

Abstract
Background
Multiple Sclerosis (MS) is a chronic inflammatory disease of the central nervous system caused by genetic and environmental factors. DNA methylation, an epigenetic mechanism that controls genome activity, may provide a link between genetic and environmental risk factors.
Objective
We sought to identify DNA methylation changes in CD4+ T cells in patients with relapsing-remitting (RR-MS) and secondary-progressive (SP-MS) disease and healthy controls (HC).
Methods
We performed DNA methylation analysis in CD4+ T cells from RR-MS, SP-MS and HC and associated identified changes with the nearby risk allele, smoking, age and gene expression.
Results
We observed significant methylation differences in the VMP1/MIR21 locus with RR-MS displaying higher methylation compared to SP-MS and HC. VMP1/MIR21 methylation did not correlate with a known MS risk variant in VMP1 or smoking but displayed a significant negative correlation with age and the levels of mature miR-21 in CD4+ T cells. Accordingly, RR-MS displayed lower levels of miR-21 compared to SP-MS, which might reflect differences in age between the groups, and healthy individuals, and a significant enrichment of up-regulated miR-21 target genes.
Conclusion
Disease-related changes in epigenetic marking of MIR21 in RR-MS lead to differences in miR-21 expression with a consequence on miR-21 target genes.
Keywords: CD4+ T cells, Autoimmunity, Epigenetics, DNA methylation, MicroRNAs, Multiple Sclerosis, Relapsing-remitting, miR-21
Introduction
Multiple Sclerosis (MS) is a chronic inflammatory disease of the CNS and one of the most common causes of neurological disability in young adults1. The majority of MS patients initially present with the relapsing-remitting disease (RR-MS) characterized by episodes of active disease and periods of clinical inactivity but most will eventually convert to a secondary-progressive form (SP-MS) characterized by a continuous worsening1.
MS results from an interplay between genes and environmental factors. The HLA-DRB1*15:01 haplotype has been consistently associated with MS suggesting critical role of the HLA class II molecules in presenting antigens to CD4+ T cells2. The importance of CD4+ T cells has further been supported by their ability to induce MS-like pathology in rodents3. More than 100 non-HLA loci have been associated with MS, jointly pointing to importance of immune-mediated functions such as lymphocyte activation and differentiation2, 4. Smoking has also been consistently associated with an increased risk of MS and carriers of HLA-DRB1*15:01 who are smokers have 14-fold higher risk compared to non-smokers and non-carriers5.
Such gene-environment interactions may be mediated through epigenetic mechanisms, by which environment modulates gene expression in a manner that is heritable through cell division. DNA methylation, histone modifications, nuclear complexes and non-coding RNAs (ncRNAs) comprise main epigenetic mechanisms. DNA methylation, where a methyl group is added to cytosine in the CpG context, is the most studied epigenetic mechanism critical for processes like genomic imprinting and X chromosome inactivation6 but also T cell activation and differentiation7–10. Two recent studies suggest subtle but numerous DNA methylation changes in CD4+ T cells of RR-MS patients11, 12.
Besides DNA methylation, gene expression is regulated by ncRNAs like microRNAs (miRNAs) that act post-transcriptionally13. Mature miRNAs regulate multiple targets by recognizing a specific mRNA sequence in target mRNA. In T cells, miRNAs are involved in most processes and have important roles in T cell activation and differentiation14. A number of studies have profiled miRNAs in MS and several miRNAs are now emerging as important regulators, one such being miR-2115. Expression of miR-21 in CD4+ T cells has been shown to negatively correlate with DNA methylation at the CpGs in the MIR21 gene16.
In this study we sought to investigate DNA methylation in CD4+ T cells isolated from peripheral blood of RR-MS patients in remission, SP-MS patients and healthy controls.
Materials and Methods
Full details of experimental procedures are provided in Supplementary Methods.
Cohorts
A discovery MS cohort and an independent cohort used for validation were recruited at the Neurology clinic at Karolinska University Hospital in Stockholm. Cohort details are provided in Table 1 and in Supplementary Table 1. The Regional Ethical Review Board in Stockholm approved this study (2009/2107-31/2 and 2010/879-31/1) and methods were carried out in accordance with institutional guidelines on human subject experiments. Informed consent was obtained from all subjects.
Table 1.
Patient demographics in the discovery cohort used for 450K methylation analysis
| Status | Gender | Age | EDSS | MSSS | OCB | MRI lesions | TAS | TPS |
|---|---|---|---|---|---|---|---|---|
| RR-MS | F | 37 | 3 | 2.91 | yes | 10–20 | never Tx | |
| RR-MS | F | 40 | 0 | 0.67 | yes | >20 | never Tx | |
| RR-MS | M | 44 | 1.5 | yes | >20 | never Tx | ||
| RR-MS | F | 26 | 1 | 5.87 | no | 9 | never Tx | |
| RR-MS | F | 29 | 2 | 5.87 | yes | >20 | never Tx | |
| RR-MS | F | 35 | 1 | 1.13 | yes | >20 | no Tx | Fingolimod |
| RR-MS | F | 46 | 5 | 3.44 | yes | >20 | IVIg | |
| RR-MS | F | 37 | 1.5 | 3.34 | yes | >20 | SD | |
| RR-MS | M | 29 | 1 | 0.88 | yes | >20 | no Tx | IFN |
| RR-MS | M | 32 | 3 | 3.05 | yes | >20 | no Tx | IFN |
| RR-MS | F | 57 | 2.5 | 4.13 | yes | >20 | no Tx | SD |
| RR-MS | F | 41 | 2 | 0.71 | yes | >20 | never tx | |
|
| ||||||||
| SP-MS | M | 45 | 4 | 2.82 | yes | >20 | no Tx | IFN |
| SP-MS | M | 50 | 6.5 | 5.99 | yes | 9 | no Tx | SD |
| SP-MS | M | 56 | 6.5 | 5.99 | yes | 10–20 | never Tx | |
| SP-MS | F | 63 | 5 | yes | >20 | never Tx | ||
| SP-MS | F | 35 | 5 | 7.32 | yes | >20 | no Tx | IFN, GA |
| SP-MS | F | 60 | 5 | 5.82 | yes | >20 | no Tx | IFN, Mtx |
| SP-MS | M | 44 | 6 | 5.43 | yes | >20 | no Tx | IFN, SD |
| SP-MS | F | 50 | 3.5 | 4.55 | yes | >20 | no Tx | IFN |
|
| ||||||||
| Summarized | ||||||||
| RR-MS | 75%a | 38 (26–57)b | 2.0 (0–5.0)b | 2.9 (0.7–5.9)b | 92%c | 17%d | ||
| SP-MS | 50%a | 50 (35–63)b | 5.2 (3.5–6.5)b | 5.4 (2.8–7.3)b | 100%c | 0%d | ||
| HC | 67%a | 42 (28–62)b | N/A | N/A | N/A | N/A | ||
Percentage of females.
Mean (range).
Percentage of OCB positive patients.
Percentage of patients treated at the time of sampling.
