

Abstract
In mucosal inflammatory disorders, the protective influence of heme oxygenase-1 (HO-1) and its metabolic by-products, carbon monoxide and biliverdin, is a topic of significant interest. Mechanisms under investigation include regulation of macrophage function and mucosal cytokine expression. While there is an increasing recognition of the importance of epithelial-derived factors in the maintenance of intestinal mucosal homeostasis, the contribution of intestinal epithelial (IEC) HO-1 on inflammatory responses has not previously been investigated. We examined the influence of modulating HO-1 expression on the inflammatory response of human IECs. Engineered deficiency of HO-1 in Caco-2 and T84 IECs lead to increased proinflammatory chemokine expression in response to pathogenic bacteria and inflammatory cytokine stimulation. Crosstalk with activated leukocytes also lead to increased chemokine expression in HO-1 deficient cells in an IL-1β dependent manner. Treatment of Caco-2 cells with a pharmacological inducer of HO-1 lead to inhibition of chemokine expression. Mechanistic studies suggest that HO-1 and HO-1 related transcription factors, not its metabolic products, are partly responsible for its influence on chemokine expression. In conclusion, our data identifies HO-1 as a central regulator of IEC chemokine expression that may contribute to homeostasis in the intestinal mucosa.
Keywords: intestinal epithelium, cytokines, chemokines, inflammation, homeostasis, colitis
Background
Epithelial cells are unique regulators of the mucosal immune response related to their function serving as the barrier between the external luminal environment and the mucosal immune system. Crosstalk between the epithelium and the immune system is dictated by a complex interplay of host genetic factors, the luminal microbiota and environmental contributions. Despite exposure to high concentrations of various potentially inflammatory environmental and bacterial antigens on their apical surface, intestinal epithelial cells contribute to the immune tolerant state of the intestinal mucosa through dampened homeostatic responses, in the absence of barrier disruption. Intestinal epithelial cells additionally have the complicated role of regulating uptake of nutritionally derived macromolecules from the intestinal lumen. One of these molecules is heme, which is preferentially absorbed by the proximal small intestine. Heme is metabolized by the heme oxygenase group of enzymes (HO) into biliverdin, carbon monoxide and ferrous iron. HO-1 is the inducible isoform of HO, which is present in most tissues of the body but highly expressed in the liver, spleen and the intestines [1]. HO-1 and its metabolites have been demonstrated to have anti-inflammatory, anti-apoptotic and anti-oxidative properties [2, 3]. HO-1 induction and CO exposure are protective in multiple murine models of inflammatory disease, including models of experimental colitis, through the regulation of pro- and anti-inflammatory cytokine expression, and promotion of macrophage antibacterial function [4–7].
While an anti-inflammatory role has been ascribed to immune cell HO-1/CO signaling, the influence of HO-1 in intestinal epithelial cells has not been well explored. There is an increased appreciation of the role of epithelial derived factors in mucosal immunity [8]. While the epithelium may not be the major source of cytokines produced in the intestinal mucosa, it is a rich source of various homeostatic cytokines. For example, epithelial derived IL-8 and TGF-beta are critically important for recruiting the resident mucosal macrophage population [9]. The epithelium also plays a critical role in the response to pro- and anti-inflammatory cytokine signaling [8]. Disruption of the epithelial barrier results in bacterial invasion and an organized innate immune response including the elaboration of several chemoattractants from the epithelium. Several epithelial derived chemokines are implicated in the pathogenesis of inflammatory bowel disease [10–13]. Recruitment to and trafficking of neutrophils across the epithelium is a consistent feature in conditions like human ulcerative colitis, where findings such as crypt abscesses are common manifestations.
We have previously shown that transmigration of neutrophils across epithelial cells molds epithelial gene expression, most prominently through localized oxygen depletion and stabilization of hypoxia-inducible factor (HIF) [14]. This signaling promotes protective/restorative epithelial responses in the setting of inflammation [15]. One of the genes upregulated in this setting is the HO-1 gene HMOX1. Here we have evaluated the influence of epithelial HO-1 on immune responses from IECs and our findings suggest that HO-1 contributes a regulatory check on epithelial inflammatory responses and may contribute to maintaining a homeostatic balance in the mucosa.
Results
HO-1 deficiency enhances chemokine release from human IECs
During active mucosal inflammation, the epithelium coordinates site specific recruitment of leukocytes to the intestinal mucosa through the elaboration of chemokines [16]. This is often augmented by signaling from resident immune cells like macrophages and their proinflammatory cytokines such as IL-1β and TNF [8]. To determine whether HO-1 influences IEC inflammatory responses, HO-1 was knocked down in the Caco-2 cells (HO-1 KD) using lentiviral transduced vectors expressing HMOX1 shRNA, with a corresponding non-targeting control (NTC) shRNA vector expressing line. A significant reduction in HMOX1 mRNA and protein was determined by qPCR (Supplemental Fig 1A) and ELISA (Fig 1A) compared to the NTC shRNA transduced cells. Knockdown of HO-1 was also achieved using the same methods in T84 epithelial cells (Supplemental Fig 1B), with comparable success.
Figure 1. HO-1 regulates chemokine expression in Caco-2 IECs.
