoe KA, Fraser C. New insights into the evolutionary rate of HIV-1 at the within-host and epidemiological levels. Proceedings of the Royal Society of London B: Biological Sciences. 2012;279:3367–3375. doi: 10.1098/rspb.2012.0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiden MCJ, Rensburg MJJV, Bray JE, et al. Europe PMC Funders Group Europe PMC Funders author manuscripts MLST revisited: the gene-by-gene approach to bacterial genomics. Nature Reviews Microbiology. 2014;11:728–736. doi: 10.1038/nrmicro3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malachowa N, Deleo FR. Mobile genetic elements of Staphylococcus aureus. Cellular and Molecular Life Sciences. 2010;67:3057–3071. doi: 10.1007/s00018-010-0389-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margot NA, Lu B, Cheng A, Miller MD. Resistance development over 144 weeks in treatment naive patients receiving tenofovir disoproxil fumarate or stavudine with lamivudine and efavirenz in study 903. HIV Medicine. 2006;7:442–450. doi: 10.1111/j.1468-1293.2006.00404.x. [DOI] [PubMed] [Google Scholar]
- Martens EA, Hallatschek O. Interfering waves of adaptation promote spatial mixing. Genetics. 2011;189:1045–1060. doi: 10.1534/genetics.111.130112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslanova I, Dosskar J, Varga M, et al. Bacteriophages of Staphylococcus aureus efficiently package various bacterial genes and mobile genetic elements including SCCmec with different frequencies. Environmental Microbiology Reports. 2013;5:66–73. doi: 10.1111/j.1758-2229.2012.00378.x. [DOI] [PubMed] [Google Scholar]
- Maynard Smith J, Haigh J. The hitch-hiking effect of a favourable gene. Genetical Research. 1974;23:23–35. [PubMed] [Google Scholar]
- Mediavilla JR, Chen L, Mathema B, Kreiswirth BN. Global epidemiology of community-associated methicillin resistant Staphylococcus aureus (CA-MRSA) Current Opinion in Microbiology. 2012;15:588–595. doi: 10.1016/j.mib.2012.08.003. [DOI] [PubMed] [Google Scholar]
- Meric G, Miragaia M, de Been M, et al. Ecological overlap and horizontal gene transfer in Staphylococcus aureus and Staphylococcus epidermidis. Genome Biology and Evolution. 2015;7:1313–1328. doi: 10.1093/gbe/evv066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milheiriço C, Oliveira DC, de Lencastre H. Multiplex PCR strategy for subtyping the staphylococcal cassette chromosome mec type IV in methicillin-resistant Staphylococcus aureus: ‘SCCmec IV multiplex’. The Journal of Antimicrobial Chemotherapy. 2007;60:42–48. doi: 10.1093/jac/dkm112. [DOI] [PubMed] [Google Scholar]
- Miotto O, Almagro-Garcia J, Manske M, et al. Multiple populations of artemisinin-resistant Plasmodium falciparum in Cambodia. Nature Genetics. 2013;45:648–655. doi: 10.1038/ng.2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miotto O, Amato R, Ashley EA, et al. Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nature Genetics. 2015;47:226–234. doi: 10.1038/ng.3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miralles R, Gerrish PJ, Moya A, Elena SF. Clonal interference and the evolution of RNA viruses. Science. 1999;285:1745–1747. doi: 10.1126/science.285.5434.1745. [DOI] [PubMed] [Google Scholar]
- Mita T, Venkatesan M, Ohashi J, et al. Limited geographical origin and global spread of sulfadoxine-resistant dhps alleles in Plasmodium falciparum populations. The Journal of Infectious Diseases. 2011;204:1980–1988. doi: 10.1093/infdis/jir664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Gamez S, Hill AL, Rosenbloom DIS, Petrov DA, Nowak MA, Pennings PS. Imperfect drug penetration leads to spatial monotherapy and rapid evolution of multidrug resistance. Proceedings of the National Academy of Sciences of the USA. 2015;112:E2874–E2883. doi: 10.1073/pnas.1424184112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscona A. Neuraminidase inhibitors for influenza. The New England Journal of Medicine. 2005;353:1363–1373. doi: 10.1056/NEJMra050740. [DOI] [PubMed] [Google Scholar]
- Moscona A. Global transmission of oseltamivir-resistant influenza. The New England Journal of Medicine. 2009;360:953–956. doi: 10.1056/NEJMp0900648. [DOI] [PubMed] [Google Scholar]
- Muller HJ. Some genetic aspects of sex. The American Naturalist. 1932;66:118–138. [Google Scholar]
