

Abstract
Mechanisms that contribute to Na+ influx during and immediately after 5 min anoxia were investigated in cultured rat hippocampal neurons loaded with the Na+-sensitive fluorophore sodium-binding benzofuran isophthalate. During anoxia, an influx of Na+ in the face of reduced Na+,K+-ATPase activity caused a rise in [Na+]i. After the return to normoxia, Na+,K+-ATPase activity mediated the recovery of [Na+]i despite continued Na+ entry. Sodium influx during and after anoxia occurred through multiple pathways and increased the longer neurons were maintained in culture. Under the experimental conditions used, Na+ entry during anoxia did not reflect the activation of ionotropic glutamate receptors, TTX- or lidocaine-sensitive Na+ channels, plasmalemmal Na+/Ca2+ exchange, Na+/H+ exchange, or 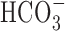 -dependent mechanisms; rather, contributions were received from a Gd3+-sensitive pathway activated by reactive oxygen species and Na+/K+/2Cl- cotransport in neurons maintained for 6-10 and 11-14 d in vitro (DIV), respectively. Sodium entry immediately after anoxia was not attributable to the activation of ionotropic glutamate receptors, voltage-activated Na+ channels, or Na+/K+/2Cl- cotransport; rather, it occurred via Na+/Ca2+ exchange, Na+/H+ exchange, and a Gd3+-sensitive pathway similar to that observed during anoxia; 11-14 DIV neurons received an additional contribution from an
-dependent mechanisms; rather, contributions were received from a Gd3+-sensitive pathway activated by reactive oxygen species and Na+/K+/2Cl- cotransport in neurons maintained for 6-10 and 11-14 d in vitro (DIV), respectively. Sodium entry immediately after anoxia was not attributable to the activation of ionotropic glutamate receptors, voltage-activated Na+ channels, or Na+/K+/2Cl- cotransport; rather, it occurred via Na+/Ca2+ exchange, Na+/H+ exchange, and a Gd3+-sensitive pathway similar to that observed during anoxia; 11-14 DIV neurons received an additional contribution from an 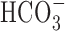 -dependent mechanism(s). The results provide insight into the intrinsic mechanisms that contribute to disturbed internal Na+ homeostasis during and immediately after anoxia in rat hippocampal neurons and, in this way, may play a role in the pathogenesis of anoxic or ischemic cell injury.
-dependent mechanism(s). The results provide insight into the intrinsic mechanisms that contribute to disturbed internal Na+ homeostasis during and immediately after anoxia in rat hippocampal neurons and, in this way, may play a role in the pathogenesis of anoxic or ischemic cell injury.
Keywords: anoxia, ischemia, intracellular sodium, Na+/K+/2Cl- cotransport, Na+/Ca2+ exchange, Na+/H+ exchange, Gd3+
Introduction
In neurons of the mammalian CNS, anoxia and ischemia lead to early changes in internal ion homeostasis that are important determinants of subsequent injury and death. Although the contribution of changes in intracellular Ca2+ (Ca2+i) has received particular attention, notably within the framework of the excitotoxic model of injury, early increases in intracellular Na+ (Na+i) also occur and contribute to the pathophysiology of neuronal death (Lipton, 1999; Martinez-Sánchez et al., 2004). Sodium influx increases the demand for cellular ATP to maintain the Na+ gradient (Chinopoulos et al., 2000) and may contribute to neuronal injury by promoting, for example, the following: membrane depolarization (Haddad and Jiang, 1993; Calabresi et al., 1999), neuronal swelling (Goldberg and Choi, 1993; Chidekel et al., 1997), reverse-mode glutamate reuptake (Roettger and Lipton, 1996), cytosolic Ca2+ accumulation via reverse-mode Na+/Ca2+ exchange and/or impaired mitochondrial Ca2+ uptake (Zhang and Lipton, 1999; Breder et al., 2000; Czyż et al., 2002), Na+i-dependent increases in NMDA receptor-mediated responses (Yu and Salter, 1998; Manzerra et al., 2001), and the activation of second-messenger pathways (Cooper et al., 1998; Hayasaki-Kajiwara et al., 1999).
Although the detrimental effects of Na+ entry are recognized, in contrast to non-neuronal cell types (e.g., cardiac myocytes) (Carmeliet, 1999), the mechanisms that mediate Na+ influx in response to anoxia or ischemia in mammalian central neurons remain relatively poorly defined. Although Na+ entry attributable to the activation of ionotropic glutamate receptors has received some attention, with conflicting results (cf. Pisani et al., 1998; Chen et al., 1999; Müller and Somjen, 2000; LoPachin et al., 2001), glutamate-mediated excitotoxicity cannot fully account for the direct actions of anoxia or ischemia on neurons, and the potential contribution from mechanisms integral to neurons (i.e., independent from glutamate receptor activation and changes in the external microenvironment) to Na+ influx during anoxia or ischemia has not been systematically addressed. Furthermore, despite indications that continued Na+ influx after reperfusion may be as damaging as Na+ entry during anoxia or ischemia (Lipton, 1999), the pathways that mediate Na+ influx after anoxia or ischemia have not been well characterized, and it remains unknown whether these pathways might differ from those active during an insult, as reported for Ca2+ (Silver and Erecińska, 1990, 1992).
We have therefore undertaken an assessment of the intrinsic mechanisms that contribute to Na+ influx during and immediately after 5 min anoxia in cultured postnatal rat hippocampal neurons. We find not only that Na+ influx during and after anoxia is positively related to the length of time that neurons are maintained in culture but also that different pathways are involved during, compared with immediately after, anoxia.
Materials and Methods
Cell preparation. Primary cultures of hippocampal neurons from 2- to 4-d-old postnatal Wistar rats (Animal Care Centre, University of British Columbia) were prepared as described previously (Diarra et al., 2001). Briefly, rat pups were anesthetized and decapitated, and the hippocampi were removed. The hippocampi were enzymatically and mechanically dissociated, and the resulting cell suspension was plated at a density of 5-8 × 105 neurons/cm2 onto glass coverslips coated with poly-d-lysine and laminin. The initial growth medium was DMEM/F-12 (Invitrogen Canada, Burlington, Ontario, Canada) supplemented with 10% fetal bovine serum. After 24 hr, this medium was fully changed to serum-free Neurobasal medium A. Cultures were fed every 4-5 d by half-changing the existing medium with fresh Neurobasal medium A. Glial proliferation was inhibited 48 hr after initial plating by adding 5-10 μm cytosine arabinoside.
Solutions and test compounds. The majority of experiments were performed under nominally 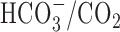 -free, HEPES-buffered conditions. Standard
-free, HEPES-buffered conditions. Standard 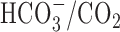 -free perfusion medium contained (mm) 136.5 NaCl, 3 KCl, 2 CaCl2, 1.5 NaH2PO4, 1.5 MgSO4, 10 d-glucose, and 10 HEPES and was titrated with 10 m NaOH to pH 7.35 at 37°C. In standard
-free perfusion medium contained (mm) 136.5 NaCl, 3 KCl, 2 CaCl2, 1.5 NaH2PO4, 1.5 MgSO4, 10 d-glucose, and 10 HEPES and was titrated with 10 m NaOH to pH 7.35 at 37°C. In standard 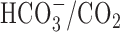 -buffered medium, HEPES was isosmotically replaced by NaCl and solutions contained 19.5 mm NaHCO3, by equimolar substitution for NaCl, together with the constituents listed above (pH 7.35 at 37°C after equilibration with 5% CO2/95% air); during perfusion with
-buffered medium, HEPES was isosmotically replaced by NaCl and solutions contained 19.5 mm NaHCO3, by equimolar substitution for NaCl, together with the constituents listed above (pH 7.35 at 37°C after equilibration with 5% CO2/95% air); during perfusion with 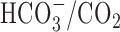 -buffered media, the atmosphere in the recording chamber contained 5% CO2/95% air. When external Na+ was reduced to 2-4 mm, N-methyl-d-glucamine (NMDG+) was used as a substitute, and solutions were titrated to pH 7.35 (37°C) with 10 m HCl. For Ca2+-free media, CaCl2 was omitted, [Mg2+] was increased to 3.5 mm, and 200 μm EGTA was added. In solutions containing Zn2+, Gd3+, or Ni2+, NaH2PO4 was omitted and MgSO4 was replaced with MgCl2. All experiments were performed at 37°C, and neurons were continuously superfused at a rate of 2 ml/min; unless noted otherwise, extracellular pH (pHo) was 7.35.
-buffered media, the atmosphere in the recording chamber contained 5% CO2/95% air. When external Na+ was reduced to 2-4 mm, N-methyl-d-glucamine (NMDG+) was used as a substitute, and solutions were titrated to pH 7.35 (37°C) with 10 m HCl. For Ca2+-free media, CaCl2 was omitted, [Mg2+] was increased to 3.5 mm, and 200 μm EGTA was added. In solutions containing Zn2+, Gd3+, or Ni2+, NaH2PO4 was omitted and MgSO4 was replaced with MgCl2. All experiments were performed at 37°C, and neurons were continuously superfused at a rate of 2 ml/min; unless noted otherwise, extracellular pH (pHo) was 7.35.
As described previously (Diarra et al., 2001), media used to calibrate sodium-binding benzofuran isophthalate (SBFI)-derived fluorescence ratio measurements contained 0.6 mm MgCl2, 0.5 mm CaCl2, 10 mm HEPES, 130 mm [Na+] plus [K+], 100 mm gluconate, 30 mm Cl-, and 4 μm gramidicin D, titrated with 10 m KOH to pH 7.35. In experiments in which seminaphthorhodafluor (SNARF) pH indicators were used, SNARF-derived fluorescence ratio measurements were calibrated with media containing the same components as the standard HEPES-buffered medium, except for the substitution of K gluconate (130.5 mm) and Na gluconate (9 mm) for NaCl and KCl and the addition of 10 μm nigericin (Sheldon et al., 2004).
Test compounds were obtained from Sigma-Aldrich Canada (Oakville, Ontario, Canada), with the exception of the Rp isomer of adenosine-3′,5′-cyclic monophosphorothioate (Rp-cAMPS) (Biolog Life Science Institute, La Jolla, CA); arachidonyltrifluoromethyl ketone (AACOCF3) (Calbiochem, San Diego, CA); and (5S, 10 R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate (MK-801), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), and 2-[2-[4-(4-nitrobenzyloxy)phenyl]ethyl]isothiourea mesylate (KBR7943) (all from Tocris Cookson, Ellisville, MO).
Induction of anoxia. Anoxia was induced by the addition of 1-2 mm sodium dithionite, an O2 scavenger, to the superfusing medium. Solutions containing Na2S2O4 were prepared immediately before use and bubbled with either 100% Ar (HEPES-buffered media) or 5% CO2/95% Ar ( -buffered media); during perfusion with these media, the atmosphere in the recording chamber was switched to 100% Ar or 5% CO2/95% Ar, respectively. We have previously reported that media containing 1-2 mm Na2S2O4 have PO2 values <1 mmHg and that the changes in pHi evoked by anoxia in rat hippocampal neurons are not secondary to any additional properties of the O2 scavenger (Diarra et al., 1999; Sheldon and Church, 2002). In the present study, we assessed the possibility that Na2S2O4 may induce changes in [Na+]i via mechanisms unrelated to its O2 scavenging property in two ways. First, solutions containing 1-2 mm Na2S2O4 were bubbled vigorously with air for 20-30 min, which elevated the PO2 in these solutions from <1 to 152 ± 6 mmHg (n = 5), as measured with an oxygen electrode (ISO2; World Precision Instruments, Sarasota, FL). The increase in [Na+]i observed during a 5 min exposure to media containing 1-2 mm Na2S2O4 equilibrated with air was 3 ± 1 mm (n = 7), significantly (p < 0.05) less than the 23 ± 3 mm (n = 23) increase observed in age-matched sister neurons exposed to media also containing 1-2 mm Na2S2O4 but equilibrated with 100% Ar (see Fig. 1 A). Second, standard HEPES-buffered medium was bubbled vigorously with ultra-pure Ar for >18 hr, reducing PO2 in the medium to <1 mmHg (Sheldon and Church, 2002). The increases in [Na+]i observed during a 5 min exposure to this medium were similar to those measured in age-matched neurons when PO2 was reduced to <1 mmHg by the addition of sodium dithionite (see Fig. 1 B). Thus, the [Na+]i changes evoked by exposure to media containing 1-2 mm Na2S2O4 primarily reflect reductions in PO2.
-buffered media); during perfusion with these media, the atmosphere in the recording chamber was switched to 100% Ar or 5% CO2/95% Ar, respectively. We have previously reported that media containing 1-2 mm Na2S2O4 have PO2 values <1 mmHg and that the changes in pHi evoked by anoxia in rat hippocampal neurons are not secondary to any additional properties of the O2 scavenger (Diarra et al., 1999; Sheldon and Church, 2002). In the present study, we assessed the possibility that Na2S2O4 may induce changes in [Na+]i via mechanisms unrelated to its O2 scavenging property in two ways. First, solutions containing 1-2 mm Na2S2O4 were bubbled vigorously with air for 20-30 min, which elevated the PO2 in these solutions from <1 to 152 ± 6 mmHg (n = 5), as measured with an oxygen electrode (ISO2; World Precision Instruments, Sarasota, FL). The increase in [Na+]i observed during a 5 min exposure to media containing 1-2 mm Na2S2O4 equilibrated with air was 3 ± 1 mm (n = 7), significantly (p < 0.05) less than the 23 ± 3 mm (n = 23) increase observed in age-matched sister neurons exposed to media also containing 1-2 mm Na2S2O4 but equilibrated with 100% Ar (see Fig. 1 A). Second, standard HEPES-buffered medium was bubbled vigorously with ultra-pure Ar for >18 hr, reducing PO2 in the medium to <1 mmHg (Sheldon and Church, 2002). The increases in [Na+]i observed during a 5 min exposure to this medium were similar to those measured in age-matched neurons when PO2 was reduced to <1 mmHg by the addition of sodium dithionite (see Fig. 1 B). Thus, the [Na+]i changes evoked by exposure to media containing 1-2 mm Na2S2O4 primarily reflect reductions in PO2.
