

Abstract
A perspective of work in our laboratory on the examination of biologically active compounds, especially natural products, is presented. In the context of individual programs and along with a summary of our work, selected cases are presented that illustrate the impact single atom changes can have on the biological properties of the compounds. The examples were chosen to highlight single heavy atom changes that improve activity, rather than those that involve informative alterations that reduce or abolish activity. The examples were also chosen to illustrate that the impact of such single atom changes can originate from steric, electronic, conformational or H-bonding effects, from changes in functional reactivity, from fundamental intermolecular interactions with a biological target, from introduction of a new or altered functionalization site, or from features as simple as improvements in stability or physical properties. Nearly all the examples highlighted represent not only unusual instances of productive deep-seated natural product modifications and were introduced through total synthesis, but are also remarkable in that they are derived from only a single heavy atom change in the structure.
TOC image
INTRODUCTION
Some years ago, I was asked to write a perspective on our work. Although the perspective is long overdue, variations on the topic highlighted by the title were digested for some time as a consequence of the invitation. Every individual that examines the interaction of a small molecule with its biological target, including proteins and nucleic acids, asks or faces the question every day on what impact a single atom can have. Whether it is in the context of drug discovery and the design of small molecules that selectively bind a therapeutic target, the delineation of a molecular mechanism of action of a natural product or chemical probe, the examination of signal transduction by endogenous signaling molecules, or the study of the interaction of substrates or inhibitors with an enzyme, the identification of structural features responsible for intermolecular ligand binding affinity and selectivity is fundamental to understanding and advancing science at the chemistry–biology interface.1–4 For chemists and medicinal chemists, the impact of not just the molecule, a substructure in the molecule, or even a substituent or functional group within the molecule, but the impact and nuanced role of even an individual atom in the molecule is fascinating, often exhibiting a remarkable influence.5 Even in fields not directly related to understanding the behavior of biologically active molecules, including reagent design, ligand development, catalysis, molecular recognition, complex molecule total synthesis, material science, and many others, the decisive role a single atom in a molecule can play is well appreciated. All those working in such fields will have their own favorite examples, whether from their own work or from that of others. At the risk of disappointing many, I have focused only on examples drawn from our own work. Hopefully no one will mistake the focus on our examples as an effort to take credit for countless other observations that lie at the heart of so much of what we all do and enjoy. Rather, it is meant to highlight the intricate details and occasional triumphs in molecular level design for those not intimately involved. In our efforts, the work has been conducted in studies typically designed to answer fundamental questions on ligand–target interactions and have been a part of our program since my career began. Thus, along with highlights of advances made in many of our long standing programs, the perspective also focuses on examples within this work where a single atom change exhibited a productive and remarkable impact.
Our work most often has been conducted with biologically active natural products.6–11 The cases presented constitute the addition, removal, or exchange of a single heavy atom. In many instances, the changes may entail more than one atom (e.g., NH vs O), but for the sake of simplicity and at the expense of accuracy, I will refer to such changes as single heavy atom changes. The examples were chosen to illustrate that the productive impact of single heavy atom changes can originate from steric, electronic, conformational or H-bonding effects, from changes in intrinsic reactivity, from intermolecular interactions with a protein or nucleic acid target, from introduction of a new functionalization site, or from effects as benign as altering stability or physical properties. The examples highlighted herein also represent single heavy atom changes that were found to substantially improve the activity, rather than those that entail informative alterations that reduced or abolished activity. Natural products display a constellation of properties and multiple functions integrated into a compact, highly functionalized molecule. This is in contrast to other biomolecules like proteins where separate functional domains are often linearly linked, rather than integrated into a more compact structure. As a result, each structural component, functional group, or substituent within a natural product is often, but not always, integral to the expression of its biological activity. When the productive properties of a natural product are directly related to its emergence in Nature where it has undergone continuous optimization by natural selection, it may not be easily subjected to structural modifications. Thus, the significant improvements highlighted herein not only represent unusual instances of productive deep-seated modifications of natural products that were accessed by total synthesis, but are also remarkable in that they are derived from only a single heavy atom change.
Vancomycin and its redesign to overcome bacterial resistance
It is likely that the work of ours that is most easily recognized as arising from single heavy atom changes or exchanges is our efforts on the redesign of vancomycin for resistant bacteria.12,13 The biological target for the glycopeptide antibiotics, including vancomycin, is bacterial cell wall precursors containing D-Ala-D-Ala.14–16 Antibiotic binding to D-Ala-D-Ala results in inhibition of cell wall maturation. Since this cell wall target is unique to bacteria and not found in mammalian hosts, it is responsible for the selectivity of the antibiotic class for bacteria. The mechanism of resistance to vancomycin first emerged in Enterococci,17,18 was co-opted from non-pathogenic bacteria and not independently evolved by the pathogenic bacteria,19 and involves a single heavy atom exchange in the biological target.20 This modification is the exchange of an ester oxygen for an amide NH. The synthesis of the bacterial cell wall precursors continue with installation of the pendant N-terminus D-Ala-D-Ala. Like the producer organisms, resistant bacteria sense the presence of the antibiotic21 and initiate an intricate late stage remodeling of their peptidoglycan termini from D-Ala-D-Ala to D-Ala-D-Lac to avoid the antibiotic.22 As a result of the single heavy atom exchange, the binding affinity of vancomycin for the resistant ligand is reduced 1000-fold. Binding studies were conducted with vancomycin (1) and the model ligands 2–4 that included examination of the ketone ligand 3, which contains a methylene that lacks both a lone pair and is incapable of H-bonding.23 Unlike the often cited origin of the diminished binding, these studies revealed that it is the introduction of a destabilizing electrostatic repulsion (100-fold), more so than a lost H-bond (10-fold), that is responsible for the majority of the 1000-fold loss in binding affinity of vancomycin for D-Ala-D-Lac (Figure 1).
Figure 1.
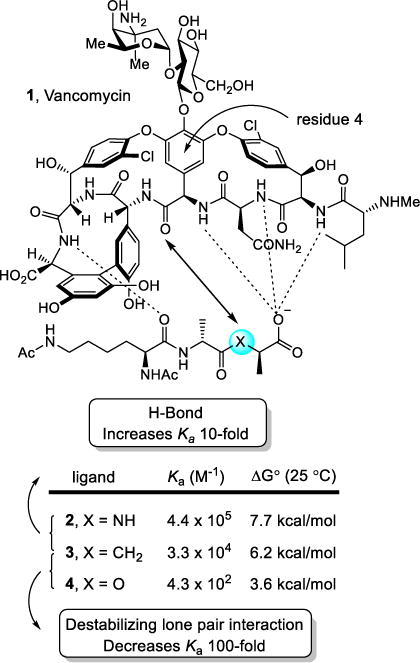
Vancomycin binding to model ligands that contain single heavy atom exchanges.
This indicated that removal of the destabilizing lone pair/lone pair interaction without even reengineering a reverse H-bond could improve affinity for the altered ligand as much as 100-fold. Thus, a binding pocket modification in the vancomycin core designed to remove the destabilizing lone pair interactions by replacement of the residue 4 amide carbonyl with an aminomethylene linkage (compound 7) was prepared by total synthesis in initial studies (Figure 2).24 This modification entailed removal of a single heavy atom from the antibiotic, the residue 4 amide carbonyl oxygen. This change provided an antibiotic analog with the balanced dual ligand binding capabilities needed for vancomycin-resistant organisms (D-Ala-D-Ala and D-Ala-D-Lac binding), while maintaining the ability to bind D-Ala-D-Ala required for vancomycin-sensitive bacteria albeit with an approximately 30-fold reduced affinity.24
Figure 2.
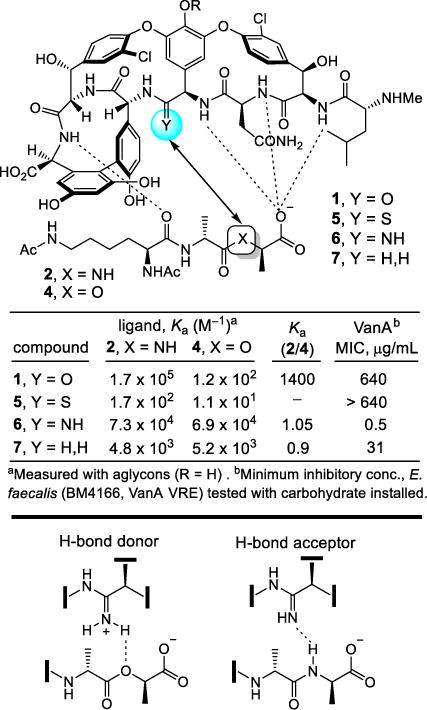
Vancomycin analogs that incorporate single heavy atom changes in the binding pocket.
In a series of subsequent studies, we reported two additional vancomycin analogs that also contained single heavy atom exchanges at this key site in its target binding pocket (residue 4 carbonyl O → S, NH), the latter of which was designed to more effectively address the underlying molecular basis of resistance to vancomycin (Figure 2).24–28 On the surface, this exchange of the vancomycin residue 4 amide carbonyl oxygen with an amidine NH would seem to be simply compensating for the target exchange of an amide NH with an ester oxygen. However, it does much more than that. Not only does the exchange reinstate full binding affinity to the ligand of the altered biological target (D-Ala-D-Lac), but it also maintains near full binding affinity for the unaltered biological target (D-Ala-D-Ala). We have suggested, and believe we have shown,29 that it displays this dual binding character with the amidine free base serving as a H-bond acceptor for binding D-Ala-D-Ala, and with the protonated amidine binding D-Ala-D-Lac, replacing the destabilizing electrostatic interaction with a stabilizing electrostatic interaction and possibly a weak reverse H-bond. This remarkable dual binding behavior could not have been easily predicted and, as a consequence, the residue 4 amidine was not the first of the modifications that we examined.
The behavior of the vancomycin residue 4 thioamide, a key synthetic intermediate in route to the corresponding amidine, was just as remarkable. Although it represents a seemingly benign single heavy atom exchange in the binding pocket, replacing an amide carbonyl oxygen with a sulfur atom to provide a thioamide, it served to completely disrupt binding to D-Ala-D-Ala (1000-fold loss in affinity)26 and resulted in a vancomycin analog devoid of antimicrobial activity.28 In retrospect, this behavior may be attributed to both the increased size of the sulfur atom and the longer C=S versus C=O bond length, which are sufficient to displace the ligand from the binding pocket. This unanticipated behavior also serves to highlight just how remarkable the properties of 6 are and how well its residue 4 amidine serves as an isosteric replacement for the vancomycin residue 4 amide in its interaction with D-Ala-D-Ala.
The two rationally designed binding pocket modifications found in 6 and 7 reinstated binding to the altered target D-Ala-D-Lac and maintained binding affinity for the unaltered target D-Ala-D-Ala. Such dual target binding compounds were found to reinstate antimicrobial activity against vancomycin-resistant organisms that employ D-Ala-D-Lac peptidoglycan precursors, and remain active against vancomycin-sensitive bacteria. Moreover, the in vitro antimicrobial potencies of such compounds correlated directly with their absolute dual binding affinities for model target ligands.
These studies were enabled by the modern techniques of total synthesis first used to prepare many of the natural products in the family of glycopeptide antibiotics,30–40 which provided the foundation on which deep-seated single atom changes or exchanges could be made in the vancomycin structure. These studies have been extended further to provide analogs that contain peripheral modifications of the pocket-modified vancomycin analogs that introduced added mechanisms of antimicrobial action independent of D-Ala-D-Ala/D-Lac binding (Figure 3).28,41 These latter efforts provided remarkable vancomycin analogs that: (1) contain synergistic binding pocket and one or two simple peripheral modifications, (2) are endowed with two or three independent mechanisms of action only one of which is dependent upon D-Ala-D-Ala/D-Lac binding, (3) display broad spectrum activity against both vancomycin-sensitive and vancomycin-resistant bacteria (e.g., MRSA, VanA/VanB VRE) at stunning potencies (MICs = 0.01–0.005 μg/mL), and (4) are more durable antibiotics than even vancomycin,41 which has been in the clinic for 60 years.
Figure 3.
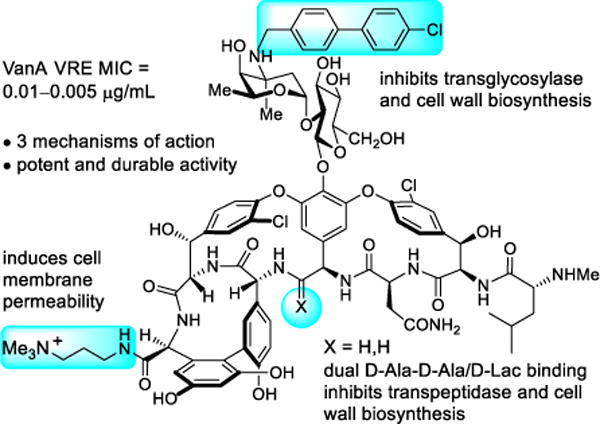
Vancomycin analog that contains a single atom change in the binding pocket, reinstating activity against vancomycin-resistant bacteria, and two peripheral modifications that add two additional independent mechanisms of action.
Ramoplanin
The ramoplanins are naturally occurring lipoglycodepsipeptides42,43 that are 2–10 fold more active than vancomycin against Gram-positive bacteria, including MRSA and vancomycin-resistant Enterococci.44,45 Ramoplanin A2 disrupts bacterial cell wall peptidoglycan biosynthesis, inhibiting the intracellular conversion of lipid intermediate I to lipid intermediate II46 and the more accessible extracellular transglycosylase-catalyzed incorporation of lipid II into the glycan strand,47 steps that precede the site of action of vancomycin. Resistance to ramoplanin has not been detected and cross resistance between ramoplanin and vancomycin has not been observed. Thus, it remains equally active against vancomycin-resistant organisms, including VanA/VanB VRE. Like vancomycin, ramoplanin acts by binding peptidoglycan precursors (lipid II > lipid I),48 sequestering these substrates from enzyme access,49,50 although the structural details of these interactions are not yet defined. In fact, ramoplanin embodies all the characteristics of vancomycin that contributes to its durability against resistance development. However, its instability derived from rapid depsipeptide hydrolysis precludes its use for systemic infections and has limited its clinical exploration.42 Our development of the first and still only convergent total synthesis of the ramoplanin A1–A3 aglycons51–53 set the stage for its use in the preparation of key analogs. In these efforts, we demonstrated that synthetic [L-Dap2]ramoplanin A2 aglycon (9), which bears a linking amide in place of the sensitive depsipeptide ester in the backbone of the 49-membered macrocycle, is roughly 2-fold more potent ramoplanin A2 and its aglycon, and stable to hydrolytic cleavage (Figure 4).54,55 Here, the single heavy atom exchange does not impact the interaction of the natural product with its biological target or substantially alter its functional activity, but it substantially improves its limiting metabolic stability.
Figure 4.
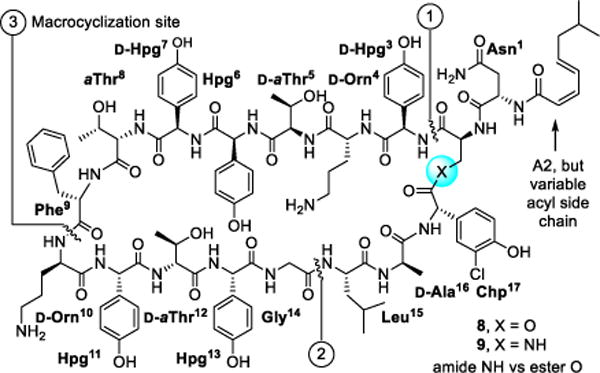
Structure of [L-Dap2]ramoplanin A2 aglycon and a single heavy atom exchange in 49-membered macrocycle that substantially improves hydrolytic stability shown to limit the clinical use of ramoplanin.
In our studies and on this stable amide template, a scan of the complete structure was conducted (Ala scan, 15 analogs prepared by total synthesis),56 establishing the impact and potential role of each residue and providing insights into the nature of its complex with lipid II.56,57 Highlights derived from the alanine scan of this amide modified ramoplanin aglycon (9) include: (1) the verification of the dominant role of Orn10 (>500-fold reduction) consistent with an integral role in lipid II diphosphate binding, (2) the surprisingly modest impact of Orn4 (44-fold), suggesting that its role in binding lipid II is not as critical, (3) the disparate importance of each of the residues in a putative lipid II recognition domain proposed58,59 in early work (residues 3–10), (4) the significant impact (>20-fold) of nearly each residue in the dimerization domain (residues 11–14) later defined by Walker60 reflective of its greater importance, and (5) the lack of importance of the hydrophobic residues 16–17 within the flexible loop that represents the membrane interacting domain (residues 15–17, 1–2). We also showed that the lipid side chain is essential for antimicrobial activity (200–800-fold reduction) and, in collaboration with Walker, showed it has no impact on lipid II binding or transglycosylase inhibition, indicating that its role is likely to anchor the antibiotic to the bacterial cell wall.54 Complementing these studies on the stable amide-modified ramoplanin 9 and other related studies,61 Suzanne Walker used inhibition kinetics and binding assays to establish that ramoplanin preferentially inhibits the transglycosylase versus MurG catalyzed reactions of their substrates lipid II versus lipid I, that it exhibits a greater affinity for lipid II (KD = 3 nM) than lipid I (KD = 170 nM), and that it binds with a 2:1 stoichiometry consistent with functional dimerization.49,50
Vinblastine
The biological properties of vinblastine were among the first to be shown to arise from tubulin binding, resulting in perturbations in microtubule dynamics that lead to inhibition of mitosis.62 In fact, it was the discovery of vinblastine that led to the identification of tubulin as an especially effective oncology drug target. As discussed below, vinblastine binds at the tubulin α/β dimer–dimer interface where it destabilizes microtubulin assembly derived from the repetitive head-to-tail tubulin binding. This action through disruption of a protein–protein interaction by vinblastine is often overlooked in discussions of such targets addressed with small molecules perhaps because the target identification preceded the contemporary interest in drugs targeting protein–protein interactions. Even by today’s standards, vinblastine and vincristine are superb clinical drugs. They, and their biological target tubulin, remain the subject of investigations because of their clinical importance in modern medicine, complex structures, low natural abundance, and unique mechanism of action.
