

Abstract
HB‐EGF, a member of the EGF family of growth factors, exerts its biological activity through activation of the EGFR and other ErbB receptors. HB‐EGF participates in diverse biological processes, including heart development and maintenance, skin wound healing, eyelid formation, blastocyst implantation, progression of atherosclerosis and tumor formation, through the activation of signaling molecules downstream of ErbB receptors and interactions with molecules associated with HB‐EGF. Recent studies have indicated that HB‐EGF gene expression is significantly elevated in many human cancers and its expression level in a number of cancer‐derived cell lines is much higher than those of other EGFR ligands. Several lines of evidence have indicated that HB‐EGF plays a key role in the acquisition of malignant phenotypes, such as tumorigenicity, invasion, metastasis and resistance to chemotherapy. Studies in vitro and in vivo have indicated that HB‐EGF expression is essential for tumor formation of cancer‐derived cell lines. CRM197, a specific inhibitor of HB‐EGF, and an antibody against HB‐EGF are both able to inhibit tumor growth in nude mice. These results indicate that HB‐EGF is a promising target for cancer therapy, and that the development of targeting tools against HB‐EGF could represent a novel type of therapeutic strategy, as an alternative to targeting ErbB receptors. (Cancer Sci 2006; 97: 341–347)
Abbreviations:
- EGF
epidermal growth factor
- EGFR
EGF receptor
- ERK
extracellular regulated kinase
- GPCR
G protein‐coupled receptor
- HB‐EGF
heparin‐binding epidermal growth factor‐like growth factor
- MAPK
mitogen‐activated protein kinase
- proHB‐EGF
membrane‐anchored form of HB‐EGF
- sHB‐EGF
soluble form of HB‐EGF
- TGF‐α
transforming growth factor‐α.
EGFR (ErbB1) and the related ErbB receptors belong to the tyrosine kinase family and comprise four members: EGFR/ErbB1, ErbB2, ErbB3 and ErbB4( 1 , 2 , 3 ) (Fig. 1). The ErbB receptors are present on the cell surface and share a common structure composed of an extracellular ligand‐binding domain, a transmembrane domain and an intracellular tyrosine kinase domain. The ErbB receptors are expressed in various tissues of epithelial, mesenchymal and neuronal origin, and their activation is controlled by the spatiotemporally regulated expression and liberation of their ligands, which are members of the EGF family of growth factors. Ligand binding induces the formation of receptor homo‐ or heterodimeric complexes and activation of the intrinsic kinase domain, resulting in phosphorylation of specific tyrosine residues that serve as docking sites for adapter molecules, which in turn lead to the activation of intracellular signaling pathways.
Figure 1.
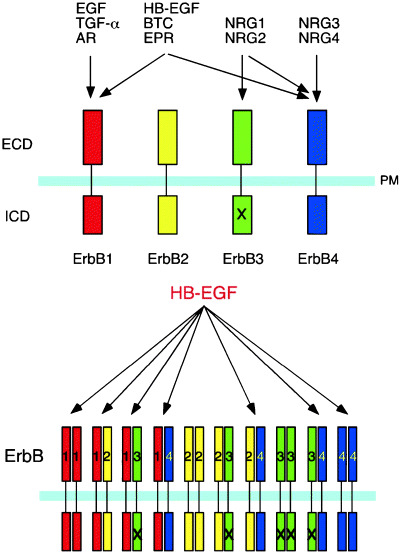
Binding specificities of members of the ErbB receptor family to EGF ligands. (a) EGF family ligands are separated into four categories according to their specificities for members of the ErbB receptor family. ErbB2 has no ligand. ErbB3 is deficient in kinase activity (X). (b) ErbB homo‐ and heterodimer combinations activated by heparin‐binding epidermal growth factor‐like growth factor (HB‐EGF). AR, amphiregulin; BTC, betacellulin; ECD, extracellular domain; EGF, epidermal growth factor; EPR, epiregulin; ICD, intracellular domain; NRG, neuregulin; PM, plasma membrane; TGF‐α, transforming growth factor‐α.
The EGF family of ligands is composed of four groups. The first group includes EGF, TGF‐α and amphiregulin, which bind specifically to EGFR. The second group includes HB‐EGF, betacellulin and epiregulin, which bind to both EGFR and ErbB4 (Fig. 1). The third and fourth groups include the neuregulins (NRG1–4). NRG‐1 and NRG‐2 bind to ErbB3 and ErbB4, whereas NRG‐3 and NRG‐4 bind to ErbB4, but not ErbB3. Although no ligands bind directly to ErbB2, ErbB2 prefers to form heterodimers with other ErbB receptors, which enhances the tyrosine kinase signals.( 2 , 3 )
The EGF family of growth factors and their cognate ErbB receptors support the proliferation, motility, differentiation and survival of various types of cells, thereby contributing to the development, morphogenesis and maintenance of homeostasis of the body. In addition to their physiological roles, ErbB receptor tyrosine kinases have important roles in human cancers.( 4 , 5 ) The ErbB family members, especially EGFR and ErbB2, are involved in many tumor cell behaviors, including excessive growth, invasive character and blood vessel formation. Enhanced expressions of ErbB receptors, the existence of constitutively active mutant forms of the receptors and the occurrence of autocrine activation are frequently observed in human cancers and, in some cases, are associated with poor disease outcomes. Accordingly, these receptors have been intensively studied to understand their roles in cancer cell biology, and a number of therapeutic strategies targeting EGFR or ErbB2 are either used in the clinic or are under development.
In contrast to their receptors, however, the ligands comprising the EGF family of growth factors have not yet been focused on as targets for cancer therapy. This is possibly due to the redundancy of ErbB ligands for each receptor, such that it is recognized that inhibiting receptor function is more effective than inhibiting multiple ligands for cancer therapy. However, recent studies have indicated that the expression level of each individual EGFR ligand varies in different cancers, and a particular ligand is often specifically expressed. For example, HB‐EGF is specifically expressed at high levels in some human cancers.( 6 , 7 ) In this review, we will describe the features of HB‐EGF as a candidate target for cancer therapy. We will also discuss the future directions for the development of therapeutic strategies targeting HB‐EGF.