Figure 1.

Changes in [Na+]i evoked by 5 min anoxia in cultured postnatal rat hippocampal neurons. A, Anoxia was imposed under nominally 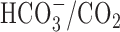 -free, HEPES-buffered conditions by exposure to medium containing 1-2 mm sodium dithionite and bubbled vigorously with 100% Ar (•). Also shown are the changes in neuronal [Na+]i evoked by anoxia in an age-matched sister culture exposed to medium containing 1-2 mm sodium dithionite and bubbled vigorously with air (○). B, Relationship between the magnitude of the increase in [Na+]i during anoxia (Δ[Na+]i(during)) and the number of days that neurons had been maintained in culture (DIV). Anoxia was imposed under HEPES-buffered conditions either by the addition of sodium dithionite to medium bubbled with 100% Ar (•; n = 21-54 for each datum point) or by the exposure to medium that had been bubbled vigorously with 100% Ar for >18 hr (○; n ≥3 for each datum point). The solid line represents a linear regression fit to the data points obtained when anoxia was imposed by the addition of sodium dithionite (correlation coefficient, 0.95; p < 0.0001). C, Under normal Na+o-containing conditions, anoxia induced an increase in [Na+]i that recovered after the return to normoxia (•). When anoxia was imposed under reduced Na+o, NMDG+-substituted conditions, the increase in [Na+]i was inhibited (○).
-free, HEPES-buffered conditions by exposure to medium containing 1-2 mm sodium dithionite and bubbled vigorously with 100% Ar (•). Also shown are the changes in neuronal [Na+]i evoked by anoxia in an age-matched sister culture exposed to medium containing 1-2 mm sodium dithionite and bubbled vigorously with air (○). B, Relationship between the magnitude of the increase in [Na+]i during anoxia (Δ[Na+]i(during)) and the number of days that neurons had been maintained in culture (DIV). Anoxia was imposed under HEPES-buffered conditions either by the addition of sodium dithionite to medium bubbled with 100% Ar (•; n = 21-54 for each datum point) or by the exposure to medium that had been bubbled vigorously with 100% Ar for >18 hr (○; n ≥3 for each datum point). The solid line represents a linear regression fit to the data points obtained when anoxia was imposed by the addition of sodium dithionite (correlation coefficient, 0.95; p < 0.0001). C, Under normal Na+o-containing conditions, anoxia induced an increase in [Na+]i that recovered after the return to normoxia (•). When anoxia was imposed under reduced Na+o, NMDG+-substituted conditions, the increase in [Na+]i was inhibited (○).
Microspectrofluorimetry. The AM ester forms of the Na+-sensitive fluorophore SBFI and the pH-sensitive fluorophores carboxy SNARF-1 and SNARF-5F were obtained from Molecular Probes (Eugene, OR). Neurons were incubated with 10 μm SBFI-AM (in the presence of 0.1% Pluronic F-127 and 5 mg/ml bovine serum albumin) for 120-180 min at 32°C. In experiments in which [Na+]i and pHi were measured concurrently (see below), SBFI-loaded neurons were incubated with 10 μm carboxy SNARF-1-AM or SNARF-5F-AM in the presence of 0.1% Pluronic F-127 for 30 min at 32°C. After dye loading, coverslips with cells attached were placed in standard HEPES-buffered medium for 20 min and then mounted in a temperature-controlled recording chamber. Neurons were superfused for 15 min with the initial experimental solution at 37°C before the start of an experiment.
In the majority of experiments, cells were loaded only with SBFI, and measurements of [Na+]i were performed using the dual-excitation ratio method using an imaging system (Atto Bioscience, Rockville, MD) in conjunction with an Axiovert 135 epifluorescence microscope (Carl Zeiss Canada, Don Mills, Ontario, Canada). As described by Diarra et al. (2001), fluorescence emissions (at >510 nm) were measured by a single intensified charge-coupled device camera from regions of interest placed on individual neuronal somata. Raw emission intensity data at each excitation wavelength (334 and 380 nm) were corrected for background fluorescence before the calculation of a ratio. Ratio pairs were acquired at 2-15 sec intervals and analyzed off-line. A one-point calibration technique was used to convert background-corrected SBFI ratio values (BI334/BI380) into [Na+]i values as described previously (Diarra et al., 2001). In brief, at the end of an experiment, neurons loaded with SBFI were exposed to a pH 7.35 medium containing 10 mm Na+ and 4 μm gramicidin D, and the resulting background-corrected ratio value at 10 mm [Na+]i was used as a normalization factor for experimentally derived BI334/BI380 ratio values. Parameters used for the calculation of [Na+]i values were derived from full in situ calibration experiments in which neurons were exposed sequentially to media containing eight different [Na+] values (Diarra et al., 2001).
In a number of experiments, carboxy SNARF-1 and SNARF-5F were used to measure pHi concurrently with changes in [Na+]i. In neurons coloaded with SBFI and a SNARF-based fluorophore, ratio pairs were collected continuously by alternating between the dual-excitation and dual-emission modes [for technical details and validation of the technique, see Sheldon et al. (2004)]. In brief, SBFI-derived fluorescence emission intensities were measured with a single camera at 550 nm during excitation at 334 nm and then at 380 nm; the excitation wavelength was then changed to 488 nm, and SNARF-derived fluorescence emissions were split by a dichroic mirror centered at 605 nm and measured by two separate cameras at 550 and 640 nm. Camera registration was confirmed before every experiment. To convert background-corrected, SNARF-derived ratio values (BI550/BI640) into pHi, neurons loaded with a SNARF derivative were exposed at the end of an experiment to a pH 7.00 high-[K+] medium containing 10 μm nigericin, and the resulting background-corrected ratio value at pHi 7.00 was used as a normalization factor for experimentally derived BI550/BI640 ratio values. The constants required to convert experimentally derived BI550/BI640 ratio values into pHi values were determined in full calibration experiments as described previously (Sheldon et al., 2004).
To limit potential cross-contamination by ionophores, perfusion lines were replaced, and the imaging chamber was decontaminated after each experiment.
Data analysis. The magnitude of the increase in [Na+]i during 5 min anoxia (Δ[Na+]i(during)) was measured as the difference between the resting [Na+]i value before anoxia and the peak [Na+]i value reached during anoxia (see Fig. 1 A). To characterize the Na+ influx pathways active immediately after anoxia, neurons were exposed to 5 min anoxia and, after the return to normoxia, Na+,K+-ATPase activity was inhibited for 7 min by exposure to K+-free medium or the application of 500 μm ouabain, as described by van Emous et al. (1998). The magnitude of the increase in [Na+]i after anoxia under these conditions (Δ[Na+]i(after)) was measured as the difference between the [Na+]i value observed at the end of 5 min anoxia and the [Na+]i value observed at the end of the 7 min exposure to 0 [K+]o or 500 μm ouabain (see Fig. 5A). In experiments in which changes in pHi and [Na+]i during and after anoxia were measured concurrently, the magnitude of the fall in pHi during anoxia was measured as the difference between the pHi value at the end of 5 min anoxia and the preanoxic pHi value (see Fig. 8 A), and the magnitude of the increase in pHi after anoxia (Na+,K+-ATPase inhibited) was measured as the difference between the pHi value at the end of 5 min anoxia and the pHi value at the end of 7 min exposure to 0 [K+]o (see Fig. 8 B).
Figure 5.

Changes in [Na+]i after anoxia. A, Na+,K+-ATPase activity was inhibited by perfusion with K+-free medium at the end of 5 min anoxia, revealing a secondary rise in [Na+]i in the immediate postanoxic period (•). Age-matched sister neurons on a different coverslip were exposed to K+- and Na+-free medium after 5 min anoxia imposed under control conditions (○); in the absence of external Na+ (NMDG+-substituted), the increase in [Na+]i after anoxia was abolished. B, Relationship between the magnitude of the increase in [Na+]i after anoxia (Δ[Na+]i(after)) and the number of days neurons had been maintained in culture (DIV). Δ[Na+]i(after) was determined as the difference between the [Na+]i value observed at the end of 5 min anoxia and the [Na+]i value observed at the end of 7 min superfusion with K+-free medium (•; n = 7-31 for each datum point). The solid line represents the linear regression fit to the data points indicated (correlation coefficient, 0.88; p < 0.0001). Also shown are Δ[Na+]i(after) values obtained when Na+, K+-ATPase activity was inhibited for 7 min with 500 μm ouabain (○; n = 3 for each datum point).
Figure 8.

Anoxia-evoked changes in pHi and [Na+]i in rat hippocampal neurons coloaded with SBFI and SNARF-5F. A, Anoxia induced an increase in [Na+]i (•) and a fall in pHi (○), both of which recovered toward resting values after the return to normoxia. B, Inhibition of Na+,K+-ATPase activity (perfusion with K+-free medium) for 7 min after anoxia failed to influence the postanoxic rise in pHi (•) but revealed an additional increase in [Na+]i (•). C, During 5 min anoxia imposed in the presence of external Na+, [Na+]i (♦) increased and pHi (⋄) fell. Immediately after anoxia, neurons were superfused with K+- and Na+-free medium (pHo constant at 7.35). Reducing Na+o (NMDG+ substitution) for the period indicated by the short bar above the records prevented the increases in pHi and [Na+]i seen after anoxia in the presence of normal Na+o (compare with B). After the return to normal Na+o (in the continued absence of external K+; arrow), both pHi and [Na+]i increased. D, Measured in 20 neurons maintained for 7-10 DIV and coloaded with SBFI and SNARF-5F, rates at which [Na+]i (•) and pHi (○) increased immediately after anoxia (Na+,K+-ATPase inhibited) exhibited an inverse dependence on absolute pHi values. Similar results were observed in 11-14 DIV neuronal cultures (E; n = 10). In D and E, rates at which pHi and [Na+]i increased after anoxia under K+o-free conditions were determined by fitting the pHi and [Na+]i records to single exponential functions; the first derivatives of these functions were used to determine rates of pHi and [Na+]i increase at 0.05 pH unit and 5 mm intervals, respectively. The pHi values at which rates of [Na+]i increase were measured were determined from obtained curve-fitted parameters.
In light of the findings that the magnitudes of the changes in [Na+]i both during and after anoxia were related to the length of time that neurons had been maintained in culture (see Results), experiments were performed routinely on cultures maintained for 6-10 d in vitro (DIV) and, where noted, were repeated using 11-14 DIV neuronal cultures. In both groups, measurements of anoxia-evoked changes in [Na+]i observed under a given test condition were normalized to measurements made under control conditions in age-matched sister neurons (yielding normalized Δ[Na+]i(during) and Δ[Na+]i(after) values) (Tables 1, 2). Student's two-tailed unpaired t tests were used to compare statistically the absolute (i.e., not normalized) Δ[Na+]i(during) and Δ[Na+]i(after) measurements made under a given test condition to the respective measurements made in age-matched sister neurons under control conditions; significance was assumed at the 5% level.
Table 1.
Effects of various treatments on increases in [Na+]i during anoxia
|
|
Normalized Δ[Na+]i(during) |
||
|---|---|---|---|
| Treatment |
6-10 DIV |
11-14 DIV |
|
| 1 μm TTX | 1.04 ± 0.11 (5) | 0.91 ± 0.17 (5) | |
| 250 μm lidocaine | 0.94 ± 0.12 (7) | n.d. | |
| 50 μm bepridil | 0.94 ± 0.17 (7) | n.d. | |
| 1 μm KB-R7943 | 1.05 ± 0.28 (5) | n.d. | |
| 10 μm KB-R7943 | 1.02 ± 0.17 (6) | n.d. | |
| 25 μm CGP-37157 | 1.02 ± 0.28 (4) | n.d. | |
| 0 Ca2+o | 0.92 ± 0.16 (5) | 1.04 ± 0.20 (4) | |
| 100 μm bumetanide | 1.12 ± 0.19 (5) | 0.61 ± 0.05 (9)* | |
| 200 μm harmalinea | 0.91 ± 0.22 (7) | 0.98 ± 0.26 (5) | |
| HCO3−/CO2 buffer | 1.11 ± 0.13 (8) | 0.79 ± 0.13 (5) | |
| HCO3−/CO2 buffer plus 200 μm DIDS |
1.18 ± 0.28 (3) |
0.78 ± 0.31 (4) |
|
Except where noted, experiments were performed under nominally HCO3−-free, HEPES-buffered conditions. To generate normalized Δ[Na+]i(during) values, measurements of Δ[Na+]i(during) under a given test condition were normalized to measurements made in control experiments performed on age-matched sister neurons. Statistical comparisons were performed by comparing absolute Δ[Na+]i(during) measurements made under a given test condition with measurements made in age-matched sister neurons under control conditions. The asterisk indicates statistical significance (p < 0.05) compared with measurements made in age-matched sister neurons in the absence of treatment. The numbers in parentheses indicate the number of neuronal populations (i.e. coverslips) from which data were obtained. n.d., Not determined.