In a study designed to probe the impact of catharanthine indole substituents on an Fe(III)-mediated coupling with vindoline,63 two new and exciting derivatives were discovered, 10′-fluorovinblastine and 10′-fluorovincristine (Figure 5).64 In addition to defining a pronounced substituent effect on the biomimetic coupling that helped refine its mechanism,65 fluorine substitution at C10′ was found to uniquely enhance the activity (IC50 = 800 pM, HCT116). This exceptional activity was confirmed with the comparative examination of vinblastine and 10′-fluorovinblastine in a more comprehensive human cancer cell line panel graciously conducted at Bristol-Myers Squibb (Figure 5).66 10′-Fluorovinblastine exhibited a remarkable potency (avg. IC50 = 300 pM), being on average 30-fold more potent than vinblastine (avg. IC50 = 10 nM).
Figure 5.
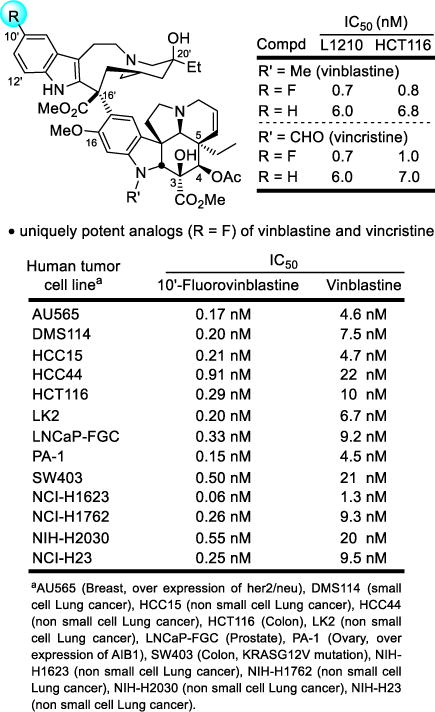
10′-Fluorovinblastine and 10′-fluorovincristine, unique impact of an added single heavy atom substituent that improved target (tubulin) binding affinity and functional activity (30-fold).
As depicted in the X-ray structure of vinblastine bound to tubulin,67,68 the C10′ site resides at one corner of a T-shaped conformation of the tubulin-bound molecule, where we have suggested the 10′-fluorine substituent makes critical contacts with the protein at a hydrophobic site sensitive to steric interactions. Although a range of 10′ substituents are tolerated, the activity of the derivatives exhibited no relationship with the electronic character of the substituents. Rather, they exhibited activity that correlated with the substituent size and shape. Thus, small hydrophobic substituents were found to be tolerated, but with only one derivative exceeding (R = F) and several matching the potency of vinblastine (R = Cl, Me, Br vs H), whereas larger (R = I, SMe) or rigidly extended (R = CN) substituents substantially reduced activity (10–100 fold).64 Although the enhanced metabolic stability of the 10′-fluoro derivative could in principle contribute to the increased potency, the lack of similar effects with related substituents indicate that a feature unique to the fluorine substitution is responsible. We have suggested that this is derived from the interaction of a perfectly sized hydrophobic substituent further stabilizing compound binding with tubulin at a deeply imbedded site exquisitely sensitive to steric interactions. Comparison models of the 10′ substituent analogs built from the X-ray structure of tubulin-bound vinblastine illustrated a unique fit for 10′-fluorovinblastine (Figure 6)64 and studies disclosed later demonstrated that it alone displays a higher tubulin binding affinity.69 Here, a singularly unique added heavy atom substantially improved target (tubulin) binding affinity and the resulting functional activity (30-fold).
Figure 6.
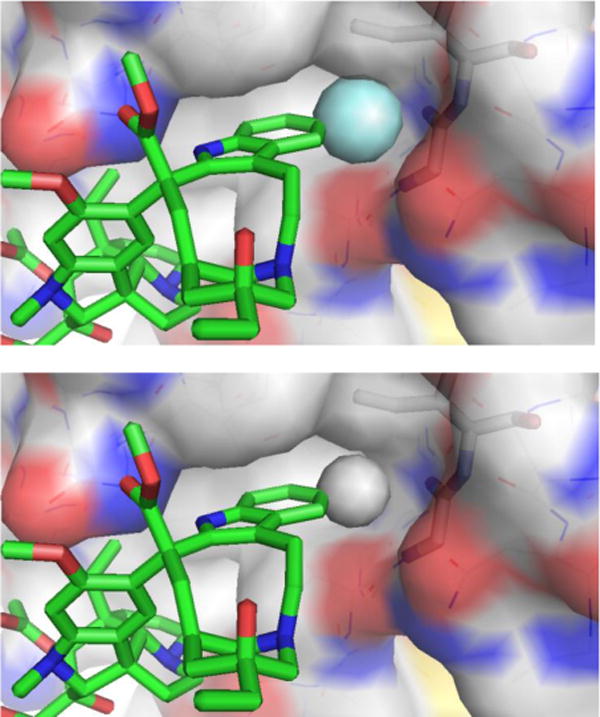
Model of the 10′-fluoro binding site of 10′-fluorovinblastine (R = F, top) generated by adding the fluorine substituent to the X-ray structure of tubulin-bound vinblastine67 (R = H, bottom).64 Modeled complexes with larger substituents (R = Cl, Me, Br, I) exhibited increasingly larger destabilizing steric interactions as the substituent size progressively increased.
These observations emerged in studies that provided a powerful approach to prepare previously inaccessible vinblastine analogs by total synthesis.70–79 It was the potential for their improvements that inspired our development of new synthetic methodology created deliberately for the intended target.70 Thus, a powerful oxadiazole tandem intramolecular [4+2]/[3+2] cycloaddition cascade was introduced80–82 that not only assembled the full pentacyclic skeleton in a single step, but also incorporated each substituent, functional group, embedded heteroatom, and all necessary stereochemistry for direct conversion of the cascade cycloadduct to vindoline.83,84 Combined with the use of a single-step Fe(III)-promoted coupling of catharanthine with vindoline and a newly developed in situ Fe(III)/NaBH4-promoted hydrogen atom transfer free radical C20′ oxidation,78,79,85,86 the approach provides vinblastine and its analogs in 8–13 steps. This was used to provide vinblastine analogs not previously accessible by semisynthetic modification of the natural products themselves that contain changes within either the lower vindoline-derived87–96 or upper catharanthine-derived subunits97–102 with the late stage divergent103 introduction of new functionality. In addition to the examination of C10′ substituents,64 we have prepared more than 400 analogs of vinblastine, defining the role of individual structural features and substituents. These studies have systematically probed the impact and role of the vindoline C16 methoxy group,79 C4 acetate,90–92 C5 ethyl substituent,93 C6–C7 double bond,94–96 and the vindoline core structure itself,96 and have systematically explored the upper subunit C20′ ethyl substituent,97,98 C16′ methyl ester,99 and added C12′ indole substituents.64 Notably, attempts at the simplification of the structure with the removal of a structural feature, a substituent, or even their subtle single heavy atom modifications have led to substantial reductions in activity. However and like the addition of a 10′-fluoro substituent, added features like that targeting the C20′ ethyl group with added benign complexity (ABC)97 can effectivity improve activity. We have shown that the single heavy atom replacement of the C20′ alcohol with an C20′ amine is possible85 and that its acylation to afford 20′ ureas or amides provides substantial100,101 and even stunning69 potency increases as much as 100-fold (IC50 = 75 pM vs 7 nM, HCT116). The ultra-potent vinblastines bind tubulin with much higher affinity and likely further disrupt the tubulin head-to-tail α/β dimer–dimer interaction by strategic placement of the conformationally well-defined, rigid, and extended 20′ urea or amide along the adjacent continuing protein–protein interface. Several 20′ amides were discovered that match or exceed the potency of vinblastine, but that are not subject to Pgp efflux and its derived vinblastine resistance.102 Within this series and reflecting an additional impact of a single heavy atom change, a benzamide substituent X was found to predictably impact activity, displaying a fundamental linear relationship between potency (−log IC50, HCT116) and the electronic character of the aryl substituent (σp) (Figure 7). All benzamides shown in Figure 7 are more potent than vinblastine and those that bear an aryl electron-donating substituent, some of which constitute single heavy atom additions, improve the H-bond acceptor ability of the added amide carbonyl that in turn proportionally increase the measured tubulin binding affinity (not shown) and functional activity (Figure 7).102 Finally, compounds in this series also displayed diminished off target activity (Pgp efflux) and diminished Pgp-derived resistance (IC50 ratio between isogenic Pgp-derived resistant and sensitive cell lines, 88-fold for vinblastine) that correlated with the increasing lipophilic character of the amide substituent.102 Here, single atom changes that simply increase lipophilic character (cLogP) were found to directly correlate with and diminish resistance derived from Pgp overexpression and drug efflux that limits vinblastine clinical use (Figure 8). The compounds emerged from the discovery of a site and functionalization strategy for the preparation of a vinblastine analog that contains a single heavy atom exchange (C20′ NH2 for OH).85 Its acylation provided the now readily accessible vinblastine analogs in three steps from commercially available materials that, unlike acylated derivatives of the alcohol itself (inactive), not only increase binding affinity to tubulin (on target affinity) and potency in cell-based assays, but also simultaneously disrupt efflux by Pgp (off target source of resistance).102 It is a tribute to the advances in organic synthesis that such detailed systematic studies can now be conducted on a natural product of a complexity as vinblastine once thought refractory to such approaches.
Figure 7.
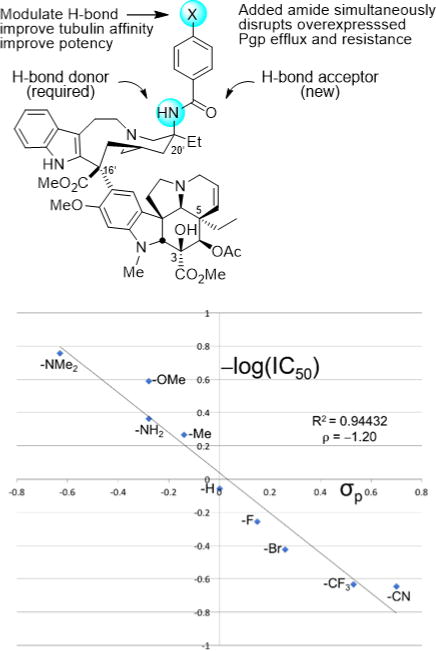
Active analogs required a single heavy atom exchange into the vinblastine structure (C20′ NH2 for OH). In a plot of −log IC50 (nM, HCT116) versus substituent σp, the analogs additionally displayed a predictable modulation of activity by a substituent (X) electronic effect, impacting benzamide carbonyl H-bonding with tubulin, some representing single heavy atom additions. All analogs shown are more active than vinblastine.
Figure 8.
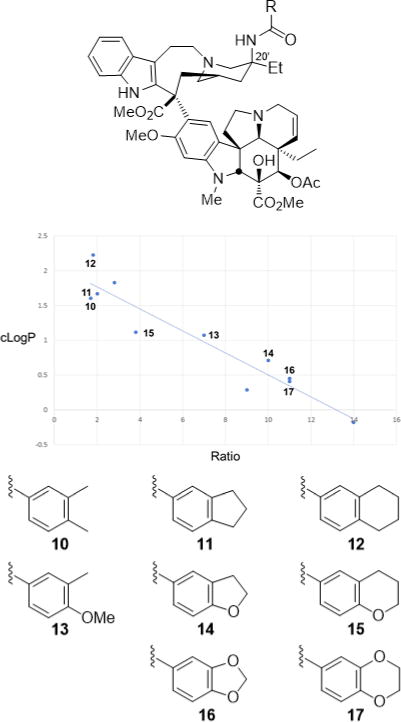
Plot of 20′ amide cLogP versus differential activity (IC50 ratio) for isogenic HCT116 resistant (Pgp overexpression) and sensitive cell lines that progressively exchange in single heavy atoms or heteroatoms. The correlation defines a linear relationship between diminished resistance (ratio) that arises from reduced/abolished Pgp efflux, and a modulated physical property of the compounds (lipophilic character, cLogP) that can be predictably impacted by single atom changes. All compounds shown are more potent than vinblastine and display less resistance (vinblastine ratio = 88).
In the course of these studies, a new and effective tubulin binding assay was necessarily developed69 to accurately measure the impact of the structural modifications, and a number of additional natural products were prepared by total synthesis104–109 with use of the newly introduced oxadiazole cycloaddition cascade.110,111
Duocarmycins, yatakemycin, and CC-1065
The first family of natural products on which we systematically examined the impact of deep-seated structural changes is composed of the duocarmycins, yatakemycin, and CC-1065, and a number of these modifications involved single atom changes in their structures. The natural products are exceptionally potent antitumor compounds that derive their activity through a sequence selective DNA alkylation.112–114 Our studies provided not only total syntheses of the natural products,115–127 but also the characterization of their DNA alkylation properties, including that of their unnatural enantiomers.128–133 In these studies, we defined their DNA alkylation selectivity, rates, and reversibility,134 isolated and characterized their adenine N3 adducts,130,132,135 and defined their stereoelectronically-controlled reaction regioselectivity.136–139 We defined the source of their alkylation selectivity as arising from their noncovalent binding selectivity preferentially in the narrower, deeper AT-rich minor groove (shape selective recognition),140–144 and identified the unusual source of catalysis for the alkylation reaction that is derived from a DNA binding induced conformation change that disrupts the stabilizing vinylogous amide conjugation (shape dependent catalysis).145–151 We demonstrated and quantified the fundamental role the hydrophobic character of the compounds plays in the expression of the biological activity, driving the intrinsically reversible DNA alkylation reaction, and defined the stunning magnitude of its effect (hydrophobic binding-driven-bonding).152 In collaboration with Walter Chazin, we provided high-resolution NMR-derived structures of the natural products and their unnatural enantiomers bound to DNA (Figure 9),153–155 and established that they are subject to an exquisite “target-based activation”.156 In the course of these studies, we introduced a convenient M13-derived alternative to 32P-end-labeling of restriction fragments for DNA cleavage studies.157 Fundamental relationships between structure and reactivity or structure and activity,158 and their contributions to the DNA alkylation properties and biological activity of the natural products, were established through the examination of more than 2000 analogs of the natural products that contained deep-seated structural changes (e.g., CBI).159–202 A compilation of the data derived from more than 30 deep-seated modifications, many of which entailed single heavy atom changes,187–202 resulted in the establishment of a predictive parabolic relationship between the alkylation subunit reactivity and the resulting cytotoxic potency that spanned a 104–106 range of reactivity and activity (Figure 10).203–206 Presumably, this fundamental relationship reflects the fact that the compound must be sufficiently stable to reach its biological target yet remain sufficiently reactive to alkylate DNA once it does. The parabolic relationship defined this optimal balance between reactivity and stability, providing a fundamental design feature that was used to improve the potency of CC-1065 by a single heavy atom exchange.
Figure 9.
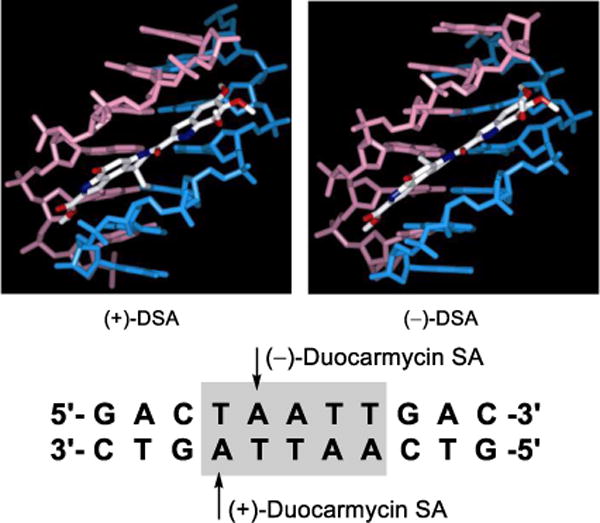
NMR structures of natural (+)- and ent-(−)-duocarmycin SA bound in the same AT-rich site of a deoxyoligonucleotide, illustrating the alkylation sites on complementary DNA stands offset by one base pair. Only the binding region of DNA is shown.
Figure 10.
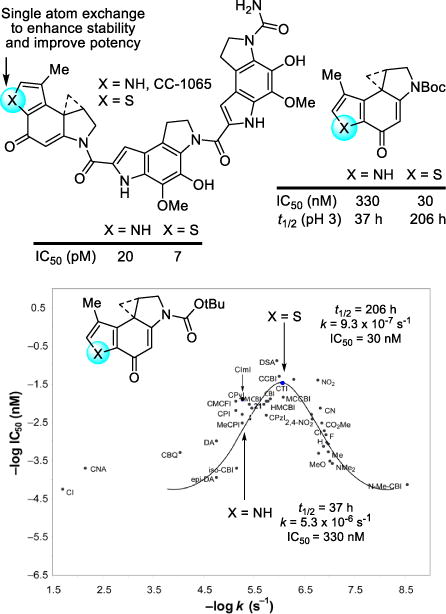
Single heavy atom exchange in the CC-1065 alkylation subunit that improves potency through a predicable reduction in intrinsic reactivity, placing it at an optimal point on a parabolic relationship between functional reactivity and activity.
The replacement of the CC-1065 alkylation subunit pyrrole NH with a sulfur atom was examined and represents the exchange of a single heavy atom.207 Its exploration rested with expectations that it would be substantially more stable than the alkylation subunit found in CC-1065, leading to a more potent CC-1065 analog. Intuitively, this was expected to arise from the strain release provided by a fused thiophene versus pyrrole, which in turn may further benefit from the greater electron-withdrawing character of a thiophene. More quantitatively, this increased stability could be approximated using semiempirical calculations (AM1, MNDO) where the thiophene analog was selected among several candidate alkylation subunits as being more stable. Analogs with the altered alkylation subunit, which lies at the pinnacle of the parabolic relationship, proved to be both 6-fold more stable (solvolysis) and 3–10-fold more potent (IC50, L1210) than those that contained the CC-1065 alkylation subunit, and they displayed an unaltered DNA alkylation selectivity but greater efficiency than CC-1065. Here, a single heavy atom exchange in the core structure of the CC-1065 alkylation subunit provided a near optimal increase in biological potency predictably derived from improvements in the stability of the reacting DNA alkylation subunit (Figure 10).207
When incorporated into an even further simplified structure, this modification of the alkylation subunit provided a potent and efficacious antitumor compound when examined in a rodent tumor model (Figure 11).207 Unlike many natural products, members of this class not only tolerate such simplifying structural changes,161,208 but their physical (solubility) and biological properties (in vivo efficacy) can be improved through such changes. An additional instructive example that highlights the productive changes derived from a single heavy atom change was the removal of the CC-1065 alkylation subunit C8 methyl substituent. Its removal increased both the rate and efficiency of DNA alkylation by removing a steric impediment to reaction with adenine and increased the biological potency of resulting analogs (Figure 11).192 Here, the modification represents an unusual example of the improvement in the biological potency of a natural product by removal of a seemingly benign single heavy atom from its structure. Finally and in a culmination of our own efforts, a unique reductively activated prodrug design that maps seamlessly onto the compound class was developed, which bears multiple structural simplifications (Figure 11).209–213 Selected members of this prodrug class are remarkably efficacious and exhibit a much wider dose range for efficacy in animal tumor models without dose-limiting toxicity.209 Thus, the exceptional potency of this drug class was tamed by a unique reductive activation prodrug design especially suited for this class of candidate drugs. Efforts with this class of molecules may represent one of the most extensive cases of molecular modification of biologically active natural products by total synthesis conducted with the intention of improving their properties, defining fundamental features of their mechanism of action, or in the development of clinical candidates.214–219
Figure 11.
