

Abstract
Z-K8 (2), the racemic form of isochaihulactone (1), previously showed significant antitumor effects in A549 and LNCaP tumor-bearing mice. In the present study, 17 derivatives of 2, were designed, synthesized and evaluated for anti-proliferative activity against four human tumor cell lines. All new derivatives exhibited high potency against A549 and P-glycoprotein (P-gp)-overexpressing KBvin. One of our new derivative exhibited greater activity against three tested tumor cells (A549, KB, and KB-VIN) than 2, and induced cell cycle arrest in the G2/M phase. Moreover, SAR conclusions were first established for this series of compounds. Our study clearly identified a structural feature that should be retained for good activity and also a moiety that can tolerate various modifications and, thus, is ideal for further changes.
Graphical Abstract
Seventeen new derivatives of (±)-isochaihulactone were synthesized. SAR conclusions were first established for isochaihulactone-type compounds.
1. Introduction
Bupleurum scorzonerifolium (Nan-Chai-Hu), a well-known Chinese herbal medicine, has a long history in the treatment of influenza, fever, cancer, and menstrual disorders.1 Isochaihulactone (1, Figure 1), a dibenzylbutyrolactone lignan, is the main active constituent found in the root of B. scorzonerifolium. Compound 1 showed potent antitumor activity against various tumor cells as well as inhibition of tubulin polymerization, resulting in induction of cell cycle arrest at G2/M and apoptosis.2,3 Especially, 1 exhibited potent antitumor activity against A549 (lung adenocarcinoma) cells in vitro and in vivo, inhibiting A549 xenograft growth in nude mice at doses of 30 and 50 mg/kg. Moreover, in histology examinations and serology testing, no significant adverse effects were found on heart, liver, brain bone marrow, or kidney upon treatment with 1, even at a high dose of 50 mg/kg.2 Further study indicated that 1 induces growth inhibition and apoptosis in A549 cells via activation of early growth response gene 1 (EGR-1) and non-steroidal anti-inflammatory drug-activated gene 1 (NAG-1) through an extracellular signal-regulated kinase 1/2 (ERK 1/2)-dependent pathway, without involving PI3K signaling.4 Furthermore, 1 enhanced paclitaxel-induced apoptotic death in human lung cells by increasing NAG-1 expression.5 Compound 1 also induced cell death in prostate cancer cells, in addition to lung cancer cells, through activation of EGR-1 and NAG-1 via a JNK-dependent pathway, without activation of ERK signaling.6 However, the molecular basis for the interaction mechanism of 1 with tubulin or EGR-1 or NAG-1 remains unclear.
Figure 1.

Structures of isochaihulactone (1), Z-K8 (2), E-K8 (3), etoposide (4), yatein (5), and (−)-parabenzlactone (6).
Harn’s group synthesized (±)-isochaihulactone (Z-K8, 2, Figure 1) and the E form of 2 (E-K8, 3, Figure 1), which possesses an E-configured alkene at C-2(5), via eight steps.7 Furthermore, they found that 2 was more potent than 1 against various tumor cells in vitro, acting by a similar mechanism of action to that of 1. Moreover, 2 also exhibited significant antitumor effects in A549 and LNcaP tumor-bearing mice. On the other hand, E-K8 (3) showed significantly decreased antitumor activity compared with its Z-form (2) in A549 or LNcaP (androgen-sensitive human prostate adenocarcinoma) cell lines.7–9 These studies indicate that isochaihulactone (1) and its racemic form 2 have promising therapeutic potential for cancer treatment.
Despite their potent in vitro and in vivo antitumor activity and novel mechanism of action, studies on new derivatives of 1 and its racemic product 2 are rather limited. Only one paper reported the synthesis of six C-5 hydroxylated analogues of 1. Four of the six analogues showed activity against breast cancer with over 50% inhibition at concentrations less than 50 μg/mL.10
As part of our continuing effort to discover and develop new natural-product inspired anticancer drug candidates, we initiated a study on the design and synthesis of new derivatives of 2. The structures of 1 and 2 feature three rings including a piperonyl ring (ring A), γ-butyrolactone (ring B), and 3,4,5-trimethoxybenzyl moiety (ring C), one Z-configured alkene at C-2(5), and a stereogenic center at C-3 (Figure 2). Among these features, the piperonyl group and γ-butyrolactone can be found in many bioactive agents, including etoposide (4, Figure 1), yatein (5, Figure 1) and (−)-parabenzlactone (6, Figure 1),11,12 and thus, may be considered as conserved domains for the bioactivity of this compound type. Meanwhile, benzyl rings with different substituents are found in 4–6, indicating a potential for further modification of the ring C moiety. Furthermore, based on the lower activity of 3 versus 2,7–9 the Z-configured alkene at C-2(5) is likely critical for the potent activity. Meanwhile, the configuration of C-3 could be a less important factor, based on the higher potency of 2 (racemic) versus 1 (non-racemic).
Figure 2.

Design rationale
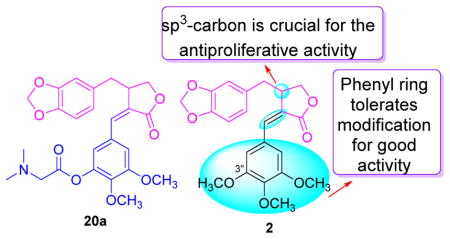
On the basis of the above background data, we designed two series of new derivatives (Figure 2) retaining the piperonyl moeity, γ-butyrolactone, and Z-configured alkene at C-2(5). In series A, the phenyl ring C was substituted with various groups with different steric and electronic properties as well as hydrophobicity. In series B, in addition to analogous modifications on ring C, the stereogenic center at C-3 was converted to an alkene at C-3(6) to give derivatives with a more simplified and rigid structure. We expected this design strategy to lead to the discovery of new derivatives with good activity. Moreover, we also explored the following two questions regarding the structure-activity relationship (SAR) of 1-type compounds. First, whether the benzyl moiety (ring C) will tolerate modification to produce new derivatives with improved drug profiles. Second, whether the sp3-carbon at C-3 could be removed to produce new simplified derivatives with good activity. Herein, we report the design, synthesis, anti-proliferative screening, and SAR study of 1-type compounds.
