

Abstract
The PutA flavoprotein from Escherichia coli plays multiple roles in proline catabolism by functioning as a membrane-associated bi-functional enzyme and a transcriptional repressor of the proline utilization genes, while the human homologue of the PutA proline dehydrogenase (PRODH) domain plays critical roles in p53-mediated apoptosis and schizophrenia. We report the crystal structure of a 669-residue truncated form of PutA that exhibits both PRODH and DNA-binding activities, which represents the first structure of a PutA protein and the first structure of a PRODH enzyme from any organism. The structure is a domain-swapped dimer with each subunit comprising three domains: a helical dimerization arm, a 120-residue domain containing a three-helix bundle similar to that found in the helix-turn-helix superfamily of DNA-binding proteins, and a beta/alpha barrel PRODH domain with bound lactate inhibitor. Analysis of the structure provides insight into the mechanism of proline oxidation to pyrroline-5-carboxylate, and functional studies of a mutant protein suggest the DNA-binding domain is located within the N-terminal 261 residues of E. coli PutA.
Enteric bacteria possess a remarkable multifunctional flavoprotein, called PutA for proline utilization A, that functions as both a membrane-associated proline catabolic enzyme and a transcriptional repressor of proline utilization (put) genes. Peripherally membrane-bound PutA catalyzes the two-step oxidation of proline to glutamate1-3 (Fig. 1) while cytoplasmic PutA regulates proline catabolism by repressing the transcription of its own gene and the gene for the PutP Na(+)/proline transporter4-6. Together, PutA and PutP enable enteric bacteria to use proline as a source of carbon, nitrogen, and electrons5,6.
Figure 1.

Reactions catalyzed by the E. coli PutA protein. PutA669 catalyzes only the first reaction, the conversion of proline to P5C.
Proline availability is the environmental cue that dictates PutA’s intracellular location and function7-10. In the absence of proline, PutA accumulates in the cytoplasm and represses the divergent transcription of the put regulon by binding to multiple sites in the 419 base-pair putP-putA intergenic region. In the presence of proline, PutA associates with the membrane where it performs its enzymatic functions. Proline is oxidized to Δ1-pyrroline-5-carboxylate (P5C) in the first, or proline dehydrogenase (PRODH) step of catalysis, which is coupled to the 2-electron reduction of the noncovalently bound FAD (Fig. 1). Electrons from the reduced FAD are subsequently transferred to an acceptor in the electron transport chain. Since proline reduces the FAD in the PRODH step of catalysis, the FAD redox state is implicated as a regulatory signal for the association of PutA with the membrane. In the second step of catalysis, glutamic semialdehyde, presumably the product of nonenzymatic hydrolysis of P5C, is oxidized to glutamate, reducing NAD+ to NADH (Fig. 1).
While the functions of PutA are well documented, this protein remains enigmatic due in large part to a complete lack of three-dimensional structural information for PutA and its domains. Thus, we have initiated X-ray crystallographic studies of the E. coli protein in order to establish a structural framework for investigating PutA’s catalytic and repressor functions, and to elucidate the conformational changes caused by substrate binding and flavin reduction.
In a larger context, the two enzymatic reactions depicted in Fig. 1 are common to all organisms, however, in eukaryotes, they appear as distinct enzymes encoded by separate genes. The human homologue of the PutA PRODH domain, known variously as hsPRODH2, PIG6 (for p53-induced gene 611), and proline oxidase12, is a mitochondrial enzyme of significant interest due to its roles in p53-mediated apoptosis11, proline-dependent reactive oxygen species generation12, and schizophrenia13. The involvement of hsPRODH2 in the devastating mental illness schizophrenia is particularly compelling. Recent molecular genetics studies based on patients with adult or childhood onset schizophrenia strongly suggest that genetic variations in the hsPRODH2 gene increase the risk of schizophrenia susceptibility13. Several of these variations result in missense changes at conserved amino acid residues; thus the encoded enzyme may not be fully functional. The precise role of hsPRODH2 in schizophrenia is yet to be determined, but one intriguing possibility is that proline may serve as a direct modulator of glutamatergic transmission in the brain, and that elevated proline levels caused by impaired hsPRODH2 activity contribute to the onset of schizophrenia13,14.
