

Abstract
Small molecule tool compounds have enabled profound advances in life science research. These chemicals are potent, cell active, and selective, and, thus, are suitable for interrogating biological processes. For these chemicals to be useful they must be correctly characterized and researchers must be aware of them. We mined the ChEMBL bioactivity database to identify high quality tool compounds in an unbiased way. We identified 407 best-in-class compounds for 278 protein targets, and these are reported in an annotated data set. Additionally, we developed informatics functions and a web application for data visualization and automated pharmacological hypothesis generation. These functions were used to predict inhibitors of the Chromobox Protein Homologue 5 (CBX5) mediated gene repression pathway that currently lacks appropriate inhibitors. The predictions were subsequently validated by a highly specific cell based assay, revealing new chemical modulators of CBX5-mediated heterochromatin formation. This data set and associated functions will help researchers make the best use of these valuable compounds.
Graphical abstract
INTRODUCTION
Drug-like small molecules can treat disease and also be valuable reagents for life science research. Small molecules are great research tools in part because they are easy to use, and their experimental use typically requires little optimization. The value of a molecule as a tool to catalyze research is related to its bioactivity and selectivity, and it must give a robust, on-target response in cells. If a molecule is promiscuous or generally reactive, the induced phenotype will not necessarily be linked to a specific biological target, and the conclusions are spurious.1 Small molecule tool compounds, or chemical probes, are high-quality research tools with potent, selective, and on-target cellular effects. Some chemical probes, such as JQ-1 and rapamycin, have transformed our understanding of epigenetic regulation of gene expression and the molecular target of rapamycin (mTOR) signaling pathway, respectively.2–5
For a tool compound to be useful, its activity and selectivity must be suitable for use in research, its function must be easily queried, and researchers must know its existence. Crowd-sourcing initiatives allow users to share information about chemical probes.6–9 One of these efforts, the Chemical Probes Portal, is a community-curated web resource that provides information on many known chemical probes, and serves to increase awareness of available probes.1 These crowd-sourced projects offer valuable practical information, have brought attention to high-quality probes, and use user feedback to identify the most valuable chemical probes.
These projects typically focus on targets that are the subject of current research, such as epigenetic targets. Our aim for this work was to supplement these databases with a database of chemical tool compounds for targets from medicinal chemistry literature and patents. The ChEMBL database contains bioactivity records for millions of chemicals from the medicinal chemistry literature, but no efforts to specifically mine these databases for chemical probes have been reported.10 We used a data-driven approach to uncover best-in-class tool compounds from this source and created informatics functions to help researchers make the best use of these chemicals and to easily locate useful functional inhibitors for a given biological pathway.
RESULTS AND DISCUSSION
Data Analysis
We mined publicly available bioactivity data to identify tool compounds in an unbiased way, where classification as a probe depends only on the data meeting these criteria. We used the ChEMBL database for the bioactivity data source because it is publicly available in SQL format, is one of the largest and highest-quality bioactivity databases, and contains sufficient annotation for classification of probes.10,11 We did not use data from PubChem because it cannot be accessed with SQL.12 ChEMBL and PubChem share much of their data. A formal definition of a chemical probe was made by Arrowsmith et al.1 A chemical probe meets the following criteria: (1) in vitro potency of <100 nM at the protein target, (2) >30-fold selectivity against other protein targets, and (3) demonstration of on-target effect in cells at <1 μM. We used a modified set of rules that enabled data mining of ChEMBL: (1) potency ≤ 100 nM for one primary protein target, (2) >30-fold selectivity against at least one other protein target, and (3) cellular activity < 1 μM at the primary target.
ChEMBL version 22, released in October 2016, contains 1 686 695 compounds and 14 371 219 activities, with the majority of data coming from medicinal chemistry literature. ChEMBL also contains data from patents, Pubchem bioassays, and other databases. The data mining was performed using R with extensive use of the dplyr package for data analysis.13 The data mining script can be run on any future version of ChEMBL, provided no changes are made to the schema that would invalidate the analysis. The script identifies one primary probe and one orthogonal, structurally dissimilar probe for each target. The primary probe for each target is the compound that meets chemical probe criteria and is selective against the greatest number of other proteins, and the orthogonal probe is the most selective compound meeting chemical probe criteria with a Tanimoto similarity score < 0.7 from the primary probe.14 For receptor targets, the script identifies both the best agonist (Emax > 25%) and the best nonagonist. Potentially reactive compounds were filtered using a pan-assay interference compounds (PAINS) substructure filter.15
Judging selectivity is a challenge for classification of chemical probes. It is unlikely that any chemical is truly selective for one target against all others, and apparent selectivity may vanish when a compound is tested at more targets. We reduced bias against compounds that were tested against many off-targets in our data mining script: For each protein target, before filtering out compounds that did not meet the selectivity criteria, we found the compound that had been tested against the most off targets and removed any potential probes for that protein target that was not tested against more than 1/2 that maximum number of off targets. For example, if for protein target X, the best-characterized compound was tested against eight off targets, we removed potential probes for X that were tested against four or fewer off targets.
The data mining identified 407 putative chemical probes targeting a total of 278 protein targets. (Figure 1A). Most targets were excluded because they did not have any high affinity ligands. Ninety-eight targets have both a primary and an orthogonal probe, and 50 probes are agonists. The median number of non-ADMET bioactivity observations per probe compound was 11, and the median number of proteins that probes were tested against was 4 (Figure 1B and C). The most selective compound is the TGF-beta receptor ligand GW693481X, which is selective against 188 other proteins. Here, 156 of the targets are receptors and 130 have transferase activity.