Structure and physiological functions of HB‐EGF
HB‐EGF is initially synthesized as a transmembrane precursor protein, similar to other members of the EGF family of growth factors.( 8 , 9 ) The membrane‐anchored form of HB‐EGF (proHB‐EGF) is composed of a pro domain followed by heparin‐binding, EGF‐like, juxtamembrane, transmembrane and cytoplasmic domains( 8 ) (Fig. 2). Subsequently, proHB‐EGF is cleaved at the cell surface by a protease to yield the soluble form of HB‐EGF (sHB‐EGF) using a mechanism known as ectodomain shedding.( 10 ) sHB‐EGF is a potent mitogen and chemoattractant for a number of different cell types.( 11 ) Studies of mice expressing non‐cleavable HB‐EGF have indicated that the major functions of HB‐EGF are mediated by the soluble form.( 12 )
Figure 2.
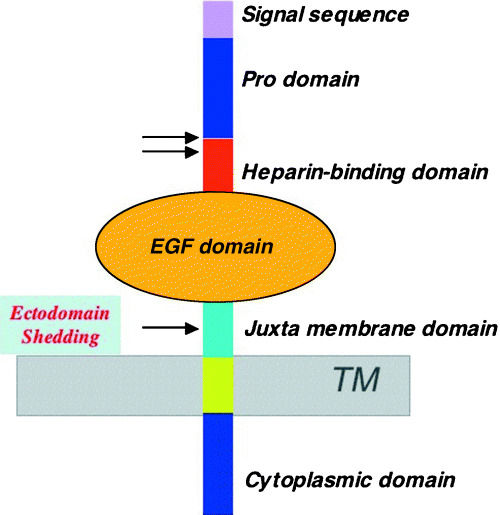
Structure of heparin‐binding epidermal growth factor‐like growth factor (HB‐EGF). The membrane‐anchored form (proHB‐EGF) possesses pro, heparin‐binding, EGF‐like, juxtamembrane, transmembrane and cytoplasmic domains. Proteolytic cleavage occurs at the positions indicated by the arrows. Cleavage at the juxtamembrane region by ‘ectodomain shedding’ results in the release of the soluble active growth factor (sHB‐EGF). TM, transmembrane.
Although the physiological function of proHB‐EGF remains obscure, this form has some unique features. ProHB‐EGF can transmit biological signals to neighboring cells by a juxtacrine mechanism, either positively or negatively.( 13 , 14 ) Furthermore, this form is able to associate with several molecules. For instance, proHB‐EGF forms a complex with CD9 and integrin α3β1 in some cells,( 15 ) and heparan‐sulfate proteoglycan can associate with proHB‐EGF and modulate its biological activity.( 16 ) In addition, proHB‐EGF functions as the sole receptor for diphtheria toxin.( 17 , 18 )
Recent studies have suggested that the cytoplasmic domain of proHB‐EGF plays some roles in HB‐EGF function. A multifunctional protein, BAG‐1, was reported to bind to the cytoplasmic domain of HB‐EGF and increase HB‐EGF secretion.( 19 ) Recently, it has been suggested that the C‐terminal fragment of HB‐EGF generated by ectodomain shedding translocates to the nucleus and promotes S‐phase entry by sequestering promyelocytic leukemia zinc finger protein, a transcriptional repressor of cyclin A.( 20 ) We recently found that the cytoplasmic domain of HB‐EGF is phosphorylated by external stimuli, and that the phosphorylation site is involved in HB‐EGF‐dependent tumorigenesis. (21)
HB‐EGF gene expression has been demonstrated in a variety of tissues, predominantly the lung, heart, brain and skeletal muscle.( 22 ) HB‐EGF participates in a variety of physiological and pathological processes,( 11 ) including wound healing,( 23 ) eyelid formation,( 24 ) blastocyst implantation,( 25 ) retinoic acid‐induced epidermal hyperplasia( 26 ) and atherosclerosis.( 27 ) Recently, HB‐EGF knockout mice have been established.( 28 , 29 ) HB‐EGF null mice show quite severe phenotypes, including an enlarged and dysfunctional heart, heart valve malformation with enlarged semilunar and atrioventricular valves, and thickened mesenchymal tissue and alveolar immaturity in the lungs, and most of the animals die at the neonatal stage. In contrast, no cardiovascular phenotypes are detected in triple null mice lacking three other EGF family members, namely TGF‐α, EGF and amphiregulin.( 30 ) Betacellulin null mice are viable and display no overt defects,( 30 ) whereas epiregulin null mice are viable but develop chronic dermatitis.( 31 ) Thus, HB‐EGF null mice show the most severe phenotypes among the mice with disrupted ligand binding to EGFR. In contrast to TGF‐α, EGF and amphiregulin, HB‐EGF binds to and activates both EGFR and ErbB4.( 28 , 32 ) Therefore, the severe phenotypes of HB‐EGF null mice might be due, in part, to the binding specificity of this ligand to ErbB receptors. Homozygous mice with disrupted ErbB2, ErbB4 or neuregulin‐1 genes all die around embryonic day 10 with defects in the formation of the heart ventricle projections known as trabeculae.( 33 , 34 , 35 ) These phenotypes are quite similar to the phenotypes observed in HB‐EGF knockout mice, suggesting that HB‐EGF serves as a ligand for ErbB4 in heart chamber formation.