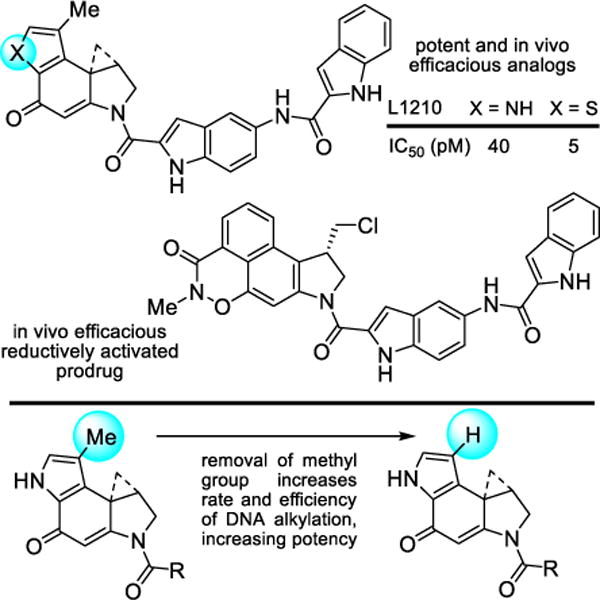
Additional simplifying structural modifications, an example of removal of a single heavy atom (Me group) that improves potency by making the underlying DNA alkylation reaction sterically more accessible, and a recent efficacious prodrug design on a simplified structure.
Bleomycin
Bleomycin A2 is a clinically employed antitumor drug that derives its properties through the sequence-selective cleavage of DNA in a process that is both metal-ion and O2 dependent.220–224 We developed a modular total synthesis of bleomycin A2 that permitted the total synthesis and examination of nearly 70 analogs of the natural product,225–242 probing each subunit and substituent in the structure.223 These studies confirmed the origin of DNA cleavage selectivity derived from G triplex-like H-bonding in the minor groove,239 defined fundamental conformational properties of bleomycin that contribute to the efficiency of DNA cleavage,240,241 clarified the functional roles of the individual subunits and their substituents,223 and in collaboration with JoAnne Stubbe provided a NMR-derived high resolution structure of DNA bound deglycobleomycin A2.243 In the course of these studies, we introduced the powerful fluorescent intercalator displacement (FID) assay for comprehensively establishing DNA binding selectivity or affinity.244,245 These combined studies, in conjunction with then emerging structural models,246 defined a remarkable combination of functional, structural, and conformational properties integrated into the natural product structure and served to underscore that it represents a natural product in which each subunit, each functional group, and nearly each substituent productively contribute to the expression of its biological properties.223 Two exceptions to this generalization are the pyrimidoblamic acid C5 methyl group that could be removed and replaced with an H atom without impacting its activity,230 and the histidine imidazole N1 atom, which could be exchanged for an oxygen atom (oxazole vs imidazole) but not removed (pyrrole versus imidazole).237 These represented cases where a single heavy atom could be removed or exchanged in the structure without impacting activity. Important among our observations was the experimental demonstration of the role the pyrimidine C4 amino group plays in H-bonding and DNA recognition, and as the source of the DNA cleavage selectivity (Figure 12).239
Figure 12.
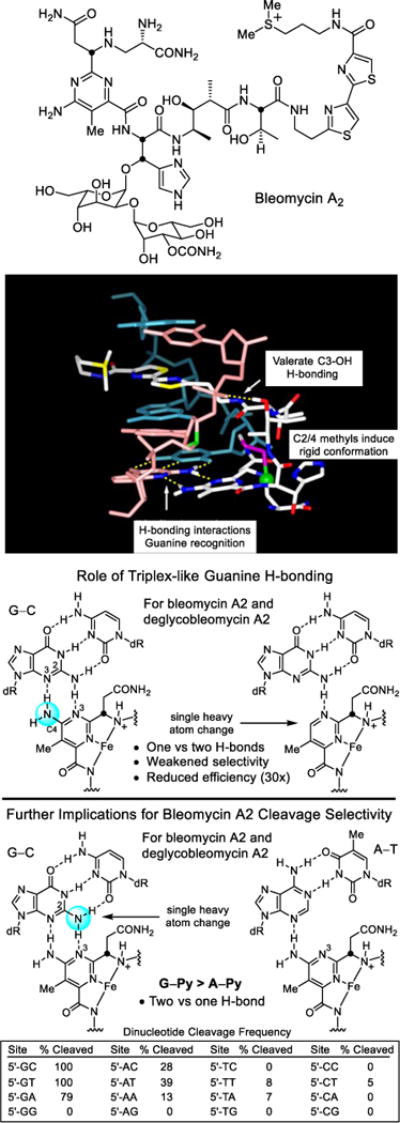
Structure of bleomycin A2, NMR structure of bleomycin bound to a DNA cleavage site (full deoxyoligonucleotide and bleomycin disaccharide removed for clarity), key H-bonding role the pyrimidine C4 amine plays in guanine recognition, and role the minor groove guanine C2 amine plays in the recognition of bleomycin.
The feature I want to highlight for the purpose of this perspective was the unrecognized subtle impact that the valerate methyl and threonine linker substituents play in preorganization and stabilization of a compact conformation implicated in DNA cleavage.240,241 Their individual or combined removal do not alter the metal chelation, O2 activation, or DNA cleavage selectivity of bleomycin, but they do progressively reduce the DNA cleavage efficiency. Predicable from first principles of conformational analysis, the heavy atom substituents combine to restrict the flexible linker region to a single dominant compact versus extended conformation, preorganizing the functional components of the molecule into a rigid conformation productive for DNA cleavage by the bound complex (Figure 13).
Figure 13.
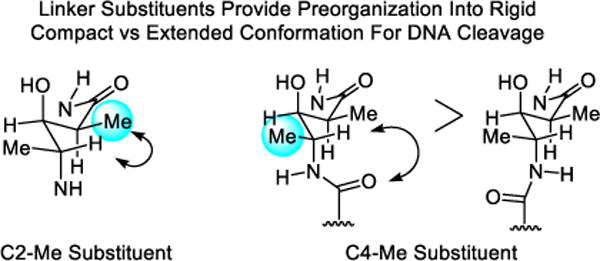
The C2 and C4 methyl groups of the valerate linker in bleomycin induce a rigid, compact versus extended conformation productive for DNA cleavage, see Figure 12. Each heavy atom substituent independently increases the efficiency of DNA cleavage without impacting metal chelation, O2 activation, or the cleavage reaction and without making direct contact with the target.
Analogous observations were made in efforts targeting the protein phosphatase inhibitors fostreicin, cytostatin, and phostriecin (sultreicin)247–255 where the presence and stereochemistry of benign methyl substituents on an aliphatic chain substantially impact the biological activity through conformational restriction of an otherwise flexible chain.252,255
Enzyme inhibitors, fatty acid amide hydrolase
A number of additional instructive examples of the impact of a single atom change can be illustrated in a program that emerged from the discovery of oleamide as an endogenous signaling molecule promoting physiological sleep.256–259 Even small changes in the simple structure of the signaling molecule oleamide (e.g., saturation of the double bond, its relocation by a single atom, and trans vs cis configuration) results in a loss in activity.257 The discovery of the physiological role of oleamide represented the delineation of the first of a growing class of endogenous signaling fatty acid primary amides,260 and was disclosed shortly after the identification of anandamide,261 a fatty acid ethanolamide, as the endogenous ligand for the cannabinoid receptors. This work led to the discovery and characterization of the enzymes responsible for the release (PAM) of signaling fatty acid primary amides262 and the degradation of signaling fatty acid amides (fatty acid amide hydrolase, FAAH).263–266 It provided orally active, long acting, potent, and selective α-ketoheterocycle inhibitors of serine hydrolases including FAAH, used a powerful proteome-wide activity-based protein profiling (ABPP)-based selectivity assessment for reversible enzyme inhibitors,267–269 characterized inhibitor bound FAAH X-ray structures,270–272 and provided the first in vivo validation of FAAH as a candidate therapeutic target.273–284 This work showed that preventing the enzymatic hydrolysis of an endocannabinoid (anandamide) provides an effective approach for the treatment of pain that avoids the side effects of a traditional blunt force agonist acting on the target receptors (CB1 and CB2).273,274 Since this only potentiates an activated signaling pathway by increasing the concentration and duration of action of the released signaling molecule at its site of action, it provides a temporal and spatial pharmacological control not available to a receptor agonist. It is the work that has been conducted as part of an extraordinary collaboration with Ben Cravatt, Richard Lerner, Aron Lichtman and many others, and has inspired efforts to target other enzymes controlling endocannabinoid signaling for the treatment of pain and inflammation,280 for the modulation of other GPCR targets, and provided the foundation for FAAH inhibitors that progressed into the clinic.282–284
Our systematic examination of α-ketoheterocycles285–289 as inhibitors of FAAH290–304 was initiated at a time when only a handful of articles on α-ketoheterocycles had been published. Representative of all efforts in medicinal chemistry directed at enzyme or receptor targets, single atom changes in the ligand had remarkable impacts on target affinity, target selectivity, PK properties, and in vivo efficacy. Optimization of candidate α-ketoheterocycles led to the identification of OL-135273 and later CE-12299 as potent, selective, and efficacious in vivo inhibitors of FAAH (Figure 14).
Figure 14.
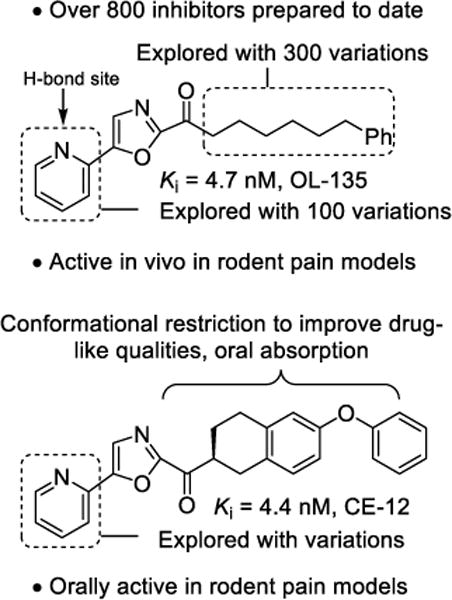
FAAH inhibitors.
The examination of OL-135273 included a systematic exploration of the central activating heterocycle.298 Several activating heterocycles were found to improve the inhibitor potency relative to the oxazole found in OL-135 and representative examples are presented in Figure 15. In short, the introduction of an additional heteroatom at position 4 (oxazole numbering, potency: N > O > CH) substantially increased inhibitory activity that may be attributed to a combination of the increased electron-withdrawing properties of the activating heterocycle as well as a reduced destabilizing steric interaction302 at the active site observed in the X-ray of the complex of FAAH with OL-135.
Figure 15.
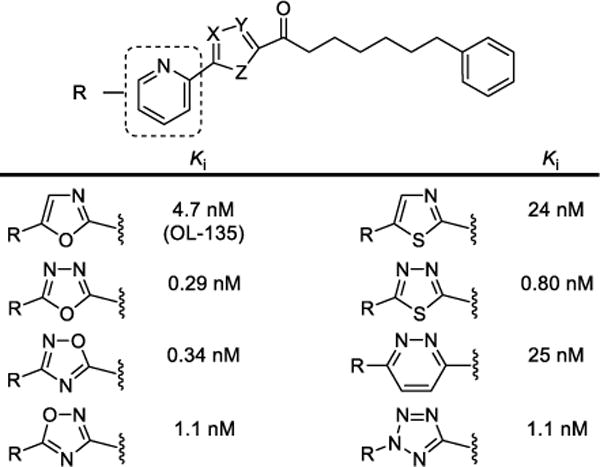
Representative OL-135 analogs containing iterative single heavy atom changes or exchanges in the activating heterocycle. Reduction in the steric size of the heterocycle position 4 heavy atom (potency: N > O > CH) contributes to increased inhibitor potency.
We also defined a role for the central activating heterocycle distinct from that observed with serine proteases287,288 that explained pronounced substituent effects. The work illustrated the importance of the electrophilic character of the ketone in driving FAAH inhibition. A well-defined linear correlation between the Hammett σp constant of the α-ketooxazole C5 or C4 substituent and FAAH inhibition was established that is of a magnitude to dominate the behavior of inhibitors (ρ = 3.0–3.4), indicating that a unit increase in σp results in a stunning 1000-fold increase in Ki.293,294 This provided a predictive tool for the rational design of α-ketoheterocycle-based serine hydrolase inhibitors beyond FAAH where modulation of the inhibitory potency could be accomplished by substitution of an oxazole, in some instances by an added single heavy atom, predictably modulating the intrinsic reactivity of the electrophilic carbonyl (Figure 16).
Figure 16.
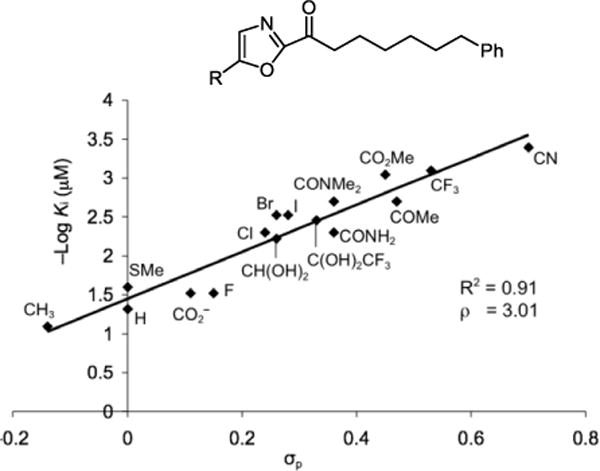
−Log Ki (μM) for FAAH versus Hammett σp, defining and quantitating a linear correlation between enzyme inhibition and the electronic impact of oxazole substituents on intrinsic reactivity of the electrophilic carbonyl of an α-ketoheterocycle (ρ = 3.0), some arising from single heavy atom substitution.
The first X-ray structures of the α-ketoheterocycle-based inhibitors bound to FAAH were disclosed in 2009 in collaboration with Ray Stevens.270 These co-crystal structures of OL-135 and its isomer with FAAH confirmed that the catalytic Ser241 is covalently bound to the inhibitor electrophilic carbonyl, providing a deprotonated hemiketal mimicking the enzymatic tetrahedral intermediate (Figure 17). It also represents an unusual case of exchanging the location of two complementary heteroatoms in the core structure of the inhibitor, each of which is essential to the activity. Neither heteroatom can be replaced with a CH, both heteroatoms are required, but their locations can exchanged. Additional co-crystal structures of key α-ketoheterocycles271,272 systematically probed the three active site regions central to substrate or inhibitor binding: (1) the conformationally mobile acyl chain-binding pocket and membrane access channel, (2) the active site catalytic residues and surrounding oxyanion hole that covalently binds the α-ketoheterocycle inhibitors, and (3) the cytosolic port and a newly identified anion binding site. These structures, including a representative member of the inhibitors containing a conformationally constricted C2 acyl side chain,299 confirmed covalent attachment through nucleophilic addition of Ser241 on the inhibitor electrophilic carbonyl and they captured the catalytic residues in an “in action” state. They also revealed an unusual Ser217 OH-π H-bond to the activating heterocycle and defined a prominent role that bound water in the cytosolic port plays in stabilizing inhibitor binding through interaction with the pyridyl nitrogen of the OL-135 substituent. These studies established that the dominant role of the activating heterocycle is its intrinsic electron-withdrawing properties and identified the key role of an ordered cytosolic port water in mediating the stabilizing hydrogen bonding of optimized oxazole substituents.
Figure 17.
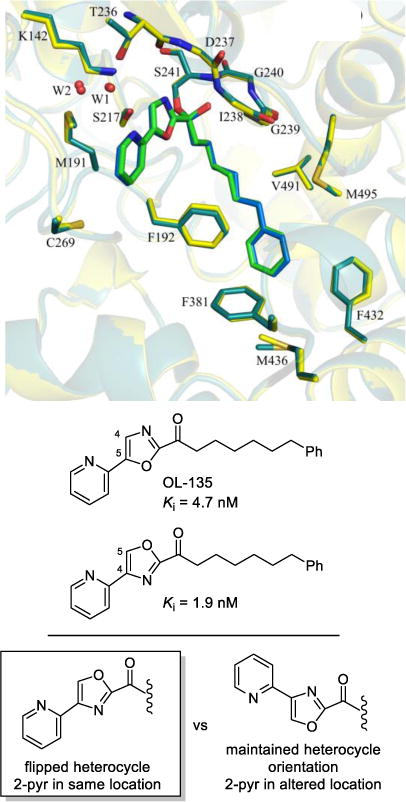
Superimposition of the X-ray structures of OL-135 (green) and its isomer (blue) bound to FAAH that illustrates the compensating impact of exchanging the location of two complementary heteroatoms.