Figure 1.
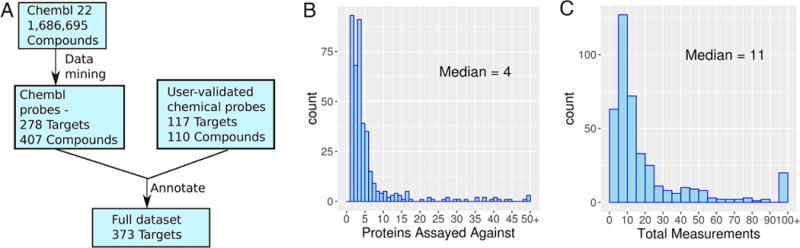
Chemical probe data mining. (A) Data flow for construction of the data set. (B) Number of proteins targets tested for probes found in ChEMBL. (C) Total bioactivity observations for probes found in ChEMBL.
We combined the probes discovered in ChEMBL with probes listed in the Chemical Probes Portal (as of August 2016) to create a unified data set. Only 22 targets are common to both data sources. The main reason for the lack of overlap is that data from many user-submitted probes are not included in the ChEMBL database, as ChEMBL is composed of data from medicinal chemistry literature. Thus, our data set is complementary to data sets of more contemporary probe molecules and highlights the best tools to interrogate a different set of targets. The data set was annotated in a target-centric way, with the KEGG pathways, gene ontology (GO) terms (molecular function (MF), biological process (BP), and cellular compartment (CC)), reactome pathways, Entrez gene IDs, and Uniprot IDs for each protein target (see supplemental file, PROBELIST.csv).16,17
Associating the targets with GO and KEGG pathways allowed us to quantitatively measure how well biological pathways are associated with chemical probes, revealing gaps and concentrations in our ability to target biological pathways. We analyzed the enrichment of probe-associated genes for GO-MF and GO-BP terms, and for KEGG pathways (Table 1).18 Besides the expected high occurrence of receptor pathway terms, probes also preferentially target proteins involved in the FOXO signaling pathway. The list of the least common GO and KEGG pathways confirms the difficulty in targeting proteins with bridging protein binding or nucleoside-triphosphatase regulator activity.
Table 1.
Selected Common and Uncommon Gene Ontology and KEGG Pathway Terms for Proteins Targeted by Chemical Probes
| source | most common terms | least common terms |
|---|---|---|
| GO–molecular function | G-protein coupled peptide receptor activity; peptide receptor activity; steroid hormone receptor activity | structural constituent of ribosome; protein binding, bridging; nucleoside-triphosphatase regulator activity; Ras guanyl-nucleotide exchange factor activity |
| GO–biological process | positive regulation of blood circulation; digestive system process; regulation of circadian rhythm; feeding behavior | humoral immune response; electron transport chain; mitochondrial translation; spermatid differentiation; natural killer cell mediated cytotoxicity |
| KEGG | neuroactive ligand–receptor interaction; FoxO signaling pathway; calcium signaling pathway | primary immunodeficiency; one carbon pool by folate; bile secretion |
Network Visualization
We also developed functions to help researchers make the best use of existing chemical probes. Many “-omics” experiments are interpreted with pathway analysis, where the data is linked to known biological pathways. Likewise, complex diseases like cancer are commonly analyzed at a network level. We leveraged the “pathview” and “ReactomePA” packages to create functions that visualize how biological pathways can be targeted with chemical probes.19–21 The functions display Reactome and KEGG pathways and highlight the targetable nodes and can be accessed through the scripts or web application associated with this publication. Output from the KEGG visualization function for the Ras signaling pathway is shown in Figure 2. One use of this graph is to generate hypotheses for combination therapies that target multiple arms of the pathway or suppress feedback loops. Combination of either AKT or PI3K inhibitors with MEK inhibitors are promising treatments for Ras-driven cancers, and it is clear on the pathway graph how these agents target separate arms of the Ras signaling pathway.22–25 Overall, 32 proteins involved in the KEGG Ras signaling pathway can be targeted by chemical probes.
Figure 2.
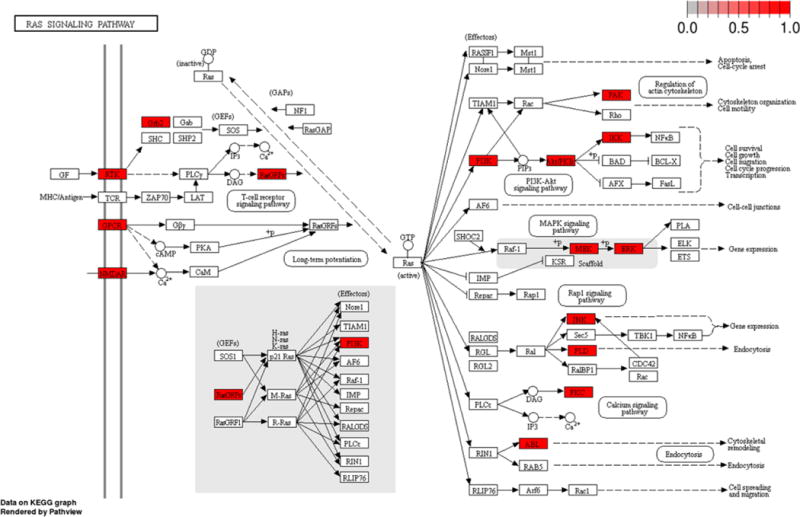
KEGG Ras signaling pathway with targetable nodes highlighted in red.