Ectodomain shedding and transactivation of EGFR
Ectodomain shedding from proHB‐EGF is critical for HB‐EGF growth factor activity. Mutant mice expressing a non‐cleavable form of proHB‐EGF by gene targeting show severe heart failure, with similar phenotypes to HB‐EGF null mice, indicating that cleavage of proHB‐EGF into the soluble form is necessary for normal cardiomyocyte function.( 12 ) However, mice carrying a transmembrane domain‐truncated form, which therefore secrete sHB‐EGF without ectodomain shedding, develop severe hyperplasia in both the skin and heart. These results indicate that unregulated release of sHB‐EGF results in severe lethal abnormalities in mice.( 12 )
Ectodomain shedding from proHB‐EGF can be stimulated by various physiological and pharmacological stimuli, including 12‐O‐tetradecanoylphorbol‐13‐acetate,( 10 ) calcium ionophores,( 36 ) and GPCR ligands such as lysophosphatidic acid.( 37 , 38 , 39 ) Cellular stresses caused by inflammatory cytokines, reactive oxygen and osmotic shock can also induce ectodomain shedding of HB‐EGF.( 40 ) The stimuli for ectodomain shedding activate intracellular signaling molecules, including protein kinase C, Ras, ERK and p38 MAPK. Activation of these molecules results in proteolytic cleavage of proHB‐EGF by the ADAM family or other metalloproteases.
GPCR ligands, including lysophosphatidic acid, angiotensin II and endothelin, have been shown to stimulate EGFR tyrosine phosphorylation, referred to as “transactivation”.( 38 ) Transactivation of EGFR is a general function of GPCR signaling and is critical for the mitogenic activity of many GPCR ligands.( 29 , 30 ) Transactivation of EGFR is achieved by ectodomain shedding of EGFR ligands, and HB‐EGF is often preferentially shed among the EGFR ligands.( 38 ) Ectodomain shedding from proHB‐EGF results in EGFR activation and subsequent ERK/MAPK activation. In turn, activation of the EGFR–MEK–ERK/MAPK pathway promotes proHB‐EGF shedding.( 39 ) EGFR signals also induce the transcription of proHB‐EGF and other EGFR ligands,( 41 ) resulting in repeated activation of proHB‐EGF shedding and EGFR activation. Thus, stimuli that induce proHB‐EGF shedding trigger multiple positive feedback loops that ensure substantial EGFR activation, a potential cause of oncogenesis and cancer progression.
Involvement of HB‐EGF in the malignant phenotypes of tumors
Cell proliferation
Emerging evidence has implicated HB‐EGF in the proliferative potential of tumor cells. Transfection of HB‐EGF into normal rat kidney cells or chicken embryo fibroblasts induces oncogenic transformation and anchorage‐independent growth.( 42 , 43 ) Overexpression of HB‐EGF in the mouse pancreas leads to fibrosis and epithelial metaplasia.( 44 ) Helicobacter pylori, which has a major etiological role in human gastric carcinogenesis, induces the activation of EGFR signaling mediated by the cleavage of proHB‐EGF to promote enhanced gastric epithelial proliferation and gastric neoplasia.( 45 , 46 , 47 ) Furthermore, in vitro experiments have shown that addition of an anti‐HB‐EGF blocking antibody to human glioblastoma cells reduces proliferation by 30–40% compared with the addition of normal goat IgG.( 48 ) Inhibition of HB‐EGF expression in myeloma cells with CRM197 or an anti‐HB‐EGF antibody results in the suppression of cell proliferation and induction of cell apoptosis.( 49 )
We recently showed that expression of HB‐EGF is critical for cell proliferation of human ovarian cancer cells in nude mice. The human ovarian cancer cell lines SKOV3 and RMG1 form tumors in nude mice when the cells are injected subcutaneously. The tumor formation of these cells is strongly enhanced by exogenous expression of proHB‐EGF, and completely blocked by inhibition of HB‐EGF gene expression by siRNA.( 6 ) HB‐EGF or other EGFR ligands are required for cell growth, particularly under 3D culture conditions, such as cell cultures in collagen gels or soft agar and in vivo (H. Mizushima, E. Mekada, unpublished observation, 2005). However, cell growth of most cultured cells under common 2D culture conditions remains largely unaffected by the presence of EGFR ligands compared with the growth under 3D conditions.
In ovarian cancer, tumor growth is characterized by local extension into the peritoneal cavity, following the circulatory pathway of the peritoneal fluid produced by the peritoneal epithelium and cancer cells. Accumulating evidence from many studies has revealed that ascites from patients with ovarian cancer represent a rich source of growth factor activity for ovarian cancer cells, and these molecules are termed ovarian cancer‐activating factors.( 50 ) It has been demonstrated that the presence of HB‐EGF in peritoneal fluid is sufficient for the survival and proliferation of ovarian cancer cells, and that HB‐EGF may play central roles in the cell survival and proliferation of ovarian cancer, as one of the ovarian cancer‐activating factors.( 51 )
Cell migration and invasion
HB‐EGF has been shown to have strong activity for enhancing cell motility.( 52 ) In eyelid formation in the mouse embryo and skin wound healing, HB‐EGF plays a role in cell motility, but not in cell proliferation.( 23 , 24 ) In MDCK cells, expression of sHB‐EGF, but not non‐cleavable proHB‐EGF, increases cell migration, decreases cell‐matrix and cell–cell interactions and promotes the development of long unbranched tubular structures, suggesting that proHB‐EGF, and sHB‐EGF give rise to distinct effects on cell–cell and cell–extracellular matrix interactions.( 53 ) It has been shown that HB‐EGF binds to N‐arginine dibasic convertase, a metalloendopeptidase of the M16 family, and that N‐arginine dibasic convertase is expressed on the cell surface of MDA‐MB451 cells, where it modulates HB‐EGF‐induced cell migration.( 54 ) In addition, HB‐EGF is downregulated in the placenta of women with pre‐eclampsia, a disorder associated with deficient trophoblast invasion, and the addition of HB‐EGF to explant cultures of first‐trimester chorionic villi enhances extravillous trophoblast differentiation and invasive activity.( 55 , 56 ) Thus, sHB‐EGF plays key roles in various physiological processes, including cell motility, cell migration and invasion.