CONCLUSIONS
Central to science at the chemistry–biology interface is the ability of small molecules to selectively bind a unique protein or nucleic acid target and elicit a response in a biological milieu. Especially informative are studies on small molecules found in Nature, biologically active natural products. Beyond their importance in modern medicine, unraveling the role their structure features play in the expression of their functional biological activity is molecular level science at its finest. This can include not only the identification of structural features that convey the affinity and selectivity for a biological target or those that are central to their molecular mechanism of action, but also how these features are intimately integrated into a complex, compact structure that simultaneously displays a constellation of functions. It is even more remarkable that a single heavy atom in such compounds can impact the underlying intermolecular interactions or functions in such pronounced ways. In the context of our work, examples were highlighted herein in which single heavy atom changes or exchanges substantially and atypically improve the activity, rather than those that informatively reduce or abolish activity. The examples illustrate that their productive impact can originate from steric, electronic, conformational or H-bonding effects, from changes in intrinsic reactivity, from fundamental intermolecular interactions with a protein or nucleic acid target, from introduction of a new functionalization site, or from effects as simple as altering stability or physical properties. It is, I believe, an instructive series of examples where modifications that entail even a single heavy atom change can have remarkable impacts on the expression of the biological properties of the natural products. In a field where the simplification of complex structures has been perceived as the most expeditious path forward and where single heavy atom removal might fall into the complex end of this category, rarely does one contemplate the addition or even exchange of a single heavy atom. Yet, as shown herein, such small changes can have predicable and especially productive effects. Key to exploring such nuanced structural modifications is the total synthesis of the modified compounds, the development of synthetic strategies and methodology suitable for systematic structural exploration, and a commitment to their implementation in such studies. It is a tribute to the advances in organic synthesis that natural products of the complexity of vancomycin and others highlighted herein can now be rationally, though not yet routinely, subjected to systematic probes of their structure and function with deep-seated structural modifications, even those involving single atom changes. Perhaps we are approaching a time when such complex structures, like the typically simpler compounds of traditional drug discovery, can be routinely embraced with confidence that their already remarkable constellation of properties can be just as effectively improved by subtle, impactful structural modifications. Finally, throughout the course of my career and in each of our programs, the questions about such molecules have also progressed from how do we identify them and can we make them, to can we understand them and can we improve on them? Similarly, the question of the impact of single atoms in such structures has progressed from “what difference can a single atom make?”, to “can a single atom make a difference?”, and now to “what single atom can make a difference?”. Hopefully, a sense of that journey is also summarized in this perspective.305
Acknowledgments
The work summarized herein has been the collective effort of a spirited, talented, and gifted group of graduate students and postdoctoral colleagues with whom I have had the opportunity and good fortune to work with. It is their personal and professional progression as scientists that has provided the true pleasure and satisfaction in the conduct of the work. I would also like to acknowledge the unconditional support of cherished colleagues at Scripps and earlier at the University of Kansas. I am especially grateful to NIH for the financial support of our studies (CA041101, CA042056, CA208669, DA015648).
Biography

Dale Boger received his BSc in Chemistry from the University of Kansas, his PhD in Chemistry from Harvard University with EJ Corey, and served on the faculty at the University of Kansas (Medicinal Chemistry) and Purdue University (Chemistry) before joining The Scripps Research Institute in 1990. He is presently the Richard and Alice Cramer Professor of Chemistry and currently serves as the Chairman of the Department of Chemistry for the TSRI La Jolla campus.
Footnotes
Notes
The author declares no competing financial interests.
References
- 1.Jorgensen WL. The many roles of computation in drug discovery. Science. 2004;303:1813–1818. doi: 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]
- 2.Schreiber SL. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science. 2000;287:1964–1969. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- 3.Wu Xu, Schultz PG. Synthesis at the interface of chemistry and biology. J Am Chem Soc. 2009;131:12497–12515. doi: 10.1021/ja9026067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dervan PB. Molecular recognition of DNA by small molecules. Bioorg Med Chem. 2001;9:2215–2235. doi: 10.1016/s0968-0896(01)00262-0. [DOI] [PubMed] [Google Scholar]
- 5.Meanwell NA. Synopsis of some recent tactical application of bioisosteres in drug design. J Med Chem. 2011;54:2529–2591. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 6.Hong J. Natural products at the interface of chemistry and biology. Chem Eur J. 2014;20:10204–10212. doi: 10.1002/chem.201402804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilson RM, Danishefsky SJ. Small molecule natural products in the discovery of therapeutic agents: The synthesis connection. J Org Chem. 2006;71:8329–8351. doi: 10.1021/jo0610053. [DOI] [PubMed] [Google Scholar]
- 8.Wender PA, Verma VA, Paxton TJ, Pillow TH. Function-oriented synthesis, step economy, and drug design. Acc Chem Res. 2008;41:40–49. doi: 10.1021/ar700155p. [DOI] [PubMed] [Google Scholar]
- 9.Szpilman AM, Carreira EM. Probing the biology of natural products: molecular editing by diverted total synthesis. Angew Chem, Int Ed. 2010;49:9592–9628. doi: 10.1002/anie.200904761. [DOI] [PubMed] [Google Scholar]
- 10.Bebbington MWP. Natural product analogues: towards a blueprint for analogue-focused synthesis. Chem Soc Rev. 2017;46:5059–5109. doi: 10.1039/c6cs00842a. [DOI] [PubMed] [Google Scholar]
- 11.Wright PM, Seiple IB, Myers AG. The evolving role of chemical synthesis in antibacterial drug discovery. Angew Chem, Int Ed. 2014;53:8840–8869. doi: 10.1002/anie.201310843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boger DL. Vancomycin, teicoplanin, and ramoplanin: synthetic and mechanistic studies. Med Res Rev. 2001;21:356–381. doi: 10.1002/med.1014. [DOI] [PubMed] [Google Scholar]
- 13.James RC, Pierce JG, Okano A, Xie J, Boger DL. Redesign of glycopeptide antibiotics: back to the future. ACS Chem Biol. 2012;7:797–804. doi: 10.1021/cb300007j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perkins HR. Vancomycin and related antibiotics. Pharmacol Ther. 1982;16:181–197. doi: 10.1016/0163-7258(82)90053-5. [DOI] [PubMed] [Google Scholar]
- 15.Kahne D, Leimkuhler C, Lu W, Walsh CT. Glycopeptide and lipoglycopeptide antibiotics. Chem Rev. 2005;105:425–448. doi: 10.1021/cr030103a. [DOI] [PubMed] [Google Scholar]
- 16.Nicolaou KC, Boddy CNC, Brase S, Winssinger N. Chemistry, biology, and medicine of the glycopeptide antibiotics. Angew Chem, Int Ed. 1999;38:2096–2152. doi: 10.1002/(sici)1521-3773(19990802)38:15<2096::aid-anie2096>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 17.Leclercq R, Derlot E, Duval J, Courvalin P. Plasmid-mediated resistance to vancomycin and teicoplanin in Enterococcus faecium. N Engl J Med. 1988;319:157–161. doi: 10.1056/NEJM198807213190307. [DOI] [PubMed] [Google Scholar]
- 18.Weigel LM, Clewell DB, Gill SR, Clark NC, McDougal LK, Flannagan SE, Kolonay JF, Shetty J, Killgore GE, Tenover FC. Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus. Science. 2003;302:1569–1571. doi: 10.1126/science.1090956. [DOI] [PubMed] [Google Scholar]
- 19.Marshall CG, Lessard IA, Park I, Wright GD. Glycopeptide antibiotic resistance genes in glycopeptide-producing organisms. Antimicrob Agents Chemother. 1998;42:2215–2220. doi: 10.1128/aac.42.9.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bugg TDH, Wright GD, Dutka-Malen S, Arthur M, Courvalin P, Walsh CT. Molecular basis of vancomycin resistance in Enterococcus faecium BM4147: biosynthesis of a depsipeptide peptidoglycan precursor by vancomycin resistance proteins VanH and VanA. Biochemistry. 1991;30:10408–10415. doi: 10.1021/bi00107a007. [DOI] [PubMed] [Google Scholar]
- 21.Hong HJ, Hutchings MI, Buttner MJ. Vancomycin resistance VanS/VanR two-component systems. Adv Exp Med Biol. 2008;631:200–213. doi: 10.1007/978-0-387-78885-2_14. [DOI] [PubMed] [Google Scholar]
- 22.Walsh CT. Vancomycin resistance: decoding the molecular logic. Science. 1993;261:308–309. doi: 10.1126/science.8392747. [DOI] [PubMed] [Google Scholar]
- 23.McComas CC, Crowley BM, Boger DL. Partitioning the loss in vancomycin binding affinity for D-Ala-D-Lac into lost H-bond and repulsive lone pair contributions. J Am Chem Soc. 2003;125:9314–9315. doi: 10.1021/ja035901x. [DOI] [PubMed] [Google Scholar]
- 24.Crowley BM, Boger DL. Total synthesis and evaluation of [Ψ[CH2NH]Tpg4]vancomycin aglycon: reengineering vancomycin for dual D-Ala-D-Ala and D-Ala-D-Lac binding. J Am Chem Soc. 2006;128:2885–2892. doi: 10.1021/ja0572912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie J, Pierce JG, James RC, Okano A, Boger DL. A redesigned vancomycin engineered for dual D-Ala-D-Ala and D-Ala-D-Lac binding exhibits potent antimicrobial activity against vancomycin-resistant bacteria. J Am Chem Soc. 2011;133:13946–13949. doi: 10.1021/ja207142h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie J, Okano A, Pierce JG, James RC, Stamm S, Crane CM, Boger DL. Total synthesis of [Ψ[C(=S)NH]Tpg4]vancomycin aglycon, [Ψ[C(=NH)NH]Tpg4]vancomycin aglycon and related key compounds: reengineering vancomycin for dual D-Ala-D-Ala and D-Ala-D-Lac binding. J Am Chem Soc. 2012;134:1284–1297. doi: 10.1021/ja209937s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okano A, Nakayama A, Schammel AW, Boger DL. Total synthesis of [Ψ[C(=NH)NH]Tpg4]vancomycin and its (4-chlorobiphenyl)methyl derivative: impact of peripheral modifications on vancomycin analogs redesigned for dual D-Ala-D-Ala and D-Ala-D-Lac binding. J Am Chem Soc. 2014;136:13522–13525. doi: 10.1021/ja507009a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okano A, Nakayama A, Wu K, Lindsey EA, Schammel AW, Feng Y, Collins KC, Boger DL. Total synthesis and initial examination of [Ψ[C(=S)NH]Tpg4]vancomycin, [Ψ[C(=NH)NH]Tpg4]vancomycin, [Ψ[CH2NH]Tpg4]vancomycin and their (4-chlorobiphenyl)methyl derivatives: synergistic binding pocket and peripheral modifications for the glycopeptide antibiotics. J Am Chem Soc. 2015;137:3693–3704. doi: 10.1021/jacs.5b01008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Okano A, James RC, Pierce JG, Xie J, Boger DL. Silver(I)-promoted conversion of thioamides to amidines: divergent synthesis of a key series of vancomycin aglycon residue 4 amidines that clarify binding behavior to model ligands. J Am Chem Soc. 2012;134:8790–8793. doi: 10.1021/ja302808p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okano A, Isley NA, Boger DL. Total synthesis of vancomycin related glycopeptide antibiotics and key analogues. Chem Rev. 2017:117. doi: 10.1021/acs.chemrev.6b00820. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boger DL, Miyazaki S, Kim SH, Wu JH, Loiseleur O, Castle SL. Diastereoselective total synthesis of the vancomycin aglycon with ordered atropisomer equilibrations. J Am Chem Soc. 1999;121:3226–3227. [Google Scholar]
- 32.Boger DL, Miyasaki S, Kim SH, Wu JH, Castle SL, Loiseleur O, Jin Q. Total synthesis of the vancomycin aglycon. J Am Chem Soc. 1999;121:10004–10011. [Google Scholar]
- 33.Nakayama A, Okano A, Feng Y, Collins JC, Collins KC, Walsh CT, Boger DL. Enzymatic glycosylation of vancomycin aglycon: completion of the total synthesis of vancomycin and N- and C-termini substituent effects of the aglycon substrate. Org Lett. 2014;16:3572–3575. doi: 10.1021/ol501568t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boger DL, Kim SH, Miyazaki S, Strittmatter H, Weng J-H, Mori Y, Rogel O, Castle SL, McAtee JJ. Total synthesis of the teicoplanin aglycon. J Am Chem Soc. 2000;122:7416–7417. doi: 10.1021/ja003835i. [DOI] [PubMed] [Google Scholar]
- 35.Boger DL, Kim SH, Mori Y, Weng J-H, Rogel O, Castle SL, McAtee JJ. First and second generation total synthesis of the teicoplanin aglycon. J Am Chem Soc. 2001;123:1862–1871. doi: 10.1021/ja003835i. [DOI] [PubMed] [Google Scholar]
- 36.Mori Y, McAtee JJ, Rogel O, Boger DL. Alternative synthesis and thermal atropisomerism of a fully functionalized DEFG ring system of teicoplanin. Tetrahedron Lett. 2001;42:6061–6064. [Google Scholar]
- 37.Crowley BM, Mori Y, McComas CC, Tang D, Boger DL. Total synthesis of the ristocetin aglycon. J Am Chem Soc. 2004;126:4310–4317. doi: 10.1021/ja039795a. [DOI] [PubMed] [Google Scholar]
- 38.Garfunkle J, Kimball FS, Trzupek JD, Takazawa S, Shimamura H, Tomishima M, Boger DL. Total synthesis of chloropeptin II (complestatin) and chloropeptin I. J Am Chem Soc. 2009;131:16036–16038. doi: 10.1021/ja907193b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimamura H, Breazzano SP, Garfunkle J, Kimball FS, Trzupek JD, Boger DL. Total synthesis of complestatin: development of a Pd(0)-mediated indole annulation for macrocyclization. J Am Chem Soc. 2010;132:7776–7783. doi: 10.1021/ja102304p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Breazzano SP, Boger DL. Synthesis and stereochemical determination of complestatin A and B (neuroprotection A and B) J Am Chem Soc. 2011;133:18495–18502. doi: 10.1021/ja208570q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okano A, Isley NA, Boger DL. Peripheral modifications of [Ψ[CH2NH]Tpg4]vancomycin with added synergistic mechanisms of action provide durable and potent antibiotics. Proc Natl Acad Sci USA. 2017;114:E5052–E5061. doi: 10.1073/pnas.1704125114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parenti F, Ciabatti R, Cavalleri B, Kettenring J. Ramoplanin: a review of its discovery and its chemitstry. Drugs Exptl Clin Res. 1990;16:451–455. [PubMed] [Google Scholar]
- 43.Walker S, Helm J, Hu Y, Chen L, Rew Y, Shin D, Boger DL. Chemistry and biology of ramoplanin: a lipoglycodepsipeptide with potent antibiotic activity. Chem Rev. 2005;105:449–476. doi: 10.1021/cr030106n. [DOI] [PubMed] [Google Scholar]
- 44.O’Hare MD, Felmingham D, Gruneberg RN. The in vitro activity of ramoplanin (A-16686/MDL-62,198), vancomycin, and teicoplanin against methicillin-susceptible and methicillin-resistant Staphylococcus sp. Drugs Exptl Clin Res. 1988;14:617–619. [PubMed] [Google Scholar]
- 45.Johnson CC, Taylor S, Pitsakis P, May P, Levison ME. Bactericidal activity of ramoplanin against antibiotic-resistant Enterococci. Antimicrob Agents Chemother. 1992;36:2342–2345. doi: 10.1128/aac.36.10.2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Somner EA, Reynolds PE. Inhibition of peptidoglycan biosynthesis by ramoplanin. Antimicrob Agents Chemother. 1990;34:413–419. doi: 10.1128/aac.34.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lo M-p, Men H, Branstrom A, Helm JS, Yao N, Goldman R, Walker S. A new mechanism of action proposed for ramoplanin. J Am Chem Soc. 2000;122:3540–3541. [Google Scholar]
- 48.Helm JS, Chen L, Walker S. Rethinking ramoplanin: the role of substrate binding in inhibition of peptidoglycan biosynthesis. J Am Chem Soc. 2002;124:13970–13971. doi: 10.1021/ja021097n. [DOI] [PubMed] [Google Scholar]
- 49.Fang X, Tiyanont K, Zhang Y, Wanner J, Boger DL, Walker S. The mechanism of action of ramoplanin and enduracidin. Mol BioSystems. 2006;2:69–76. doi: 10.1039/b515328j. [DOI] [PubMed] [Google Scholar]
- 50.Hu Y, Helm JS, Chen L, Ye X-Y, Walker S. Ramoplanin inhibits bacterial transglycosylases by binding as a dimer to Lipid II. J Am Chem Soc. 2003;125:8736–8737. doi: 10.1021/ja035217i. [DOI] [PubMed] [Google Scholar]
