

Abstract
Cytidine triphosphate synthetases (CTPSs) produce CTP from UTP and glutamine, and regulate intracellular CTP levels through interactions with the four ribonucleotide triphosphates. We solved the 2.3-Å resolution crystal structure of Escherichia coli CTPS using Hg-MAD phasing. The structure reveals a nearly symmetric 222 tetramer, in which each bifunctional monomer contains a dethiobiotin synthetase-like amidoligase N-terminal domain and a Type 1 glutamine amidotransferase C-terminal domain. For each amidoligase active site, essential ATP- and UTP-binding surfaces are contributed by three monomers, suggesting that activity requires tetramer formation, and that a nucleotide-dependent dimer–tetramer equilibrium contributes to the observed positive cooperativity. A gated channel that spans 25 Å between the glutamine hydrolysis and amidoligase active sites provides a path for ammonia diffusion. The channel is accessible to solvent at the base of a cleft adjoining the glutamine hydrolysis active site, providing an entry point for exogenous ammonia. Guanine nucleotide binding sites of structurally related GTPases superimpose on this cleft, providing insights into allosteric regulation by GTP. Mutations that confer nucleoside drug resistance and release CTP inhibition map to a pocket that neighbors the UTP-binding site and can accommodate a pyrimidine ring. Its location suggests that competitive feedback inhibition is affected via a distinct product/drug binding site that overlaps the substrate triphosphate binding site. Overall, the E. coli structure provides a framework for homology modeling of other CTPSs and structure-based design of anti-CTPS therapeutics.
Cytidine triphosphate synthetases (CTPSs,1 E. C. 6.4.3.2, uridine triphosphate ammonia lyases, 525–630 amino acid residues) are essential ubiquitous enzymes that catalyze the rate-limiting step of de novo CTP biosynthesis (1, 2). Genes pyrG (bacteria), ctrA (some gram positive bacteria), URA7 and URA8 (Saccharomyces cerevisiae), and CTPS1 and CTPS2 (human) encode CTP synthetase enzymes, with most eukaryotes expressing two isoforms. CTPSs not only provide a key precursor for synthesis of DNA, RNA, and phospholipid (3, 4), they also control the intracellular CTP concentrations that limit these processes (5–9).
CTP is derived from UTP in three reaction steps catalyzed by CTPSs (Figure 1a). In one active site, the UTP O4 oxygen is activated by Mg-ATP-dependent phosphorylation, followed by displacement of the resulting 4-phosphate moiety by ammonia (10, 11). In a separate site, ammonia is generated via rate-limiting glutamine hydrolysis (glutaminase) activity (12). CTPS activity was first reported in 1955 by Liebermann, who established the requirements for UTP, ATP, GTP, and glutamine in Escherichia coli extracts (1). Chakraborty and Hulbert demonstrated that ammonia could be utilized directly in vitro (13). The E. coli enzyme, EcCTPS (545 residues, Mr ~ 60 kDa), was subsequently purified by Long and Pardee (14), who quantified the apparent allosteric behavior induced by substrates, inhibition by CTP, and allosteric activation by GTP. The glutamine hydrolysis reaction, substrate-dependent oligomerization, and the kinetic effects of GTP were studied by Long, Levitzki, and Koshland (12, 15–18). Pyrimidine nucleotide substrate specificity was investigated by Scheit and Linke (19). The role of ATP in UTP activation was established by Villafranca, Anderson, and co-workers (10, 11). Anderson and co-workers improved the purification and further characterized the oligomerization response to substrates (20, 21). CTPS activity is sensitive to the levels of each ribonucleotide. At subsaturating ATP or UTP, with EcCTPS KM values of 150 and 200 μM, respectively (14), activity exhibits positive cooperativity (14, 17, 20, 22). GTP binding, with a KD value of 80 μM (19), stimulates glutaminase activity by 45-fold in kcat/KM (18), primarily through increased transition state stabilization (23). The product CTP provides negative feedback by competing with UTP (6, 9, 14, 24) with a Ki value of 110 μM (14). These properties are conserved, and mammalian (6, 13, 25, 26) and yeast enzymes (22, 24) behave similarly to EcCTPS. In S. cerevisiae, regulatory phosphorylation further modulates URA7 activity (7, 27, 28).
Figure 1.

Reactions and regulation of CTPS and related DTBS. (a) Kinase, glutaminase, and ligase reactions catalyzed by CTPS. UTP is phosphorylated by ATP, and both bind with positive cooperativity. Glutamine hydrolysis, allosterically activated by GTP binding, generates ammonia in a separate active site. Nucleophilic attack of 4-phospho-UTP by ammonia displaces the phosphate, yielding CTP. The CTP product is a feedback inhibitor competitive with UTP substrate. Thus, all four ribonucleotides regulate CTPS activity. (b) Analogous ligase reaction catalyzed by dethiobiotin synthetase (DTBS), a structural homologue of the CTPS N-terminal domain. The precursor DAPA is first carboxylated with CO2 in an ATP-dependent manner (not shown). DAPA carbamate is phosphorylated by ATP, and the product DAPA-carbamoyl phosphate cyclizes by intramolecular nucleophilic attack of the vicinal amino group and elimination of phosphate to yield dethiobiotin.
The requirement for de novo CTP makes CTPS activity key to the viability of a number of cancers (29–31), viruses (32–34), pathogenic bacteria (35, 36), and protozoan parasites (37–39). As a result, CTPSs are both the targets for therapeutics and responsible for resistance to them by selected mutations. CTPS inhibitors are promising treatments for leukemias and neuroblastomas, which exhibit elevated intracellular CTP levels (31, 40–42). Pyrimidine drugs cyclopentenyl cytosine (CPEC (43–45)) and 3-deazauridine (46), and the glutamine analogue acivicin (47), inhibit CTPS and reduce CTP levels, thereby slowing or arresting proliferation of tumor cells, sometimes inducing their differentiation (48). Further, CPEC and 3-deazauridine deplete intracellular CTP pools and increase cytosine arabinoside uptake in neuroblastoma (49), leukemia (50), and HIV-infected (51, 52) cells, suggesting that CTPS inhibitors are potentially useful in combination drug therapy. Conversely, cancers have become resistant to anti-CTPS drugs (53–55), and resistance mutations typically cause loss of CTP inhibition (56, 57). Similar CTPS mutations also mediate cancer resistance to cytosine arabinoside and 5-fluorouracil, drugs that do not target this enzyme, by dramatically increasing intracellular CTP concentrations (57, 58). These observations suggest that CTPSs are master regulators of CTP levels and that resistance-evading anti-CTPS drugs could be effective therapeutics.
In addition to cancers, Trypanosoma brucei, the infectious agent in African sleeping sickness, is dependent on its own CTPS, and its viability in vitro was reduced by acivicin (37). The malarial parasite Plasmodium falciparum (38), intestinal parasite Giardia intestinalis (39), intracellular bacterial pathogen Chlamydia trachomatis (36), and hemorrhagic fever viruses (32) are also potentially treatable by anti-CTPS therapies. The relevance to human diseases make CTPSs attractive targets for structure-based drug discovery. CTPS amino acid sequences are typically 35–40% conserved between species and isoforms, down to ~20% identity between distant orthologs (59). Given this level of conservation, a representative structure should provide a reasonable framework for modeling other CTPSs, while sufficient structural diversity exists to allow for species- or isoform-specific drug targeting.
To address functional questions and to provide a starting point for ligand design and homology modeling, we solved the apo-EcCTPS structure to 2.3-Å resolution using Hg-MAD methods. Bioinformatic analysis indicated the key ligand binding surfaces and provided suggestions for how each of the four nucleotides regulates enzyme activity.
EXPERIMENTAL PROCEDURES
Crystallization and Data Collection
Highly purified EcCTPS was obtained using a previously published procedure (31), except that cell pellets were derived from an EcCTPS-overproducing strain (60), and the second hydrophobic interaction step employed a buffer of 20 mM potassium phosphate, pH 7.7, 1 mM EDTA, 2 mM glutamine, 0.7 mM GTP, 2 mM ATP, 2 mM UTP, and 10 mM MgCl2. Ammonium sulfate pellets were dialyzed against 2 mM TCEP, 20 mM Tris-HCl, pH 8.5, and then concentrated to 15 mg/mL using a Centricon 10 concentrator. EcCTPS crystallized using vapor diffusion methods from molar concentrations of ammonium sulfate, ammonium phosphate, and sodium citrate over a pH range of 6–8.5. Orthorhombic crystals, 1 × 0.7 × 0.4 mm in size, appeared within 1–2 weeks. They diffract anisotropically and belong to space group P21212 with cell dimensions a ~ 165 Å, b ~ 105–108 Å, and c ~ 130 Å, which suggests two or three EcCTPS 60 kDa monomers per asymmetric unit (VM = 4.7 vs 3.1 Å3/Da).
For MAD phasing, Hg-CTPS cocrystals were grown at 4 °C from drops containing 3 μL of 15 mg/mL CTPS in 2 mM TCEP, 20 mM Tris-Cl, pH 8.5, mixed with 3 μL of 1.26 M (NH4)2SO4, 5 mM MgCl2, 1 mM HgCl2, and inverted over a reservoir of 1.26 M (NH4)2SO4 and 5 mM MgCl2. For cryocooling, crystals were equilibrated to ~20% (v/v) MPD within 45 s by adding 100% MPD directly to the drop, mounted on a cryoloop, dried of excess mother liquor, and flash-cooled in liquid nitrogen. MAD data were collected at 100 K at SSRL beamline 9-2 (Hg-MAD data set, see Table 1). A second data set to 2.6-Å resolution was collected at the peak wavelength with longer exposure times (Native-Hg data set). Prior to synchrotron data collection, a highly redundant 3.2-Å resolution home-source data set was collected from this crystal using Cu–Kα radiation (Home-Hg data set).
Table 1.
Data Collection, MAD Phasing, and Refinement Statisticsa
| Data Sets | |||||
|---|---|---|---|---|---|
| Native-I | Native-Hg | F11500 | F12293 | F12312 | |
| wavelength, Å | 1.08 | 1.0085 | 1.0781 | 1.0085 | 1.0070 |
| resolution, Å (outer bin) | 2.3 (2.34–2.3) | 2.6 (2.72–2.6) | 3.0 (3.16–3.0) | 3.0 (3.16–3.0) | 3.0 (3.16–3.0) |
| R-sym, % | 7.7 (38) | 6.7 (35) | 7.2 (23) | 6.7 (22) | 6.3 (20) |
| coverage, % | 99.2 (99.1) | 97.0 (96.8) | 99.6 (99.6) | 99.8 (99.7) | 99.8 (99.6) |
| multiplicity (outer bin) | 3.7 (3.6) | 6.7 (6.5) | 3.6 (3.5) | 7.1 (6.9) | 7.1 (6.9) |
| I/SD (outer bin) | 23.8(2.1) | 25.2 (3.3) | 15.4 (4.5) | 22.6 (7.4) | 23.4 (8.1) |
| Phasing | |||||
| SHELX (phasing power) | 4 Hg sites at 3.5 Å (0.71) | ||||
| MLPHARE FOM | 0.31 (3 Å) | ||||
| DM FOM | 0.75 (3 Å) | ||||
| DM Phase-extension FOM | 0.90 (2.6 Å) | ||||
| Refinement | |||||
| resolution range (outer bin) | 20–2.3 (2.34–2.30) | ||||
| R-work, % | 21.4 (36.0) | ||||
| R-free, % | 28.1 (38.0) | ||||
| no. reflections | |||||
| working | 97,650 | ||||
| free | 4,051 | ||||
| no. atoms | 8,992 | ||||
| protein | 8,334 | ||||
| solvent | 532 | ||||
| iodide | 16 | ||||
| sulfate | 20 | ||||
| magnesium | 4 | ||||
| MPD | 16 | ||||
| RMS from ideality | |||||
| bonds (Å) | 0.009 | ||||
| angles (°) | 1.95 | ||||
| B-factors (Å2) | 3.4 | ||||
| average B factors (Å2) | |||||
| protein | 59.4 | ||||
| solvent | 65.8 | ||||
| iodide ions | 74.2 | ||||
| sulfate | 76.3 | ||||
| magnesium ions | 73.8 | ||||
| MPD | 78.6 | ||||
| Wilson B | 57 | ||||
Hg-MAD and Native-Hg data were collected from a single crystal at SSRL beamline 9–2 using an ADSC CCD detector. The Native-I data were collected at SSRL beamline 7–1 using a MAR Image Plate. The data were integrated by MOSFLM and merged with SCALEA (62) assisted by Wedger Elves (61). The merging R values were based on intensities for all of the data calculated by SCALEA. Rfree and Rfactor values were calculated by TNT release 5F (68) using the following scaling parameters: Ksol = 0.82, Bsol = 185, K = 0.82; and anisotropic thermal corrections: B11 = −12.78, B22 = 18.43 B33 = −5.65, as described in Experimental Procedures. Model deviations from ideal geometry were calculated by TNT using Engh and Huber parameters (69) and the BCORRELS library (70). Average model B factors were calculated by EDPDB (74). The Wilson B was calculated by TNT using the linear region of the Wilson plot (5–2.4 Å).