In cancer, HB‐EGF can contribute to the acquisition of tumor aggressiveness, including invasion, metastasis and chemoresistance, through interactions with molecules bound to HB‐EGF. It has been reported that HB‐EGF is one of the target candidates for metastasis, following comparisons of non‐metastatic pancreatic cancer cells with highly metastatic pancreatic cancer cells.( 57 ) In response to a variety of pathophysiologically relevant GPCR ligands, metalloproteinase‐mediated transactivation of EGFR by ectodomain shedding of EGF family members promotes tumor cell migration and invasion as well as MAPK activation.( 58 ) Lidocaine, which blocks ectodomain shedding from proHB‐EGF at the cell surface, inhibits the invasive ability of human cancer cells, such as HT1080, HOA and RPMI‐7951.( 59 ) In addition, a neutralizing antibody against HB‐EGF blocks not only transactivation of EGFR and cell proliferation, but also the migration mediated by IL‐8 in human colon carcinoma cells.( 60 ) The soluble form of HB‐EGF and wild‐type HB‐EGF each induce the expression and activities of the metalloproteases MMP9 and MMP3, leading to enhanced cell migration in a human bladder carcinoma cell line.( 61 ) Furthermore, it has been suggested that the production of HB‐EGF by human mesothelial cells might play a significant role in postoperative peritoneal tumor recurrence.( 62 ) Taken together, the present evidence shows that HB‐EGF is involved in the migration, invasion and metastasis of cancer cells as well as their proliferation.
Expression of HB‐EGF in human cancers
There is growing evidence of increased HB‐EGF expression in tumors, including pancreatic, liver, esophageal, melanoma, colon, gastric, ovarian and bladder cancers as well as glioblastoma, compared with normal tissues( 6 , 11 , 48 , 63 , 64 ) (S. Miyamoto et al., unpublished observation, 2005). The relationship between HB‐EGF expression and human cancers has been minutely investigated in human ovarian cancer. A number of tumors isolated from ovarian cancer patients express high levels of HB‐EGF, and ascitic fluid obtained from ovarian cancer patients contains higher concentrations of HB‐EGF protein than the concentrations found in normal control subjects and patients with benign tumors.( 6 , 7 ) Furthermore, the expression levels of other EGFR ligands in ovarian cancer samples and the protein levels in ascitic fluid from ovarian cancer patients are one or two orders of magnitude lower than those of HB‐EGF. It has also been shown that HB‐EGF expression is significantly associated with the clinical outcome in ovarian cancer( 7 ) (Fig. 3). Similar to ovarian cancer, HB‐EGF is also highly expressed in bladder cancer, by at least 10–100‐fold of the expression levels of other EGFR ligands.( 63 ) Thus, HB‐EGF is the only abundantly expressed molecule among the EGFR ligands, indicating that HB‐EGF possibly contributes to tumor growth signals through EGFR activation in both ovarian and bladder cancer, whereas the other EGFR ligands do not.
Figure 3.
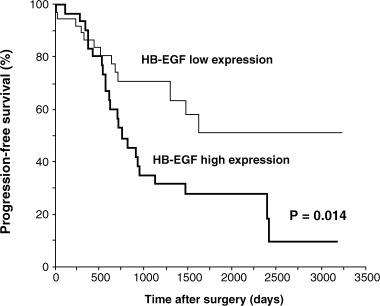
Clinical significance of heparin‐binding epidermal growth factor‐like growth factor (HB‐EGF) expression in ovarian cancer. Progression‐free survival of patients with ovarian cancer in relation to the tumor HB‐EGF expression status. (Reproduced from Tanaka et al.,( 7 ) with permission.)
In situ hybridization and immunostaining analyses have revealed that HB‐EGF gene expression is elevated in human hepatocellular carcinoma biopsies, compared with the surrounding liver tissues which only show faint positivity in normal hepatocytes.( 65 ) However, in colon and pancreatic cancers, expression of HB‐EGF is associated with the early stage.( 66 ) In ovarian cancer, positive signals for HB‐EGF are found abundantly in interstitial tissues, but not in cancer tissues, by immunostaining, and most cells, except for stromal cells, are positively stained using in situ hybridization, indicating that most of the HB‐EGF is shed from the cell membrane in cancer cells and deposited in interstitial tissues.( 7 ) Therefore, it may not be informative to evaluate the association between the clinical outcome in cancer and HB‐EGF expression by only measuring the amount of HB‐EGF using immunostaining.
HB‐EGF as a target molecule for cancer therapy
Low molecular weight inhibitors and antagonists or neutralizing monoclonal antibodies against HB‐EGF are not yet available. CRM197, a non‐toxic mutant of diphtheria toxin,( 67 ) is a protein of approximately 58 kDa. CRM197 binds to the sHB‐EGF, as well as to proHB‐EGF, and inhibits the mitogenic action of HB‐EGF by inhibiting its binding to ErbB receptors.( 68 ) As significant amounts of CRM197 are available through large‐scale cultures, CRM197 is currently the only inhibitor that can be used for animal studies to evaluate HB‐EGF as a target for cancer therapy.
Ovarian cancer cells, in which HB‐EGF is predominantly expressed among the EGFR ligands, form tumors on xenografted mice. Administration of CRM197 into the peritoneal cavity was found to completely suppress the tumor growth in these mice( 6 ) (Fig. 4). In principle, no inflammatory reactions occur in xenografted mice. Therefore, the antitumor effects mediated by the administration of CRM197 can be considered to directly block the tumor formation induced by HB‐EGF expression.
Figure 4.