Because of its fluorescence properties, neurons were pretreated with harmaline for 60 min before the start of an experiment.
Table 2.
Effects of various treatments on increases in [Na+]i after anoxia (Na+, K+-ATPase blocked)
|
|
Normalized Δ[Na+]i(after) |
||
|---|---|---|---|
| Treatment |
6-10 DIV |
11-14 DIV |
|
| 1 μm TTX | 0.98 ± 0.20 (5) | 0.92 ± 0.02 (3) | |
| 100 μm TTX | 0.83 ± 0.13 (4) | n.d. | |
| 250 μm lidocaine | n.d. | 0.89 ± 0.29 (3) | |
| 100 μm bumetanide | 0.91 ± 0.27 (3) | 1.21 ± 0.25 (4) | |
| 50 μm bepridil | 0.43 ± 0.12 (6)* | n.d. | |
| 1 μm KB-R7943 | 1.56 ± 0.25 (4)* | n.d. | |
| 0 Ca2+o | 1.75 ± 0.31 (3)* | n.d. | |
| 10 μm KB-R7943 |
0.67 ± 0.09 (5)*
|
n.d. |
|
Na+, K+-ATPase activity was blocked immediately after anoxia by perfusion with K+-free medium, except in the case of bumetanide treatment in which Na+, K+-ATPase was inhibited immediately after anoxia by exposure to 500 μm ouabain. To generate normalized Δ[Na+]i(after) values, measurements of Δ[Na+]i(after) under a given test condition were normalized to measurements made in control experiments performed on age-matched sister neurons. Statistical comparisons were performed by comparing absolute Δ[Na+]i(after) measurements made under a given test condition with measurements made in age-matched sister neurons under control conditions. The asterisk indicates statistical significance (p < 0.05) compared with measurements made in age-matched sister neurons in the absence of treatment. The numbers in parentheses denote the number of neuronal populations (i.e., coverslips) from which the data were obtained. n.d., Not determined.
Data are reported as means ± SEM, with the accompanying n value referring to the number of neuronal populations (i.e., coverslips) from which data were obtained. In studies in which neurons were coloaded with SBFI and either carboxy SNARF-1 or SNARF-5F, experiments were performed on at least three (usually more than five) coverslips obtained from at least three (usually more than four) different batches of neuronal cultures, and the accompanying n value refers to the number of neurons from which data were analyzed.
Intracellular ATP measurements. Cellular ATP content was determined from luciferin-luciferase luminescence, using the ATP determination kit (Molecular Probes). Anoxia was induced in neuronal cultures under conditions identical to those used for the great majority of the microspectrofluorimetric studies (i.e., conditions of constant superfusion at 37°C, pHo 7.35). Before and at fixed time points during and after anoxia, neurons were lysed by the addition of 40 μl of 10 mm Tris buffer, pH 7.5, 0.1 m NaCl, 1 mm EDTA, and 0.01% Triton X-100 in the presence of a protease inhibitor mixture (Roche Diagnostics, Laval, Quebec, Canada). Aliquots (10 μl) were then removed, and sample luminescence was detected with a Berthold LB9507 Lumat luminometer (Fisher Scientific, Ottawa, Ontario, Canada). ATP measurements were made in triplicate on a minimum of four samples at each time point and are reported as percentage declines compared with measurements made before anoxia in paired samples.
Results
Changes in [Na+]i during 5 min anoxia
Characterization of baseline response
Before anoxia, resting [Na+]i was 11 ± 1 mm (n = 444), a value similar to those reported previously in mammalian central neurons both in culture (Rose and Ransom, 1997; Chen et al., 1999; Diarra et al., 2001) and in slice preparations (Pisani et al., 1998; Calabresi et al., 1999). Five-minute anoxia evoked an increase in [Na+]i that began ∼90 sec after the start of anoxia and recovered to resting levels within ∼6-10 min of the return to normoxia (Fig. 1A). As illustrated in Figure 1B, there was a positive correlation between the magnitude of the increase in [Na+]i observed during anoxia and the length of time neurons had been maintained in culture. The increases in [Na+]i seen during anoxia are broadly consistent with those observed previously in a variety of mammalian central neurons in response to anoxia or oxygen-glucose deprivation in vitro (Friedman and Haddad, 1994; Pisani et al., 1998; Calabresi et al., 1999; Diarra et al., 2001) and in CA1 neurons in response to 8 min global ischemia in vivo (Erecińska and Silver, 2001). When anoxia was imposed under reduced [Na+]o, NMDG+-substituted conditions, the increase in [Na+]i was prevented (n = 8) (Fig. 1C), indicating a requirement for Na+ entry. In neurons maintained for either 6-10 or 11-14 DIV, ionotropic glutamate receptor activation did not contribute to the increase in [Na+]i induced by anoxia under the constant superfusion conditions of the present experiments (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Consistent with previous findings that the maintenance of resting [Na+]i in rat hippocampal neurons reflects a balance between Na+,K+-ATPase activity and Na+ influx driven by a steep inward electrochemical gradient for Na+ (Rose and Ransom, 1997), 5 min applications of K+-free medium or 500 μm ouabain under normoxic conditions evoked increases in [Na+]i of 26 ± 5 and 27 ± 4 mm, respectively (n = 6 neuronal cultures at 6-10 DIV in each case) (Fig. 2A). The accumulation of Na+i in neurons during anoxia also reflects a balance between reduced Na+,K+-ATPase activity and ongoing/increased Na+ influx. In the present experiments, increases in [Na+]i during anoxia occurred at times at which ATP levels were reduced; after 3 min anoxia, internal ATP had fallen by 66 ± 7%, with an additional decrease to 24 ± 8% of preanoxic values at the end of 5 min anoxia (Fig. 2B). When neuronal cultures were incubated with 10 mm creatine for 2 hr to increase intracellular phosphocreatine levels (Balestrino et al., 2002), the fall in ATP observed after 3 min anoxia under control conditions was attenuated (Fig. 2B) (Sheldon and Church, 2004) and the increase in [Na+]i observed at this time was reduced by ∼55% compared with that seen in age-matched sister neurons not pretreated with creatine (Fig. 2C). Creatine pretreatment failed to significantly limit either the fall in internal ATP (Fig. 2 B) or the increase in [Na+]i (Fig. 2C) seen after 5 min anoxia. For each of these experimental series, qualitatively similar effects were observed in neurons maintained for 11-14 DIV.
Figure 2.

Contribution of reduced Na+,K+-ATPase activity to the increase in [Na+]i during anoxia. A, Superimposed records of the changes in [Na+]i observed under normoxic conditions in response to 5 min exposures to either K+-free medium (♦) or normal medium (3 mm K+) containing 500 μm ouabain (⋄), as indicated by the bar above the traces. B, Internal ATP levels were determined after 3 and 5 min anoxia induced by exposure to sodium dithionite-containing medium in 6-10 DIV neurons under control conditions (•) or after pretreatment with 10 mm creatine for 2 hr (○). In each experimental group, ATP levels were normalized to preanoxic measurements made in age-matched sister neuronal cultures. The fall in internal ATP levels evoked by 3 min, but not 5 min, anoxia under control conditions was significantly attenuated by creatine pretreatment (*p < 0.05). The numbers in parentheses indicate the number of neuronal populations examined under each experimental condition. C, In neurons pretreated with 10 mm creatine for 2 hr (□), the increase in [Na+]i measured 3 min after the start of anoxia was significantly less than that observed under control conditions (▪; *p < 0.05; n = 6 neuronal cultures at 6-10 DIV in each case). The increases in [Na+]i measured after 5 min anoxia under control conditions and in creatine-treated, age-matched sister neurons were not significantly different (p = 0.57).
Together, these results indicate that the increase in [Na+]i during anoxia under the present experimental conditions reflects reduced Na+,K+-ATPase activity in the face of an ongoing/increased entry of Na+ ions that is not dependent on ionotropic glutamate receptor activation. Therefore, in subsequent experiments, we were able to assess the role of pathways intrinsic to hippocampal neurons in Na+ influx during anoxia.
Voltage-activated Na+ channels
In whole-cell recordings obtained using the perforated patch (amphotericin B) configuration, the resting membrane potential of cultured postnatal rat hippocampal neurons before anoxia was -63 ± 2 mV, and 5 min anoxia evoked a depolarization of 24 ± 4 mV (n = 6), similar to depolarizations observed by others in a variety of isolated mammalian central neurons in response to 5 min anoxia (Haddad and Jiang, 1993) or 30 min metabolic inhibition (Aarts et al., 2003). However, although 1 μm tetrodotoxin (TTX) reduced the increases in [Na+]i evoked by 60 sec applications of 50 mm K+o under normoxic conditions from 14 ± 3 to 2 ± 1 mm (n = 6; p < 0.05) (Rose and Ransom, 1997), it failed to affect the magnitudes of anoxia-induced increases in [Na+]i in neurons maintained for either 6-10 or 11-14 DIV (Table 1, Fig. 3A). Lidocaine (250 μm) also failed to significantly affect the rise in [Na+]i during anoxia (Table 1, Fig. 3A), suggesting that TTX-resistant Na+ channels do not contribute to Na+ influx at this time.
Figure 3.

Contribution of voltage-activated Na+ channels, Na+/Ca2+ exchange, and Na+/K+/2Cl- cotransport to the increase in [Na+]i during anoxia. Shown are superimposed records of the changes in [Na+]i observed in response to 5 min anoxia imposed in age-matched sister neurons under control conditions (•) and in the presence of an experimental treatment (open symbols); pharmacological inhibitors were present throughout the duration of the traces shown. A, Neither 1 μm TTX nor 250 μm lidocaine affected the increase in [Na+]i during anoxia. B, Neither 1 μm KB-R7943 nor 50 μm bepridil exerted a significant effect on the increase in [Na+]i during anoxia. C, A concentration of 100 μm bumetanide significantly reduced the rise in [Na+]i during anoxia in 14 DIV neurons. See also Table 1.
Plasmalemmal Na+/Ca2+ exchange and Na+/K+/2Cl- cotransport
Although the increase in [Ca2+]i observed in cultured postnatal rat hippocampal neurons during 5 min anoxia (Diarra et al., 1999) could activate forward-mode Na+/Ca2+ exchange and thereby contribute to Na+ influx, a rise in [Na+]i could promote reverse-mode operation of the exchanger and thus Na+ efflux. Forward- and reverse-mode operation of the plasmalemmal Na+/Ca2+ exchanger can be inhibited with bepridil (50 μm) and KB-R7943 (1 μm), respectively, whereas higher concentrations of KB-R7943 (10 μm) inhibit both forward and reverse Na+/Ca2+ exchange in rat hippocampal neurons (Breder et al., 2000). In the present study, neither bepridil (50 μm) nor KB-R7943 (1 or 10 μm) significantly influenced the magnitude of the increase in [Na+]i during anoxia (Table 1, Fig. 3B). 7-Chloro-5-(2-chlorophenyl)-1,5-dihydro-4,1-benzothiazepin-2(3H)-one (CGP-37157) (25 μm), which has been reported to inhibit both mitochondrial and plasmalemmal Na+/Ca2+ exchange (Blaustein and Lederer, 1999; Czyż and Kiedrowski, 2003), or the removal of external Ca2+ similarly failed to modulate the increase in [Na+]i during anoxia (Table 1).
To assess the potential contribution of Na+/K+/2Cl- cotransport, neurons were pretreated for 10 min with 100 μm bumetanide, which has previously been found to inhibit Na+/K+/2Cl- cotransport in cultured rat cortical neurons (Sun and Murali, 1999). Given that Na+/K+/2Cl- cotransporter expression increases with time in rat brain (Plotkin et al., 1997) and in cultured neurons (Sun and Murali, 1999), the effects of bumetanide were examined in 6-10 and 11-14 DIV neurons. Bumetanide did not affect resting [Na+]i in either 6-10 (n = 5) or 11-14 (n = 9) DIV neurons (Rose and Ransom, 1997) and failed to significantly influence the increase in [Na+]i during anoxia in 6-10 DIV neurons (Table 1). In contrast, in 11-14 DIV neurons, bumetanide caused an ∼40% reduction in the rise in [Na+]i during anoxia (Table 1, Fig. 3C). Although 100 μm bumetanide may inhibit  exchange and K+/2Cl- cotransport (Russell, 2000), there are no indications that these mechanisms contribute to changes in Na+i homeostasis during brief periods of anoxia or ischemia (Diarra et al., 1999; Sheldon and Church, 2002; Payne et al., 2003).
exchange and K+/2Cl- cotransport (Russell, 2000), there are no indications that these mechanisms contribute to changes in Na+i homeostasis during brief periods of anoxia or ischemia (Diarra et al., 1999; Sheldon and Church, 2002; Payne et al., 2003).
pHi-regulating mechanisms
Proton-equivalent efflux by the major acid extrusion mechanisms present in rat hippocampal neuron somata, Na+/H+ exchange, and Na+-dependent  exchange (Raley-Susman et al., 1991; Schwiening and Boron, 1994; Baxter and Church, 1996) is accompanied by Na+ influx (Canzoniero et al., 1996; Rose and Ransom, 1997; Sheldon et al., 2004).
exchange (Raley-Susman et al., 1991; Schwiening and Boron, 1994; Baxter and Church, 1996) is accompanied by Na+ influx (Canzoniero et al., 1996; Rose and Ransom, 1997; Sheldon et al., 2004).