A higher quality 2.3-Å resolution data set was obtained from another crystal grown by vapor diffusion at 4 °C from drops containing 3 μL of 15 mg/mL CTPS in 1 mM TCEP, 20 mM Tris-Cl, pH 8.5 mixed with 3 μL of 1.26 M (NH4)2-SO4, 2 mM UTP, 7 mM MgCl2, 120 mM Tris-Cl, pH 8.5 (Native-I). Crystals were briefly soaked in well solution containing ~0.5 M sodium iodide before cryocooling, as described above.
Structure Solution
Data reduction and phasing were performed with the assistance of the ELVES package (61). Data were integrated with MOSFLM, and scaled using SCALA (62). Heavy atom sites were located using SHELX at 3.5-Å resolution (63). Inclusion of higher resolution data did not lead to self-consistent solutions, which we attribute to photodetachment of mercury during data collection (data not shown). Initial phases were calculated from MLPHARE refinement (62) of Hg-MAD data at 3-Å resolution, resulting in a figure of merit of 0.31. After solvent flattening with DM (62) was performed, the low-quality electron density map was used to generate a rough backbone trace for one monomer through model building assisted by the O program (64).
Comparisons to homologous structures of E. coli BioD, or dethiobiotin synthetase (DTBS PDB# 1BS1, (65)) and the CarA subunit of E. coli carbamoyl phosphate synthetase (CPS, PDB# 1CS0, (66)) resolved ambiguities of chain direction and connectivity. Molecular replacement with EPMR (67) defined the noncrystallographic symmetry. Twofold averaging, solvent flattening, and phase extension using DM generated the high-quality 2.6-Å resolution map used to trace more than 95% of the structure. Refinement was performed with TNT (68) using geometric (69) and B-factor (70) restraints with isotropic temperature scaling. Five percent of the available data were used for the free R calculation (71). The final 5–2.6 Å resolution model had an R-factor of 19% and an R-free of 27%.
The 2.6-Å resolution model was refined against the 2.3-Å Native-I data set with TNT release 5F. Anisotropic temperature factor scaling greatly improved map quality and refinement behavior. At no time during refinement were NCS restraints employed. There was no visible electron density for bound UTP, nor for residues 423–437 within loop L13 and C-terminal residue 545 from chain A, and 429–437 from chain B, which are omitted from the model. Solvent molecules were added and deleted by manual inspection. Strong difference features were observed that resembled sulfate, iodide ion, or MPD. The identity of the sulfates is supported by the coincidence of the sulfur locations and peaks in 3.2-Å resolution F+ – F− difference maps calculated from the Home-Hg data set and phased with the 2.6-Å resolution model. Iodide positions were indicated by strong anomalous difference peaks calculated using the Native-I set and the refined 2.3-Å resolution model phases. The iodide occupancies were refined separately, and varied between 0.4 and 1. A strong feature in each active site was modeled as a magnesium ion because it is coordinated by four negative charges, Asp72,2 Asp140, and sulfates 602 and 603, and occupies the same position as a magnesium ion in the DTBS structures. A persistent feature, ~ +5 sigma, near the amide of Arg468 in both subunits, corresponds to the position of a water molecule proposed to resolve the acyl-enzyme intermediate in CPS (66). However, multiple attempts to place a water molecule in this location resulted in a B-factor greater than 100 Å2 and 2Fo − Fc density that is inconsistent with a water molecule.
The final model contains residues 1–427 and 438–544 for chain A and residues 1–428 and 438–545 for chain B, two magnesium ions, four sulfate molecules, 16 iodides, two MPD molecules, and 532 water molecules. Ninety-nine percent of the residues are in the allowed Ramachandran regions, and those with “disallowed” angles had unambiguous electron density to define their conformations. For the 8390 nonsolvent atoms of the model, 608 have B-factors of 100 Å2, while 89 out of 532 solvent molecules have B-factors between 80 and 90 Å2. These atoms were included because persistent difference features greater than +3 sigma and 2Fo − Fc density greater than 0.7 sigma after omission and refinement defined their positions. Model coordinates and structure factors for the 2.3 Å data set were deposited in the Protein Data Bank under the accession number 1S1M.
Figures and Model Calculations
Models used for figure generation were the EcCTPS model, the structure of CPS in complex with the covalent transition-state analogue glutamyl γ-semialdehyde (GSA) (PDB# 1CS0, (66)), the structure of DTBS in a pseudotransition state complex with ADP, magnesium ion, carboxylated diaminoperlargonic acid (DAPA), and AlF3 (PDB# 1BS1, (65)), or the elongation factor G-GDP complex (PDB# 1DAR, (72)). Overall superpositions were obtained from the DALI website (73). Various comparisons and other superpositions were aided by EDPDB (74). All figures depict the A chain, which is essentially identical to the B chain in the regions shown. Solvent molecules, where shown, are those present in both subunits. Figures were generated with MOLSCRIPT (75) and rendered with RASTER3D (76). The surface points in Figure 6b were calculated using GRASP (77), with a 1.2-Å probe.
Figure 6.

Stereo diagrams of the glutaminase active site and the putative ammonia diffusion channel in the A subunit GATase domain. (a) Active site comparison of EcCTPS (cyan backbone coils and colored ball-and-sticks) to the CPS-GSA 1CS0 transition state analogue complex (beige backbone coils and sticks). The CPS was superimposed on EcCTPS using Cys379, Leu380, Gly351 and Gly352 main chain atoms. Covalently bound GSA from 1CS0 is indicated by the pink carbon atoms (“Gln”). Conserved residues are indicated by ball-and-sticks, with black carbon atoms for those suggested to be directly involved with substrate binding. Hydrogen bonds between CPS and GSA are indicated by purple dotted lines, and those between EcCTPS and bound solvent molecules (red balls) are indicated by yellow dotted lines. Two solvent molecules, present in both subunits, occupy the expected positions of the amide nitrogen and carbonyl oxygen of the Gln acylation transition state. The structures differ significantly in the conformation of the L11 “lid”, and the positioning of catalytic His and Glu residues. (b) A solvent-containing vestibule and adjacent tubular channel connects the glutaminase and ALase active sites. Backbone positions from the A subunit (yellow) or A′ subunit (green) are indicated by ribbons or coils. The dot molecular surface was generated by GRASP using a 1.2-Å probe (77). The hypothetical positions of bound GTP, UTP, and glutamine are indicated by translucent sticks. Seven buried solvent molecules that are present in both subunits define a possible ammonia trajectory, and are shown as cyan balls, with their associated hydrogen bonds indicated by yellow dotted lines. Conserved residues that are suggested to be catalytically important (black), that line the proposed UTP (purple) or GTP (red-purple) binding sites, or that line the proposed ammonia channel (gray bonds) are indicated by sticks. Potential hydrogen-bond acceptors are provided by the carbonyl oxygens of Met52, His57, Gly58, Glu59, Val 60, Asp70, Ala304, Arg468, and the carboxylate oxygens of Glu59, Asp70, and Glu517 (red balls). Hydrogen-bond donors are furnished by the amides of Val60, Leu71, and Glu351, and the Arg468 NH1 atom (blue balls), with Tyr298 and His57 side chains contributing additional hydrogen-bonding potential. Both side chain conformations for His57 are shown. The arrow indicates the suggested location of the ultimate or penultimate position of the conducted ammonia based on hydrogen bonding environment. A 3-Å opening at the base of the proposed GTP-binding site may provide entry for exogenous ammonia.
The ligand positions in the various figures were generated as follows. The glutamine substrate position was taken directly from the superposition of the CPS 1CS0 model without adjustment. A number of CPS-ligand active site structures (66) were very similar except directly adjacent to the substrate δ-carbon. The ADP, magnesium ion, and AlF3 positions were generated from the superposition of the 1BS1 model using the equivalent main chain positions of Gly17, Lys18, Gly19, Asp70, Glu140, Gly143 from EcCTPS. The exception is Figure 3a in which the DALI transformation was used. UTP was manually docked into the EcCTPS using the following constraints: (i) the UTP O4 atom was placed near the carboxy-DAPA carboxylate oxygen that formed the Lewis acid–base complex with AlF3 in 1BS1, (ii) the γ-phosphate was placed in the anion-binding site formed by B subunit residues 187–189, using the sulfate ion positions from the 2.6-Å resolution structure as a guide, (iii) the UTP torsion angles were manually adjusted to approximate a good steric and electrostatic fit while minimizing torsional strain. Steric constraints suggested an end-on placement for the uracil ring. The fact that 5-BrUTP, but not 5-methyl-UTP, is an inhibitor made it difficult to resolve which edge faces into the active site (19). We chose the orientation with O2 faced into the cleft for recognition by protein groups.
Figure 3.