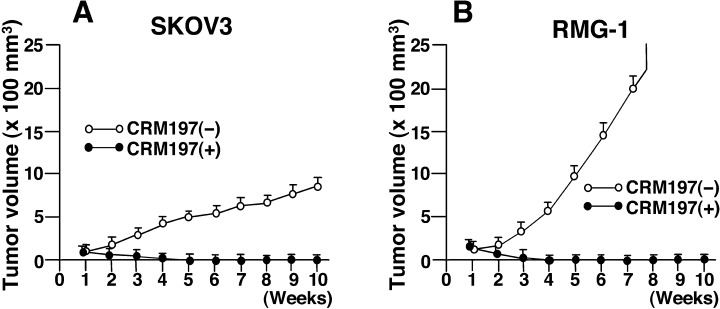
Suppression of tumor growth following administration of CRM197. The human ovarian cancer cell lines SKOV3 (A) and RMG‐1 (B) were injected subcutaneously into nude mice, followed by intraperitoneal injection of CRM197 or control saline once a week for 10 weeks. (Reproduced from Miyamoto et al.,( 6 ) with permission.)
In the past, diphtheria toxin was considered to be a potential antitumor drug, although it was not then known that the toxin could bind to HB‐EGF. Thus, clinical trials of the use of small amounts of diphtheria toxin were carried out in patients with advanced cancer.( 69 ) It was reported that 24 of 50 patients showed a partial or complete response after this treatment, although reversible side‐effects were detected in 17 patients. In the same line of trials using a diphtheria‐related protein and expecting an inflammatory‐immunological reaction, CRM197 was injected subcutaneously into the abdominal wall of 25 advanced cancer outpatients, who were refractory to standard therapies or had refused conventional therapies.( 70 ) Two patients showed a complete response, one showed a partial response and six patients showed disease stability. The toxicities were minimal, as only one patient had skin irritation at the injection sites and a flu‐like syndrome with fever. These results suggest that CRM197‐mediated inhibition of HB‐EGF function contributes to the loss of in vitro and in vivo tumor formation in cancer and that HB‐EGF is a potential target for cancer therapy. Further clinical trials of CRM197 involving cancer patients with HB‐EGF overexpression could provide powerful support for improving the clinical outcome.
Future directions
It has been widely accepted that the expressions or activations of EGFR and ErbB2 are altered in many epithelial tumors, and that these molecules play important roles in tumor etiology and progression. Accordingly, EGFR and ErbB2 have been intensely studied as therapeutic targets, and many ErbB inhibitors and ErbB‐targeted antibodies are now being either used in the clinic or assessed in clinical trials.( 4 , 71 ) In contrast, EGFR ligands have not been focused on as therapeutic targets for cancer. One reason why EGFR ligands have not been considered as therapeutic targets is partly due to the redundancy of EGFR ligands for the ErbB receptors. However, as described in this review, HB‐EGF is predominantly expressed, compared to other EGFR ligands, in cases of ovarian cancer, and suppression of HB‐EGF gene expression by siRNA or inhibition of its mitogenic activity by CRM197 inhibits tumor formation by ovarian cancer cells in xenografted mice. These results indicate that EGFR ligands are also potential therapeutic targets for cancer. There is a potential advantage in targeting an EGFR ligand compared to an ErbB receptor, as HB‐EGF binds to EGFR and ErbB4, and eventually activates EGFR, ErbB2 and ErbB4. Thus, targeting HB‐EGF seems to be more effective than targeting EGFR for tumors that predominantly express HB‐EGF. Furthermore, recent studies have suggested that HB‐EGF is a major ligand among the EGFR ligands in some other types of cancer,( 6 , 7 , 63 ) (S. Miyamoto et al, unpublished observation, 2005). Therefore HB‐EGF would be a promising target for the therapy of these cancers. In this context, amphiregulin, TGF‐α and epiregulin could also represent potential targets for other cancers, as cases in which these EGFR ligands are the major EGFR ligand have also been reported.( 30 )
CRM197 is remarkably effective for nude mice bearing human ovarian cancer cells. There is a positive tendency for chemoresistance of ovarian cancer relative to the HB‐EGF expression level.( 51 ) CRM197 would be equally effective for tumors that express high amounts of HB‐EGF, and it is hoped that this drug will be useful for chemoresistant patients. Regarding the toxicity of CRM197, mouse experiments do not give any information regarding the side‐effects of CRM197 injection, because CRM197 does not bind to mouse HB‐EGF.( 68 ) However, as mentioned above, the clinical trial in human patients in Italy provides some evidence regarding its safety if it is used within a limited dose.( 70 ) CRM197 is now in the process of being developed as a therapeutic agent for ovarian cancer and a clinical trial is expected in the near future. In addition, although antiHB‐EGF neutralizing antibodies and low molecular weight inhibitors of HB‐EGF actions have not yet been reported, these molecules are also potentially hopeful therapeutic molecules. In the future, we hope that, through continuing basic and clinical studies, we will have a variety of therapeutic molecules, including both receptor‐targeted and ligand‐targeted agents, and will be able to select the best one for a particular cancer or an individual patient.