To provide a structural foundation for understanding the multifunctional E. coli PutA protein, as well as PRODH enzymes from other organisms, we determined the crystal structure of a protein (PutA669) corresponding to the N-terminal 669 residues of the 1320 amino acid E. coli PutA polypeptide chain. PutA669 binds FAD, exhibits the PRODH activity of the full-length PutA protein, and binds specifically to the put intergenic DNA with a dissociation constant of 15 nM15. Thus, PutA669 is a good model system for understanding the PRODH and DNA-binding functions of PutA. In contrast, PutA669 does not contain the P5C dehydrogenase domain, which is located within residues 650-1130 based on sequence alignments16, nor does it exhibit the membrane association activity of PutA15. Therefore, the PutA669 structure provides little insight into other aspects of PutA function, such as the mechanism by which flavin reduction induces membrane binding, and the structural basis of PutA/membrane association.
Overall description of the PutA669 structure
The PutA669 crystal structure is the first structure of a PutA protein and the first structure of a PRODH enzyme from any organism. PutA669 is a homodimer of interlocking subunits with overall dimensions 97 Å × 57 Å × 50 Å (Fig. 2a). The dimeric model was generated from the subunit in the asymmetric unit by a crystallographic 2-fold rotation around the c-axis. This particular mode of dimerization was identified by virtue of its extraordinarily large interfacial buried surface area (9002 Å2). Note that PutA669, like PutA, forms a dimer in solution according to gel filtration studies15.
Figure 2.



Overall structure of PutA669. a, Stereo-view of the homodimer, with domain I colored yellow (87-139), domain II blue (140-260), and domain III red/green (261-612). Selected residue numbers of one subunit are indicated. b, Cylinder drawing of domain II. The helices are labeled D-I, and the dashed curves represent disordered residues. c, Domain III, the PRODH domain, looking into the barrel. The helices are colored red and labeled 0-11, while the strands are colored green and labeled 1-8. Note that strands 5-8 are hidden by α-helix 8. The FAD cofactor is drawn as a ball-and-stick model in yellow. This and other figures were prepared with Molscript 37 and Raster3d 38.
Each subunit comprises three structurally distinct domains. Domain I (residues 87-139) consists of three α-helices (αA, αB, αC) that form an arm that reaches out from one subunit to embrace the other subunit, thus creating an interlocking, or three-dimensional domain swapped17, dimer (Fig. 2a). Domain II (140-260) is a cluster of six α-helices labelled αD-αI (Fig. 2b). This domain does not make significant intersubunit contacts, nor does it contribute resides to the active site. Thus, the function of domain II is not immediately apparent from its structure. Domain III (residues 261-612) is a β8α8 barrel that binds FAD at the carboxyl ends of the β-strands of the barrel (Fig. 2c), and thus domain III performs the PRODH function of PutA669.
The proline dehydrogenase domain
The PutA669 structure is only the second example of a FAD cofactor bound to an β8α8 barrel, with the other example being methylenetetrahydrofolate reductase (MTHFR)18. The FAD binds with its pyrimidine carbonyls directed toward β3 and the dimethyl benzene edge of the isoalloxazine near β7 (Fig. 2c), which is similar to the orientation of FAD in the barrel of MTHFR. Strands 4-6 support the re face of the isoalloxazine, while the ribityl and pyrophosphate groups insert between β5 and β6. The adenine sits above the rim of the barrel where it is wedged between α5a and α5.
The fortuitous discovery of the competitive inhibitor L-lactate bound to the enzyme provides insight into the identity of active site residues. Lactate binds near the pyrimidine portion of the si face of the FAD isoalloxazine (Fig. 3a), with its carboxylate bound by three basic residues: Lys329, Arg555, and Arg556 (Fig. 3b). These three residues are stabilized by an ion pair network involving Glu559, Glu289, Asp370, and Arg431. Residues Arg556 and Glu559 hydrogen bond to the flavin ribityl, while Arg431 donates a hydrogen bond to the FAD N5. The lactate hydroxyl forms hydrogen bonds to Asp370, Tyr540 and a water molecule (Wat1). Wat1, in turn, hydrogen bonds to Tyr437. Finally, the lactate methyl group forms van der Waals interactions with Leu513.
Figure 3.