- 51.Jiang W, Wanner J, Lee RJ, Bounaud P-Y, Boger DL. Total synthesis of the ramoplanin A2 and ramoplanose aglycon. J Am Chem Soc. 2002;124:5288–5290. doi: 10.1021/ja020237q. [DOI] [PubMed] [Google Scholar]
- 52.Jiang W, Wanner J, Lee RJ, Bounaud P-Y, Boger DL. Total synthesis of the ramoplanin A2 and ramoplanose aglycon. J Am Chem Soc. 2003;125:1877–1887. doi: 10.1021/ja0212314. [DOI] [PubMed] [Google Scholar]
- 53.Shin D, Rew Y, Boger DL. Total synthesis and structure of the ramoplanin A1 and A3 aglycons: two minor components of the ramoplanin complex. Proc Natl Acad Sci USA. 2004;101:11977–11979. doi: 10.1073/pnas.0401419101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rew Y, Shin D, Hwang I, Boger DL. Total synthesis of three key analogs of ramoplanin: a lipoglycodepsipeptide with potent antibiotic activity. J Am Chem Soc. 2004;126:1041–1043. doi: 10.1021/ja039671y. [DOI] [PubMed] [Google Scholar]
- 55.Chen L, Yuan Y, Helm JS, Hu Y, Rew Y, Shin D, Boger DL, Walker S. Dissecting ramoplanin: mechanistic analysis of synthetic ramoplanin analogs as a guide to the design of improved antibiotics. J Am Chem Soc. 2004;126:7462–7463. doi: 10.1021/ja047879t. [DOI] [PubMed] [Google Scholar]
- 56.Nam J, Shin D, Rew Y, Boger DL. Alanine scan of [L-Dap2]ramoplanin A2 aglycon: assessment of the importance of each residue. J Am Chem Soc. 2007;129:8747–8755. doi: 10.1021/ja068573k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fang X, Nam J, Shin D, Rew Y, Boger DL, Walker S. Functional and biochemical analysis of a key series of ramoplanin analogues. Bioorg Med Chem Lett. 2009;19:6189–6191. doi: 10.1016/j.bmcl.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cudic P, Kranz JK, Behenna DC, Kruger RG, Tadesse H, Wand AJ, Veklich YI, Weisel JW, McCafferty DG. Complexation of peptidoglycan intermediates by the lipoglycodepsipeptide antibiotic ramoplanin: minimal structural requirements for intermolecular complexation and fibril formation. Proc Natl Acad Sci USA. 2002;99:7384–7389. doi: 10.1073/pnas.102192099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cudic P, Behenna DC, Kranz JK, Kruger RG, Wand AJ, Veklich YI, Weisel JW, McCafferty DG. Functional analysis of the lipoglycodepsipeptide antibiotic ramoplanin. Chem Biol. 2002;9:897–906. doi: 10.1016/s1074-5521(02)00191-6. [DOI] [PubMed] [Google Scholar]
- 60.Lo M-C, Helm JS, Sarngadharan G, Pelczer I, Walker S. A new structure for the substrate-binding antibiotic ramoplanin. J Am Chem Soc. 2001;123:8640–8641. doi: 10.1021/ja011080p. [DOI] [PubMed] [Google Scholar]
- 61.Hamburger JB, Hoertz AJ, Lee A, Senturla RJ, McCafferty DG, Loll PJ. A crystal structure of a dimer of the antibiotic ramoplanin illustrates membrane positioning and a potential Lipid II docking interface. Proc Natl Acad Sci USA. 2009;106:13759–13764. doi: 10.1073/pnas.0904686106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brossi A, Suffness M, editors. The Alkaloids. Vol. 37 Academic; San Diego, CA: 1990. [Google Scholar]
- 63.Vukovic J, Goodbody AE, Kutney JP, Misawa M. Production of 3′,4′-anhydrovinblastine: a unique chemical synthesis. Tetrahedron. 1988;44:325–331. [Google Scholar]
- 64.Gotoh H, Duncan KK, Robertson WM, Boger DL. 10′-Fluorovinblastine and 10′-fluorovincristine: synthesis of a key series of modified Vinca alkaloids. ACS Med Chem Lett. 2011;2:948–952. doi: 10.1021/ml200236a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gotoh H, Sears JE, Eschenmoser A, Boger DL. New insights into the mechanism and an expanded scope of the Fe(III)-mediated vinblastine coupling reaction. J Am Chem Soc. 2012;134:13240–13243. doi: 10.1021/ja306229x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.We thank Gregory Vite and Robert Borzilleri for arranging and overseeing this assessment and Craig Fairchild, Kathy Johnson, and Russell Peterson for conducting the testing at Bristol–Myers Squibb
- 67.Gigant B, Wang C, Ravelli RBG, Roussi F, Steinmetz MO, Curmi PA, Sobel A, Knossow M. Structural basis for the regulation of tubulin by vinblastine. Nature. 2005;435:519–522. doi: 10.1038/nature03566. [DOI] [PubMed] [Google Scholar]
- 68.Waight AB, Bargsten K, Doronina S, Steinmetz MO, Sussman D, Prota AE. Structural basis of microtubule destabilization by potent auristatin antimitotics. Plos One. 2016;11:e0160890. doi: 10.1371/journal.pone.0160890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carney DW, Lukesh JC, III, Brody DM, Brutsch MM, Boger DL. Ultrapotent vinblastines in which added molecular complexity further disrupts the target tubulin dimer–dimer interface. Proc Natl Acad Sci USA. 2016;113:9691–9698. doi: 10.1073/pnas.1611405113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sears JE, Boger DL. Total synthesis of vinblastine, related natural products, key analogues, and development of inspired methodology suitable for the systematic study of their structure–function properties. Acc Chem Res. 2015;48:653–662. doi: 10.1021/ar500400w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Langlois N, Gueritte F, Langlois Y, Potier P. Application of a modification of the Polonovski reaction to the synthesis of vinblastine-type alkaloids. J Am Chem Soc. 1976;98:7017–7024. doi: 10.1021/ja00438a046. [DOI] [PubMed] [Google Scholar]
- 72.Kutney JP, Hibino T, Jahngen E, Okutani T, Ratcliffe AH, Treasurywala AM, Wunderly S. Total synthesis of indole and dihydroindole alkaloids. IX. Studies on the synthesis of bisindole alkaloids in the vinblastine-vincristine series. The biogenetic approach. Helv Chim Acta. 1976;59:2858–2882. doi: 10.1002/hlca.19760590824. [DOI] [PubMed] [Google Scholar]
- 73.Kuehne ME, Matson PA, Bornmann WG. Enantioselective syntheses of vinblastine, leurosidine, vincovaline and 20′-epi-vincovaline. J Org Chem. 1991;56:513–528. [Google Scholar]
- 74.Bornmann WG, Kuehne ME. A common intermediate providing syntheses of ψ-tabersonine, coronaridine, iboxyphylline, ibophyllidine, vinamidine, and vinblastine. J Org Chem. 1992;57:1752–1760. [Google Scholar]
- 75.Yokoshima S, Ueda T, Kobayashi S, Sato A, Kuboyama T, Tokuyama H, Fukuyama T. Stereocontrolled total synthesis of (+)-vinblastine. J Am Chem Soc. 2002;124:2137–2139. doi: 10.1021/ja0177049. [DOI] [PubMed] [Google Scholar]
- 76.Kuboyama T, Yokoshima S, Tokuyama H, Fukuyama T. Stereocontrolled total synthesis of (+)-vincristine. Proc Natl Acad Sci USA. 2004;101:11966–11970. doi: 10.1073/pnas.0401323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Magnus P, Stamford A, Ladlow M. Synthesis of the antitumor bisindole alkaloid vinblastine: diastereoselectivity and solvent effect on the stereochemistry of the crucial C-15-C-18′ bond. J Am Chem Soc. 1990;112:8210–8212. [Google Scholar]
- 78.Ishikawa H, Colby DA, Boger DL. Direct coupling of catharanthine and vindoline to provide vinblastine: total synthesis of (+)- and ent-(−)-vinblastine. J Am Chem Soc. 2008;130:420–421. doi: 10.1021/ja078192m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ishikawa H, Colby DA, Seto S, Va P, Tam A, Kakei H, Rayl TJ, Hwang I, Boger DL. Total synthesis of vinblastine, vincristine, related natural products, and key structural analogues. J Am Chem Soc. 2009;131:4904–4916. doi: 10.1021/ja809842b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wilkie GD, Elliott GI, Blagg BSJ, Wolkenberg SE, Soenen DB, Miller MM, Pollack S, Boger DL. Intramolecular Diels–Alder and tandem intramolecular Diels–Alder/1,3-dipolar cycloaddition reactions of 1,3,4-oxadiazoles. J Am Chem Soc. 2002;124:11292–11294. doi: 10.1021/ja027533n. [DOI] [PubMed] [Google Scholar]
- 81.Elliott GI, Fuchs JR, Blagg BSJ, Ishikawa H, Tao H, Yuan Z, Boger DL. Intramolecular Diels–Alder/1,3-dipolar cycloaddition cascade of 1,3,4-oxadiazoles. J Am Chem Soc. 2006;128:10589–10595. doi: 10.1021/ja0612549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sears JE, Boger DL. Tandem intramolecular Diels-Alder/1,3-dipolar cycloaddition cascade of 1,3,4-oxadiazoles: initial scope and applications. Acc Chem Res. 2016;49:241–251. doi: 10.1021/acs.accounts.5b00510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ishikawa H, Elliott GI, Velcicky J, Choi Y, Boger DL. Total synthesis of (−)- and ent-(+)-vindoline and related alkaloids. J Am Chem Soc. 2006;128:10596–10612. doi: 10.1021/ja061256t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Choi Y, Ishikawa H, Velcicky J, Elliott GI, Miller MM, Boger DL. Total synthesis of (−)- and ent-(+)-vindoline. Org Lett. 2005;7:4539–4542. doi: 10.1021/ol051975x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Leggans EK, Barker TJ, Duncan KK, Boger DL. Iron(III)/NaBH4-mediated additions to unactivated alkenes: synthesis of novel 20′-vinblastine analogues. Org Lett. 2012;14:1428–1431. doi: 10.1021/ol300173v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Barker TJ, Boger DL. Fe(III)/NaBH4-mediated free radical hydrofluorination of unactivated alkenes. J Am Chem Soc. 2012;134:13588–13591. doi: 10.1021/ja3063716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yuan Z, Ishikawa H, Boger DL. Total synthesis of natural (−)- and ent-(+)-4-desacetoxy-6,7-dihydrovindorosine and natural and ent-minovine: oxadiazole tandem intramolecular Diels–Alder/1,3-dipolar cycloaddition reaction. Org Lett. 2005;7:741–744. doi: 10.1021/ol050017s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Elliott GI, Velcicky J, Ishikawa H, Li Y, Boger DL. Total synthesis of (−)- and ent-(+)-vindorosine: tandem intramolecular Diels–Alder/1,3-dipolar cycloaddition reaction of 1,3,4-oxadiazoles. Angew Chem, Int Ed. 2006;45:620–622. doi: 10.1002/anie.200503024. [DOI] [PubMed] [Google Scholar]
- 89.Ishikawa H, Boger DL. Total synthesis of natural (−)- and ent -(+)-4-desacetoxy-5-desethylvindoline. Heterocycles. 2007;72:95–102. [Google Scholar]
- 90.Sears JE, Barker TJ, Boger DL. Total synthesis of (−)-vindoline and (+)-4-epi-vindoline based on a 1,3,4-oxadiazole tandem intramolecular [4+2]/[3+2] cycloaddition cascade initiated by an allene dienophile. Org Lett. 2015;17:5460–5463. doi: 10.1021/acs.orglett.5b02818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Campbell EL, Skepper CK, Sankar K, Duncan KK, Boger DL. Transannular Diels–Alder/1,3-dipolar cycloaddition cascade of 1,3,4-oxadiazoles: total synthesis of a unique set of vinblastine analogues. Org Lett. 2013;15:5306–5309. doi: 10.1021/ol402549n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yang S, Sankar K, Skepper CK, Barker TJ, Lukesh JC, III, Brody DM, Brutsch MM, Boger DL. Total synthesis of a key series of vinblastines modified at C4 that define the importance and surprising trends in activity. Chem Sci. 2017;8:1560–1569. doi: 10.1039/c6sc04146a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Va P, Campbell EL, Robertson WM, Boger DL. Total synthesis and evaluation of a key series of C5-substituted vinblastine derivatives. J Am Chem Soc. 2010;132:8489–8495. doi: 10.1021/ja1027748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sasaki Y, Kato D, Boger DL. Asymmetric total synthesis of vindorosine, vindoline, and key vinblastine analogues. J Am Chem Soc. 2010;132:13533–13544. doi: 10.1021/ja106284s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kato D, Sasaki Y, Boger DL. Asymmetric total synthesis of vindoline. J Am Chem Soc. 2010;132:3685–3687. doi: 10.1021/ja910695e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schleicher KD, Sasaki Y, Tam A, Kato D, Duncan KK, Boger DL. Total synthesis and evaluation of vinblastine analogues containing systematic deep-seated modifications in the vindoline subunit ring system: core redesign. J Med Chem. 2013;56:483–495. doi: 10.1021/jm3014376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Allemann O, Brütsch M, Lukesh JC, III, Brody DM, Boger DL. Synthesis of a potent vinblastine: rationally designed added benign complexity. J Am Chem Soc. 2016;138:8376–8379. doi: 10.1021/jacs.6b04330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Allemann O, Cross RM, Brütsch MM, Radakovic A, Boger DL. Key analogs of a uniquely potent synthetic vinblastine that contain modifications of the C20′ ethyl substituent. Bioorg Med Chem Lett. 2017;27:3055–3059. doi: 10.1016/j.bmcl.2017.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tam A, Gotoh H, Robertson WM, Boger DL. Catharanthine C16 substituent effects on the biomimetic coupling with vindoline: preparation and evaluation of a key series of vinblastine analogues. Bioorg Med Chem Lett. 2010;20:6408–6410. doi: 10.1016/j.bmcl.2010.09.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Leggans EK, Duncan KK, Barker TJ, Schleicher KD, Boger DL. A remarkable series of vinblastine analogues displaying enhanced activity and an unprecedented tubulin binding steric tolerance: C20′ urea derivatives. J Med Chem. 2013;56:628–639. doi: 10.1021/jm3015684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Barker TJ, Duncan KK, Otrubova K, Boger DL. Potent vinblastine C20′ ureas displaying additionally improved activity against a vinblastine-resistant cancer cell line. ACS Med Chem Lett. 2013;4:985–988. doi: 10.1021/ml400281w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lukesh JC, III, Carney DW, Dong H, Cross RM, Shukla V, Duncan KK, Yang S, Brody DM, Brütsch MM, Radakovic A, Boger DL. Vinblastine 20′ amides: synthetic analogs that maintain or improve potency and simultaneously overcome Pgp-derived efflux and resistance. J Med Chem. 2017;60:7591–7604. doi: 10.1021/acs.jmedchem.7b00958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Boger DL, Brotherton CE. Total synthesis of azafluoranthene alkaloids: rufescine and imelutine. J Org Chem. 1984;49:4050–4055. [Google Scholar]
- 104.Wolkenberg SE, Boger DL. Total synthesis of anhydrolycorinone utilizing sequential intramolecular Diels–Alder reactions of a 1,3,4-oxadiazole. J Org Chem. 2002;67:7361–7364. doi: 10.1021/jo020437k. [DOI] [PubMed] [Google Scholar]
- 105.Lajiness JP, Jiang W, Boger DL. Divergent total syntheses of (−)-aspidospermine and (+)-spegazzinine. Org Lett. 2012;14:2078–2081. doi: 10.1021/ol300599p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Campbell EL, Zuhl AM, Lui CM, Boger DL. Total synthesis of (+)-fendleridine (aspidoalbidine) and (+)-1-acetylaspidoalbidine. J Am Chem Soc. 2010;132:3009–3012. doi: 10.1021/ja908819q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xie J, Wolfe AL, Boger DL. Total synthesis of kopsinine. Org Lett. 2013;15:868–870. doi: 10.1021/ol303573f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lee K, Boger DL. Total syntheses of (−)-kopsifoline D and (−)-deoxoapodine: divergent total synthesis via late-stage key strategic bond formation. J Am Chem Soc. 2014;136:3312–3317. doi: 10.1021/ja500548e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lee K, Boger DL. Total synthesis of (−)-kopsinine and ent-(+)-kopsinine. Tetrahedron. 2015;71:3741–3746. doi: 10.1016/j.tet.2014.07.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Boger DL. Diels–Alder reactions of azadienes. Tetrahedron. 1983;34:2869–2939. [Google Scholar]
- 111.Boger DL. Diels–Alder cycloaddition reactions of heterocyclic azadienes: scope and applications. Chem Rev. 1986;86:781–793. [Google Scholar]
- 112.Boger DL. The duocarmycins: synthetic and mechanistic studies. Acc Chem Res. 1995;28:20–29. [Google Scholar]
- 113.Boger DL, Johnson DS. CC-1065 and the duocarmycins: unraveling the keys to a new class of sequence selective DNA alkylating agents. Proc Natl Acad Sci USA. 1995;92:3642–3649. doi: 10.1073/pnas.92.9.3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Boger DL, Johnson DS. CC-1065 and the duocarmycins: understanding their biological function through mechanistic studies. Angew Chem, Int Ed Engl. 1996;35:1439–1474. [Google Scholar]
- 115.Boger DL, Boyce CW, Garbaccio RM, Goldberg JA. CC-1065 and the duocarmycins: synthetic studies. Chem Rev. 1997;97:787–828. doi: 10.1021/cr960095g. [DOI] [PubMed] [Google Scholar]
- 116.Boger DL, Coleman RS. Diels–Alder cycloaddition of heterocyclic azadienes: total synthesis of PDE-II methyl ester. J Org Chem. 1986;51:3250–3252. [Google Scholar]
- 117.Boger DL, Coleman RS. Diels–Alder reactions of heterocyclic azadienes: total synthesis of PDE-I, PDE-II, and PDE-I dimer methyl ester. J Am Chem Soc. 1987;109:2717–2727. [Google Scholar]
- 118.Boger DL, Coleman RS. Total Synthesis of (+)-CC-1065 and ent-(−)-CC-1065. J Am Chem Soc. 1988;110:1321–1323. [Google Scholar]
- 119.Boger DL, Coleman RS. Total synthesis of (±)-N2-phenylsulfonyl-CPI, (+)-CC-1065, ent-(−)-CC-1065, and the precise, functional agents: (+)-CPI-CDPI2, and (−)-CPI-CDPI2. J Am Chem Soc. 1988;110:4796–4807. [Google Scholar]
