

Abstract
Zika, dengue, and chikungunya viruses are transmitted by mosquitoes, causing diseases with similar patient symptoms. However, they have different downstream patient-to-patient transmission potentials, and require very different patient treatments. Thus, recent Zika outbreaks make it urgent to develop tools that rapidly discriminate these viruses in patients and trapped mosquitoes, to select the correct patient treatment, and to understand and manage their epidemiology in real time.
Unfortunately, current diagnostic tests, including those receiving 2016 emergency use authorizations and fast-track status, detect viral RNA by reverse transcription polymerase chain reaction (RT-PCR), which requires instrumentation, trained users, and considerable sample preparation. Thus, they must be sent to “approved” reference laboratories, requiring time. Indeed, in August 2016, the Center for Disease Control (CDC) was asking pregnant women who had been bitten by a mosquito and developed a Zika-indicating rash to wait an unacceptable 2 to 4 weeks before learning whether they were infected. We very much need tests that can be done on site, with few resources, and by trained but not necessarily licensed personnel.
This video demonstrates an assay that meets these specifications, working with urine or serum (for patients) or crushed mosquito carcasses (for environmental surveillance), all without much sample preparation. Mosquito carcasses are captured on paper carrying quaternary ammonium groups (Q-paper) followed by ammonia treatment to manage biohazards. These are then directly, without RNA isolation, put into assay tubes containing freeze-dried reagents that need no chain of refrigeration. A modified form of reverse transcription loop-mediated isothermal amplification with target-specific fluorescently tagged displaceable probes produces readout, in 30 min, as a three-color fluorescence signal. This is visualized with a handheld, battery-powered device with an orange filter. Forward contamination is prevented with sealed tubes, and the use of thermolabile uracil DNA glycosylase (UDG) in the presence of dUTP in the amplification mixture.
Keywords: Point-of-care diagnostics, multiplexed isothermal amplification, loop-mediated isothermal amplification, Zika detection, fluorescence read-out, no RNA extraction, mosquito surveillance, virus detection
INTRODUCTION
Mosquito-borne virus infections, including dengue, chikungunya, and Zika viruses are on the rise and demand immediate management strategies. Dengue and chikungunya viruses are already endemic in many of the tropical regions where Zika is now spreading in the Western Hemisphere1. Zika virus, like dengue, is a member of the Flaviviridae family and is native to Africa with one Asian and two African genetic lineages2. Even though the identification of the Zika virus dates back to 1947, Zika infection in humans remained sporadic for a half century before emerging in the Pacific and the Americas. The first reported outbreak of Zika fever occurred on the island of Yap in the Federated States of Micronesia in 2007, followed by French Polynesia in 2013 and 2014. The first major outbreak in the Americas occurred in 2015 in Brazil.
Zika, chikungunya, and dengue viruses are primarily transmitted by Aedes aegypti and Aedes albopictus. However, Zika has additional downstream human-to-human transmission possibilities, likely being spread through sexual contact, mother-to-fetus interaction, and via breast-feeding3–5. Zika fever was first believed to cause only mild illness. However, it was later associated with Guillain-Barré syndrome in adults, microcephaly in neonates, and chronic musculoskeletal diseases that may last months to years. Diagnosis of Zika illness can be challenging, since the symptoms of a Zika infection are similar to those of other mosquito-spread viruses6. Common co-infections of these viruses make differential diagnosis even more challenging7,8. Therefore, rapid and reliable detection of the nucleic acids from Zika and other viruses is needed to understand epidemiology in real time, to initiate control and preventive measures, and to manage patient care9.
Current diagnostic tests for these viruses include serological tests, virus isolation, virus sequencing, and reverse-transcription PCR (RT-PCR). Standard serological approaches often suffer from inadequate sensitivity and results can be complicated by cross-reactivity in patients who have previously been infected by other flaviviruses.
Therefore, nucleic acid testing remains the most reliable way to detect and differentiate these viruses. Detection of Zika and other mosquito-borne viruses is usually performed using RT-PCR or real-time RT-PCR in variety of biological fluids, such as serum, urine, saliva, semen, breast milk, and cerebral fluid10,11. Urine and saliva samples are generally preferred over blood, since they exhibit less PCR-inhibition, higher viral loads, virus presence for longer periods of time, and increased ease of collection and handling12,13. RT-PCR-based diagnostic tests, however, comprise extensive sample preparation steps and expensive thermal cycling equipment, making it less optimal for the point-of-care.
Reverse transcription loop-mediated isothermal amplification (RT-LAMP) has emerged as a powerful RT-PCR alternative due to its high sensitivity and specificity14, its tolerance for inhibitory substances in biological samples15, and operation on single temperature, which significantly lowers assay complexity and associated costs, making it suitable for low resource environments. RT-LAMP, as it is classically implemented, comprises six primers that bind to eight distinct regions within the target RNA. It runs at constant temperatures between 60 °C and 70 °C, and uses a reverse transcriptase and a DNA polymerase with strong strand displacing activity.
During the initial stages of RT-LAMP, forward and backward internal primers (FIP and BIP, Figure 1A) along with outer forward and backward primers (F3 and B3) form a dumbbell structure, the seed structure of exponential LAMP amplification. Amplification is further accelerated by the loop forward and backward primers (LF and LB), which are designed to bind the single stranded regions of the dumbbell, and results in the formation of concatemers with multiple repeating loops16. Classical LAMP assays based on turbidity or readout by DNA intercalating dyes is not entirely suited for point-of-care detection of Zika, where some level of multiplexing is desired17–19. Multiplexing is not easily obtained in these systems, as they are prone to generate false-positives due to off-target amplifications.
Figure 1. LAMP Assay Design.
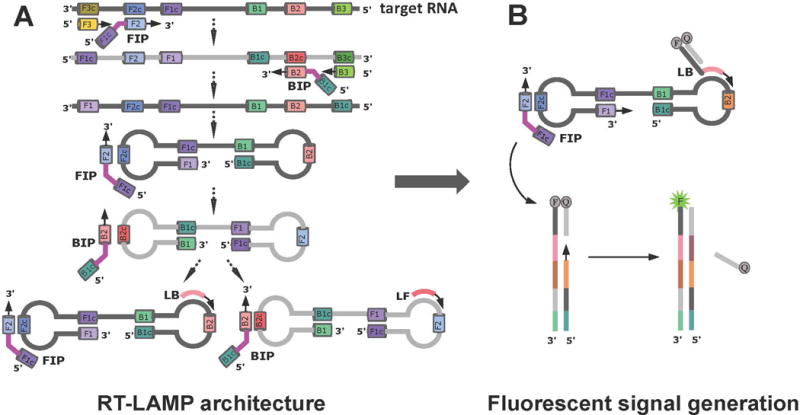
(A) RT-LAMP primers and formation of a dumbbell structure by a strand displacing DNA polymerase. (B) Introduction of strand-displacing probes to allow multiplexing and fluorescence readout, and to monitor RT-LAMP progress in real-time. Reprinted with permission from Yaren et al. 201720.
To manage these issues, the literature adds an additional component in the form of a “strand-displacing probe” to the classical RT-LAMP architecture20–22. Each probe has a sequence-specific double-strand region and a single-stranded priming region. The probe with the single-stranded region is tagged with a 5′-fluorophore, and the complementary probe is modified with a 3′-end quencher. In the absence of a target, no fluorescence is observed due to hybridization of the complementary probe strands, which brings the fluorophore and quencher into close proximity. In the presence of a target, the single-stranded portion of the fluorescent probe binds to its complement on the target, and is then extended by a strand displacing polymerase. Further polymerase extension by reverse primers causes the separation of the quencher strand from its complementary fluorescently labeled strand, allowing emission of fluorescence (Figure 1B). With this design, the signal is generated after the dumbbell formation, reducing the chances of false-positive signals.