Because most proteins cannot be directly targeted by currently available chemical probes, we created a network visualization function to help find probes that may indirectly modulate a target of interest. This could occur through a number of mechanisms including interfering with protein–protein interactions, disrupting signaling pathways, or modulating post-translational modifications. The network used by the function is a subset of the IntAct database of protein–protein interactions and contains all proteins that are separated from a probe target by one or two edges.26,27 Given a protein target, the function displays all nodes that can be targeted by probes in the local network. We used the function to generate the probe-target network for c-MYC, with an IntAct confidence score cutoff of 0.5 (Figure 3). Many known ways to pharmacologically modulate c-MYC are found in the graph, including BRD3, GSK3B, EGFR, and PLK1 inhibition.5,28–30 Because we used the IntAct database to construct the network, the linkages between c-MYC and these proteins are not based upon pharmacological evidence, but upon proteomics and coimmunoprecipitation experiments. Therefore, the network identifies known pharmacological interactions from independent data. The network also shows a connection between CARM1 and c-MYC through SMRC1. In a recent report, genetic knockdown of CARM1 blocked methylation of SMRC1, reducing the localization of SMRC1 at c-MYC pathway genes.31 It is very likely that pharmacological inhibition of CARM1 will also suppress transcription of genes driven by c-MYC.
Figure 3.
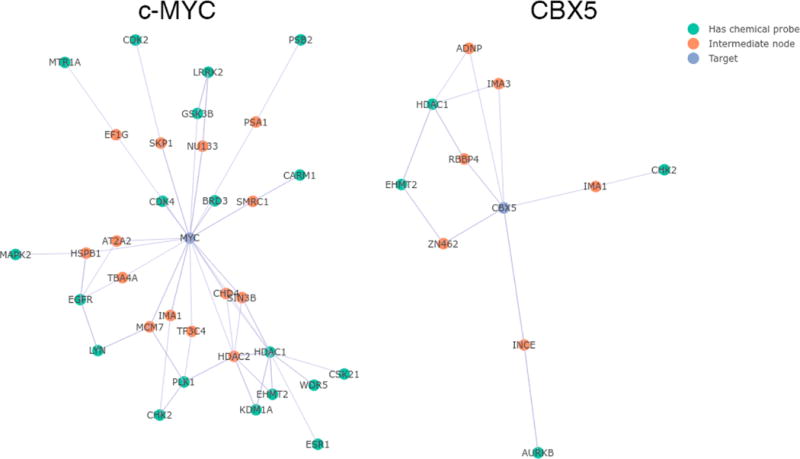
Probe–target network visualization. Output from the probe–target network visualization function for c-MYC (left) and CBX5 (right), with an IntAct confidence score cutoff of 0.5.
Discovery of Modulators of CBX5 Function
The CBX5 chromodomain-containing protein has no reported direct or indirect pharmacological modulators. CBX5 binds to methylated lysine 9 on histone H3 (H3K9me3) and facilitates the spreading of H3K9me3 to nearby nucleosomes via recruitment of additional methyltransferase enzymes. H3K9me3 marks lead to a repressive, condensed heterochromatin state of DNA with decreased gene expression.32,33 We generated the probe–target network for CBX5 with a confidence score cutoff of 0.5 (Figure 3). Aurora kinase B (AURKB), checkpoint kinase 2 (CHK2), histone deacetylase 1 (HDAC1), and euchromatic histone-lysine N-methyltransferase 2 (EHMT2) all appear in the network. To validate the predictive ability of the function, we tested chemical probes for these proteins in an assay for CBX5-mediated gene repression.
In the Chromatin in vivo Assay at Oct4 (CiA:Oct4) cell line, one allele of the haplosufficient pluripotency factor, Oct4, is replaced with a Gal4 and Zinc finger DNA binding array upstream of a nuclear eGFP reporter gene.34 CBX5 is rapidly recruited to the locus using chemical induced proximity (CIP) upon the addition of rapamycin. CBX5 recruitment facilitates the deposition of H3K9me3 marks on the histones leading to DNA silencing and a decrease in GFP expression. Inhibition of CBX5-mediated heterochromatin upon addition of chemical probe results in a larger GFP positive population due to a failure to repress the reporter gene (Figure 4A).
Figure 4.