2. Result and discussion
2.1 Chemistry
A concise synthetic route featuring the Stobbe reaction as a key step was developed to obtain all target compounds. As shown in Scheme 1, a Stobbe condensation13 between diethyl succinate (8) and piperonal (7) gave 9, in which the ester group non-adjacent to the reacting carbon was selectively removed via a typical Stobbe condensation mechanism to leave a carboxylic acid. With Super-H as the reducing reagent, the ester group of 9 was selectively reduced to a hydroxyl group, which then was cyclized with the carboxylic acid in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) to form the key γ-butyrolactone product 10. The methodology was modified from the first synthesis of this known intermediate of podophyllotoxin analogs performed by Lewis and Popper.14 Aldol addition of various substituted benzaldehydes to 10, followed by Pd/C catalyzed hydrogenation, and dehydration using methanesulfonyl chloride (MsCl), produced compounds 2 and 12a–14a (Series A). Compounds 11b–14b (Series B) were obtained via Aldol condensation and dehydration, without the hydrogenation step. Subsequently, the methoxymethyl (MOM) group in 13a–14a and 13b–14b was removed with pyridinium p-toluenesulfonate (PPTS) to produce compounds 15a–16a and 15b–16b, respectively.
Scheme 1.

Synthesis of 2 and its derivatives including 12a–16a (series A) and 11b–16b (series B). (a) tBuOK; (b) (i) LiBHEt3, THF; (ii) EDCI, DMF; (c). (i) CH3ONa, CH3OH, substituted benzaldehyde (ii) H2/Pd-C (only for 2, 12a–14a); (iii) MsCl, pyr (only for 11b–14b); (d) PPTS, THF
Furthermore, condensation of 15a and 16a with Boc-L-phenylalanine, followed by reaction with 2.0 M HCl in diethyl ether gave 17a and 18a, respectively. The coupling of 15b and 16b with Boc-L-phenylalanine followed by deprotection with TFA yielded 17b and 18b, respectively. Compounds 19a–20a, and 19b–20b were obtained from condensation of N,N-dimethylglycine with 15a–16a, and 15b–16b, respectively. Further treatment of 19a–20a with 2M HCl gave products 21a and 22a (Scheme 2).
Scheme 2.

Synthesis of new derivatives 17a–22a (Series A) and 17b–20b (Series B). (a) (i) Boc-L-phenylalanine (17a-b and 18a-b) or N,N-dimethylglycine (for 19a–22a and 19b–20b), EDCI, DMAP; (ii) 2M HCl in diethyl ether (for 17a–18a and 21a–22a) or TFA, DCM (for 17b–18b)
The configurations of the generated alkenes were determined from the NOESY spectroscopic data of 2 from Series A and 11b from series B. In compound 2, the NOESY correlations of H-5 (δ 6.30 ppm) to H-6 (δ 2.52, 2.39 ppm), and H-5 to H-3 (δ 2.76 ppm) clearly indicated a Z configuration at C2(5). In compound 11b, the NOESY correlation of H-6 (δ 5.81 ppm) to H-4 (δ 3.81 ppm) suggested a Z configuration of the alkene at C3(6). Furthermore, NOE correlations of H-5 (δ 6.01 ppm) with H-6′ (δ 6.71–6.70 ppm) indicated a Z configuration of the alkene at C2(5).
2.2 Antiproliferative activity and SAR study
The 17 designed target compounds together with 2 were screened for their anti-proliferative activity against four tumor cell lines, A549 (lung adenocarcinoma), MDA-MB-231[triple-negative ER-/PgR-/erbB2 (HER2)− breast cancer], KB (originally isolated as a nasopharyngeal carcinoma), and multidrug-resistant (MDR) KB subline KB-VIN overexpressing P-glycoprotein (P-gp), by the sulforhodamine B (SRB) colorimetric assay with 0.2% DMSO as a (negative) control (0% growth inhibition with 0.2% DMSO).15 Paclitaxel was used as the positive reference. In previous studies,6 compound 2 exhibited potent activity in vitro and in vivo against A549 and LNCaP cells. Consistent with these results, compound 2 also showed potent activity in our study against the four tested cell lines with IC50 values ranging from 5.50 to 9.90 μM (Table 1). Moreover, compound 2 was more potent against A549, MDA-MB-231, and KB-VIN compared with KB.
Table 1.
Cytotoxicity of new derivatives of 21
| Compound | IC50 (μM)
|
|||
|---|---|---|---|---|
| A549 | MDA-MB-231 | KB | KB-VIN | |
| 2 | 5.50 ± 0.064 | 5.88 ± 0.10 | 9.90 ± 0.133 | 5.51 ± 0.16 |
| 12a | 5.72 ± 0.66 | 8.81 ± 0.46 | 6.47 ± 0.421 | 5.51 ± 0.29 |
| 15a | 6.17 ± 0.17 | > 40 | 11.5 ±1.94 | 6.78 ± 1.46 |
| 16a | 4.32 ± 0.093 | 8.55 ± 0.72 | 5.18 ± 0.160 | 4.84 ± 0.28 |
| 17a | 5.97 ±0.45 | 9.73 ± 0.084 | 9.20 ± 1.05 | 7.59 ± 0.44 |
| 18a | 4.80 ± 0.51 | 8.80 ± 0.16 | 5.40 ± 0.366 | 4.95 ± 0.35 |
| 19a | 5.93 ± 0.35 | 9.37 ± 0.29 | 8.71 ± 1.31 | 7.06 ± 0.39 |
| 20a | 3.32 ± 1.03 | 9.04 ± 1.32 | 4.98 ± 0.269 | 4.51 ± 0.50 |
| 21a | 6.15 ± 0.72 | 29.00 ± 2.87 | 18.10 ± 2.06 | 17.60 ± 1.06 |
| 22a | 5.02 ± 0.37 | 8.91 ± 0.44 | 5.53 ± 0.041 | 5.34 ± 0.37 |
| 11b | 35.17 ± 0.99 | > 40 | 34.0 ± 0.569 | 28.30 ± 2.04 |
| 12b | > 40 | > 40 | > 40 | > 40 |
| 15b | > 40 | > 40 | > 40 | > 40 |
| 16b | 31.24 ± 0.045 | > 40 | 34.0 ± 0.674 | 29.04 ± 2.07 |
| 17b | > 40 | > 40 | > 40 | > 40 |
| 18b | > 40 | > 40 | > 40 | > 40 |
| 19b | > 40 | > 40 | > 40 | > 40 |
| 20b | > 40 | > 40 | > 40 | > 40 |
| paclitaxel (nM) | 5.82 ± 0.19 | 5.34 ± 0.068 | 7.52 ± 0.020 | 1650 ± 55.70 |
Data are presented as the mean ± standard deviation (SD) determined in three separate experiments with duplicate assays
The structures of the nine derivatives in series A featured a modified phenyl C ring and a sp3-carbon at C-3. As shown in Table 1, seven (12a, 16a–20a, 22a) derivatives in series A exhibited significant to moderate activity (IC50 3.32–9.73 μM) against the four tested cell lines, while two (15a, 21a) showed lower activity (IC50 > 10 μM) against at least one cell line. All new series A derivatives were more or as potent against KB-VIN compared with KB and, unlike 2, were least potent against MDA-MB-231 among the four cell lines tested. Generally, all derivatives in Series A exhibited the following general rank order of activity: A549 > KB-VIN > KB > MDA-MB-231, which is different from that of 2: A549 > KB-VIN > MDA-MB-231> KB. Moreover, the new compounds generally displayed greater, equivalent, or slightly lower potency compared with 2 against A549, KB, or KB-VIN, which indicated that the phenyl C ring tolerates modification for good activity against these cell lines.