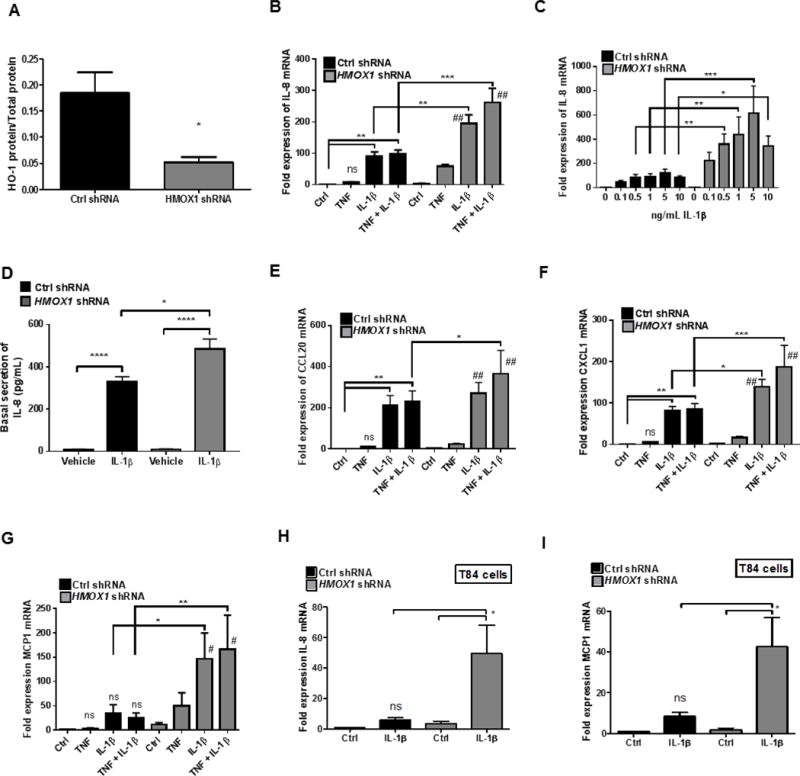
Lentiviral transduction of HMOX1 shRNA and NTC shRNA (Ctrl) in WT Caco-2 IECs was performed and HO-1 protein was analyzed by ELISA (A). Expression of chemokines by confluent monolayers 4h after stimulation with IL-1β (10 ng/mL, unless otherwise stated) and/or TNF was compared to vehicle (Ctrl) in HMOX1 shRNA and Ctrl shRNA transduced Caco-2 IECs as measured by qPCR and ELISA for IL-8 (B–D), CCL20 (E), CXCL1 (F) and MCP1 (G). IL-1β (5 ng/mL) induced expression of IL-8 (H) and MCP1 (I) was also compared to vehicle in T84 HMOX1 shRNA and Ctrl shRNA IECs. Data represent combined results from at least 3 independent experiments. *, p <0.05; **, p <0.01; ***, p <0.001; ****, p <0.0001 compared to untreated ctrl or other comparison shown on graph. #, p <0.01 and ## p <0.0001 vs untreated ctrl HMOX1 shRNA group.
To define the contribution of IEC HO-1 to stimulated chemokine expression, we modeled inflammation by exposing IECs to recombinant IL-1β, TNFα or both and expression of various epithelial chemokines was assessed by qPCR and ELISA. TNα and especially IL-1β, induced chemokine expression in both Caco-2 and T84 cells. Targeted knockdown of HO-1 resulted in a significant increase in cytokine-induced expression of the chemokines IL-8 (CXCL8, Fig 1B–D), CCL20 (MIP3A, Fig 1E), CXCL1 (Fig 1F) and MCP-1 (CCL2, Fig 1G). Indeed, the loss of HO-1 enhanced mRNA expression of IL-8 by as much as 5 times over similarly stimulated NTC shRNA cells (Fig 1C, p<0.001). To a lesser extent than mRNA, such increases were also reflected at the protein level (p<0.05, Fig 1D).
To demonstrate that these results were not cell line specific, similar analysis inT84 cells revealed that the loss of HO-1 resulted in significantly increased expression of IL-1β-stimulated IL-8 and MCP-1 (p<0.05, Fig 1H and I, respectively). Such findings strongly implicate the HO-1 pathway in the control of epithelial chemokine expression.
Epithelial-leukocyte crosstalk is regulated by HO-1
To extend these results and explore the influence of epithelial HO-1 on leukocyte-epithelial crosstalk, we utilized a co-culture model of IECs and monocytes/macrophages to simulate interactions that may occur in vivo. Monolayers of polarized Caco-2 cells were grown on the upper side of Transwell inserts and co-cultured with THP-1 monocytes in the lower chamber. To activate co-cultures, we infected THP-1 cells (2×10^5) with the invasive pathogen Salmonella Typhimurium (2×106), then harvested epithelial mRNA and profiled chemokine expression (see model shown in Fig 2A).
Figure 2. Epithelial HO-1 regulates leukocyte-epithelial crosstalk.
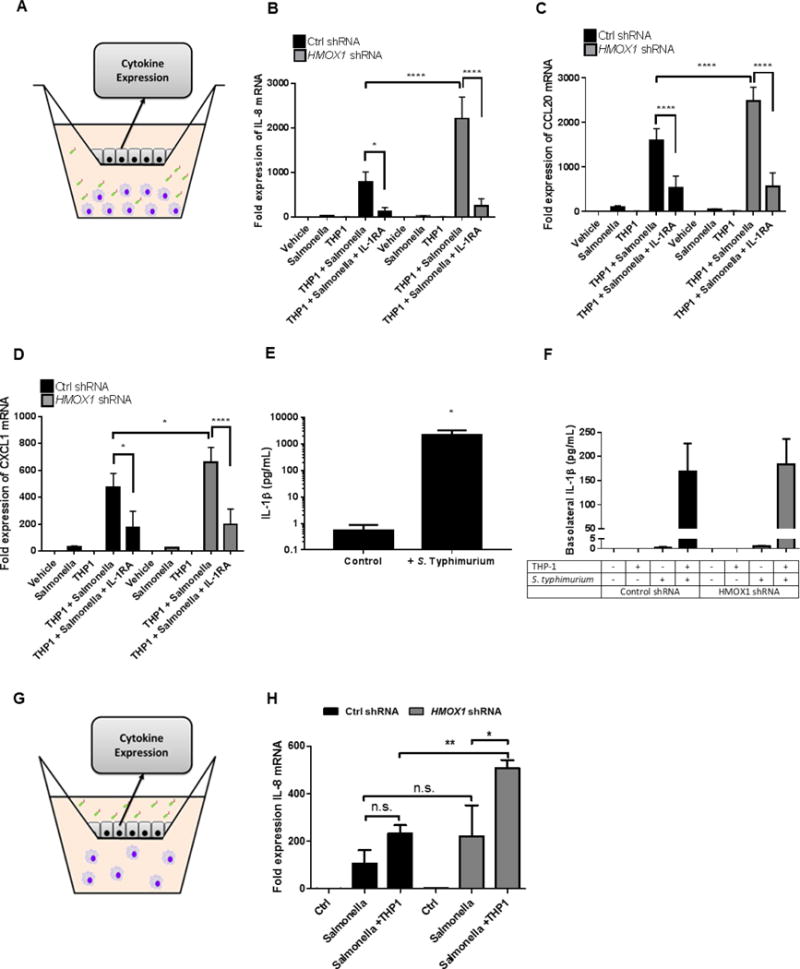
(A) Model to examine epithelial-leukocyte interactions using polarized Caco-2 IECs in the upper chamber of a Transwell insert with THP-1 monocytes in the lower chamber, activated with S. Typhimurium for 6h for mRNA or 24h for protein. (B–D) Expression of IL-8, CCL20 and CXCL1 mRNA from Caco-2 IECs during co-culture with activated THP-1 cells. (E) IL-1β secretion from THP-1 cell supernatants after 24h, as measured by ELISA. (F) IL-1 β secretion from THP-1 cells co-cultured with Control shRNA and HMOX1 shRNA transduced Caco-2 IECs after 24h. Data examine physiologic bacterial-epithelial activation using polarized IECs in the upper chamber of a Transwell insert exposed to enteric bacteria in the presence (or absence) or leukocytes in the lower chamber, evaluating IEC chemokine expression. (H) Expression of IL-8 mRNA from Caco-2 IECs 6h after exposure of apical surface to pathogenic bacteria in presence or absence of THP-1 monocytes. Combined results from 3 independent experiments *, p <0.05; ***, p <0.001; ****, p <0.0001.