The double-stranded portion of the strand-displacing probe can be any sequence, and when multiplexing is applied, the same sequence may be used with different fluorophore-quencher pairs. With this architecture, virus-contaminated urine, serum, or mosquito samples squished on paper were directly introduced to the assay without sample preparation. Three-color fluorescence read-outs visible to human eye were generated within 30-45 min, and signals were visualized by a 3D-printed observation box that uses a blue LED and an orange filter. Freeze-drying the RT-LAMP reagents enabled deployment of this kit to lower resource settings without a need for refrigeration.
PROTOCOL
Note: Mosquitoes were the only animals directly used in this study. The procedures to manage chickens, whose blood was used to feed the infected mosquitoes, were approved as IACUC Protocol #201507682 by the University of Florida Institutional Animal Care and Use Committee. Virus propagation and mosquito infection studies were performed at the BSL-3 facility of the Florida Medical Entomology Laboratory in Vero Beach, FL. RT-LAMP experiments were performed in the BSL-2 laboratory shared by FfAME and Firebird Biomolecular Sciences LLC in Alachua, FL.
1. Design of Primers and Strand Displacing Probes
1.1
Extract viral sequences for Dengue 1 from the Broad Institute database23; sequences for Zika and chikungunya from the NIAID Virus Pathogen Database and Analysis Resource24.
1.1.1
Create multiple sequence alignments (MSAs) for sequences of interest using software MUSCLE v3.8.3125. Use generated MSAs to search for LAMP primers sets within a conserved region of the target, and avoid non-relevant or unintended targets, such as distinctions between subtypes of a target.
1.2
Follow the design rules for LAMP primers as described online (http://loopamp.eiken.co.jp/e/lamp/), and allow the use of mixed bases in primers to cover the virus divergence26. To avoid cross-reactivity of primer sets, compare generated LAMP primers to the NCBI RNA virus database using NCBI BLAST27. Compare each set to eliminate primers that would dimerize in a multiplexed assay by using NCBI BLAST as well.
1.3
Screen the double-stranded portion of the strand-displacing probe against any viral genome sequence and mosquito genomic sequence. Modify 5′-ends of target binding probe with fluorescein amidite (FAM), HEX dye, and 5-carboxytetramethylrhodamine (TAMRA) dye for Zika, chikungunya, and Dengue 1, respectively.
1.3.1
Include a positive control primer set for urine samples. Design LAMP primers to target human mitochondrial DNA. Label strand-displacing probe with TET on 5′-end. Label quencher probes that are partially complementary to each fluorescently labeled probe with quencher (e.g., Iowa Black-FQ) on 3′-end (Table 1).
Table 1. Primers and strand displaceable probes.
Reprinted with permission from Yaren et al. 201720. Sequences in bold represent the double-stranded region of strand displacing probes. FAM-labeled (seen as green fluorescence), HEX-labeled (seen as light green-yellow fluorescence), TAMRA-labeled (seen as orange-red fluorescence), and TET-labeled (seen as yellow fluorescence) probes were assigned to detect Zika, Chikungunya, Dengue 1, and mitochondrial DNA in urine, respectively. FAM has excitation and emission maxima at 495 nm and 520 nm, respectively. HEX has excitation and emission maxima at 538 nm and 555 nm, respectively. TAMRA has excitation and emission maxima at 559 nm and 583 nm, respectively. TET has the excitation and emission maxima at 522 nm and 539 nm, respectively. To enable the use of a single quencher for all fluorophores, Iowa Black-FQ was used due to its wide absorption range (420-620 nm).
| Target virus | Name | Sequence (5′-3′) | Length | Start Position | End Position |
|---|---|---|---|---|---|
| Zika | ZV-F3 | GAGACTGCTTGCCTAG | 16 | 9905 | 9920 |
| ZV-B3 | CTGGGGTCTTGTCTTC | 16 | 10145 | 10130 | |
| ZV-LF | CAGTTGGAACCCAGTCAAC | 19 | 10028 | 10010 | |
| ZV-LB | GTGGAACAGAGTGTGGATTG | 20 | 10093 | 10112 | |
| ZV-FIP | CCATGGATTGACCAGGTAGTTTTTTCGACTGATGGCCAATG | 41 | 9974 | 10053 | |
| ZV-BIP | ACCACTGARGACATGCTTGTTTTTCATGTGGTCGTTYTCC | 40 | 10070 | 10129 | |
| ZV-LB_NatTail | FAM-CGGGTTTGCGCTCAGCCATCCGTTCAGTCCGTCAGGTCAGGTGGAACAGAGTGTGGATTG | 60 | 10093 | 10112 | |
| Chikungunya | CH-F3 | CGTCAACGTACTCCTAAC | 18 | 2891 | 2908 |
| CH-B3 | ACGTTGGCTTTRTTTTGG | 18 | 3094 | 3077 | |
| CH-LF | AGCGTCTTTATCCACGGG | 18 | 2968 | 2951 | |
| CH-LB | AYGCATCRATAATGGCGGG | 19 | 3025 | 3043 | |
| CH-FIP | GAAGTTTCCTTTCGGTGGGTTTTTGGAAGACACTYTCYGG | 40 | 2932 | 2993 | |
| CH-BIP | AAGGAGTGGGAGGTGGATTTTTTCAYTTGGTGACTGCAG | 39 | 3006 | 3063 | |
| CH-LF_NatTail | HEX-CGGGTTTGCGCTCAGCCATCCGTTCAGTCCGTCAGGTCAGAGCGTCTTTATCCACGGG | 58 | 2968 | 2951 | |
| Dengue-1 | D1-F3 | ACAGCYCTGAATGAYATGG | 19 | 9583 | 9601 |
| D1-B3 | GCAGTTTCTCTCAGGC | 16 | 9803 | 9788 | |
| D1-LF | CACTTGYTGCCARTCATTCC | 20 | 9666 | 9647 | |
| D1-LB | CCATGCCGYAACCAAG | 16 | 9727 | 9742 | |
| D1-FIP | CTGGTGGAARTGGTGTGATTTTTTGGGAACCTTCAAAAGG | 40 | 9628 | 9693 | |
| D1-BIP | GAAGGAYGGGAGGGAAATAGTTTTTTTAGCCCTRCCCACAAG | 42 | 9702 | 9763 | |
| D1-LB_NatTail | TAMRA-CGGGTTTGCGCTCAGCCATCCGTTCAGTCCGTCAGGTCAGCCATGCCGYAACCAAG | 56 | 9727 | 9742 | |
| Mitochondrial DNA | MtDNA-F3 | AGCCTACGTTTTCACAC | 17 | 9183 | 9199 |
| MtDNA-B3 | GCGCCATCATTGGTAT | 16 | 9410 | 9395 | |
| MtDNA-LB | GCCTAGCCATGTGATTTCAC | 20 | 9322 | 9341 | |
| MtDNA-LF | GGCATGTGATTGGTGGGT | 18 | 9254 | 9237 | |
| MtDNA-FIP | GTCATGGGCTGGGTTTTACTTTTTCTACCTGCACGACAAC | 40 | 9213 | 9228 | |
| MtDNA-BIP | CTCAGCCCTCCTAATGACCTTTTTGAGCGTTATGGAGTGG | 40 | 9359 | 9344 | |
| MtDNA-LB_NatTail | TET-CGGGTTTGCGCTCAGCCATCCGTTCAGTCCGTCAGGTCAGGCCTAGCCATGTGATTTCAC | 60 | 9322 | 9341 | |
| Common quencher | CTGACCTGACGGACTGAACGGATGGCTGAGCGCAAACCCG-lowa Black FQ | 40 |
2. Virus Isolates and Infected Mosquito Samples
2.1
Use isolates of Zika virus (Puerto Rico strain), Chikungunya virus (La Réunion or British Virgin Islands strain), and Dengue serotype-1 virus (Key West strain). Determine viral titers using plaque assay28 or quantitative RT-PCR29. Extract viral RNAs using commercially available RNA extraction kits.
Note: Table 2 represents the viral titers of each virus isolate used in this study.
