

Abstract
Background:
Identifying the neural correlates of ketamine treatment may facilitate and expedite the development of novel, robust, and safe rapid-acting antidepressants. Prefrontal cortex (PFC) global brain connectivity with global signal regression (GBCr) was recently identified as a putative biomarker of major depressive disorder (MDD). Accumulating evidence have repeatedly shown reduced PFC GBCr in MDD, an abnormality which appears to normalize following ketamine treatment.
Methods:
Fifty-six unmedicated participants with MDD were randomized to intravenous placebo (normal saline; n = 18), ketamine (0.5mg/kg; n = 19) or lanicemine (100mg; n = 19). PFC GBCr was computed using time series from functional magnetic resonance imaging (fMRI) scans that were completed at baseline, during infusion, and 24h post-treatment.
Results:
Compared to placebo, ketamine significantly increased average PFC GBCr during infusion (p = 0.01) and 24h post-treatment (p = 0.02). Lanicemine had no significant effects on GBCr during infusion (p = 0.45) and 24h post-treatment (p = 0.23), compared to placebo. Average delta PFC GBCr (during minus baseline) showed a pattern of positively predicting depression improvement in participants receiving ketamine (r = 0.44; p = 0.06; d = 1.0) or lanicemine (r = 0.55; p = 0.01; d = 1.3), but not those receiving placebo (r = –0.1; p = 0.69; d = 0.02). Follow-up vertex-wise analyses showed ketamine-induced GBCr increases in the dorsolateral, dorsomedial, and frontomedial PFC during infusion, and in the dorsolateral and dorsomedial PFC 24h post-treatment (corrected p < 0.05). Exploratory vertex-wise analyses examining the relationship with depression improvement showed positive correlation with GBCr in the dorsal PFC during infusion and 24h post-treatment, but negative correlation with GBCr in the ventral PFC during infusion (uncorrected p < 0.01).
Conclusions:
In a randomized placebo-controlled approach, the results provide the first evidence in MDD of ketamine-induced increases in PFC GBCr during infusion, and suggests that ketamine’s rapid-acting antidepressant properties are related to its acute effects on prefrontal connectivity. Overall, the study findings underscore the similarity and differences between ketamine and another N-methyl-D-aspartate receptor (NMDAR) antagonist, while proposing a pharmacoimaging paradigm for optimization of novel rapid-acting antidepressants prior to testing in costly clinical trials.
Introduction
The discovery of the rapid-acting antidepressant effects of ketamine has generated considerable interest in academia and industry about the prospect of targeting N-methyl-D-aspartate receptors (NMDAR) for the treatment of refractory depression 1, 2. However, the results from clinical trials failing to achieve their primary outcomes 3, 4, combined with recent preclinical evidence proposing NMDAR-independent mechanisms of ketamine 5, 6, have cast doubts about the potential utility of this line of research 1, 7. Therefore, it is essential to establish a rigorous and reproducible in vivo biomarker of the rapid-acting antidepressant ketamine. First, to better understand the underlying mechanisms of action in humans and second, perhaps most importantly, to facilitate and expedite the development of novel, robust, and safe rapid-acting antidepressants (RAADs). Central nervous system (CNS) drug development is increasingly challenging with high failure rates in large clinical trials and many stakeholders abandoning their CNS division or significantly reducing their clinical trials funding 8. High failure rates are especially relevant to depression research considering the urgent need for RAADs and the failure of many clinical trials 9.
A significant barrier to the development of psychotropic agents is the lack of reliable, clinically relevant biomarkers that could guide early stages of drug development 10. For example, the low trapping NMDAR antagonist lanicemine (AZD6765) has shown promise in early small studies 11, but failed to reach primary antidepressant endpoints in follow-up large, costly clinical trials using 50mg and 100mg doses 3, leading to the termination of its development. Establishing rigorous and reproducible human biomarkers of RAADs may play a critical role in these early stages of drug development 1, 2; (1) by expediting the in-human-screening of novel potential agents prior to conducting expensive trials, and (2) by determining the therapeutic window and identifying the optimal drug administration regimen, which could ultimately reduce the failure rate of clinical trials. In the current study, we investigated a novel biomarker of RAADs, termed global brain connectivity with global signal regression (GBCr). Prior studies have shown reduced prefrontal cortex (PFC) GBCr in depression 12–14, and open-label trials showed significant increases of PFC GBCr 24h post-ketamine administration in depressed patients 13, 14. However, the 24h effects were not previously tested in randomized placebo-controlled depression studies, and the effects of ketamine on PFC GBCr during infusion in depressed patients remain unknown.