HC = healthy controls; F = Female; M = Male; EDSS = Expanded Disability Status Scale; MSSS = Multiple Sclerosis Severity Score; OCB = Oligoclonal bands; MRI = Magnetic resonance imaging; TAS = Treatment at sampling; TPS = Treatment prior to sampling; Tx = Treatment; never Tx = treatment naïve; no Tx = sampling after 6 month wash out period; SD = Study Drug; IFN = interferon beta; GA = glatiramer acetate; Mtx = mitotrexate; N/A = not applicable.
The Genetics of Lipid Lowering Drugs and Diet Network (GOLDN) cohort recruited families with at least two siblings from the National Heart, Lung, and Blood Institute Family Heart Study sites at Minneapolis and Salt Lake City17–19. The Institutional Review Board (IRB) approved the GOLDN study (E160405007).
Preparation of CD4+ T cells
For discovery and validation cohorts peripheral blood mononuclear cells (PBMCs) were isolated using a standard Ficoll (GE Healthcare) and sodium citrate-containing preparation tubes (Becton Dickinson) procedures, respectively. Sorting of the CD4+ T cells was performed on a MoFlo™ cell sorter (Beckman Coulter) and an autoMACS® cell separator (Miltenyi Biotec), respectively. Extraction of genomic DNA was carried out using GenElute Mammalian Genomic DNA Miniprep kit (Sigma-Aldrich). RNA was isolated using standard TRIzol protocol (Invitrogen) and Allprep Total RNA/DNA Kit (Qiagen), respectively.
DNA methylation analysis
DNA methylation was profiled using the Infinium HumanMethylation450 BeadChip (Illumina) arrays at the bioinformatics and expression analysis core facility (BEA) at Karolinska Institutet. Methylation data was individually analyzed using the minfi and ChAMP package and normalized using quantile normalization and BMIQ. Differentially methylated positions were determined using the limma package applying a linear modelling that included MS status, age and sex as co-variates. A representative CpG was validated using pyrosequencing.
Transcriptome analysis
Total RNA was subjected to the Illumina TruSeq Stranded mRNA Library Protocol with Dual Indexes (Illumina) and sequenced on the Illumina HiSeq 2500 to generate 75 bp Paired End Data with an average of 10M reads above Q30. Differential expression analysis was conducted using the limma package.
Expression of miR-21 was determined using TaqMan MicroRNA Assay Kit (No: 000397, Applied Biosystems) and VMP1 expression was assessed by qPCR using SYBR green chemistry (Bio-Rad).
Genotyping
Allelic discrimination was performed using a predesigned TaqMan SNP Genotyping Assay (No: 4351379, Applied Biosystems).
Association, correlation and meta-analysis
In the GOLDN cohort we used the lmekin function of the R kinship package to fit linear mixed effects models with the methylation β-value at each of the interrogated CpG sites as the outcome and different predictors.
In the MS cohort we fit linear regression model in Rcmdr with the methylation β-value at each of the interrogated CpG sites as the outcome and different predictors.
We used two different meta-analysis methodologies: (1) the “summation of p value” method for combining p-values, and (2) the effect-size based meta-analysis using a fixed effect model (estimated by restricted maximum-likelihood) since there was no evidence of heterogeneity.
Target gene enrichment analysis
Targets of miR-21 identified in Jurkat T cells by RIP-Chip (Czepiel et al., ISBN 978-90-367-6788-0) and predicted by TarBase7.0 were selected for analysis. Deviation of up- and down-regulated miR-21 target genes from the expected ratio was calculated using the Chi-squared test. Enrichment of differentially expressed targets among up- and down-regulated genes was calculated using the Fisher’s exact test.
Ingenuity Pathways Analysis
Upstream regulators and biological functions were determined in the Ingenuity Pathways Analysis platform (Qiagen) using Fisher’s exact test and right-tailed Fisher’s exact test, followed by Benjamini-Hochberg correction, respectively.
Results
CD4+ T cells from RR-MS patients display higher DNA methylation in the VMP1/MIR21 locus
We investigated DNA methylation in CD4+ T cells from a Swedish cohort comprising RR-MS, SP-MS and matched healthy controls (HC). Analysis of 432 874 probes from the Infinium Human Methylation 450 BeadChip (referred as 450K hereinafter), did not yield genome-wide significant changes. However, among the top most significant hits we observed multiple probes mapping to last two exons of VMP1 and the MIR21 gene (Figure 1A, Table 2). RR-MS displayed significantly higher methylation compared to HC and SP-MS at the 11 consecutive CpG probes. The cg07181702 in MIR21, which was validated using pyrosequencing (Figure 1A), had the largest effect size of 14% methylation change between RR-MS and HC.
Figure 1. CD4+ T cells from RR-MS patients display higher DNA methylation levels in the VMP1/MIR21 locus.
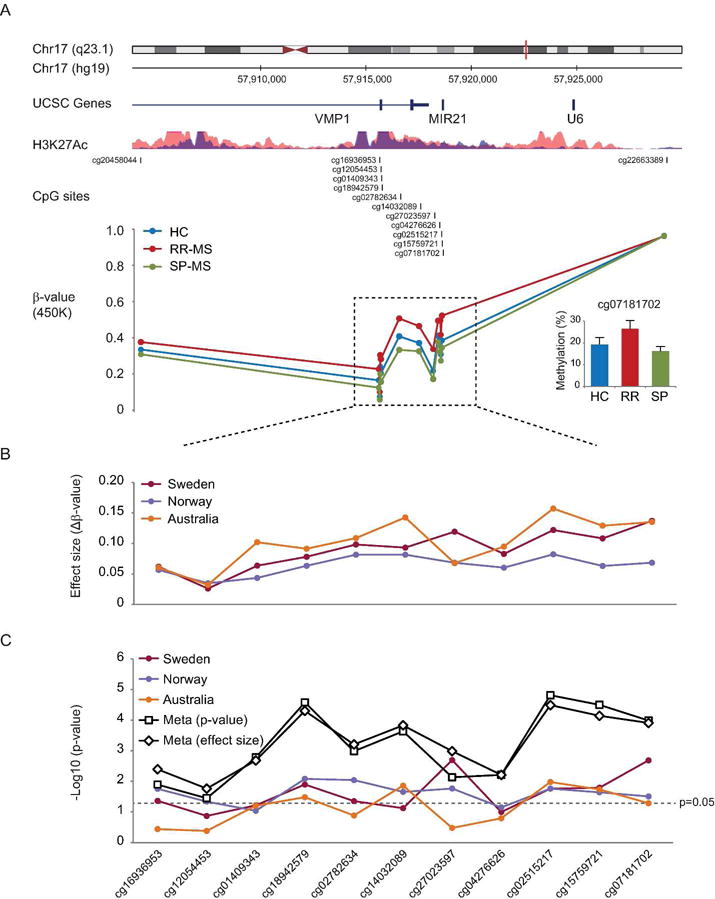
(A) DNA methylation was measured using 450K arrays in CD4+ T cells sorted from peripheral blood of RR-MS (N=12) and SP-MS (N=8) patients and healthy controls (HC, N=12). Eleven consecutive CpG sites (dashed box) displayed significantly higher methylation in RR-MS compared to SP-MS and HC. Detailed analysis of the locus is given in Table 1. The CpGs map to the VMP1 gene (last two exons and intron) and MIR21 gene, the region that is enriched for active histone mark (H3K27Ac) in two immune cell lines (ENCODE: GM12878 and K562). The methylation difference at a selected CpG, cg07181702 in the MIR21 gene, was technically validated using pyrosequencing (right lower panel). (B) The effect size that represents a difference in β-values between RR-MS and HC at a given CpG displayed the same pattern of increased DNA methylation in RR-MS compared to HC at the 11 consecutive CpG sites in the Swedish (N=24) cohort and two additional cohorts from Australia (N=40)11 and Norway (N=30)12. (C) Comparison of RR-MS with HC was performed on β-values and corrected for age in the Norwegian and Australian cohort (comprising females only), and age and sex in the Swedish cohort. Meta-analysis performed combining p-values (using the “summation of p value” method) and effect sizes (using fixed effect model) yielded significant p-values for all 11 CpGs. Significance levels for each CpG in different analyses are given on the y-axis as a −log10(p-value).