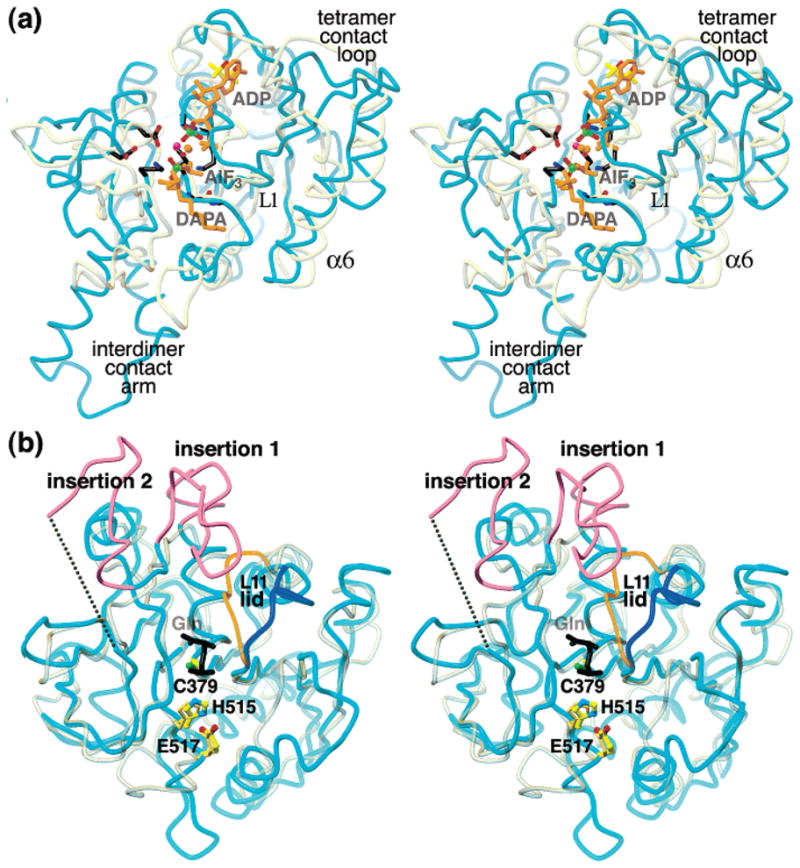
Backbone superpositions of EcCTPS domains with homologues. Transformations for superposition were generated by the DALI server (73). (a) EcCTPS N-terminal domain (cyan, residues A1-A264) superimposed on DTBS (yellow-gray, 1BS1 residues A1-A224). The sulfate ions (green and red sticks) from the EcCTPS structure, the DTBS substrate ligands ADP, AlF3, and carboxy-DAPA (orange sticks), and six key residues conserved between the two enzymes (black sticks), are shown. The EcCTPS tetramer interface mediated by loop L1 and helix α6 corresponds to the DTBS dimer interface, but is augmented in CTPS by the “tetramer contact loop”. (b) Superposition of the EcCTPS C-terminal domain (cyan, residues A288-A544) on CPS (yellow-gray, 1CS0 residues B191–B380). The two insertions relative to other GATase proteins are indicated (magenta). The dotted line connects residues 427–438 in lieu of the disordered loop that is omitted from the structure. The Cys–His–Glu catalytic triads are shown (CTPS, atom-colored ball-and-sticks, CPS, beige sticks). The covalently bound GSA transition state analogue from 1CS0 (black sticks, labeled “Gln”) defines the glutamine-binding site. The L11 “lid” segment of EcCTPS (blue) is folded away from the substrate site compared to the corresponding segment in CPS (orange).
We chose the CTP ring position based on the locations of mutations that release CTP inhibition ((56, 57), see Figure 8c Legend for details), which line a deep ovular pocket that faces away from the UTP site. CTP was docked without steric clash into this cleft with N4 close to Glu155.
Figure 8.

Stereo diagrams showing the atomic details of the proposed (a) catalytic, (b) ATP, and (c) UTP/CTP subsites in the EcCTPS A subunit. The carbon atoms of conserved residues are colored-coded in ball-and-stick representations by their proposed functions: ATP binding (orange); UTP binding (purple); CTP binding (cyan); or catalysis (black). Hypothetical bound ligands, indicated by similarly color-coded translucent sticks, were placed as described in Experimental Procedures. Solvent molecules present in both A and B subunits are shown (red balls). Hydrogen bonds are indicated by the dotted lines. (a) The proposed catalytic subsite. For clarity, only the UTP uracil ring and C1′ atoms (purple), the ADP α- and β-phosphates, and AlF3 are shown (orange). Nonconserved residues (white) are shown are shown in ball-and-stick. Proposed catalytic residues Gly17, Lys18, Lys40, Asp70, Glu140, and Gly143 are conserved from DTBS. Asp72 occupies a similar position to Asn56 in DTBS. Asp42 is positioned to interact with the uracil O4 position. Hydrogen bonds for γ-phosphate binding and phospho/aminotransfer transition-state stabilization are potentially donated by the Lys18 and Lys40 amino and Gly143 amide nitrogen atoms. A putative magnesium ion (cyan ball), chelated by Asp72 and Glu140 and bridging the bound sulfate ions, directly overlaps the proposed activating magnesium ion in DTBS. The α- and β-phosphates would be recognized by the amides in P-loop residues 17–19, and bridging solvent interactions with Asp72 and His75. Conserved Tyr76 buttresses the active site through interactions with Glu140. (b) Proposed subsite for ATP. For the A (yellow) and B (red) subunits, backbone positions are indicated by ribbons and the nonconserved side chains are indicated by yellow ball-and-sticks. Additional conserved side chains are shown in ball-and-stick with white bonds. Positions of the magnesium ion (cyan), solvent molecules (red), and nearby iodine ions (“I”, orange) are indicated by balls. Hydrogen bonds are indicated by black dotted lines. ADP from the DTBS 1BS1 structure (translucent orange sticks) overlaps the EcCTPS-bound sulfate ions (green and red ball-and-sticks) and MPD molecule (gray and red sticks). By analogy with DTBS, the adenine recognition site is formed by the loop L8 (residues 240–245). (c) Proposed overlapping subsites for UTP substrate and CTP feedback inhibitor. The A (yellow) A′ (green), and B (red) subunit backbone positions are indicated by ribbons. Conserved side chains which line the proposed ligand binding surfaces are shown in color-coded ball-and-stick. For clarity, only the UTP subsite-proximal His57 conformation is shown. Positions of solvent molecules (red) and the iodine ion (“I–”, orange) are indicated by balls. Italicized labels indicate sites for which known change-of-function mutations exist (see text for details). The Lys187-Ala substitution abolishes ALase activity in EcCTPS. For the CTP site, some mutations, selected for resistance to CPEC or 5-fluorouracil, resulted in loss of CTP inhibition in CTPSs from C. trachomatis, Asp147-Glu, (56)), and hamster, Ile/Val116-Phe, Gly146-Glu, Ile148-Thr, Leu/Met151-Ile, Glu155-Lys, Arg158-His, and Asn/His229-Lys (57), where bold indicates a conserved residue for CTPSs, and the hamster residue is underlined. Residues Gly146, Ile151, and Asn229 are not shown for clarity. The sulfate/iodide site provide a potential site for binding the UTP or CTP γ-phosphates.
The GTP positioning was derived from the GDP ligand in 1DAR after superposition on EcCTPS. Initially, the GDP overlapped residues that form the entrance to the vestibule. After the γ-phosphate was added, a linear 3-Å translation away from the protein interior and into the cleft was applied, followed by an approximate 40° rotation and minor torsional adjustments to minimize steric clash and to position the phosphates for interaction with Lys297 and residues 354–356.
RESULTS
Crystal Packing, Oligomeric Structure, and Domain Folds
The apo-ECTPS structure was traced from a 2.6-Å resolution density map generated by Hg-MAD phasing and density modification. It was refined against 2.3-Å resolution data obtained from iodide-soaked crystals, to yield the final model discussed here (PDB# 1S1M, see Experimental Procedures and Table 1). In these crystals, EcCTPS forms a dimer of dimers with near 222 symmetry (Figure 2b). The high protein concentrations likely promoted tetramer formation in the absence of bound substrates (20, 21). The tetramer is generated from the noncrystallographic AB dimer by the 2-fold crystallographic rotation about the c axis. The approximate 2-fold axis that relates the A monomer to B (178° rotation) and the near-perfect 2-fold axis relating A and B′3 are nearly parallel to the 21 screw axes of the crystal. The A and B monomers differ mainly by a 1.7° hinging motion of the N- and C-terminal domains that reduces the width of the putative CTP synthesis active site cleft by ~1 Å. After accounting for the hinge, the main chain root-mean squared deviations (rmsd) after superposition are 0.4 and 0.6 Å for the N- and C-terminal domains, respectively.
Figure 2.

Oligomeric and monomeric structure of E. coli CTPS (a) Models and typical electron densities from experimental and model-phased maps for the highly conserved ERHRHRYE sequence, residues 465–472. The best-phased solvent-flattened 3.0-Å resolution map (upper left, see Experimental Procedures) was greatly improved by 2-fold averaging and phase extension to 2.6-Å resolution (upper right). Starting with the 2.6-Å model (gray sticks), several rounds of model building and refinement yielded the final 2.3-Å resolution model (yellow sticks), shown enclosed by the final 2Fo − Fc density map (lower center). (b) Three views of the EcCTPS tetramer, along the crystallographic b axis (left), a axis (right), and c axis (bottom). The unique molecules in the asymmetric unit, A and B, are related to their A′ and B′ symmetry mates by the crystallographic 2-fold axis along c. The tight soluble dimers, A–A′ and B–B′, tetramerize via the A–B and A′–B′ interfaces in response to ATP and UTP binding. (c) Space-filling model of the EcCTPS A monomer (yellow) showing the contact regions, defined as atoms that are within 5 Å of the atoms in the other monomers, colored for the interacting monomer from (b). Conserved residues (see Figure 4) are indicated by the translucent surface. Space-filling models of the bound sulfate ions (purple and white), iodide ion (orange), and MPD (black and white) are shown. The entrance to the glutaminase active site (purple surface in upper domain) is also shown. (d) Ribbon diagram of the EcCTPS A monomer, color coded by sequence position, from the N-terminus (purple) to the C-terminus (red). The colors correspond to those in Figure 4.
Each EcCTPS monomer contacts the others, primarily via the N-terminal domain (Figure 2c). The most extensive contact occurs between the crystallographic dimers (A–A′ and B–B′ interfaces), which buries ~1300 Å2 of solvent-accessible-surface per monomer, about 60% of which is nonpolar. Symmetry contacts between the helix–loop–helix extension formed by residues 93–130 (the interdimer contact loop, Figure 2d), account for half of the buried hydrophobic surface. This interface is much larger than the other subunit interfaces (A–B interface, ~800 Å2, 60% nonpolar; A–B′ interface, ~320 Å2, 75% nonpolar). On the basis of this comparison, A–A′ and B–B′ pairs comprise the tightly associated soluble dimers (20, 78), while substrate-induced tetramer formation occurs via the A–B and A′–B′ interfaces.
The EcCTPS monomer consists of three distinct segments. The N-terminal amidoligase (ALase) domain, residues 1–266 (Figure 2d), is similar to the structure of BioD, the E. coli dethiobiotin synthetase (DTBS, PDB# 1BYI (79), 3.2 Å rmsd for 194 equivalent Cα atoms, Figure 3a). This domain mediates oligomerization and contains the ALase active site where ATP and UTP substrates bind. An interrupted helical segment (the interdomain linker, residues 267–286) connects the ALase domain to the Type I glutamine amidotransferase (GATase) domain, residues 287–544 (Figure 2d), which generates ammonia via glutamine hydrolysis. This domain is similar to other GATase structures, most notably the carbamoyl phosphate synthetase CarA subunit (CPS, PDB# 1C3O (66), 2.0 Å rmsd for 175 equivalent Cα atoms, Figure 3b). The ALase domain, interdomain linker, and GATase domain share an extensive interface of hydrophobic residues and buried hydrogen-bonded networks. This two-domain arrangement is analogous to that suggested by the sequences of the CbiA/CobQ family of phosphotrans-acetylases (80).
The ALase and GATase domains both contain Rossmann folds and structurally resemble each other, with 111 structurally similar residues that superimpose with an rmsd of 4.1 Å. This structural homology was underscored by DALI search results, which identified a similar overlapping group of nucleotide binding proteins, particularly NarL, MinD, SRP, ArsA, and elongation factors. These related proteins belong to the GTPase/ATPase superfamily based on three conserved sequence blocks (81) (Figure 4).
Figure 4.