Over the past decade, GBCr and other graph theory functional connectivity strength (a.k.a. degree) measures have been used extensively in neuroscience research to investigate normal brain functions, as well as pathophysiology 15. In major depressive disorder (MDD), studies have repeatedly shown reduced global connectivity within the PFC, using GBCr 12–14 or other functional connectivity strength measures 16, 17. The reduction in PFC GBCr was shown to be due to a dysconnectivity between the PFC and the rest of the brain in MDD subjects 13. Open-label ketamine enhanced the connectivity between the PFC and the rest of the brain, as well as normalized average PFC GBCr (24h post-administration) – particularly in responders to treatment 13. Considering glutamate models of MDD linking depression to prefrontal synaptic loss and dysconnectivity, and relating the RAAD effects of ketamine to increased synaptic density and enhanced connectivity 18, the pilot human data suggested a temporal relationship between glutamate synaptic connectivity and PFC GBCr changes in MDD and following ketamine infusion 13. Supporting the putative relationship between GBCr and glutamate synaptic connectivity is the known coupling between brain energy needs and glutamate neurotransmission 19, combined with the positive correlation between GBCr and indices of brain energy consumption 20. This relationship is also supported by the recent evidence from nonhuman primates showing a strong relationship between functional and invasive electrophysiological and anatomical connectivity 21, 22. Recent mechanistic investigation in healthy volunteers has provided additional evidence directly relating PFC GBCr to glutamate neurotransmission 14. First by showing reduced PFC GBCr following the administration of an inhibitor of glutamate transmission. Second by demonstrating a ketamine-induced surge in PFC GBCr during infusion, which was modulated by the glutamate transmission inhibitor 14. The latter finding, combined with substantial preclinical evidence relating the RAAD effect of ketamine to its ability to induce an acute glutamate surge 23, 24, raised the question whether ketamine produces an acute PFC GBCr surge in MDD patients.
The primary aim of the current report is to determine whether ketamine induces a PFC GBCr surge in MDD patients during infusion. In addition, we aimed to provide confirmatory evidence that the previously reported PFC GBCr changes following open-label ketamine would remain significant compared to placebo in a randomized controlled design. To accomplish these aims, we conducted a secondary analysis on previously collected data 25. In contrast to the current report aims, which are focused on ketamine and recent GBCr findings, the original trial was a pharmaco-MRI study investigating the blood oxygen level dependent (BOLD) activation during lanicemine compared to ketamine and placebo. It was found that ketamine evoked activations of BOLD signal in the anterior cingulate cortex (ACC) greater than lanicemine but both predicted improvement in mood 24 hours later 25.
To expand the scientific benefit and to inform future studies, we conducted secondary analyses examining the effects of lanicemine on PFC GBCr. These analyses are conducted separately, deemed secondary, and their results should be regarded as pilot in nature. Finally, we performed exploratory analyses investigating the GBCr correlates of ketamine-induced symptoms improvement, compared to placebo.
Methods
All imaging data and analyses, at the 24h time point, are new and have not been previously published. The imaging study during the infusion day is a novel connectivity analysis of a previously published pharmaco-MRI data set 25. None of the reported connectivity measures and analyses in the current report overlap with the previous study. The details of the clinical trial and the behavioral effects of the study drugs have been reported previously 25 and are not included in the current paper.
Participants
The parent clinical trial randomized 58 participants to normal saline (n = 19; mean age = 25.7), 0.5 mg/kg ketamine (n = 21; mean age = 27.1), or 100 mg lanicemine (n = 20; mean age = 26.7) infused intravenously over 60 minutes (Table S1). Study procedures were conducted at two sites (Manchester, UK & Oxford, UK), approved by institutional review boards, and informed consent were completed before participation (ClinicalTrials.gov identifier: NCT01046630). The study criteria were previously reported 25. Briefly, unmedicated depressed patients between the age of 18 and 45 (mean 27 years, 24 males) were randomized, provided they met criteria for a current MDD diagnosis (Structured Clinical Interview for DSM IV), were not taking psychoactive drugs for at least 2 weeks, had no lifetime history of psychosis, bipolar disorder, or alcohol/substance use disorder, had no unstable medical condition, were all left handed, females were not pregnant, were not consuming more than 10 cigarettes or 8 cups of caffeinated drinks per day, had a negative drug screen, and had no MR contraindications. Beck Depression Inventory (BDI) was used to assess depression severity correlates of the neuroimaging measures 25.
Neuroimaging
The details of the neuroimaging acquisition were previously reported 25. Briefly, resting-state functional magnetic resonance imaging (fMRI) scans (voxel size = 3 × 3 × 2.5 mm; TR = 3000 ms; TE = 30 ms) were acquired for 5 min. prior to the infusion, for 40 min. during the infusion, and for 25 min. 24h following the infusion. High resolution structural scans (voxel size = 1 × 1 × 1 mm) were acquired for coregistration. The Human Connectome Pipeline was adapted to conduct surface based preprocessing and optimize registration 26. Details of the image processing are provided in the Supplemental Information (SI). GBCr calculation followed our previous reports 13, 14, 27, i.e., they were computed as the average of the correlations between each voxel and all other voxels in the brain gray matter (see SI).