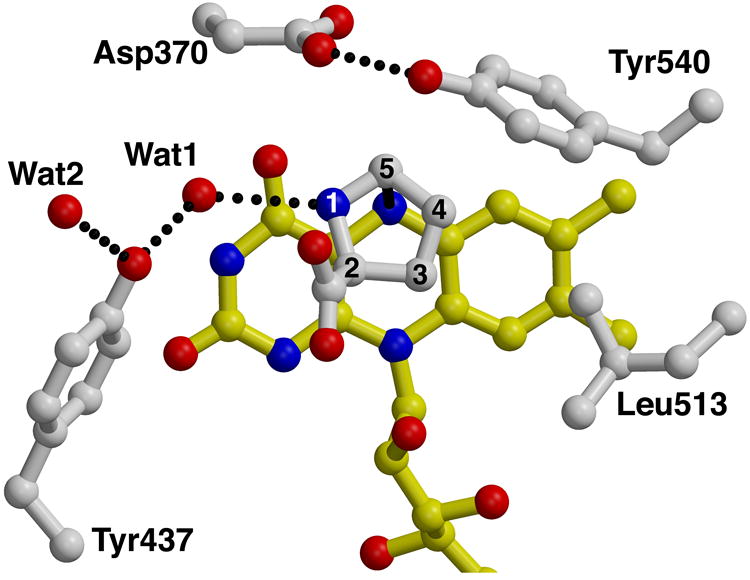
The active site of PutA669. a, Stereo-view of a σA-weighted39, simulated annealing, Fo-Fc omit electron density map (2.5 σ) covering the bound L-lactate inhibitor (white). FAD is drawn in yellow. This figure was prepared with Bobscript40. b, Stereo-view of the active site with FAD shown in yellow, and amino acid side chains and lactate inhibitor in white. The dotted lines represent hydrogen bonds and ion pairs. Helix α8 is shown in red. c, d, Two orthogonal views of a model of the substrate proline bound to the active site. Proline appears in white with its atom numbered. The dotted lines represent hydrogen bonds, and the solid line denotes a distance of 3.2 Å between hydride transfer partners C5 and N5. Interactions with the proline carboxylate are identical to those shown for the lactate carboxylate in panel b, and thus are omitted for clarity.
All the aforementioned active site residues are highly conserved in sequences of bacterial PutA proteins and eukaryotic PRODH enzymes from various organisms with overall sequence identities to E. coli PutA of 19-90%. Thus, the PutA669 structure serves as the prototype for FAD-dependent proline dehydrogenase domains. In particular, the active site residues of PutA669 display a high degree of sequence conservation with hsPRODH2. Of the 40 residues within 5 Å of FAD or lactate, 19 are identical in the hsPRODH2 sequence, and 6 residues display conservative substitutions. Such extensive conservation (47% identity, 62% similarity) argues that PutA669 and hsPRODH2 share a common active site and possibly a common fold, and that the PutA669 PRODH domain structure provides a three-dimensional framework for understanding the function of hsPRODH2 and its role in disease.
The bound inhibitor was used to model the binding of the substrate proline to PutA669 in order to identify residues that could contact the substrate, and to generate hypotheses about the catalytic mechanism. Our model indicates that the proline C5-H bond points toward the flavin N5 with a C5-N5 distance of 3.2 Å (Fig. 3c), which is consistent with a direct hydride transfer mechanism. For example, similar ligand-FAD geometries are observed in structures of D-amino acid oxidase (DAAO), which catalyzes a reaction analogous to that of PutA669, the oxidation of D-amino acids to imino acids19,20. The direct hydride transfer mechanism has been implicated in DAAO based on structural19,20 and kinetic21,22 data.
Asp370, Tyr540, and Leu513 interact with the methylene groups of proline (Fig. 3d). Asp370 and Tyr540 appear to form the ceiling of the active site, and they are held in place by a hydrogen bond between them, while Leu513 forms the right-hand wall of the active site. These three side-chains might play a role in catalysis by enforcing size and shape requirements on the substrate. Wat1 is located 2.9 Å from the proline N atom, and thus could stabilize the substrate and P5C product through hydrogen bonding (Fig. 3d). Presumably, a base in the active site serves to accept a proton from the substrate N, assuming the zwitterionic form of proline binds in the active site (pKa=10.5). There are several possible residues that could potentially play this role, including Asp370, Tyr437, and Wat1 (Fig. 3d). Mutagenesis studies and crystal structures of PutA669 complexed with proline and proline analogues should provide insight into this aspect of the catalytic mechanism.
Conventional wisdom holds that hydrolysis of P5C to glutamic semi-aldehyde occurs non-enzymatically after the P5C product exits the active site. The structure suggests this step of catalysis might occur within the active site based on the proximity of Wat1 to proline in the model. Note that attack of Wat1 on P5C would require a counterclockwise rotation of P5C relative to the orientation of proline in Fig. 3d to bring C5 close to Wat1. Performing the hydrolysis reaction within the active site could offer a catalytic advantage in that the increase in molecular volume and conformational entropy due to conversion of P5C to glutamic semi-aldehyde facilitates product release. Formation of the glutamic semi-aldehyde product in the PRODH active site raises the possibility of channeling between the PRODH and P5C dehydrogenase active sites, which has been proposed23. Since PutA669 lacks the P5C dehydrogenase domain, it is difficult to speculate which residues might be involved in channeling. But, the structure does suggest that the issue of channeling deserves more study.