In the absence of a selective pharmacological inhibitor of Na+/H+ exchange in rat hippocampal neurons (Raley-Susman et al., 1991; Schwiening and Boron, 1994; Baxter and Church, 1996), the transport mechanism was inhibited with harmaline, a nonselective agent that has previously been found to inhibit not only functional Na+/H+ exchange activity in rat hippocampal neurons (Raley-Susman et al., 1991) but also the activities of the Na+/H+ exchanger (NHE) isoforms likely to mediate this activity under conditions of normal osmolarity (i.e., NHE1 and NHE5) (Orlowski, 1993; Attaphitaya et al., 1999; Szabó et al., 2000; Douglas et al., 2001). Initially, we confirmed that pretreatment with 200 μm harmaline inhibited Na+/H+ exchange activity under our experimental conditions by measuring rates of pHi recovery from NH4+-induced internal acid loads imposed under normoxic nominally  -free, HEPES-buffered conditions (see Fig. 7A, inset). However, consistent with previous suggestions (made on the basis of pHi measurements) that Na+/H+ exchange activity in rat hippocampal neurons becomes inhibited soon after the onset of anoxia (Diarra et al., 1999; Sheldon and Church, 2004), harmaline pretreatment failed to limit the rise in [Na+]i during 5 min anoxia in either 6-10 or 11-14 DIV neurons (Table 1).
-free, HEPES-buffered conditions (see Fig. 7A, inset). However, consistent with previous suggestions (made on the basis of pHi measurements) that Na+/H+ exchange activity in rat hippocampal neurons becomes inhibited soon after the onset of anoxia (Diarra et al., 1999; Sheldon and Church, 2004), harmaline pretreatment failed to limit the rise in [Na+]i during 5 min anoxia in either 6-10 or 11-14 DIV neurons (Table 1).
Figure 7.

Effects of maneuvers that modulate Na+/H+ exchange activity on increases in [Na+]i after anoxia (Na+,K+-ATPase inhibited). A, At the end of 5 min anoxia, neurons were exposed to K+-free medium for 7 min. Compared with the increase in [Na+]i seen under control conditions (•), the increase in [Na+]i after anoxia was reduced by 60 min pretreatment with 200 μm harmaline (○). Inset, Rates of pHi recovery from internal acid loads imposed under nominally 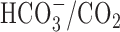 -free, HEPES-buffered normoxic control conditions (•; n = 10) or after 60 min pretreatment with 200 μm harmaline (○; n = 6). Harmaline pretreatment reduced rates of pHi recovery, consistent with inhibition of Na+/H+ exchange activity. B, Compared with the [Na+]i changes evoked by 5 min anoxia under control conditions (•), the increase in [Na+]i after anoxia in age-matched sister neurons was augmented by exposure to 100 μm Zn2+ (□). Exposure to Zn2+ began immediately after the end of 5 min anoxia and continued for the duration of the record shown. C, Summary of the effects of maneuvers that modulate Na+/H+ exchange activity on the increase in [Na+]i after anoxia in 6-10 and 11-14 DIV neurons. The asterisk indicates statistical significance (p < 0.05) compared with measurements made in age-matched sister neurons under control conditions.
-free, HEPES-buffered normoxic control conditions (•; n = 10) or after 60 min pretreatment with 200 μm harmaline (○; n = 6). Harmaline pretreatment reduced rates of pHi recovery, consistent with inhibition of Na+/H+ exchange activity. B, Compared with the [Na+]i changes evoked by 5 min anoxia under control conditions (•), the increase in [Na+]i after anoxia in age-matched sister neurons was augmented by exposure to 100 μm Zn2+ (□). Exposure to Zn2+ began immediately after the end of 5 min anoxia and continued for the duration of the record shown. C, Summary of the effects of maneuvers that modulate Na+/H+ exchange activity on the increase in [Na+]i after anoxia in 6-10 and 11-14 DIV neurons. The asterisk indicates statistical significance (p < 0.05) compared with measurements made in age-matched sister neurons under control conditions.
The potential contribution of 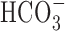 -dependent mechanisms to anoxia-induced increases in [Na+]i was examined by measuring changes in [Na+]i during anoxia under
-dependent mechanisms to anoxia-induced increases in [Na+]i was examined by measuring changes in [Na+]i during anoxia under  -containing conditions. As reported previously (Rose and Ransom, 1997), the transition from a
-containing conditions. As reported previously (Rose and Ransom, 1997), the transition from a  -free, HEPES-buffered medium to a
-free, HEPES-buffered medium to a 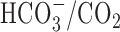 -buffered medium (pHo constant at 7.35) caused a small (∼3 mm) increase in [Na+]i, consistent with the activation of Na+-dependent
-buffered medium (pHo constant at 7.35) caused a small (∼3 mm) increase in [Na+]i, consistent with the activation of Na+-dependent  exchange. However, the increases in [Na+]i during 5 min anoxia imposed in the presence of
exchange. However, the increases in [Na+]i during 5 min anoxia imposed in the presence of  were not significantly different from those observed in age-matched sister neurons at 6-10 or 11-14 DIV under
were not significantly different from those observed in age-matched sister neurons at 6-10 or 11-14 DIV under  -free conditions (Table 1). In addition, the application of 200 μm DIDS under
-free conditions (Table 1). In addition, the application of 200 μm DIDS under 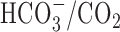 -buffered conditions failed to limit the rise in [Na+]i during anoxia (Table 1), further suggesting that
-buffered conditions failed to limit the rise in [Na+]i during anoxia (Table 1), further suggesting that 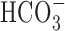 -dependent, pHi-regulating transport mechanisms do not contribute significantly to the increase in [Na+]i during anoxia in rat hippocampal neurons.
-dependent, pHi-regulating transport mechanisms do not contribute significantly to the increase in [Na+]i during anoxia in rat hippocampal neurons.
Nonselective cation channels
During anoxia or ischemia, increases in [Ca2+]i or free radical production can activate nonselective cation channels (NSCCs) that in turn may contribute to the pathogenesis of injury (Chen et al., 1999; Barros et al., 2001; Aarts et al., 2003). To examine the contribution of Na+ influx through NSCCs to the rise in [Na+]i during anoxia, we applied Gd3+ (Caldwell et al., 1998).
Five to 20 min exposures to 30 or 100 μm Gd3+ did not affect resting [Na+]i but reduced the magnitude of the increase in [Na+]i during anoxia in 6-10 DIV neurons by ∼40% (Fig. 4A,B). The finding presented above that the increase in [Na+]i during anoxia was not attenuated in the absence of external Ca2+ suggests that the effect of Gd3+ is unlikely to be mediated via an inhibition of NSCCs activated by increases in [Ca2+]i (we were unable to assess the effect of flufenamate, an inhibitor of Ca2+i-activated NSCCs, on the increase in [Na+]i during anoxia because it evoked variable increases in resting [Na+]i). In addition, neither the broad spectrum voltage-activated Ca2+ channel blocker Ni2+ nor the L-type Ca2+ channel blockers verapamil and nifedipine significantly influenced the increase in [Na+]i during anoxia (Fig. 4B). CNQX (20 μm), applied alone (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) or in the presence of the broad spectrum high-voltage-activated Ca2+ channel blocker loperamide (50 μm) (Church et al., 1994) (Fig. 4B), also failed to influence the increase in [Na+]i during anoxia. Together, these findings indicate that the effect of Gd3+ to limit Na+ influx during anoxia is likely independent of its ability to block voltage-activated Ca2+ channels (Boland et al., 1991) or AMPA/kainate receptors (Lei and MacDonald, 2001) (see also Caldwell et al., 1998).
Figure 4.

Effect of Gd3+ on the increase in [Na+]i during anoxia. A, Compared with the increase observed in age-matched sister neurons under control conditions (•), the increase in [Na+]i during anoxia was reduced in the presence of 30 μm Gd3+ (○). B, Summary of the effects of Gd3+ and the other treatments indicated on the figure on the increase in [Na+]i during anoxia in 6-10 DIV neuronal cultures. C, After 2 hr pretreatment with 1 mm trolox, the increase in [Na+]i during anoxia was reduced (○) compared with that observed in age-matched sister neurons not pretreated with the antioxidant (•). In sister neurons pretreated with 1 mm trolox, 30 μm Gd3+ failed to attenuate the residual increase in [Na+]i during anoxia (⋄). D, Summary of the effects of the test treatments indicated on the figure on the increases in [Na+]i during anoxia in 6-10 DIV neurons. Neurons were exposed to l-NAME or AACOCF3 for 20 min before the start of anoxia. In B and D, the asterisk indicates statistical significance (p < 0.05) compared with measurements made in age-matched sister neurons under control conditions. In neurons pretreated with trolox, there was no statistically significant difference (N.S.; p = 0.54) between the increases in [Na+]i observed during anoxia in the presence and absence of Gd3+.
To examine the potential role of NSCCs activated by reactive oxygen species (ROS) in the effect of Gd3+ to reduce Na+ influx during anoxia, neurons were treated with the antioxidant 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylate (trolox) for 2-3 hr before anoxia. After pretreatment with 1 mm trolox (Chow et al., 1994; Brookes et al., 1998; Vergun et al., 2001; Aarts et al., 2003), the magnitude of the increase in [Na+]i during anoxia in 6-10 DIV neurons was reduced by ∼40% (Fig. 4C). Pretreatment with 200 μm trolox elicited similar effects in both 6-10 DIV neurons (Fig. 4D) and 11-14 DIV neurons (the normalized Δ[Na+]i(during) value after pretreatment with 200 μm trolox in 11-14 DIV neurons was 0.51 ± 0.18; n = 4; p < 0.05). In neurons pretreated with trolox, Gd3+ failed to exert an additional inhibitory effect (Fig. 4C,D), supporting the possibility that NSCCs activated by ROS may contribute to Na+ influx during anoxia. However, neither 20 min pretreatment with 500 μm
NG-nitro-l-arginine methyl ester hydrochloride (l-NAME) or 15-30 μm AACOCF3 significantly affected the increase in [Na+]i (Fig. 4D), suggesting a lack of involvement of nitric oxide synthase (NOS) and cytosolic phospholipase A2 (cPLA2), respectively, in the generation of the ROS involved. We were unable to examine the potential effects of manganese (III) tetrakis (4-benzoic acid) porphyrin (an 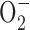 scavenger) or mepacrine (a non-specific PLA2 inhibitor), which were highly fluorescent during excitation at both 334 and 380 nm.
scavenger) or mepacrine (a non-specific PLA2 inhibitor), which were highly fluorescent during excitation at both 334 and 380 nm.
Changes in [Na+]i immediately after 5 min anoxia
Characterization of baseline response
The rise in [Na+]i observed during 5 min anoxia recovered rapidly to resting levels after the return to normoxia (Fig. 1A), presumably reflecting the resumption of Na+,K+-ATPase activity (Ekholm et al., 1993). In the present experiments, internal ATP levels 5 min after the return to normoxia increased by ∼33% (n = 4) from those measured at the end of 5 min anoxia. In addition, inhibition of Na+,K+-ATPase activity (K+o-free conditions for 7 min, starting at reoxygenation) prevented the recovery of [Na+]i seen under control conditions and revealed a secondary increase in [Na+]i that, as for the increase in [Na+]i during anoxia, was positively related to the length of time neurons had been maintained in culture (Fig. 5A,B). Similar results were observed when 500 μm ouabain was used to inhibit Na+,K+-ATPase after anoxia (Fig. 5B). When Na+,K+-ATPase activity was reestablished by perfusion with standard medium containing 3 mm K+, [Na+]i recovered to preanoxic resting values (Fig. 5A). The secondary increase in [Na+]i observed during Na+,K+-ATPase inhibition after anoxia was blocked when NMDG+ was substituted for external Na+ (n = 4) (Fig. 5A). Together, these results indicate that, in the period immediately after 5 min anoxia, Na+,K+-ATPase activity acts to lower [Na+]i to resting levels despite continued Na+ entry. The positive relationship between the rise in [Na+]i (Na+,K+-ATPase blocked) after anoxia and the length of time that neurons had been maintained in culture could reflect, for example, developmental upregulation of the pathways that contribute to Na+ influx after anoxia (see below) or the intracellular signaling cascades that regulate their activities (Durkin et al., 1997). In subsequent experiments, to characterize the pathways that contribute to Na+ influx after 5 min anoxia, Na+,K+-ATPase was blocked for 7 min immediately after the return to normoxia (van Emous et al., 1998).
Ionotropic glutamate receptor-operated and voltage-activated Na+ channels
Analogous to findings made during anoxia, 2 μm MK-801 and 20 μm CNQX failed to significantly affect the increase in [Na+]i observed after anoxia under conditions of Na+,K+-ATPase inhibition (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Similarly, 1 μm TTX failed to reduce the increase in [Na+]i after anoxia in 6-10 and 11-14 DIV neurons (Table 2). TTX (100 μm) and lidocaine (250 μm) were also without significant effects in 6-10 and 11-14 DIV neurons, respectively (Table 2), suggesting that TTX-resistant Na+ channels do not contribute to Na+ influx in the immediate postanoxic period.