- 120.Boger DL, Machiya K. Total synthesis of (+)-duocarmycin SA. J Am Chem Soc. 1992;114:10056–10058. [Google Scholar]
- 121.Boger DL, Machiya K, Hertzog DL, Kitos PA, Holmes D. Total synthesis and preliminary evaluation of (+)- and ent-(−)-duocarmycin SA. J Am Chem Soc. 1993;115:9025–9036. [Google Scholar]
- 122.Boger DL, McKie JA, Nishi T, Ogiku T. Enantioselective total synthesis of (+)-duocarmycin A, epi-(+)-duocarmycin A and their unnatural enantiomers. J Am Chem Soc. 1996;118:2301–2302. [Google Scholar]
- 123.Boger DL, McKie JA, Nishi T, Ogiku T. Total synthesis of (+)-duocarmycin A, epi-(+)-duocarmycin A and their unnatural enantiomers: assessment of chemical and biological properties. J Am Chem. 1997;119:311–325. [Google Scholar]
- 124.Tichenor MS, Kastrinsky DB, Boger DL. Total synthesis, structure revision, and absolute configuration of (+)-yatakemycin. J Am Chem Soc. 2004;126:8396–8398. doi: 10.1021/ja0472735. [DOI] [PubMed] [Google Scholar]
- 125.Tichenor MS, Trzupek JD, Kastrinsky DB, Shiga F, Hwang I, Boger DL. Asymmetric total synthesis of (+)- and ent-(−)-yatakemycin and duocarmycin SA: evaluation of yatakemycin key partial structures and its unnatural enantiomer. J Am Chem Soc. 2006;128:15683–15696. doi: 10.1021/ja064228j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tichenor MS, MacMillan KS, Trzupek JD, Rayl TJ, Hwang I, Boger DL. Systematic exploration of the structural features of yatakemycin impacting DNA alkylation and biological activity. J Am Chem Soc. 2007;129:10858–10869. doi: 10.1021/ja072777z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tichenor MS, Boger DL. Yatakemycin: total synthesis, DNA alkylation, and biological properties. Natural Prod Rep. 2008;25:220–226. doi: 10.1039/b705665f. [DOI] [PubMed] [Google Scholar]
- 128.Boger DL, Ishizaki T, Zarrinmayeh H, Munk SA, Kitos PA, Suntornwat O. Duocarmycin DNA alkylation properties and identification, synthesis and evaluation of agents incorporating the pharmacophore of the duocarmycin alkylation subunit: identification of the CC-1065/duocarmycin common pharmacophore. J Am Chem Soc. 1990;112:8961–8971. [Google Scholar]
- 129.Boger DL, Yun W, Terashima S, Fukuda Y, Nakatani K, Kitos PA, Jin Q. DNA alkylation properties of the duocarmycins: (+)-duocarmycin A, epi-(+)-duocarmycin A, ent-(−)-duocarmycin A, and epi,ent-(−)-duocarmycin A. BioOrg Med Chem Lett. 1992;2:759–765. [Google Scholar]
- 130.Boger DL, Johnson DS, Yun W. (+)- and ent-(−)-Duocarmycin SA and (+)- and ent-(−)-N-Boc-DSA DNA alkylation properties. Alkylation site models that accommodate the offset AT-rich adenine N3 alkylation selectivities of the enantiomeric agents. J Am Chem Soc. 1994;116:1635–1656. [Google Scholar]
- 131.Boger DL, Johnson DS, Yun W, Tarby CM. Molecular basis for sequence selective DNA alkylation by (+)- and ent-(−)-CC-1065 and related agents: alkylation site models that accommodate the offset AT-rich adenine N3 alkylation selectivity. BioOrg Med Chem. 1994;2:115–135. doi: 10.1016/s0968-0896(00)82007-6. [DOI] [PubMed] [Google Scholar]
- 132.Parrish JP, Kastrinsky DB, Wolkenberg SE, Igarashi Y, Boger DL. DNA alkylation properties of yatakemycin. J Am Chem Soc. 2003;125:10971–10976. doi: 10.1021/ja035984h. [DOI] [PubMed] [Google Scholar]
- 133.Trzupek JD, Gottesfeld JM, Boger DL. Sequence-selective alkylation of duplex DNA in nucleosome core particles with duocarmycin SA and yatakemycin. Nat Chem Biol. 2006;2:79–82. doi: 10.1038/nchembio761. [DOI] [PubMed] [Google Scholar]
- 134.Boger DL, Yun W. Reversibility of the duocarmycin A and SA DNA alkylation reaction. J Am Chem Soc. 1993;115:9872–9873. [Google Scholar]
- 135.Boger DL, Ishizaki T, Zarrinmayeh H. Isolation and characterization of the duocarmycin–adenine DNA adduct. J Am Chem Soc. 1991;113:6645–6649. [Google Scholar]
- 136.Boger DL, Mesini P, Tarby CM. Chemical and structural comparison of N-Boc-CBQ and N-Boc-CBI: identification and structural origin of an unappreciated but productive stability of the CC-1065 and duocarmycin SA alkylation subunits. J Am Chem Soc. 1994;116:6461–6462. [Google Scholar]
- 137.Boger DL, Mesini P. Design, synthesis and evaluation of CC-1065 and duocarmycin analogs incorporating the 2,3,10,10a-tetrahydro-1H-cyclopropa[d]benzo[f]quinol-5-one (CBQ) alkylation subunit: identification and structural origin of subtle stereoelectronic features that govern reactivity and regioselectivity. J Am Chem Soc. 1994;116:11335–11348. [Google Scholar]
- 138.Boger DL, Mesini P. DNA alkylation properties of CC-1065 and duocarmycin analogs incorporating the 2,3,10,10a-tetrahydrocyclopropa[d]benzo[f]quinol-5-one (CBQ) alkylation subunit: identification of subtle structural features that contribute to the regioselectivity of the adenine N3 alkylation reaction. J Am Chem Soc. 1995;117:11647–11655. [Google Scholar]
- 139.Boger DL, Turnbull P. Synthesis and evaluation of CC-1065 and duocarmycin analogues incorporating the 1,2,3,4,11,11a-hexahydrocyclopropa[c]naphtho[2,1-b]azepin-6-one (CNA) alkylation subunit: structural features that govern reactivity and reaction regioselectivity. J Org Chem. 1997;62:5849–5863. [Google Scholar]
- 140.Boger DL, Coleman RS, Invergo BJ, Zarrinmayeh H, Kitos PA, Thompson SC, Leong T, McLaughlin LW. A demonstration of the intrinsic importance of stabilizing hydrophobic binding and noncovalent van der Waals contacts dominant in the noncovalent CC-1065 DNA binding. Chem-Biol Interactions. 1990;73:29–52. doi: 10.1016/0009-2797(90)90107-x. [DOI] [PubMed] [Google Scholar]
- 141.Boger DL, Coleman RS, Invergo BJ, Sakya SM, Ishizaki T, Munk SA, Zarrinmayeh H, Kitos PA, Thompson SC. Synthesis and evaluation of aborted and extended CC-1065 functional analogs: (+)- and (−)-CPI-PDE1; (+)- and (−)-CPI-CDPI1; and (+)- and (−)-CPI-CDPI3. Preparation of key partial structures and definition of an additional functional role of the CC-1065 central and right-hand subunits. J Am Chem Soc. 1990;112:4623–4632. [Google Scholar]
- 142.Boger DL, Zarrinmayeh H, Munk SA, Kitos PA, Suntornwat O. Demonstration of a pronounced effect on noncovalent binding selectivity on the sequence-selectivity of the (+)-CC-1065 DNA alkylation and identification of the pharmacophore of the alkylation subunit. Proc Natl Acad Sci USA. 1991;88:1431–1435. doi: 10.1073/pnas.88.4.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Boger DL, Munk SA, Zarrinmayeh H. The (+)-CC-1065 DNA alkylation: key studies demonstrating a noncovalent binding selectivity contribution to the alkylation selectivity. J Am Chem Soc. 1991;113:3980–3983. [Google Scholar]
- 144.Boger DL, Johnson DS. Second definitive test of proposed models for the origin of the CC-1065 and duocarmycin DNA alkylation selectivity. J Am Chem Soc. 1995;117:1443–1444. [Google Scholar]
- 145.Boger DL, Garbaccio RM. Shape-dependent catalysis: insights into the source of catalysis for the CC-1065 and duocarmycin DNA alkylation reaction. Acc Chem Res. 1999;32:1043–1052. [Google Scholar]
- 146.Boger DL, Garbaccio RM. Catalysis of the CC-1065 and duocarmycin DNA alkylation reaction: DNA binding induced conformational change in the agent results in activation. BioOrg Med Chem. 1997;5:263–276. doi: 10.1016/s0968-0896(96)00238-6. [DOI] [PubMed] [Google Scholar]
- 147.Boger DL, Hertzog DL, Bollinger B, Johnson DS, Cai H, Goldberg J, Turnbull P. Duocarmycin SA shortened, simplified, and extended agents: a systematic examination of the role of the DNA binding subunit. J Am Chem Soc. 1997;119:4977–4986. [Google Scholar]
- 148.Boger DL, Bollinger B, Hertzog DL, Johnson DS, Cai H, Mésini P, Garbaccio RM, Jin Q, Kitos PA. Reversed and sandwiched analogs of duocarmycin SA: establishment of the origin of the sequence selective alkylation of DNA and new insights into the source of catalysis. J Am Chem Soc. 1997;119:4987–4998. [Google Scholar]
- 149.Boger DL, Turnbull P. Synthesis and evaluation of a carbocyclic analog of the CC-1065 and duocarmycin alkylation subunits: role of the vinylogous amide and implications on DNA alkylation catalysis. J Org Chem. 1998;63:8004–8011. [Google Scholar]
- 150.Boger DL, Garbaccio RM. Are the duocarmycin and CC-1065 DNA alkylation reactions acid-catalyzed? Solvolysis pH rate profiles suggest they are not. J Org Chem. 1999;64:5666–5669. doi: 10.1021/jo990762g. [DOI] [PubMed] [Google Scholar]
- 151.Ambroise Y, Boger DL. The DNA phosphate backbone is not involved in catalysis of the duocarmycin and CC-1065 DNA alkylation reaction. BioOrg Med Chem Lett. 2002;12:303–306. doi: 10.1016/s0960-894x(01)00740-5. [DOI] [PubMed] [Google Scholar]
- 152.Wolfe AL, Duncan KK, Lajiness JP, Zhu K, Deurfeldt AS, Boger DL. A fundamental relationship between hydrophobic properties and biological activity for the duocarmycin class of DNA alkylating antitumor drugs: hydrophobic binding-driven-bonding. J Med Chem. 2013;56:6845–6857. doi: 10.1021/jm400665c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Eis PG, Smith JA, Rydzewski MJ, Case DA, Boger DL, Chazin WJ. High resolution structure of a DNA duplex alkylated by the antitumor agent duocarmycin SA. J Mol Biol. 1997;272:237–252. doi: 10.1006/jmbi.1997.1223. [DOI] [PubMed] [Google Scholar]
- 154.Schnell JR, Ketchem RR, Boger DL, Chazin WJ. DNA binding-induced alkylation: insights from the structure of a DNA duplex alkylated by the indole derivative of duocarmycin SA. J Am Chem Soc. 1999;121:5645–5652. [Google Scholar]
- 155.Smith JA, Bifulco G, Case DA, Boger DL, Gomez-Paloma L, Chazin WJ. The structural basis for in situ activation of DNA alkylation by duocarmycin SA. J Mol Biol. 2000;300:1195–1204. doi: 10.1006/jmbi.2000.3887. [DOI] [PubMed] [Google Scholar]
- 156.Wolkenberg SE, Boger DL. Mechanisms of in situ activation of DNA targeting antitumor agents. Chem Rev. 2002;102:2477–2496. doi: 10.1021/cr010046q. [DOI] [PubMed] [Google Scholar]
- 157.Boger DL, Munk SA, Zarrinmayeh H, Ishizaki T, Haught J, Bina M. An alternative and convenient strategy for generation of substantial quantities of singly 5′-32P-end-labeled double-stranded DNA for binding studies: development of a protocol for the examination of the functional features of (+)-CC-1065 and the duocarmycins that contribute to their sequence-selective DNA binding properties. Tetrahedron. 1991;47:2661–2682. [PubMed] [Google Scholar]
- 158.MacMillan KS, Boger DL. Fundamental relationships between structure, reactivity, and biological activity for the duocarmycins and CC-1065. J Med Chem. 2009;52:5771–5780. doi: 10.1021/jm9006214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Boger DL, Ishizaki T, Wysocki RJ, Jr, Munk SA, Kitos PA, Suntornwat O. Total synthesis and evaluation of (±)-N-(tert-butyloxycarbonyl)-CBI, (±)-CBI-CDPI1, and (±)-CBI-CDPI2: CC-1065 functional analogs incorporating the equivalent 1,2,9,9a-tetrahydrocycloprop[1,2-c]benz[1,2-e]indol-4-one (CBI) left-hand subunit. J Am Chem Soc. 1989;111:6461–6463. [Google Scholar]
- 160.Boger DL, Ishizaki T, Kitos PA, Suntornwat O. Synthesis of N-(tert-butyloxycarbonyl)-CBI, CBI, CBI-CDPI1, and CBI-CDPI2: enhanced functional analogs incorporating the 1,2,9,9a-tetrahydrocycloprop[1,2-c]benz[1,2-e]indol-4-one (CBI) left-hand subunit. J Org Chem. 1990;55:5823–5832. [Google Scholar]
- 161.Boger DL, Ishizaki T, Sakya SM, Munk SA, Kitos PA, Jin Q, Besterman JM. Synthesis and preliminary evaluation of (+)-CBI-indole2: an enhanced functional analog of (+)-CC-1065. BioOrg Med Chem Lett. 1991;1:115–120. [Google Scholar]
- 162.Boger DL, Munk SA. DNA alkylation properties of CBI-based agents: enhanced functional analogs of CC-1065 incorporating the 1,2,9,9a-tetrahydrocycloprop[1,2-c]benz[1,2-e]indol-4-one (CBI) alkylation subunit. J Am Chem Soc. 1992;114:5487–5496. [Google Scholar]
- 163.Boger DL, Yun W, Cai H, Han N. CBI-CDPBO1 and CBI-CDPBI1: CC-1065 analogs containing deep-seated modifications in the DNA binding subunits. BioOrg Med Chem. 1995;3:761–775. doi: 10.1016/0968-0896(95)00066-p. [DOI] [PubMed] [Google Scholar]
- 164.Boger DL, Yun W, Han N. 1,2,9,9a-Tetrahydrocyclopropa[c]benz[e]indol-4-one (CBI) analogs of CC-1065 and the duocarmycins: synthesis and evaluation. BioOrg Med Chem. 1995;3:1492–1453. doi: 10.1016/0968-0896(95)00130-9. [DOI] [PubMed] [Google Scholar]
- 165.Boger DL, Yun W, Teegarden BR. An improved synthesis of 1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one (CBI): a simplified analog of the CC-1065 alkylation subunit. J Org Chem. 1992;57:2873–2876. [Google Scholar]
- 166.Boger DL, McKie JA. An efficient synthesis of CBI: an enhanced and simplified analog of the CC-1065 and duocarmycin alkylation subunits. J Org Chem. 1995;60:1271–1275. [Google Scholar]
- 167.Boger DL, McKie JA, Boyce CW. Asymmetric synthesis of the CBI alkylation subunit of the CC-1065 and duocarmycin analogs. Synlett. 1997:515–517. [Google Scholar]
- 168.Boger DL, Boyce CW, Garbaccio RM, Searcey M. Synthesis of CC-1065/duocarmycin analogs via intramolecular aryl radical cyclization of a tethered vinyl chloride. Tetrahedron Lett. 1998;39:2227–2230. [Google Scholar]
- 169.Kastrinsky DB, Boger DL. Effective asymmetric synthesis of CBI. J Org Chem. 2004;69:2284–2289. doi: 10.1021/jo035465x. [DOI] [PubMed] [Google Scholar]
- 170.Lajiness JP, Boger DL. Asymmetric synthesis of 1,2,9,9a-tetrahydrocyclopropa[c]benzo-[e]indol-4-one (CBI) J Org Chem. 2011;76:583–587. doi: 10.1021/jo102136w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Boger DL, Coleman RS. Total Synthesis of (+)- and (−)-CPI-CDPI2: (+)-3bR,4aS- and (−)-3bS,4aR-deoxy CC-1065. J Org Chem. 1988;53:695–698. [Google Scholar]
- 172.Boger DL, Wysocki RJ., Jr Total synthesis of (±)-N-benzenesulfonyl- and (±)-N-(tert-butyloxycarbonyl)-CI, (±)-CI-CDPI1, and (±)-CI-CDPI2: CC-1065 functional analogs incorporating the parent 1,2,7,7a-tetrahydrocycloprop[1,2-c]indol-4-one (CI) left-hand subunit. J Org Chem. 1989;54:1238–1240. [Google Scholar]
- 173.Boger DL, Wysocki RJ, Jr, Ishizaki T. Synthesis of N-phenylsulfonyl-CI, N-(tert-butyloxycarbonyl)-CI, CI-CDPI1, and CI-CDPI2: CC-1065 functional analogs incorporating the parent 1,2,7,7a-tetrahydrocycloprop[1,2-c]indol-4-one (CI) left-hand subunit. J Am Chem Soc. 1990;112:5230–5240. [Google Scholar]
- 174.Boger DL, Ishizaki T, Zarrinmayeh H, Kitos PA, Suntornwat O. Synthesis and preliminary evaluation of agents incorporating the pharmacophore of the duocarmycin/pyrindamycin alkylation subunit: Identification of the CC-1065/duocarmycin common pharmacophore. J Org Chem. 1990;55:4499–4502. [Google Scholar]
- 175.Boger DL, Palanki MS. Functional analogs of CC-1065 and the duocarmycins incorporating the 9a-chloromethyl-1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one (C2BI) alkylation subunit: Synthesis and preliminary DNA alkylation studies. J Am Chem Soc. 1992;114:9318–9327. doi: 10.1016/s0968-0896(00)82100-8. [DOI] [PubMed] [Google Scholar]
- 176.Boger DL, Johnson DS, Palanki MSS, Kitos PA, Chang J, Dowell P. Evaluation of functional analogs of CC-1065 and the duocarmycins incorporating the cross-linking 9a-chloromethyl-1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indole-4-one (C2BI) alkylation subunit. BioOrg Med Chem. 1993;1:27–38. doi: 10.1016/s0968-0896(00)82100-8. [DOI] [PubMed] [Google Scholar]
- 177.Boger DL, McKie JA, Cai H, Cacciari B, Baraldi PG. Synthesis and properties of substituted CBI analogs of CC-1065 and the duocarmycins incorporating the 7-methoxy-1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one (MCBI) alkylation subunit: magnitude of electronic effects on the functional reactivity. J Org Chem. 1996;61:1710–1729. doi: 10.1021/jo952033g. [DOI] [PubMed] [Google Scholar]
- 178.Boger DL, Han N, Tarby CM, Boyce CW, Cai H, Jin Q, Kitos PA. Synthesis, chemical properties, and preliminary evaluation of substituted CBI analogs of CC-1065 and the duocarmycins incorporating the 7-cyano-1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one (CCBI) alkylation subunit: Hammett quantitation of the magnitude of electronic effects on functional reactivity. J Org Chem. 1996;61:4894–4912. doi: 10.1021/jo952033g. [DOI] [PubMed] [Google Scholar]
- 179.Boger DL, Hughes TV, Hedrick MP. Synthesis, chemical properties, and biological evaluation of CC-1065 and duocarmycin analogues incorporating the 5-methoxycarbonyl-1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one alkylation subunit. J Org Chem. 2001;66:2207–2216. doi: 10.1021/jo001772g. [DOI] [PubMed] [Google Scholar]
- 180.Boger DL, Jenkins TJ. Synthesis, X-ray structure, and properties of fluorocyclopropane analogs of the duocarmycins incorporating the 9,9-difluoro-1,2,9,9a-tetrahydrocyclopropa-[c]benz[e]indol-4-one (F2CBI) alkylation subunit. J Am Chem Soc. 1996;118:8860–8870. [Google Scholar]