- Musser JM, Kapur V. Clonal analysis of methicillin-resistant Staphylococcus aureus strains from intercontinental sources: association of the mec gene with divergent phylogenetic lineages implies dissemination by horizontal transfer and recombination. Journal of Clinical Microbiology. 1992;30:2058–2063. doi: 10.1128/jcm.30.8.2058-2063.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musser JM, Amin A, Ramaswamy S. Negligible genetic diversity of Mycobacterium tuberculosis host immune system protein targets: evidence of limited selective pressure. Genetics. 2000;155:7–16. doi: 10.1093/genetics/155.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musset L, Le Bras J, Clain J. Parallel evolution of adaptive mutations in Plasmodium falciparum mitochondrial DNA during atovaquone-proguanil treatment. Molecular Biology and Evolution. 2007;24:1582–1585. doi: 10.1093/molbev/msm087. [DOI] [PubMed] [Google Scholar]
- Nair S, Williams JT, Brockman A, et al. A selective sweep driven by pyrimethamine treatment in Southeast Asian malaria parasites. Molecular Biology and Evolution. 2003;20:1526–1536. doi: 10.1093/molbev/msg162. [DOI] [PubMed] [Google Scholar]
- Namouchi A, Didelot X, Scho U, et al. After the bottleneck: genome-wide diversification of the Mycobacterium tuberculosis complex by mutation, recombination, and natural selection. Genome Research. 2012;22:721–734. doi: 10.1101/gr.129544.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro Y, Herranz M, Pérez-Lago L, et al. Systematic survey of clonal complexity in tuberculosis at a populational level and detailed characterization of the isolates involved. Journal of Clinical Microbiology. 2011;49:4131–4137. doi: 10.1128/JCM.05203-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negroni M, Buc H. Copy-choice recombination by reverse transcriptases: reshuffling of genetic markers mediated by RNA chaperones. Proceedings of the National Academy of Sciences of the USA. 2000;97:6385–6390. doi: 10.1073/pnas.120520497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher RA. Genetic draft, selective interference, and population genetics of rapid adaptation. 2013. arXiv preprint arXiv:1302.1148. [Google Scholar]
- Neher RA, Russell CA, Shraiman BI. Predicting evolution from the shape of genealogical trees. eLife. 2014;3:e03568. doi: 10.7554/eLife.03568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson KE, Clayton RA, Gill SR, et al. Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritima. Nature. 1999;399:323–329. doi: 10.1038/20601. [DOI] [PubMed] [Google Scholar]
- Nielsen R, Williamson S, Kim Y, Hubisz MJ, Clark AG, Bustamante C. Genomic scans for selective sweeps using SNP data. Genome Research. 2005;15:1566–1575. doi: 10.1101/gr.4252305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noedl H, Se Y, Schaecher K, Smith BL, Socheat D, Fukuda MM. Evidence of artemisinin-resistant malaria in western Cambodia. The New England Journal of Medicine. 2008;359:2619–2620. doi: 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- Nübel U, Roumagnac P, Feldkamp M, et al. Frequent emergence and limited geographic dispersal of methicillin-resistant Staphylococcus aureus. Proceedings of the National Academy of Sciences of the USA. 2008;105:14130–14135. doi: 10.1073/pnas.0804178105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuma K, Kozue I, Turnidge JD, et al. Dissemination of new methicillin-resistant Staphylococcus aureus clones in the community. Journal of Clinical Microbiology. 2002;40:4289–4294. doi: 10.1128/JCM.40.11.4289-4294.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira DC, Tomasz A, De Lencastre H. Secrets of success of a human pathogen: molecular evolution of pandemic clones of meticillin-resistant Staphylococcus aureus. Lancet Infectious Diseases. 2002;2:180–189. doi: 10.1016/s1473-3099(02)00227-x. [DOI] [PubMed] [Google Scholar]
- van Opijnen T, Berkhout B. The host environment drives HIV-1 fitness. Reviews in Medical Virology. 2005;15:219–233. doi: 10.1002/rmv.472. [DOI] [PubMed] [Google Scholar]