References
- 1. Riese DJ 2nd, Stern DF. Specificity within the EGF family/ErbB receptor family signaling network. Bioessays 1998; 20: 41–8. [DOI] [PubMed] [Google Scholar]
- 2. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001; 2: 127–37. [DOI] [PubMed] [Google Scholar]
- 3. Schlessinger J. Common and distinct elements in cellular signaling via EGF and FGF receptors. Science 2004; 306: 1506–7. [DOI] [PubMed] [Google Scholar]
- 4. Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 2005; 5: 341–54. [DOI] [PubMed] [Google Scholar]
- 5. Mosesson Y, Yarden Y. Oncogenic growth factor receptors: implications for signal transduction therapy. Semin Cancer Biol 2004; 14: 262–70. [DOI] [PubMed] [Google Scholar]
- 6. Miyamoto S, Hirata M, Yamazaki A et al. Heparin‐binding EGF‐like growth factor is a promising target for ovarian cancer therapy. Cancer Res 2004; 64: 5720–7. [DOI] [PubMed] [Google Scholar]
- 7. Tanaka Y, Miyamoto S, Suzuki SO et al. Clinical significance of heparin‐binding epidermal growth factor‐like growth factor and a disintegrin and metalloprotease 17 expression in human ovarian cancer. Clin Cancer Res 2005; 11: 4783–92. [DOI] [PubMed] [Google Scholar]
- 8. Higashiyama S, Abraham JA, Miller J, Fiddes JC, Klagsbrun M. A heparin‐binding growth factor secreted by macrophage‐like cells that is related to EGF. Science 1991; 251: 936–9. [DOI] [PubMed] [Google Scholar]
- 9. Massague J, Pandiella A. Membrane‐anchored growth factors. Annu Rev Biochem 1993; 62: 515–41. [DOI] [PubMed] [Google Scholar]
- 10. Goishi K, Higashiyama S, Klagsbrun M et al. Phorbol ester induces the rapid processing of cell surface heparin‐binding EGF‐like growth factor: conversion from juxtacrine to paracrine growth factor activity. Mol Biol Cell 1995; 6: 967–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Raab G, Klagsbrun M. Heparin‐binding EGF‐like growth factor. Biochim Biophys Acta 1997; 1333: F179–99. [DOI] [PubMed] [Google Scholar]
- 12. Yamazaki S, Iwamoto R, Saeki K et al. Mice with defects in HB‐EGF ectodomain shedding show severe developmental abnormalities. J Cell Biol 2003; 163: 469–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Higashiyama S, Iwamoto R, Goishi K et al. The membrane protein CD9/DRAP 27 potentiates the juxtacrine growth factor activity of the membrane‐anchored heparin‐binding EGF‐like growth factor. J Cell Biol 1995; 128: 929–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Iwamoto R, Handa K, Mekada E. Contact‐dependent growth inhibition and apoptosis of epidermal growth factor (EGF) receptor‐expressing cells by the membrane‐anchored form of heparin‐binding EGF‐like growth factor. J Biol Chem 1999; 274: 25906–12. [DOI] [PubMed] [Google Scholar]
- 15. Nakamura K, Iwamoto R, Mekada E. Membrane‐anchored heparin‐binding EGF‐like growth factor (HB‐EGF) and diphtheria toxin receptor‐associated protein (DRAP27)/CD9 form a complex with integrin alpha 3 beta 1 at cell‐cell contact sites. J Cell Biol 1995; 129: 1691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shishido Y, Sharma KD, Higashiyama S, Klagsbrun M, Mekada E. Heparin‐like molecules on the cell surface potentiate binding of diphtheria toxin to the diphtheria toxin receptor/membrane‐anchored heparin‐binding epidermal growth factor‐like growth factor. J Biol Chem 1995; 270: 29578–85. [DOI] [PubMed] [Google Scholar]
- 17. Naglich JG, Metherall JE, Russell DW, Eidels L. Expression cloning of a diphtheria toxin receptor: identity with a heparin‐binding EGF‐like growth factor precursor. Cell 1992; 69: 1051–61. [DOI] [PubMed] [Google Scholar]
- 18. Iwamoto R, Higashiyama S, Mitamura T, Taniguchi N, Klagsbrun M, Mekada E. Heparin‐binding EGF‐like growth factor, which acts as the diphtheria toxin receptor, forms a complex with membrane protein DRAP27/CD9, which up‐regulates functional receptors and diphtheria toxin sensitivity. EMBO J 1994; 13: 2322–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lin J, Hutchinson L, Gaston SM, Raab G, Freeman MR. BAG‐1 is a novel cytoplasmic binding partner of the membrane form of heparin‐binding EGF‐like growth factor: a unique role for proHB‐EGF in cell survival regulation. J Biol Chem 2001; 276: 30127–32. [DOI] [PubMed] [Google Scholar]
- 20. Nanba D, Mammoto A, Hashimoto K, Higashiyama S. Proteolytic release of the carboxy‐terminal fragment of proHB‐EGF causes nuclear export of PLZF. J Cell Biol 2003; 163: 489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang X, Mizushima H, Adachi S, Ohishi M, Iwamoto R, Makada E. Cytoplasmic domain phosphorylation of heparin‐binding EGF‐like growth factor. Cell Struct Funct 2006; 31: 15–27. [DOI] [PubMed] [Google Scholar]
- 22. Abraham JA, Damm D, Bajardi A, Miller J, Klagsbrun M, Ezekowitz RA. Heparin‐binding EGF‐like growth factor: characterization of rat and mouse cDNA clones, protein domain conservation across species, and transcript expression in tissues. Biochem Biophys Res Commun 1993; 190: 125–33. [DOI] [PubMed] [Google Scholar]
- 23. Shirakata Y, Kimura R, Nanba D et al. Heparin‐binding EGF‐like growth factor accelerates keratinocyte migration and skin wound healing. J Cell Sci 2005; 118: 2363–70. [DOI] [PubMed] [Google Scholar]
- 24. Mine N, Iwamoto R, Mekada E. HB‐EGF promotes epithelial cell migration in eyelid development. Development 2005; 132: 4317–26. [DOI] [PubMed] [Google Scholar]