Sequence alignments, secondary structure correspondences, and suggested roles for conserved residues. The CTPS sequences of E. coli (Ec, 545 residues, Genbank NP_417260), S. cerevisiae URA7 (URA7, 579 residues, 41% identical and 62% similar to EcCTPS, NP_009514), human CTPS1 (HUM1, 591 residues, 40% identical and 60% similar, NP_001896), and T. brucei (Tb, 589 residues, 37% identical and 57% similar, CAB95405) were aligned with BLAST and adjusted manually based on the structure. The E. coli sequence is numbered every 10 residues above the alignment. The corresponding secondary structures are shown above the alignment, color-coded as in Figure 2d. Horizontal double-arrows denote sequence blocks that constitute key structural elements (“interdomain linker”, “interdimer contact arm”, and “tetramer interface”) that are signatory of the SIMIBI NTPase family (81) (“block 1”, residues 4–46 containing the Walker A P-loop; “block 2”, residues 133–147 containing the Walker B motif; and “block 3”, residues 201–211, containing the NKxD motif), or that are CTPS-specific insertions relative to other GATase proteins (“insertion 1” and “insertion 2”). Highly conserved residues are highlighted in red (present in 41 out of 43 representative sequences) and conserved residues are highlighted in yellow (present in 34 out of 43 representative sequences) (59). The majority of conserved residues stabilize the folded structure by hydrophobic packing, hydrogen bonds, or backbone torsion preferences (labeled “f” above the sequences). The remainder are hypothesized to be involved in ligand recognition (labeled “A”, “U”, “G”, and “C”) or catalysis (labeled “c”) (see text for details). The functions of four residues could not be ascertained (labeled “?”). Some conserved residue labels are shaded according to their proximities to the A′ (green), B (red), or B′ (blue) subunits, relative to A, or because they line the putative ammonia channel (gray). Phosphorylated URA7 Ser residues are highlighted (black). The structure-based alignments of DTBS with EcCTPS residues 2–257 and CPS with EcCTPS residues 289–544 are also shown (bottom line). Underlined residues are identical to EcCTPS. Insertions are indicated as “><” and the inserted residues are shown below. Residues that lack structural homologues in EcCTPS are indicated (lower-case letters).
From comparisons of 43 sequences within the CTPS cluster of orthologous groups, COG0504 (59), CTPSs share 105 highly conserved residues, present in 41 out of 43 representative sequences, and 169 conserved residues, present in 34 out of 43 sequences (Figures 2c and 4). The majority of conserved residues, 108 out of 169, contribute to fold stabilization or quaternary interactions. Of the remaining conserved residues, based on structural comparisons detailed below, 45 are apparently involved in ligand recognition, and 12 are involved in catalysis. The function of four conserved residues could not be ascertained because they were disordered or were exposed to solvent away from apparent active surfaces.
Surface Features
Concave surfaces are abundant in the EcCTPS tetramer, suggesting potential ligand-binding sites. The most prominent feature is a large trench, 35 Å long × 8–10 Å wide × 15–20 Å deep, created by the interface between the A, B, and A′ subunits (Figure 5a). This trench contains the highest concentration of conserved residues and harbors three anion-binding sites and an MPD-binding site. On the basis of homology comparisons, we propose this cleft to be the ALase active site (see below) that binds ATP, UTP, and CTP (Figure 5a,c). Each of the four ALase sites are connected via a set of indentations and channels. A 30 Å long × 20 Å deep canyon, lined by interfaces of diagonally related subunits (e.g., A′–B and A–B′, Figure 2b), connects the ALase “UTP/CTP subsites” (Figure 5a) of the complimentary subunit pair. A channel exits each CTP subsite and converges in a large central cavity containing the tetramer center-of-mass (not shown). A crevice created at the A–B noncrystallographic symmetry interface by the 178–186 loops connects the “ATP subsites” (Figure 5b).
Figure 5.

Surface features in the vicinity of the amidoligase (ALase) active site. (a) Space-filling view into the ALase active site groove showing the proposed locations of the four nucleotide-binding sites. The subunits A (yellow), A′ (green), and B (red) each donate crucial active site surfaces. Each active site contains two sulfate ions (purple and white), an iodide ion (orange), and an MPD molecule (black and white). Hypothetical positions for the ATP (orange), UTP (purple), CTP (cyan), and GTP (dark pink), suggested by structural comparisons, conserved residue patterns and mutation data as described in Experimental Procedures, are shown in translucent thick sticks. The putative CTP-binding site connects the ALase site to a large internal space containing the tetramer center-of-mass. (b) Close-up view looking approximately along the crystallographic b axis, showing the prominent groove that traverses the ATP-binding sites on the A and B subunits. This groove is hydrated and is lined with hydrophobic residues. (c) Summary stereoview of the proposed ALase active site showing the backbone contributions from the A (yellow), A′ (green), and B (red) subunits in coils; the positions of the sulfate ions (green and red ball-and-stick), iodide ion (orange ball), magnesium ion (gray ball), and MPD (red and white sticks); the proposed locations of substrates ATP (orange) and UTP (purple), and the feedback inhibitor CTP (cyan) in translucent sticks. The conserved residues that are suggested to interact with ATP (orange), UTP (purple), CTP (cyan), or promote catalysis (black) are represented in ball-and-stick. The proposed entry site for ammonia is indicated with the black arrow.
The glutaminase active site is indicated by the characteristic GATase Cys–His–Glu catalytic triad (82) (Figures 3b and 6a). Nearby, a cleft with dimensions 15 × 10 × 12 Å is juxtaposed between the GATase and ALase domains near the A–A′ dimer interface. We suggest this is the GTP activator binding site (Figure 5a and 7b).
Figure 7.

Proposed GTP-binding site based on structural homology to GDP/GTP binding proteins, EcCTPS surface features, and conserved residues. (a) Stereo diagram of superpositions of EcCTPS A subunit C-terminal domain (cyan coils, only residues A288–A390 A438–A473, and A499–A534 are shown for clarity) with elongation factor G/GDP complex (yellow-gray coils, 1DAR residues 12–39, 74–191, 241–280) using DALI server-generated transformation. The position of the bound GDP (black sticks) superimposes near a prominent groove located between the GATase domain and the L2 loop (dark cyan). The equivalent residues of the EF-G P-loop and the CTPS L11 “lid”, colored orange (EF-G) and blue (EcCTPS), are in different conformations. The EcCTPS P-loop would need to undergo substantial rearrangement to achieve the EF-G conformation, while the EF-G “lid” more closely resembles that in CPS. (b) Stereo diagram of the proposed GTP binding cleft. The backbones of the A (yellow) and A′ (green) subunits are represented as coils. The proposed positions of GTP (red-purple), UTP (purple), and the Gln substrate (gray) are indicated by translucent sticks (see Experimental Procedures). The positions of conserved residues proposed to line the GTP-binding (red-purple) or UTP-binding (purple) sites, or to stabilize the folded structure (white) are represented in ball-and-stick. Residues which when mutated interfere with GTP binding are marked with italicized labels.
Structure of the GATase Domain
The CTPS GATase domain sequences contain two conserved residue blocks, residues 374–383 and 513–529 (Figure 4), that are shared with other Type 1 GATase domains and participate in catalysis and substrate binding (Figure 6a). Other blocks, unique to CTPSs, may be involved in GTP activation and coupling GATase and ALase activities (18). In particular, conserved blocks 295–298, 350–360, 436–442, and 464–470 are located near the entrance to the glutaminase site. Two insertions, relative to other Type 1 GATase proteins, form a mobile loosely packed subdomain, with an average B-factor 18 Å2 higher than the conserved portion (Figure 3b). These insertions vary in length and composition in different CTPSs. In EcCTPS, insertion 1 (residues 390–411, Figure 4) extends the C-terminus of α14 by one turn and contains conserved Glu403, Arg391, Ala399, and Pro411 residues. Insertion 2 (residues 418–437, Figures 3b and 4) contains the only undefined region of the EcCTPS structure, residues 428–437, and is followed by conserved residue block 436–442. CTPS is proteolyzed by trypsin at Arg429 and Lys432, indicating that this region is solvent-accessible and unconstrained (83).
We superimposed the EcCTPS GATase domain on CPS, using residues 191–380 from the 1CS0 structure (Figure 3b), and inferred residue functions from comparisons with liganded CPS structures determined by Holden and coworkers (66) (Figure 6a). Accordingly, the triad of Cys379, His515, and Glu517 (EcCTPS numbering) generates ammonia through formation of a labile acyl-enzyme intermediate (82). The tetrahedral transition state is stabilized by interactions with the main chain amides of residues Gly 352 and Leu380, forming the “oxyanion hole”. Perturbing this region by Gly351 and Gly352 substitution greatly decreased EcCTPS activity (60).
In the empty EcCTPS GATase site, important glutamine-binding and catalytic residues are positioned differently from those in the liganded and apo-CPS structures (66) (Figure 6a). Relative to the nucleophile and oxyanion hole in the CPS–GSA complex, the main chain atoms of residues 513–517 are shifted 0.8 Å rmsd. This shift displaces the key Gln513, His515, and Glu517 side chains by 1.0, 0.8, and 1.5 Å, respectively, away from Cys379. In addition, the proposed attacking water in 1CS0 (66), which would be hydrogen bonded to the Arg468 amide, Gln513 carbonyl oxygen, and His515 side chain, is not well-defined (See Experimental Procedures). EcCTPS recognition of the glutamine substrate α-amino acid moiety also differs from that of other GATases. In CPS, imidazole glycerol phosphate synthetase, and GMP synthetase, hydrogen bonds from the Arg470 and Tyr471 main chain and the Gln383 side chain amides are positioned to recognize the glutamine carboxylate (Figure 6a). Residues equivalent to EcCTPS 351–356 further sequester the substrate by forming a “lid” over the glutaminase site (the “L11 lid”), with main chain carbonyl atoms of residues 352 and 354 providing hydrogen-bond acceptors for the substrate α-amino group. In EcCTPS, residues 353–357 are shifted away, primarily via backbone rotations at Phe353 and Val358, opening one end of the active site and preventing substrate α-amino group recognition by Gly354 (Figure 6a). However, the conserved Glu403 carboxylate occupies the analogous position, perhaps providing an alternative hydrogen bond. The conserved Phe353 side chain forms a nonpolar lid that would both sterically hinder the amino group and prevent solvent from accepting its third hydrogen bond. This conformation may not be competent for glutamine binding, but the conserved differences between CTPSs and other GATases suggest that the structural differences are functionally relevant.
A solvent-filled vestibule (~230 Å3) connects the GATase active site and the GATase/ALase interface (Figure 6b). This volume is occupied by polypeptide in CPS but a conserved insertion, EcCTPS residues 297–301, diverts the chain and forms part of the vestibule wall (Figure 3b). The vestibule contains triad residues His515 and Glu517, and is lined by conserved residues Tyr298, Tyr305, Arg468, L11 residues 350–352, and ALase domain L2 residues 49–60. The vestibule interior contains a number of hydrogen bonding groups and six solvent molecules that are common to both subunits. Two solvents that are hydrogen bonded to the main chain of residues 352 and 468 correspond to substrate carbonyl oxygen and amide nitrogen positions in CPS (66) (Figure 6a). The vestibule is connected to a tubular passage that exits into the ALase active site adjacent to the expected location for the 4-phospho-UTP intermediate (see below, Figures 5c and 6b). The narrowest point, 0.8–1.2 Å between van der Waals surfaces of the Pro54 and Val60 side chains, forms a “gate” between the vestibule and the “exit”. We propose the vestibule and the tubular passage provide a path for ammonia diffusion between GATase and ALase active sites (see Discussion). In addition, bulk solvent can access the vestibule through a 3-Å wide opening to the surface (Figure 6b) into the proposed GTP-binding site (Figure 7b, see below). We suggest that this opening provides entry for exogenous ammonia.