- Orr HA, Betancourt AJ. Haldane’s sieve and adaptation from the standing genetic variation. Genetics. 2001;157:875–884. doi: 10.1093/genetics/157.2.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit A, de Boer RJ. Reliable reconstruction of HIV-1 whole genome haplotypes reveals clonal interference and genetic hitchhiking among immune escape variants. Retrovirology. 2014;11:56. doi: 10.1186/1742-4690-11-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes R, Lalama CM, Ribaudo HJ, et al. Pre-existing minority drug-resistant HIV-1 variants, adherence, and risk of antiretroviral treatment failure. The Journal of Infectious Diseases. 2010;201:662–671. doi: 10.1086/650543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SC, Krug J. Clonal interference in large populations. Proceedings of the National Academy of Sciences of the USA. 2007;104:18135–18140. doi: 10.1073/pnas.0705778104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennings PS. Standing genetic variation and the evolution of drug resistance in HIV. PLoS Computational Biology. 2012;8:e1002527. doi: 10.1371/journal.pcbi.1002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennings PS, Hermisson J. Soft sweeps II-molecular population genetics of adaptation from recurrent mutation or migration. Molecular Biology and Evolution. 2006;23:1076–1084. doi: 10.1093/molbev/msj117. [DOI] [PubMed] [Google Scholar]
- Pennings PS, Kryazhimskiy S, Wakeley J. Loss and recovery of genetic diversity in adapting populations of HIV. PLoS Genetics. 2014;10:e1004000. doi: 10.1371/journal.pgen.1004000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepin KM, Wichman HA. Experimental evolution and genome sequencing reveal variation in levels of clonal interference in large populations of bacteriophage phiX174. BMC Evolutionary Biology. 2008;8:85. doi: 10.1186/1471-2148-8-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piret J, Boivin G. Antiviral drug resistance in herpesviruses other than cytomegalovirus. Reviews in Medical Virology. 2014;24:186–218. doi: 10.1002/rmv.1787. [DOI] [PubMed] [Google Scholar]
- Rajon E, Masel J. Compensatory evolution and the origins of innovations. Genetics. 2013;193:1209–1220. doi: 10.1534/genetics.112.148627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A, Pybus OG, Nelson MI, Viboud C, Taubenberger JK, Holmes EC. The genomic and epidemiological dynamics of human influenza A virus. Nature. 2008;453:615–619. doi: 10.1038/nature06945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read AF, Huijben S. Perspective: evolutionary biology and the avoidance of antimicrobial resistance. Evolutionary Applications. 2009;2:40–51. doi: 10.1111/j.1752-4571.2008.00066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renzette N, Kowalik TF, Jensen JD. On the relative roles of background selection and genetic hitchhiking in shaping human cytomegalovirus genetic diversity. Molecular Ecology. 2015 doi: 10.1111/mec.13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SY, Blanco JL, Jordan MR, et al. Geographic and temporal trends in the molecular epidemiology and genetic mechanisms of transmitted HIV-1 drug resistance: an individual-patient- and sequence-level meta-analysis. PLoS Medicine. 2015;12:e1001810. doi: 10.1371/journal.pmed.1001810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D, Enright M. Evolutionary models of the emergence of methicillin-resistant Staphylococcus aureus. Antimicrobial Agents and Chemotherapy. 2003;47:3926–3934. doi: 10.1128/AAC.47.12.3926-3934.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson DA, Enright MC. Evolution of Staphylococcus aureus by large chromosomal replacements. Journal of Bacteriology. 2004;186:1060–1064. doi: 10.1128/JB.186.4.1060-1064.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers MB, Song T, Sebra R, et al. Intrahost dynamics of antiviral resistance in influenza A virus reflect complex patterns of segment linkage, reassortment, and natural selection. mBio. 2015;6:e02464–14. doi: 10.1128/mBio.02464-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper C, Pearce R, Nair S, Sharp B, Nosten F, Anderson T. Intercontinental spread of pyrimethamine-resistant malaria. Science. 2004;305:1124. doi: 10.1126/science.1098876. [DOI] [PubMed] [Google Scholar]
- Rouzine IM, Wakeley J, Coffin JM. The solitary wave of asexual evolution. Proceedings of the National Academy of Sciences of the USA. 2003;100:587–592. doi: 10.1073/pnas.242719299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti PC, Reich DE, Higgins JM, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- Sabeti PC, Varilly P, Fry B, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449:913–918. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandgren A, Strong M, Muthukrishnan P, Weiner BK, Church GM, Murray MB. Tuberculosis drug resistance mutation database. PLoS Medicine. 2009;6:e1000002. doi: 10.1371/journal.pmed.1000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutten M. Antiretroviral Resistance in Clinical Practice. Chapter 5. Mediscript; London: 2006. Resistance assays. [Google Scholar]