Regarding SAR correlations identified among series A derivatives, 4″-fluoro-3″-methyl substituted compound 12a, as compared with 2, exhibited equivalent activity against A549 and KB-VIN cells, obviously improved activity against KB, and decreased activity against MDA-MB-231. Replacement of the 3″-OCH3 or 4″-OCH3 of 2 with 3″-OH or 4″-OH yielded 16a and 15a, respectively. Compared with 2, compound 16a exhibited improved activity against A549, KB, and KB-VIN with IC50 values between 4.32 and 5.18 μM. However, compound 15a showed somewhat decreased potency compared with 2 against A549, KB, and KB-VIN. Esterification of the 3″-OH of 16a with N,N-dimethylglycine yielded 20a, which showed the greatest potencies (IC50 3.32–4.98 μM) against A549, KB, and KB-VIN among all tested compounds. Moreover, the tertiary amine present in 20a can readily form quaternary amine salts. Accordingly, reaction of 20a with 2M HCl produced 22a, the hydrochloride salt of 20a, which exhibited improved activity compared with 2 against A549, KB, and KB-VIN cells (IC50 5.02–5.35 μM), although 22a was slightly less potent than 20a. Moreover, compound 18a with L-phenylalanine hydrochloride as the C-3″ ester also exhibited better activity than 2 against A549, KB, and KB-VIN. Conjugation of L-phenylalanine or N,N-dimethylglycine to the 4″-OH of 15a gave 17a, 19a, and 21a, which exhibited somewhat decreased activity (IC50 5.93–29.0 μM) against the four tested cell lines, particularly when compared with the corresponding 3″-esters (compare 17a vs 18a, 19a vs 20a, 21a vs 22a). These results clearly indicated that various structural variations on the phenyl C ring are tolerated for good activity against A549, KB, and KB-VIN. Moreover, regarding the trimethoxybenzyl moiety, which is often found in many bioactive lignans,11 modification on C-3″ is more favorable than modification on C-4″. Generally, the phenyl C ring is a promising moiety that can be modified to produce new derivatives with good drug profiles (Figure 3).
Figure 3.

Structure-activity relationship of 1-type derivatives. * indicates the following conclusion was summarized from the previous reported study.6–8
In contrast to the general potency of all compounds in series A, which generally exhibited significant antiproliferative potency against multiple cell lines, six out of eight compounds in series B showed no activity against the four tested tumor cell lines (IC50 >40 μM), and two derivatives (11b and 16b) exhibited only marginal activity (IC50 29.0 to >40 μM). Converting the sp3-carbon at C-3 of 2 to an sp2-carbon [alkene at C3(6)] produced 11b with a more rigid simplified structure. However, 11b exhibited only weak activity against the tested cell lines (IC50 28.3 to > 40 μM). The same result was also observed with other compounds (compare 12a vs 12b, 15a vs 15b, 16a vs 16b, 19a vs 19b, and 20a vs 20b). These results strongly indicated that the sp3-carbon at C-3 is crucial for the antiproliferative activity (Figure 3). Moreover, it should be noted that, although 11b and 16b exhibited only marginal activity, they also showed a similar tumor type profile to that of the series A derivatives with greater activity against A549, KB, and KB-VIN cells compared with MDA-MB-231.
2.3 Effect on cell cycle progression by a flow cytometry analysis
Because compound 20a was highly potent against A549, KB and KB-VIN cell lines, a flow cytometry analysis15 was performed to evaluate its effect on cell cycle progression and probe its mode of action. DMSO and combretastatin A-4 (CA-4), a tubulin polymerization inhibitor inducing cell cycle arrest in G2/M, were used as controls in this experiment. Moreover, the effect of 2 on cell cycle progression was also assayed. As shown in Figure 4, when A549 cells were treated with 20a at the IC50 concentration (3.32 μM) for 24 h, the population of cells in the G2/M phase clearly increased. This trend was even more obvious, when the concentration of 20a was increased to 9.96 μM (three-fold increase). Prior studies reported that compound 2 caused A549 and LNCaP G2/M cell cycle arrest,1,6–8 and this study also showed very clear G2/M arrest with 2. Altogether, we found that, similarly to CA-4, both 20a and 2 can induce cell cycle arrest at the G2/M phase in a dose-dependent manner.
Figure 4.
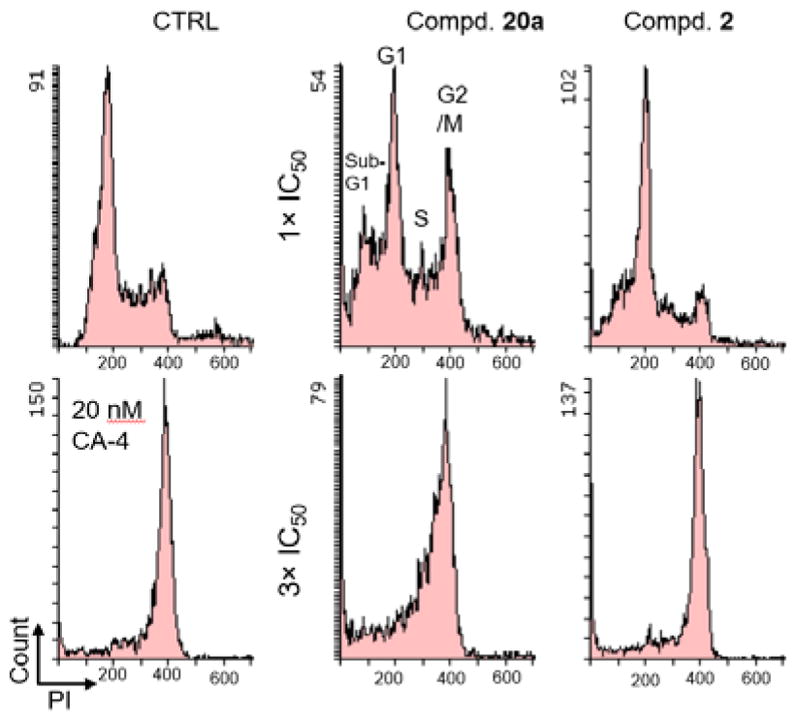
Effect of compounds on cell cycle progression. A549 cells were treated with compound for 24 h at the IC50 concentration (1× IC50) or three-fold of the IC50 concentration (3× IC50). Fixed and propidium iodide (PI)-stained cells were analyzed by flow cytometry. DMSO and 20 nM CA-4 were used as controls.