Table 2. Viral titers in cell cultures and infected mosquito.
pfu: plaque-forming unit. Reprinted with permission from Yaren et al. 201720.
| Virus, Strain (GenBank accession number) | Family/Genus | Viral titers |
|---|---|---|
| Zika virus (ZV), Puerto Rico (PRVABC59, KU501215.1) | Flaviviridae/Flavivirus Group IV, positive, ssRNA | 2.85 × 108 pfu/mL |
| Chikungunya virus (CH), British Virgin | Togaviridae/Alphavirus Group | 2.42 × 108 |
| Islands (Asian lineage, KJ451624) | IV, positive, ssRNA | genomes/mL |
| Chikungunya virus (CH), La Reunion (Indian Ocean lineage, LR2006-OPY1, KT449801) | Togaviridae/Alphavirus Group IV, positive, ssRNA | 1.89 × 108 genomes/mL |
| Chikungunya virus (CH), La Reunion extracted total NA from Aedes aegypti female (Indian Ocean lineage, LR2006- OPY1, KT449801) | Togaviridae/Alphavirus Group IV, positive, ssRNA | 3.85 × 105 genomes/mL |
| Dengue serotype 1 (D-l), Key West (FL) (JQ675358) | Flaviviridae/Flavivirus Group IV, positive, ssRNA | 1.22 × 106 pfu/mL |
|
| ||
| Zika (ZV) Mosquito identity, Strain | Leg titer pfu/mL | |
|
|
||
| ZV 9, Puerto Rico | 4.76 × 103 | |
2.2
Follow the article published by Yaren et al. for a detailed protocol about the infection of Ae. aegypti females with Zika and chikungunya viruses20. See Table 2 for the viral titer of the mosquito sample infected with Zika virus.
3. RT-LAMP Coupled with Thermolabile Uracil DNA Glycosylase
3.1. 10X primer mix preparation
3.1.1
Prepare 100 μM stock solution of each primer and probe (see step 1) by adding nuclease free water. Vortex well and store at −20 °C until use.
3.1.2
For 10X Primer mix for each Zika, Dengue 1, or mitochondrial DNA target, mix 16 μL of FIP, 16 μL BIP, 2 μL of F3, 2 μL of B3, 5 μL of LF, 2 μL of LB, 3 μL of LB-fluorescent labeled probe, 4 μL of quencher probe, and 50 μL of nuclease-free water to give a total of 100 μL volume.
3.1.3
For 10X Primer mix for Chikungunya, mix 16 μL of FIP, 16 μL BIP, 2 μL of F3, 2 μL of B3, 5 μL of LB, 2 μL of LF, 3 μL of LF-fluorescent labeled probe, 4 μL of quencher probe, and 50 μL of nuclease-free water to give a total of 100 μL volume.
Note: The probe concentration described above uses 300 nM of final fluorescent probe and 400 nM of quencher probe. Alternatively, use 80 nM of fluorescently labeled probe and 200 nM of its quencher probe for real-time analysis.
3.2
Add 5 μL of 10X primer mix (For 3-plex, add 5 μL of each 10X primer mix), 5 μL of 10X isothermal amplification buffer (1X buffer composition: 20 mM Tris-HCl buffer pH 8.8, 50 mM KCl, 10 mM (NH4)2SO4, 8 mM MgSO4, 0.1% Tween-20, 1 mM DTT), 7 μL of dNTP mixture (10 mM of dATP, dCTP and dGTP; 5 mM of dTTP and dUTP), 16 units of DNA Polymerase, 15 units of Reverse Transcriptase, 80 units of recombinant ribonuclease inhibitor, 2 units of thermolabile UDG, 1-2 μL of varying amounts of viral RNAs, and water to bring the total volume up to 50 μL in a 0.25 mL PCR tube (see Table of Materials).
3.3
Include negative control assays at this stage by replacing viral RNA volume with water. Incubate samples between 65-68 °C for 45 min to 1 h, then analyze by running 5 μL of each sample on 2.5% agarose gel in 1X TBE buffer containing ethidium bromide (0.4 μg/mL). Use a 25 bp or 50 bp DNA ladder as marker on the gel.
Note: Addition of a non-target specific template is optional.
3.4
To detect the presence of pathogenic RNA, test each primer set and its no-template control by varying the temperature and/or magnesium concentration, since each RT-LAMP primer set was designed to work in varying temperatures (65 °C to 68 °C) or magnesium concentrations (6 to 10 mM final). Prepare the experiments at room temperature.
4. RT-LAMP Using Viral RNA-Spiked Urine
4.1
Test RT-LAMP primers in varying final concentrations of urine (0%, 10%, 20%, and 50%) in 50 μL of total reaction volume. For 10% final urine concentration, add 1 μL of viral RNA to 5 μL of urine. For 20% final urine concentration, add 1 μL of viral RNA to 10 μL of urine. For 50% final urine concentration, add 1 μL of viral RNA to 25 μL of urine.
4.2
For real-time monitoring of RT-LAMP in 10% of urine, use a real-time PCR instrument (see Table of Materials) with different fluorophore filters.
4.2.1
To read fluorescence signals from FAM-labeled amplicons for Zika, use a filter ranging from 483 to 533 nm; for HEX-labeled amplicons for chikungunya, use a filter ranging from 523 to 568 nm; for TAMRA-labeled amplicons for Dengue 1, use a filter ranging from 558 to 610 nm; and for TET-labeled amplicons for mitochondrial DNA, use a filter ranging from 523 to 568 nm.
Note: FAM has excitation and emission maxima of 495 nm and 520 nm, respectively. HEX has excitation and emission maxima of 538 nm and 555 nm, respectively. TAMRA has excitation and emission maxima of 559 nm and 583 nm, respectively. TET has excitation and emission maxima of 522 nm and 539 nm, respectively.
4.3
Use 96-well plates and seal the plate with a clear plastic sheet, then incubate samples at 65-68 °C for 60-90 min and record the fluorescence every 30 s using the light cycler. Initially use 80 nM of fluorescently labeled probes, and then test the 300 nM of fluorescently labeled probes.
4.3.1
Determine limit of detection for each target by adding serially diluted viral RNAs in final urine concentration of 10%.
Note: Use real-time RT-LAMP to determine optimum temperature, magnesium concentration, fluorescently labeled probe concentration, and reaction time.
4.4
To visualize the fluorescence generated by strand displaceable probes by eye, use a blue LED light source from a light cycler with excitation at 470 nm (any gel electrophoresis box with built-in blue light source will work, see table of materials), and record the image with a cell phone camera at room temperature in the dark.
Note: Use 300 nM of fluorescently labeled probes, when visualization of all three colors by eye using blue LED is needed.
5. RT-LAMP on Detection of Zika-Infected Mosquitoes Using Q-Paper Technology
5.1
Prepare quaternary ammonium-modified filter paper (Q-paper), according to previously published methods with slight modifications30.
5.1.1
Immerse 1 g of cellulose filter paper circles (see Table of Materials) with diameters of 3.5 cm in 50 mL of 1.8% of aqueous NaOH solution for 15 min for activation.
5.1.2
Collect activated paper circles by vacuum filtration and then immerse them in 40 mL of aqueous solution of (2,3-epoxypropyl) trimethylammonium chloride (0.28 g) overnight at room temperature.
Note: Keep the mass ratio of (2,3-epoxypropyl) trimethylammonium chloride to filter paper at 0.28 to 1.
5.1.3
Collect the resulting cationic paper by vacuum filtration, and neutralize with 50 mL of 1% of acetic acid.
5.1.4
Wash the paper three times with ethanol and air-dry under a hood with constant airflow.
5.2
Cut Q-paper sheets into small rectangles (3 mm × 4 mm) using a paper punch or scissors. Crush Ae. aegypti female mosquitoes potentially infected with Zika on each paper with a micro pestle.
5.3
Add to each paper 20 μL of 1 M aqueous ammonia solution (pH ≈ 12) and wait for 5 min. Then wash each paper once with 20 μL of 50% EtOH and once with 20 μL of nuclease-free water. Air-dry the paper in BSL-2 biosafety cabinet for about 1 h.
Note: At this point, samples can be stored at −20 °C overnight until use.
5.4
Using tweezers, place each paper in 100 μL of RT-LAMP reaction mixture and incubate at 65-68 °C for 45 min. Make sure to fully submerge the paper in the solution; it should not be floating. Read and record the fluorescence arising from Zika virus using blue LED light source and orange or yellow filter.