- Seybold U, Kourbatova EV, Johnson JG, et al. Emergence of community-associated methicillin resistant Staphylococcus aureus USA300 genotype as a major cause of health care-associated blood stream infections. Clinical Infectious Diseases. 2006;42:647–656. doi: 10.1086/499815. [DOI] [PubMed] [Google Scholar]
- Shapiro BJ, Polz MF. Ordering microbial diversity into ecologically and genetically cohesive units. Trends in Microbiology. 2014;22:235–247. doi: 10.1016/j.tim.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhu AB, Valderramos SG, Fidock DA. pfmdr1 mutations contribute to quinine resistance and enhance mefloquine and artemisinin sensitivity in Plasmodium falciparum. Molecular Microbiology. 2005;57:913–926. doi: 10.1111/j.1365-2958.2005.04729.x. [DOI] [PubMed] [Google Scholar]
- Simen BB, Simons JF, Hullsiek KH, et al. Low-abundance drug-resistant viral variants in chronically HIV-infected, antiretroviral treatment-naive patients significantly impact treatment outcomes. The Journal of Infectious Diseases. 2009;199:693–701. doi: 10.1086/596736. [DOI] [PubMed] [Google Scholar]
- Simon-Loriere E, Holmes EC. Why do RNA viruses recombine? Nature Reviews Microbiology. 2011;9:617–626. doi: 10.1038/nrmicro2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth DS, Wong A, Robinson DA. Cross-species spread of SCCmec IV subtypes in staphylococci. Infection, Genetics and Evolution. 2012;11:446–453. doi: 10.1016/j.meegid.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreevatsan S, Pan X, Stockbauer KE, et al. Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissemination. Proceedings of the National Academy of Sciences. 1997;94:9869–9874. doi: 10.1073/pnas.94.18.9869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockman J. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. Yearbook of Pediatrics. 2009;2009:260–263. [Google Scholar]
- Stokes HW, Gillings MR. Gene flow, mobile genetic elements and the recruitment of antibiotic resistance genes into Gram-negative pathogens. FEMS Microbiology Reviews. 2011;35:790–819. doi: 10.1111/j.1574-6976.2011.00273.x. [DOI] [PubMed] [Google Scholar]
- Strahilevitz J, Hooper DC. Dual targeting of topoisomerase IV and gyrase to reduce mutant selection: direct testing of the paradigm by using WCK-1734, a new fluoroquinolone, and ciprofloxacin. Antimicrobial Agents and Chemotherapy. 2005;49:1949–1956. doi: 10.1128/AAC.49.5.1949-1956.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straimer J, Gnadig NF, Witkowski B, et al. Drug resistance. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science. 2015;347:428–431. doi: 10.1126/science.1260867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strelkowa N, Lässig M. Clonal interference in the evolution of influenza. Genetics. 2012;192:671–682. doi: 10.1534/genetics.112.143396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stryjewski ME, Corey GR. Methicillin-resistant Staphylococcus aureus: an evolving pathogen. Clinical Infectious Diseases. 2014;58:S10–S19. doi: 10.1093/cid/cit613. [DOI] [PubMed] [Google Scholar]
- Su X, Ferdig MT, Huang Y, et al. A genetic map and recombination parameters of the human malaria parasite Plasmodium falciparum. Science. 1999;286:1351–1353. doi: 10.1126/science.286.5443.1351. [DOI] [PubMed] [Google Scholar]
- Sun G, Luo T, Yang C, et al. Dynamic population changes in Mycobacterium tuberculosis during acquisition and fixation of drug resistance in patients. The Journal of Infectious Diseases. 2012;206:1724–1733. doi: 10.1093/infdis/jis601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syvanen M. Evolutionary implications of horizontal gene transfer. Annual Review of Genetics. 2012;46:341–358. doi: 10.1146/annurev-genet-110711-155529. [DOI] [PubMed] [Google Scholar]
- Szendro IG, Schenk MF, Franke J, Krug J, de Visser JAG. Quantitative analyses of empirical fitness landscapes. Journal of Statistical Mechanics: Theory and Experiment. 2013;2013:P01005. [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takala-Harrison S, Clark TG, Jacob CG, et al. Genetic loci associated with delayed clearance of Plasmodium falciparum following artemisinin treatment in Southeast Asia. Proceedings of the National Academy of Sciences of the USA. 2013;110:240–245. doi: 10.1073/pnas.1211205110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takala-Harrison S, Jacob CG, Arze C, et al. Independent emergence of artemisinin resistance mutations among Plasmodium falciparum in Southeast Asia. The Journal of Infectious Diseases. 