- 181.Boger DL, Garbaccio RM, Jin Q. Synthesis and evaluation of CC-1065 and duocarmycin analogs incorporating the iso-CI and iso-CBI alkylation subunits: impact of relocation of the C-4 carbonyl. J Org Chem. 1997;62:8875–8891. [Google Scholar]
- 182.Boger DL, Garbaccio RM. A novel class of CC-1065 and duocarmycin analogs subject to mitomycin related reductive activation. J Org Chem. 1999;64:8350–8362. doi: 10.1021/jo991301y. [DOI] [PubMed] [Google Scholar]
- 183.Parrish JP, Kastrinsky DB, Stauffer F, Hedrick MP, Hwang I, Boger DL. Establishment of substituent effects in the DNA binding subunit of CBI analogues of the duocarmycins and CC-1065. BioOrg Med Chem. 2003;11:3815–3838. doi: 10.1016/s0968-0896(03)00194-9. [DOI] [PubMed] [Google Scholar]
- 184.Parrish JP, Kastrinsky DB, Boger DL. Synthesis and x-ray analysis of an unprecedented and stable 2-aza-4,4-spirocyclopropacyclohexadienone. Org Lett. 2003;5:2577–2579. doi: 10.1021/ol035000t. [DOI] [PubMed] [Google Scholar]
- 185.Parrish JP, Kastrinsky DB, Hwang I, Boger DL. Synthesis and evaluation of duocarmycin and CC-1065 analogues incorporating the 1,2,9,9a-tetrahydrocyclopropa[c]benz[e]-3-azaindole-4-one (CBA) alkylation subunit. J Org Chem. 2003;68:8984–8990. doi: 10.1021/jo035119f. [DOI] [PubMed] [Google Scholar]
- 186.Gauss CM, Hamasaki A, Parrish JP, MacMillan KS, Rayl TJ, Hwang I, Boger DL. Synthesis and preliminary evaluation of duocarmycin analogues incorporating the 1,2,11,11a-tetrahydrocyclopropa[c]naphtho[2,3-e]indol-4-one (CNI) and 1,2,11,11a-tetrahydrocyclopropa-[c]naphtha[1,2-e]indol-4-one (iso-CNI) alkylation subunits. Tetrahedron. 2009;65:6591–6599. doi: 10.1016/j.tet.2009.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Mohamadi F, Spees MM, Staten GS, Marder P, Kipka JK, Johnson DA, Boger DL, Zarrinmayeh H. Total synthesis and biological properties of novel antineoplastic (chloromethyl)furanoindolines: an asymmetric hydroboration mediated synthesis of the alkylation subunits. J Med Chem. 1994;37:232–239. doi: 10.1021/jm00028a005. [DOI] [PubMed] [Google Scholar]
- 188.Boger DL, Santillán A, Jr, Searcey M, Jin Q. The critical role of the linking amide in CC-1065 and the duocarmycins: implications on the source of DNA alkylation catalysis. J Am Chem Soc. 1998;120:11554–11557. [Google Scholar]
- 189.Boger DL, Santillán A, Jr, Searcey M, Jin Q. Synthesis and evaluation of duocarmycin and CC-1065 analogues containing modifications in the subunit linking amide. J Org Chem. 1999;64:5241–5244. doi: 10.1021/jo990452y. [DOI] [PubMed] [Google Scholar]
- 190.Baraldi PG, Cacciari B, Romagnoli R, Spalluto G, Boyce CW, Boger DL. Resolution of a CPzI precursor, synthesis and biological evaluation of (+)- and (−)-N-Boc-CPzI: a further validation of the relationship between chemical solvolytic stability and cytotoxicity. BioOrg Med Chem Lett. 1999;9:3087–3092. doi: 10.1016/s0960-894x(99)00533-8. [DOI] [PubMed] [Google Scholar]
- 191.Boger DL, Boyce CW. Selective metal cation activation of a DNA alkylating agent: synthesis and evaluation of methyl 1,2,9,9a-tetrahydrocyclopropa[c]pyrido[3,2-e]indol-4-one-7-carboxylate. J Org Chem. 2000;65:4088–4100. doi: 10.1021/jo000177b. [DOI] [PubMed] [Google Scholar]
- 192.Boger DL, Santillan A, Jr, Searcey M, Brunette SR, Wolkenberg SE, Hedrick MP, Jin Q. Synthesis and evaluation of 1,2,8,8a-tetrahydrocyclopropa[c]pyrrolo[3,2-e]indol-4(5H)-one, the parent alkylation subunit of CC-1065 and the duocarmycins: impact of the alkylation subunit substituents and its implications for DNA alkylation catalysis. J Org Chem. 2000;65:4101–4111. doi: 10.1021/jo000297j. [DOI] [PubMed] [Google Scholar]
- 193.Boger DL, Wolkenberg SE, Boyce CW. A new method of in situ activation for a novel class of DNA alkylating agents: tunable metal cation complexation and activation. J Am Chem Soc. 2000;122:6328–6326. [Google Scholar]
- 194.Boger DL, Ellis DA, Wolkenberg SE. Reversed CPyI analogues of CC-1065 and duocarmycin SA. Metal cation complexation and activation of a sequence selective DNA alkylating agent: partitioning the effects of binding and catalysis. J Am Chem Soc. 2001;123:9299–9306. doi: 10.1021/ja010769r. [DOI] [PubMed] [Google Scholar]
- 195.Boger DL, Brunette SR, Garbaccio RM. Synthesis and evaluation of a series of C3-substituted CBI analogues of CC-1065 and the duocarmycins. J Org Chem. 2001;66:5163–5173. doi: 10.1021/jo010309g. [DOI] [PubMed] [Google Scholar]
- 196.MacMillan KS, Boger DL. An additional spirocyclization for duocarmycin SA. J Am Chem Soc. 2008;130:16521–16523. doi: 10.1021/ja806593w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.MacMillan KS, Nguyen T, Hwang I, Boger DL. Total synthesis and evaluation of iso-duocarmycin SA and iso-yatakemycin. J Am Chem Soc. 2009;131:1187–1194. doi: 10.1021/ja808108q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.MacMillan KS, Lajiness JP, Lopez-Cara C, Romagnoli R, Robertson WM, Hwang I, Baraldi PG, Boger DL. Synthesis and evaluation of a thio analogue of duocarmycin SA. BioOrg Med Chem Lett. 2009;19:6962–6965. doi: 10.1016/j.bmcl.2009.10.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Lajiness JP, Boger DL. Synthesis and characterization of a cyclobutane duocarmycin derivative incorporating the CbBI (1,2,10,11-tetrahydro-9H-cyclobuta[c]benzo[e]indol-4-one) alkylation subunit. J Am Chem Soc. 2010;132:13936–13940. doi: 10.1021/ja106986f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Boyle KE, MacMillan KS, Ellis DA, Lajiness JP, Robertson WM, Boger DL. Synthesis and evaluation of duocarmycin SA analogs incorporating the methyl 1,2,8,8a-tetrahydrocyclopropa[c]oxazolo[2,3-e]indole-4-one-6-carboxylate (COI) alkylation subunit. BioOrg Med Chem Lett. 2010;20:1854–1857. doi: 10.1016/j.bmcl.2010.01.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Robertson WM, Kastrinsky DB, Hwang I, Boger DL. Synthesis and evaluation of a series of C5′-substituted duocarmycin SA analogs. BioOrg Med Chem Lett. 2010;20:2722–2725. doi: 10.1016/j.bmcl.2010.03.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Chanda PB, Boyle KE, Brody DM, Shukla V, Boger DL. Synthesis and evaluation of duocarmycin SA analogs incorporating the methyl 1,2,8,8a-tetrahydrocyclopropa[c]imidazole-[4,5-e]indol-4-one-6-carboxylate (CImI) alkylation subunit. BioOrg Med Chem. 2016;24:4779–4786. doi: 10.1016/j.bmc.2016.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Parrish JP, Hughes TV, Hwang I, Boger DL. Establishing the parabolic relationship between reactivity and activity for derivatives and analogues of the duocarmycin and CC-1065 alkylation subunits. J Am Chem Soc. 2004;126:80–81. doi: 10.1021/ja038162t. [DOI] [PubMed] [Google Scholar]
- 204.Boger DL, Yun W. Role of the CC-1065 and duocarmycin N2 substituent: validation of a direct relationship between solvolysis chemical stability and in vitro biological potency. J Am Chem Soc. 1994;116:5523–5524. [Google Scholar]
- 205.Boger DL, Munk SA, Ishizaki T. The (+)-CC-1065 DNA alkylation: observation of an unexpected relationship between cyclopropane electrophile reactivity and the intensity of DNA alkylation. J Am Chem Soc. 1991;113:2779–2780. [Google Scholar]
- 206.Boger DL, Yun W. CBI-TMI: synthesis and evaluation of a key analog of the duocarmycins. Validation of a direct relationship between chemical solvolytic stability and cytotoxic potency and confirmation of the structural features responsible for the distinguishing behavior of enantiomeric pairs of agents. J Am Chem Soc. 1994;116:7996–8006. [Google Scholar]
- 207.Tichenor MS, MacMillan KS, Stover JS, Wolkenberg SE, Pavani MG, Zanella L, Zaid AN, Spalluto G, Rayl TJ, Hwang I, Baraldi PG, Boger DL. Rational design, synthesis, and evaluation of key analogues of CC-1065 and the duocarmycins. J Am Chem Soc. 2007;129:14092–14099. doi: 10.1021/ja073989z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Warpehoski MA, Gebhard I, Kelly RC, Krueger WC, Li LH, McGovren JP, Prairie MD, Wicnienski N, Wierenga W. Stereoelectronic factors influencing the biological activity and DNA interaction of synthetic antitumor agents modeled on CC-1065. J Med Chem. 1988;31:590–603. doi: 10.1021/jm00398a017. [DOI] [PubMed] [Google Scholar]
- 209.Wolfe AL, Duncan KK, Parelkar NK, Brown D, Vielhauer GA, Boger DL. Efficacious cyclic N-acyl O-amino phenol duocarmycin prodrugs. J Med Chem. 2013;56:4104–4115. doi: 10.1021/jm400413r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Lajiness JP, Robertson WM, Dunwiddie I, Broward MA, Vielhauer GA, Weir SJ, Boger DL. Design, synthesis, and evaluation of CC-1065 and duocarmycin O-aminophenol prodrugs subject to tunable reductive activation. J Med Chem. 2010;53:7731–7738. doi: 10.1021/jm1010397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Jin W, Trzupek JD, Rayl TJ, Broward MA, Vielhauer GA, Weir SJ, Hwang I, Boger DL. A unique class of duocarmycin and CC-1065 analogues subject to reductive activation. J Am Chem Soc. 2007;129:15391–15397. doi: 10.1021/ja075398e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Uematsu M, Boger DL. Asymmetric synthesis of a CBI-based cyclic N-acyl O-amino phenol duocarmycin prodrug. J Org Chem. 2014;79:9699–9703. doi: 10.1021/jo501839x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Wolfe AL, Duncan KK, Parelkar N, Weir SJ, Vielhauer GA, Boger DL. A novel, unusually efficacious duocarmycin carbamate prodrug that releases no residual byproduct. J Med Chem. 2012;55:5878–5886. doi: 10.1021/jm300330b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Li LH, Kelly RC, Warpehoski MA, McGovren JP, Gebhard I, DeKoning TF. Adozelesin, a selected lead among cyclopropylpyrroloindole analogs of the DNA-binding antibiotic, CC-1065. Invest New Drugs. 1991;9:137–148. doi: 10.1007/BF00175081. [DOI] [PubMed] [Google Scholar]
- 215.Wierenga W, Bhuyan BK, Kelly RC, Krueger WC, Li LH, McGovren JP, Swenson DH, Warpehoski M. Antitumor activity and biochemistry of novel analogs of CC-1065. Adv Enzyme Reg. 1986;25:141–155. doi: 10.1016/0065-2571(86)90012-9. [DOI] [PubMed] [Google Scholar]
- 216.Amishiro N, Nagamura S, Kobayashi E, Gomi K, Saito H. New water soluble duocarmycin derivatives: synthesis and antitumor activity of A-ring pyrrole compounds bearing β-heteroarylacryloyl groups. J Med Chem. 1999;42:669–676. doi: 10.1021/jm980559y. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 217.Li LH, DeKoning TF, Kelly RC, Krueger WC, McGovren JP, Padbury GE, Petzold GL, Wallace TL, Ouding RJ, Prairie MD, Gebhard I. Cytotoxicity and antitumor activity of carzelesin, a prodrug cyclopropylpyrroloindole analogue. Cancer Res. 1992;52:4904–4913. [PubMed] [Google Scholar]
- 218.Walker DL, Reid JM, Ames MM. Preclinical pharmacology of bizelesin, a potent bifunctional analog of the DNA-binding antibiotic CC-1065. Cancer Chemother Pharmacol. 1994;34:317–322. doi: 10.1007/BF00686039. [DOI] [PubMed] [Google Scholar]
- 219.Kobayashi E, Okamoto A, Asada M, Okabe M, Nagamura S, Asai A, Saito H, Gomi K, Hirata T. Characteristics of antitumor activity of KW-2189, a novel water-soluble derivative of duocarmycin, against murine and human tumors. Cancer Res. 1994;54:2404–2410. [PubMed] [Google Scholar]
- 220.Hecht SM, editor. Bleomycin: Chemical, Biochemical, and Biological Aspects. Springer-Verlag; New York: 1979. [Google Scholar]
- 221.Hecht SM. The chemistry of activated bleomycin. Acc Chem Res. 1986;19:383–391. [Google Scholar]
- 222.Stubbe J, Kozarich JW. Mechanisms of bleomycin-induced DNA degradation. Chem Rev. 1987;87:1107–1136. [Google Scholar]
- 223.Boger DL, Cai H. Bleomycin: synthetic and mechanistic studies. Angew Chem, Int Ed. 1999;38:448–476. doi: 10.1002/(SICI)1521-3773(19990215)38:4<448::AID-ANIE448>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 224.Tse WC, Boger DL. Sequence-selective DNA recognition: natural products and Nature’s lessons. Chem Biol. 2004;11:1607–1617. doi: 10.1016/j.chembiol.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 225.Boger DL, Menezes RF, Honda T. Total synthesis of (−)-pyrimidoblamic acid and deglycobleomycin A2. Angew Chem, Int Ed Engl. 1993;32:273–275. [Google Scholar]
- 226.Boger DL, Honda T, Menezes RF, Colletti SL, Dang Q, Yang W. Total synthesis of (+)-P-3A, epi-(−)-P-3A, and (−)-desacetamido P-3A. J Am Chem Soc. 1994;116:82–92. [Google Scholar]
- 227.Duerfeldt AS, Boger DL. Total synthesis of (−)-pyrimidoblamic acid and P-3A. J Am Chem Soc. 2014;136:2119–2125. doi: 10.1021/ja412298c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Boger DL, Colletti SL, Honda T, Menezes RF. Total synthesis of bleomycin A2 and related agents. 1. Synthesis and DNA binding properties of the extended C-terminus: tripeptide S, tetrapeptide S, pentapeptide S, and related agents. J Am Chem Soc. 1994;116:5607–5618. [Google Scholar]
- 229.Boger DL, Honda T, Dang Q. Total synthesis of bleomycin A2 and related agents. 2. Synthesis of (−)-pyrimidoblamic acid, epi-(+)-pyrimidoblamic acid, (+)-desacetamidopyrimidoblamic acid, and (−)-descarboxamidopyrimidoblamic acid. J Am Chem Soc. 1994;116:5619–5630. [Google Scholar]
- 230.Boger DL, Honda T, Menezes RF, Colletti SL. Total synthesis of bleomycin A2 and related agents. 3. Synthesis and comparative evaluation of deglycobleomycin A2, epi-deglycobleomycin A2, deglycobleomycin A1, desacetamido, descarboxamido, desmethyl and desimidazole deglycobleomycin A2. J Am Chem Soc. 1994;116:5631–5646. [Google Scholar]
- 231.Boger DL, Honda T. Total synthesis of bleomycin A2 and related agents. 4. Synthesis of the disaccharide subunit: 2-O-(3-O-carbamoyl-α-D-mannopyranosyl)-L-gulopyranose and completion of the total synthesis of bleomycin A2. J Am Chem Soc. 1994;116:5647–5656. [Google Scholar]
- 232.Boger DL, Menezes RF, Dang Q. Synthesis of desacetamidopyrimidoblamic acid and deglyco desacetamidobleomycin A2. J Org Chem. 1992;57:4333–4336. [Google Scholar]
- 233.Boger DL, Teramoto S, Honda T, Zhou J. Synthesis and evaluation of the fully functionalized bleomycin A2 metal binding domain containing the 2-O-(3-O-carbamoyl-α-D-mannopyranosyl)-α-L-gulopyranosyl disaccharide. J Am Chem Soc. 1995;117:7338–7343. [Google Scholar]
- 234.Boger DL, Teramoto S, Zhou J. Key synthetic analogs of bleomycin A2 that directly address the effect and role of the disaccharide: demannosylbleomycin A2 and α-D-mannopyranosyldeglycobleomycin A2. J Am Chem Soc. 1995;117:7344–7356. [Google Scholar]
- 235.Boger DL, Colletti SL, Teramoto S, Ramsey TM, Zhou J. Synthesis of key analogs of bleomycin A2 that permit a systematic evaluation of the linker region: identification of an exceptionally prominent role for the L-threonine substituent. BioOrg Med Chem. 1995;3:1281–1295. doi: 10.1016/0968-0896(95)00113-u. [DOI] [PubMed] [Google Scholar]
- 236.Boger DL, Teramoto S, Cai H. Synthesis and evaluation deglycobleomycin A2 analogs containing a tertiary N-methyl amide and simple ester replacement for the L-histidine secondary amide: direct functional characterization of the requirement for secondary amide metal complexation. BioOrg Med Chem. 1996;4:179–194. doi: 10.1016/0968-0896(95)00183-2. [DOI] [PubMed] [Google Scholar]
- 237.Boger DL, Ramsey TM, Cai H. Synthesis and Evaluation of potential Nπ and Nσ metal chelation sites within the β-hydroxy-L-histidine subunit of bleomycin A2: functional characterization of imidazole Nπ metal complexation. BioOrg Med Chem. 1996;4:195–208. doi: 10.1016/0968-0896(95)00184-0. [DOI] [PubMed] [Google Scholar]
- 238.Boger DL, Teramoto S, Cai H. N-Methyl-L-threonine analogs of deglycobleomycin A2: synthesis and evaluation. BioOrg Med Chem. 1997;5:1573–1590. doi: 10.1016/s0968-0896(97)00107-7. [DOI] [PubMed] [Google Scholar]
- 239.Ramsey TM, Cai H, Hoehn ST, Kozarich JW, Stubbe J, Boger DL. Assessment of the role of the bleomycin A2 pyrimidoblamic acid C4 amino group. J Am Chem Soc. 1998;120:55–65. [Google Scholar]
- 240.Boger DL, Ramsey TM, Cai H, Hoehn ST, Stubbe J. A systematic evaluation of the bleomycin A2 L-threonine side chain: its role in preorganization of a compact conformation implicated in sequence selective DNA cleavage. J Am Chem Soc. 1998;120:9139–9148. [Google Scholar]
- 241.Boger DL, Ramsey TM, Cai H, Hoehn ST, Stubbe J. Definition of the effect and role of the bleomycin A2 valerate substituents: preorganization of a rigid, compact conformation implicated in sequence selective DNA cleavage. J Am Chem Soc. 1998;120:9149–9158. [Google Scholar]
- 242.Boger DL, Aquila BM, Tse WC, Searcey M. Synthesis and evaluation of a novel bleomycin A2 analogue: continuing assessment of the linker domain. Tetrahedron Lett. 2000;41:9493–9498. [Google Scholar]