The DNA-binding function of PutA699
In addition to its enzymatic function, PutA669 exhibits the DNA-binding function of PutA15, thus the DNA-binding apparatus is located somewhere in the first 669 residues of the PutA protein. To further isolate which part of the PutA polypeptide interacts with DNA, a truncated protein corresponding to the first 261 residues of PutA (PutA261) was engineered, expressed in E. coli, and assayed for DNA-binding activity using gel shift analysis. Note that PutA261 contains domains I and II, as well as the N-terminal 86 residues of PutA, which are disordered in the current structure. Gel shift analysis clearly shows that PutA261 inhibits the migration of the 419 base-pair put intergenic DNA through an agarose/polyacrylamide gel matrix (Fig. 4a, lanes 2-5). In addition, PutA261 does not retard the migration of ladder DNA (Fig. 4a, lane 6), which shows that PutA261, like PutA and PutA669, binds specifically to the put intergenic DNA. These results show that the residues that perform the DNA-binding function of PutA are located within residues 1-261, and that the PRODH and DNA-binding activities are separable.
Figure 4.
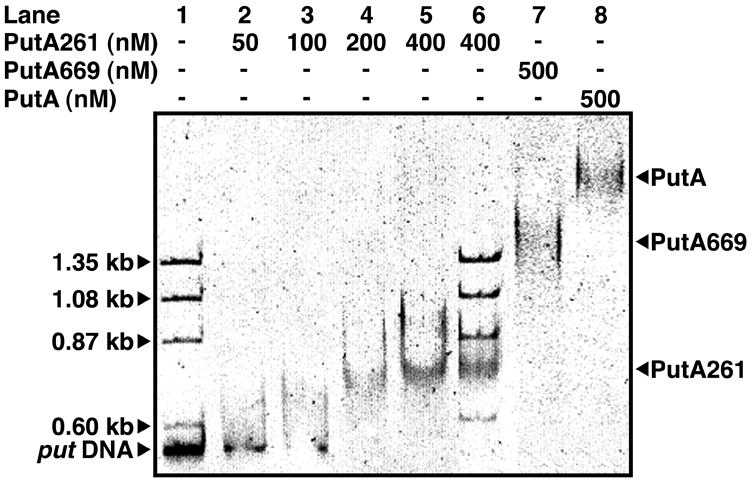

The DNA-binding function of PutA669. a, Gel mobility shift analysis of PutA proteins and the put control DNA. PutA261 (0-400 nM), PutA669 (500 nM), and PutA (500 nM each) were added to separate binding mixtures in 70 mM Tris (pH 7.5) containing put control intergenic DNA (20 nM) at 20 °C. Nonspecific ΦX174 ladder DNA (20 μg/ml) was also added to binding mixtures containing 0 nM and 400 nM PutA261. The complexes were separated using an agarose/polyacrylamide (0.5%/3%) native gel at 4 °C. The gel was stained with ethidium bromide to visualize the put DNA-protein complexes and the ΦX174 ladder DNA. b, Comparison of PutA669 domain II (blue) and the HTH three-helix bundle of Cdc6/Cdc18 (yellow, pdb id 1fnn, residues A287-A343). The orientation is identical to that of (Fig. 2b). The dashed curves represent disordered residues in PutA669.
The PutA669 structure and the PutA261 functional results together suggest three possible candidates for the DNA-binding domain of PutA: residues 1-86, domain I, and domain II. Residues 1-86 are disordered in the current crystal structure, therefore the structure does not provide insight into the functional role of these residues. Domain I is involved in dimerization by forming many intimate contacts with the barrel of the opposite subunit (Fig. 2a). Direct participation of this domain in DNA association seems unlikely due to its role in dimer formation. Domain II (Fig. 2b) is not involved in catalysis or in dimerization, thus process of elimination suggests this domain could play a role in binding DNA.