Table 2.
DNA methylation changes in the VMP1/MIR21 locus in CD4+ T cells from RR-MS and SP-MS patients and healthy controls
| Probe IDa | Genea | Feature | ALLb | RR vs. HCb | RR vs. SPb | SP vs. HCb | |||
|---|---|---|---|---|---|---|---|---|---|
| P-val (M) | Eff (Δ1β) | P-val (M) | Eff (Δβ) | P-val (M) | Eff (Δβ) | P-val (M) | |||
| cg20458044 | VMP1 | Intron | 1E-01 | 0.04 | 2E-01 | 0.07 | 4E-02 | −0.03 | 3E-01 |
| cg16936953 | VMP1 | Exon | 2E-03 | 0.06 | 6E-02 | 0.10 | 5E-04 | −0.04 | 4E-02 |
| cg12054453 | VMP1 | Exon | 4E-03 | 0.03 | 9E-02 | 0.04 | 1E-03 | −0.02 | 5E-02 |
| cg01409343 | VMP1 | Exon | 1E-02 | 0.06 | 7E-02 | 0.10 | 3E-03 | −0.04 | 2E-01 |
| cg18942579 | VMP1 | Intron | 4E-04 | 0.08 | 1E-02 | 0.12 | 1E-04 | −0.05 | 6E-02 |
| cg02782634 | VMP1 | Intron | 4E-03 | 0.10 | 5E-02 | 0.17 | 1E-03 | −0.08 | 8E-02 |
| cg14032089 | MIR21 | TSS150 | 3E-02 | 0.09 | 7E-02 | 0.14 | 1E-02 | −0.05 | 3E-01 |
| cg27023597 | MIR21 | TSS150 | 2E-04 | 0.12 | 2E-03 | 0.16 | 6E-05 | −0.04 | 1E-01 |
| cg04276626 | MIR21 | TSS200 | 7E-02 | 0.08 | 1E-01 | 0.12 | 3E-02 | −0.04 | 4E-01 |
| cg02515217 | MIR21 | TSS200 | 4E-03 | 0.12 | 2E-02 | 0.17 | 2E-03 | −0.05 | 3E-01 |
| cg15759721 | MIR21 | Body | 9E-03 | 0.11 | 4E-02 | 0.14 | 3E-03 | −0.04 | 2E-01 |
| cg07181702 | MIR21 | Body | 2E-03 | 0.14 | 7E-03 | 0.18 | 9E-04 | −0.04 | 3E-01 |
| cg22663389 | – | IGR | 6E-01 | −0.00 | 3E-01 | −0.00 | 6E-01 | −0.00 | 7E-01 |
CpG probe ID, annotated gene names and genomic features from 450K arrays.
M-values (indicated by M) were used to compare RR-MS (RR, N=12), SP-MS (SP, N=8) patients and healthy controls (HC, N=12) and p-values were derived using a linear modelling that included MS status, age and sex as co-variates. The effect size (Eff) represents a difference in β-values (Δβ) between the groups for each probe. P-val < 0.05 are indicated in bold.
We sought to replicate our findings between RR-MS and HC (n=24) in independent CD4+ T cell cohorts comprising RR-MS from Australia (n=40) and Norway (n=30)11, 12. Higher methylation in RR-MS was observed at all 11 CpGs in both cohorts (Figure 1B). Out of 11 CpGs nine and four displayed significant changes in the Norwegian and Australian cohort, respectively (Figure 1C). Meta-analysis performed combining p-values and effect sizes from all three cohorts demonstrated significant changes for all 11 CpGs (Figure 1C).
These data support higher methylation levels in the VMP1/MIR21 locus in RR-MS patients in three independent cohorts.
Investigation of association between methylation levels in VMP1/MIR21 and MS risk factors
It has been shown that genetic variants can mediate disease risk through changes in DNA methylation20 and there is a well-established genetic association between MS and the VMP1 locus4. Thus, we investigated if genetic variation in VMP1 may mediate MS risk by affecting VMP1/MIR21 methylation levels. We detected significant differences in CD4+ T cells from healthy individuals in the GOLDN cohort (n=717)17–19 for only 2/11 CpGs comparing individuals carrying none, one or two of the RS8070345 risk alleles (Figure 2A, Table 3) and no significant differences in the MS cohort (Figure 2B, Table 4).
Figure 2. MS risk factors, genetic variation in the VMP1 gene and smoking, do not affect DNA methylation in the VMP1/MIR21 locus.
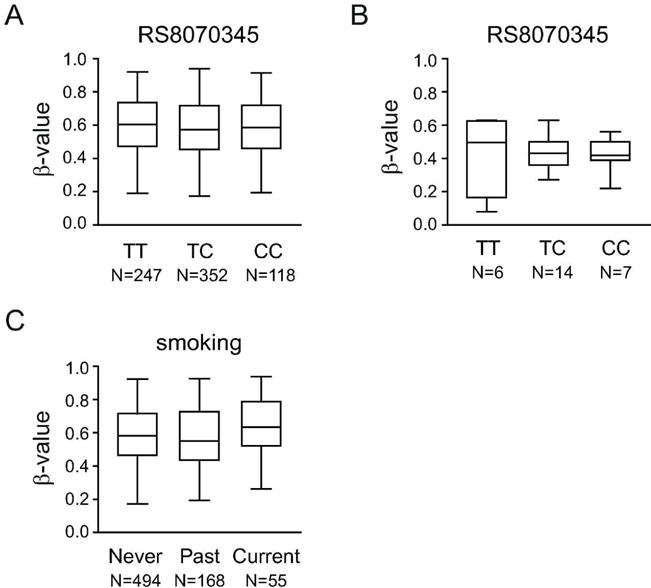
DNA methylation levels (β-values) in the VMP1/MIR21 locus, exemplified by cg07181702, do not associate with the MS risk genotype at RS8070345 (T=risk allele) in a large cohort of healthy individuals (A) or in the Swedish cohort (B). Detailed analysis of the locus is given in Table 2 and Table 3. (C) DNA methylation levels (β-values) in the VMP1/MIR21 locus, exemplified by cg07181702, are not affected by the smoking status in a large cohort of healthy individuals. Detailed analysis of the locus is given in Table 2.
Table 3.