- 243.Wu W, Vanderwall DE, Teramoto S, Lui SM, Hoehn ST, Tang X-J, Turner CJ, Boger DL, Kozarich JW, Stubbe J. NMR studies of Co·deglycobleomycin A2 green and its complex with oligonucleotide d(CCAGGCCTGG)2. J Am Chem Soc. 1998;120:2239–2250. [Google Scholar]
- 244.Boger DL, Fink BE, Brunette SR, Tse WC, Hedrick MP. A simple, high resolution method for establishing DNA binding affinity and sequence selectivity. J Am Chem Soc. 2001;123:5878–5891. doi: 10.1021/ja010041a. [DOI] [PubMed] [Google Scholar]
- 245.Tse WC, Boger DL. A fluorescent intercalator displacement (FID) assay for establishing DNA binding selectivity and affinity. Acc Chem Res. 2004;37:61–69. doi: 10.1021/ar030113y. [DOI] [PubMed] [Google Scholar]
- 246.Stubbe J, Kozarich JW, Wu W, Vanderwall DE. Bleomycins: a structural model for specificity, binding, and double strand cleavage. Acc Chem Res. 1996;29:322–330. [Google Scholar]
- 247.Trost BM, Knopf JD, Brindle CS. Synthetic strategies employed for the construction of fostriecin and related natural products. Chem Rev. 2016;116:15035–15088. doi: 10.1021/acs.chemrev.6b00488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Lewy D, Gauss C-M, Soenen DR, Boger DL. Fostriecin: chemistry and biology. Curr Med Chem. 2002;9:2005–2032. doi: 10.2174/0929867023368809. [DOI] [PubMed] [Google Scholar]
- 249.Boger DL, Hikota M, Lewis BM. Determination of the relative and absolute stereochemistry of fostriecin. J Org Chem. 1997;62:1748–1753. [Google Scholar]
- 250.Boger DL, Ichikawa S, Zhong W. Total synthesis of fostriecin (CI-920) J Am Chem Soc. 2001;123:4161–4167. doi: 10.1021/ja010195q. [DOI] [PubMed] [Google Scholar]
- 251.Buck SB, Hardouin C, Ishikawa S, Soenen DR, Gauss C-M, Hwang I, Swingle MR, Bonness KM, Honkanen RE, Boger DL. Fundamental role of the fostriecin unsaturated lactone and implications for selective protein phosphatase inhibition. J Am Chem Soc. 2003;125:15694–15695. doi: 10.1021/ja038672n. [DOI] [PubMed] [Google Scholar]
- 252.Lawhorn BG, Boga SB, Wolkenberg SE, Colby DA, Gauss C-M, Swingle MR, Amable L, Honkanen RE, Boger DL. Total synthesis and evaluation of cytostatin, its C10–C11 diastereomers, and additional key analogues: impact on PP2A inhibition. J Am Chem Soc. 2006;128:16720–16732. doi: 10.1021/ja066477d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Swingle MB, Amable L, Lawhorn BG, Buck SB, Burke CP, Ratti P, Fisher KL, Boger DL, Honkanen RE. Structure-activity relationship studies of fostriecin, cytostatin, and key analogs with PP1, PP2A, PP5 and (β12 – β13)-chimeras (PP1/PP2A and PP5/PP2A) provide further insight into inhibitory actions of fostriecin family inhibitors. J Pharmacol Exp Ther. 2009;331:45–53. doi: 10.1124/jpet.109.155630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Burke CP, Haq N, Boger DL. Total synthesis, assignment of the relative and absolute stereochemistry, and structural reassignment of phostriecin (aka sultriecin) J Am Chem Soc. 2010;132:2157–2159. doi: 10.1021/ja9097252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Burke CP, Swingle MR, Honkanen RE, Boger DL. Total synthesis and evaluation of phostriecin and key structural analogues. J Org Chem. 2010;75:7505–7513. doi: 10.1021/jo1010203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Cravatt BF, Lerner RA, Boger DL. Structure determination of an endogenous sleep-inducing lipid, cis-9-octadecenoamide (oleamide): a synthetic approach to the chemical analysis of trace quantities of a natural product. J Am Chem Soc. 1996;118:580–590. [Google Scholar]
- 257.Cravatt BF, Prospero–Garcia O, Suizdak G, Gilula NB, Henriksen SJ, Boger DL, Lerner RA. Chemical characterization of a family of brain lipids that induce sleep. Science. 1995;268:1506–1509. doi: 10.1126/science.7770779. [DOI] [PubMed] [Google Scholar]
- 258.Lerner RA, Siuzdak G, Prospero–Garcia O, Henriksen SJ, Boger DL, Cravatt BF. Cerebrodiene: a brain lipid isolated from sleep-deprived cats. Proc Natl Acad Sci USA. 1994;91:9505–9508. doi: 10.1073/pnas.91.20.9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Boger DL, Henriksen SJ, Cravatt BF. Oleamide: an endogenous sleep-inducing lipid and prototypical member of a new class of biological signaling molecules. Curr Pharm Des. 1998;4:303–314. [PubMed] [Google Scholar]
- 260.Ezzili C, Otrubova K, Boger DL. Fatty acid amide signaling molecules. BioOrg Med Chem Lett. 2010;20:5959–5968. doi: 10.1016/j.bmcl.2010.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 262.Wilcox BJ, Ritenour–Rodgers KJ, Asser AS, Baumgart LE, Baumgart MA, Boger DL, Patterson JE, DeBlassio JL, deLong MA, Glufke U, Henz ME, King L, III, Merkler KA, Robleski JJ, Vederas JC, Merkler DJ. N-Acylglycine amidation: implications for the biosynthesis of fatty acid primary amides. Biochemistry. 1999;38:3235–3245. doi: 10.1021/bi982255j. [DOI] [PubMed] [Google Scholar]
- 263.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 264.Giang DK, Cravatt BF. Molecular characterization of human and mouse fatty acid amide hydrolase. Proc Natl Acad Sci USA. 1997;94:2238–2242. doi: 10.1073/pnas.94.6.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.McKinney MK, Cravatt BF. Structure and function of fatty acid amide hydrolase. Ann Rev Biochem. 2005;74:411–432. doi: 10.1146/annurev.biochem.74.082803.133450. [DOI] [PubMed] [Google Scholar]
- 266.Boger DL, Fecik RA, Patterson JE, Miyauchi H, Patricelli MP, Cravatt BF. Fatty acid amide hydrolase substrate specificity. BioOrg Med Chem Lett. 2000;10:2613–2616. doi: 10.1016/s0960-894x(00)00528-x. [DOI] [PubMed] [Google Scholar]
- 267.Leung D, Hardouin C, Boger DL, Cravatt BF. Discovering potent and selective reversible inhibitors of enzymes in complex proteomes. Nat Biotechnol. 2003;21:687–691. doi: 10.1038/nbt826. [DOI] [PubMed] [Google Scholar]
- 268.Liu Y, Patricelli MP, Cravatt BF. Activity-based protein profiling: the serine hydrolases. Proc Natl Acad Sci USA. 1999;96:14694–14699. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Kidd D, Liu Y, Cravatt BF. Profiling serine hydrolase activity in complex proteomes. Biochemistry. 2001;40:4005–4015. doi: 10.1021/bi002579j. [DOI] [PubMed] [Google Scholar]
- 270.Mileni M, Garfunkle J, DeMartino JK, Cravatt BF, Boger DLStevens RC. Binding and inactivation mechanism of a humanized fatty acid amide hydrolase by α-ketoheterocycle inhibitors revealed from cocrystal structures. J Am Chem Soc. 2009;131:10497–10506. doi: 10.1021/ja902694n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Mileni M, Garfunkle J, Ezzili C, Kimball FS, Cravatt BF, Stevens RC, Boger DL. X-ray crystallographic analysis of α-ketoheterocycle inhibitors bound to a humanized variant of fatty acid amide hydrolase. J Med Chem. 2010;53:230–240. doi: 10.1021/jm9012196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Mileni M, Garfunkle J, Ezzili C, Cravatt BF, Stevens R C, Boger DL. Fluoride-mediated capture of a noncovalent bound state of a reversible covalent enzyme inhibitor: X-ray crystallographic analysis of an exceptionally potent α-ketoheterocycle inhibitor of fatty acid amide hydrolase. J Am Chem Soc. 2011;133:4092–4100. doi: 10.1021/ja110877y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Boger DL, Miyauchi H, Du W, Hardouin C, Fecik RA, Cheng H, Hwang I, Hedrick MP, Leung D, Acevedo O, Guimaráes CRW, Jorgensen WL, Cravatt BF. Discovery of a potent, selective, and efficacious class of reversible α-ketoheterocycle inhibitors of fatty acid amide hydrolase as analgesics. J Med Chem. 2005;48:1849–1856. doi: 10.1021/jm049614v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Lichtman AH, Leung D, Shelton CC, Saghatelian A, Hardouin C, Boger DL, Cravatt BF. Reversible inhibitors of fatty acid amide hydrolase that promote analgesia: evidence for an unprecedented combination of potency and selectivity. J Pharmacol Exp Ther. 2004;311:441–448. doi: 10.1124/jpet.104.069401. [DOI] [PubMed] [Google Scholar]
- 275.Chang L, Luo L, Palmer JA, Sutton S, Wilson SJ, Barbier AJ, Breitenbucher JG, Chaplan SR, Webb M. Inhibition of fatty acid amide hydrolase produces analgesia by multiple mechanisms. Br J Pharmacol. 2006;148:102–113. doi: 10.1038/sj.bjp.0706699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Kinsey SG, Long JZ, O’Neal ST, Abdulla RA, Poklis JL, Boger DL, Cravatt BF, Lichtman AH. Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. J Pharmacol Exp Ther. 2009;330:902–910. doi: 10.1124/jpet.109.155465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Schlosburg JE, Boger DL, Cravatt BF, Lichtman AH. Endocannabinoid modulation of scratching response in an acute allergenic model: new prospective neural therapeutic target for pruritus. J Pharmacol Exp Ther. 2009;329:314–323. doi: 10.1124/jpet.108.150136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Booker L, Kinsey SG, Abdullah RA, Blankman JL, Long JZ, Ezzili C, Boger DL, Cravatt BF, Lichtman AH. The FAAH inhibitor PF-3845 acts in the nervous system to reverse lipopolysaccharide-induced tactile allodynia in mice. Br J Pharmacol. 2012;165:2485–2496. doi: 10.1111/j.1476-5381.2011.01445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Cravatt BF, Lichtman AH. Fatty acid amide hydrolase: an emerging therapeutic target in the endocannabinoid system. Curr Opin Chem Biol. 2003;7:469–775. doi: 10.1016/s1367-5931(03)00079-6. [DOI] [PubMed] [Google Scholar]
- 280.Ahn K, McKinney MK, Cravatt BF. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem Rev. 2008;108:1687–1707. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Ahn K, Johnson DS, Cravatt BF. Fatty acid amide hydrolase as a potential therapeutic target for the treatment of pain and CNS disorders. Exp Opin Drug Discov. 2009;4:763–784. doi: 10.1517/17460440903018857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Seierstad M, Breitenbucher JG. Discovery and development of fatty acid amide hydrolase (FAAH) inhibitors. J Med Chem. 2008;51:7327–7343. doi: 10.1021/jm800311k. [DOI] [PubMed] [Google Scholar]
- 283.Deng HF. Fatty acid amide hydrolase as a therapeutic target. Exp Opin Drug Discov. 2010;5:961–970. doi: 10.1517/17460441.2010.513378. [DOI] [PubMed] [Google Scholar]
- 284.Otrubova K, Ezzili C, Boger DL. The discovery and development of inhibitors of fatty acid amide hydrolase. BioOrg Med Chem Lett. 2011;21:4674–4685. doi: 10.1016/j.bmcl.2011.06.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Patterson JE, Ollmann IR, Cravatt BF, Boger DL, Wong C-H, Lerner RA. Inhibition of oleamide hydrolase catalyzed hydrolysis of the endogenous sleep-inducing lipid:cis-9-octadecenamide. J Am Chem Soc. 1996;118:5938–5945. [Google Scholar]
- 286.Boger DL, Sato H, Lerner AE, Austin BJ, Patterson JE, Patricelli MP, Cravatt BF. Trifluoromethyl ketone inhibitors of fatty acid amide hydrolase: a probe of structural and conformational features contributing to inhibition. BioOrg Med Chem Lett. 1999;9:265–270. doi: 10.1016/s0960-894x(98)00734-3. [DOI] [PubMed] [Google Scholar]
- 287.Edwards PD, Meyer EFJ, Vijayalakshmi J, Tuthill PA, Andisik DA, Gomes B, Strimpler A. Design, synthesis, and kinetic evaluation of a unique class of elastase inhibitors, the peptidyl α-ketooxazoles, the x-ray crystal structure of the covalent complex between porcine pancreatic elastase and Ac-Ala-Pro-Val-2-benzoxazole. J Am Chem Soc. 1992;114:1854–1863. [Google Scholar]
- 288.Maryanoff BE, Costanzo MJ. Inhibitors of proteases and amide hydrolases that employ an alpha-ketoheterocycle as a key enabling functionality. BioOrg Med Chem. 2008;16:1562–1595. doi: 10.1016/j.bmc.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 289.Otrubova K, Boger DL. α-Ketoheterocycle-based inhibitors of fatty acid amide hydrolase. ACS Chem Neurosci. 2012;3:340–348. doi: 10.1021/cn2001206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Boger DL, Sato H, Lerner AE, Hedrick MP, Fecik RA, Miyauchi H, Wilkie GD, Austin BJ, Patricelli MP, Cravatt BF. Exceptionally potent inhibitors of fatty acid amide hydrolase: the enzyme responsible for degradation of endogenous oleamide and anandamide. Proc Natl Acad Sci USA. 2000;97:5044–5049. doi: 10.1073/pnas.97.10.5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Boger DL, Miyauchi H, Hedrick MP. α-Keto heterocycle inhibitors of fatty acid amide hydrolase: carbonyl group modification and α-substitution. BioOrg Med Chem Lett. 2001;11:1517–1520. doi: 10.1016/s0960-894x(01)00211-6. [DOI] [PubMed] [Google Scholar]
- 292.Leung D, Du W, Hardouin C, Cheng H, Hwang I, Cravatt BF, Boger DL. Discovery of an exceptionally potent and selective class of fatty acid amide hydrolase inhibitors enlisting proteome-wide selectivity screening: concurrent optimization of enzyme inhibitor potency and selectivity. BioOrg Med Chem Lett. 2005;15:1423–1428. doi: 10.1016/j.bmcl.2004.12.085. [DOI] [PubMed] [Google Scholar]
- 293.Romero FA, Hwang I, Boger DL. Delineation of a fundamental α-ketoheterocycle substituent effect for use in the design of enzyme inhibitors. J Am Chem Soc. 2006;68:14004–14005. doi: 10.1021/ja064522b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.DeMartino JK, Garfunkle J, Hochstatter DG, Cravatt BF, Boger DL. Exploration of a fundamental substituent effect of α-ketoheterocycle enzyme inhibitors: potent and selective inhibitors of fatty acid amide hydrolase. BioOrg Med Chem Lett. 2008;18:5842–5846. doi: 10.1016/j.bmcl.2008.06.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Romero FA, Du W, Hwang I, Rayl TJ, Kimball FS, Leung D, Hoover HS, Apodaca RL, Breitenbucher JG, Cravatt BF, Boger DL. Potent and selective α-ketoheterocycle-based inhibitors of the anandamide and oleamide catabolizing enzyme, fatty acid amide hydrolase. J Med Chem. 2007;50:1058–1068. doi: 10.1021/jm0611509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Hardouin C, Kelso MJ, Romero FA, Rayl TJ, Leung D, Hwang I, Cravatt BF, Boger DL. Structure–activity relationships of α-ketooxazole inhibitors of fatty acid amide hydrolase. J Med Chem. 2007;50:3359–3368. doi: 10.1021/jm061414r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Kimball FS, Romero FA, Ezzili C, Garfunkle J, Rayl TJ, Hochstatter DG, Hwang I, Boger DL. Optimization of α-ketooxazole inhibitors of fatty acid amide hydrolase. J Med Chem. 2008;51:937–947. doi: 10.1021/jm701210y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Garfunkle J, Ezzili C, Rayl TJ, Hochstatter DG, Hwang I, Boger DL. Optimization of the central heterocycle of α-ketoheterocycle inhibitors of fatty acid amide hydrolase. J Med Chem. 2008;51:4393–4403. doi: 10.1021/jm800136b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Ezzili C, Mileni M, McGlinchey N, Long JZ, Kinsey SG, Hochstatter DG, Stevens RC, Lichtman AH, Cravatt BF, Bilsky EJ, Boger DL. Reversible competitive α-ketoheterocycle inhibitors of fatty acid amide hydrolase containing additional conformational constraints in the acyl side chain: orally active, long acting analgesics. J Med Chem. 2011;54:2805–2822. doi: 10.1021/jm101597x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Duncan KK, Otrubova K, Boger DL. α-Ketoheterocycle inhibitors of fatty acid amide hydrolase: exploration of conformational contraints in the acyl side chain. Bioorg Med Chem. 2014;22:2763–2770. doi: 10.1016/j.bmc.2014.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Otrubova K, Brown M, McCormick MS, Han GW, O’Neal ST, Cravatt BF, Stevens RC, Lichtman AH, Boger DL. Rational design of fatty acid amide hydrolase inhibitors that act by covalently bonding to two active site residues. J Am Chem Soc. 2013;135:6289–6299. doi: 10.1021/ja4014997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Otrubova K, Cravatt BF, Boger DL. Design, synthesis and characterization of α-ketoheterocycles additionally targeting the cytosolic port Cys269 of fatty acid amide hydrolase. J Med Chem. 2014;57:1079–1089. doi: 10.1021/jm401820q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Guimaráes CRW, Boger DL, Jorgensen WL. Elucidation of fatty acid amide hydrolase inhibition by potent α-ketoheterocycle derivatives from Monte Carlo simulations. J Am Chem Soc. 2005;127:17377–17384. doi: 10.1021/ja055438j. [DOI] [PubMed] [Google Scholar]
- 304.Janssen FJ, Baggelaar MP, Hummel JJA, Overkleeft HS, Cravatt BF, Boger DL, van der Stelt M. Comprehensive analysis of structure activity relationships of α-ketoheterocycles as sn-1-diacylglycerol lipase α inhibitors. J Med Chem. 2015;58:9742–9753. doi: 10.1021/acs.jmedchem.5b01627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Some destinations are incapable of being reached. J Sollenberger. 2017 on El SoSo, NM (aka el 5050) [Google Scholar]