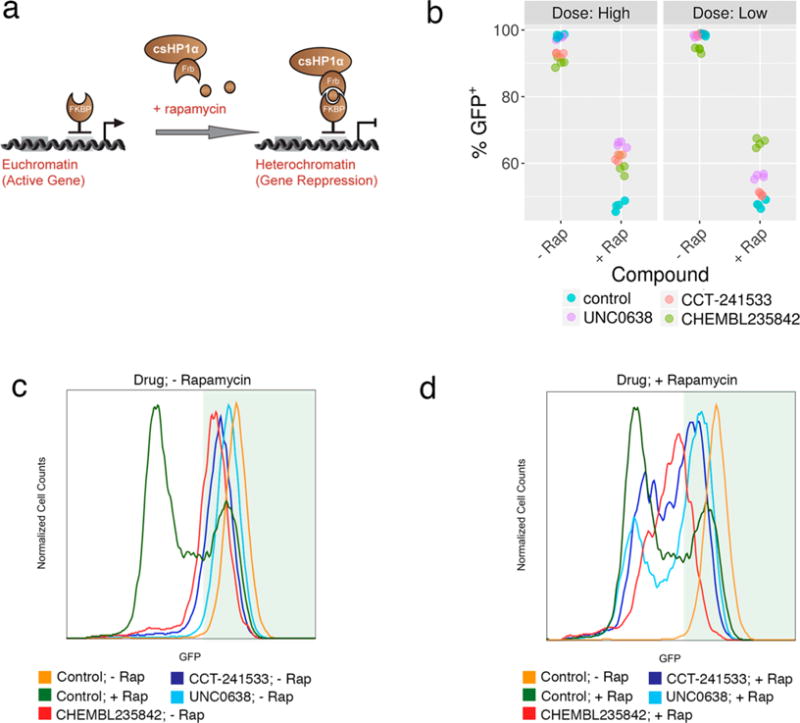
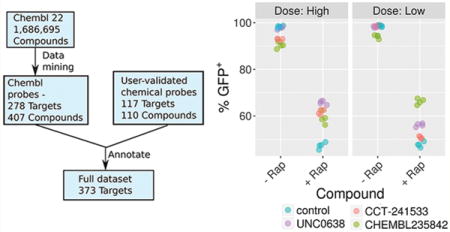
Cellular assays of CBX5 function. (a) CBX5 (HP1α) was recruited to the engineered CiA:Oct4 locus using chemical induced proximity in a mouse embryonic stem (ES) cell line. (b) Chemical probes were added in low and high doses to the ± rapamycin containing samples for 2 days in triplicate. Samples were analyzed by flow cytometry and GFP levels were quantified using FlowJo software. (c) DMSO (−Rap) or (d) 6 nM rapamycin (+ Rap) was added along with high doses of compounds for 3 days, leading to normal or decreased GFP expression, respectively. Four biological replicates are shown (n = 4).
We tested chemical probes for targets in the CBX5 network at two doses in the CiA assay: HDAC1/2 inhibitor CHEMBL235842 (1 μM and 5 μM); AURKB inhibitor AMG-900 (50 nM and 100 nM); CHK2 inhibitor CCT-241533 (1 μM and 5 μM); and EHMT2 inhibitor UNC0638 (300 nM and 1 μM).35–38 Upon addition of rapamycin to the CiA cells, the percent of GFP positive cells decreased from 99% to 47%, indicating that 52% of cells have a decrease in gene expression due to CBX5 recruitment. Little to no decrease in GFP expression was observed in the low dose samples without rapamycin, though small decreases in GFP expression were observed in the high doses for CHK2 and HDAC inhibitors (Figure 4B). This is likely due to cell toxicity at high doses leading to cell differentiation, and the Oct4 locus being silenced independent of the chemical probe. The HDAC inhibitor was effective at blocking heterochromatin formation at both doses resulting in 66% (low) and 59% (high) GFP+ cells. Histone acetylation correlates with active gene expression. Inhibiting the enzymes that remove the active mark leads to the acetyl mark remaining on the histone, which may compete against the deposition of repressive H3K9me3 upon CBX5 recruitment.39 Similarly, the EHMT2 inhibitor was also effective at both doses resulting in 56% and 66% GFP+ populations. EHMT2 is recruited by CBX5 and adds H3K9 methyl and dimethyl marks.40 Inhibiting this enzyme directly prevents the increase in H3K9me3 repressive marks. The CHK2 inhibitor was effective at the highest dose resulting in 62% GFP+ population. CHK2 phosphorylates the CBX5 binding pocket of KAP-1 at S473. A mutation to S473A resulted in a mobilization defect in CBX5 during DNA damage response.41 It is plausible that the CHK2 inhibitor may similarly decrease CBX5 recruitment to chromatin and compromise its ability to form heterochromatin. The AURKB inhibitor was toxic at all doses tested in this cell line so the data could not be interpreted.
The KEGG and probe–target network functions, along with other functions, can also be accessed through an R Shiny web application, located at chemicalprobesapp.shinyapps.io/chemicalprobesapp.
DISCUSSION
The potential for tool compounds to accelerate life science research has been fettered by a lack of information. By identifying these compounds with a data-driven classification scheme, we have identified best-in-class tool compounds for targets covered by prior medicinal chemistry research programs. This approach is complementary to community-driven efforts to identify chemical probes, as there is relatively little overlap between the two data sets, and our approach is meant to discover the best probes from targets in the medicinal chemistry literature. For the most part, the tool compounds identified through ChEMBL data mining target well-studied proteins (GPCRs, kinases), but also some targets that are less well-known, such as peregrin. Although all chemical probes we have identified are correctly classified according to the given data, researchers should thoroughly investigate all of the best modulators of a given target while planning an experiment because errors in ChEMBL data can result in misclassification. The collaborative generosity of many laboratories in the chemical biology field enables access to a subset of these compounds, but commercial availability remains a roadblock. Most of the probes identified through ChEMBL are not commercially available, but hopefully, identifying these useful research chemicals will encourage suppliers to make them available.