Influence of RS8070345, smoking and age on the DNA methylation in the VMP1/MIR21 locus in healthy individuals
| CpG probea | Genea | RS8070345b (N=717) |
Current smokerc (N=994) |
Past smokerc (N=994) |
Agec (N=994) |
||||
|---|---|---|---|---|---|---|---|---|---|
| Coeff | P-val | Coeff | P-val | Coeff | P-val | Coeff | P-val | ||
| cg16936953 | VMP1 | −0.0106 | 0.049 | −0.0098 | 0.34 | −0.0060 | 0.37 | −0.0010 | 5E-07 |
| cg12054453 | VMP1 | −0.0170 | 1E-04 | −0.0158 | 0.07 | −0.0120 | 0.031 | −0.0005 | 4E-03 |
| cg01409343 | VMP1 | −0.0046 | 0.43 | 0.0011 | 0.92 | 0.0002 | 0.97 | −0.0011 | 2E-07 |
| cg18942579 | VMP1 | −0.0086 | 0.13 | 0.0000 | 1.00 | 0.0020 | 0.78 | −0.0011 | 3E-07 |
| cg02782634 | VMP1 | 0.0016 | 0.78 | −0.0065 | 0.55 | 0.0007 | 0.92 | −0.0010 | 5E-07 |
| cg14032089 | MIR21 | 0.0057 | 0.33 | −0.0041 | 0.71 | 0.0019 | 0.79 | −0.0009 | 2E-05 |
| cg27023597 | MIR21 | −0.0058 | 0.34 | −0.0126 | 0.28 | 0.0002 | 0.98 | −0.0010 | 5E-06 |
| cg04276626 | MIR21 | 0.0032 | 0.58 | −0.0057 | 0.59 | 0.0028 | 0.68 | −0.0004 | 0.06 |
| cg02515217 | MIR21 | 0.0016 | 0.82 | 0.0022 | 0.85 | −0.0001 | 0.99 | −0.0012 | 3E-07 |
| cg15759721 | MIR21 | 0.0048 | 0.42 | −0.0017 | 0.88 | 0.0019 | 0.79 | −0.0010 | 1E-06 |
| cg07181702 | MIR21 | 0.0034 | 0.57 | 0.0011 | 0.92 | 0.0014 | 0.85 | −0.0011 | 4E-07 |
CpG probe ID and annotated gene names from 450K arrays.
Coefficients (Coeff) and p-values (P-val) from a linear mixed effects model with the methylation β-score at each CpG site as the outcome and the following predictors: genotype at RS8070345, age, sex, study site, current smoking, body mass index (BMI), principal components capturing T-cell purity and family, as previously described19. P-val < 0.05 are indicated in bold.
Coefficients (Coeff) and p-values (P-val) from a linear regression model with the methylation β-score at each CpG site as the outcome and the following predictors: age, current and past smoking, BMI and principal components capturing T-cell purity, as previously described19. P-val < 0.05 are indicated in bold.
Table 4.
Correlation of DNA methylation in the VMP1/MIR21 locus with patient characteristics
| CpG probea | Genea | RS8070345b (N=27) |
Ly#c (N=18) |
Agec (N=18) |
|||
|---|---|---|---|---|---|---|---|
| Coeff | P-val | Coeff | P-val | Coeff | P-val | ||
| cg16936953 | VMP1 | −0.0076 | 0.59 | 0.0534 | 0.10 | −0.0067 | 5E-05 |
| cg12054453 | VMP1 | −0.0061 | 0.36 | 0.0178 | 0.37 | −0.0027 | 3E-03 |
| cg01409343 | VMP1 | 0.0003 | 0.98 | 0.0788 | 0.078 | −0.0070 | 2E-04 |
| cg18942579 | VMP1 | −0.0013 | 0.94 | 0.0510 | 0.22 | −0.0055 | 6E-03 |
| cg02782634 | VMP1 | −0.0275 | 0.32 | 0.1077 | 0.12 | −0.0096 | 2E-03 |
| cg14032089 | MIR21 | −0.0111 | 0.69 | 0.0956 | 0.16 | −0.0101 | 1E-03 |
| cg27023597 | MIR21 | 0.0090 | 0.67 | 0.1034 | 0.039 | −0.0077 | 5E-04 |
| cg04276626 | MIR21 | −0.0104 | 0.72 | 0.0955 | 0.15 | −0.0103 | 8E-04 |
| cg02515217 | MIR21 | −0.0162 | 0.56 | 0.0582 | 0.38 | −0.0112 | 4E-04 |
| cg15759721 | MIR21 | −0.0134 | 0.61 | 0.1116 | 0.082 | −0.0086 | 2E-03 |
| cg07181702 | MIR21 | −0.0053 | 0.84 | 0.0664 | 0.31 | −0.0105 | 7E-04 |
CpG probe ID and annotated gene names from 450K arrays.
Coefficients (Coeff) and p-values (P-val) from a linear regression model with the methylation β-score at each CpG site as the outcome and the following predictors: age, sex and genotype at RS8070345 (N=27).
Coefficients (Coeff) and p-values (P-val) from a linear regression model with the methylation β-score at each CpG site as the outcome and the following predictors: age, sex, and lymphocyte count (Ly#) (N=18). P-val < 0.05 are indicated in bold.
We then tested if VMP1/MIR21 methylation levels mediate the effect of smoking, a well-established risk factor for MS5 also known to affect DNA methylation21. No consistent significant differences at any of the 11 CpGs between never, past or current smokers were detected in the GOLDN cohort (Figure 2C, Table 3).
There was a tendency for a positive correlation with lymphocyte counts with 1/11 and 3/11 CpGs displaying p<0.05 and p<0.1, respectively (Figure 3A, Table 4). Additionally, methylation in VMP1/MIR21 displayed a strong negative correlation with age (e.g. for cg07181702, n=32, Pearson’s r=−0.62, p<0.0001) (Figure 3B, Table 4), with higher methylation levels in CD4+ T cells of younger individuals, which was also detected in the GOLDN cohort (e.g. for cg07181702, n=994, coefficient= −0.0011, p<4×10−7) (Table 3).
Figure 3. DNA methylation in the VMP1/MIR21 locus displays strong negative correlation with age.
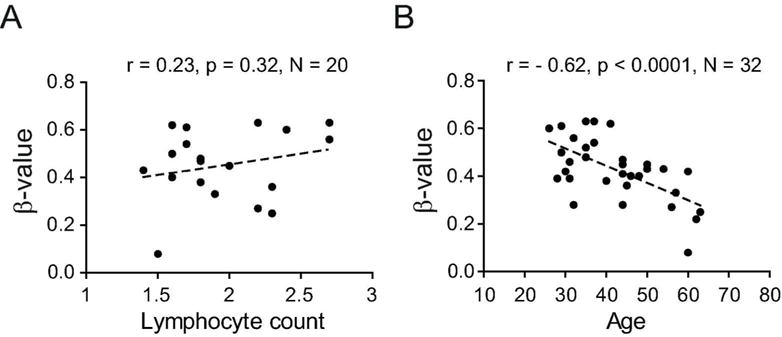
DNA methylation levels (β-values) in the VMP1/MIR21 locus, exemplified by cg07181702, do not show significant association with lymphocyte count, as a surrogate of inflammation levels (A) but display a significant strong negative correlation with age (B). Detailed analysis of the locus is given in Table 3.