Utilizing this model system, we defined whether THP-1-derived IL-1β would stimulate epithelial chemokine expression and whether IEC HO-1 regulated such responses. As shown in Figure 2, S. Typhimurium infected THP-1 cells significantly increased the expression of IL-8, CCL20 and CXCL1 mRNA in both NTC shRNA and HO-1 KD IECs (p<0.01 for each, Fig 2B, C and D, respectively). This response was confirmed to be primarily driven by THP-1-derived IL-1β since the addition of an IL-1 receptor antagonist (Anakinra) markedly diminished this chemokine response (p<0.001 for IL-8, CCL20 and CXCL1). We verified that in these defined conditions, S. Typhimurium induced prominent expression and secretion of IL-1β from THP-1 cells (Fig 2E, p<0.05) and IL-1β secretion was consistent when exposed to NTC shRNA and HO-1 KD cells (Fig 2F).
To evaluate the influence of epithelial HO-1 on responses to luminal bacteria, we exposed the apical surface of the polarized IECs to a gram-negative enteric pathogenic bacteria (S. Typhimurium) in the presence and absence of THP-1 cells (Fig 2G). After 6h of exposure, we evaluated the expression of IL-8 from our control and HO-1 KD IECs using qPCR. S. Typhimurium elicited an increase in IL-8 expression that was enhanced in the HO-1 deficient cells (Fig 2H). This was significantly increased in the presence of THP-1 cells (p<0.01). Prior work has previously demonstrated crosstalk between IECs and leukocytes can promote an inflammatory response to bacterial exposure to IEC apical membranes.[17] These results suggest that HO-1 in part, also regulates the magnitude of epithelial inflammatory responses to bacterial challenge.
Consistent with our findings of direct stimulation with recombinant IL-1β (Fig 1), the knockdown of IEC HO-1 enhanced IL-8 (p<0.0001), CCL20 (p<0.0001) and CXCL1 (p<0.05) mRNA responses to activated THP-1 cells and pathogenic bacteria (p<0.01), indicating that even in a more complex and physiologically-relevant setting, the HO-1 pathway is central to the regulated IEC expression of chemokines.
Induction of HO-1 inhibits IEC chemokine release
We next sought to determine if alternatively, increasing HO-1 could impact IEC chemokine expression. To do this, endogenous HO-1 was induced in the WT Caco-2 cell line using CoPP, a potent heme analog small-molecule inducer of HO-1 expression. Treatment with CoPP resulted in a significant increase in HO-1 mRNA (Fig 3A, p<0.01) and protein (Fig 3B) expression. Compared to vehicle controls, CoPP pretreated cells, subsequently treated with IL-1β, demonstrated diminished induction of the chemokines IL-8 and CCL20, but not CXCL1 (Fig 3C–D).
Figure 3. HO-1 induction inhibits chemokine expression.
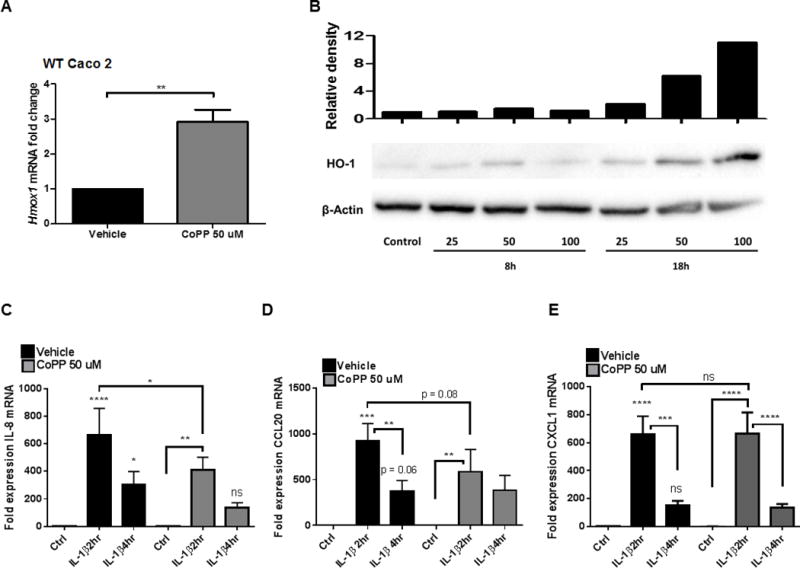
WT Caco-2 IECs were treated with 50μM of CoPP or vehicle overnight and HMOX1 expression measured by qPCR (A) and western blot (B). After vehicle or CoPP treatment, WT Caco-2 cells were treated with 10 ng/mL of IL-1β for 2 and 4h and chemokine expression assessed by qPCR (C–E). Combined results from 3 experiments. *, p <0.05; **, p <0.01; ***, p <0.001; ****, p <0.0001 compared to untreated vehicle ctrl or other comparison shown on graph.