Statistical Analyses
Statistical Package for the Social Sciences (SPSS, version 24) was used for analyses. The distribution of outcome measures was examined using probability plots and test statistics. Transformations and non-parametric tests were used as necessary. Estimates of variation are provided as the standard error of the mean (SEM). Baseline BDI differed between the study groups (SI - Table S1). Therefore, it was included as a covariate. To demonstrate the effects of ketamine on average PFC GBCr, a repeated measure general linear model (GLM) examined the effects of treatment (placebo vs ketamine), time (baseline vs during infusion vs 24h post-treatment), and treatment-by-time interaction, with study sites and baseline BDI as covariates. Secondary analyses examined the effects of lanicemine using similar GLM. Significance was set at p ≤ 0.05, with 2-tailed tests.
Vertex-wise fMRI analyses used FSL Permutation Analysis of Linear Models (PALM), with tail approximation and threshold-free cluster enhancement (TFCE) for Type I error correction (corrected α = 0.05) 28. Independent t-tests compared PFC delta GBCr (during infusion – baseline & 24h – baseline) between treatment groups (ketamine vs placebo & lanicemine vs placebo), with study sites and baseline BDI as covariates. All vertex-wise analyses were limited to the PFC, and only vertices surviving correction for multiple comparisons are reported in the main text and figures.
Exploratory analyses without correction for multiple correction examined the correlation between BDI percent improvement at 24h post-treatment and PFC GBCr during and 24h following treatment.
Results
As previously reported 25, there was significant reduction in BDI scores at 24h following ketamine (32%), placebo (27%), and lanicemine treatments (26%). However, there were no significant differences in BDI improvement between treatment groups (treatment-by-time: F(2,52) = 0.47, p = 0.63).
Effects of Study Drugs – Average Prefrontal GBCr
Primary Analysis:
The GLM examining the effects of ketamine revealed a treatment-by-time interaction (F(1,32) = 5.7; p = 0.02), showing a significant increase in PFC GBCr – in the ketamine group compared to the placebo group – during infusion (F(1,32) = 6.3; p = 0.02; Fig. 1A) and 24h post-treatment (F(1,32) = 5.7; p = 0.02; Fig. 1B). We found no treatment effect (p = 0.59), but a significant time effect (F(1,32) = 7.0; p = 0.01), reflecting a significant increase of PFC GBCr during infusion and 24h post-treatment compared to baseline (p ≤ 0.01) with no significant differences between PFC GBCr during infusion and 24h post-treatment (p = 0.15). Exploratory analyses examining the time effects within each group showed a significant increase of PFC GBCr during infusion (p = 0.001; Fig. 1A) and 24h post-treatment (p = 0.0005; Fig. 1B) in the ketamine group, but not in the placebo group (p ≥ 0.69).
Figure 1. The effects of the study drugs on average prefrontal global connectivity.

Compared to placebo, ketamine significantly increased PFC GBCr during infusion (A) and 24h post-treatment (B). PFC GBCr changes during and following lanicemine did not significantly differ from PFC GBCr changes in the placebo group (A & B). Delta PFC GBCr (time point minus baseline) values are the estimated marginal means covarying for study site and baseline depression severity; p values reflect the results of the time effect within each group (i.e., during/post infusion vs. baseline); * (p < 0.05) and n.s. (p > 0.05) reflect the comparison of delta PFC GBCr between groups (i.e., drug vs. placebo); Abbreviations: PFC = prefrontal cortex; GBCr = global brain connectivity with global signal regression.
Secondary Analysis:
The GLM of the lanicemine effects found no significant treatment-by-time interaction (p = 0.23), reflecting the absence of differences between the changes in the PFC GBCr in the lanicemine group compared to the placebo group during infusion (p = 0.45) and 24h post-treatment (p = 0.23). Also, we found no significant treatment (p = 0.57) or time effects (p = 0.43). Exploratory analyses examining the time effects within the lanicemine group showed a significant increase of PFC GBCr 24h post-treatment (p = 0.01; Fig. 1B), but not during infusion (p = 0.34; Fig. 1A). We further explored whether the changes in PFC GBCr during or following lanicemine differed from ketamine, which showed no statistically significant differences between the two drugs (p > 0.05; Fig. 1A & 1B).
Effects of Study Drugs – Vertex-wise Prefrontal GBCr
Primary Analysis:
Compared to placebo, ketamine significantly increased GBCr in multiple clusters within the PFC during infusion and at 24h post-treatment. No reduction in PFC GBCr was observed. The GBCr increases were found in the dorsolateral, dorsomedial, and frontomedial PFC during infusion (Fig. 2), and in the dorsolateral and dorsomedial PFC 24h post-treatment (Fig. 3).