Thus, increased methylation in VMP1/MIR21 is not caused by the genetic variation in the locus or smoking but instead age strongly affects VMP1/MIR21 methylation.
Hypermethylation of the VMP1/MIR21 locus associates with lower expression of miR-21 in CD4+ T cells
There was a high correlation between methylation levels at all CpGs (Supplementary Fig.1), suggesting that methylation changes in the region are co-regulated and likely have the same functional impact. We then investigated if methylation in VMP1/MIR21 has an impact on the expression of miR-21 and VMP1, analyzed by qPCR in patients and HC from the MS cohort. We observed a significant negative correlation between methylation at 7/11 CpGs and miR-21 levels, with additional 4/11 CpGs displaying the same trend (Figure 4A, Table 5), while there was no correlation with VMP1 levels (Figure 4B, Table 5). This inverse correlation between methylation and miR-21 levels was strongest for the CpGs in MIR21 (e.g. for cg 07181702, n=27, Pearson’s r=−0.51, p=0.006) and remained significant after age correction (Table 5).
Figure 4. DNA methylation in the VMP1/MIR21 locus displays significant negative correlation with miR-21 expression and RR-MS patients display lower miR-21 levels.
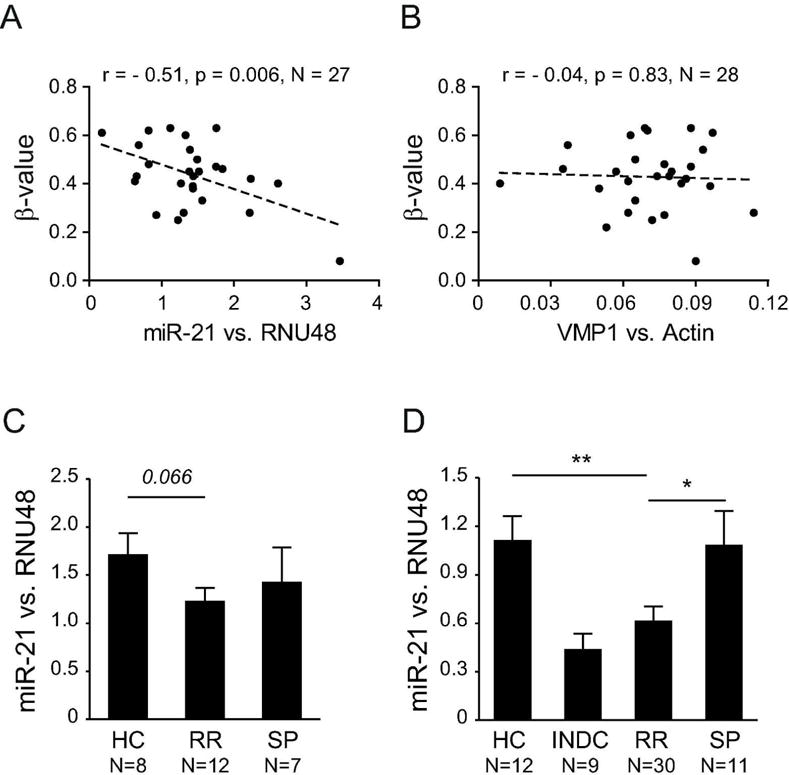
DNA methylation levels (β-values) in the VMP1/MIR21 locus, exemplified by cg07181702, display a significant strong anti-correlation with miR-21 levels (A) but not with VMP1 levels in CD4+ T cells from RR-MS, SP-MS and healthy controls (HC). Accordingly, RR-MS patients have lower levels of miR-21 compared to HC and SP-MS patients in the 450K cohort (C) and in an independent validation cohort from Sweden (D). Detailed analysis of the locus is given in Table 4.
Table 5.
Correlation of DNA methylation in the VMP1/MIR21 locus with miR-21 and VMP1 expression levels
| CpG probea | Genea | R | miR-21b (N=27) | P-val | VMP1b (N=27) | ||
|---|---|---|---|---|---|---|---|
| P-val | Coeff | R | P-val | ||||
| cg16936953 | VMP1 | −0.34 | 0.083 | −3.49 | 0.14 | −0.02 | 0.91 |
| cg12054453 | VMP1 | −0.23 | 0.24 | −3.84 | 0.42 | 0.02 | 0.90 |
| cg01409343 | VMP1 | −0.40 | 0.038 | −3.85 | 0.054 | −0.06 | 0.77 |
| cg18942579 | VMP1 | −0.32 | 0.108 | −2.53 | 0.21 | 0.09 | 0.66 |
| cg02782634 | VMP1 | −0.42 | 0.027 | −2.39 | 0.042 | −0.11 | 0.57 |
| cg14032089 | MIR21 | −0.38 | 0.047 | −2.11 | 0.081 | −0.08 | 0.70 |
| cg27023597 | MIR21 | −0.35 | 0.076 | −2.50 | 0.12 | 0.13 | 0.52 |
| cg04276626 | MIR21 | −0.54 | 0.004 | −3.08 | 0.006 | −0.04 | 0.82 |
| cg02515217 | MIR21 | −0.43 | 0.025 | −2.82 | 0.029 | 0.08 | 0.68 |
| cg15759721 | MIR21 | −0.50 | 0.008 | −3.20 | 0.013 | −0.06 | 0.76 |
| cg07181702 | MIR21 | −0.51 | 0.006 | −3.32 | 0.006 | −0.04 | 0.83 |
CpG probe ID and annotated gene names from 450K arrays.
Pearson’s (R) and regression coefficients (Coeff), with accompanying p-values (P-val), from a Pearson’s correlation test, for relative expression of miR-21 and VMP1, and a linear regression model with the relative expression of miR 21 as the outcome and methylation β-score at each CpG site and age as predictors. P-val < 0.05 are indicated in bold. Analysis was performed in CD4+ T cells from RR-MS, SP-MS and HC.
Accordingly, a tendency for decrease in expression of miR-21 was observed in RR-MS patients compared to SP-MS and HC (Figure 4C). To validate our findings we performed expression analysis in an independent MS cohort from Sweden including more cases (Supplementary Table 1). Significantly lower levels of miR-21 were detected in RR-MS patients compared to SP-MS patients and HC (Figure 4D).
These data strongly suggest that increased methylation in MIR21 associates with lower expression of mature miR-21 in CD4+ T cells of RR-MS patients.
MiR-21 target genes are enriched among genes up-regulated in CD4+ T cells of RR-MS patients
Since the best described function of miRNAs is to induce degradation of target mRNAs we explored if the observed changes in miR-21 methylation and expression lead to actual functional changes in miR-21 target genes. For that purpose, we used RNA-seq data from a cohort largely overlapping (73%) the cohort used for 450K analysis (Supplementary Table 1). We used miR-21 targets identified in Jurkat T cells using RIP-Chip and predicted by TarBase7.0 (Supplementary Table 2). We first investigated if the ratio of up-regulated miR-21 targets deviated from the expected ratio of up-regulated genes between RR-MS, SP-MS and HC (Table 6). There was a significant enrichment of miR-21 targets among the genes that were up-regulated in CD4+ T cells, both when comparing RR-MS patients with HC and when comparing RR-MS with SP-MS but not when SP-MS patients were compared with HC (Table 6). Similar results were obtained when we calculated enrichment of miR-21 targets among up- and down-regulated genes where we found significant enrichment of miR-21 targets predominantly among up-regulated, but not down-regulated, genes between RR-MS and HC (Table 7). Additionally, an unbiased IPA prediction of upstream regulators identified miR-21 as a significantly inhibited upstream regulator (z=−2.1, p=7×10−7) in RR-MS that can explain the observed pattern of differential gene expression between RR-MS and HC (p<0.05).