Na+/K+/2Cl- cotransport and plasmalemmal Na+/Ca2+ exchange
In contrast to findings made in 11-14 DIV neurons during anoxia (Table 1), 100 μm bumetanide failed to significantly reduce the increase in [Na+]i after anoxia in either 6-10 or 11-14 DIV neuronal cultures (Table 2). The tendency for bumetanide to augment the rise in [Na+]i after anoxia in 11-14, but not 6-10, DIV hippocampal neurons (Table 2) may reflect the finding in cardiac myocytes that Na+/K+/2Cl- cotransport contributes to Na+ efflux at reperfusion (Anderson et al., 1996).
Na+/Ca2+ exchange, operating in forward mode, may mediate Ca2+ efflux after anoxia or ischemia (Breder et al., 2000) and thereby contribute to Na+ influx at this time. Conversely, the present experimental conditions (in which [Na+]i after anoxia was maintained at a relatively high level) could favor reverse-mode operation of the exchange mechanism and thus Na+ efflux (Czyż et al., 2002). Applied immediately after anoxia under conditions in which Na+,K+-ATPase was inhibited, 50 μm bepridil significantly reduced the increase in [Na+]i (Table 2, Fig. 6), suggesting that forward-mode Na+/Ca2+ exchange contributes to Na+ influx immediately after anoxia. In contrast, KB-R7943 (1 μm) or the removal of external Ca2+ at reoxygenation enhanced the increase in [Na+]i (Table 2, Fig. 6), suggesting that some Na+/Ca2+ exchangers may be operating in reverse mode to promote Na+ efflux at this time. Finally, when applied at 10 μm to inhibit both forward- and reverse-mode Na+/Ca2+ exchange, KB-R7943 reduced the increase in [Na+]i after anoxia compared with the increase observed in age-matched sister neurons under control conditions (Table 2, Fig. 6). Together, these results are consistent with those of White and Reynolds (1995), who reported considerable variability in the contribution of forward-mode Na+/Ca2+ exchange to the recovery of [Ca2+]i after glutamate stimulation in rat forebrain neurons, and those of Yu and Choi (1997), who suggested the possibility that both forward and reverse Na+/Ca2+ exchange can take place concurrently in the same cell. These issues are considered further in Discussion.
Figure 6.

Effects of pharmacological modulators of Na+/Ca2+ exchange activity on increases in [Na+]i after anoxia (Na+,K+-ATPase inhibited). Neurons were exposed to K+-free medium for 7 min at the end of 5 min anoxia. Compared with the increase in [Na+]i observed under control conditions (-), the increase in [Na+]i after anoxia was reduced in the presence of 50 μm bepridil (•) or 10 μm KB-R7943 (□) and was enhanced in the presence of 1 μm KB-R7943 (○). Pharmacological treatments began at the end of 5 min anoxia and were continued for the duration of the records shown. See also Table 2.
Na+/H+ exchange
Although Na+/H+ exchange in rat hippocampal neurons is inhibited during anoxia, pHi measurements indicate that the transporter is activated immediately after reoxygenation (Diarra et al., 1999; Sheldon and Church, 2002) and thus may contribute to Na+ influx at this time. Consistent with this possibility, harmaline pretreatment significantly reduced the increase in [Na+]i after anoxia in 6-10 and 11-14 DIV neurons (Fig. 7A,C). Although harmaline also affects voltage-gated Na+ channels (Deecher et al., 1992) and Na+,K+-ATPase (Kim et al., 1988), conventional inhibitors of voltage-gated Na+ channels failed to influence the rise in [Na+]i after anoxia (see above), and harmaline reduced Na+ entry after anoxia under conditions in which Na+,K+-ATPase activity was already blocked. Intracellular pH measurements also indicate that Na+/H+ exchange activity in rat hippocampal neurons after anoxia is attenuated at pHo 6.60 or by inhibition of the cAMP-protein kinase A (PKA) pathway and is augmented at pHo 7.80 (Diarra et al., 1999; Sheldon and Church, 2002). In the present study, pHo 6.60 conditions or the PKA inhibitor Rp-cAMPS reduced the rise in [Na+]i after anoxia, whereas, at pHo 7.80, the increase in [Na+]i was enhanced (Fig. 7C). We also reported previously that a Zn2+-sensitive H+ efflux pathway contributes to acid extrusion after anoxia in rat hippocampal neurons and that inhibition of this pathway increases the observable contribution of Na+/H+ exchange to acid extrusion at this time (Diarra et al., 1999; Sheldon and Church, 2002) (see also Demaurex et al., 1995). In the present experiments, 100 μm Zn2+ failed to influence the increase in [Na+]i during anoxia [i.e., at a time when Na+/H+ exchange is inhibited; normalized Δ[Na+]i(during) values in the presence of 100 μm Zn2+ were 1.03 ± 0.18 (n = 12; p = 0.38) and 1.17 ± 0.15 (n = 15; p = 0.62) in 6-10 and 11-14 DIV neurons, respectively]. In contrast, applied immediately after anoxia under K+o-free conditions, Zn2+ significantly enhanced the increase in [Na+]i (Fig. 7B,C), lending additional support to the possibility that Na+/H+ exchange contributes to Na+ influx immediately after anoxia. Although Zn2+ can modulate the activities of a variety of ion channels and transport mechanisms (Harrison and Gibbons, 1994), under the conditions used here, anoxia-evoked changes in [Na+]i were unaffected by NMDA or AMPA receptor antagonists or blockers of voltage-activated Ca2+ channels, and the effect of Zn2+ on the increase in [Na+]i after anoxia was observed when Na+,K+-ATPase activity was already inhibited (cf. Manzerra et al., 2001).
Although the above findings are consistent with an important contribution from Na+/H+ exchange to Na+ influx in the period immediately after anoxia, they are limited by the lack of selectivity of harmaline and the other maneuvers used to modulate Na+/H+ exchange activity at this time. Therefore, to further explore the role of Na+/H+ exchange in Na+ influx after anoxia, we examined directly the relationship between the changes in [Na+]i and pHi evoked by anoxia by measuring [Na+]i and pHi concurrently in individual neurons (Sheldon et al., 2004). In neurons coloaded with SBFI and either carboxy SNARF-1 or SNARF-5F, 5 min anoxia evoked a 16 ± 2 mm increase in [Na+]i and a 0.37 ± 0.04 pH unit fall in pHi (n = 13 neurons at 7-10 DIV), both of which recovered to near resting values on the return to normoxia (Fig. 8A). Perfusion with K+-free medium for 7 min immediately after anoxia did not affect the recovery of pHi (Fig. 8, compare A, B); however, consistent with changes seen in neurons single-loaded with SBFI (Fig. 5A), inhibition of Na+,K+-ATPase revealed an additional rise in [Na+]i after anoxia of 26 ± 2 mm (Fig. 8B) (n = 49 neurons at 7-10 DIV). For both of these experimental series, qualitatively similar changes were observed in neurons maintained for 11-14 DIV (data not shown). If Na+/H+ exchange contributes to increases in [Na+]i and pHi in the immediate postanoxic period (Na+,K+-ATPase blocked), both events should be dependent on external Na+, and the rates at which pHi and [Na+]i increase should exhibit an inverse dependency on pHi. Indeed, as illustrated in Figure 8C, the increases in pHi and [Na+]i after 5 min anoxia were abolished under reduced [Na+]o, NMDG+-substituted conditions; when [Na+]o was returned to normal (in the continued absence of K+o), both pHi and [Na+]i rapidly increased (n = 6). In addition, when rates at which [Na+]i and pHi increased after anoxia were plotted as functions of absolute pHi in 7-10 (Fig. 8D) and 11-14 (Fig. 8E) DIV neurons, both parameters were faster at lower pHi values.
Finally, we used neurons coloaded with SBFI and a SNARF derivative to examine concurrently the effects on [Na+]i and pHi of maneuvers previously found (Diarra et al., 1999; Sheldon and Church, 2002) to limit the activation of Na+/H+ exchange after 5 min anoxia. Consistent with findings that Na+/H+ exchange is inhibited during anoxia, pretreatment with harmaline failed to influence the magnitudes of the increase in [Na+]i or decrease in pHi during anoxia (Fig. 9A); in contrast, harmaline significantly reduced the increases in [Na+]i and pHi after anoxia (Fig. 9B). In further support of a contribution from Na+/H+ exchange to Na+ influx (and H+ efflux) in the immediate postanoxic period, lowering pHo to 6.60 or the application of 50 μm Rp-cAMPS also reduced the increases in [Na+]i and pHi after anoxia (Fig. 9B). Although PKA regulates Na+/Ca2+ exchange activity in rat hippocampal neurons under normoxic conditions (Blaustein and Lederer, 1999), the effect of Rp-cAMPS to reduce the increase in [Na+]i after anoxia was accompanied by a concomitant reduction in the postanoxic increase in pHi and, as shown in Figure 9B, was not mimicked when anoxia was imposed under Ca2+o-free conditions. In the latter experiments (compare the 0 Ca2+o experiments reported in Table 2), Ca2+o was absent before, during, and after anoxia to prevent the rise in [Ca2+]i during anoxia (Diarra et al., 1999; Sheldon and Church, 2002) and inhibit both forward and reverse Na+/Ca2+ exchange in the immediate post-anoxic period. In addition, we have shown previously that this maneuver does not influence anoxia-evoked changes in pHi in rat hippocampal neurons (Diarra et al., 1999; Sheldon and Church, 2002).
Figure 9.

The influence of maneuvers that inhibit Na+/H+ exchange activity on anoxia-evoked changes in [Na+]i and pHi measured concurrently in neurons coloaded with SBFI and either carboxy SNARF-1 or SNARF-5F. Measurements of the changes in [Na+]i and pHi observed during (A) and after (B) anoxia under the test conditions indicated on the figure were normalized to corresponding measurements made in experiments performed on age-matched sister neurons (7-10 DIV) under control conditions. Statistical comparisons were performed by comparing absolute measurements of anoxia-evoked changes in [Na+]i and pHi made under a given test condition with corresponding measurements made in age-matched sister neurons under control conditions. The asterisk indicates statistical significance (p < 0.05) compared with measurements made in age-matched sister neurons under control conditions. The numbers in parentheses indicate the number of neurons from which data were obtained under each test condition.
 -dependent, pHi-regulating mechanisms
-dependent, pHi-regulating mechanisms
The increase in [Na+]i after anoxia (Na+,K+-ATPase inhibited) was consistently greater under 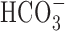 -containing versus
-containing versus  -free, HEPES-buffered conditions in 11-14, but not 6-10, DIV neurons (Fig. 10). Consistent with the possibility that
-free, HEPES-buffered conditions in 11-14, but not 6-10, DIV neurons (Fig. 10). Consistent with the possibility that  -dependent transport mechanisms(s) may contribute to Na+ influx in the immediate postanoxic period in 11-14 DIV neurons, the augmented increase in [Na+]i observed in the presence of
-dependent transport mechanisms(s) may contribute to Na+ influx in the immediate postanoxic period in 11-14 DIV neurons, the augmented increase in [Na+]i observed in the presence of  was blocked by 200 μm DIDS (Fig. 10). Interestingly, 200 μm DIDS also reduced slightly, albeit significantly, the increase in [Na+]i observed after anoxia under HEPES-buffered conditions in 11-14 DIV neurons (Fig. 10B), a finding that may reflect residual activity of
was blocked by 200 μm DIDS (Fig. 10). Interestingly, 200 μm DIDS also reduced slightly, albeit significantly, the increase in [Na+]i observed after anoxia under HEPES-buffered conditions in 11-14 DIV neurons (Fig. 10B), a finding that may reflect residual activity of 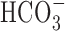 -dependent, Na+-transporting mechanisms in the nominal absence of
-dependent, Na+-transporting mechanisms in the nominal absence of 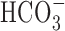 (Wu et al., 1994; Deitmer and Schneider, 1998) or the effects of DIDS on
(Wu et al., 1994; Deitmer and Schneider, 1998) or the effects of DIDS on  -independent processes potentially involved in the neuronal response to anoxia (e.g., free radical release from mitochondria) (Han et al., 2003; Tauskela et al., 2003).
-independent processes potentially involved in the neuronal response to anoxia (e.g., free radical release from mitochondria) (Han et al., 2003; Tauskela et al., 2003).
Figure 10.