HO-1 metabolic products do not suppress IL-8 expression
Consequences of HO-1 induction are frequently mediated by HO-1 metabolic products such as CO and biliverdin, which is further metabolized to bilirubin. Caco-2 IECs have been demonstrated to uptake and metabolize exogenous HO-1 metabolic products.[18–20] To determine if these molecules regulate chemokine expression we treated our control and HO-1 deficient IECs overnight with biliverdin and its product bilirubin (Fig 4A–B). Neither biliverdin nor bilirubin significantly impacted IL-1β induced IL-8 expression and there was no significant change in the relative IL-8 expression in the HO-1 deficient cells compared to controls. Alternatively, exogenous CO has been demonstrated to have an inhibitory influence on chemokine expression in IECs stimulated with a mixture of cytokines (IL-1β, TNF and IFN-γ) [18]. We exposed our Caco-2 cells briefly to CO via a CO releasing molecule (CORM-2 at 100μM) as previously described and again examined IL-8 release after stimulation with IL-1β. Contrary to prior studies we did not see a significant decrease in IL-8 secretion, instead we saw a small increase suggesting a more complicated mechanism of regulation of IL-8 in response to IL-1β alone compared to a mixture of cytokines (Fig 4C).
Figure 4. Role of HO-1 metabolic products.
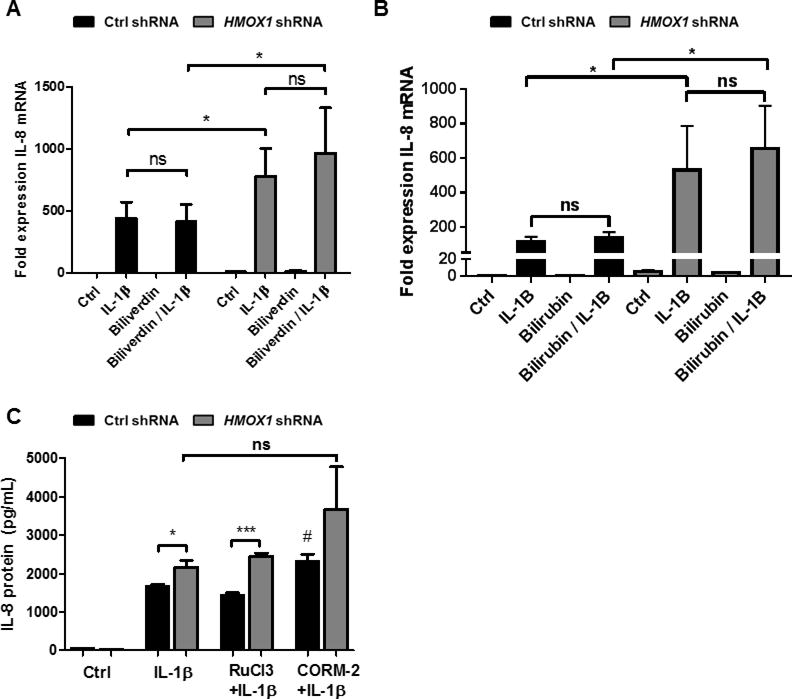
(A–B) Control and HO-1 deficient Caco2 IECs were treated with biliverdin and bilirubin (50 μM) overnight and then stimulated with IL-1β (1 ng/mL) for 3h. IL-8 was measured by qPCR. Data reflects combined results from at least three independent experiments. (C) Cells were exposed to freshly prepared CO releasing molecule, CORM-2 for 1h or its negative control RuCl3, prior to stimulation with IL-1β (1 ng/mL) for 24h. IL-8 was measured by cytokine immunoassay and normalized to mg total protein. Data are representative of results from two independent experiments *, p < 0.05, ***, p < 0.001 vs untreated control NTC shRNA cells or other comparison shown. #, p < 0.05 vs IL-1β treated NTC shRNA cells.
Mechanisms of regulation of epithelial chemokines by HO-1
To determine the role of transcriptional regulation of chemokine expression by HO-1, Actinomycin D (ActD) was used to block transcription in IL-1β treated Caco-2 cells. After 1h of IL-1β exposure, significantly higher levels of IL-8 were expressed in the HO-1 KD cells compared to NTC shRNA cells. The addition of ActD after 1h subsequently resulted in a marked and similar reduction in IL-8 expression in both controls and HO-1 KD cells at 2–4h (Fig 5C), indicating that the enhanced IL-8 expression in HO-1 KD IECs is secondary to increased transcriptional activation not stabilization of the mRNA.
Figure 5. Mechanisms of transcriptional regulation by HO-1.
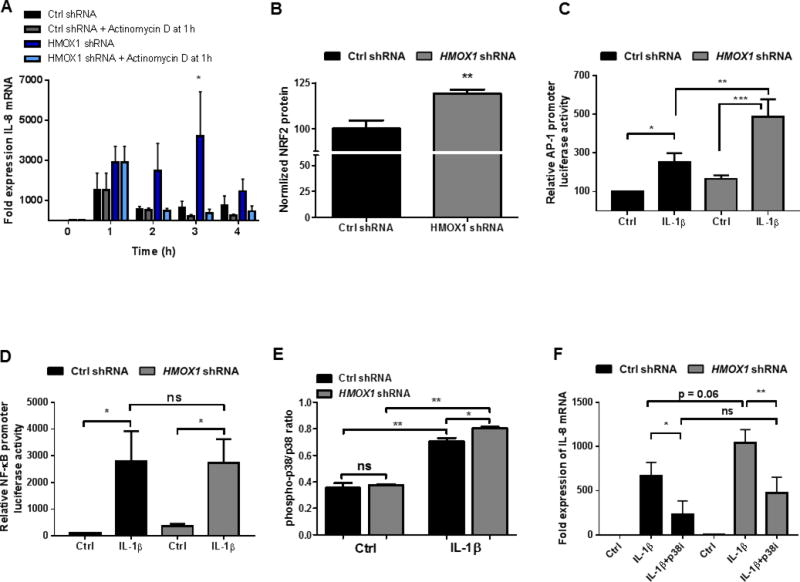
(A) Expression of IL-8 in confluent NTC shRNA (Ctrl) and HMOX1 shRNA transduced Caco-2 IECs treated with IL-1β (1 ng/mL) +/− transcriptional blockade with Actinomycin D after 1h. (B) Expression of NRF2 protein at baseline normalized to cell density via Crystal Violet staining and presented as % of control. Activation of AP-1 (C) and NF-κB (D) transcription factors by IL-1β (1 ng/mL) was measured using luciferase reporter plasmid transfection in NTC shRNA and HMOX1 shRNA transduced Caco-2 IECs. (E) Activation (phosphorylation) of p38 MAPK was assessed at baseline and in the setting of IL-1β treatment (1 ng/mL × 30 minutes) by cell-based ELISA. (F) The influence of inhibition of p38 MAPK on the increased expression of IL-8 in HMOX1 shRNA Caco-2 cells was assessed by pretreatment with a p38 MAPK inhibitor (SB202190 at 40 μM) 1h prior to IL-1β (1 ng/mL) exposure for 2h. Data represent combined results from at least 3 independent experiments. *, p <0.05; **, p <0.01.