Table 6.
Deviation of the miR-21 target genes from the expected ratio of the up-regulated genes in CD4+ T cells from RR-MS and SP-MS patients and healthy controls
| Predictiona | Comparisona | DEb (P-val) |
Groupb | UP obsb (%) |
UP expb (%) |
P-valb |
|---|---|---|---|---|---|---|
| Jurkat (N=64) |
RR vs. HC | 0.01 | RR | 100 | 53 | 0.1 |
| 0.05 | RR | 80 | 44 | 0.02 | ||
| 0.1 | RR | 86 | 43 | 0.001 | ||
|
| ||||||
| RR vs. SP | 0.01 | RR | – | 35 | ||
| 0.05 | RR | 100 | 34 | 0.006 | ||
| 0.1 | RR | 78 | 37 | 0.01 | ||
|
| ||||||
| SP vs. HC | 0.01 | SP | 100 | 63 | 0.4 | |
| 0.05 | SP | 100 | 58 | 0.1 | ||
| 0.1 | SP | 78 | 56 | 0.2 | ||
|
| ||||||
| TarBase (N=433) |
RR vs. HC | 0.01 | RR | 90 | 53 | 0.02 |
| 0.05 | RR | 65 | 44 | 0.004 | ||
| 0.1 | RR | 61 | 43 | 0.002 | ||
|
| ||||||
| RR vs. SP | 0.01 | RR | 75 | 35 | 0.09 | |
| 0.05 | RR | 64 | 34 | 0.004 | ||
| 0.1 | RR | 68 | 37 | 0.00001 | ||
|
| ||||||
| SP vs. HC | 0.01 | SP | 50 | 63 | 0.5 | |
| 0.05 | SP | 52 | 58 | 0.5 | ||
| 0.1 | SP | 52 | 56 | 0.6 | ||
Target genes of miR-21 identified in Jurkat T cells (n=64, 100% present in RNAseq data) and genes predicted and experimentally validated to be miR-21 targets by TarBase7.0 (n=433, 88% present in RNAseq data) were selected for analysis.
Differentially expressed (DE) genes between RR-MS (RR, N=12), SP-MS (SP, N=10) patients and healthy controls (HC, N=12) were identified using RNAseq (significance for calling DE is indicated by DE (P-val)). For each comparison a deviation of the percentage of the observed up-regulated miR-21 target genes (UP obs), in a given group (Group), from the expected percentage of the up-regulated genes (UP exp) was calculated using a Chi-squared test (P-val). P-val < 0.05 are indicated in bold.
Table 7.
Overlap of miR-21 target genes with up- and down-regulated genes in CD4+ T cells from RR-MS and SP-MS patients and healthy controls
| Predictiona | Comparisona | DE (P-val)b | P-val (UP)b | P-val (DOWN)b |
|---|---|---|---|---|
| Jurkat (N=64) |
RR vs. HC | 0.01 | 0.06 | 0.8 |
| 0.05 | 0.003 | 0.6 | ||
| 0.1 | 0.0009 | 0.1 | ||
|
| ||||
| RR vs. SP | 0.01 | – | – | |
| 0.05 | 0.1 | 0.1 | ||
| 0.1 | 0.06 | 0.2 | ||
|
| ||||
| SP vs. HC | 0.01 | 0.9 | 0.7 | |
| 0.05 | 0.4 | 0.4 | ||
| 0.1 | 0.3 | 0.6 | ||
|
| ||||
| TarBase (N=433) |
RR vs. HC | 0.01 | 0.09 | 0.2 |
| 0.05 | 0.0005 | 0.4 | ||
| 0.1 | 0.003 | 0.1 | ||
|
| ||||
| RR vs. SP | 0.01 | 0.8 | 0.7 | |
| 0.05 | 0.2 | 0.02 | ||
| 0.1 | 0.003 | 0.002 | ||
|
| ||||
| SP vs. HC | 0.01 | 0.8 | 0.6 | |
| 0.05 | 0.9 | 0.3 | ||
| 0.1 | 0.3 | 0.1 | ||
Target genes of miR-21 identified in Jurkat T cells (n=64, 100% present in RNAseq data) and genes predicted and experimentally validated to be miR-21 targets by TarBase7.0 (n=433, 88% present in RNAseq data) were selected for analysis.
Differentially expressed (DE) genes between RR-MS (RR, N=12), SP-MS (SP, N=10) patients and healthy controls (HC, N=12) were identified using RNAseq (significance for calling DE is indicated by DE (P-val)). For each comparison an overlap of miR-21 target genes with either up-regulated (UP) or down-regulated (DOWN) genes was calculated using a Fisher’s exact test (P-val). P-val < 0.05 and < 0.1 are indicated in bold and italic bold, respectively.
In order to identify biological functions that are regulated by miR-21 in CD4+ T cells we performed IPA on TarBase7.0-predicted miR-21 targets that were up-regulated in RR-MS compared to HC (p<0.05, n=30) (Supplementary Table 2). Despite a limited list of genes several functions were significant after Benjamini-Hochberg correction including “Apoptosis” (z=−1.624) and “Proliferation of cells” (z=1.007).
These analyses strongly suggest a link between lower miR-21 expression and up-regulation of a set of miR-21 target genes in CD4+ T cells that potentially influence apoptosis and cell proliferation.
Discussion
We performed DNA methylation analysis in CD4+ T cells from RR-MS, SP-MS and healthy individuals and subsequently characterized the relationship between MIR21 methylation and expression of miR-21 and its target genes. MIR21 displayed increased methylation levels that correlated with a lower expression of mature miR-21 and an enrichment of up-regulated miR-21 target genes in RR-MS patients.
In order to assess the relevance of MIR21 hypermethylation in RR-MS we investigated both the methylation of this locus in previously published MS cohorts, and assessed potential functional impact of MIR21 methylation using RNA-seq data. A similar pattern as in our study, i.e. hypermethylation of all 11 CpGs in RR-MS, was observed in the Norwegian and Australian cohorts11, 12, with nine and four significant probes (p<0.05), respectively (two additional probes from each cohort demonstrated p<0.1). The most significant changes were observed in the Norwegian cohort, which included untreated MS patients with relatively benign disease course12 that can potentially explain a more modest effect size of 6–8% change in MIR21. The effect size was similar in the Swedish and Australian cohorts ranging from 8–14% and 7–16%, respectively, but the changes were less significant in the Australian cohort potentially due to higher variability caused by treatments11. It is important to consider that all studies investigated bulk CD4+ T cells that comprises subsets with distinct methylomes16, 22 and methylation changes could reflect differences in their cellular composition. However, as MIR21 is hypomethylated in Th1/Th222 and regulatory T cells (Tregs)16 compared to naïve CD4+ T cells, and naive CD4+ T cells display the lowest expression of miR-21 compared to other CD4+ subsets14, the differences in frequency are less likely to explain hypermethylation in RR-MS.