3. Conclusion
New derivatives of 2 were designed and synthesized via a concise route. The anti-proliferative activities of these new derivatives were determined against four tumor cell lines, A549, MDA-MB-231, KB, and KB-VIN. All active derivatives exhibited the following general rank order of activity: A549 > KB-VIN > KB > MDA-MB-231. Furthermore, our SAR study indicated that the sp3-carbon at C-3 is critical for activity, although its configuration is less important. Modification on the phenyl C ring tolerated for good activity against A549, KB, and KB-VIN cells. Furthermore, regarding the trimethoxybenzyl group, modification at C-3″ is more favorable than modification at C-4″. Therefore, we expect that further modification of this moiety will produce additional new derivatives with good potency. In our present study, among all tested compounds including 2, compound 20a showed the most potent activity against A549, KB, and KB-VIN tumor cells and induced the cell cycle arrest at the G2/M phase. Furthermore, 20a can be readily converted to its water-soluble salts with various inorganic or organic acids, due to the presence of a tertiary amine. Thus, better therapeutic potential has been achieved through structural modification on the phenyl C ring. Overall our present results may shed light on further optimization and development of new 1-derivatives as potential anticancer drugs.
4. Experimental section
General Chemistry
All reagents and solvents were used as received from Sigma-Aldrich or other commercial source. 1D and 2D NMR spectra were measured on an Inova 400 MHz spectrometer with Me4Si (TMS) as internal standard. Unless otherwise indicated, the solvent used was CDCl3. Thin-layer chromatography (TLC) was performed on Merck percolated silica gel 60 F-254 plates. To purify all synthetic compounds, silica gel chromatography was carried out on an ISCO CombiFlash Rf flash chromatography system with prepacked Redi Sep Rf Si gel column (Teledyne ISCO). HPLC purity determinations were conducted using a Shimadzu LCMS-2010 with Shimadzu SPD-M20A detector at 254 or 220 nm wavelength and a Grace Alltima 2.1 mm × 150 mm HP C18 5 μm column. A linear gradient of 35% acetonitrile in water to 100% acetonitrile with a flow rate of 0.2 mL/min was used. All compounds used for biological assay were at least 95% pure.
Synthesis of Compound 9
To a solution of tBuOK (7.85 g, 69.9 mmol) in tBuOH (80 mL), was added dropwise mixture of diethyl succinate (8, 11.7 mL, 69.9 mmol) and piperonal (7, 10 g, 66.6 mmol) in tBuOH (50 mL) under reflux. The reaction was stirred under reflux for additional 4 h. After cooling to rt, the reaction was acidified with 3 N HCl and concentrated in vacuo. The residue was diluted with H2O and DCM. The aqueous layer was extracted twice with DCM. The combined organic layer was washed with brine and dried over Na2SO4. The residue was chromatographed using a silica gel column (40% ethyl acetate in hexane) to yield 9 (12.6 g, 68%) as a yellow amorphous solid. 1H NMR (400 MHz, CDCl3): δ 7.82 (1H, s, CH=), 6.92–6.84 (3H, overlap, 2,5,6-Ph,), 6.01 (2H, s, -OCH2O-), 4.29 (2H, dd, J = 7.2, 14.2 Hz, -OCH2), 3.59 (2H, s, CH2-COOH), 1.34 (3H, t, J = 7.2 Hz, -OCH2CH3). ESI-MS m/z: 279.15 [M+H]+.
Synthesis of Compound 10
LiBHEt3 (1 M in THF, 58.4 mL) was added dropwise to a suspension of 9 (3.61 g, 13.0 mmol) in THF (30 mL) under N2 at 0 °C. The reaction was stirred at 0 to 10 °C for 3 h and then quenched by 1M HCl (10 mL) and H2O. The mixture was extracted twice with EtOAc. The combined organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was dissolved in DCM. To the solution, EDCI (4.98 g, 26.0 mmol) and DMAP (159 mg, 1.30 mmol) were added. After being stirred overnight, the mixture was diluted with DCM, washed with brine, dried over Na2SO4, and concentrated. The residue was chromatographed on a silica gel column (8% ethyl acetate in hexane) to yield 10 (1.52 g, 54%) as a white amorphous solid. 1H NMR (400 MHz, CDCl3): δ 6.83 (1H, d, J = 7.6 Hz, 6-Ph), 6.75–6.73 (2H, overlap, 2, 5-Ph), 6.37 (1H, s, CH=), 5.99 (2H, s, -OCH2O-), 5.00 (2H, dd, J = 2.0, 4.0 Hz, -OCH2-), 3.44 (2H, dd, J = 2.4, 4.6 Hz, -CH2-CO-). ESI-MS m/z: 218.25 [M+H]+.
General Procedure for Synthesis of 2, 12a–14a
A solution of 10 (1 mmol) and the appropriate aldehyde (1.1 mmol) in anhydrous MeOH was stirred for 30 min at 0 °C. To the stirred mixture, CH3ONa (25% w.t. in MeOH, 1.1 mmol) was added. The reaction was stirred at 0 °C for 30 min and then quenched by adding 2M HCl. The mixture was extracted three times with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated. The crude product was used in the next step without further modification.
To the solution of crude product in MeOH, Pd-C (10%) was added. The mixture was hydrogenated under H2 at 30 psi until all starting material was consumed. The catalyst was filtered and the filtrate was concentrated. The residue was dissolved in pyridine, and methanesulfonyl chloride (5 mmol) was added. The reaction was stirred and monitored until all starting material was consumed. The solution was then diluted with EtOAc, acidified with 2M HCl, and washed with brine. The organic layer was dried over Na2SO4 and concentrated. The residue was chromatographed on a silica gel column to yield the pure compound (2, 12a–14a).