Contribution of 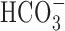 -dependent mechanisms to increases in [Na+]i after anoxia (Na+,K+-ATPase inhibited). A, Under HEPES-buffered (•) or
-dependent mechanisms to increases in [Na+]i after anoxia (Na+,K+-ATPase inhibited). A, Under HEPES-buffered (•) or  -buffered (○) conditions, neurons were exposed to 5 min anoxia, followed by 7 min perfusion with K+-free medium. The augmented increase in [Na+]i observed after anoxia in the presence of
-buffered (○) conditions, neurons were exposed to 5 min anoxia, followed by 7 min perfusion with K+-free medium. The augmented increase in [Na+]i observed after anoxia in the presence of 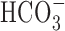 was inhibited by 200 μm DIDS (⋄), which was added at the start of anoxia and remained present throughout the rest of the record shown. B, Summary of the effects of external buffering conditions and DIDS on the increase in [Na+]i after anoxia in 6-10 and 11-14 DIV neurons. In contrast to results obtained in 6-10 DIV neurons, the increase in [Na+]i after anoxia in 11-14 DIV neurons was enhanced in the presence of
was inhibited by 200 μm DIDS (⋄), which was added at the start of anoxia and remained present throughout the rest of the record shown. B, Summary of the effects of external buffering conditions and DIDS on the increase in [Na+]i after anoxia in 6-10 and 11-14 DIV neurons. In contrast to results obtained in 6-10 DIV neurons, the increase in [Na+]i after anoxia in 11-14 DIV neurons was enhanced in the presence of  and, under this condition, DIDS significantly reduced (†p < 0.05) the magnitude of the rise in [Na+]i to a value not significantly different (p = 0.66) to that seen under
and, under this condition, DIDS significantly reduced (†p < 0.05) the magnitude of the rise in [Na+]i to a value not significantly different (p = 0.66) to that seen under 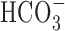 -free, HEPES-buffered conditions. In 11-14 DIV neurons, DIDS also significantly (*p < 0.05) reduced the increase in [Na+]i observed after anoxia under nominally
-free, HEPES-buffered conditions. In 11-14 DIV neurons, DIDS also significantly (*p < 0.05) reduced the increase in [Na+]i observed after anoxia under nominally  -free, HEPES-buffered conditions. There was no significant difference (p = 0.15) between the increases in [Na+]i after anoxia observed in the presence of DIDS under
-free, HEPES-buffered conditions. There was no significant difference (p = 0.15) between the increases in [Na+]i after anoxia observed in the presence of DIDS under  -containing versus nominally
-containing versus nominally  -free conditions.
-free conditions.
Nonselective cation channels
As detailed above, Gd3+ significantly reduced Na+ influx during anoxia via a mechanism(s) that appeared dependent on the production of ROS. Because ROS production is enhanced after reoxygenation, we applied Gd3+ immediately after anoxia under K+o-free conditions. As illustrated in Figure 11, 30 μm Gd3+ significantly reduced the magnitude of the increase in [Na+]i after anoxia in 6-10 DIV neurons; a similar effect was observed in 11-14 DIV neurons, in which 30 μm Gd3+ reduced the normalized Δ[Na+]i(after) value to 0.73 ± 0.08 (n = 6; p < 0.05). Similar to findings made during anoxia, the effect of Gd3+ to limit increases in [Na+]i after anoxia was occluded by 2 hr pretreatment with 1 mm trolox (Fig. 11B). Although 500 μm l-NAME was without effect, 15-30 μm AACOCF3 significantly reduced the increase in [Na+]i after anoxia (Fig. 11B), suggesting that ROS generated via the cPLA2-arachidonic acid pathway may play a role in regulating Na+ influx in the immediate postanoxic period, possibly by modulating the activity of a Gd3+-sensitive NSCC.
Figure 11.

Effects of Gd3+ on increases in [Na+]i after anoxia (Na+,K+-ATPase inhibited). A, Exposure to 30 μm Gd3+ immediately after anoxia under K+o-free conditions (○) reduced the increase in [Na+]i after anoxia compared with that observed in age-matched sister neurons under control conditions (•). B, Summary of the effects of the test treatments indicated on the figure on the increases in [Na+]i after anoxia in 6-10 DIV neuronal cultures. Neurons were pretreated with trolox for 2 hr before the start of anoxia and with l-NAME or AACOCF3 for 20 min before the start of anoxia. The asterisk indicates statistical significance (p < 0.05) compared with measurements made in age-matched sister neurons under control conditions. N.S., No statistically significant difference (p = 0.67) between the increase in [Na+]i observed after anoxia in the absence and presence of Gd3+ in neurons pretreated with trolox.
Discussion
In the present study, we used isolated neurons to characterize pathways independent from glutamate receptor activation that, together with excitotoxic mechanisms (see Introduction), contribute to Na+ influx during and after anoxia. Not only were different complements of mechanisms involved during versus after anoxia but also Na+ influx during and after anoxia became more pronounced the longer that neurons were maintained in culture. The latter observations correspond with previous findings (Jiang et al., 1992) that anoxia-induced falls in [Na+]o are smaller in slices taken from neonatal versus adult rats and suggest that differences in Na+, as well as Ca2+ (Rothman, 1983; Di Loreto and Balestrino, 1997), entry may contribute to the enhanced vulnerability of more phenotypically mature neurons.
We found no evidence to suggest that voltage-activated Na+ channels contribute to Na+ influx during or after 5 min anoxia (compare prolonged hypoxia) (Banasiak et al., 2004). Although the relatively small depolarization that occurred during anoxia may have limited the contribution of Na+ channels, even in studies in which more severe insults have been imposed and more marked depolarizations have occurred, TTX has failed to limit increases in [Na+]i (Pisani et al., 1998; Calabresi et al., 1999; Chen et al., 1999; Müller and Somjen, 2000). Thus, our findings support suggestions that the protective actions of Na+ channel blockers may be mediated presynaptically and that much of the Na+ that enters neurons during anoxia or ischemia must do so through a TTX-insensitive mechanism(s) (Kimura et al., 1998; Müller and Somjen, 2000).
Although Na+/K+/2Cl- cotransport did not play a role in regulating resting [Na+]i (see also Rose and Ransom, 1997), it contributed to Na+ influx during anoxia in neurons expected to express significant levels of functional transporters (i.e., neurons ≥11 DIV) (Sun and Murali, 1999) (see also Plotkin et al., 1997). Similarly, Na+/K+/2Cl- cotransport contributes to Na+i accumulation induced by metabolic inhibition in astrocytes (Lenart et al., 2004) and cardiac myocytes (Anderson et al., 1996). In contrast, bumetanide failed to affect Na+ influx after anoxia in 6-10 and 11-14 DIV neurons. Although the basis for the differential contribution of Na+/K+/2Cl- cotransport to Na+ influx during versus after anoxia in 11-14 DIV neurons remains unknown, inward Na+/K+/2Cl- cotransport after anoxia may be limited by the high [Na+]i and, presumably, [Cl-]i seen at this time as well as anoxia-induced changes in the activities of second-messenger systems that in turn regulate the activity of the transport mechanism and that are themselves developmentally regulated (Durkin et al., 1997; Flatman, 2002; Lenart et al., 2004). Together, our observations parallel findings that bumetanide reduces infarct size when applied during but not after ischemia (Yan et al., 2003) and suggest that the protective effects of bumetanide may in part reflect a reduction in Na+ influx during the insult.
In contrast to Na+/K+/2Cl- cotransport, Na+/Ca2+ exchange, Na+/H+ exchange, and the 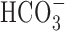 -dependent mechanism(s) did not contribute to Na+ influx during anoxia. These observations are consistent with findings that Na+/Ca2+ exchange does not contribute to [Na+]i and/or [Ca2+]i changes during metabolic inhibition in rat neocortical (Pisani et al., 1998) or cerebellar granule (Chen et al., 1999) neurons and that Na+/H+ exchange and Na+-dependent
-dependent mechanism(s) did not contribute to Na+ influx during anoxia. These observations are consistent with findings that Na+/Ca2+ exchange does not contribute to [Na+]i and/or [Ca2+]i changes during metabolic inhibition in rat neocortical (Pisani et al., 1998) or cerebellar granule (Chen et al., 1999) neurons and that Na+/H+ exchange and Na+-dependent  exchange do not contribute to pHi changes during anoxia in rat hippocampal neurons (Diarra et al., 1999; Sheldon and Church, 2004). The lack of involvement of Na+/Ca2+, Na+/H+, and Na+-dependent
exchange do not contribute to pHi changes during anoxia in rat hippocampal neurons (Diarra et al., 1999; Sheldon and Church, 2004). The lack of involvement of Na+/Ca2+, Na+/H+, and Na+-dependent  exchange in Na+,Ca2+ and/or pHi fluxes during anoxia contrasts with their activation by glutamate or NMDA (Hartley and Dubinsky, 1993; Canzoniero et al., 1996; Yu and Choi, 1997) and may reflect the inhibitory effects of reductions in internal ATP and/or phosphatidylinositol 4,5-bisphosphate levels on the activities of each of these mechanisms (Blaustein and Lederer, 1999; Romero et al., 2004; Sheldon and Church, 2004).
exchange in Na+,Ca2+ and/or pHi fluxes during anoxia contrasts with their activation by glutamate or NMDA (Hartley and Dubinsky, 1993; Canzoniero et al., 1996; Yu and Choi, 1997) and may reflect the inhibitory effects of reductions in internal ATP and/or phosphatidylinositol 4,5-bisphosphate levels on the activities of each of these mechanisms (Blaustein and Lederer, 1999; Romero et al., 2004; Sheldon and Church, 2004).
Counter to findings made during anoxia, Na+/Ca2+ exchange, Na+/H+ exchange, and the  -dependent mechanism(s) contributed to Na+ fluxes immediately after anoxia. With regard to Na+/Ca2+ exchange, measurements of [Na+]i (present study) and [Ca2+]i (Diarra et al., 1999) suggest that the reversal potential of 3Na+/1Ca2+ exchange at the return to normoxia is approximately -90 mV [i.e., more negative than the membrane potential at the end of 5 min anoxia (approximately -40 mV)]. Also consistent with a contribution from reverse Na+/Ca2+ exchange to Na+ efflux after anoxia, 1 μm KB-R7943 enhanced the increase in [Na+]i at this time. However, when KB-R7943 was applied at 10 μm, a concentration that inhibits forward and reverse Na+/Ca2+ exchange, the increase in [Na+]i after anoxia was reduced, an effect that was mimicked by bepridil. These observations are consistent with the possibility (Yu and Choi, 1997) that forward and reverse Na+/Ca2+ exchange can take place concomitantly in the same cell, perhaps reflecting differences in the distribution of Ca2+ and/or Na+ in submembrane microdomains. Simultaneous measurements of [Na+]i and [Ca2+]i with appropriate spatial resolution in combination with the control of membrane potential will be required to substantiate or refute this possibility. The present results also extend previous findings, made on the basis of pHi measurements, that Na+/H+ exchange in rat hippocampal neurons is activated immediately after anoxia (Diarra et al., 1999; Sheldon and Church, 2002) (see also Vornov et al., 1996; Kintner et al., 2004) and indicate that Na+/H+ exchange is an important determinant of Na+ influx at this time. In cardiac myocytes, activation of Na+/H+ exchange immediately after anoxia or ischemia promotes reverse Na+/Ca2+ exchange and Ca2+i loading (Avkiran, 2001). Not only might a similar sequence of events occur in rat hippocampal neurons but also the rapid rise in pHi consequent on the activation of Na+/H+ exchange after anoxia stimulates PLA2 (Phillis and O'Regan, 2004) that in turn may promote additional Na+ entry via the production of ROS (see below). Our findings support the possibility (Vornov et al., 1996) that the neuroprotective effects of Na+/H+ exchange inhibitors are mediated, not during anoxia but by limiting Na+ influx and/or H+ efflux in the immediate postanoxic period, and parallel reports that Na+/H+ exchange inhibitors are neuroprotective in vivo only when administered before reperfusion (Phillis et al., 1999; Horikawa et al., 2001a,b). Finally, although an
-dependent mechanism(s) contributed to Na+ fluxes immediately after anoxia. With regard to Na+/Ca2+ exchange, measurements of [Na+]i (present study) and [Ca2+]i (Diarra et al., 1999) suggest that the reversal potential of 3Na+/1Ca2+ exchange at the return to normoxia is approximately -90 mV [i.e., more negative than the membrane potential at the end of 5 min anoxia (approximately -40 mV)]. Also consistent with a contribution from reverse Na+/Ca2+ exchange to Na+ efflux after anoxia, 1 μm KB-R7943 enhanced the increase in [Na+]i at this time. However, when KB-R7943 was applied at 10 μm, a concentration that inhibits forward and reverse Na+/Ca2+ exchange, the increase in [Na+]i after anoxia was reduced, an effect that was mimicked by bepridil. These observations are consistent with the possibility (Yu and Choi, 1997) that forward and reverse Na+/Ca2+ exchange can take place concomitantly in the same cell, perhaps reflecting differences in the distribution of Ca2+ and/or Na+ in submembrane microdomains. Simultaneous measurements of [Na+]i and [Ca2+]i with appropriate spatial resolution in combination with the control of membrane potential will be required to substantiate or refute this possibility. The present results also extend previous findings, made on the basis of pHi measurements, that Na+/H+ exchange in rat hippocampal neurons is activated immediately after anoxia (Diarra et al., 1999; Sheldon and Church, 2002) (see also Vornov et al., 1996; Kintner et al., 2004) and indicate that Na+/H+ exchange is an important determinant of Na+ influx at this time. In cardiac myocytes, activation of Na+/H+ exchange immediately after anoxia or ischemia promotes reverse Na+/Ca2+ exchange and Ca2+i loading (Avkiran, 2001). Not only might a similar sequence of events occur in rat hippocampal neurons but also the rapid rise in pHi consequent on the activation of Na+/H+ exchange after anoxia stimulates PLA2 (Phillis and O'Regan, 2004) that in turn may promote additional Na+ entry via the production of ROS (see below). Our findings support the possibility (Vornov et al., 1996) that the neuroprotective effects of Na+/H+ exchange inhibitors are mediated, not during anoxia but by limiting Na+ influx and/or H+ efflux in the immediate postanoxic period, and parallel reports that Na+/H+ exchange inhibitors are neuroprotective in vivo only when administered before reperfusion (Phillis et al., 1999; Horikawa et al., 2001a,b). Finally, although an 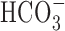 -dependent mechanism(s) contributed to Na+ influx after anoxia in 11-14, but not 6-10, DIV neurons, its identity remains unclear. The sensitivity of the mechanism(s) to DIDS and the inability of TTX and lidocaine to inhibit Na+ influx after anoxia appear to exclude an effect of
-dependent mechanism(s) contributed to Na+ influx after anoxia in 11-14, but not 6-10, DIV neurons, its identity remains unclear. The sensitivity of the mechanism(s) to DIDS and the inability of TTX and lidocaine to inhibit Na+ influx after anoxia appear to exclude an effect of 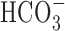 ions on Na+ currents (Bruehl and Witte, 2003). In addition, although Na+-dependent
ions on Na+ currents (Bruehl and Witte, 2003). In addition, although Na+-dependent  exchange and electroneutral or inward electrogenic
exchange and electroneutral or inward electrogenic 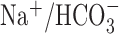 cotransport are sensitive to DIDS, mediate Na+ influx, and are developmentally upregulated (Douglas et al., 2001; Giffard et al., 2003; Romero et al., 2004), they act to increase pHi, which is at odds with previous findings that
cotransport are sensitive to DIDS, mediate Na+ influx, and are developmentally upregulated (Douglas et al., 2001; Giffard et al., 2003; Romero et al., 2004), they act to increase pHi, which is at odds with previous findings that  limits H+ efflux from rat hippocampal neurons after 5 min anoxia (Diarra et al., 1999; Sheldon and Church, 2002). Rather, the
limits H+ efflux from rat hippocampal neurons after 5 min anoxia (Diarra et al., 1999; Sheldon and Church, 2002). Rather, the 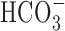 -induced increase in Na+ influx at reoxygenation may reflect the net effect of the activities of multiple Na+- and
-induced increase in Na+ influx at reoxygenation may reflect the net effect of the activities of multiple Na+- and 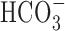 -dependent, DIDS-sensitive transport mechanisms, including outward electrogenic
-dependent, DIDS-sensitive transport mechanisms, including outward electrogenic  cotransport. Additional experiments will be required to establish precisely which mechanisms are involved and to assess whether their inhibition contributes to the neuroprotective properties of DIDS (Tauskela et al., 2003).