We further demonstrated a functional impact of the observed methylation on the expression of miR-21. The higher methylation levels in VMP1/MIR21 correlated strongly with lower expression of mature miR-21 in CD4+ T cells whereas no correlation was observed with VMP1. The negative correlation with miR-21 levels was the strongest at the CpGs in the MIR21 gene. Methylation at the same CpGs has previously been shown to negatively correlate with miR-21 expression in naïve CD4+ T cells upon activation16. Accordingly, we observed lower expression of mature miR-21 in RR-MS patients compared to SP-MS and controls and this difference became significant in a larger independent cohort. The difference between RR-MS and SP-MS, however, may reflect differences in age between the groups since MIR21 methylation displays a strong negative correlation with age.
Consistent with the function of miRNAs in down-regulating their target genes we found miR-21 targets enriched among up-regulated genes in RR-MS patients. The enrichment was significant irrespective of the target prediction tool, the cut-off levels for differential expression and the enrichment analysis approach used. Importantly, the enrichment was detected only in RR-MS, which is in line with the observed higher methylation and lower expression of miR-21 in RR-MS compared to HC or SP-MS and no difference between HC and SP-MS. The most prominent biological functions that associated with the up-regulated targets imply possible anti-apoptotic and pro-proliferative effects. Representatives for such genes are DEGS1, PTPN13 and CD47 that have been shown to inhibit apoptosis and increase proliferation in different cells. For instance DEGS1, enzyme involved in the ceramide synthesis, has been shown to increase proliferation and decrease apoptosis in mammalian cells23. In CD4+ T cells, ceramides have been implicated in regulating T cell proliferation and differentiation into Th124. Overexpression of PTPN13, a protein tyrosine phosphatase, leads to increased resistance to FAS-induced apoptosis in immortalized T cells25. The ligation of CD47, a plasma membrane protein, leads in primary and Jurkat T cells to an increase in IL-2 production and more proliferation26. Gene deletion or blocking of CD47 protects against development of MS-like disease in mice through a failure of immune cell activation27. In relation to these observations, our data suggest a potential role of methylation-dependent miR-21 expression in modulating proliferation and apoptosis of CD4+ T cells in RR-MS.
Expression of miR-21 is increased in many inflammatory diseases and elevated miR-21 levels represent a sign of inflammation16, 28. In MS, miR-21 has been found up-regulated in PBMCs of RR-MS patients in relapse suggesting that hypermethylation of MIR21 may be a mechanism to down-regulate miR-21 that may contribute to remission29. However, another longitudinal study did not report differences in miR-21 expression in peripheral blood leukocytes from RR-MS patients in relapse and remission, but did observe down-regulation of miR-21 in RR-MS patients in remission compared to controls30, which is similar to our observation. Regarding T cells, miR-21 has been shown to both promote and inhibit T cell activation31, 32. Overexpression of miR-21 in naïve mouse T cells promotes expression of Th2 and Treg genes33 and expression of FoxP3 in human Tregs34. Accordingly, lower expression of miR-21 has been reported in PBMCs and CD4+ T cells of rheumatoid arthritis patients compared to controls, with low miR-21 showing strong correlation with an increased ratio of Th17/Treg cells35. Arguably miR-21 may play different roles in different stages of disease depending on the stage of CD4+ T cell activation and the cell subset.
We also investigated potential causes of the observed changes in DNA methylation. Since MIR21 resides in the locus that associates with susceptibility to MS4, we first investigated if methylation of MIR21 is genetically regulated. However, we found no consistent evidence of association of the risk variant with the levels of MIR21 methylation. Similarly, we found no influence of smoking, which is known to induce DNA methylation changes21 and is an established lifestyle MS risk factor5. One possibility is that MIR21 methylation and expression are regulated by the inflammation itself. The fact that miR-21 expression does not differ in SP-MS but shows low expression in RR-MS and inflammatory neurological disease controls supports this scenario. This is further supported by the observed trend for positive correlation between methylation and lymphocyte counts, as a surrogate of inflammation. However, none of the known proinflammatory regulators of miR-2128 were differentially expressed between RR-MS and SP-MS patients or healthy controls suggesting that maybe differential methylation in their binding sites in MIR21 may lead to differences in miR-21 expression. Whether this is the case and the exact mechanisms of how inflammatory signals induce epigenetic repression in the locus need to be established.
To our knowledge this is the first study that delivers a detailed description of the methylation status of MIR21 in CD4+ T cells of MS patients and associates it with the nearby risk allele, smoking, age, and changes in expression of miR-21 and its target genes. Our study sheds further light on a role of miR-21 in inflammatory diseases, in particular MS, demonstrating that disease-related changes in an epigenetic marking have the potential to lead to differences in the expression of miR-21 with a consequence on miR-21 target genes.
Supplementary Material
Acknowledgments
We would like to thank Hanne Flinstad Harbo and Steffan Daniël Bos-Haugen for providing the Norwegian methylation data, and Moira C Graves, Jeanette Lechner-Scott, Rodney A Lea, Andreas Tjärnberg and Mika Gustafsson for providing the Australian methylation data.
FP has received unrestricted academic research grants from Biogen, Genzyme and Novartis, and travel support and/or compensation for lectures and/or participation in advisory boards from Biogen, Genzyme, Merckserono, Novartis, Roche and Teva, which have been exclusively used for the support of research activities.
Funding
This work was supported by grants from the Swedish Research Council, the Swedish Association for Persons with Neurological Disabilities, the Swedish Brain Foundation, Petrus and Augusta Hedlunds Foundation, the regional agreement on medical training and clinical research (ALF) between Stockholm County Council and Karolinska Institutet, and AstraZeneca (AstraZeneca-Science for Life Laboratory collaboration) and grant R01HL104135 from the National Institutes of Health/National Heart, Lung and Blood Institute (GOLDN). S.S-B. was funded by a contract from Instituto de Salud Carlos III FEDER [IFI14/00007] and Daniel Bravo Andreu Private Foundation.