The compounds in this data set are precise chemical tools that can be used to study the covered targets. Compounds within the set could be included in a phenotypic screening collection, so that any hits could be immediately linked to a target likely to be responsible for the phenotype. Because small molecules are easy to use, they are powerful agents for phenotypic screening.
Judging selectivity is a challenge for chemical probe classification. New targets are constantly being found for established drugs, and it is likely that most bioactive molecules potently bind to multiple targets.42,43 Requiring chemical probes to be >30 fold selective against other targets is a bias against compounds that have been tested at many proteins. But, this requirement does exclude promiscuous compounds and gives confidence to the association of a phenotype with the modulation of a target. More sophisticated measurements of selectivity will help the identification of useful chemical research tools.44
In our data set, chemical probes are linked to target proteins, and those target proteins are linked to pathways. Many big data experiments end in pathway analysis, and we provide an easy way to link those pathways to chemical probes. By profiling the target proteins at GO and KEGG identifiers, we see the limits of known probes to target important molecular functions. We hope that identifying the molecular functions that cannot be pharmacologically controlled will encourage development of chemical modulators of these functions. Connecting the probes with pathways also suggests future experiments. For example, it would be useful to test all probes involved in the KEGG Ras signaling pathway pairwise, to investigate synergy.
Life science research could benefit from more straightforward, visually appealing research tools for hypothesis generation. To this end, we have merged protein interaction and pathway databases with our chemical probe data set to create an integrated informatics toolkit for automated pharmacological hypothesis generation and visualization. The probe–target network function was used to discover chemical probes that block CBX5-mediated formation of heterochromatin. Pharmacological control of CBX5 function has never been reported, and the information used to inform the prediction of CBX5 inhibitors came solely from a database of reported protein–protein interactions. The identification of three previously unknown modulators of CBX5 activity by this computing tool supports the use of this tool to rapidly identify chemical modulators of a protein function of interest. An advantage of using chemicals rather than genetic methods to manipulate protein function and quantity is that chemicals can be used immediately, with little optimization. Thus, the probe–target network allowed us to use chemicals to quickly identify three proteins likely to be involved in CBX5 function. The mechanism by which CBX5 depends upon either CHK2 or HDAC2 is unclear and will be characterized in future work.
Our chemical probe data set provides researchers with an exhaustive list of tool compounds. This work also circumscribes the set of existing tool compounds, and identifies deficits in our ability to pharmacologically target certain molecular functions. This data set, together with the computing tools presented here, will help researchers get the most use out of these valuable chemicals.
METHODS
Chemicals Used
AMG-900 and CCT-241533 were purchased from MedChemExpress. UNC0638 was prepared as described.37 CHEMBL235842 was prepared as described.35
Data Analysis
The data was analyzed in the following way: a short list of compounds with activity ≤100 nM in a “SINGLE PROTEIN” assay and activity <1000 nM in a cell-based assay was compiled. Each compound was annotated with the total number of bioactivity observations, to identify well-characterized compounds, and also with the total number of protein targets it was assayed against, to measure compound selectivity. Compounds were excluded if they did not have bioactivity data against at least two different proteins. Each compound was associated with the protein target for which it has the greatest affinity in a dose response assay. To prevent the data mining from being biased for compounds that were selective against fewer off targets, we found the compound with the most selectivity data for each target, and then removed potential probes for the target that were not tested against more than 0.5 times the highest number of off targets. Next, compounds were excluded if they did not meet the following selectivity criteria: no dose–response curve assay values for off targets <30 times the value for the main protein target, no fold-selectivity or IC50 ratio values <30, no activities >50% in single point activity assays for off targets, no values <50% in off-target single point inhibition assays, and no off-target delta Tm >5.0 in thermal melting shift assays. Compounds also must have activity <1000 nM in a cell based assay for the main target. The cellular assay condition and the ≤100 nM potency condition can be satisfied by the same bioactivity observation. Compounds were designated as agonists if they had an efficacy value >25% for the main target. Many popular targets were associated with many possible probes, so for each protein target, we designated a primary probe as the probe with the greatest number of selectivity observations, with a tie break on the total number of bioactivity observations. For targets that have a probe noted to be an agonist, we kept the best agonist and the best nonagonist in the final list, giving researchers access to agonists and antagonists at receptor targets. It is useful to have multiple orthogonal, or structurally dissimilar, probes for a target. To find orthogonal probes, for compounds with the same main protein target we calculated the Tanimoto similarity of each compound from the primary probe and kept the compound with the greatest number of selectivity/total observations and a Tanimoto similarity of T < 0.7. We chose 0.7 because it was an inflection point on the density plot of all calculated Tanimoto scores, representing the shift between a population centered around T = 1 (compounds very similar to the primary probe) and 0.5 (compounds dissimilar to the primary probe). We then applied a pan-assay interference compound (PAINS) filter to remove reactive and promiscuous compounds.15 Compounds with the following functional groups were exluded: catechols, push–pull fluorophores, Michael-acceptor rhodanines, phenolic mannich bases, 2-hydroxy-phenyl-hydrazones, and compounds with unsubstituted saturated carbon chains >6 atoms. This removes only the most offensive substructures, because some PAINS-contianing compounds can be valuable research tools.