cotransport. Additional experiments will be required to establish precisely which mechanisms are involved and to assess whether their inhibition contributes to the neuroprotective properties of DIDS (Tauskela et al., 2003).
Finally, Gd3+ attenuated Na+ influx during and after anoxia. Although the effects of Gd3+ could not be ascribed to inhibition of AMPA/kainate receptors or Ca2+i-activated NSCCs, they were occluded by pretreatment with trolox, suggesting that the Gd3+-sensitive pathway mediating Na+ influx may be activated by ROS. Although Gd3+ also limits Ca2+ influx induced by 5 min anoxia under conditions identical to those used here (A. Diarra and J. Church, unpublished observations) (see also Diarra et al., 1999), the pathway involved in the production of ROS in the present experiments differs from that implicated in the activation of the Gd3+-sensitive NSCC that contributes to Ca2+ influx during prolonged (>30 min) O2-glucose deprivation in murine cortical neurons (Aarts et al., 2003) in that it does not appear to depend on NOS. Although additional experiments are required to identify the pathway(s) involved in the generation of ROS during anoxia (including possible contributions from NO generated via NOS-independent pathways), cPLA2 may be involved after anoxia [compare rat cerebellar granule neurons in which PLA2, acting through the production of ROS, is an important determinant of Na+ influx during metabolic inhibition (Chen et al., 1999)].
In summary, Na+ influx occurs during and immediately after a short period of anoxia in rat hippocampal neurons. Sodium influx during anoxia may contribute to acute neuronal injury, whereas Na+ influx after anoxia may be a factor in the progression of injury that occurs in the reperfusion period. Our findings emphasize that multiple pathways, the relative contributions of which vary during versus immediately after anoxia, contribute to Na+ influx induced by anoxia in rat hippocampal neurons and suggest that combination therapies may prove more useful than strategies directed toward individual pathways in reducing the potentially detrimental Na+ influx that occurs in response to metabolic inhibition.
Footnotes
This work was supported by a grant-in-aid from the Heart and Stroke Foundation of British Columbia and Yukon. C.S. is a recipient of the Michael Smith Foundation for Health Research Doctoral Research Trainee and the Canadian Institutes of Health Research MD/PhD Studentship awards. Y.M.C. is a recipient of the Michael Smith Foundation for Health Research Junior Graduate Studentship and Natural Sciences and Engineering Research Council of Canada Graduate Scholarship awards.
Correspondence should be addressed to Dr. John Church, Department of Anatomy and Cell Biology, University of British Columbia, 2177 Wesbrook Mall, Vancouver, British Columbia, Canada V6T 1Z3. E-mail: jchurch@interchange.ubc.ca.
A. Diarra's present address: Marine Biological Laboratory, 7 MBL Street, Woods Hole, MA 02543.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2411057-13$15.00/0
References
- Aarts M, Iihara K, Wei W-L, Xiong K-G, Arundine M, Cerwinski W, MacDonald JF, Tymianski M (2003) A key role for TRPM7 channels in anoxic neuronal death. Cell 115: 863-877. [DOI] [PubMed] [Google Scholar]
- Anderson SE, Dickinson CZ, Liu H, Cala PM (1996) Effects of Na-K-2Cl cotransport inhibition on myocardial Na and Ca during ischemia and reperfusion. Am J Physiol 270: C608-C618. [DOI] [PubMed] [Google Scholar]
- Attaphitaya S, Park K, Melvin JE (1999) Molecular cloning and functional expression of a rat Na+/H+ exchanger (NHE5) highly expressed in brain. J Biol Chem 274: 4383-4388. [DOI] [PubMed] [Google Scholar]
- Avkiran M (2001) Protection of the ischemic myocardium by Na+/H+ exchange inhibitors: potential mechanisms of action. Basic Res Cardiol 96: 306-311. [DOI] [PubMed] [Google Scholar]
- Balestrino M, Lensman M, Parodi M, Perasso L, Rebaudo R, Melani R, Polenov S, Cupello A (2002) Role of creatine and phosphocreatine in neuronal protection from anoxic and ischemic damage. Amino Acids 23: 221-229. [DOI] [PubMed] [Google Scholar]
- Banasiak KJ, Burenkova O, Haddad GG (2004) Activation of voltage-sensitive sodium channels during oxygen deprivation leads to apoptotic neuronal death. Neuroscience 126: 31-44. [DOI] [PubMed] [Google Scholar]
- Barros LF, Stutzin A, Calixto A, Catalán M, Castro J, Hetz C, Hermosilla T (2001) Nonselective cation channels as effectors of free radical-induced rat liver cell necrosis. Hepatology 33: 114-122. [DOI] [PubMed] [Google Scholar]
- Baxter KA, Church J (1996) Characterization of acid extrusion mechanisms in cultured fetal rat hippocampal neurons. J Physiol (Lond) 23: 457-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein MP, Lederer WJ (1999) Sodium/calcium exchange: its physiological implications. Physiol Rev 79: 763-854. [DOI] [PubMed] [Google Scholar]
- Boland LM, Brown TA, Dingledine R (1991) Gadolinium block of calcium channels: influence of bicarbonate. Brain Res 563: 142-150. [DOI] [PubMed] [Google Scholar]
- Breder J, Sabelhau CF, Opitz T, Reymann KG, Schröder UH (2000) Inhibition of different pathways influencing Na+ homeostasis protects organotypic hippocampal slice cultures from hypoxic/hypoglycemic injury. Neuropharmacology 39: 1779-1787. [DOI] [PubMed] [Google Scholar]
- Brookes PS, Land JM, Clark JB, Heales SJR (1998) Peroxynitrite and brain mitochondria: evidence for increased proton leak. J Neurochem 70: 2195-2202. [DOI] [PubMed] [Google Scholar]
- Bruehl G, Witte OW (2003) Relation between bicarbonate concentration and voltage dependence of sodium currents in freshly isolated CA1 neurons of the rat. J Neurophysiol 89: 2489-2498. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Marfia GA, Centonze D, Pisani A, Bernardi G (1999) Sodium influx plays a major role in membrane depolarization induced by oxygen and glucose deprivation in rat striatal spiny neurons. Stroke 30: 171-178. [DOI] [PubMed] [Google Scholar]
- Caldwell RA, Clemo HF, Baumgarten CM (1998) Using gadolinium to identify stretch-activated channels: technical considerations. Am J Physiol 275: C619-C621. [DOI] [PubMed] [Google Scholar]
- Canzoniero LM, Sensi SL, Choi DW (1996) Recovery from NMDA-induced intracellular acidification is delayed and dependent on extracellular bicarbonate. Am J Physiol 270: C593-C599. [DOI] [PubMed] [Google Scholar]
- Carmeliet E (1999) Cardiac ionic currents and acute ischemia: from channels to arrhythmias. Physiol Rev 79: 917-1017. [DOI] [PubMed] [Google Scholar]
- Chen W-H, Chu K-C, Wu S-J, Wu J-C, Shui H-A, Wu M-L (1999) Early metabolic inhibition-induced intracellular sodium and calcium increase in rat cerebellar granule cells. J Physiol (Lond) 515: 133-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chidekel AS, Friedman JE, Haddad GG (1997) Anoxia-induced neuronal injury: role of Na+ entry and Na+-dependent transport. Exp Neurol 146: 403-413. [DOI] [PubMed] [Google Scholar]
- Chow HS, Lynch III JJ, Rose K, Chow DW (1994) Trolox attenuates cortical neuronal injury induced by iron, ultraviolet light, glucose deprivation, or AMPA. Brain Res 639: 102-108. [DOI] [PubMed] [Google Scholar]
- Chinopoulos C, Tretter L, Rozsa A, Adam-Vizi V (2000) Exacerbated responses to oxidative stress by an Na+ load in isolated nerve terminals: the role of ATP depletion and rise of [Ca2+]i J Neurosci 20: 2094-2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church J, Fletcher EJ, Abdel-Hamid K, MacDonald JF (1994) Loperamide blocks high-voltage-activated calcium channels and N-methyl-d-aspartate-evoked responses in rat and mouse cultured hippocampal pyramidal neurons. Mol Pharmacol 45: 747-757. [PubMed] [Google Scholar]
- Cooper DMF, Schell MJ, Thorn P, Irvine RF (1998) Regulation of adenylyl cyclase by membrane potential. J Biol Chem 273: 27703-27707. [DOI] [PubMed] [Google Scholar]
- Czyż A, Kiedrowski L (2003) Inhibition of plasmalemmal Na+/Ca2+ exchange by mitochondrial Na+/Ca2+ exchange inhibitor 7-chloro-5-(2-chlorophenyl)-1,5-dihydro-4,1-benzothiazepin-2(3H)-one (CGP-37157) in cerebellar granule cells. Biochem Pharmacol 66: 2409-2411. [DOI] [PubMed] [Google Scholar]
- Czyż A, Baranauskas G, Kiedrowski L (2002) Instrumental role of Na+ in NMDA excitotoxicity in glucose-deprived and depolarized cerebellar granule cells. J Neurochem 81: 379-389. [DOI] [PubMed] [Google Scholar]
- Deecher DC, Teitler M, Soderlund DM, Bornmann WG, Kuehne ME, Glick SD (1992) Mechanisms of action of ibogaine and harmaline congeners based on radioligand binding studies. Brain Res 571: 242-247. [DOI] [PubMed] [Google Scholar]
- Deitmer JW, Schneider H-P (1998) Acid/base transport across the leech giant glial cell membrane at low external bicarbonate concentration. J Physiol (Lond) 512: 459-469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaurex N, Orlowski J, Brisseau G, Woodside M, Grinstein S (1995) The mammalian Na+/H+ antiporters NHE-1, NHE-2 and NHE-3 are electroneutral and voltage independent, but can couple to an H+ conductance. J Gen Physiol 106: 85-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Loreto S, Balestrino M (1997) Development of vulnerability to hypoxic damage in in vitro hippocampal neurons. Int J Dev Neurosci 15: 225-230. [DOI] [PubMed] [Google Scholar]
- Diarra A, Sheldon C, Brett CL, Baimbridge KG, Church J (1999) Anoxia-evoked intracellular pH and Ca2+ concentration changes in cultured postnatal rat hippocampal neurons. Neuroscience 93: 1003-1016. [DOI] [PubMed] [Google Scholar]
- Diarra A, Sheldon C, Church J (2001) In situ calibration and [H+] sensitivity of the fluorescent Na+ indicator, SBFI. Am J Physiol 280: C1623-C1633. [DOI] [PubMed] [Google Scholar]
- Douglas RM, Schmitt BM, Xia Y, Bevensee MO, Biemesderfer D, Boron WF, Haddad GG (2001) Sodium-hydrogen exchangers and sodium-bicarbonate co-transporters: ontogeny of protein expression in the rat brain. Neuroscience 102: 217-228. [DOI] [PubMed] [Google Scholar]
- Durkin JP, Tremblay R, Chakravarthy B, Mealing G, Morley P, Small D, Song D (1997) Evidence that the early loss of membrane protein kinase C is a necessary step in the excitatory amino acid-induced death of primary cortical neurons. J Neurochem 68: 1400-1412. [DOI] [PubMed] [Google Scholar]
- Ekholm A, Katsura K, Kristián T, Liu M, Folbergrová J, Siesjö BK (1993) Coupling of cellular energy state and ion homeostasis during recovery following brain ischemia. Brain Res 604: 185-191. [DOI] [PubMed] [Google Scholar]
- Erecińska M, Silver IA (2001) Tissue oxygen tension and brain sensitivity to hypoxia. Respir Physiol 128: 263-276. [DOI] [PubMed] [Google Scholar]
- Flatman PW (2002) Regulation of Na-K-2Cl cotransport by phosphorylation and protein-protein interactions. Biochim Biophys Acta 1566: 140-151. [DOI] [PubMed] [Google Scholar]