Compound 2
Column chromatography (15% ethyl acetate in hexane); white amorphous powder; (33% yield over three steps). 1H NMR (400 MHz, CDCl3): δ 7.25 (2H, s, 2″,6″-Ph), 6.70 (1H, d, J = 7.6 Hz, 5″-Ph), 6.55–6.49 (2H, overlap, 2′,6′-Ph), 6.30 (1H, s, H-5), 5.93 (2H, s, -OCH2O-), 3.84 (2H, t, J = 4.8 Hz, H-4), 3.83 (9H, s, 3×OCH3), 2.76 (1H, m, H-3), 2.52 and 2.39 (1H×2, m, H-6). NOESY correlation: 2″,6″-H to H-5; H-5′ to H-6′; 2′,6′-H to H-6, H-5, and H-5′; H-5 to H-3, H-6 and 2″,6″-H; H-4 to H-6, and H-3; H-3 to H-6, H-5, H-4; H-6 to H-3, 2′,6′-H; ESI-MS m/z: 399.12 [M+H]+.
Compound 12a
Column chromatography (12% ethyl acetate in hexane); white amorphous; Colorless oil; (47% yield over two steps). 1H NMR (400 MHz, CDCl3): δ 7.14 (1H, m, 6″-Ph), 7.04–6.94 (2H, overlap, 2″,5″-Ph), 6.63 (1H, d, J = 8.0 Hz, 5′-Ph), 6.49–6.38 (2H, overlap, 2′,6′-Ph), 6.28 (1H, s, H-5), 5.94 (2H, s, -OCH2O-), 4.45 (2H, m, H-4), 2.67 (1H, m, H-3), 2.57 and 2.47 (1H×2, m, H-6), 2.24 (3H, s, CH3). ESI-MS m/z: 341.15 [M+H]+.
Compound 13a
Column chromatography (15% ethyl acetate in hexane); Colorless oil; (31% yield over two steps). 1H NMR (400 MHz, CDCl3): δ 6.72 (1H, br s, 5′-Ph), 6.68 (2H, s, 2″,6″-Ph), 6.39–6.36 (2H, overlap, 2′,6′-Ph), 6.30 (1H, s, H-5), 5.90 (2H, s, -OCH2O-), 5.27 (2H, -OCH2-O-), 4.08 and 3.92 (1H×2, d, J = 6.5Hz, H-4), 3.85 (6H, s, 2×OCH3), 3.54 (3H, s, OCH3), 2.89 (1H, m, H-3), 2.61 and 2.39 (1H×2, m, H-6). ESI-MS m/z: 356.05 [M+H]+.
Compound 14a
Column chromatography (15% ethyl acetate in hexane); Colorless oil; (38% yield over two steps). 1H NMR (400 MHz, CDCl3): δ 6.82 (1H, s, 2″-Ph), 6.70 (1H, br s, 5′-Ph), 6.61 (1H, s, 6″-Ph), 6.42 (1H, d, J = 2.0 Hz, 2′-Ph), 6.37–6.33 (2H, overlap, 6′-Ph, H-5 ), 5.90 (2H, s, -OCH2O-), 5.26 (2H, -OCH2-O-), 3.90 and 3.91 (1H×2, d, J = 6.0 Hz, H-4), 3.90 (3H, s, OCH3), 3.84 (3H, s, OCH3) 3.55 (3H, s, OCH3), 2.91 (1H, m, H-3), 2.53 and 2.37 (1H×2, m, H-6). ESI-MS m/z: 429.00 [M+H]+.
General Procedure for Synthesis of 11b–14b
A solution of 10 (1 mmol) and the appropriate aldehyde (1.1 mmol) in anhydrous MeOH was stirred for 30 min at 0 °C. To the stirred mixture, CH3ONa (25% w.t. in MeOH, 1.1 mmol) was added. The reaction was stirred at 0 °C for 30 min and then quenched by adding 2M HCl. The mixture was extracted three times with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated. The crude product was used in the next step without further purification.
To a solution of crude product in pyridine, methanesulfonyl chloride (5 mmol) was added. The reaction was stirred and monitored until all starting material was consumed. The solution was then diluted with EtOAc, acidified with 2M HCl, and washed with brine. The organic layer was dried over Na2SO4 and concentrated. The residue was chromatographed on a silica gel column to yield the pure compound (12a-d).
Compound 11b
Column chromatography (15% ethyl acetate in hexane); yellow amorphous powder; (49% yield). 1H NMR (400 MHz, CDCl3): δ 7.04 (2H, s, 2″,6″-Ph), 6.80 (1H, d, J = 8.0 Hz, 5′-Ph), 6.71–6.70 (2H, overlap, 2′,6′-Ph), 6.01 (1H, s, H-5), 5.97 (2H, s, -OCH2O-), 5.81 (1H, s, H-6), 3.91 (6H, s, 2×OCH3), 3.89 (3H, s, OCH3), 3.81 (2H, s, H-4). NOESY correlation: 2″,6″-H to OCH3, and H-5; H-5′ to H-6′; 2′,6′-H to H-5, and H-5′; H-5 to 2′,6′-H and 2″,6″-H; H-6 to H-4 and 2′,6′-H; OCH3 to 2″,6″-H; H-4 to H-6; ESI-MS m/z: 397.05 [M+H]+.
Compound 12b
Column chromatography (10% ethyl acetate in hexane); Yellow amorphous powder; (53% yield). 1H NMR (400 MHz, CDCl3): δ 7.12 (1H, m, 6″-Ph), 7.03–6.94 (2H, overlap, 2″,5″-Ph), 6.76 (1H, d, J = 7.8 Hz, 5′-Ph), 6.53–6.39 (2H, overlap, 2′,6′-Ph), 6.05 (1H, s, H-5), 5.97 (2H, s, -OCH2O-), 5.88 (1H, s, H-6), 3.87(2H, s, H-4), 2.67 (1H, m,) 2.23 (3H, s, CH3). ESI-MS m/z: 339.05 [M+H]+.
Compound 13b
Column chromatography (15% ethyl acetate in hexane); Yellow oil; (33% yield). 1H NMR (400 MHz, CDCl3): δ 7.16 (2H, s, 2″,6″-Ph), 6.78 (1H, d, J = 8.0 Hz, 5′-Ph), 6.70–6.67 (2H, overlap, 2′,6′-Ph), 6.11 (1H, s, H-5), 5.99 (2H, s, -OCH2O-), 5.77 (1H, s, H-6), 5.24 (2H, -OCH2-O-), 3.95 (6H, s, 2×OCH3), 3.76 (2H, s, H-4), 3.54 (3H, s, OCH3). ESI-MS m/z: 378.15 [M+Na]+.