The probe–target network is an annotated subset of the IntAct database. The network includes all nodes connected to a probe-targetable protein by one or two edges. The database includes all interactions between the proteins in that set. The database was accessed with the RefNet package in R/Bioconductor. Construction of the probe–target network was accomplished using the script: createProbeTargetNetwork.R
Please see the readme.html for instructions on using the network visualization functions. Network visualization and data search functions can also be found at chemicalprobesapp.shinyapps.io/chemicalprobesapp.
All computer code is freely availabile at https://github.com/KyleVButler/ChemicalProbesDataMining
HP1-Mediated Heterochromatin Formation Assay
CiA:Oct4 mouse embryonic stem cells (ES) containing both viral integrations of N118 and N163 plasmids (N118-nLV EF-1a-Gal-FKBPx1-HA-PGK-Blast, N163-nLV EF-1a-Hp1a(CS)-Frbx2(Frb+FrbWobb)-V5-PGK-Puro) were grown in DMEM supplemented with 4.5 g/L glucose, 15% FBS, L-glutamate, sodium pyruvate, HEPES buffer, NEAA, 2-mercaptoethanol, and penicillin/streptomycin. On day 0, cells were seeded into 96 well plate formats with 10 000 cells per well with a minimum of three replicates. Day 1, media was aspirated and replaced with fresh culture media containing appropriate chemical probe doses and ±6 nM rapamycin to recruit HP1 to the Oct4 locus and initiate heterochromatin formation. Day 2, culture media was aspirated and fresh media containing chemical probes and ± rapamycin was added as on day 1. Day 3, media was aspirated and the cells were washed with PBS and trypsinized using 0.25% trypsin-EDTA. Trypsin was quenched with serum and the cells were prepared for flow cytometry analysis. The CiA ES cell line is the same as previously established, has not been authenticated, and tested negative for mycoplasma contamination.34
Flow Cytometry
Flow cytometry analysis was performed using an iQue Screener by Intellicyt and FCS data files were exported and analyzed with FlowJo software. Cell populations were gated based on forward and side scatter. Single cell populations were isolated using forward scatter area by forward scatter height gating. Autofluorescent cells were omitted, and remaining cells were then analyzed for GFP levels. Percent GFP was calculated by the FlowJo software.
Supplementary Material
Acknowledgments
K.V.B. was supported by a postdoctoral fellowship from the American Cancer Society (PF-14-021-01-CDD). N.A.H. acknowledges support of this work from an American Association of Colleges of Pharmacy new investigator award and an Eshelman Institute for Innovation award. J.J. acknowl edges the support of grants R01GM122749, R01CA218600, and R01HD088626 (to J.J.) from the U.S. National Institutes of Health. We thank Heather King and Amy Donner of the Chemical Probes Portal for discussions regarding the work.
Footnotes
ORCID
Jian Jin: 0000-0002-2387-3862
Notes
The authors declare no competing financial interest.
The main data set of chemical probes is available as PROBELIST.csv. Data visualization functions can also be accessed at chemicalprobesapp.shinyapps.io/chemicalprobesapp or by downloading files at https://github.com/KyleVButler/ChemicalProbesDataMining.
References
- 1.Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J, Bountra C, Brennan PE, Brown PJ, Bunnage ME, Buser-Doepner C, Campbell RM, Carter AJ, Cohen P, Copeland RA, Cravatt B, Dahlin JL, Dhanak D, Edwards AM, Frye SV, Gray N, Grimshaw CE, Hepworth D, Howe T, Huber KVM, Jin J, Knapp S, Kotz JD, Kruger RG, Lowe D, Mader MM, Marsden B, Mueller-Fahrnow A, Müller S, O’Hagan RC, Overington JP, Owen DR, Rosenberg SH, Roth B, Ross R, Schapira M, Schreiber SL, Shoichet B, Sundström M, Superti-Furga G, Taunton J, Toledo-Sherman L, Walpole C, Walters MA, Willson TM, Workman P, Young RN, Zuercher WJ, Frederiksen M. The promise and peril of chemical probes. Nat Chem Biol. 2015;11(8):536–541. doi: 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ballou LM, Lin RZ. Rapamycin and mTOR kinase inhibitors. J Chem Biol. 2008;1:27–36. doi: 10.1007/s12154-008-0003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li J, Kim SG, Blenis J. Rapamycin: One drug, many effects. Cell Metab. 2014;19:373. doi: 10.1016/j.cmet.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, Heffernan TP, Vakoc CR, Bergsagel PL, Ghobrial IM, Richardson PG, Young RA, Hahn WC, Anderson KC, Kung AL, Bradner JE, Mitsiades CS. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bunnage ME, Chekler ELP, Jones LH. Target validation using chemical probes. Nat Chem Biol. 2013;9:195–199. doi: 10.1038/nchembio.1197. [DOI] [PubMed] [Google Scholar]