To provide support for the idea that domain II is involved in DNA association, the program DALI24 was used to determine whether domain II bears any structural similarity to known DNA-binding proteins. DALI failed to find a domain in the PDB with high global structural similarity to domain II, which suggests that domain II might represent a novel protein fold. On the other hand, DALI did find a significant local structural similarity between the helix-turn-helix (HTH) superfamily of DNA-binding proteins and helices αE, αG, and αH of domain II. In particular, DALI identified the following HTH proteins as the top three matches with the structural database: the sporulation response regulator protein Spo0A25 (Z-score = 3.7, RMSD = 3.6 Å), the nitrate response regulator protein NarL26 (Z-score = 3.7, RMSD = 4.9 Å), and the cell division control protein Cdc6/Cdc1827 (Z-score = 3.6, RMSD = 3.4 Å). Alignment of these proteins onto domain II revealed that αE, αG, and αH form a three-helix bundle similar to that found in the HTH superfamily. Note, for example, that the HTH three-helix bundle of Cdc6/Cdc18 superimposes almost perfectly with helices E, G, and H of PutA669 (Fig. 4b). This comparison suggests that αE is structurally analogous to the first helix of the HTH three-helix bundle, while αG and αH are analogous to the HTH scaffolding and recognition helices28.
While PutA contains a three-helix bundle resembling those found in HTH DNA-binding proteins, the larger question of how PutA uses its HTH substructure, if at all, to perform its repressor function remains unanswered. The structure presented here provides a foundation for designing biochemical experiments, such as mutagenesis, that will help answer this question. Ultimately, the crystal structures of PutA proteins complexed with DNA will be needed to fully understand how E. coli PutA functions as both an enzyme and a transcriptional repressor.
Methods
Crystallization and heavy atom derivative preparation
PutA669 was expressed in E. coli, purified, and crystallized as previously described29. Crystals were grown in hanging drops over a reservoir of 21-26% (w/v) PEG 3000, 0.1 M citrate buffer, pH 6-7. They occupy space group I222 with unit cell dimensions a=72.62 Å, b=140.43 Å, c=145.85 Å, and the asymmetric unit contains one PutA669 monomer29. Halide derivatives for phase determination were prepared by soaking native crystals for 30 s in the cryobuffer (24% PEG 3000 (w/v), 10% PEG 200 (v/v), 0.1 M citrate buffer, pH 6.6) supplemented with either 0.5 M NaBr or 0.25 M KI. A mercury derivative was obtained from a four-hour soak in cryobuffer containing 4 mM thimerosal.
Data collection and structure determination
The structure was solved to 2.0 Å resolution by multiple isomorphous replacement with anomalous scattering (Table 1). Data from native and KI derivative crystals were collected at NSLS beamline X8-C using a wavelength of 1.0070 Å. The NaBr data were collected at APS beamline SBC19-ID using a wavelength of 0.91965 Å, which corresponded to the experimentally determined maximum of f”. Data from the thimerosal derivative were obtained using a copper rotating anode source and an R-axis IV detector. All data collections were performed using cryocooled crystals, and the data were processed with HKL30. MIRAS phasing calculations were performed using CCP431. Initial phases were determined from two Hg sites located from isomorphous difference Patterson maps, while the remaining Hg sites and the halide sites were located by cross-Fourier maps calculated from these initial phases. The heavy atom parameters were refined using MLPHARE and the phases were improved by density modification using DM 31.
Table 1.
Data collection and refinement statistics
| Parameter | Native | Thimerosal | NaBr | KI |
|---|---|---|---|---|
| Resolution (Å) | 30.0 – 2.0 | 30.0 - 2.5 | 30.0 - 2.2 | 30.0 - 2.0 |
| Observations | 292,513 | 99,491 | 322,794 | 131,057 |
| Unique reflections | 50,208 | 25,894 | 35,783 | 50,716 |
| Completeness (%) | 99.0 (97.9)1 | 98.2 (94.0)1 | 91.4 (93.9)1 | 98.2 (99.8)1 |
| I/σ(I) | 20.1 (2.7)1 | 19.0 (2.7)1 | 25.2 (4.5)1 | 15.3 (2.3)1 |
| Rsym2 | 0.060 (0.40)1 | 0.075 (0.43)1 | 0.089 (0.57)1 | 0.051 (0.43)1 |
| Riso3 | 0.111 | 0.122 | 0.152 | |
| Number of sites | 6 | 7 | 7 | |
| Phasing power (20-2.5 Å)4 | 0.74 | 1.06 | 1.11 | |
| Rcullis(20-2.5 Å)5 | 0.87 | 0.83 | 0.82 |
| Model and Refinement Statistics | |||
| Resolution (Å) | 30.0 – 2.0 | Average B-value, protein (Å2) | 38 |
| Number of reflections | 50,206 | Average B-value, water (Å2) | 42 |
| Rcrys / Rfree | 0.206 / 0.244 | RMSD bond lengths (Å) | 0.006 |
| Number of protein residues | 514 | RMSD bond angles (°) | 1.3 |
| Number of water molecules | 476 | RMSD dihedral angles (°) | 25 |
Values for the outer resolution shell of data are given in parenthesis.