The transcription factor, nuclear factor erythroid 2-related factor 2 (NRF2) is activated under cellular stress and inflammation and is largely responsible for activation of HMOX1 transcription in mice and humans.[21] NRF2 has also been shown to promote IL-8 transcription in certain cells [22, 23] A likely compensatory increase in NRF2 in the setting of HO-1 deficiency was felt to potentially play a role in IL-8 expression. Therefore we measured NRF2 protein in our control and HO-1 KD Caco-2 IECs by indirect ELISA (Fig 5B). We found significantly increased NRF2 protein in our HO-1 KD cells. Previous studies have also shown that the transcription factors AP-1 and NF-κB are critical to activation of IL-8 transcription by IL-1β [24, 25]. Therefore, we examined the relationship between HO-1 expression and activity of these transcription factors using luciferase reporter constructs. Caco-2 cells were transiently transfected with AP-1 and NF-κB reporter plasmids and treated with IL-1β (1 ng/mL for 6h), as shown in Figure 5C and D. HO-1 deficiency increased IL-1β-induced AP-1 (p<0.05), but not NF-κB (p = not significant) activity, implicating a role for AP-1 in HO-1-dependent increases in chemokine expression. The relationship between MAPKs, AP-1 and induction of IL-8 transcription has been previously described [25]. In addition, p38 MAPK has also been demonstrated to regulate HO-1 via NRF2, while inhibition of HO-1 increases p38 MAPK activation.[26, 27] Therefore, we assessed the level of activated (phosphorylated) p38 MAPK (controlled for total p38) in our HO-1 deficient cells compared to controls and found that after IL-1β treatment there was a small but significant increase in phospho-p38 in the HO-1 deficient IECs (Fig 5E). We then inhibited p38 MAPK to determine if it played a role in the increased IL-8 expression seen in our HO-1 KD cells in response to IL-1β treatment. As shown in Figure 5F, inhibition of p38 MAPK significantly inhibited the HO-1-dependent enhancement of IL-8 expression at 2h. Such results implicate, at least in part, p38 MAPK activation in the increased chemokine induction in the setting of HO-1 deficiency.
Discussion
The intricate maintenance of an effective mucosal immune system in the GI tract is composed of numerous components working in concert [28]. Induction of HO-1 in the intestine and delivery of its metabolite, CO, has previously been shown to contribute to the maintenance of homeostasis in various murine models of colitis [4–6]. This has been associated with augmented macrophage function including a balance towards increased anti-inflammatory cytokine and decreased pro-inflammatory cytokine expression [6, 29]. The role of the intestinal epithelium in the protective influence of the HO-1 pathway has not been evaluated.
The increasingly understood importance of epithelial derived factors and responses has expanded our view of the epithelium as an important contributor to the immune response, and regulator of mucosal homeostasis [8, 9, 30]. Seemingly dysregulated expression of epithelial derived chemokines has been strongly associated with the pathogenesis of IBD [10, 12, 31, 32]. The result of increased chemokine expression in the intestine typically includes increased recruitment and activation of immune cells and increased pro-inflammatory cytokine expression in the mucosa [8] and can lead to a loss of epithelial barrier integrity, increased bacterial invasion and perpetuation of inflammation [30]. Our results identify a prominent role for HO-1 in the regulation of epithelial chemokine expression. Using HO-1 loss and gain of function approaches, we have found that the epithelial HO-1 pathway regulates epithelial chemokine production and serves as an endogenous “braking” mechanism to the mucosal inflammatory response.
As part of these studies, we developed a co-culture model system to define soluble factor cross-talk between monocyte/macrophage and intestinal epithelial cells. We previously identified the induction of epithelial HO-1 in a cohort of genes upregulated during neutrophil transepithelial migration [14]. In this setting, neutrophils induce HIF [14], and given that HO-1 is a well-established HIF target gene [33], it is likely that HIF contributes epithelial HO-1 expression in this co-culture system. Consequently, we have shown that HO-1 deficiency in IECs leads to an elevated inflammatory response demonstrated by markedly increased pro-inflammatory chemokine expression. This appears to be the case both in response to macrophage derived cytokines and exposure to pathogenic bacteria. Similar findings have recently been demonstrated for alveolar epithelial cells in a murine model of acute lung injury [34]. Diminished levels of LPS-induced chemokine expression and neutrophil migration was observed after HO-1 induction and this was found to be dependent on non-myeloid HO-1 including pulmonary epithelial cells and endothelial cells.
The regulation of inflammatory responses by HO-1 has been demonstrated in other cell types as well [35]. Induction of HO-1 in astrocytes, for example, has been reported to inhibit NO production in response to IL-1β treatment through modulation of p38 MAPK activation. Helicobacter pylori infection induced secretion of IL-8 by gastric epithelial cells also appears to be inhibited by HO-1 induction [36]. The findings of HO-1 regulation of IL-8 secreted by IECs is particularly important given prior evidence that IECs and intestinal mast cells are the main source of basal intestinal IL-8 secretion [9]. Such basal secretion is responsible for the recruitment and protective retention of intestinal macrophages, but in excess, can contribute to IBD pathogenesis [11]. We propose that in this setting, HO-1 sub-serves the mucosal inflammatory response by functioning as an endogenous braking mechanism to uncontrolled chemokine production.