GTP-Binding Site
We located a candidate GTP activation site by comparing the GATase domain with structurally related GTP binding proteins identified by DALI, such as EF-Tu and EF-G (PDB# 1DAR (84), 4.1 Å rmsd for 145 equivalent Cα atoms). The EF-G GTP/GDP-binding site superimposes near the deep cleft adjacent to the entrance to the glutaminase site, formed by residues 297–301, 353–356, 438–441, 468, and 470, and residues 50–55 from the ALase domain (Figure 7a). A number of conserved residues define the cleft surface (Figure 7b). The Leu301 amide and the Lys297 and Arg356 side chains provide potential interaction with the GTP phosphates in the cleft. In Lactococcus lactis CTPS, substitution at the equivalent position to Arg356 with Met and Pro significantly reduced GTP binding and the degree of activation at saturation (Willemoës and Mølgaard, personal communication) (Figure 7b). L. lactis CTPS substitutions equivalent to T438V and R440M in the CTPS-specific sequence GGTMRLV had similar although smaller effects (84), perhaps by perturbing Met439, since Arg440 points away from the cleft. The cleft rim is extended by A′ subunit loop L4 residues 105–110, which pack against A subunit loop L2 residues 50–55. Loop L4 is sandwiched between the GATase and ALase active sites of the same subunit, and is connected via polypeptide to the symmetry-related subunit. This segment may mediate the coupling between ATP binding and glutamine hydrolysis (18). Alanine substitution of Glu103, Arg104, or Gly110, which would be expected to disrupt the internal structure of this loop, severely reduces both GATase and ALase activities.
Structure of the ALase Domain
A DALI search with the EcCTPS ALase domain yielded 23 similar folds with Z > 6. In addition to closely resembling DTBS (Figure 3a), the canonical dinucleotide binding fold belongs to the ATP/GTPase superfamily, as previously predicted (81), which includes GTP binding proteins SRP and MinD, kinases such as APS kinase, and nitrogenase iron protein. We utilized comparisons with DTBS to infer substrate-binding sites since it is the best structural match with EcCTPS and catalyzes a similar chemical transformation (Figure 1b). The most notable structural differences between these enzymes are insertions that form the EcCTPS subunit interfaces (Figures 3a and 4). The CTPS tetramer (A–B) interface is provided by helix α6 and loop L1, similar to the DTBS dimer, but is augmented by the inserted β8–L6–β9 loop. More strikingly, the tight dimer (A–A′) interface is mediated by the novel “interdimer contact arm” that arises from a conformational rearrangement of 19 residues and an associated seven-residue insertion.
The putative ALase active site, in which UTP phosphorylation and amino displacement occur (Figure 1a), resides in a large trench that overlaps the DTBS active site cleft (Figures 2c and 3a). The walls are contributed by three different subunits (Figure 5a,c). Subunit A provides the majority of the ATP and UTP binding surfaces and all the catalytic groups. Subunit B augments the ATP and UTP subsites via residues 182–196 and 219–227, and subunit A′ residues 112–120 and 155–163 complete the UTP and CTP subsites. The trench contains three anion-binding sites, two of which are occupied by sulfate, and one by iodide. In the Native-Hg structure, the iodide was replaced by sulfate (data not shown).
Sulfates 602 and 603 define the catalytic and ATP-binding subsites (Figure 8a,b). This assemblage precisely overlaps the positions of the ADP β-phosphate and AlF3 moieties in DTBS complex structure 1BS1 (65) (Figures 3a and 8a), and two sulfate ions are similarly located in the apo-DTBS structure (PDB# 1DBS (85)). Conservation of six residues surrounding this subsite between DTBS and CTPS, Gly17, Lys18, Lys40, Asp72, Glu140, and Gly143 further supports the idea that they share a common mechanism (65). A magnesium ion bridges two sulfate oxygen atoms and is also chelated by Asp72 and Glu140 carboxylates, analogously to one bridging magnesium in DTBS complexes. However, the second DTBS magnesium ion is absent, perhaps because serine replaces glutamate at position 14. Sulfate 602 makes direct hydrogen bonds to the main chain amides of residues 17–19 of the P-loop, while 603 is contacted by the amide of 143 and the side chains of Lys18 and Lys40. Bound magnesium would activate the ATP γ-phosphate for transfer to UTP O4, while the Gly143 amide and Lys18 and Lys40 side chains would stabilize the anionic transition state. A key role for Lys18 is supported by loss of CTP synthesis activity from Ala-substitution (83).
ATP recognition by CTPS is likely analogous to that for DTBS (Figure 8b). The α-phosphate would be ligated by the Ile20 amide, and the ribose hydroxyl group would form hydrogen bonds with conserved Asp303. On the basis of DTBS and kinase structures, the loop L8 (residues 240–245) defines the adenine-binding subsite, which is occupied by MPD. Main chain hydrogen bonds provide recognition of adenine base N6 and N1 atoms via the Lys239 carbonyl oxygen (not shown) and the amide of Val241. Another surface involved in adenine recognition is formed by the conserved salt bridge between Arg211 and Asp240. In a structurally related APS kinase/ADP/APS complex (PDB# 1M7G, (86)), the adenine base stacks on the Arg148 guanidinium moiety, which occupies an analogous position to EcCTPS Arg211. The adenosine cleft is further buttressed by the B subunit L6 loop with Ala182 packing one edge (Figure 8b). Met180 packs against the P-loop, while conserved Lys187 forms a hydrogen bond with the Ser15 side chain (not shown). A groove adjacent to the adenine subsite is lined with conserved side chains of Arg78, Phe79, Tyr245, and Glu310 (Figure 8b).
The UTP binding subsite is suggested by the carboxy-DAPA location in DTBS (Figure 8c). EcCTPS is highly specific for the uracil, ribose, and 5′-triphosphate moieties of UTP, and no alternate substrates have been identified (14, 19). The uracil O4 atom would be expected to be close to the carboxylate oxygen of the carboxy-DAPA (Figure 3a). Conserved residues Pro43, Tyr44, and His57 are positioned to line the uracil pocket. Given the shape of the cleft, the uracil ring plane would be positioned “edge-on” (Figure 8a,c), but the existing data do not easily resolve the two possible ring orientations (19). Either uracil ring placement immediately suggests other residues involved in UTP recognition. A P-loop-like structure, composed of conserved residues Lys187, Thr188, and Lys189, forms the iodide/sulfate-binding site and is reasonably placed to interact with the UTP γ-phosphate (Figure 8c). Consistent with this idea, Lys187 is protected from proteolysis in the presence of UTP and the Lys187-Ala substituted enzyme does not synthesize CTP (83). The importance of the γ-phosphate-binding site is supported by the lack of reactivity of UDP and γ-phosphoryl methyl diester UTP (14, 19). The UTP β-phosphate would be proximal to side chains of Asp147, Lys189, Gln192, and Lys223.4 Gln149, Thr112(A′), and Gln114(A′) would be positioned to recognize the ribose moiety. The His118(A′) side chain reinforces the Thr112(A′) and Gln114-(A′) positions through main chain hydrogen bonds (not shown), and substituting it with alanine eliminates ALase activity (87). Interestingly, URA7 and L. lactis CTPSs convert dUTP to dCTP and are nearly identical to EcCTPS in this region, but have different hydrophobic residues at positions 115 and 116, Ile–Val and Met–Val, respectively, versus Val-Ile in CTPS. These substitutions may shift Thr112 or Gln114 to obviate recognition of the 2′ hydroxyl group (88, 89). Consistent with this idea, T. brucei CTPS would also be expected to utilize dUTP (Figure 4).
A tubular “exit” channel connects the ALase site to the GATase domain vestibule (Figures 5c and 6b). For His57 in the A subunit, electron density corresponding to two equally populated side chain conformers is apparent. One conformation places the side chain adjacent to the UTP site, while the other blocks the channel exit. The exit-blocking rotamer is dominant in the B subunit. Near the exit, a single solvent donates three hydrogen bonds to the His57 and Glu59 carbonyl and the Asp70 carboxylate oxygen atoms, and accepts one hydrogen bond from the Leu71 amide nitrogen. This solvent may occupy the ultimate or penultimate position of the attacking ammonia molecule. Residues Asp42, Asp70, and Tyr44 likely orient the ammonia and perform acid–base chemistry for displacing the 4-phosphate group.
CTP-Binding Site
Negative feedback regulation of CTPS has been proposed to occur via competitive CTP binding to the UTP site (9, 14). However, spontaneous mutations that render Chlamydia trachomatis (56) and Chinese hamster (57) CTPSs insensitive to CPEC and 3-deazauridine exhibit decreased CTP inhibition but increased UTP amination activity. These mutation sites line a pocket at the rear of the proposed UTP site (italicized residues, Figure 8c). One of these substitutions, replacement of conserved Glu155 to lysine (EcCTPS numbering) also induced CPEC resistance, higher activity, and reduced CTP inhibition in URA7 and URA8 (7). Glu155 is located 20 Å from the amino/phosphotransfer site, making it unlikely that it could interact with the UTP substrate directly. Simple modeling yielded a chemically reasonable CTP position in the pocket formed by residues 146–149 from subunit A, 227–228 from subunit B, and 114–117 from subunit A′, that placed the N4 atom proximal to the Glu155 carboxylate. Most of the surrounding residues are conserved (Figure 8c), except consensus Cys228, which is limited to small residues Cys, Ser, Thr, or Ala. Conserved Asp147, Glu149, and Gln192(B) are positioned to interact with the ribose or phosphates, and the cytidine ring would pack against conserved Ile148, Phe227(B), and Ile116(A′), with conserved Lys196 available for hydrogen bonding (not shown) with the cytosine ring. As with UTP, the proposed CTP positioning would allow the γ-phosphate to interact with the sulfate/iodide site at the A–B interface. This arrangement implies that UTP and CTP both utilize the same triphosphate-binding sites.
DISCUSSION
The comprehensive structural elucidation of pyrimidine de novo biosynthetic pathway enzymes, starting with aspartate transcarbamoylase by Lipscomb and co-workers (90), is completed by the EcCTPS structure. Although Thermus thermophilus CTPS crystals were previously reported (91), the work reported here provides the first representative structure for this enzyme family. A bioinformatic analysis combining homologue comparisons, residue conservation patterns, and mapping of functional mutations provided insights into substrate recognition, catalysis, and regulation. The EcCTPS structure may also provide a framework for modeling related bifunctional CbiA/CobQ enzymes involved in cobalamin biosynthesis.
As predicted by sequence comparisons, the two CTPS domains strongly resemble NTPases and GATase enzymes. The similarity between the GATase and ALase domains might suggest that CTPS arose from a gene duplication. However, while the EcCTPS GATase domain is quite similar to those in other two-domain amidoligase enzymes, the ammonia utilization domains are not obviously related (92). This discontinuity in homology suggests that these proteins more likely acquired their GATase domains from a common ancestor through gene fusion.
Comparisons with DTBS readily revealed the ATP-binding site. The UTP site was inferred by structural analogy to the DAPA substrate, residue conservation within CTPSs, steric restrictions, and the location of the interfacial anion-binding site that is well-placed to interact with the UTP γ-phosphate. The ATP- and UTP-binding sites span the dissociable A–B interface, providing strong evidence that the dimer is inactive and that tetramer formation is required for activity. This observation provides support for the idea that the apparent positive cooperativity (14, 17) is induced via a substrate-dependent association–dissociation mechanism (20). Substrate binding at one site would stabilize the tetramer through a linkage equilibrium, which would in turn promote binding and catalysis at the symmetry-related site. The estimated concentrations of EcCTPS in vivo, which are near or below the estimated KD for tetramer formation without substrate (20, P. Anderson, unpublished analysis), suggest that this regulatory model is relevant. However, our structure does not rule out allosteric contributions from ATP–UTP interactions in the same cleft, since the subsites share a contiguous polypeptide segment, residues 180–192.