6. Lyophilization of RT-LAMP Reagents for 100 μL of Reaction Volume
6.1
Prepare 10X LAMP primer mixture according to section 3.1, and prepare dNTP mixture containing dUTP according to section 3.2. Store primer mixture and dNTPs at −20 °C until use.
6.2
Prepare 10 mL of enzyme dialysis buffer to remove glycerol, by mixing 100 μL of 1 M Tris-HCl pH 7.5, 500 μL of 1 M KCl, 2 μL of 0.5 M EDTA, 100 μL of 10% Triton X-100, 100 μL of 0.1 M DTT and 9.198 mL of nuclease-free water. Store the dialysis buffer at 4 °C.
Note: Make sure to filter (0.2-micron filters) all buffer reagent solutions prior to use and store them at 4 °C.
6.2.1
Use an ultrafiltration membrane with 10 kDa cut-off limit (see Table of Materials). Place 350 μL of dialysis buffer, 4 μL of 32 units of DNA Polymerase, 2 μL of 30 units of Reverse Transcriptase, and 2 μL of 80 units of RNase inhibitor into ultrafiltration membrane and centrifuge at 14,000 × g for 5 min at 4 °C.
6.2.2
Add another 350 μL of dialysis buffer to the ultrafiltration membrane and centrifuge (14,000 × g) for another 5 min. Empty the collection tube and repeat this step, then centrifuge (14,000 × g) for 3 min.
6.2.3
For elution, invert the membrane and place in a new collection tube. Spin at 1,000 × g for 2 min. Measure the elution volume; if less than 8 μL, bring the volume up to 8 μL by adding dialysis buffer. Store the elution at 4 °C, and immediately proceed to next step.
Note: This protocol is for single tube lyophilization, but prepare at least 10 reaction tubes at a time using the same ultrafiltration membrane and use the same amount of dialysis buffer.
6.3
Mix 10 μL of 10X LAMP primer if singleplex (for 3-plex, add 10 μL of each 10X primer mix), 14 μL of dNTP mixture, 8 μL of dialyzed enzyme mix, 2 μL of 2 units of thermolabile UDG, and 66 μL of nuclease free water (for 3-plex, use 46 μL) in a 0.5 mL microcentrifuge tube and leave the lid open. Instead, add another lid punctured with a needle to allow vapor to escape.
6.4
Immediately freeze the tubes at −80 °C for 2 h, and lyophilize the tube for 4 h. Cover the lyophilization chamber with aluminum foil for protection from light. When reagents are dried, remove the punctured lids, close the original lid, and store the tubes with lyophilized reagents at 4 °C in the dark.
Note: Reagents can be lyophilized overnight, if necessary.
7. Testing Lyophilized RT-LAMP Reagents on Urine and Mosquito Samples Containing Zika
7.1
Use the following items from the kit (see the Table of Materials) for Zika: A 3D-printed observing box with orange filter (long pass filter, cut-on ~540 nm), 2 AAA batteries for observing box, lyophilized reaction tubes labeled ZV for Zika detection, and 1.1X rehydration Buffer in 1.5 mL screw top tubes.
7.2
Prepare 50 mL of 1.1X rehydration buffer by mixing 5.5 mL of 10X isothermal amplification buffer that comes along with DNA Polymerase (see the Table of Materials), 3.3 mL of 100 mM MgSO4, 110 μL of 0.5 M DTT, and 41.09 mL of nuclease-free water. Mix well, aliquot 1 mL of this solution to 1.5 mL screw top tubes, and store at 4 °C until use.
7.3
Add 90 μL of 1.1X rehydration buffer to each reaction tube (0.5 mL). Ensure that the liquid fills the bottom of the tube containing the lyophilized reagents (dissolve them with mixing by shaking, then briefly spin down (1,000 × g)). Add 10 μL of urine sample spiked with viral RNA to the reaction tubes (see section 4.1 for details).
7.3.1
If testing mosquitoes, add a rectangle of Q-paper holding mosquito carcasses (as prepared in the steps described in sections 5.2 and 5.3), along with 10 μL purified water instead of urine samples.
Note: Alternatively, add 10 μL of saliva or serum samples instead of urine. If the samples are extracted DNA or RNA, add 2-5 μL of the sample into rehydrated mixture and complete with nuclease-free water to 10 μL of final sample volume.
7.4
Close the lid of the reaction tubes. Place them in a heat block/bath (or incubator) pre-set at 65-68 °C. Incubate for 30-45 min, but no more than 1 h. After incubation, remove the tube from the heat. To prevent contamination of future assays, ensure that these tubes remain closed.
7.5
Place reaction tubes in the observing box; the temperature will fall to the ambient temperature (preferably 25 °C). Turn on the switch on the back of the observing box. Observe sample through the orange filter and capture the image by a cell phone camera which is held at a distance where the image fills 80% of the field of view.
Note: Better visualization is achieved in low light environments.
7.6
After visualization is completed, turn off the switch. Store the sealed tubes in the dark, preferably with refrigeration, if later visualization is needed.
REPRESENTATIVE RESULTS
Initially, the performances of each RT-LAMP primer (Table 1) with its corresponding viral RNA substrate as well as negative controls were assessed by gel electrophoresis. RT-LAMP primers were designed to target NS5 region (RNA-dependent RNA polymerase) for Zika and Dengue 1, and nsP2 region (non-structural protein P2) for Chikungunya. Templates were total RNA extracted from viral stocks cultured in African green monkey kidney (Vero) cells. In one case, total nucleic acid (DNA and RNA) was extracted from a Chikungunya-infected female Ae. aegypti (Table 2).
Figure 2A shows some representative results with RT-LAMP performed with these samples. In both singleplex and multiplexed (3-plexed) cases, RT-LAMP products appear as a ladder of concatemers when run on agarose gel. Negative control samples containing water as sample gave only the bands for primers themselves, including the non-specific target control where total nucleic acid extracted from a non-infected Ae. aegypti female mosquito was used as template.
Figure 2. Gel electrophoresis analysis of amplicons.
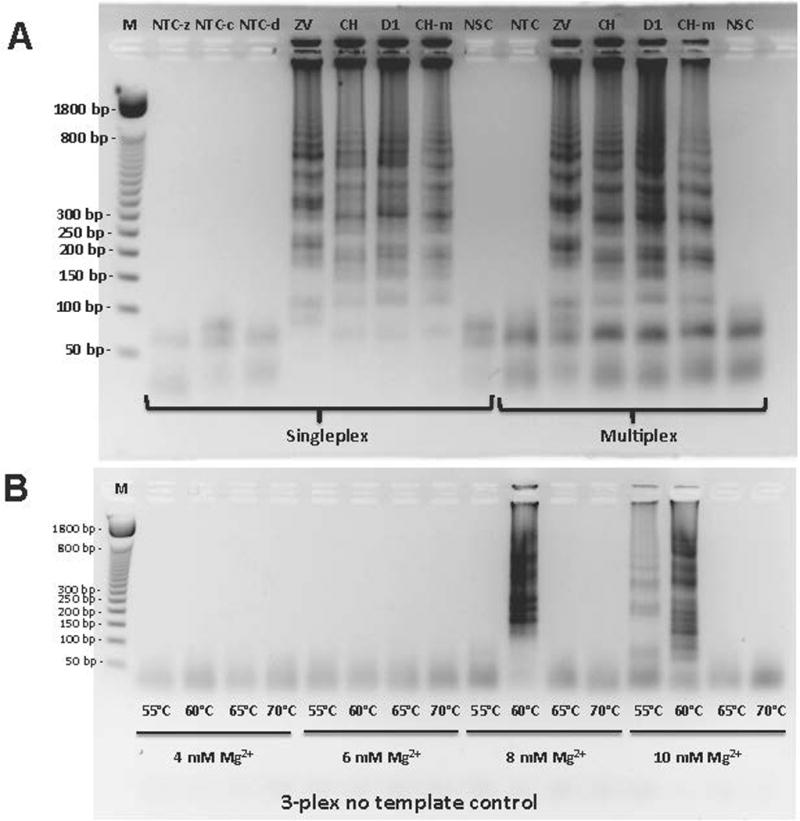
(A) Testing LAMP primers in singleplex or multiplex (3-plex) format with proper positive and negative controls. Positive controls include purified viral RNA with following titers: 2.85 pfu for Zika (ZV), 242 genomic copies for Chikungunya (CH), and 1.22 pfu for Dengue 1 (D1). NTC-z, NTC-c and NTC-d stand for no template control for Zika, Chikungunya, and Dengue 1 primers in singleplex format, respectively. NSC stands for non-specific target control where total xNA (DNA and RNA) is extracted from infection-free Ae. aegypti. M stands for marker (50 base pair DNA ladder). (B) Optimization of assay conditions by testing a range of RT-LAMP incubation temperatures (from 55 °C to 70 °C) and MgSO4 concentrations (from 4 mM to 10 mM) in multiplex format in the absence of target RNA. Reprinted with permission from Yaren et al. 201720.