Prior studies have shown that the HO-1 metabolic by-product CO likely plays an intermediate step in regulation of cytokine expression in macrophages by modulating MAPKs and AP-1 [37, 38]. In addition, exogenous CO (in the form of CORM-2) inhibits the inflammatory response in Caco-2 cells to a mix of inflammatory cytokines.[18] This response included inhibition of IL-8 expression in Caco-2 cells and the contribution of AP-1, NF-κB and MAPK family signaling. We have extended these findings by showing the importance of endogenous HO-1 levels on IEC responses to pathogenic bacteria, on IEC-leukocyte signaling, and on a broader range of IEC chemokines associated with IBD. Additionally, we have shown that the HO-1 metabolic by-product, biliverdin, which has anti-inflammatory properties in other settings, does not appear to be responsible for the regulation of IEC chemokines we have demonstrated. However, in contrast to prior studies we did not see inhibition of IL-8 expression with CO exposure when stimulating with a single cytokine, IL-1β in the presence of CORM-2 in either our control or HO-1 deficient cells. Instead we saw slightly increased IL-8 relative to controls. The influence of CO may be specific to the mix of cytokines used; here we used only IL-1β alone in contrast to the prior study which stimulated cells with a combination of three different inflammatory cytokines.[18] Alternative mechanisms for our findings include a role for HO-1 protein independent of its metabolites which has been demonstrated in other settings.[39–41] In this regard, there is evidence that HO-1 protein helps directly coordinate the transcriptional activity of NRF2 and related oxidative stress genes. We demonstrated a likely compensatory increase in NRF2 protein in our HO-1 deficient cells which if dysregulated may contribute to increased chemokine transcription (such as IL-8).[22] Not surprisingly, p38 MAPK and CO are both activators of NRF2 which may explain the contrasting responses we found to inhibiting p38 and CO treatment.[42–44]
In conclusion, we identify HO-1 as a central regulator of epithelial chemokine expression. Using loss and gain of function approaches, we demonstrate that HO-1, through regulation of transcriptional mechanisms, controls the expression of multiple IEC-derived chemokines. In the absence of such control, as occurs in diseases such as IBD, mucosal inflammation may proceed in an unchecked manner. These results further suggest that administration of HO-1 inducing agents (e.g. certain heme analogs) and/or promoting HO-1 gene expression (e.g. HIF stabilizers) may be of therapeutic benefit.
Methods
Reagents
Cobalt(III) protoporphyrin IX chloride (CoPP) was from Frontier Scientific (Logan, Utah). Human TNF and IL-1β cytokines were from Invitrogen (Carlsbad, CA). Bacterial strains used were Salmonella enterica serovar Typhimurium ATCC: 700408. Actinomycin D, Tricarbonyldichlororuthenium (II) dimer (CORM-2) and its negative control Ruthenium (II) chloride hydrate (RuCl3) were obtained from Sigma-Aldrich (St. Louis, MO). An inhibitor of the p38 mitogen activated protein kinase, SB202190 was obtained from Santa Cruz Biotechnology (Dallas, Texas). Compounds were tested for cytotoxicity using the Cell Counting Kit-8 (Sigma-Aldrich) with all compounds used demonstrating >90% viability within the experimental parameters compared to untreated cells.
Cell Culture
Human Caco-2 and T84 intestinal epithelial cells as well as the monocytic THP-1 cell line were obtained from the American Type Culture Collection (Manassas, VA) and cultured according to standard protocols in 95% air with 5% CO2 at 37°C [45]. Lentiviral transduction of Sigma MISSION® shRNA against HMOX1 (TRCN0000290436) and a control non-targeting shRNA (SHC216) was performed using established protocols (University of Colorado Functional Genomics Facility, Aurora, CO) [46]. THP-1 cells were exposed to bacteria at an MOI of 10:1. IL-1 receptor antagonist (Anakinra, Amgen, Thousand Oaks, CA) was used at a concentration of 100 ng/mL. Polarized IECs were grown on Transwell inserts with 0.4 μm pore size (Corning, Tewksbury, MA) and cultured at least overnight with THP-1 cells for co-culture experiments.
Western Blot
Western immunoblot analyses were performed on whole-cell extracts from Caco-2 cells in RIPA buffer. Blots were incubated overnight with anti-HO-1 (1:1000; ADI-SPA-895, ENZO, Farmingdale, NY) and anti-beta actin (ab8227; Abcam, Cambridge, MA). Immunoreactive proteins were visualized using Gel-Doc imager with Image Lab™ Software (Bio-Rad, Hercules, CA).
Cytokine Immunoassay
Human IL-8 immunoassay (BioLegend, San Diego, CA) was used according to manufacturer’s instructions. Human IL-1β immunoassay (MSD Diagnostics, Rockville, MD) was performed according to manufacturer’s instructions. Human NRF2 Colorimetric Cell-Based ELISA (Assay Biotechnology Company, Fremont, CA) was performed according to manufacturer’s instructions. Results were normalized to cell density as determined by Crystal Violet staining and confirmed with GAPDH as an internal positive control. Activation of human p38 MAPK was assessed using cell-based ELISA to measure levels of phosphorylated Thr180 and Tyr182 compared to total p38 MAPK (antibodies #9211 and #9212, Cell Signaling Technology, Danvers, MA).
Luciferase Assay
Promoter constructs for p-NF-κB-luciferase and p-AP-1 have been described previously [47]. Transfection of Caco-2 cells was performed using Lipofectamine™ LTX with PLUS™ Reagent (ThermoFisher Scientific, Waltham, MA). Following overnight transfection, cells were assayed for luciferase activity at 6h after stimulation, respectively, with IL-1β at 1 ng/mL using the Luciferase Reporter Assay system (Promega, Madison, WI) on a GloMax-Multi Microplate Multimode Reader (Promega, Madison, WI). Luciferase signal was controlled using total protein via Bradford assay or a control renilla luciferase expressing promoter.