Rsym =Σh (Σj | Ij,h- < Ih > | / ΣIj,h), where h=set of Miller indices and j=set of observations of reflection h.
Riso=Σh| |Fder| - |Fnat| | / Σh |Fnat|, Fder =observed derivative structure factor amplitude, Fnat =observed native structure factor amplitude.
Phasing power=root mean square (fh / residual)=(Σfh2 / Σ (Fder-FPH)2)1/2, fh=calculated heavy atom structure factor, Fder =calculated derivative structure factor.
Rcullis =Σ | |fh| - (|Fder| - |Fnat|) | / Σ ∥Fder| - |Fnat∥.
An initial model was built into density modified maps using O32, and refinement was performed with X-PLOR33 using all data from 30.0 to 2.0 Å. The electron density for the main chain was continuous throughout the protein, except for residues 1-86, 194-199, 218-223, and 613-669, which apparently are disordered, and therefore omitted from the model.
Electron density maps calculated at every stage of chain tracing and refinement clearly indicated the presence of a ligand bound near the pyrimidine portion of the si face of the FAD isoalloxazine. The shape of this feature matched that of lactate, and L-lactate is known to be a competitive inhibitor of PRODH with KI of 1.4 mM3. The PEG 3000 solution used in crystallization and cryoprotection appears to be the source of L-lactate. Using a L-lactate detection assay based on NAD-linked lactate dehydrogenase and alanine aminotransferase34, a 50% PEG 3000 (Fluka) solution was found to contain 3-4 mM L-lactate (M. Zhang and J. Tanner, unpublished results). Thus, we concluded that the electron density feature represents a bound L-lactate molecule, and L-lactate was modeled into the density and refined (Fig. 3a).
The refined model includes 514 of the expected 669 amino acid residues, one FAD, one L-lactate molecule, and 476 water molecules. Stereochemical analysis of the final structure revealed that 87% of the non-glycine residues lie in the most favorable regions of the Ramachandran plot with no residues in the disallowed regions35. The stereochemistry meets or surpasses all the main chain and side chain tests of PROCHECK35.
Modelling
A simple model of L-proline was constructed to identify which residues could potentially contact the substrate. A model of L-proline was superimposed onto the L-lactate molecule bound in the active site using a least squares fit based on the coordinates of the carboxylate, C2, and C3 carbon atoms. The system was then minimized without restraints using the bonded and van der Waals energy terms of CNS36. The only change in the active site necessitated by the introduction of proline was a 0.5 Å movement of Tyr540 to avoid a steric clash with the proline C5.
Preparation of and functional analysis of PutA261
The gene encoding a truncated E. coli PutA protein containing the first 261 residues (PutA261) was engineered by positioning an EagI site immediately after codon 261 in the pUT669 construct (putA669 gene in a pET23b vector) using the QuikChange (Stratagene) site-directed mutagenesis kit. The product of the QuikChange reaction was cut with EagI to remove codons 262-669 and then ligated to generate putA261 in a pET23b vector carrying a C-terminal hexahistidine tag (Novagen). The resulting construct, pUT261, was introduced into the E. coli expression strain BL21(DE3) pLysS. Nucleotide sequencing of pUT261 showed no mutations were introduced during the QuikChange protocol.
PutA261 with the C-terminal hexahistidine tag was expressed in E. coli by induction with 0.5 mM IPTG at 0.8-1.0 OD for 5 h at 28 °C. PutA261 was purified by Ni2+ NTA affinity chromatography (Novagen) using procedures similar to those reported previously for PutA66929, except that 1 M urea was added to the dialysis buffer used after Ni2+ NTA chromatography to reduce protein loss due to precipitation. The urea concentration was then reduced to less than 0.5 mM by dialysis in 70 mM Tris (pH 7.5) containing 10% (v/v) glycerol. PutA261 retained the C-terminal hexahistidine tag after purification.
The binding of PutA261 to put control intergenic DNA was measured by nondenaturing gel mobility shift assays as previously described15. PutA261 (0-400 nM monomer) was incubated with put control intergenic DNA (20 nM) in a total volume of 25 μL in 70 mM Tris (pH 7.5) containing 10% (v/v) glycerol and 2 mM EDTA for 20 min prior to electrophoresis. Nonspecific ΦX174 ladder DNA was also added to PutA261 binding mixtures to demonstrate the change in migration of the put control DNA upon complexation with PutA261 and the specificity of PutA261 DNA binding. For comparison, binding reactions were performed with PutA (500 nM monomer) and PutA669 (500 nM monomer) under identical conditions. PutA protein concentrations were determined spectrophotometrically at 451 and by the BCA method (Pierce) using bovine serum albumin as the standard.