Compound 14b
Column chromatography (15% ethyl acetate in hexane); Yellow oil; (37% yield). 1H NMR (400 MHz, CDCl3): δ 7.33 (1H, s, 6″-Ph), 7.17 (1H, s, 2″-Ph), 6.73 (1H, d, J = 8.0 Hz, 5′-Ph), 6.70–6.68 (2H, overlap, 2′,6′-Ph), 6.32 (1H, s, H-5), 6.03 (2H, s, -OCH2O-), 5.91 (1H, s, H-6), 5.20 (2H, -OCH2-O-), 4.07 and 4.04 (3H each, 2s, 2×OCH3), 3.99 (2H, s, H-4), 3.54 (3H, s, OCH3). ESI-MS m/z: 378.05 [M+Na]+.
General Procedure for Synthesis of 15a–16a, and 15b–16b
The appropriate compound 13a, 14a, 13b, or 14b (1 equiv) and PPTs (1.5 equiv) were dissolved in tBuOH (5 mL). The mixture was stirred at reflux temperature overnight. The reaction was then cooled to rt, diluted with EtOAc, and washed with brine. The organic layer was dried over Na2SO4 and concentrated. The residue was chromatographed on a silica gel column to yield the pure compound (15a, 16a, 15b, and 16b, respectively.
Compound 15a
Column chromatography (25% ethyl acetate in hexane); Colorless oil (78% yield). 1H NMR (400 MHz, CDCl3): δ 6.71 (1H, br s, 5′-Ph), 6.43 (2H, s, 2″,6″-Ph), 6.39–6.38 (2H, overlap, 2′,6′-Ph), 6.33 (1H, s, H-5), 5.90 (2H, s, -OCH2O-), 4.02 and 3.93 (1H×2, d, J = 7.2 Hz, H-4), 3.85 (6H, s, 2×OCH3), 2.90 (1H, m, H-3), 2.55 and 2.42 (1H×2, m, H-6). ESI-MS m/z: 385.15 [M+H]+.
Compound 16a
Column chromatography (25% ethyl acetate in hexane); Colorless oil (83% yield). 1H NMR (400 MHz, CDCl3): δ 6.70 (1H, br s, 5′-Ph), 6.60 (1H, s, 2″-Ph), 6.44 (1H, s, 6″-Ph), 6.41 (1H, d, J = 2.0 Hz, 2′-Ph), 6.36–6.30 (2H, overlap, 6′-Ph, H-5 ), 5.91 (2H, s, -OCH2O-), 3.95 and 3.93 (1H×2, d, J = 4.8 Hz, H-4), 3.91 (3H, s, OCH3), 3.82 (3H, s, OCH3) 2.88 (1H, m, H-3), 2.51 and 2.38 (1H×2, m, H-6). ESI-MS m/z: 385.25 [M+H]+.
Compound 15b
Column chromatography (25% ethyl acetate in hexane); Yellow amorphous powder (63% yield). 1H NMR (400 MHz, CDCl3): δ 7.06 (2H, s, 2″,6″-Ph), 6.80 (1H, d, J = 8.8 Hz, 5′-Ph), 6.71–6.70 (2H, overlap, 2′,6′-Ph), 6.00 (1H, s, H-5), 5.97 (2H, s, -OCH2O-), 5.79 (1H, s, H-6), 3.95 (6H, s, 2×OCH3), 3.80 (2H, s, H-4). ESI-MS m/z: 383.00 [M+H]+.
Compound 16b
Column chromatography (25% ethyl acetate in hexane); Yellow amorphous powder (76% yield). 1H NMR (400 MHz, CDCl3): δ 7.16 (1H, s, 6″-Ph), 7.05 (1H, s, 2″-Ph), 6.93 (1H, d, J = 8.0 Hz, 5′-Ph), 6.89–6.83 (2H, overlap, 2′,6′-Ph), 6.19 (1H, s, H-5), 6.11 (2H, s, -OCH2O-), 5.94 (1H, s, H-6), 4.04 and 4.03 (3H each, 2s, 2×OCH3), 3.96 (2H, s, H-4). ESI-MS m/z: 383.10 [M+H]+.
General Procedure for Synthesis of 17a–18a and 17b–18b
To a solution of compound 15a, 16a, 15b, or 16b (1 eq) in dry DCM was added Boc-L-phenylalanine (1.1 eq), EDCI (2.0 eq), and DMAP (0.1 eq). The reaction was stirred at rt until the starting material was consumed. The solution was diluted with DCM and washed three times with brine. The organic layer was dried over Na2SO4 and concentrated to give the crude product.
For compound 17a and 18a, the crude product was dissolved in 10 mL HCl (2 M in diethyl ether). The mixture was stirred at rt overnight. Then the solution was concentrated in vacuo. The residue was triturated with diethyl ether (10 mL) to precipitate the crude product. The resulting solid was washed with diethyl ether to afford 17a and 18a as white powder.
For compounds 17b and 18b, the crude product (1 eq) was dissolved in TFA (25 eq) and dichloromethane (DCM/TFA 10/1). The reaction was stirred for 2 h. Then the mixture was concentrated to remove TFA. The residue was dissolved in DCM. The organic layer was washed twice with saturated sodium bicarbonate, dried over Na2SO4 and concentrated. The crude product was purified by column chromatography (5% MeOH in DCM) to produce the target compound, 17b and 18b, respectively.
Compound 17a
White amorphous powder (68% yield) 1H NMR (400 MHz, CDCl3): δ 7.32–7.16 (5H, overlap, C6H5), 6.81 (1H, br s, 5′-Ph), 6.40 (2H, s, 2″,6″-Ph), 6.42–6.37 (2H, overlap, 2′,6′-Ph), 6.30 (1H, s, H-5), 5.91 (2H, s, -OCH2O-), 4.42 (1H, m, CH), 4.12 and 3.96 (1H×2, d, J = 6.8 Hz, H-4), 3.30 and 3.18 (1H each, m, CH2), 3.86 (6H, s, 2×OCH3), 2.67 (1H, m, H-3), 2.41 and 2.33 (1H×2, m, H-6). ESI-MS m/z: 569.10 [M+H]+.