We propose that the CTP feedback inhibitor binds at a distinct subsite that overlaps the UTP subsite, with the cytosine ring residing in a pocket whose walls are lined by drug-resistance mutation sites, and the β- and γ-phosphates bound similarly to the UTP phosphates (Figures 5c and 8c). Presumably, the drugs CPEC and 3-deazauridine also bind this subsite. The proposed placement is consistent with competitive binding of CTP and UTP, and the fact that CTP-and drug-resistance mutations do not disrupt CTP synthesis. Similarly, in pantothenate kinase (93), the adenine rings of the ATP substrate and co-enzyme A inhibitor also occupy widely separated pockets while the phosphate positions overlap. The novel aspect is that CTPS is directly inhibited by its product, rather than by a regulator that is produced downstream in the pathway.
The majority of catalytic and glutamine-binding residues are structurally conserved between CPS and EcCTPS GATase domains. However, several differences suggest that the EcCTPS is not optimally configured for glutaminase activity, which is perhaps expected given the lack of bound GTP. The candidate GTP-binding cleft lies directly adjacent to the glutaminase site and superimposes near the nucleotide-binding site in structurally related GTPases. The idea that GTP activation is associated with a substantial conformational change in this region is supported by the reduced CTP synthesis rates and GTP binding of EcCTPS mutants in which loop L4 residues Arg105 and Leu109 (87) or conserved disordered loop L13 residue Arg429 are substituted with alanine (83). These side chains make no significant contact with other residues in apo-EcCTPS (Figure 7b). Although the GATase domain contains a potential P-loop-like GKY sequence near loop L10, it does not fold into a P-loop structure (Figure 7a). Interestingly, adjacent loops in EF-Tu and EFG, analogous to CTPS loop L11, resemble the “lid” structure in CPS and other GATases (compare Figures 3b and 7a). Perhaps, coupled rearrangements of loops L10 and L11, to form a P-loop and a closed active site “lid”, mediate GTP activation. GTP binding may also shift catalytic His515 and Glu517 residues closer to the position observed in CPS.
In GATase enzymes, glutamine hydrolysis sites and ammonia utilization sites are widely separated and require a conducting channel to prevent ammonia escape into bulk solvent (92). Such channels are readily apparent in some structures, such as the prototypical 45 Å tunnel in CPS (95), while in others, their formation requires conformational changes induced by substrate, effector, or protein docking (92, 94). In CTPS, the most apparent ammonia diffusion path is defined by two voids that span the 25 Å distance between GATase and ALase active sites (Figure 6b). The channel is distinct from that in CPS, although they share a common entrance from the glutaminase site. Unlike the ammonia channels in some enzymes, it contains a number of solvent molecules and polar groups (92). The structure suggests the possibility that ammonia passage is coordinated by ligand-induced conformational changes or Brownian fluctuations. The constriction between the vestibule and exit channel, and the two His57 conformations, suggest potential gating points (Figures 5c and 6b). One His57 conformation places the side chain adjacent to the UTP site, while the other blocks the exit, suggesting that UTP binding might trigger “opening” of the gate. In imidazole glycerol phosphate synthetase, a rearrangement of the oxyanion hole and the ammonia conduction channel induced by cosubstrate binding is suggested to couple glutamine hydrolysis activity and ammonia transfer to the utilization site (94).
The vestibule interior is freely accessible through a 3-Å wide entrance at the base of the GTP-binding site. Exogenously added ammonia, which can be utilized for CTP synthesis (13, 23), may also enter through this opening. This idea is supported by inhibition of ammonia-dependent CTP synthesis when GTP is present in combination with glutamine, the glutamine analogue affinity label DON (18, 96) or the covalent transition-state analogue GSA (23). Together, these ligands would block ammonia entry through both the glutaminase site and the vestibule opening. This idea also provides an explanation for why GTP increases the utilization of ammonia derived from glutamine hydrolysis in L. lactis CTPS (96), which, unlike in EcCTPS, is only loosely coupled to CTP synthesis. In this case, GTP binding may prevent ammonia escape and divert it to the ALase site.
The locations of URA7 protein kinase A and C phosphorylation sites in the EcCTPS structure lend few clues on how this modification controls activity (27, 28) (Figure 4). Although four of five positions are proximal to binding or catalytic sites, the regulatory mechanisms are not obvious. Particularly, positions equivalent to URA7 Ser36, Ser330, and Ser424 are buried or occluded in EcCTPS, but nearby insertions at positions 134, 270, and 408 (EcCTPS numbering), respectively, may expose these residues for modification. On the other hand, Ser354 would be located far from either active site. The generality of phosphoregulation of CTPS also remains to be established. In other CTPSs, Ser454 is conserved as serine or threonine in most CTPSs, serine or threonine occur in the majority of enzymes that do not contain glycine at position 330, and Ser36 also occurs in human isoform1 and T. brucei CTPSs. In contrast, Ser354 is located in a variable-length loop, and Ser424 replaces a conserved alanine.
Control of CTP production has been an objective of anticancer, antiviral, and antiparasitic therapies. CTPSs would seem a prime target for specific designed inhibitors, but such efforts have been previously hampered by lack of structural information. The EcCTPS structure provides a framework for structure-assisted drug design of agents that are more specific and less toxic than existing nucleoside or glutamine analogues. Indeed, there are several cavities and crevices adjacent to ligand sites that could be utilized by elaborated substrate chimeras. For example, novel triphosphates could be targeted to the conserved crevice adjacent to the adenine site, or glutamine analogues could constructed to interact favorably with functionality within the proposed ammonia tunnel. Design of species-targeted anti-CTPS therapeutics would be enhanced by obtaining the structures of the human and pathogen enzymes, and the presence of species-specific insertions, deletions, and polymorphisms near ligand-binding sites, indicated by structure-sequence comparisons, may provide unique ligand interaction surfaces.
NOTE ADDED IN PROOF
We note that the coordinates for apo-, sulfate-, and glutamine-bound T. thermophilus CTPS were deposited in the Protein Data Bank as entries 1VCM, 1VCN, and 1VCO, respectively.
Acknowledgments
The authors would like to thank Martin Willemoës (University of Copenhagen) for providing us with manuscripts ahead of publication. We would also like to thank Dave King (U.C. Berkeley) for providing mass spectra of dissolved EcCTPS crystals. Protein purifications, home-source data collections, and all computations were carried out in the W.M. Keck Protein Expression and X-ray Crystallographic Facilities at U.C. Davis. Synchrotron data were obtained at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences.
Footnotes
This work was funded by the UC Systemwide Biotechnology Training Program Grant #2002-07, and the National Institutes of Health, General Medical Sciences, #GM63109.
Protein Data Bank accession number 1S1M.
Abbreviations: CTPS, cytidine triphosphate synthetase; EcCTPS, Escherichia coli cytidine triphosphate synthetase; CPEC, cyclopentenyl cytosine; MAD, multiwavelength anomalous dispersion; TCEP, tricarboxyethyl-phosphine; MPD, 2,4-2-methylpentanediol; DTBS, E. coli dethiobiotin synthetase (BioD) enzyme; CPS, E. coli carbamoyl phosphate synthetase enzyme, CarA subunit; 10 GSA, glutamyl γ-semialdehyde; DAPA, diaminoperlargonic acid; rmsd, root-mean-squared difference in position between atom sets, in angstrom units; ALase, amidoligase reaction, formation of a carbon–nitrogen bond by displacement of a phosphorylated oxygen; GATase, glutamine amidotransferase, refers to the conserved glutamine hydrolysis domain that provides ammonia for a number of diverse reactions.
Specific protein residues are referred to by their three-letter residue type and residue number.
Symmetry chains are denoted by a “ ′” mark, for example, the symmetry mate of chain B is “B′”.
In mentions of residues that are donated by other subunits, the chain designation occurs in parentheses, after the residue designations described in Footnote 2. For example, lysine 187 from the symmetry mate of the B subunit is denoted as “Lys187(B′)”.
References
- 1.Liebermann I. Enzymatic Amination of uridine triphosphate to cytidine triphosphate. J Am Chem Soc. 1955;55:2661–2662. [PubMed] [Google Scholar]
- 2.Jones ME. Pyrimidine nucleotide biosynthesis in animals: genes, enzymes, and regulation of UMP biosynthesis. Annu Rev Biochem. 1980;49:253–279. doi: 10.1146/annurev.bi.49.070180.001345. [DOI] [PubMed] [Google Scholar]
- 3.Kent C, Carman GM, Spence MW, Dowhan W. Regulation of eukaryotic phospholipid metabolism. FASEB J. 1991;5:2258–2266. doi: 10.1096/fasebj.5.9.1860617. [DOI] [PubMed] [Google Scholar]
- 4.Kent C, Carman GM. Interactions among pathways for phosphatidylcholine metabolism, CTP synthesis and secretion through the Golgi apparatus. Trends Biochem Sci. 1999;24:146–150. doi: 10.1016/s0968-0004(99)01365-1. [DOI] [PubMed] [Google Scholar]
- 5.West TP, O’Donovan GA. Repression of cytidine triphosphate synthetase in Salmonella typhimurium by pyrimidines during uridine nucleotide depletion. J Gen Microbiol. 1982;128:895–899. doi: 10.1099/00221287-128-4-895. [DOI] [PubMed] [Google Scholar]
- 6.Aronow B, Ullman B. In situ regulation of mammalian CTP synthetase by allosteric inhibition. J Biol Chem. 1987;262:5106–5112. [PubMed] [Google Scholar]
- 7.Ostrander DB, O’Brien DJ, Gorman JA, Carman GM. Effect of CTP synthetase regulation by CTP on phospholipid synthesis in Saccharomyces cerevisiae. J Biol Chem. 1998;273:18992–19001. doi: 10.1074/jbc.273.30.18992. [DOI] [PubMed] [Google Scholar]
- 8.Hatch GM, McClarty G. Regulation of cardiolipin biosynthesis in H9c2 cardiac myoblasts by cytidine 5′-triphosphate. J Biol Chem. 1996;271:25810–25816. doi: 10.1074/jbc.271.42.25810. [DOI] [PubMed] [Google Scholar]