Footnotes
Declaration of Conflicting Interests
SR, EE, EP, LK, JCCL, SF, SA, HM, SSB, SA, JT, DGC and MJ declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–17. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 2.International Multiple Sclerosis Genetics C. Wellcome Trust Case Control C. Sawcer S, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–9. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goverman J. Autoimmune T cell responses in the central nervous system. Nature reviews Immunology. 2009;9:393–407. doi: 10.1038/nri2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.International Multiple Sclerosis Genetics C. Beecham AH, Patsopoulos NA, et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nature genetics. 2013;45:1353–60. doi: 10.1038/ng.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olsson T, Barcellos LF, Alfredsson L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nature reviews Neurology. 2017;13:25–36. doi: 10.1038/nrneurol.2016.187. [DOI] [PubMed] [Google Scholar]
- 6.Li E, Zhang Y. DNA methylation in mammals. Cold Spring Harbor perspectives in biology. 2014;6:a019133. doi: 10.1101/cshperspect.a019133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agarwal S, Rao A. Modulation of chromatin structure regulates cytokine gene expression during T cell differentiation. Immunity. 1998;9:765–75. doi: 10.1016/s1074-7613(00)80642-1. [DOI] [PubMed] [Google Scholar]
- 8.Mullen AC, Hutchins AS, High FA, et al. Hlx is induced by and genetically interacts with T-bet to promote heritable T(H)1 gene induction. Nature immunology. 2002;3:652–8. doi: 10.1038/ni807. [DOI] [PubMed] [Google Scholar]
- 9.Bruniquel D, Schwartz RH. Selective, stable demethylation of the interleukin-2 gene enhances transcription by an active process. Nature immunology. 2003;4:235–40. doi: 10.1038/ni887. [DOI] [PubMed] [Google Scholar]
- 10.Floess S, Freyer J, Siewert C, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS biology. 2007;5:e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graves MC, Benton M, Lea RA, et al. Methylation differences at the HLA-DRB1 locus in CD4+ T-Cells are associated with multiple sclerosis. Multiple sclerosis. 2014;20:1033–41. doi: 10.1177/1352458513516529. [DOI] [PubMed] [Google Scholar]
- 12.Bos SD, Page CM, Andreassen BK, et al. Genome-wide DNA methylation profiles indicate CD8+ T cell hypermethylation in multiple sclerosis. PloS one. 2015;10:e0117403. doi: 10.1371/journal.pone.0117403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–5. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 14.Rossi RL, Rossetti G, Wenandy L, et al. Distinct microRNA signatures in human lymphocyte subsets and enforcement of the naive state in CD4+ T cells by the microRNA miR-125b. Nature immunology. 2011;12:796–803. doi: 10.1038/ni.2057. [DOI] [PubMed] [Google Scholar]
- 15.Ma X, Zhou J, Zhong Y, et al. Expression, regulation and function of microRNAs in multiple sclerosis. International journal of medical sciences. 2014;11:810–8. doi: 10.7150/ijms.8647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Maksimovic J, Naselli G, et al. Genome-wide DNA methylation analysis identifies hypomethylated genes regulated by FOXP3 in human regulatory T cells. Blood. 2013;122:2823–36. doi: 10.1182/blood-2013-02-481788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aslibekyan S, Kabagambe EK, Irvin MR, et al. A genome-wide association study of inflammatory biomarker changes in response to fenofibrate treatment in the Genetics of Lipid Lowering Drug and Diet Network. Pharmacogenetics and genomics. 2012;22:191–7. doi: 10.1097/FPC.0b013e32834fdd41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhi D, Aslibekyan S, Irvin MR, et al. SNPs located at CpG sites modulate genome-epigenome interaction. Epigenetics. 2013;8:802–6. doi: 10.4161/epi.25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Irvin MR, Zhi D, Joehanes R, et al. Epigenome-wide association study of fasting blood lipids in the Genetics of Lipid-lowering Drugs and Diet Network study. Circulation. 2014;130:565–72. doi: 10.1161/CIRCULATIONAHA.114.009158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y, Aryee MJ, Padyukov L, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nature biotechnology. 2013;31:142–7. doi: 10.1038/nbt.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao X, Jia M, Zhang Y, Breitling LP, Brenner H. DNA methylation changes of whole blood cells in response to active smoking exposure in adults: a systematic review of DNA methylation studies. Clinical epigenetics. 2015;7:113. doi: 10.1186/s13148-015-0148-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nestor CE, Lentini A, Hagg Nilsson C, et al. 5-Hydroxymethylcytosine Remodeling Precedes Lineage Specification during Differentiation of Human CD4(+) T Cells. Cell reports. 2016;16:559–70. doi: 10.1016/j.celrep.2016.05.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Devlin CM, Lahm T, Hubbard WC, et al. Dihydroceramide-based response to hypoxia. The Journal of biological chemistry. 2011;286:38069–78. doi: 10.1074/jbc.M111.297994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan G, Ochi A. Sphingomyelin-ceramide turnover in CD28 costimulatory signaling. European journal of immunology. 1995;25:1999–2004. doi: 10.1002/eji.1830250730. [DOI] [PubMed] [Google Scholar]
- 25.Li Y, Kanki H, Hachiya T, et al. Negative regulation of Fas-mediated apoptosis by FAP-1 in human cancer cells. International journal of cancer. 2000;87:473–9. doi: 10.1002/1097-0215(20000815)87:4<473::aid-ijc3>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 26.Reinhold MI, Lindberg FP, Kersh GJ, Allen PM, Brown EJ. Costimulation of T cell activation by integrin-associated protein (CD47) is an adhesion-dependent, CD28-independent signaling pathway. The Journal of experimental medicine. 1997;185:1–11. doi: 10.1084/jem.185.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han MH, Lundgren DH, Jaiswal S, et al. Janus-like opposing roles of CD47 in autoimmune brain inflammation in humans and mice. The Journal of experimental medicine. 2012;209:1325–34. doi: 10.1084/jem.20101974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang S, Wan X, Ruan Q. The MicroRNA-21 in Autoimmune Diseases. International journal of molecular sciences. 2016;17 doi: 10.3390/ijms17060864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fenoglio C, Cantoni C, De Riz M, et al. Expression and genetic analysis of miRNAs involved in CD4+ cell activation in patients with multiple sclerosis. Neuroscience letters. 2011;504:9–12. doi: 10.1016/j.neulet.2011.08.021. [DOI] [PubMed] [Google Scholar]
- 30.Munoz-Culla M, Irizar H, Saenz-Cuesta M, et al. SncRNA (microRNA &snoRNA) opposite expression pattern found in multiple sclerosis relapse and remission is sex dependent. Scientific reports. 2016;6:20126. doi: 10.1038/srep20126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang L, He L, Zhang R, et al. Regulation of T lymphocyte activation by microRNA-21. Molecular immunology. 2014;59:163–71. doi: 10.1016/j.molimm.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 32.Carissimi C, Carucci N, Colombo T, et al. miR-21 is a negative modulator of T-cell activation. Biochimie. 2014;107(Pt B):319–26. doi: 10.1016/j.biochi.2014.09.021. [DOI] [PubMed] [Google Scholar]
- 33.Sawant DV, Wu H, Kaplan MH, Dent AL. The Bcl6 target gene microRNA-21 promotes Th2 differentiation by a T cell intrinsic pathway. Molecular immunology. 2013;54:435–42. doi: 10.1016/j.molimm.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rouas R, Fayyad-Kazan H, El Zein N, et al. Human natural Treg microRNA signature: role of microRNA-31 and microRNA-21 in FOXP3 expression. European journal of immunology. 2009;39:1608–18. doi: 10.1002/eji.200838509. [DOI] [PubMed] [Google Scholar]
- 35.Dong L, Wang X, Tan J, et al. Decreased expression of microRNA-21 correlates with the imbalance of Th17 and Treg cells in patients with rheumatoid arthritis. Journal of cellular and molecular medicine. 2014;18:2213–24. doi: 10.1111/jcmm.12353. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.