Compound 18a
White amorphous powder (77% yield) 1H NMR (400 MHz, CDCl3): δ 7.27–7.12 (5H, overlap, C6H5), 6.58–6.51 (3H, overlap, 2″,6″-Ph, 2′-Ph), 6.29–6.25 (3H, overlap, 5′,6′-Ph), 5.78 (2H, s, -OCH2O-), 4.56 (1H, m, CH), 3.83 to 3.66 (8H, overlap, H-4, 2×OCH3), 3.39 and 3.16 (1H each, m, CH2), 2.75 (1H, m, H-3 ), 2.38 and 2.18 (1H×2, m, H-6). ESI-MS m/z: 569.15 [M+H]+.
Compound 17b
Colorless oil (82% yield) 1H NMR (400 MHz, CDCl3): δ 7.29–7.14 (5H, overlap, C6H5), 6.99 (2H, s, 2″,6″-Ph), 6.84 (1H, d, J = 8.4 Hz, 5′-Ph), 6.64–6.58 (2H, overlap, 2′,6′-Ph), 5.93 (1H, s, H-5), 5.90 (2H, s, -OCH2O-), 5.71 (1H, s H-6), 4.26 (1H, m, CH), 3.87 (6H, s, 2×OCH3), 3.73 (2H, s, H-4), 3.31 and 3.17 (1H each, m, CH2). ESI-MS m/z: 530.05 [M+H]+.
Compound 18b
Yellow amorphous powder (88% yield) 1H NMR (400 MHz, CDCl3): δ 7.43–7.10 (7H, overlap, 2″,6″-Ph, C6H5), 6.73 (1H, d, J = 8.4 Hz, 5′-Ph), 6.64–6.63 (2H, overlap, 2′,6′-Ph), 5.91–5.90 (3H, overlap, -OCH2O-, H-5), 5.74 (1H, s, H-6), 4.20 (1H, m, CH), 3.85 and 3.82 (3H each, 2s, 2×OCH3), 3.73 (2H, s, H-4), 3.43 and 3.29 (1H each, m, CH2). ESI-MS m/z: 530.20 [M+H]+.
General Procedure for the Synthesis of 19a–20a and 19b–20b
To a solution of 15a, 16a, 15b, or 16b (1 eq) in dry DCM was added N,N-dimethylglycine (1.5 eq), EDCI (2.0 eq), and DMAP (0.1 eq). The reaction was stirred for 24 h. Then the solution was diluted with DCM and washed three times with brine. The organic layer was dried over Na2SO4 and concentrated to give the crude product, which was chromatographed on a silica gel column to yield the pure compound (19a, 20a, 19b, 20b, respectively). For compounds 21a and 22a, the crude product was dissolved in 10 mL HCl (2 M in diethyl ether). The mixture was stirred at r.t overnight. Then the solution was concentrated in vacuo. The resulted residue was triturated with diethyl ether (10 mL) to precipitate the crude product. The resulting solid was washed with diethyl ether to afford 21a and 22a as white powder.
Compound 19a
Column chromatography (35% ethyl acetate in hexane); Colorless oil (77% yield) 1H NMR (400 MHz, CDCl3): δ 6.71 (1H, br s, 5′-Ph), 6.44–6.33 (5H, overlap, 2″,6″-Ph, and 2′,6′-Ph, H-5), 5.93 (2H, s, -OCH2O-), 4.02 and 3.90 (2H, J = 6.0 Hz, H-4), 3.76 (6H, s, 2×OCH3), 3.58 (2H, s, N-CH2), 2.89 (1H, m, H-3), 2.58 and 2.40 (1H×2, m, H-6), 2.54 (6H, s, 2×CH3). ESI-MS m/z: 470.20 [M+H]+.
Compound 20a
Column chromatography (35% ethyl acetate in hexane); White amorphous powder (83% yield) 1H NMR (400 MHz, CDCl3): δ 6.69 (1H, s, 5′-Ph), 6.43–6.32 (5H, overlap, 2″,6″-Ph, and 2′,6′-Ph, H-5), 5.90 (2H, s, -OCH2O-), 3.95–3.89 (8H, CH2, 2×OCH3), 3.49 (2H, s, N-CH2), 2.87 (1H, m, H-3), 2.85 and 2.36 (1H×2, m, H-6), 2.51 (6H, s, 2×CH3). ESI-MS m/z: 470.20 [M+H]+.
Compound 19b
Column chromatography (35% ethyl acetate in hexane); Yellow oil (76% yield) 1H NMR (400 MHz, CDCl3): δ 7.46 (2H, s, 2″,6″-Ph), 6.99 (1H, d, J = 8.0 Hz, 5′-Ph), 6.91–6.89 (2H, overlap, 2′,6′-Ph), 6.20 (1H, s, H-5), 6.17 (2H, s, -OCH2O-), 5.98 (1H, s, H-6), 4.14 (6H, s, 2×OCH3), 4.00 (2H, s, H-4), 3.80 (2H, s, N-CH2), 2.76 (6H, s, 2×CH3). ESI-MS m/z: 468.10 [M+H]+.
Compound 20b
Column chromatography (25% ethyl acetate in hexane); yellow oil (69% yield) 1H NMR (400 MHz, CDCl3): δ 7.04 (1H, s, 6″-Ph), 6.94 (1H,s, 2″-Ph), 6.78 (1H, d, J = 8.0 Hz, 5′-Ph), 6.70–6.68 (2H, overlap, 2′,6′-Ph), 5.96 (2H, s, -OCH2O-), 5.95 (1H, s, H-5), 5.80 (1H, s, H-6), 3.99 and 3.91 (3H each, 2s, 2×OCH3), 3.79 (2H, s, H-4), 3.59 (2H, s, N-CH2), 2.54 (6H, s, 2×CH3). ESI-MS m/z: 468.20 [M+H]+.
Compound 21a
White amorphous powder (54% yield) 1H NMR (400 MHz, CDCl3): δ 6.71 (1H, br s, 5′-Ph), 6.49–6.37 (5H, overlap, 2″,6″-Ph, and 2′,6′-Ph, H-5), 5.91 (2H, s, -OCH2O-), 3.93 and 3.87 (1H×2, d, J = 6.8 Hz, H-4), 3.85 (6H, s, 2×OCH3), 3.80 (2H, s, N-CH2), 3.01 (1H, m, H-3), 2.87 (6H, s, 2×CH3 ), 2.58 and 2.38 (1H×2, m, H-6), 2.93 (6H, s, 2×CH3). ESI-MS m/z: 506.10 [M+H]+.