Coordinates
Coordinates and structure factors have been deposited with the Protein Data Bank (accession code 1K87).
Acknowledgments
This research was supported by NIH grants GM065546 and GM061068 (J.J.T. and D.F.B) and NSF MCB-0340912 (D.F.B.). We thank L. Beamer for collecting the NaBr derivative data, L. Beamer and P. Tipton for helpful comments on the manuscript, and the beamline personnel at APS SBC19-ID, NSLS X8C, and NSLS X12B for technical assistance.
References and Notes
- 1.Brown E, Wood JM. Redesigned Purification Yields a Fully functional PutA Protein Dimer from Escherichia coli. J Biol Chem. 1992;267:13086–13092. [PubMed] [Google Scholar]
- 2.Menzel R, Roth J. Purification of the putA gene Product. J Biol Chem. 1981;256:9755–9761. [PubMed] [Google Scholar]
- 3.Scarpulla RC, Soffer RL. Membrane-Bound Proline Dehydrogenase from Escherichia coli. J Biol Chem. 1978;253:5997–6001. [PubMed] [Google Scholar]
- 4.Maloy S, Roth JR. Regulation of Proline Utilization in Salmonella typhimurium: Characterization of put:Mu L(Ap, lac) Operon Fusions. J Bacteriol. 1983;154:561–568. doi: 10.1128/jb.154.2.561-568.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Menzel R, Roth J. Regulation of genes for Proline Utilization in Salmonella typhimurium: Autogenous Repression by the putA gene Product. J Mol Biol. 1981;148:21–44. doi: 10.1016/0022-2836(81)90233-3. [DOI] [PubMed] [Google Scholar]
- 6.Wood JM. Genetics of L-proline Utilization in Eschericia coli. J Bacteriol. 1981;146:895–901. doi: 10.1128/jb.146.3.895-901.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Becker DF, Thomas EA. Redox Properties of the PutA Protein from Escherichia coli and the Influence of the Flavin Redox State on PutA-DNA Interactions. Biochemistry. 2001;40:4714–4722. doi: 10.1021/bi0019491. [DOI] [PubMed] [Google Scholar]
- 8.Ostrovsky De Spicer P, Maloy S. PutA Protein, a Membrane-Associated Flavin Dehydrogenase, Acts as a Redox-Dependent Transcriptional Regulator. Proc Natl Acad Sci USA. 1993;90:4295–4298. doi: 10.1073/pnas.90.9.4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Surber MW, Maloy S. Regulation of Flavin Dehydrogenase Compartmentalization: Requirements for PutA-Membrane Association in Salmonella typhimurium. Biochim Biophys Acta. 1999;1421:5–18. doi: 10.1016/s0005-2736(99)00104-2. [DOI] [PubMed] [Google Scholar]
- 10.Wood J. Membrane Association of Proline Dehydrogenase in Escherichia coli is Redox Dependent. Proc Natl Acad Sci USA. 1987;84:373–377. doi: 10.1073/pnas.84.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–5. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 12.Donald SP, et al. Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species. Cancer Res. 2001;61:1810–5. [PubMed] [Google Scholar]
- 13.Liu H, et al. Genetic variation at the 22q11 PRODH2/DGCR6 locus presents an unusual pattern and increases susceptibility to schizophrenia. Proc Natl Acad Sci USA. 2002;99:3717–22. doi: 10.1073/pnas.042700699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chakravarti A. A compelling genetic hypothesis for a complex disease: PRODH2/DGCR6 variation leads to schizophrenia susceptibility. Proc Natl Acad Sci USA. 2002;99:4755–6. doi: 10.1073/pnas.092158299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vinod MP, Bellur P, Becker DF. Electrochemical and functional characterization of the proline dehydrogenase domain of the PutA flavoprotein from Escherichia coli. Biochemistry. 2002;41:6525–6532. doi: 10.1021/bi025706f. [DOI] [PubMed] [Google Scholar]
- 16.Ling M, Allen SW, Wood JM. Sequence Analysis Identifies the Proline Dehydrogenase and Pyrroline-5-Carboxylate Dehydrogenase Domains of the Multifunctional Escherichia coli PutA Protein. J Mol Biol. 1994;245:950–956. doi: 10.1006/jmbi.1994.1696. [DOI] [PubMed] [Google Scholar]