- Friedman JE, Haddad GG (1994) Anoxia induces an increase in intracellular sodium in rat central neurons in vitro. Brain Res 663: 329-334. [DOI] [PubMed] [Google Scholar]
- Giffard RG, Lee Y-S, Ouyang YB, Murphy SL, Monyer H (2003) Two variants of the rat brain sodium-driven chloride bicarbonate exchanger (NCBE): developmental expression and addition of a PDZ motif. Eur J Neurosci 18: 2935-2945. [DOI] [PubMed] [Google Scholar]
- Goldberg MP, Choi DW (1993) Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci 13: 3510-3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad GG, Jiang C (1993) Mechanisms of anoxia-induced depolarization in brainstem neurons: in vitro current and voltage clamp studies in the adult rat. Brain Res 625: 261-268. [DOI] [PubMed] [Google Scholar]
- Han D, Antunes F, Canali R, Rettori D, Cadenas E (2003) Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem 278: 5557-5563. [DOI] [PubMed] [Google Scholar]
- Harrison NL, Gibbons SJ (1994) Zn2+: an endogenous modulator of ligand- and voltage-gated ion channels. Neuropharmacology 33: 935-952. [DOI] [PubMed] [Google Scholar]
- Hartley Z, Dubinsky JM (1993) Changes in intracellular pH associated with glutamate excitotoxicity. J Neurosci 13: 4690-4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayasaki-Kajiwara Y, Kitano Y, Iwasaki T, Shimamura T, Naya N, Iwaki K, Nakajima M (1999) Na+ influx via Na+/H+ exchange activates protein kinase C isozymes δ and ϵ in cultured neonatal cardiac myocytes. J Mol Cell Cardiol 31: 1559-1572. [DOI] [PubMed] [Google Scholar]
- Horikawa N, Kuribayashi Y, Matsui K, Ohashi N (2001a) Relationship between the neuroprotective effect of Na+/H+ exchange inhibitor SM-20220 and the timing of its administration in a transient middle cerebral artery occlusion model of rats. Biol Pharm Bull 24: 767-771. [DOI] [PubMed] [Google Scholar]
- Horikawa N, Nishioka M, Itoh N, Kuribayashi Y, Matsui K, Ohashi N (2001b) The Na+/H+ exchanger SM-20220 attenuates ischemic injury in in vitro and in vivo models. Pharmacology 63: 76-81. [DOI] [PubMed] [Google Scholar]
- Jiang C, Aqulian S, Haddad GG (1992) Cl- and Na+ homeostasis during anoxia in rat hypoglossal neurons: intracellular and extracellular in vitro studies. J Physiol (Lond) 448: 697-708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Park YS, Lee SH (1988) Effect of harmaline on organic ion transport in rabbit renal cortical slices. Arch Int Pharmacodyn Ther 294: 259-272. [PubMed] [Google Scholar]
- Kimura M, Sawada K, Miyagawa T, Kuwada M, Katayama K, Nishizawa Y (1998) Role of glutamate receptors and voltage-dependent calcium and sodium channels in the extracellular glutamate/aspartate accumulation and subsequent neuronal injury induced by oxygen/glucose deprivation in cultured hippocampal neurons. J Pharmacol Exp Ther 285: 178-185. [PubMed] [Google Scholar]
- Kintner DB, Su G, Lenart BA, Ballard AJ, Meyer JW, Ng LL, Shull GE, Sun D (2004) Increased tolerance to oxygen and glucose deprivation in astrocytes from Na+/H+ exchanger isoform 1 null mice. Am J Physiol 287: C12-C21. [DOI] [PubMed] [Google Scholar]
- Lei S, MacDonald JF (2001) Gadolinium reduces AMPA receptor desensitization and deactivation in hippocampal neurons. J Neurophysiol 86: 173-182. [DOI] [PubMed] [Google Scholar]
- Lenart B, Kintner DB, Shull GE, Sun D (2004) Na-K-Cl cotransporter-mediated intracellular Na+ accumulation affects Ca2+ signaling in astrocytes in an in vitro ischemic model. J Neurosci 24: 9585-9597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton P (1999) Ischemic cell death in brain neurons. Physiol Rev 79: 1431-1568. [DOI] [PubMed] [Google Scholar]
- LoPachin RM, Gaughan CL, Lehning EJ, Weber ML, Taylor CP (2001) Effects of ion channel blockage on the distribution of Na, K, Ca and other elements in oxygen-glucose deprived CA1 hippocampal neurons. Neuroscience 103: 971-983. [DOI] [PubMed] [Google Scholar]
- Manzerra P, Behrens MM, Canzoniero LMT, Wang XQ, Heidinger V, Ichinose T, Yu SP, Choi DW (2001) Zinc induces a Src family kinase-mediated up-regulation of NMDA receptor activity and excitotoxicity. Proc Natl Acad Sci USA 98: 11055-11061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Sánchez M, Striggow F, Schröder UH, Kahlert S, Reymann KG, Reiser G (2004) Na+ and Ca2+ homeostasis pathways, cell death and protection after oxygen-glucose deprivation in organotypic hippocampal slice cultures. Neuroscience 128: 729-740. [DOI] [PubMed] [Google Scholar]
- Müller M, Somjen GG (2000) Na+ and K+ concentrations, extra- and intracellular voltages, and the effect of TTX in hypoxic rat hippocampal slices. J Neurophysiol 83: 735-745. [DOI] [PubMed] [Google Scholar]
- Orlowski J (1993) Heterologous expression and functional properties of amiloride high affinity (NHE-1) and low affinity (NHE-3) isoforms of the rat Na/H exchanger. J Biol Chem 268: 16369-16377. [PubMed] [Google Scholar]
- Payne JA, Rivera C, Voipio J, Kaila K (2003) Cation-chloride cotransporters in neuronal communication, development and trauma. Trends Neurosci 26: 199-206. [DOI] [PubMed] [Google Scholar]
- Phillis JW, O'Regan MH (2004) A potentially critical role of phospholipases in central nervous system ischemic, traumatic and neurodegenerative disorders. Brain Res Rev 44: 13-47. [DOI] [PubMed] [Google Scholar]
- Phillis JW, Estevez AY, Guyot LL, O'Regan MH (1999) 5-(N-ethyl-N-isopropyl)-amiloride, an Na+-H+ exchange inhibitor, protects gerbil hippocampal neurons from ischemic injury. Brain Res 839: 199-202. [DOI] [PubMed] [Google Scholar]
- Pisani A, Calabresi P, Tozzi A, Bernardi G, Knopfel T (1998) Early sodium elevations induced by combined oxygen and glucose deprivation in pyramidal cortical neurons. Eur J Neurosci 10: 3572-3584. [DOI] [PubMed] [Google Scholar]
- Plotkin MD, Snyder EY, Hebert SC, Delpire E (1997) Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA's excitatory role in immature brain. J Neurobiol 33: 781-795. [DOI] [PubMed] [Google Scholar]
- Raley-Susman KM, Cragoe Jr EJ, Sapolsky RM, Kopito RR (1991) Regulation of intracellular pH in cultured hippocampal neurons by an amiloride-insensitive Na+/H+ exchanger. J Biol Chem 266: 2739-2745. [PubMed] [Google Scholar]
- Roettger V, Lipton P (1996) Mechanism of glutamate release from rat hippocampal slices during in vitro ischemia. Neuroscience 75: 677-685. [DOI] [PubMed] [Google Scholar]
- Romero MF, Fulton CM, Boron WF (2004) The SLC4 family of HCO3- transporters. Pflügers Arch 447: 495-509. [DOI] [PubMed] [Google Scholar]
- Rose CR, Ransom BR (1997) Regulation of intracellular sodium in cultured rat hippocampal neurones. J Physiol (Lond) 499: 573-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman SM (1983) Synaptic activity mediates death of hypoxic neurons. Science 220: 536-537. [DOI] [PubMed] [Google Scholar]
- Russell JM (2000) Sodium-potassium-chloride cotransport. Physiol Rev 80: 211-276. [DOI] [PubMed] [Google Scholar]
-
Schwiening CJ, Boron WF (1994) Regulation of intracellular pH in pyramidal neurones from the rat hippocampus by Na+-dependent
 exchange. J Physiol (Lond)
475: 59-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
exchange. J Physiol (Lond)
475: 59-67. [DOI] [PMC free article] [PubMed] [Google Scholar] - Sheldon C, Church J (2002) Intracellular pH response to anoxia in acutely dissociated adult rat hippocampal CA1 neurons. J Neurophysiol 87: 2209-2224. [DOI] [PubMed] [Google Scholar]
- Sheldon C, Church J (2004) Reduced contribution from Na+/H+ exchange to acid extrusion during anoxia in adult rat hippocampal CA1 neurons. J Neurochem 88: 594-603. [DOI] [PubMed] [Google Scholar]
- Sheldon C, Cheng YM, Church J (2004) Concurrent measurements of the free cytosolic concentrations of H+ and Na+ ions with fluorescent indicators. Pflügers Arch, in press. [DOI] [PubMed]
- Silver IA, Ereciñska M (1990) Intracellular and extracellular changes in [Ca2+]i in hypoxia and ischemia in rat brain in vivo. J Gen Physiol 95: 837-866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver IA, Erecińska M (1992) Ion homeostasis in rat brain in vivo: intraand extracellular [Ca2+] and [H+] in the hippocampus during recovery from short-term, transient ischemia. J Cereb Blood Flow Metab 12: 759-772. [DOI] [PubMed] [Google Scholar]
- Sun D, Murali SG (1999) Na+-K+-2Cl- cotransporter in immature cortical neurons: a role in intracellular Cl- regulation. J Neurophysiol 81: 1939-1948. [DOI] [PubMed] [Google Scholar]
- Szabó EZ, Numata M, Shull GE, Orlowski J (2000) Kinetic and pharmacological properties of human brain Na+/H+ exchanger isoform 5 stably expressed in chinese hamster ovary cells. J Biol Chem 275: 6302-6307. [DOI] [PubMed] [Google Scholar]
- Tauskela JS, Mealing G, Comas R, Brunette E, Monette R, Small DL, Morley P (2003) Protection of cortical neurons against oxygen-glucose deprivation and N-methyl-d-aspartate by DIDS and SITS. Eur J Pharmacol 464: 17-25. [DOI] [PubMed] [Google Scholar]
- van Emous JG, Schreur JHM, Ruigrok TJC, Van Echteld CJA (1998) Both Na+/K+ ATPase and Na+/H+ exchanger are immediately active upon post-ischemic reperfusion in isolated rat hearts. J Mol Cell Cardiol 30: 337-348. [DOI] [PubMed] [Google Scholar]
- Vergun O, Sobolevsky AI, Yelshansky MV, Keelan J, Khodorov BI, Duchen MR (2001) Exploration of the role of reactive oxygen species in glutamate neurotoxicity in rat hippocampal neurones in culture. J Physiol (Lond) 531: 147-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vornov JJ, Thomas AG, Jo D (1996) Protective effects of extracellular acidosis and blockade of sodium/hydrogen ion exchange during recovery from metabolic inhibition in neuronal tissue culture. J Neurochem 67: 2379-2389. [DOI] [PubMed] [Google Scholar]
- White RJ, Reynolds IJ (1995) Mitochondria and Na+/Ca2+ exchange buffer glutamate-induced calcium loads in cultured cortical neurons. J Neurosci 15: 1318-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
Wu ML, Tsai ML, Tseng YZ (1994) DIDS-sensitive pHi regulation in single rat cardiac myocytes in nominally
 -free conditions. Circ Res
75: 123-132. [DOI] [PubMed] [Google Scholar]
-free conditions. Circ Res
75: 123-132. [DOI] [PubMed] [Google Scholar] - Yan Y, Dempsey RJ, Flemmer A, Forbush B, Sun D (2003) Inhibition of Na+-K+-Cl- cotransporter during focal cerebral ischemia decreases edema and neuronal damage. Brain Res 961: 22-31. [DOI] [PubMed] [Google Scholar]
- Yu SP, Choi DW (1997) Na+-Ca2+ exchange currents in cortical neurons: concomitant forward and reverse operation and effect of glutamate. Eur J Neurosci 9: 1273-1281. [DOI] [PubMed] [Google Scholar]
- Yu X-M, Salter MW (1998) Gain control of NMDA-receptor currents by intracellular sodium. Nature 396: 469-474. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Lipton P (1999) Cytosolic Ca2+ changes during in vitro ischemia in rat hippocampal slices: major roles for glutamate and Na+-dependent Ca2+ release from mitochondria. J Neurosci 19: 3307-3315. [DOI] [PMC free article] [PubMed] [Google Scholar]