- 9.Kizaki H, Ohsaka F, Sakurada T. CTP synthetase from Ehrlich ascites tumor cells. Subunit stoichiometry and regulation of activity. Biochim Biophys Acta. 1985;829:34–43. doi: 10.1016/0167-4838(85)90065-2. [DOI] [PubMed] [Google Scholar]
- 10.von der Saal W, Anderson PM, Villafranca JJ. Mechanistic investigations of Escherichia coli cytidine-5′-triphosphate synthetase. Detection of an intermediate by positional isotope exchange experiments. J Biol Chem. 1985;260:14993–14997. [PubMed] [Google Scholar]
- 11.Lewis DA, Villafranca JJ. Investigation of the mechanism of CTP synthetase using rapid quench and isotope partitioning methods. Biochemistry. 1989;28:8454–8459. doi: 10.1021/bi00447a027. [DOI] [PubMed] [Google Scholar]
- 12.Levitzki A, Koshland DE., Jr Cytidine triphosphate synthetase. Covalent intermediates and mechanisms of action. Biochemistry. 1971;10:3365–3371. doi: 10.1021/bi00794a008. [DOI] [PubMed] [Google Scholar]
- 13.Charkraborty KP, Hurlbert RB. Role of glutamine in the biosynthesis of cytidine nucleotides in Escherichia coli. Biochim Biophys Acta. 1961;47:607–609. doi: 10.1016/0006-3002(61)90563-7. [DOI] [PubMed] [Google Scholar]
- 14.Long CW, Pardee AB. Cytidine triphosphate synthetase of Escherichia coli B. I. Purification and kinetics. J Biol Chem. 1967;242:4715–4721. [PubMed] [Google Scholar]
- 15.Long C, Koshland DE., Jr Cytidine triphosphate synthetase. Methods Enzymol. 1978;51:79–83. doi: 10.1016/s0076-6879(78)51014-8. [DOI] [PubMed] [Google Scholar]
- 16.Levitzki A, Stallcup WB, Koshland DE., Jr Half-of-the-sites reactivity and the conformational states of cytidine triphosphate synthetase. Biochemistry. 1971;10:3371–3378. doi: 10.1021/bi00794a009. [DOI] [PubMed] [Google Scholar]
- 17.Levitzki A, Koshland DE., Jr Ligand-induced dimer-to-tetramer transformation in cytosine triphosphate synthetase. Biochemistry. 1972;11:247–253. doi: 10.1021/bi00752a016. [DOI] [PubMed] [Google Scholar]
- 18.Levitzki A, Koshland DE., Jr Role of an allosteric effector. Guanosine triphosphate activation in cytosine triphosphate synthetase. Biochemistry. 1972;11:241–246. doi: 10.1021/bi00752a015. [DOI] [PubMed] [Google Scholar]
- 19.Scheit KH, Linke HJ. Substrate specificity of CTP synthetase from Escherichia coli. Eur J Biochem. 1982;126:57–60. doi: 10.1111/j.1432-1033.1982.tb06745.x. [DOI] [PubMed] [Google Scholar]
- 20.Anderson PM. CTP synthetase from Escherichia coli: an improved purification procedure and characterization of hysteretic and enzyme concentration effects on kinetic properties. Biochemistry. 1983;22:3285–3292. doi: 10.1021/bi00282a038. [DOI] [PubMed] [Google Scholar]
- 21.Robertson JG. Determination of subunit dissociation constants in native and inactivated CTP synthetase by sedimentation equilibrium. Biochemistry. 1995;34:7533–7541. doi: 10.1021/bi00022a029. [DOI] [PubMed] [Google Scholar]
- 22.Pappas A, Yang WL, Park TS, Carman GM. Nucleotide-dependent tetramerization of CTP synthetase from Saccharomyces cerevisiae. J Biol Chem. 1998;273:15954–15960. doi: 10.1074/jbc.273.26.15954. [DOI] [PubMed] [Google Scholar]
- 23.Bearne SL, Hekmat O, Macdonnell JE. Inhibition of Escherichia coli CTP synthase by glutamate gamma-semial-dehyde and the role of the allosteric effector GTP in glutamine hydrolysis. Biochem J. 2001;356:223–232. doi: 10.1042/0264-6021:3560223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang WL, McDonough VM, Ozier-Kalogeropoulos O, Adeline MT, Flocco MT, Carman GM. Purification and characterization of CTP synthetase, the product of the URA7 gene in Saccharomyces cerevisiae. Biochemistry. 1994;33:10785–10793. doi: 10.1021/bi00201a028. [DOI] [PubMed] [Google Scholar]
- 25.Hurlbert RB, Kammen HO. Formation of cytidine nucleotides from uridine nucleotides by soluble mammalian enzymes: requirements for glutamine and guanosine nucleotides. J Biol Chem. 1960;235:443–449. [Google Scholar]
- 26.Weinfeld H, Savage CR, Jr, McPartland RP. CTP synthetase of bovine calf liver. Methods Enzymol. 1978;51:84–90. doi: 10.1016/s0076-6879(78)51015-x. [DOI] [PubMed] [Google Scholar]
- 27.Choi MG, Park TS, Carman GM. Phosphorylation of Saccharomyces cerevisiae CTP synthetase at Ser424 by protein kinases A and C regulates phosphatidylcholine synthesis by the CDP-choline pathway. J Biol Chem. 2003;278:23610–23616. doi: 10.1074/jbc.M303337200. [DOI] [PubMed] [Google Scholar]
- 28.Park TS, O’Brien DJ, Carman GM. Phosphorylation of CTP synthetase on Ser36, Ser330, Ser354, and Ser454 regulates the levels of CTP and phosphatidylcholine synthesis in Saccharomyces cerevisiae. J Biol Chem. 2003;278:20785–20794. doi: 10.1074/jbc.M301394200. [DOI] [PubMed] [Google Scholar]
- 29.Ellims PH, Gan TE, Medley G. Cytidine triphosphate synthetase activity in lymphoproliferative disorders. Cancer Res. 1983;43:1432–1435. [PubMed] [Google Scholar]
- 30.Marijnen YM, de Korte D, Roos D, van Gennip AH. Purine and pyrimidine metabolism of normal and leukemic lymphocytes. Adv Exp Med Biol. 1989;253A:433–438. doi: 10.1007/978-1-4684-5673-8_71. [DOI] [PubMed] [Google Scholar]
- 31.Verschuur AC, van Gennip AH, Muller EJ, Voute PA, van Kuilenburg AB. Increased activity of cytidine triphosphate synthetase in pediatric acute lymphoblastic leukemia. Adv Exp Med Biol. 1998;431:667–671. doi: 10.1007/978-1-4615-5381-6_129. [DOI] [PubMed] [Google Scholar]
- 32.Andrei G, De Clercq E. Molecular approaches for the treatment of hemorrhagic fever virus infections. Antiviral Res. 1993;22:45–75. doi: 10.1016/0166-3542(93)90085-w. [DOI] [PubMed] [Google Scholar]
- 33.De Clercq E, Murase J, Marquez VE. Broad-spectrum antiviral and cytocidal activity of cyclopentenylcytosine, a carbocyclic nucleoside targeted at CTP synthetase. Biochem Pharmacol. 1991;41:1821–1829. doi: 10.1016/0006-2952(91)90120-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neyts J, Clercq ED. Therapy and short-term prophylaxis of poxvirus infections: historical background and perspectives. Antiviral Res. 2003;57:25–33. doi: 10.1016/s0166-3542(02)00197-3. [DOI] [PubMed] [Google Scholar]
- 35.McClarty G, Qin B. Pyrimidine metabolism by intracellular Chlamydia psittaci. J Bacteriol. 1993;175:4652–4661. doi: 10.1128/jb.175.15.4652-4661.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tipples G, McClarty G. Cloning and expression of the Chlamydia trachomatis gene for CTP synthetase. J Biol Chem. 1995;270:7908–7914. doi: 10.1074/jbc.270.14.7908. [DOI] [PubMed] [Google Scholar]
- 37.Hofer A, Steverding D, Chabes A, Brun R, Thelander L. Trypanosoma brucei CTP synthetase: a target for the treatment of African sleeping sickness. Proc Natl Acad Sci USA. 2001;98:6412–6416. doi: 10.1073/pnas.111139498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hendriks EF, O’Sullivan WJ, Stewart TS. Molecular cloning and characterization of the Plasmodium falci-parum cytidine triphosphate synthetase gene. Biochim Biophys Acta. 1998;1399:213–218. doi: 10.1016/s0167-4781(98)00108-0. [DOI] [PubMed] [Google Scholar]
- 39.Jimenez BM, O’Sullivan WJ. CTP synthetase and enzymes of pyrimidine ribonucleotide metabolism in Giardia intestinalis. Int J Parasitol. 1994;24:713–718. doi: 10.1016/0020-7519(94)90125-2. [DOI] [PubMed] [Google Scholar]
- 40.Williams JC, Kizaki H, Weber G, Morris HP. Increased CTP synthetase activity in cancer cells. Nature. 1978;271:71–73. doi: 10.1038/271071a0. [DOI] [PubMed] [Google Scholar]
- 41.Maehara Y, Moriguchi S, Emi Y, Watanabe A, Kohnoe S, Tsujitani S, Sugimachi K. Comparison of pyrimidine nucleotide synthetic enzymes involved in 5-fluorouracil metabolism between human adenocarcinomas and squamous cell carcinomas. Cancer. 1990;66:156–161. doi: 10.1002/1097-0142(19900701)66:1<156::aid-cncr2820660128>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 42.Hansel DE, Rahman A, Hidalgo M, Thuluvath PJ, Lillemoe KD, Shulick R, Ku JL, Park JG, Miyazaki K, Ashfaq R, Wistuba II, Varma R, Hawthorne L, Geradts J, Argani P, Maitra A. Identification of novel cellular targets in biliary tract cancers using global gene expression technology. Am J Pathol. 2003;163:217–229. doi: 10.1016/S0002-9440(10)63645-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lim MI, Moyer JD, Cysyk RI, Marquez VE. Cyclopentenyluridine and cyclopentenylcytidine analogues as inhibitors of uridine-cytidine kinase. J Med Chem. 1984;27:1536–1538. doi: 10.1021/jm00378a002. [DOI] [PubMed] [Google Scholar]
- 44.Verschuur AC, van Gennip AH, Brinkman J, Voute PA, van Kuilenburg AB. Cyclopentenyl cytosine induces apoptosis and secondary necrosis in a T-lymphoblastic leukemic cell-line. Adv Exp Med Biol. 2000;486:319–325. doi: 10.1007/0-306-46843-3_62. [DOI] [PubMed] [Google Scholar]
- 45.Gharehbaghi K, Zhen W, Fritzer-Szekeres M, Szekeres T, Jayaram HN. Studies on the antitumor activity and biochemical actions of cyclopentenyl cytosine against human colon carcinoma HT-29 in vitro and in vivo. Life Sci. 1999;64:103–112. doi: 10.1016/s0024-3205(98)00540-2. [DOI] [PubMed] [Google Scholar]
- 46.Brockman RW, Shaddix SC, Williams M, Nelson JA, Rose LM, Schabel FM., Jr The mechanism of action of 3-deazauridine in tumor cells sensitive and resistant to arabinosylcytosine. Ann N Y Acad Sci. 1975;255:501–521. doi: 10.1111/j.1749-6632.1975.tb29254.x. [DOI] [PubMed] [Google Scholar]
- 47.Lui MS, Kizaki H, Weber G. Biochemical pharmacology of acivicin in rat hepatoma cells. Biochem Pharmacol. 1982;31:3469–3473. doi: 10.1016/0006-2952(82)90628-1. [DOI] [PubMed] [Google Scholar]
- 48.Huang M, Wang Y, Collins M, Mitchell BS, Graves LM. A77 1726 induces differentiation of human myeloid leukemia K562 cells by depletion of intracellular CTP pools. Mol Pharmacol. 2002;62:463–472. doi: 10.1124/mol.62.3.463. [DOI] [PubMed] [Google Scholar]
- 49.Bierau J, Van Gennip AH, Leen R, Helleman J, Caron HN, Van Kuilenburg AB. Cyclopentenyl cytosine primes SK-N-BE(2)c neuroblastoma cells for cytarabine toxicity. Int J Cancer. 2003;103:387–392. doi: 10.1002/ijc.10858. [DOI] [PubMed] [Google Scholar]
- 50.Verschuur AC, Van Gennip AH, Leen R, Voute PA, Brinkman J, Van Kuilenburg AB. Cyclopentenyl cytosine increases the phosphorylation and incorporation into DNA of 1-beta-D-arabinofuranosyl cytosine in a human T-lymphoblastic cell line. Int J Cancer. 2002;98:616–623. doi: 10.1002/ijc.10211. [DOI] [PubMed] [Google Scholar]