Compound 22a
White amorphous powder (63% yield) 1H NMR (400 MHz, CDCl3): δ 6.89 (1H, s, 5′-Ph), 6.65–6.52 (2H, overlap, 2″, 6″-Ph), 6.37–6.26 (3H, overlap, 2′,6′-Ph, H-5), 5.85 (2H, -OCH2O-), 3.94–3.78 (8H, H-4, 2×OCH3), 3.76 (2H, s, N-CH2), 3.03 (6H, s, 2×CH3), 2.81 (1H, m, H-3), 2.55 and 2.29 (1H×2, m, H-6), 2.51 (6H, s, 2×CH3). ESI-MS m/z: 506.15 [M+H]+.
Antiproliferative Activity Assay
All cell lines were obtained from the Lineberger Comprehensive Cancer Center (UNC-CH) or from ATCC (Manassas, VA, USA), except KB-VIN, which was a generous gift of Professor Y.-C. Cheng (Yale University), and all cell lines were maintained in T-75 flasks at 37 °C with 5% CO2 in air. Cells were cultured in RPMI-1640 medium supplemented with 2 mM L-glutamine and 25 mM HEPES (Gibco), supplemented with 10% fetal bovine serum (Corning), 100 μg/mL streptomycin, 100 IU/mL penicillin, and 0.25 μg/mL amphotericin B (Gibco). KB-VIN cells were maintained in the presence of 100 nM vincristine. Antiproliferative activity of compounds were determined by sulforhodamine B (SRB) assay as previously described.15 In brief, freshly trypsinized cell suspensions were seeded in 96-well microtiter plates at densities of 4,000–7,500 cells per well (bases on the doubling time of the cell line) with compounds. After 72 h in culture with test compounds, cells were fixed in 10% trichloroacetic acid and then stained with 0.04% SRB. After solubilizing the protein-bound dye with 10 mM Tris base, absorbance was measured at 515 nm using a microplate reader (ELx800, BioTek) with Gen5 software (BioTek). The mean IC50 is the concentration of agent that reduced cell growth by 50% compared with vehicle (DMSO) control under the experimental conditions used and is the average from at least three independent experiments with duplicate samples.
Cell Cycle Analysis
The effect of compound on cell cycle progression was evaluated by measurement of the cellular DNA content by propidium iodide (PI) staining.15 Briefly, freshly trypsinized A549 cells were seeded in 12-well plates at densities of 75,00 cells per well 24 h prior to treatment with compounds. After a 24 h treatment, supernatants and trypsinized cells were collected together, followed by centrifugation for 10 min at 1,000 rpm. The pellet was resuspended with PBS and fixed in 70% EtOH overnight at −20 °C, followed by staining with PI (BD Biosciences) for 30 min at 37 °C. Stained cells were analyzed by flow cytometry (LSRFortessa, BD Biosciences). Experiments were repeated a minimum of three times.
Statement of significance.
New derivatives of (±)-isochaihulactone were designed, synthesized, and evaluated for antiproliferative activity. SAR conclusions were first established for this series of compounds. The present results may shed light on the design and development of new derivatives of isochaihulactone-type compounds as drug candidates for cancer treatment.
Acknowledgments
This investigation was supported by NIH grant CA177584 from the National Cancer Institute awarded to K.-H.L. Support was also due in part to grants from Eshelman Institute for Innovation, Chapel Hill, North Carolina awarded to Y. Z. and M. G.
Footnotes
Conflict of Interests
The authors declare no competing interest
References
- 1.Chen YL, Lin SZ, Chang WL, YL, Harn HJ. Life Sci. 2005;76:2409–2420. doi: 10.1016/j.lfs.2004.09.044. [DOI] [PubMed] [Google Scholar]
- 2.Chen YL, Lin SZ, Chang JY, Cheng YL, Tsai NM, Chen SP, Chang WL, Harn HJ. Biochem Pharmacol. 2006;72:308–319. doi: 10.1016/j.bcp.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 3.Cheng YL, Lee SC, Lin SZ, Chang WL, Chen YL, Tsai NM, Liu YC, Tzao C, Yu DS, Harn HJ. Cancer Lett. 2005;222:183–193. doi: 10.1016/j.canlet.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 4.Chen YL, Lin PC, Chen SP, Lin CC, Tsai NM, Cheng YL, Chang WL, Chang L, Lin SZ, Harn HJ. J Pharm Exp Ther. 2007;323:746–756. doi: 10.1124/jpet.107.126193. [DOI] [PubMed] [Google Scholar]
- 5.Yu YL, Su KJ, Chen CJ, Wei CW, Lin CJ, Yiang GT, Lin SZ, Harn HJ, Chen YL. J Cell Physiol. 2012;227:213–222. doi: 10.1002/jcp.22719. [DOI] [PubMed] [Google Scholar]
- 6.Chiu SC, Wang MJ, Yang HH, Chen SP, Huang SY, Chen YL, Lin SZ, Harn HJ, Pang CY. BMC Cancer. 2011;11:46. doi: 10.1186/1471-2407-11-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ou JP, Lin HY, Su KY, Yu SL, Tseng IH, Chen CJ, Hsu HC, Chan DC, Chen YLS. Evid Based Complement Alternat Med. 2012;809204 doi: 10.1155/2012/809204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen YL, Harn HJ, Lin SZ, Chiou TW, Lin CT. 201113008597. US Pat. 2010
- 9.Liu PY, Lin SZ, Sheu JJ, Lin CT, Lin PC, Chou YW, Huang MH, Chiou TW, Harn HJ. Prostate. 2013;73:531–541. doi: 10.1002/pros.22593. [DOI] [PubMed] [Google Scholar]
- 10.Alizadeh BH, Foroumadi A, Emami S, Khoobi M, Panah F, Ardestani SK, Shafiee A. Eur J Med Chem. 2010;45:5979–5984. doi: 10.1016/j.ejmech.2010.09.064. [DOI] [PubMed] [Google Scholar]
- 11.Chang WL, Chiu LW, Lai JH, Lin HC. Phytochemistry. 2003;64:1375–1379. doi: 10.1016/j.phytochem.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 12.Lee KH, Xiao Z. Phytochem Rev. 2003;2:341–362. [Google Scholar]
- 13.Mizufune H, Nakamura M, Mitsudera H. Tetrahedron. 2006;62:8539–8549. [Google Scholar]
- 14.Lewis SN, Popper TL. Tetrahedron. 1967;23:4197–4208. doi: 10.1016/s0040-4020(01)88817-7. [DOI] [PubMed] [Google Scholar]
- 15.Nakagawa-Goto K, Oda A, Hamel E, Ohkoshi E, Lee KH, Goto M. J Med Chem. 2015;58:2378–2389. doi: 10.1021/jm501859j. [DOI] [PMC free article] [PubMed] [Google Scholar]