Figure 2. The effects of ketamine on prefrontal global connectivity during infusion.

Compared to placebo, ketamine significantly increased GBCr in the red/yellow clusters. Abbreviations: GBCr = global brain connectivity with global signal regression.
Figure 3. The effects of ketamine on prefrontal global connectivity 24h post-treatment.

Compared to placebo, ketamine significantly increased GBCr in the red/yellow clusters. Abbreviations: GBCr = global brain connectivity with global signal regression.
Secondary Analysis:
There were no clusters with significant changes in PFC GBCr in the lanicemine group compared to the placebo group.
Relationship to Treatment Response – Exploratory Analyses
Exploring the relationship between delta PFC GBCr during infusion and percent improvement of depression, we found a pattern of positively predicting depression improvement in participants receiving ketamine (r = 0.44; p = 0.06; d = 1.0) or lanicemine (r = 0.55; p = 0.01; d = 1.3), but not those receiving placebo (r = –0.1; p = 0.69; d = 0.02). We found no significant correlations between depression improvement and average delta PFC GBCr 24h post-treatment (p ≥ 0.60). However, vertex-level exploratory analyses examining the relationship with depression improvement showed positive correlation with GBCr in the dorsal PFC during infusion (Fig. 4) and 24h post-treatment (Fig. 5), but negative correlation with GBCr in the ventral PFC during infusion (Fig. 5; uncorrected p < 0.01).
Figure 4. Statistical maps examining the correlation between percent improvement of depression and GBCr changes during infusion of ketamine (uncorrected p ≤ 0.01).
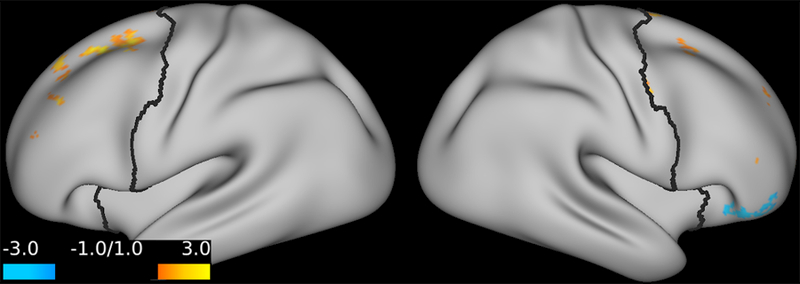
Compared to placebo, treatment response – as measured by the Beck Depression Inventory at 24h post-treatment – is associated with increased GBCr in the dorsal PFC (red/yellow), but reduced GBCr in the ventral PFC (blue). Abbreviations: PFC = prefrontal cortex; GBCr = global brain connectivity with global signal regression.
Figure 5. Statistical maps examining the correlation between percent improvement of depression and GBCr changes 24h post-treatment with ketamine (uncorrected p ≤ 0.01).
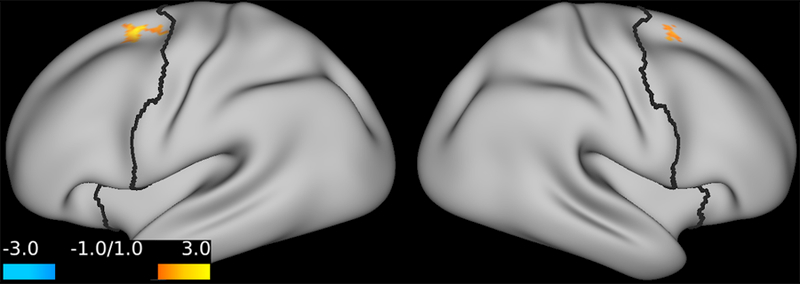
Compared to placebo, treatment response – as measured by the Beck Depression Inventory at 24h post-treatment – is associated with increased GBCr in the dorsal PFC (red/yellow). Abbreviations: PFC = prefrontal cortex; GBCr = global brain connectivity with global signal regression.
Discussion
Consistent with the study hypotheses, the results provide evidence of a prefrontal global connectivity surge during the infusion of an antidepressant dose of ketamine in MDD patients. Furthermore, the prefrontal connectivity changes were sustained at 24h post-ketamine administration, a finding that reproduced prior open-label evidence while providing strong support of the rigor and reproducibility of the PFC GBCr biomarker in the context of ketamine treatment. Follow-up vertex-wise analyses have localized the GBCr changes to prefrontal clusters primarily overlapping with the central executive and salience networks 29–31. Of note, the corrected statistical maps (Fig. 2 & 3) showed hemispheric lateralization and limited overlap between the significant clusters during infusion and at 24h post-ketamine. However, it is important to note that this lateralization and time-dependent localization may be due to varying effect size, considering the significant effects on average prefrontal connectivity (Fig. 1) and the bilateral effects on the uncorrected maps (Fig. S1).