- 17.Bennett MJ, Schlunegger MP, Eisenberg D. 3D domain swapping: a mechanism for oligomer assembly. Protein Sci. 1995;4:2455–68. doi: 10.1002/pro.5560041202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guenther BD, et al. The structure and properties of methylenetetrahydrofolate reductase from Escherichia coli suggest how folate ameliorates human hyperhomocysteinemia. Nat Struct Biol. 1999;6:359–65. doi: 10.1038/7594. [DOI] [PubMed] [Google Scholar]
- 19.Umhau S, et al. The x-ray structure of D-amino acid oxidase at very high resolution identifies the chemical mechanism of flavin-dependent substrate dehydrogenation. Proc Natl Acad Sci USA. 2000;97:12463–8. doi: 10.1073/pnas.97.23.12463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mattevi A, et al. Crystal structure of D-amino acid oxidase: a case of active site mirrorimage convergent evolution with flavocytochrome b2. Proc Natl Acad Sci USA. 1996;93:7496–501. doi: 10.1073/pnas.93.15.7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pollegioni L, Blodig W, Ghisla S. On the mechanism of D-amino acid oxidase. Structure/linear free energy correlations and deuterium kinetic isotope effects using substituted phenylglycines. J Biol Chem. 1997;272:4924–34. doi: 10.1074/jbc.272.8.4924. [DOI] [PubMed] [Google Scholar]
- 22.Kurtz KA, Rishavy MA, Cleland WW, Fitzpatrick PF. Nitrogen Isotope Effects As Probes of the Mechanism of D-Amino Acid Oxidase. J Am Chem Soc. 2000:12896–12897. [Google Scholar]
- 23.Surber MW, Maloy S. The PutA Protein of Salmonella typhimurium Catalyzes the Two Steps of Proline Degradation via a Leaky Channel. Arch Biochem Biophys. 1998;354:281–287. doi: 10.1006/abbi.1998.0697. [DOI] [PubMed] [Google Scholar]
- 24.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 25.Lewis RJ, et al. The trans-activation domain of the sporulation response regulator Spo0A revealed by X-ray crystallography. Mol Microbiol. 2000;38:198–212. doi: 10.1046/j.1365-2958.2000.02134.x. [DOI] [PubMed] [Google Scholar]
- 26.Baikalov I, et al. Structure of the Escherichia coli response regulator NarL. Biochemistry. 1996;35:11053–61. doi: 10.1021/bi960919o. [DOI] [PubMed] [Google Scholar]
- 27.Liu J, et al. Structure and function of Cdc6/Cdc18: implications for origin recognition and checkpoint control. Mol Cell. 2000;6:637–48. doi: 10.1016/s1097-2765(00)00062-9. [DOI] [PubMed] [Google Scholar]
- 28.Brennan RG, Matthews BW. Structural basis of DNA-protein recognition. Trends Biochem Sci. 1989;1989:286–290. doi: 10.1016/0968-0004(89)90066-2. [DOI] [PubMed] [Google Scholar]
- 29.Nadaraia S, Lee YH, Becker DF, Tanner JJ. Crystallization and Preliminary Crystallographic Analysis of the Proline Dehydrogenase Domain of the Multifunctional PutA Flavoprotein from Escherichia coli. Acta Crystallogr. 2001;D57:1925–1927. doi: 10.1107/s0907444901017140. [DOI] [PubMed] [Google Scholar]
- 30.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 31.CCP4. The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 32.Jones TA, Zou J-Y, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 33.Brünger AT. X-PLOR version 3.1 A system for x-ray crystallography and NMR. Vol. 382. Yale University Press; New Haven: 1992. [Google Scholar]
- 34.Noll F. L-(+)-Lactate. Chap. 3.12. In: Bergmeyer J, Grassl M, editors. Methods of Enzymatic Analysis. Vol. 6. Verlag Chemie, Weinheim; Deerfield Beach, Fla: 1983. pp. 582–588. [Google Scholar]
- 35.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 36.Brünger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 37.Kraulis PJ. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 38.Merritt EA, Murphy MEP. Raster3D Version 2.0: A program for photorealistic molecular graphics. Acta Crystallogr. 1994;D50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]
- 39.Read RJ. Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Crystallogr A. 1986;42:140–149. [Google Scholar]
- 40.Esnouf RM. An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J Mol Graphics Modell. 1997;15:132–134. doi: 10.1016/S1093-3263(97)00021-1. [DOI] [PubMed] [Google Scholar]