Quantitative Polymerase Chain Reaction (qPCR)
RNA was extracted using TRIzol reagent (Invitrogen), and first-strand complementary DNA synthesis was performed with 1μg of total RNA using iScript reverse transcription supermix (Bio-Rad, Hercules, CA). Real-time qPCR was performed using SYBR green (Applied Biosystems, Carlsbad, CA) on an ABI 7900HT Fast Real-Time PCR System (Applied Biosystems). Beta-actin (ACTB) was used as the housekeeping gene. Primer sequences were: ACTB, sense 5′-CACTCTTCCAGCCTTCCTTCC-3′, antisense 5′-CAGGTCTTTGCGGATGTCCACG-3′; HMOX1, sense 5′-CATGACACCAAGGACCAGA-3′, antisense 5′-AGTGTAAGGACCCATCGGAG-3′; IL-8, sense 5′-CTGGCCGTGGCTCTCTTG-3′, antisense 5′-CCTTGGCAAAACTGCACCTT-3′; MIP3a (CCL20), sense 5′-CTGGCTGCTTTGATGTCAGT-3′, antisense 5′-CGTGTGAAGCCCACAATAAA-3′; CXCL1, sense 5′-AACCGAAGTCATAGCCACAC-3′, antisense 5′-GTTGGATTTGTCACTGTTCAGC-3′; MCP1, sense 5′-ATCAATGCCCCAGTCACC-3′, antisense 5′-AGTCTTCGGAGTTTGGG-3′.
Statistical Analysis
Unless otherwise indicated, values are given as means +/− standard error of the mean. Statistical analysis was performed using GraphPad Prism 7 for Windows (GraphPad Software, San Diego, CA). Differences between groups were evaluated using the unpaired Student’s t-test or one-way analysis of variance with Fisher’s LSD where appropriate. Two-tailed P < 0.05 was considered to be significant.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants (DK103639, DK50189, DK95491) and the US Department of Veterans Affairs (Merit Award BX002182).
Footnotes
The authors declare no financial interests in any of the work submitted here.
References
- 1.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86(2):583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 2.Otterbein LE, et al. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol. 2003;24(8):449–55. doi: 10.1016/s1471-4906(03)00181-9. [DOI] [PubMed] [Google Scholar]
- 3.Ryter SW, Choi AM. Targeting heme oxygenase-1 and carbon monoxide for therapeutic modulation of inflammation. Transl Res. 2016;167(1):7–34. doi: 10.1016/j.trsl.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hegazi RA, et al. Carbon monoxide ameliorates chronic murine colitis through a heme oxygenase 1-dependent pathway. J Exp Med. 2005;202(12):1703–13. doi: 10.1084/jem.20051047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sheikh SZ, et al. An anti-inflammatory role for carbon monoxide and heme oxygenase-1 in chronic Th2-mediated murine colitis. J Immunol. 2011;186(9):5506–13. doi: 10.4049/jimmunol.1002433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Onyiah JC, et al. Carbon monoxide and heme oxygenase-1 prevent intestinal inflammation in mice by promoting bacterial clearance. Gastroenterology. 2013;144(4):789–98. doi: 10.1053/j.gastro.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takagi T, et al. Inhalation of carbon monoxide ameliorates TNBS-induced colitis in mice through the inhibition of TNF-alpha expression. Dig Dis Sci. 2010;55(10):2797–804. doi: 10.1007/s10620-009-1112-x. [DOI] [PubMed] [Google Scholar]
- 8.Onyiah JC, Colgan SP. Cytokine responses and epithelial function in the intestinal mucosa. Cell Mol Life Sci. 2016 doi: 10.1007/s00018-016-2289-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smythies LE, et al. Mucosal IL-8 and TGF-beta recruit blood monocytes: evidence for cross-talk between the lamina propria stroma and myeloid cells. J Leukoc Biol. 2006;80(3):492–9. doi: 10.1189/jlb.1005566. [DOI] [PubMed] [Google Scholar]
- 10.Kwon JH, et al. Colonic epithelial cells are a major site of macrophage inflammatory protein 3alpha (MIP-3alpha) production in normal colon and inflammatory bowel disease. Gut. 2002;51(6):818–26. doi: 10.1136/gut.51.6.818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hommes DW, et al. Production and cellular source of interleukin-8 in ulcerative colitis. Inflamm Bowel Dis. 1995;1(2):108–16. [PubMed] [Google Scholar]
- 12.Izutani R, et al. Increased expression of interleukin-8 mRNA in ulcerative colitis and Crohn’s disease mucosa and epithelial cells. Inflamm Bowel Dis. 1995;1(1):37–47. [PubMed] [Google Scholar]
- 13.Atreya R, Neurath MF. Chemokines in inflammatory bowel diseases. Dig Dis. 2010;28(3):386–94. doi: 10.1159/000320392. [DOI] [PubMed] [Google Scholar]
- 14.Campbell EL, et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity. 2014;40(1):66–77. doi: 10.1016/j.immuni.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colgan SP, Campbell EL, Kominsky DJ. Hypoxia and Mucosal Inflammation. Ann Rev Pathol. 2016;11:77–100. doi: 10.1146/annurev-pathol-012615-044231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szabady RL, McCormick BA. Control of neutrophil inflammation at mucosal surfaces by secreted epithelial products. Front Immunol. 2013;4:220. doi: 10.3389/fimmu.2013.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haller D, et al. Non-pathogenic bacteria elicit a differential cytokine response by intestinal epithelial cell/leucocyte co-cultures. Gut. 2000;47(1):79–87. doi: 10.1136/gut.47.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Megias J, Busserolles J, Alcaraz MJ. The carbon monoxide-releasing molecule CORM-2 inhibits the inflammatory response induced by cytokines in Caco-2 cells. Br J Pharmacol. 2007;150(8):977–86. doi: 10.1038/sj.bjp.0707184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cable JW, Cable EE, Bonkovsky HL. Induction of heme oxygenase in intestinal epithelial cells: studies in Caco-2 cell cultures. Mol Cell Biochem. 1993;129(1):93–8. doi: 10.1007/BF00926580. [DOI] [PubMed] [Google Scholar]
- 20.Uc A, McDonagh AF, Stokes JB. Metabolism of haem in Caco-2 cells. Exp Physiol. 2010;95(2):296–303. doi: 10.1113/expphysiol.2009.050203. [DOI] [PubMed] [Google Scholar]
- 21.Alam J, et al. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem. 1999;274(37):26071–8. doi: 10.1074/jbc.274.37.26071. [DOI] [PubMed] [Google Scholar]