Conversely, the study failed to demonstrate a significant overall or localized effect of lanicemine on PFC GBCr compared to placebo. However, two observations are worth considering in future studies. First, the acute prefrontal connectivity surge positively correlated with depression improvement post-lanicemine treatment. Second, the PFC GBCr changes following the administration of lanicemine (100mg) appeared to be numerically in between those of placebo and ketamine, raising the question whether future optimization of the lanicemine dose or administration regimen could lead to successful modulation of prefrontal global connectivity. Additionally, lanicemine was administer as fixed dose, while ketamine dose was based on body weight. Thus, considering the larger error bars in the lanicemine vs. ketamine (Fig. 1), it is plausible that administering lanicemine dose based on weight may reduce the variance across subjects and improve its effect on prefrontal global connectivity.
In our previous studies 13, 14, we have demonstrated both reduced depression severity and increased prefrontal GBCr following ketamine treatment. These findings raised at least two possibilities: (1) ketamine increases prefrontal synaptic plasticity and global connectivity, leading to reduction in depression (i.e., consistent with the preclinical models), or (2) reduction of depression, regardless of treatment modality (i.e., placebo or ketamine), leads to an increase in prefrontal GBCr. While it is unfortunate that the current clinical trial failed to show significant behavioral differences between ketamine and placebo, it did offer a unique mechanistic opportunity to distinguish the pharmacodynamic effects of ketamine from the non-specific behavioral effects of reducing depression symptoms (i.e., to the rule out hypothesis #2). In fact, the current results provide strong evidence against hypothesis #2, by demonstrating that the GBCr increases are only evident after ketamine, but not placebo; despite the fact that depression improvement was comparable in both arms. This is further supported by the lack of correlation between GBCr changes and depression improvement in the placebo arm. Together, the results are consistent with the preclinical hypothesis that ketamine induces its antidepressant response through enhancing prefrontal synaptic and functional connectivity 1, 2, while the antidepressant effects of placebo are through unknown mechanisms – independent of prefrontal global connectivity. In summary, there is extensive evidence in the literature that both placebo and ketamine significantly reduce depression severity. The current study results provide mechanistic evidence that, the neural mechanisms underlying the placebo antidepressant effects are not related to prefrontal connectivity. In contrast, the results show a pattern of positive association between improvement of depression symptoms and increases in prefrontal connectivity following ketamine and lanicemine treatment.
Based on extensive preclinical evidence and suggestive clinical findings, it was proposed that prefrontal synaptic loss and dysconnectivity underlie the pathophysiology of depression 1, 18, 23, 32, 33. In this model, the altered synaptic connectivity affects large-scale networks precipitating and perpetuating depressive symptomatology 34. Moreover, the reversal of synaptic dysconnectivity appears to be a necessary step to exert antidepressant effects 18. Ketamine has been found to robustly reverse the prefrontal synaptic dysconnectivity in animal models of depression, and to normalize related network disturbances in humans 18, 34, 35. At the molecular level, it is believed that ketamine and other RAADs (e.g. scopolamine) exert their effects primarily through induction of AMPA-dependent glutamate neurotransmission surge and subsequent enhancement of neurotrophic factors and synaptogenesis 2, 36, 37. Yet the role of NMDAR antagonism in inducing this glutamatergic synaptogenesis remains contested 1, 6, 7, 38, 39. The current study findings show a remarkable temporal parallel between the ketamine-induced acute glutamate surge and sustained synaptogenesis in rodents, and the ketamine-induced acute and sustained PFC GBCr in depressed patients.
In this context, and considering prior evidence linking GBCr to glutamate transmission and neuroenergetics 14, 20, we interpret the acute increases in PFC GBCr during ketamine infusion and its positive relationship to treatment response, as a supportive human evidence, (1) that the ketamine-induced acute glutamate surge is putatively present in humans 40–42, (2) that it contributes to the RAAD of the drug 43, 44, and (3) that PFC GBCr is a clinically relevant rigorous and reproducible biomarker of the underlying glutamate surge 14. The sustained PFC GBCr increases, 24h post-ketamine, are hypothesized to reflect the underlying ketamine-induced glutamatergic synaptogenesis reported in animal models. Consistent with our previous findings 13, the current study provided exploratory evidence associating depression improvement with increased GBCr in the dorsal PFC 24h post-ketamine. However, it is important to caution the reader that these findings would require replication in future studies, considering the trend level of significance during infusion (i.e., r = 0.44; p = 0.06; d = 1.0), the lack of correction for multiple comparisons and the lack of correlation with overall PFC GBCr. The latter could be due to the low response rate in the parent clinical trial, which reduced the sample power to detect positive correlation between improvement and 24h GBCr increases. Moreover, as noted in the vertex-wise analysis (Fig. 5), it is plausible that at the 24h time point a more localized PFC GBCr effect would better map to the antidepressant response. Hence, this exploratory analysis supports the need for larger samples to robustly identify the localized GBCr correlates of treatment response at the 24h time point.