To find optimal RT-LAMP conditions, different magnesium concentrations and incubation temperatures were tested. It was found that higher magnesium concentrations and lower temperatures could generate non-specific amplicons, giving rise to false positives. Therefore, 8 mM of magnesium and incubation at 65 °C were used in all experiments from then on (Figure 2B).
Gel electrophoresis requires the opening of the reaction tube, which may give rise to the contamination of the next set of experiments by previously generated amplicons, leading to false positives. To prevent the forward contamination, dTTP was replaced by equal ratios of dTTP and dUTP, so that dUTP would be incorporated into RT-LAMP amplicons. This turns the possible contaminating amplicon into a substrate for uracil-DNA glycosylase (UDG) during the preparation of any subsequent RT-LAMP reactions31,32. When a thermolabile version of UDG is used, dU-containing amplicons can be digested at room temperature as the RT-LAMP samples are being set up, and the UDG will be inactivated during isothermal amplification. Moreover, the reaction tube can be opened for a downstream analysis, when needed. In addition to the use of UDG digestion, an architecture using displaceable probes generates fluorescence that can be read without the need to open the reaction tube.
Figure 3A delivers the result of the limit of detection (LOD) for Zika using fluorescent probes targeting Zika RNA. Serial dilution studies showed that as low as 1.42 pfu could be detected within 30 min in a singleplex assay or a 3-plexed assays. When samples were further diluted to 0.71 pfu, a fluorescent signal was observed after 40 min for a singleplex assay, and after 50 min for 3-plexed mixtures. In addition to real-time RT-LAMP, fluorescence was visualized after 35 min through an orange filter, after samples were exposed to blue LED light with an excitation wavelength of 470 nm (Figure 3B). Similarly, limits of detection were measured at 37.8 copies and 1.22 pfu for Chikungunya and Dengue 1, respectively (Figure 3C-D).
Figure 3. RT-LAMP in real-time.
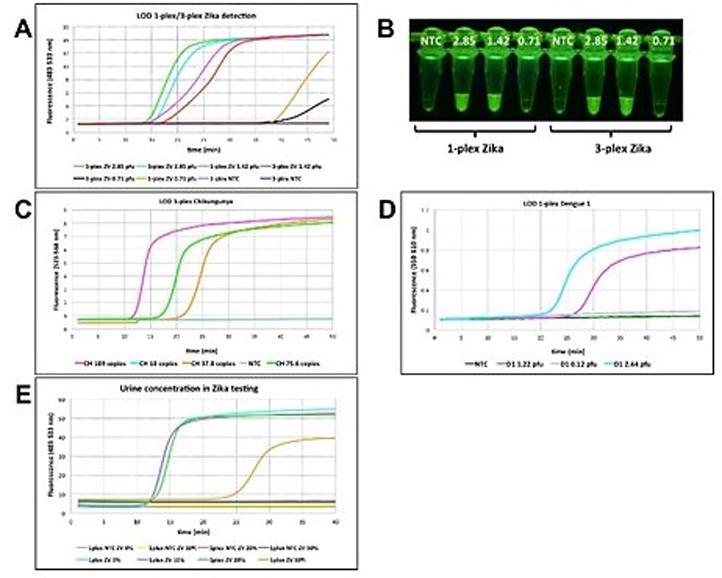
(A) Singleplex and multiplexed limit of detection (LOD) for Zika (ZV) using 80 nM FAM-labeled strand-displacing probe in real-time using real-time PCR instrument (channel 483-533 nm). Samples were incubated at 65°C for about 50 min. (B) Fluorescence was observed though an orange filter followed by the excitation of the samples at 470 nm with blue light. Samples from A were used for visualization. (C) Real-time analysis for LOD on Chikungunya (CH) target using 80 nM of HEX-bearing LAMP probes in singleplex format using fluorescence channel of 523-568 nm on a real-time PCR instrument. (D) Real-time analysis for LOD on Dengue 1 (D1) targets using 80 nM of TAMRA-bearing LAMP probes in singleplex format using fluorescence channel of 558-610 nm on a real-time PCR instrument. (E) Determination of maximum urine content in RT-LAMP reaction. A range of final urine concentrations (0 %-50 %) were tested using 80 nM FAM-labeled probe in the presence of 2.85 pfu of Zika vRNA. NTC = No template control on all panels. Reprinted with permission from Yaren et al. 201720.
To determine the optimum urine concentration, Zika viral RNA (2.85 pfu) was added to varying concentrations of an authentic human urine sample (0%, 10%, 20% and 50%). Presumably because of its electrolytes, the RT-LAMP signal was delayed from 15 to 35 min when 50% urine was used in the assay. 10% urine was determined to be optimal due to presenting a lower background. In the absence of Zika targets, no amplification was observed at any concentration, but higher background fluorescence was observed in samples with higher urine concentration (Figure 3E).
To allow multiplexing with read-outs of different fluorescence colors, 80 nM of strand displaceable probes were labeled with different fluorophores (FAM for Zika, HEX for Chikungunya, and TAMRA for Dengue 1), but accompanied by the same quencher (Iowa Black-FQ) with a concentration of 200 nM. Cell culture-extracted viral RNAs (2.85 pfu for Zika, 242 viral RNA copies for Chikungunya, and 1.22 pfu for Dengue 1) were used as targets, diluted in 10% urine. Real time analysis of the fluorescence data showed a delay in signal generation (about 10 min) for 3-plexed assays where primers for all targets were present. In singleplex cases, on the other hand, the reaction was completed within 10-15 min (Figure 4A-C). When fluorescence was visualized by using a blue LED and orange filter, differences in signal strength were observed for different fluorophores, where FAM-labeled amplicons were the most visible. This is expected, since excitation at 470 nm is closest to the maximum of the FAM excitation spectrum (Figure 4D).
Figure 4. Singleplex and multiplex virus detection in urine using 80 nM probe.
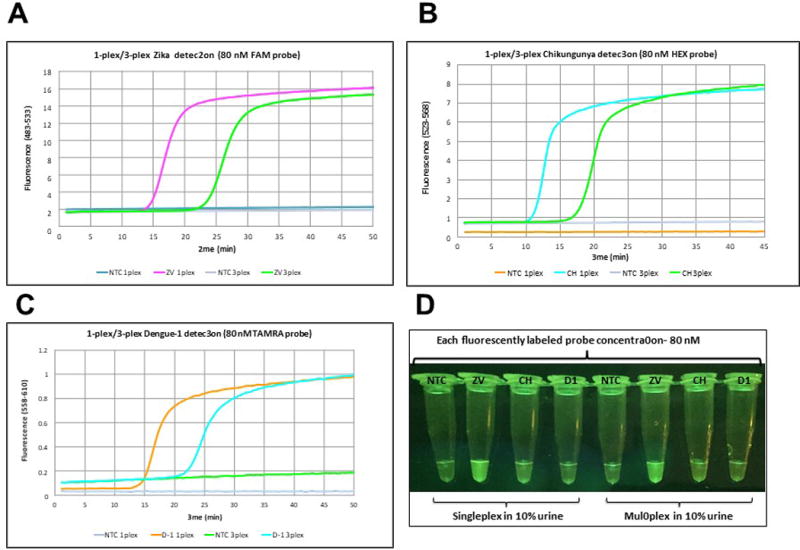
(A) Zika detection in 10% urine in singleplex and multiplex format using 2.85 pfu of Zika (ZV) viral RNA with FAM-labeled probe (80 nM). (B) Chikungunya detection in 10% urine in singleplex and multiplex format using 242 copies of Chikungunya (CH) viral RNA with HEX-labeled probe (80 nM). (C) Dengue 1 detection in 10% urine in singleplex and multiplex format using 1.22 pfu of Dengue 1 (D1) viral RNA with TAMRA-labeled probe (80 nM). (D) Visualization of RT-LAMP reactions through orange filter after excitation at 470 nm with blue LED light. Reprinted with permission from Yaren et al. 201720. NTC = No template control on all panels.