- 25. Paria BC, Elenius K, Klagsbrun M, Dey SK. Heparin‐binding EGF‐like growth factor interacts with mouse blastocysts independently of ErbB1: a possible role for heparan sulfate proteoglycans and ErbB4 in blastocyst implantation. Development 1999; 126: 1997–2005. [DOI] [PubMed] [Google Scholar]
- 26. Kimura R, Iwamoto R, Mekada E. Soluble form of heparin‐binding EGF‐like growth factor contributes to retinoic acid‐induced epidermal hyperplasia. Cell Struct Funct 2006; 30: 35–42. [DOI] [PubMed] [Google Scholar]
- 27. Miyagawa J, Higashiyama S, Kawata S et al. Localization of heparin‐binding EGF‐like growth factor in the smooth muscle cells and macrophages of human atherosclerotic plaques. J Clin Invest 1995; 95: 404–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Iwamoto R, Yamazaki S, Asakura M et al. Heparin‐binding EGF‐like growth factor and ErbB signaling is essential for heart function. Proc Natl Acad Sci USA 2003; 100: 3221–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jackson LF, Qiu TH, Sunnarborg SW et al. Defective valvulogenesis in HB‐EGF and TACE‐null mice is associated with aberrant BMP signaling. EMBO J 2003; 22: 2704–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Luetteke NC, Qiu TH, Fenton SE et al. Targeted inactivation of the EGF and amphiregulin genes reveals distinct roles for EGF receptor ligands in mouse mammary gland development. Development 1999; 126: 2739–50. [DOI] [PubMed] [Google Scholar]
- 31. Shirasawa S, Sugiyama S, Baba I et al. Dermatitis due to epiregulin deficiency and a critical role of epiregulin in immune‐related responses of keratinocyte and macrophage. Proc Natl Acad Sci USA 2004; 101: 13921–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Elenius K, Paul S, Allison G, Sun J, Klagsbrun M. Activation of HER4 by heparin‐binding EGF‐like growth factor stimulates chemotaxis but not proliferation. EMBO J 1997; 16: 1268–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Meyer D, Birchmeier C. Multiple essential functions of neuregulin in development. Nature 1995; 378: 386–90. [DOI] [PubMed] [Google Scholar]
- 34. Gassmann M, Casagranda F, Orioli D et al. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature 1995; 378: 390–4. [DOI] [PubMed] [Google Scholar]
- 35. Lee KF, Simon H, Chen H, Bates B, Hung MC, Hauser C. Requirement for neuregulin receptor ErbB2 in neural and cardiac development. Nature 1995; 378: 394–8. [DOI] [PubMed] [Google Scholar]
- 36. Dethlefsen SM, Raab G, Moses MA, Adam RM, Klagsbrun M, Freeman MR. Extracellular calcium influx stimulates metalloproteinase cleavage and secretion of heparin‐binding EGF‐like growth factor independently of protein kinase C. J Cell Biochem 1998; 69: 143–53. [DOI] [PubMed] [Google Scholar]
- 37. Hirata M, Umata T, Takahashi T et al. Identification of serum factor inducing ectodomain shedding of proHB‐EGF and studies of noncleavable mutants of proHB‐EGF. Biochem Biophys Res Commun 2001; 283: 915–22. [DOI] [PubMed] [Google Scholar]
- 38. Prenzel N, Zwick E, Daub H et al. EGF receptor transactivation by G‐protein‐coupled receptors requires metalloproteinase cleavage of proHB‐EGF. Nature 1999; 402: 884–8. [DOI] [PubMed] [Google Scholar]
- 39. Umata T, Hirata M, Takahashi T et al. A dual signaling cascade that regulates the ectodomain shedding of heparin‐binding epidermal growth factor‐like growth factor. J Biol Chem 2001; 276: 30475–82. [DOI] [PubMed] [Google Scholar]
- 40. Takenobu H, Yamazaki A, Hirata M, Umata T, Mekada E. The stress‐ and inflammatory cytokine‐induced ectodomain shedding of heparin‐binding epidermal growth factor‐like growth factor is mediated by p38 MAPK, distinct from the 12‐O‐tetradecanoylphorbol‐13‐acetate‐ and lysophosphatidic acid‐induced signaling cascades. J Biol Chem 2003; 278: 17255–62. [DOI] [PubMed] [Google Scholar]
- 41. Hashimoto K, Higashiyama S, Asada H et al. Heparin‐binding epidermal growth factor‐like growth factor is an autocrine growth factor for human keratinocytes. J Biol Chem 1994; 269: 20060–6. [PubMed] [Google Scholar]
- 42. Fu S, Bottoli I, Goller M, Vogt PK. Heparin‐binding epidermal growth factor‐like growth factor, a v‐Jun target gene, induces oncogenic transformation. Proc Natl Acad Sci USA 1999; 96: 5716–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Harding PA, Davis‐Fleischer KM, Crissman‐Combs MA et al. Induction of anchorage independent growth by heparin‐binding EGF‐like growth factor (HB‐EGF). Growth Factors 1999; 17: 49–61. [DOI] [PubMed] [Google Scholar]
- 44. Means AL, Ray KC, Singh AB et al. Overexpression of heparin‐binding EGF‐like growth factor in mouse pancreas results in fibrosis and epithelial metaplasia. Gastroenterology 2003; 124: 1020–36. [DOI] [PubMed] [Google Scholar]
- 45. Busiello I, Acquaviva R, Di Popolo A et al. Helicobacter pylori gamma‐glutamyltranspeptidase upregulates COX‐2 and EGF‐related peptide expression in human gastric cells. Cell Microbiol 2004; 6: 255–67. [DOI] [PubMed] [Google Scholar]
- 46. Keates S, Sougioultzis S, Keates AC et al. cag+Helicobacter pylori induce transactivation of the epidermal growth factor receptor in AGS gastric epithelial cells. J Biol Chem 2001; 276: 48127–34. [DOI] [PubMed] [Google Scholar]
- 47. Wallasch C, Crabtree JE, Bevec D, Robinson PA, Wagner H, Ullrich A. Helicobacter pylori‐stimulated EGF receptor transactivation requires metalloprotease cleavage of HB‐EGF. Biochem Biophys Res Commun 2002; 295: 695–701. [DOI] [PubMed] [Google Scholar]