In the parent pharmaco-MRI trial, ketamine evoked activations of blood oxygen level dependent (BOLD) signal in the anterior cingulate cortex (ACC) greater than lanicemine but both predicted improvement in mood 24 hours later25. Evidence suggests that ACC BOLD responses to ketamine also reflect increased glutamate release, considering its modulation by a glutamate release inhibitor45. Thereforre, the pharmaco-MRI and connectivity analyses reinforce a primary role for a glutamate surge as a mechanism of the observed rapid antidepressant effects. Since BOLD signal is linked to neuronal metabolism46, it is possible that the glutamate surge occurs in ACC and not in frontal cortex which did not show BOLD responses to ketamine. The major reciprocal innervation between frontal cortex and ACC could then initiate the increased frontal GBCr connectivity which mediates the antidepressant response.
Limitations and Strengths:
A main limitation is the low response rate in the parent trial which may have reduced our ability to detect significant association between depression improvement and 24h PFC GBCr. It also limits the generalizability of the results to the larger population of ketamine treated patients who show, on average, superior response rate following ketamine treatment. Additionally, the study could have benefited from a follow-up time point at 10–14 days post-treatment to determine the longevity of the PFC GBCr changes and its relationship to relapse. Of note, the ketamine-induced synaptogenesis lasts for approximately 10 days in rodents, and the majority of depressed patients relapse within 2 weeks of single ketamine infusion 47. Another limitation is the use of a self-report measure of depression and the enrollment of non-treatment resistance population in contrast to previous ketamine studies. Additionally, the pre-treatment fMRI acquisition was only 5 minutes, future connectivity studies may benefit from longer acquisition time. Finally, future studies may particularly benefit from more substantial sample and comprehensive standardized assessments of depression-related constructs, e.g. anhedonia and suicidality, to better understand the relationship between alterations in these constructs and better localize the underlying prefrontal GBCr changes. The strengths of the study include, (1) a randomized placebo-controlled design, (2) a relative large sample compared to previous ketamine neuroimaging studies, and (3) the use of GBCr a well validated robust biomarker that is based on established graph theory models with extensive prior evidence in health and disease to facilitate the interpretation of the study findings. The robustness of the GBCr measure is further supported by the lack of any notable changes in PFC GBCr in the placebo group in the current study in MDD and in previous studies in healthy subjects 14.
Conclusions
The study provides evidence of ketamine-induced prefrontal global connectivity during infusion in depressed patients. It also replicates, in a placebo-controlled design, previous open-label evidence of ketamine-induced prefrontal global connectivity 24h post-treatment. Lanicemine (100mg) failed to induce significant prefrontal global connectivity increases during and 24h post-treatment, compared to placebo. However, it is evident that numerically the lanicemine-induced connectivity changes appear to be in the same direction as ketamine, which raises the question for future studies whether optimization of the lanicemine administration regimen could lead to significant prefrontal connectivity normalization – especially considering the positive relationship between prefrontal global connectivity increases and treatment response.
Supplementary Material
Acknowledgments
Funding and Disclosure
The authors would like to thank the subjects who participated in this study for their invaluable contribution.
Funding support was provided by AstraZeneca, NIMH (K23MH101498), the VA National Center for PTSD, the Brain & Behavior Foundation (NARSAD), and the Robert E. Leet and Clara Guthrie Patterson Trust. The content of this report is solely the responsibility of the authors and does not necessarily represent the official views of the sponsors, the Department of Veterans Affairs, NIH, or the U.S. Government. We are grateful for the expert assistance of the staff of the Clinical Research Facilities in Manchester and Oxford in which the study was conducted. P1vital managed the original study on behalf of AstraZeneca.
CG Abdallah has served as a consultant and/or on advisory boards for Genentech and Janssen, and as editor for Chronic Stress by Sage Publications, Inc. JFW Deakin currently advises or carries out research funded by Autifony, Sunovion, Lundbeck, AstraZeneca and Servier. A.D., C.L.A., S.M., T.J.A., and L.A.A. report no financial disclosure.