To enable the visualization of all three colors with a single LED excitation wavelength (470 nm), the concentrations of displaceable probes were increased to 300 nM and the complementary quencher probe to 400 nM. In singleplex cases, signal generation was completed within 10 min for Zika, 20 min for Chikungunya, and 40 min for Dengue 1. In 3-plexed assays, however, it was further delayed by 20 min in all assays (Figure 5A-C). Nevertheless, sacrifice in time to signal generation enabled the visualization of all three colors using blue LED light with orange filter. Green fluorescence is observed for Zika using probes 5′-end labeled with FAM, light green-yellow fluorescence is assigned to Chikungunya where the probe is 5′-end labeled with HEX, and orange-red fluorescence is used for Dengue 1, where its probe is 5′-end labeled with TAMRA (Figure 5D).
Figure 5. Singleplex and multiplex virus detection in urine using 300 nM probe.
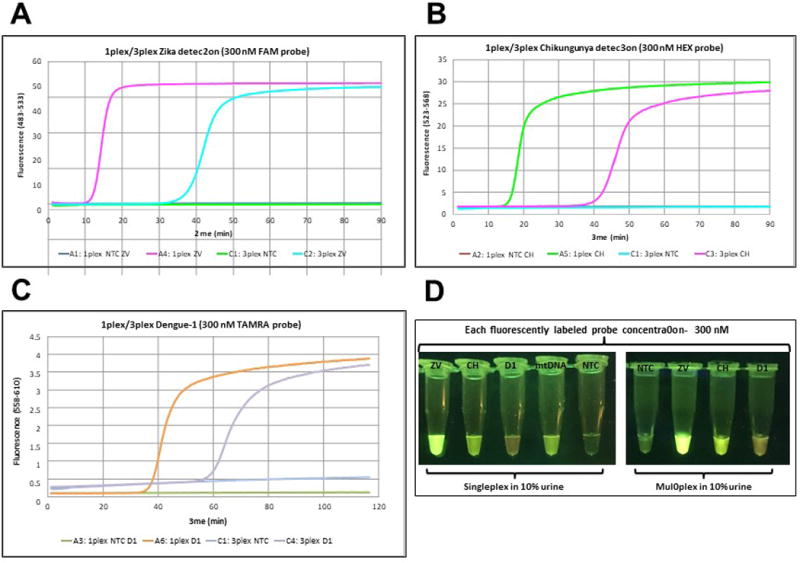
(A) Zika detection in 10% urine in singleplex and multiplex format using 2.85 pfu of Zika (ZV) viral RNA with FAM-labeled probe (300 nM). (B) Chikungunya detection in 10% urine in singleplex and multiplex format using 242 copies of Chikungunya (CH) viral RNA with HEX-labeled probe (300 nM). (C) Dengue 1 detection in 10% urine in singleplex and multiplex format using 1.22 pfu of Dengue1 (D1) viral RNA with TAMRA-labeled probe (300 nM). (D) Visualization of RT-LAMP reactions through orange filter after excitation at 470 nm with blue LED light. As a positive control in urine experiments, a LAMP primer set targeting human mitochondrial DNA (mtDNA) was used with TET-labeled probe (300 nM). NTC = No template control on all panels. Reprinted with permission from Yaren et al. 201720.
For testing mosquito samples with possible Zika infection, laboratory-infected Ae. aegypti female mosquitos (Table 2) were crushed on a rectangle of Q-paper, followed by ammonia treatment to sterilize the virus and to bind the exposed viral RNA onto positively charged Q-paper. The paper was briefly washed with ethanol to remove pterin compounds present in mosquito carcasses that might interfere with fluorescence readout33,34. Q-papers containing mosquitos were then directly submerged in the RT-LAMP mixture, and after incubation at 65 °C for 30 min, bright green fluorescence was observed in the presence of Zika virus (Figure 6).
Figure 6. Workflow for Zika detection of infected mosquito samples using Q-paper technology with healthy and Zika-infected Ae. aegypti (ZV 9, Table 2).
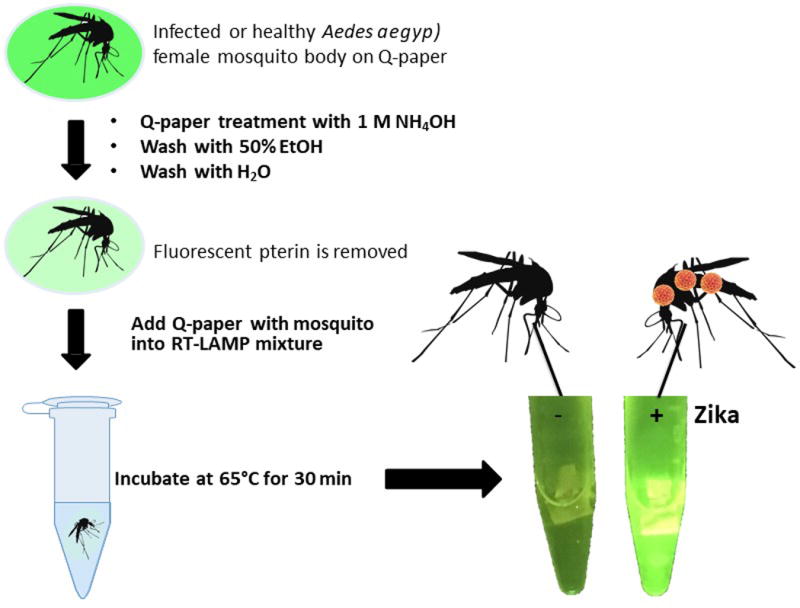
Reprinted with permission from Yaren et al. 201720.
To deliver the detection kit without a chain of refrigeration, and to make the assay easy to use, RT-LAMP reagents were lyophilized and accompanied by a rehydration buffer and a 3D-printed observation box that uses a 470 nm emitting LED and an orange filter (Figure 7A). Using 9 volumes of rehydration buffer and 1 volume of urine sample, visible green fluorescence can be generated within 30 min and the image can be captured by any cell phone camera (Figure 7B). Alternatively, mosquito samples on Q-paper can be used by the same assay by adding 1 volume of water instead of urine sample.
Figure 7. The Observing Box.

(A) The observing box uses two AAA batteries in the base. These batteries power four blue LED lights that illuminate the samples held in 0.5 mL tubes placed in the four holes. The LED lights are turned on with a switch. The samples are visualized through the orange filter. (B) Fluorescence in observation box. From left to right: negative, positive for Zika, negative, and positive for Zika.
DISCUSSION
Mosquito-borne viruses including Zika, chikungunya, and dengue threaten the public health and recent Zika outbreaks highlight the need for low-cost point-of-care detection alternatives for patient diagnostics, as well as for mosquito surveillance. Isothermal amplification methods were developed as affordable alternatives to PCR-based systems. Particularly, RT-LAMP-based platforms have been applied to detect a wide range of pathogens. However, the use of isothermal platforms has been mainly limited to single target detection. The method reported here uses a modified RT-LAMP to allow simultaneous detection of three different targets in a single reaction mixture, which runs at a range of moderate temperatures (65-68 °C), proven to be especially convenient because temperatures higher than 60 °C render Zika and other viruses non-infectious30,35HYPERLINK \l “_ENREF_33” \o “Janis, 2016 #430” .