- 48. Mishima K, Higashiyama S, Asai A et al. Heparin‐binding epidermal growth factor‐like growth factor stimulates mitogenic signaling and is highly expressed in human malignant gliomas. Acta Neuropathol (Berl) 1998; 96: 322–8. [DOI] [PubMed] [Google Scholar]
- 49. Wang YD, De Vos J, Jourdan M et al. Cooperation between heparin‐binding EGF‐like growth factor and interleukin‐6 in promoting the growth of human myeloma cells. Oncogene 2002; 21: 2584–92. [DOI] [PubMed] [Google Scholar]
- 50. Xu Y, Shen Z, Wiper DW et al. Lysophosphatidic acid as a potential biomarker for ovarian and other gynecologic cancers. JAMA 1998; 280: 719–23. [DOI] [PubMed] [Google Scholar]
- 51. Yagi H, Miyamoto S, Tanaka Y et al. Clinical significance of heparin‐binding epidermal growth factor‐like growth factor in peritoneal fluid of ovarian cancer. Br J Cancer 2005; 92: 1737–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Higashiyama S, Abraham JA, Klagsbrun M. Heparin‐binding EGF‐like growth factor stimulation of smooth muscle cell migration: dependence on interactions with cell surface heparan sulfate. J Cell Biol 1993; 122: 933–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Singh AB, Tsukada T, Zent R, Harris RC. Membrane‐associated HB‐EGF modulates HGF‐induced cellular responses in MDCK cells. J Cell Sci 2004; 117: 1365–79. [DOI] [PubMed] [Google Scholar]
- 54. Nishi E, Prat A, Hospital V, Elenius K, Klagsbrun M. N‐arginine dibasic convertase is a specific receptor for heparin‐binding EGF‐like growth factor that mediates cell migration. EMBO J 2001; 20: 3342–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Leach RE, Kilburn B, Wang J, Liu Z, Romero R, Armant DR. Heparin‐binding EGF‐like growth factor regulates human extravillous cytotrophoblast development during conversion to the invasive phenotype. Dev Biol 2004; 266: 223–37. [DOI] [PubMed] [Google Scholar]
- 56. Leach RE, Romero R, Kim YM et al. Pre‐eclampsia and expression of heparin‐binding EGF‐like growth factor. Lancet 2002; 360: 1215–9. [DOI] [PubMed] [Google Scholar]
- 57. Tarbe N, Losch S, Burtscher H, Jarsch M, Weidle UH. Identification of rat pancreatic carcinoma genes associated with lymphogenous metastasis. Anticancer Res 2002; 22: 2015–27. [PubMed] [Google Scholar]
- 58. Schafer B, Gschwind A, Ullrich A. Multiple G‐protein‐coupled receptor signals converge on the epidermal growth factor receptor to promote migration and invasion. Oncogene 2004; 23: 991–9. [DOI] [PubMed] [Google Scholar]
- 59. Mammoto T, Higashiyama S, Mukai M et al. Infiltration anesthetic lidocaine inhibits cancer cell invasion by modulating ectodomain shedding of heparin‐binding epidermal growth factor‐like growth factor (HB‐EGF). J Cell Physiol 2002; 192: 351–8. [DOI] [PubMed] [Google Scholar]
- 60. Itoh Y, Joh T, Tanida S et al. IL‐8 promotes cell proliferation and migration through metalloproteinase‐cleavage proHB‐EGF in human colon carcinoma cells. Cytokine 2005; 29: 275–82. [DOI] [PubMed] [Google Scholar]
- 61. Ongusaha PP, Kwak JC, Zwible AJ et al. HB‐EGF is a potent inducer of tumor growth and angiogenesis. Cancer Res 2004; 64: 5283–90. [DOI] [PubMed] [Google Scholar]
- 62. Jayne DG, Perry SL, Morrison E, Farmery SM, Guillou PJ. Activated mesothelial cells produce heparin‐binding growth factors: implications for tumour metastases. Br J Cancer 2000; 82: 1233–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Thogersen VB, Sorensen BS, Poulsen SS, Orntoft TF, Wolf H, Nexo E. A subclass of HER1 ligands are prognostic markers for survival in bladder cancer patients. Cancer Res 2001; 61: 6227–33. [PubMed] [Google Scholar]
- 64. Kobrin MS, Funatomi H, Friess H, Buchler MW, Stathis P, Korc M. Induction and expression of heparin‐binding EGF‐like growth factor in human pancreatic cancer. Biochem Biophys Res Commun 1994; 202: 1705–9. [DOI] [PubMed] [Google Scholar]
- 65. Ito Y, Takeda T, Higashiyama S et al. Expression of heparin binding epidermal growth factor‐like growth factor in hepatocellular carcinoma: an immunohistochemical study. Oncol Rep 2001; 8: 903–7. [DOI] [PubMed] [Google Scholar]
- 66. Ito Y, Higashiyama S, Takeda T, Yamamoto Y, Wakasa KI, Matsuura N. Expression of heparin‐binding epidermal growth factor‐like growth factor in pancreatic adenocarcinoma. Int J Pancreatol 2001; 29: 47–52. [DOI] [PubMed] [Google Scholar]
- 67. Uchida T, Gill DM, Pappenheimer AM Jr. Mutation in the structural gene for diphtheria toxin carried by temperate phage. Nat New Biol 1971; 233: 8–11. [DOI] [PubMed] [Google Scholar]
- 68. Mitamura T, Higashiyama S, Taniguchi N, Klagsbrun M, Mekada E. Diphtheria toxin binds to the epidermal growth factor (EGF)‐like domain of human heparin‐binding EGF‐like growth factor/diphtheria toxin receptor and inhibits specifically its mitogenic activity. J Biol Chem 1995; 270: 1015–9. [DOI] [PubMed] [Google Scholar]
- 69. Buzzi S. Diphtheria toxin treatment of human advanced cancer. Cancer Res 1982; 42: 2054–8. [PubMed] [Google Scholar]
- 70. Buzzi S, Rubboli D, Buzzi G, Buzzi AM, Morisi C, Pironi F. CRM197 (nontoxic diphtheria toxin): effects on advanced cancer patients. Cancer Immunol Immunother 2004; 53: 1041–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. El‐Rayes BF, LoRusso PM. Targeting the epidermal growth factor receptor. Br J Cancer 2004; 91: 418–24. [DOI] [PMC free article] [PubMed] [Google Scholar]