References
- 1.Abdallah CG, Sanacora G, Duman RS and Krystal JH. The neurobiology of depression, ketamine and rapid-acting antidepressants: Is it glutamate inhibition or activation? Pharmacology & Therapeutics. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murrough JW, Abdallah CG and Mathew SJ. Targeting glutamate signalling in depression: progress and prospects. Nat Rev Drug Discov. 2017; 16: 472–86. [DOI] [PubMed] [Google Scholar]
- 3.Sanacora G, Johnson MR, Khan A, et al. Adjunctive Lanicemine (AZD6765) in Patients with Major Depressive Disorder and History of Inadequate Response to Antidepressants: A Randomized, Placebo-Controlled Study. Neuropsychopharmacology. 2017; 42: 844–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zarate CA, Singh JB Jr., Quiroz JA, et al. A double-blind, placebo-controlled study of memantine in the treatment of major depression. Am J Psychiatry. 2006; 163: 153–5. [DOI] [PubMed] [Google Scholar]
- 5.Zanos P, Moaddel R, Morris PJ, et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature. 2016; 533: 481–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abdallah CG. What’s the Buzz About Hydroxynorketamine? Is It the History, the Story, the Debate, or the Promise? Biol Psychiatry. 2017; 81: e61–e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aleksandrova LR, Wang YT and Phillips AG. Hydroxynorketamine: Implications for the NMDA Receptor Hypothesis of Ketamine’s Antidepressant Action. Chronic Stress. 2017; 1: 2470547017743511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wegener G and Rujescu D. The current development of CNS drug research. Int J Neuropsychopharmacol. 2013; 16: 1687–93. [DOI] [PubMed] [Google Scholar]
- 9.Hendrie C, Pickles A, Stanford SC and Robinson E. The failure of the antidepressant drug discovery process is systemic. J Psychopharmacol. 2013; 27: 407–13; discussion 13–6. [DOI] [PubMed] [Google Scholar]
- 10.Paul SM, Mytelka DS, Dunwiddie CT, et al. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat Rev Drug Discov. 2010; 9: 203–14. [DOI] [PubMed] [Google Scholar]
- 11.Sanacora G, Smith MA, Pathak S, et al. Lanicemine: a low-trapping NMDA channel blocker produces sustained antidepressant efficacy with minimal psychotomimetic adverse effects. Mol Psychiatry. 2014; 19: 978–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murrough JW, Abdallah CG, Anticevic A, et al. Reduced global functional connectivity of the medial prefrontal cortex in major depressive disorder. Hum Brain Mapp. 2016; 37: 3214–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abdallah CG, Averill LA, Collins KA, et al. Ketamine Treatment and Global Brain Connectivity in Major Depression. Neuropsychopharmacology. 2017; 42: 1210–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abdallah CG, Averill CL, Salas R, et al. Prefrontal Connectivity and Glutamate Transmission: Relevance to Depression Pathophysiology and Ketamine Treatment. Biol Psychiatry Cogn Neurosci Neuroimaging. 2017; 2: 566–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bullmore E and Sporns O. Complex brain networks: graph theoretical analysis of structural and functional systems. Nat Rev Neurosci. 2009; 10: 186–98. [DOI] [PubMed] [Google Scholar]
- 16.Wang L, Dai Z, Peng H, et al. Overlapping and segregated resting-state functional connectivity in patients with major depressive disorder with and without childhood neglect. Hum Brain Mapp. 2014; 35: 1154–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scheinost D, Holmes SE, DellaGioia N, et al. Multimodal Investigation of Network Level Effects Using Intrinsic Functional Connectivity, Anatomical Covariance, and Structure-to-Function Correlations in Unmedicated Major Depressive Disorder. Neuropsychopharmacology. 2018; 43: 1119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duman RS and Aghajanian GK. Synaptic dysfunction in depression: potential therapeutic targets. Science. 2012; 338: 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hyder F, Rothman DL and Bennett MR. Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc Natl Acad Sci U S A. 2013; 110: 3549–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang X, Zou Q, He Y and Yang Y. Coupling of functional connectivity and regional cerebral blood flow reveals a physiological basis for network hubs of the human brain. Proc Natl Acad Sci U S A. 2013; 110: 1929–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen LM, Yang PF, Wang F, et al. Biophysical and neural basis of resting state functional connectivity: Evidence from non-human primates. Magn Reson Imaging. 2017; 39: 71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Z, Chen LM, Negyessy L, et al. The relationship of anatomical and functional connectivity to resting-state connectivity in primate somatosensory cortex. Neuron. 2013; 78: 1116–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abdallah CG, Sanacora G, Duman RS and Krystal JH. Ketamine and rapid-acting antidepressants: a window into a new neurobiology for mood disorder therapeutics. Annu Rev Med. 2015; 66: 509–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abdallah CG, Adams TG, Kelmendi B, Esterlis I, Sanacora G and Krystal JH. Ketamine’s Mechanism of Action: A Path to Rapid-Acting Antidepressants. Depress Anxiety. 2016; 33: 689–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Downey D, Dutta A, McKie S, et al. Comparing the actions of lanicemine and ketamine in depression: key role of the anterior cingulate. Eur Neuropsychopharmacol. 2016; 26: 994–1003. [DOI] [PubMed] [Google Scholar]