- 22.Zhang X, et al. Activation of the Nrf2/antioxidant response pathway increases IL-8 expression. Eur J Immunol. 2005;35(11):3258–67. doi: 10.1002/eji.200526116. [DOI] [PubMed] [Google Scholar]
- 23.Loboda A, et al. HIF-1 induction attenuates Nrf2-dependent IL-8 expression in human endothelial cells. Antioxid Redox Signal. 2009;11(7):1501–17. doi: 10.1089/ars.2008.2211. [DOI] [PubMed] [Google Scholar]
- 24.Campbell LM, Maxwell PJ, Waugh DJ. Rationale and Means to Target Pro-Inflammatory Interleukin-8 (CXCL8) Signaling in Cancer. Pharmaceuticals (Basel) 2013;6(8):929–59. doi: 10.3390/ph6080929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hwang YS, et al. Interleukin-1beta stimulates IL-8 expression through MAP kinase and ROS signaling in human gastric carcinoma cells. Oncogene. 2004;23(39):6603–11. doi: 10.1038/sj.onc.1207867. [DOI] [PubMed] [Google Scholar]
- 26.Naidu S, et al. Inhibition and genetic deficiency of p38 MAPK up-regulates heme oxygenase-1 gene expression via Nrf2. J Immunol. 2009;182(11):7048–57. doi: 10.4049/jimmunol.0900006. [DOI] [PubMed] [Google Scholar]
- 27.Al-Huseini LM, et al. Heme oxygenase-1 regulates dendritic cell function through modulation of p38 MAPK-CREB/ATF1 signaling. J Biol Chem. 2014;289(23):16442–51. doi: 10.1074/jbc.M113.532069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abraham C, Medzhitov R. Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology. 2011;140(6):1729–37. doi: 10.1053/j.gastro.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Onyiah JC, et al. Heme oxygenase-1 and carbon monoxide regulate intestinal homeostasis and mucosal immune responses to the enteric microbiota. Gut Microbes. 2014;5(2):220–4. doi: 10.4161/gmic.27290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luissint AC, Parkos CA, Nusrat A. Inflammation and the Intestinal Barrier: Leukocyte-Epithelial Cell Interactions, Cell Junction Remodeling, and Mucosal Repair. Gastroenterology. 2016;151(4):616–32. doi: 10.1053/j.gastro.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mitsuyama K, et al. IL-8 as an important chemoattractant for neutrophils in ulcerative colitis and Crohn’s disease. Clin Exp Immunol. 1994;96(3):432–6. doi: 10.1111/j.1365-2249.1994.tb06047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Puleston J, et al. A distinct subset of chemokines dominates the mucosal chemokine response in inflammatory bowel disease. Aliment Pharmacol Ther. 2005;21(2):109–20. doi: 10.1111/j.1365-2036.2004.02262.x. [DOI] [PubMed] [Google Scholar]
- 33.Lee PJ, et al. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J Biol Chem. 1997;272(9):5375–81. [PubMed] [Google Scholar]
- 34.Konrad FM, et al. Tissue heme oxygenase-1 exerts anti-inflammatory effects on LPS-induced pulmonary inflammation. Mucosal Immunol. 2016;9(1):98–111. doi: 10.1038/mi.2015.39. [DOI] [PubMed] [Google Scholar]
- 35.Sheng WS, et al. Hemin inhibits NO production by IL-1beta-stimulated human astrocytes through induction of heme oxygenase-1 and reduction of p38 MAPK activation. J Neuroinflammation. 2010;7:51. doi: 10.1186/1742-2094-7-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gobert AP, et al. Disruption of nitric oxide signaling by Helicobacter pylori results in enhanced inflammation by inhibition of heme oxygenase-1. J Immunol. 2011;187(10):5370–9. doi: 10.4049/jimmunol.1102111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morse D, et al. Suppression of inflammatory cytokine production by carbon monoxide involves the JNK pathway and AP-1. J Biol Chem. 2003;278(39):36993–8. doi: 10.1074/jbc.M302942200. [DOI] [PubMed] [Google Scholar]
- 38.Otterbein LE, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. 2000;6(4):422–8. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- 39.Biswas C, et al. Nuclear heme oxygenase-1 (HO-1) modulates subcellular distribution and activation of Nrf2, impacting metabolic and anti-oxidant defenses. J Biol Chem. 2014;289(39):26882–94. doi: 10.1074/jbc.M114.567685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin Q, et al. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J Biol Chem. 2007;282(28):20621–33. doi: 10.1074/jbc.M607954200. [DOI] [PubMed] [Google Scholar]
- 41.Lin QS, et al. Catalytic inactive heme oxygenase-1 protein regulates its own expression in oxidative stress. Free Radic Biol Med. 2008;44(5):847–55. doi: 10.1016/j.freeradbiomed.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma L, et al. p38 MAPK-dependent Nrf2 induction enhances the resistance of glioma cells against TMZ. Med Oncol. 2015;32(3):69. doi: 10.1007/s12032-015-0517-y. [DOI] [PubMed] [Google Scholar]
- 43.Sun Z, Huang Z, Zhang DD. Phosphorylation of Nrf2 at multiple sites by MAP kinases has a limited contribution in modulating the Nrf2-dependent antioxidant response. PLoS One. 2009;4(8):e6588. doi: 10.1371/journal.pone.0006588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang B, et al. Carbon monoxide-activated Nrf2 pathway leads to protection against permanent focal cerebral ischemia. Stroke. 2011;42(9):2605–10. doi: 10.1161/STROKEAHA.110.607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lanis JM, et al. Tryptophan metabolite activation of the aryl hydrocarbon receptor regulates IL-10 receptor expression on intestinal epithelia. Mucosal Immunol. 2017 doi: 10.1038/mi.2016.133. In press. Epub, Jan 18, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Glover LE, et al. Control of creatine metabolism by HIF is an endogenous mechanism of barrier regulation in colitis. Proc Natl Acad Sci U S A. 2013;110(49):19820–5. doi: 10.1073/pnas.1302840110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ehrentraut SF, et al. Perturbation of neddylation-dependent NF-kappaB responses in the intestinal epithelium drives apoptosis and inhibits resolution of mucosal inflammation. Mol Biol Cell. 2016 doi: 10.1091/mbc.E16-05-0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.