Further, data presented here shows that RT-LAMP is capable of tolerating inhibiting substances that might be present in various biological fluids15. This allows viral RNAs to be amplified without an extra step of RNA purification by directly adding the sample into the reaction mixture. Depending on the type of sample, the maximum volume of sample allowed needs to be determined as to not inhibit the reaction and not cause background fluorescence (false positive). The study presented here focuses on the use of 10% final volume for urine samples. However, saliva and serum samples can also be applied to the assay in the same manner20. Limits of detection were found to be well above the reported viral loads in patient urine13, as well as virus-infected mosquitoes20,36,37.
Mosquito surveillance usually has a lower priority for public health resources, so an inexpensive multiplexed kit to survey a household or a public area would have special value. Therefore, this kit was modified by the addition of Q-paper to allow the detection of viruses in mosquito carcasses. Q-paper contains high levels of covalently attached quaternary ammonium groups, which allows the capture of viral nucleic acids on its surface through coulombic interactions. Even though it was a concern that Q-paper might inhibit RT-LAMP, it was found that by adding Q-paper carrying mosquito carcasses directly into RT-LAMP mixture, successful virus detection could be achieved within 30 min. For successful signal generation, Q-paper needs to be small enough (e.g., sized as 3 × 4 mm for 100 μL reaction volume) to be submerged in the RT-LAMP reaction mixture. If it is too big for the reaction tube, or starts floating instead of remaining immersed in the liquid, the reaction may not take place and results may appear as false negatives.
For successful detection and differentiation of each virus, each primer set needs to follow the guidelines for RT-LAMP primer design, the design rules for displacing probes, and include a choice of fluorophores for multiplexing. Each primer set needs to be screened against different targets to avoid cross-reactivity. Additionally, magnesium concentration and incubation temperature need to be optimized for each set. Methods presented here use fluorescence readouts visible by eye, which was accomplished by introducing strand displaceable probes to RT-LAMP architecture. Three distinct colors can be visualized in a multiplexed assay when different fluorophores were assigned to each target virus. This provides advantages over existing isothermal Zika detection methods, since most of the published methods rely on single target detection and the use of intercalating or fluorescence dyes, which are not sequence-specific and are prone to generating non-specific amplicons.
A critical step to consider is the determination of the final concentration of fluorescently labeled displacing probes. It was found that 80 to 100 nM of displacing probes could generate fluorescence signals within 15-20 min, whereas time to signal was delayed when 300 nM of probe concentration was used. Reaction time was further delayed when the assays were run in a multiplexed fashion. The reason for switching from 80 nM to 300 nM was to enable visualization of all three fluorophores using a single LED source. Blue LED with excitation at 470 nm is suitable for FAM-labeled probes, but is less suitable for HEX- or TAMRA-labeled probes. Therefore, a sacrifice was made on incubation time to be able to visualize all three targets.
One of the problems with RT-LAMP is forward contamination. RT-LAMP generates large amounts of amplicon in a short incubation time, and amplicons could be easily released as aerosols. To mitigate this problem, dUTP was included along with other dNTPs followed by UDG digestion during assay set-up. It is important to use a heat-labile version of UDG, since it is necessary to inactivate UDG to prevent the digestion of newly generated RT-LAMP amplicons.
To allow the distribution of the kit without refrigeration, all the components of the assay needed to be lyophilized and the kit supplemented with a rehydration buffer. Therefore, any glycerol present in commercially available enzymes was removed by ultrafiltration because glycerol cannot be removed by lyophilization. At a high concentration in a mixture, glycerol may interfere with enzyme activity. The only enzyme not included in the glycerol removal process was the thermolabile UDG. UDG is a small enzyme with molecular weight of 24.6 kDa, making it unsuitable for ultrafiltration experiments. Therefore, it was added to the lyophilization mixture as purchased. The residual glycerol did not negatively affect the assay’s outcome.
One of the limitations of this fluorescent RT-LAMP technique is the use of the single LED source. When assays were run in singleplex or multiplex format, higher concentrations of displacing probes were needed for signal visualization of all targets. However, this change causes delays in time-to-signal generation. One possible way to overcome this could be the use of two LED sources to better accommodate the other fluorophores (HEX and TAMRA). This way, lower probe concentration could be used to generate a signal with shorter incubation times.
Another limitation that needs consideration is judging the fluorescence color by eye in a dimly lit environment. It is easier if a single target is present in a singleplex or multiplex format, since primers were designed not to interact with each other. The assay would be harder to interpret if multiple targets were present in a single assay tube, such as a pool of mosquitoes or a patient with more than one viral infection. This would require a change of read-out architecture, such as a digital display rather than judging by eye.
A possible future approach would be to increase the level of multiplexing by creating a larger panel of mosquito-borne arboviruses. This requires careful primer design to avoid primer-primer interactions, and may require the use of modified nucleic acid analogs such as self-avoiding molecular recognition systems (SAMRS) and artificially expanded genetic information system (AEGIS) to better accommodate high level of multiplexing38. Accordingly, the solution for signal read-out in a higher multiplex system would be to use a single fluorophore for each displacing probe, and to assign a unique displaced sequence for each target and capture the displaced probes on a solid surface by DNA hybridization. This would create an array-formatted platform with a single fluorophore and LED to excite, but judging the presence of each target would be based on its position in the array39.
Acknowledgments
The work was supported in part by FDOH-7ZK15 and NIAID 1R21AI128188-01. Research reported in this publication was supported in part by the National Institutes of Allergy and Infectious Diseases, and in part by Biomedical Research Program of Florida Department of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or Florida Department of Health. Dynamic Combinatorial Chemistry LLC is acknowledged for their support and contribution to this project.
Dengue 1 virus (strain BOL-KW010) was kindly provided by the Florida Department of Health Bureau of Laboratories. Zika virus and the Asian lineage of chikungunya virus were graciously provided by the Centers for Disease Control and Prevention. The Indian Ocean lineage of chikungunya virus was kindly provided by Robert Tesh (World Reference Center for Emerging Viruses and Arboviruses, through the University of Texas Medical Branch in Galveston, Texas) to the UF-FMEL. We thank S. Bellamy, B. Eastmond, S. Ortiz, D. Velez, K. Wiggins, R. ZimLer, and K. Zirbel for assistance with the infection studies. We also thank M. S. Kim for providing Q-paper.
Footnotes
A complete version of this article that includes the video component is available at http://dx.doi.org/10.3791/57051.
DISCLOSURES:
Several of the authors and their institutions own intellectual property associated with this assay.
Contributor Information
Ozlem Yaren, Email: oyaren@ffame.org.
Barry W Alto, Email: bwalto@ufl.edu.
Kevin M Bradley, Email: kbradley@firebirdbio.com.
Patricia Moussatche, Email: pmoussatche@firebirdbio.com.
Lyudmyla Glushakova, Email: lglushakova@firebirdbio.com.
Steven A Benner, Email: manuscripts@ffame.org.