- 7.Workman P, Collins I. Probing the Probes: Fitness Factors For Small Molecule Tools. Chem Biol. 2010;17:561. doi: 10.1016/j.chembiol.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oprea TI, Bologa CG, Boyer S, Curpan RF, Glen RC, Hopkins AL, Lipinski CA, Marshall GR, Martin YC, Ostopovici-Halip L, Rishton G, Ursu O, Vaz RJ, Waller C, Waldmann H, Sklar LA. A crowdsourcing evaluation of the NIH chemical probes. Nat Chem Biol. 2009;5:441–7. doi: 10.1038/nchembio0709-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frye SV. The art of the chemical probe. Nat Chem Biol. 2010;6:159–161. doi: 10.1038/nchembio.296. [DOI] [PubMed] [Google Scholar]
- 10.Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40:D1100. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bento AP, Gaulton A, Hersey A, Bellis LJ, Chambers J, Davies M, Krüger FA, Light Y, Mak L, McGlinchey S, Nowotka M, Papadatos G, Santos R, Overington JP. The ChEMBL bioactivity database: An update. Nucleic Acids Res. 2014;42:D1083. doi: 10.1093/nar/gkt1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu G, Batchelor C, Dumontier M, Hastings J, Willighagen E, Bolton E. PubChemRDF: Towards the semantic annotation of PubChem compound and substance databases. J Cheminf. 2015 doi: 10.1186/s13321-015-0084-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wickham H, Francois R. dplyr: A Grammar of Data Manipulation. R Packag version 0.4.2.3. 2015 [Google Scholar]
- 14.Willett P, Barnard JM, Downs GM. Chemical Similarity Searching. J Chem Inf Comput Sci. 1998;38:983–996. [Google Scholar]
- 15.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 16.Wu C, MacLeod I, Su AI. BioGPS and MyGene.info: Organizing online, gene-centric information. Nucleic Acids Res. 2013;41:D561. doi: 10.1093/nar/gks1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xin J, Mark A, Afrasiabi C, Tsueng G, Juchler M, Gopal N, Stupp GS, Putman TE, Ainscough BJ, Griffith OL, Torkamani A, Whetzel PL, Mungall CJ, Mooney SD, Su AI, Wu C. High-performance web services for querying gene and variant annotation. Genome Biol. 2016;17:91. doi: 10.1186/s13059-016-0953-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shannon PT, Grimes M, Kutlu B, Bot JJ, Galas DJ. RCytoscape: tools for exploratory network analysis. BMC Bioinf. 2013;14:217. doi: 10.1186/1471-2105-14-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo W, Brouwer C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics. 2013;29:1830–1831. doi: 10.1093/bioinformatics/btt285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu G, He Q-Y. ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Mol BioSyst. 2016;12:477–479. doi: 10.1039/c5mb00663e. [DOI] [PubMed] [Google Scholar]
- 22.Saini KS, Loi S, de Azambuja E, Metzger-Filho O, Saini ML, Ignatiadis M, Dancey JE, Piccart-Gebhart MJ. Targeting the PI3K/AKT/mTOR and Raf/MEK/ERK pathways in the treatment of breast cancer. Cancer Treat Rev. 2013;39:935. doi: 10.1016/j.ctrv.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 23.Meng J, Dai B, Fang B, Bekele B, Bornmann WG, Sun D, Peng Z, Herbst RS, Papadimitrakopoulou V, Minna JD, Peyton M, Roth JA. Combination treatment with MEK and AKT inhibitors is more effective than each drug alone in human non-small cell lung cancer in vitro and in vivo. PLoS One. 2010;5:e14124. doi: 10.1371/journal.pone.0014124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Temraz S, Mukherji D, Shamseddine A. Dual inhibition of MEK and PI3K pathway in KRAS and BRAF mutated colorectal cancers. Int J Mol Sci. 2015;16:22976. doi: 10.3390/ijms160922976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jokinen E, Laurila N, Koivunen JP. Alternative dosing of dual PI3K and MEK inhibition in cancer therapy. BMC Cancer. 2012;12:612. doi: 10.1186/1471-2407-12-612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kerrien S, Aranda B, Breuza L, Bridge A, Broackes-Carter F, Chen C, Duesbury M, Dumousseau M, Feuermann M, Hinz U, Jandrasits C, Jimenez RC, Khadake J, Mahadevan U, Masson P, Pedruzzi I, Pfeiffenberger E, Porras P, Raghunath A, Roechert B, Orchard S, Hermjakob H. The IntAct molecular interaction database in 2012. Nucleic Acids Res. 2012;40:D841. doi: 10.1093/nar/gkr1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hermjakob H, Montecchi-Palazzi L, Lewington C, Mudali S, Kerrien S, Orchard S, Vingron M, Roechert B, Roepstorff P, Valencia A, Margalit H, Armstrong J, Bairoch A, Cesareni G, Sherman D, Apweiler R. IntAct: an open source molecular interaction database. Nucleic Acids Res. 2004;32:452D–5. doi: 10.1093/nar/gkh052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye S, Tan L, Yang R, Fang B, Qu S, Schulze EN, Song H, Ying Q, Li P. Pleiotropy of glycogen synthase kinase-3 inhibition by CHIR99021 promotes self-renewal of embryonic stem cells from refractory mouse strains. PLoS One. 2012;7:e35892. doi: 10.1371/journal.pone.0035892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diersch S, Wirth M, Schneeweis C, Jörs S, Geisler F, Siveke JT, Rad R, Schmid RM, Saur D, Rustgi AK, Reichert M, Schneider G. Kras(G12D) induces EGFR-MYC cross signaling in murine primary pancreatic ductal epithelial cells. Oncogene. 2016;35:3880–3886. doi: 10.1038/onc.2015.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiao D, Yue M, Su H, Ren P, Jiang J, Li F, Hu Y, Du H, Liu H, Qing G. Polo-like Kinase-1 Regulates Myc Stabilization and Activates a Feedforward Circuit Promoting Tumor Cell Survival. Mol Cell. 2016;64:493–506. doi: 10.1016/j.molcel.2016.09.016. [DOI] [PubMed] [Google Scholar]