2015;211:670–679. doi: 10.1093/infdis/jiu491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toprak E, Veres A, Michel JB, Chait R, Hartl DL, Kishony R. Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nature Genetics. 2012;44:101–105. doi: 10.1038/ng.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tun KM, Imwong M, Lwin KM, et al. Spread of artemisinin-resistant Plasmodium falciparum in Myanmar: a cross-sectional survey of the K13 molecular marker. The Lancet Infectious Diseases. 2015;15:415–421. doi: 10.1016/S1473-3099(15)70032-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UK Collaborative Group on HIV Drug Resistance and UK CHIC Study Group and Others. Long term probability of detecting drug-resistant HIV in treatment-naive patients initiating combination antiretroviral therapy. Clinical Infectious Diseases. 2010;50:1275–1285. doi: 10.1086/651684. [DOI] [PubMed] [Google Scholar]
- Veyrier F, Pletzer D, Turenne C, Behr MA. Phylogenetic detection of horizontal gene transfer during the step-wise genesis of Mycobacterium tuberculosis. BMC Evolutionary Biology. 2009;9:196. doi: 10.1186/1471-2148-9-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinayak S, Alam MT, Mixson-Hayden T, et al. Origin and evolution of sulfadoxine resistant Plasmodium falciparum. PLoS Pathogens. 2010;6:e1000830. doi: 10.1371/journal.ppat.1000830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitti JJ, Grossman SR, Sabeti PC. Detecting natural selection in genomic data. Annual Review of Genetics. 2013;47:97–120. doi: 10.1146/annurev-genet-111212-133526. [DOI] [PubMed] [Google Scholar]
- Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biology. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter MJ, Shen D, Ding L, et al. Clonal architecture of secondary acute myeloid leukemia. The New England Journal of Medicine. 2012;366:1090–1098. doi: 10.1056/NEJMoa1106968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreich DM, Delaney NF, DePristo MA, Hartl DL. Darwinian evolution can follow only very few mutational paths to fitter proteins. Science. 2006;312:111–114. doi: 10.1126/science.1123539. [DOI] [PubMed] [Google Scholar]
- Wensing AM, Calvez V, Gunthard HF, et al. 2014 update of the drug resistance mutations in HIV-1. Topics in Antiviral Medicine. 2014;22:642–650. [PMC free article] [PubMed] [Google Scholar]
- Wheeler WH, Ziebell RA, Zabina H, et al. Prevalence of transmitted drug resistance associated mutations and HIV-1 subtypes in new HIV-1 diagnoses, US-2006. AIDS. 2010;24:1203–1212. doi: 10.1097/QAD.0b013e3283388742. [DOI] [PubMed] [Google Scholar]
- WHO. [accessed 26 May 2015];Influenza A (H1N1) virus resistance to oseltamivir— 2008/2009 influenza season, northern hemisphere. 2009 Available from http://www.who.int/influenza/resources/documents/H1N1webupdate20090318_ed_ns.pdf.
- WHO. Global Tuberculosis Control: Surveillance, Planning, Financing. World Health Organization; 2014a. [accessed 23 May 2015]. Available from http://www.who.int/tb/publications/global_report/en/ [Google Scholar]
- WHO. [accessed 26 May 2015];Influenza fact sheet no. 211. 2014b Available from http://www.who.int/mediacentre/factsheets/fs211/en/
- WHO. World Malaria Report 2014. World Health Organization; 2014c. [Google Scholar]
- WHO. Antimicrobial Resistance: Global Report on Surveillance. World Health Organization; 2014d. [Google Scholar]
- Woolhouse ME, Haydon DT, Antia R. Emerging pathogens: the epidemiology and evolution of species jumps. Trends in Ecology & Evolution. 2005;20:238–244. doi: 10.1016/j.tree.2005.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootton JC, Feng X, Ferdig MT, et al. Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum. Nature. 2002;418:320–323. doi: 10.1038/nature00813. [DOI] [PubMed] [Google Scholar]
- Wu S, Piscitelli C, de Lencastre H, Tomasz A. Tracking the evolutionary origin of the methicillin resistance gene: cloning and sequencing of a homologue of mecA from a methicillin susceptible strain of Staphylococcus sciuri. Microbial Drug Resistance. 1996;2:435–441. doi: 10.1089/mdr.1996.2.435. [DOI] [PubMed] [Google Scholar]
- Yu G, Lam TTY. ggtree: a phylogenetic tree viewer for different types of tree annotations 2015 [Google Scholar]
- Zanini F, Neher RA. Quantifying selection against synonymous mutations in HIV-1 env evolution. Journal of Virology. 2013;87:11843–11850. doi: 10.1128/JVI.01529-13. [DOI] [PMC free article] [PubMed] [Google Scholar]