References
- 1.Patterson J, Sammon M, Garg M. Dengue, Zika and Chikungunya: Emerging Arboviruses in the New World. Western Journal of Emergency Medicine. 2016;17(6):671–679. doi: 10.5811/westjem.2016.9.30904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Faye O, et al. Molecular Evolution of Zika Virus during Its Emergence in the 20th Century. PLOS Neglected Tropical Diseases. 2014;8(1):e2636. doi: 10.1371/journal.pntd.0002636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dick GWA, Kitchen SF, Haddow AJ. Zika virus (I). Isolations and serological specificity. Trans R Soc Trop Med Hyg. 1952;46 doi: 10.1016/0035-9203(52)90042-4. [DOI] [PubMed] [Google Scholar]
- 4.Musso D, Gubler DJ. Zika Virus. Clin Microbiol Rev. 2016;29 doi: 10.1128/CMR.00072-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Musso D. Zika Virus Transmission from French Polynesia to Brazil. Emerging Infectious Diseases. 2015;21(10):1887–1887. doi: 10.3201/eid2110.151125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gasque P, Bandjee MCJ, Reyes MM, Viasus D. Chikungunya Pathogenesis: From the Clinics to the Bench. The Journal of Infectious Diseases. 2016;214(suppl_5):S446–S448. doi: 10.1093/infdis/jiw362. [DOI] [PubMed] [Google Scholar]
- 7.Villamil-Gómez WE, et al. Zika, dengue, and chikungunya co-infection in a pregnant woman from Colombia. International Journal of Infectious Diseases. 2016;51:135–138. doi: 10.1016/j.ijid.2016.07.017. doi: http://dx.doi.org/10.1016/j.ijid.2016.07.017. [DOI] [PubMed] [Google Scholar]
- 8.Sardi SI. Coinfections of Zika and Chikungunya viruses in Bahia, Brazil, identified by metagenomic next-generation sequencing. J Clin Microbiol. 2016;54 doi: 10.1128/JCM.00877-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shukla S, Hong SY, Chung SH, Kim M. Rapid Detection Strategies for the Global Threat of Zika Virus: Current State, New Hypotheses, and Limitations. Frontiers in Microbiology. 2016;7:1685. doi: 10.3389/fmicb.2016.01685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jesse JW, et al. Single-Reaction Multiplex Reverse Transcription PCR for Detection of Zika, Chikungunya, and Dengue Viruses. Emerging Infectious Disease journal. 2016;22(7):1295. doi: 10.3201/eid2207.160326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pabbaraju K, et al. Simultaneous detection of Zika, Chikungunya and Dengue viruses by a multiplex real-time RT-PCR assay. Journal of Clinical Virology. 2016;83:66–71. doi: 10.1016/j.jcv.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 12.Musso D, et al. Detection of Zika virus in saliva. Journal of Clinical Virology. 2015;68:53–55. doi: 10.1016/j.jcv.2015.04.021. [DOI] [PubMed] [Google Scholar]
- 13.Gourinat AC, O’Connor O, Calvez E, Goarant C, Dupont-Rouzeyrol M. Detection of zika virus in urine. Emerg Infect Dis. 2015;21 doi: 10.3201/eid2101.140894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Notomi T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000;28 doi: 10.1093/nar/28.12.e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edwards T, Burke PA, Smalley HB, Gillies L, Hobbs G. Loop-mediated isothermal amplification test for detection of Neisseria gonorrhoeae in urine samples and tolerance of the assay to the presence of urea. J Clin Microbiol. 2014;52 doi: 10.1128/JCM.00314-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagamine K, Hase T, Notomi T. Accelerated reaction by loop-mediated isothermal amplification using loop primers. Mol Cell Probes. 2002;16 doi: 10.1006/mcpr.2002.0415. [DOI] [PubMed] [Google Scholar]
- 17.Tian B, et al. Attomolar Zika virus oligonucleotide detection based on loop-mediated isothermal amplification and AC susceptometry. Biosensors and Bioelectronics. 2016;86:420–425. doi: 10.1016/j.bios.2016.06.085. doi: http://dx.doi.org/10.1016/j.bios.2016.06.085. [DOI] [PubMed] [Google Scholar]
- 18.Wang X, et al. Rapid and sensitive detection of Zika virus by reverse transcription loop-mediated isothermal amplification. Journal of Virological Methods. 2016;238:86–93. doi: 10.1016/j.jviromet.2016.10.010. doi: http://dx.doi.org/10.1016/j.jviromet.2016.10.010. [DOI] [PubMed] [Google Scholar]
- 19.Song J, et al. Instrument-Free Point-of-Care Molecular Detection of Zika Virus. Analytical Chemistry. 2016;88(14):7289–7294. doi: 10.1021/acs.analchem.6b01632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yaren O, et al. Point of sampling detection of Zika virus within a multiplexed kit capable of detecting dengue and chikungunya. BMC Infectious Diseases. 2017;17(1):293. doi: 10.1186/s12879-017-2382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kubota K, Jenkins DM, Alvarez AM, Su WW. Fret-based assimilating probe for sequence-specific real-time monitoring of loop-mediated isothermal amplification (LAMP) Biol Eng Trans. 2011;4:81–100. [Google Scholar]
- 22.Kubota R, Jenkins DM. Real-Time Duplex Applications of Loop-Mediated AMPlification (LAMP) by Assimilating Probes. Int J Mol Sci. 2015;16(3):4786–4799. doi: 10.3390/ijms16034786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J, Macdonald J. Advances in isothermal amplification: novel strategies inspired by biological processes. Biosens Bioelectron. 2015;64 doi: 10.1016/j.bios.2014.08.069. [DOI] [PubMed] [Google Scholar]
- 24.Pickett BE. ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012;40 doi: 10.1093/nar/gkr859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32 doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boonham N. Methods in virus diagnostics: from ELISA to next generation sequencing. Virus Res. 2014;186 doi: 10.1016/j.virusres.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 27.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215 doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 28.Baer A, Kehn-Hall K. Viral Concentration Determination Through Plaque Assays: Using Traditional and Novel Overlay Systems. Journal of visualized experiments : JoVE. 2014;(93):e52065–e52065. doi: 10.3791/52065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reiskind MH, Pesko K, Westbrook CJ, Mores CN. Susceptibility of Florida Mosquitoes to Infection with Chikungunya Virus. The American journal of tropical medicine and hygiene. 2008;78(3):422–425. [PMC free article] [PubMed] [Google Scholar]
- 30.Glushakova LG, et al. Detection of chikungunya viral RNA in mosquito bodies on cationic (Q) paper based on innovations in synthetic biology. Journal of Virological Methods. 2017;246:104–111. doi: 10.1016/j.jviromet.2017.04.013. doi: https://doi.org/10.1016/j.jviromet.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsieh K, Mage PL, Csordas AT, Eisenstein M, Tom Soh H. Simultaneous elimination of carryover contamination and detection of DNA with uracil-DNA-glycosylase-supplemented loop-mediated isothermal amplification (UDG-LAMP) Chemical Communications. 2014;50(28):3747–3749. doi: 10.1039/C4CC00540F. [DOI] [PubMed] [Google Scholar]
- 32.Tang Y, Chen H, Diao Y. Advanced uracil DNA glycosylase-supplemented real-time reverse transcription loop-mediated isothermal amplification (UDG-rRT-LAMP) method for universal and specific detection of Tembusu virus. 2016;6:27605. doi: 10.1038/srep27605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thomas AH. Fluorescence of pterin, 6-formylpterin, 6-carboxypterin and folic acid in aqueous solution: pH effects. Photochem Photobiol Sci. 2002;1 doi: 10.1039/b202114e. [DOI] [PubMed] [Google Scholar]
- 34.Wu D, Lehane MJ. Pteridine fluorescence for age determination of anopheles mosquitoes. Med Vet Entomol. 1999;13 doi: 10.1046/j.1365-2915.1999.00144.x. [DOI] [PubMed] [Google Scholar]
- 35.Janis AM. Inactivation and environmental stability of Zika virus. Emerg Infect Dis J. 2016;22 doi: 10.3201/eid2209.160664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poloni TR, et al. Detection of dengue virus in saliva and urine by real time RT-PCR. Virology Journal. 2010;7(1):22. doi: 10.1186/1743-422X-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones P, Okeoma C. Detection of Chikungunya virus (CHIKV) in urine of infected mice: a potential non-invasive diagnostic tool for CHIKV. J Infect Dis Ther. 2015;3(4):1000226. [Google Scholar]
- 38.Glushakova LG, et al. High-throughput multiplexed xMAP Luminex array panel for detection of twenty two medically important mosquito-borne arboviruses based on innovations in synthetic biology. J Virol Methods. 2015;214:60–74. doi: 10.1016/j.jviromet.2015.01.003. doi: http://dx.doi.org/10.1016/j.jviromet.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yaren O, Benner SA. Loop Mediated Amplifications with Nucleoside Analogs. US Provisional patent application no 62381759. 2016 [Google Scholar]