- 31.Wang L, Zhao Z, Meyer M, Saha S, Yu M, Guo A, Wisinski K, Huang W, Cai W, Pike JW, Yuan M, Ahlquist P, Xu W. CARM1Methylates Chromatin Remodeling Factor BAF155 to Enhance Tumor Progression and Metastasis. Cancer Cell. 2014;25:21–36. doi: 10.1016/j.ccr.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vermaak D, Malik HS. Multiple Roles for Heterochromatin Protein 1 Genes in Drosophila. Annu Rev Genet. 2009;43:467–492. doi: 10.1146/annurev-genet-102108-134802. [DOI] [PubMed] [Google Scholar]
- 34.Hathaway NA, Bell O, Hodges C, Miller EL, Neel DS, Crabtree GR. Dynamics and memory of heterochromatin in living cells. Cell. 2012;149:1447–1460. doi: 10.1016/j.cell.2012.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Methot JL, Chakravarty PK, Chenard M, Close J, Cruz JC, Dahlberg WK, Fleming J, Hamblett CL, Hamill JE, Harrington P, Harsch A, Heidebrecht R, Hughes B, Jung J, Kenific CM, Kral AM, Meinke PT, Middleton RE, Ozerova N, Sloman DL, Stanton MG, Szewczak AA, Tyagarajan S, Witter DJ, Paul Secrist J, Miller TA. Exploration of the internal cavity of histone deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1:2) Bioorg Med Chem Lett. 2008;18:973–978. doi: 10.1016/j.bmcl.2007.12.031. [DOI] [PubMed] [Google Scholar]
- 36.Payton M, Bush TL, Chung G, Ziegler B, Eden P, McElroy P, Ross S, Cee VJ, Deak HL, Hodous BL, Nguyen HN, Olivieri PR, Romero K, Schenkel LB, Bak A, Stanton M, Dussault I, Patel VF, Geuns-Meyer S, Radinsky R, Kendall RL. Preclinical evaluation of AMG 900, a novel potent and highly selective pan-aurora kinase inhibitor with activity in taxane-resistant tumor cell lines. Cancer Res. 2010;70:9846–9854. doi: 10.1158/0008-5472.CAN-10-3001. [DOI] [PubMed] [Google Scholar]
- 37.Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V, Wigle TJ, Dimaggio PA, Wasney GA, Siarheyeva A, Dong A, Tempel W, Wang S-C, Chen X, Chau I, Mangano TJ, Huang X-P, Simpson CD, Pattenden SG, Norris JL, Kireev DB, Tripathy A, Edwards A, Roth BL, Janzen WP, Garcia BA, Petronis A, Ellis J, Brown PJ, Frye SV, Arrowsmith CH, Jin J. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat Chem Biol. 2011;7:566–74. doi: 10.1038/nchembio.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anderson VE, Walton MI, Eve PD, Boxall KJ, Antoni L, Caldwell JJ, Aherne W, Pearl LH, Oliver AW, Collins I, Garrett MD. CCT241533 is a potent and selective inhibitor of CHK2 that potentiates the cytotoxicity of PARP inhibitors. Cancer Res. 2011;71:463–472. doi: 10.1158/0008-5472.CAN-10-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nightingale KP, Gendreizig S, White DA, Bradbury C, Hollfelder F, Turner BM. Cross-talk between histone modifications in response to histone deacetylase inhibitors: MLL4 links histone H3 acetylation and histone H3K4 methylation. J Biol Chem. 2007;282:4408–4416. doi: 10.1074/jbc.M606773200. [DOI] [PubMed] [Google Scholar]
- 40.Salton M, Voss TC, Misteli T. Identification by high-throughput imaging of the histone methyltransferase EHMT2 AS an epigenetic regulator of VEGFA alternative splicing. Nucleic Acids Res. 2014;42:13662–13672. doi: 10.1093/nar/gku1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bolderson E, Savage KI, Mahen R, Pisupati V, Graham ME, Richard DJ, Robinson PJ, Venkitaraman AR, Khanna KK. Kr??ppel-associated box (KRAB)-associated co-repressor (KAP-1) Ser-473 phosphorylation regulates heterochromatin protein 1?? (HP1-??) mobilization and DNA repair in heterochromatin. J Biol Chem. 2012;287:28122–28131. doi: 10.1074/jbc.M112.368381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Connor KA, Roth BL. Finding new tricks for old drugs: an efficient route for public-sector drug discovery. Nat Rev Drug Discovery. 2005;4:1005–1014. doi: 10.1038/nrd1900. [DOI] [PubMed] [Google Scholar]
- 43.Lounkine E, Keiser MJ, Whitebread S, Mikhailov D, Hamon J, Jenkins JL, Lavan P, Weber E, Doak AK, Côté S, Shoichet BK, Urban L. Large-scale prediction and testing of drug activity on side-effect targets. Nature. 2012;486:361–7. doi: 10.1038/nature11159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y, Cornett A, King FJ, Mao Y, Nigsch F, Paris CG, McAllister G, Jenkins JL. Evidence-Based and Quantitative Prioritization of Tool Compounds in Phenotypic Drug Discovery. Cell Chem Biol. 2016;23:862–874. doi: 10.1016/j.chembiol.2016.05.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.