- 51.Dereuddre-Bosquet N, Roy B, Routledge K, Clayette P, Foucault G, Lepoivre M. Inhibitors of CTP biosynthesis potentiate the anti-human immunodeficiency virus type 1 activity of 3TC in activated peripheral blood mononuclear cells. Antiviral Res. 2004;61:67–70. doi: 10.1016/j.antiviral.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 52.Gao WY, Johns DG, Mitsuya H. Potentiation of the anti-HIV activity of zalcitabine and lamivudine by a CTP synthase inhibitor, 3-deazauridine. Nucleosides Nucleotides Nucleic Acids. 2000;19:371–377. doi: 10.1080/15257770008033015. [DOI] [PubMed] [Google Scholar]
- 53.Zhang H, Cooney DA, Zhang MH, Ahluwalia G, Ford H, Jr, Johns DG. Resistance to cyclopentenylcytosine in murine leukemia L1210 cells. Cancer Res. 1993;53:5714–5720. [PubMed] [Google Scholar]
- 54.Blaney SM, Grem JL, Balis FM, Cole DE, Adamson PC, Poplack DG. Mechanism of resistance to cyclopentenyl cytosine (CPE-C) in Molt-4 lymphoblasts. Biochem Pharmacol. 1993;45:1493–1501. doi: 10.1016/0006-2952(93)90050-7. [DOI] [PubMed] [Google Scholar]
- 55.Whelan J, Smith T, Phear G, Rohatiner A, Lister A, Meuth M. Resistance to cytosine arabinoside in acute leukemia: the significance of mutations in CTP synthetase. Leukemia. 1994;8:264–265. [PubMed] [Google Scholar]
- 56.Wylie JL, Wang LL, Tipples G, McClarty G. A single point mutation in CTP synthetase of Chlamydia trachomatis confers resistance to cyclopentenyl cytosine. J Biol Chem. 1996;271:15393–15400. doi: 10.1074/jbc.271.26.15393. [DOI] [PubMed] [Google Scholar]
- 57.Whelan J, Phear G, Yamauchi M, Meuth M. Clustered base substitutions in CTP synthetase conferring drug resistance in Chinese hamster ovary cells. Nat Genet. 1993;3:317–322. doi: 10.1038/ng0493-317. [DOI] [PubMed] [Google Scholar]
- 58.Aronow B, Ullman B. Mammalian mutants genetically altered in CTP synthetase activity. Adv Exp Med Biol. 1986;195(Pt B):263–269. doi: 10.1007/978-1-4684-1248-2_42. [DOI] [PubMed] [Google Scholar]
- 59.Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22–8. doi: 10.1093/nar/29.1.22. ( http://ww-w.ncbi.nlm.nih.gov/COG/aln/COG0504.aln) [DOI] [PMC free article] [PubMed]
- 60.Weng ML, Zalkin H. Structural role for a conserved region in the CTP synthetase glutamine amide transfer domain. J Bacteriol. 1987;169:3023–3028. doi: 10.1128/jb.169.7.3023-3028.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Holton J, Alber T. Automated protein crystal structure determination using ELVES. Proc Natl Acad Sci USA. 2004;101:1537–1542. doi: 10.1073/pnas.0306241101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Collaborative Computational Project Number 4. The CCP4 suite programs for protein crystallography. Acta Crystallogr Sect D: Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 63.Sheldrick G, Schneider T. SHELXL: high-resolution refinement. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 64.Jones TA, Zou JY, Cowan SW, Kjeldgaard Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr Sect A: Found Crystallogr. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 65.Kack H, Sandmark J, Gibson KJ, Schneider G, Lindqvist Y. Crystal structure of two quaternary complexes of dethiobiotin synthetase, enzyme-MgADP–AlF3-diaminopel-argonic acid and enzyme-MgADP-dethiobiotin-phosphate; implications for catalysis. Protein Sci. 1998;7:2560–2566. doi: 10.1002/pro.5560071209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thoden JB, Huang X, Raushel FM, Holden HM. The small subunit of carbamoyl phosphate synthetase: snapshots along the reaction pathway. Biochemistry. 1999;38:16158–16166. doi: 10.1021/bi991741j. [DOI] [PubMed] [Google Scholar]
- 67.Kissinger CR, Gehlhaar DK, Fogel DB. Rapid automated molecular replacement by evolutionary search. Acta Crystallogr Sect D: Biol Crystallogr. 1999;55:484–491. doi: 10.1107/s0907444998012517. [DOI] [PubMed] [Google Scholar]
- 68.Tronrud DE. TNT refinement package. Methods Enzymol. 1997;277:306–319. doi: 10.1016/s0076-6879(97)77017-4. [DOI] [PubMed] [Google Scholar]
- 69.Engh R, Huber R. Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallogr Sect A: Found Crystallogr. 1991;47:392–400. [Google Scholar]
- 70.Tronrud D. Knowledge-based B-factor restraints for the refinement of proteins. J Appl Crystallogr. 1996;29:100–104. [Google Scholar]
- 71.Brunger A. Assessment of phase accuracy by cross validation: the free R value. Methods and Applications. Acta Crystallogr, Sect D: Biol Crystallogr. 1993;49:24–36. doi: 10.1107/S0907444992007352. [DOI] [PubMed] [Google Scholar]
- 72.al-Karadaghi S, Aevarsson A, Garber M, Zheltonosova J, Liljas A. The structure of elongation factor G in complex with GDP: conformational flexibility and nucleotide exchange. Structure. 1996;4:555–565. doi: 10.1016/s0969-2126(96)00061-5. [DOI] [PubMed] [Google Scholar]
- 73.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. ( http://www.ebi.ac.uk./dali) [DOI] [PubMed]
- 74.Zhang XJ, Matthews BW. EDPDB: A multi-functional tool for protein structure analysis. J Appl Crystallogr. 1995;28:624–630. [Google Scholar]
- 75.Esnouf RM. An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J Mol Graphics Model. 1997;15:132–134. 112–113. doi: 10.1016/S1093-3263(97)00021-1. [DOI] [PubMed] [Google Scholar]
- 76.Merritt EA, Bacon DJ. Raster3D: Photorealistic Molecular Graphics. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 77.Nicholls A, Sharp KA, Honig B. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 78.Long CW, Levitzki A, Koshland DE., Jr The subunit structure and subunit interactions of cytidine triphosphate synthetase. J Biol Chem. 1970;245:80–87. [PubMed] [Google Scholar]
- 79.Sandalova T, Schneider G, Kack H, Lindqvist Y. Structure of dethiobiotin synthetase at 0.97 Å resolution. Acta Crystallogr Sect D: Biol Crystallogr. 1999;55:610–624. doi: 10.1107/s090744499801381x. [DOI] [PubMed] [Google Scholar]
- 80.Galperin MY, Grishin NV. The synthetase domains of cobalamin biosynthesis amidotransferases cobB and cobQ belong to a new family of ATP-dependent amidoligases, related to dethiobiotin synthetase. Proteins. 2000;41:238–247. doi: 10.1002/1097-0134(20001101)41:2<238::aid-prot80>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 81.Leipe DD, Wolf YI, Koonin EV, Aravind L. Classification and evolution of P-loop GTPases and related ATPases. J Mol Biol. 2002;317:41–72. doi: 10.1006/jmbi.2001.5378. [DOI] [PubMed] [Google Scholar]
- 82.Tesmer JJ, Klem TJ, Deras ML, Davisson VJ, Smith JL. The crystal structure of GMP synthetase reveals a novel catalytic triad and is a structural paradigm for two enzyme families. Nat Struct Biol. 1996;3:74–86. doi: 10.1038/nsb0196-74. [DOI] [PubMed] [Google Scholar]
- 83.Simard D, Hewitt KA, Lunn F, Iyengar A, Bearne SL. Limited proteolysis of Escherichia coli cytidine 5′-triphosphate synthase. Identification of residues required for CTP formation and GTP-dependent activation of glutamine hydrolysis, Eur J Biochem. 2003;270:2195–2206. doi: 10.1046/j.1432-1033.2003.03588.x. [DOI] [PubMed] [Google Scholar]
- 84.Willemoës M. Thr-431 and Arg-433 are part of a conserved sequence motif of the glutamine amidotransferase domain of CTP synthases and are involved in GTP activation of the Lactococcus lactis enzyme. J Biol Chem. 2003;278:9407–9411. doi: 10.1074/jbc.M212995200. [DOI] [PubMed] [Google Scholar]
- 85.Alexeev D, Baxter RL, Sawyer L. Mechanistic implications and family relationships from the structure of dethiobiotin synthetase. Structure. 1994;2:1061–1072. doi: 10.1016/s0969-2126(94)00109-x. [DOI] [PubMed] [Google Scholar]
- 86.Lansdon EB, Segel IH, Fisher AJ. Ligand-induced structural changes in adenosine 5′-phosphosulfate kinase from Penicillium chrysogenum. Biochemistry. 2002;41:13672–13680. doi: 10.1021/bi026556b. [DOI] [PubMed] [Google Scholar]
- 87.Iyengar A, Bearne SL. Aspartate-107 and leucine-109 facilitate efficient coupling of glutamine hydrolysis to CTP synthesis by Escherichia coli CTP synthase. Biochem J. 2003;369:497–507. doi: 10.1042/BJ20021110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pappas A, Park TS, Carman GM. Characterization of a novel dUTP-dependent activity of CTP synthetase from Saccharomyces cerevisiae. Biochemistry. 1999;38:16671–16677. doi: 10.1021/bi9920127. [DOI] [PubMed] [Google Scholar]
- 89.Wadskov-Hansen SL, Willemoes M, Martinussen J, Hammer K, Neuhard J, Larsen S. Cloning and verification of the Lactococcus lactis pyrG gene and characterization of the gene product, CTP synthase. J Biol Chem. 2001;276:38002–38009. doi: 10.1074/jbc.M100531200. [DOI] [PubMed] [Google Scholar]
- 90.Ladner JE, Kitchell JP, Honzatko RB, Ke HM, Volz KW, Kalb AJ, Ladner RC, Lipscomb WN. Gross quaternary changes in aspartate carbamoyltransferase are induced by the binding of N-(phosphonacetyl)-L-aspartate: A 3.5-Å resolution study. Proc Natl Acad Sci USA. 1982;79:3125–3128. doi: 10.1073/pnas.79.10.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Goto M, Omi R, Hoseki J, Nakagawa N, Miyahara I, Hirotsu K. Expression, purification and preliminary X-ray characterization of CTP synthetase from Thermus thermophilus HB8. Acta Crystallogr Sect D: Biol Crystallogr. 2003;59:551–553. doi: 10.1107/s0907444903000106. [DOI] [PubMed] [Google Scholar]
- 92.Raushel FM, Thoden JB, Holden HM. Enzymes with molecular tunnels. Acc Chem Res. 2003;36:539–548. doi: 10.1021/ar020047k. [DOI] [PubMed] [Google Scholar]
- 93.Yun M, Park CG, Kim JY, Rock CO, Jackowski S, Park HW. Structural basis for the feedback regulation of Escherichia coli pantothenate kinase by coenzyme A. J Biol Chem. 2000;275:28093–28099. doi: 10.1074/jbc.M003190200. [DOI] [PubMed] [Google Scholar]
- 94.Chaudhuri BN, Lange SC, Myers RS, Davisson VJ, Smith JL. Toward understanding the mechanism of the complex cyclization reaction catalyzed by imidazole glycerolphosphate synthase: crystal structures of a ternary complex and the free enzyme. Biochemistry. 2003;42:7003–7012. doi: 10.1021/bi034320h. [DOI] [PubMed] [Google Scholar]
- 95.Thoden JB, Holden HM, Wesenberg G, Raushel FM, Rayment I. Structure of carbamoyl phosphate synthetase: a journey of 96 Å from substrate to product. Biochemistry. 1997;36:6305–6316. doi: 10.1021/bi970503q. [DOI] [PubMed] [Google Scholar]
- 96.Willemoës M, Sigurskjold BW. Steady-state kinetics of the glutaminase reaction of CTP synthase from Lactococcus lactis. The role of the allosteric activator GTP in coupling between glutamine hydrolysis and CTP synthesis. Eur J Biochem. 2002;269:4772–4779. doi: 10.1046/j.1432-1033.2002.03175.x. [DOI] [PubMed] [Google Scholar]