- 26.Glasser MF, Sotiropoulos SN, Wilson JA, et al. The minimal preprocessing pipelines for the Human Connectome Project. Neuroimage. 2013; 80: 105–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abdallah CG, Wrocklage KM, Averill CL, et al. Anterior hippocampal dysconnectivity in posttraumatic stress disorder: a dimensional and multimodal approach. Translational psychiatry. 2017; 7: e1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Winkler AM, Ridgway GR, Webster MA, Smith SM and Nichols TE. Permutation inference for the general linear model. Neuroimage. 2014; 92: 381–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akiki TJ, Averill CL and Abdallah CG. A Network-Based Neurobiological Model of PTSD: Evidence From Structural and Functional Neuroimaging Studies. Curr Psychiatry Rep. 2017; 19: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yeo BT, Krienen FM, Sepulcre J, et al. The organization of the human cerebral cortex estimated by intrinsic functional connectivity. J Neurophysiol. 2011; 106: 1125–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akiki TJ and Abdallah CG. Determining the Hierarchical Architecture of the Human Brain Using Subject-Level Clustering of Functional Networks. bioRxiv. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McEwen BS. Neurobiological and Systemic Effects of Chronic Stress. Chronic Stress. 2017; 1: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis MT, Holmes SE, Pietrzak RH and Esterlis I. Neurobiology of Chronic Stress-Related Psychiatric Disorders: Evidence from Molecular Imaging Studies. Chronic Stress. 2017; 1: 2470547017710916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lener MS, Niciu MJ, Ballard ED, et al. Glutamate and Gamma-Aminobutyric Acid Systems in the Pathophysiology of Major Depression and Antidepressant Response to Ketamine. Biol Psychiatry. 2017; 81: 886–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murrough JW, Collins KA, Fields J, et al. Regulation of neural responses to emotion perception by ketamine in individuals with treatment-resistant major depressive disorder. Translational psychiatry. 2015; 5: e509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hare B, Ghosal S and Duman R. Rapid acting antidepressants in chronic stress models: molecular and cellular mechanisms. Chronic Stress. 2017; 1: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Machado-Vieira R, Henter ID and Zarate CA, Jr. New targets for rapid antidepressant action. Prog Neurobiol. 2017; 152: 21–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Collingridge GL, Lee Y, Bortolotto ZA, Kang H and Lodge D. Antidepressant Actions of Ketamine Versus Hydroxynorketamine. Biol Psychiatry. 2017; 81: e65–e7. [DOI] [PubMed] [Google Scholar]
- 39.Zanos P, Moaddel R, Morris PJ, et al. Reply to: Antidepressant Actions of Ketamine Versus Hydroxynorketamine. Biol Psychiatry. 2017; 81: e69–e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abdallah CG, De Feyter HM, Averill LA, et al. The effects of ketamine on prefrontal glutamate neurotransmission in healthy and depressed subjects. Neuropsychopharmacology. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DeLorenzo C, DellaGioia N, Bloch M, et al. In Vivo Ketamine-Induced Changes in [(11)C]ABP688 Binding to Metabotropic Glutamate Receptor Subtype 5. Biol Psychiatry. 2015; 77: 266–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Javitt DC, Carter CS, Krystal JH, et al. Utility of Imaging-Based Biomarkers for Glutamate-Targeted Drug Development in Psychotic Disorders: A Randomized Clinical Trial. JAMA psychiatry. 2018; 75: 11–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Esterlis I, DellaGioia N, Pietrzak RH, et al. Ketamine-induced reduction in mGluR5 availability is associated with an antidepressant response: an [(11)C]ABP688 and PET imaging study in depression. Mol Psychiatry. 2018; 23: 824–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Milak MS, Proper CJ, Mulhern ST, et al. A pilot in vivo proton magnetic resonance spectroscopy study of amino acid neurotransmitter response to ketamine treatment of major depressive disorder. Mol Psychiatry. 2016; 21: 320–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deakin JF, Lees J, McKie S, Hallak JE, Williams SR and Dursun SM. Glutamate and the neural basis of the subjective effects of ketamine: a pharmaco-magnetic resonance imaging study. Arch Gen Psychiatry. 2008; 65: 154–64. [DOI] [PubMed] [Google Scholar]
- 46.Bonvento G, Sibson N and Pellerin L. Does glutamate image your thoughts? Trends Neurosci. 2002; 25: 359–64. [DOI] [PubMed] [Google Scholar]
- 47.Abdallah CG, Averill LA and Krystal JH. Ketamine as a promising prototype for a new generation of rapid-acting antidepressants. Ann N Y Acad Sci. 2015; 1344: 66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.