

Abstract
From a normal human brain phage display library screen we identified the gamma (A)-globin chain of fetal hemoglobin (Hb F) as a protein that bound strongly to Aβ1-42. We showed the oxidized form of adult Hb (metHb A) binds with greater affinity to Aβ1-42 than metHb F. MetHb is more toxic than oxyhemoglobin because it loses its heme group more readily. Free Hb and heme readily damage vascular endothelial cells similar to Alzheimer's disease (AD) vascular pathology. The XmnI polymorphism (C→T) at −158 of the gamma (G)-globin promoter region can contribute to increased Hb F expression. Using family-based association testing, we found a significant protective association of this polymorphism in the NIMH sibling dataset (n=489) in families, with at least two affected and one unaffected sibling (p=0.006), with an age of onset >50 years (p=0.010) and >65 years (p=0.013), and families not homozygous for the APOE4 allele (p=0.041). We hypothesize that Hb F may be less toxic than adult Hb in its interaction with Aβ and may protect against the development of AD.
Keywords: fetal hemoglobin, gamma globin, methemoglobin, heme, neurological, vascular disease, polymorphism, amyloid
1. Introduction
1.1. Aβ and Cerebral Amyloid Angiopathy
Alzheimer's Disease (AD), a neurodegenerative disorder with a complex etiology and pathogenesis, is characterized by progressive loss of memory and cognitive functions. Beta amyloid peptide (Aβ) appears to be central to the pathogenesis of AD. Derived from the amyloid protein precursor, Aβ aggregates in plaques in the brain and in cerebral vessels are a diagnostic feature of AD [37]. Aβ deposited in plaques in the walls of cerebral blood vessels causes cerebral amyloid angiopathy (CAA) [100], and is available to bind to circulating cells including erythrocytes [65, 71, 76]. Amyloid deposits in cerebral vessels can obliterate the lumens of cerebral arteries and damage the endothelium and basal lamina, causing a breakdown of the blood brain barrier (BBB), ischemia, and neurodegeneration [51, 71, 102]. Interestingly, vascular changes, including hypoperfusion, appear to precede evidence of neuronal injury [21]. Morphological and inflammatory changes are consistently observed in the brain vasculature in AD [21, 95].
1.2. Aβ Binding with Hb
Proteins that interact with Aβ may activate or enhance its deposition or pathogenicity, and thus contribute to the development of AD. While isolating specific proteins with strong affinity for Aβ, one group of investigators reported the α chain of hemoglobin (Hb) as one of only a few proteins recovered and identified from rat brain homogenates [72]. In addition, Hb has been co-immunoprecipitated with Aβ from both AD brains and plasma [53, 72, 105].
1.3. Hb and Heme in AD Brains and its Vasculature
Abnormal levels of Hb and heme have been associated with brain and vascular tissue in AD. Hb, Hb derived peptides, and Hb mRNA levels have been reported to be increased in AD brains relative to the brains of non-demented control subjects [17, 75, 85, 91, 105]. Brain Hb levels in AD were highest in the hippocampus and parietal gray and white matter and lowest in the cerebellum, and there was co-localization of Hb with senile plaques and CAA [105]. The increased presence of Hb and its breakdown products in the brain is probably derived from erythrocytes in the circulation as a result of injury to the endothelium and the BBB with subsequent leakage of plasma or blood components into the perivascular space where additional heme iron-mediated damage may occur [17].
1.4. Aβ Damage to Erythrocytes
Circulating blood cells are exposed to both soluble Aβ1-40/42 and to Aβ aggregates associated with the luminal surfaces of cerebral microvessels (e.g., CAA) [76, 79]. In vitro, Aβ25-35 induces rapid lysis of human erythrocytes, whereas Aβ1-42 induces delayed lysis of erythrocytes that can be attenuated by antioxidants [63]. Human red blood cells (RBC) that bind fibrillar amyloid in vitro and in vivo show increased RBC hemoglobin oxidative modification and endothelial adhesion [27, 42, 76, 79]. AD patients have increased RBC membrane injury suggesting the increased potential for erythrocyte lysis and liberation of Hb when the cells are exposed to Aβ1-42 [11, 36, 63, 92].
1.5 Oxidized Heme and Damage to Vascular Cells
Extracellular or free Hb released from lysed erythrocytes (oxyHb; Fe+2) causes injury to endothelial cells [5, 59, 81] and death of cultured neurons [24, 77, 83]. It also undergoes spontaneous oxidation to methemoglobin (metHb, Fe+3) which loses its heme group more readily than oxyHb [24, 69]. Oxidized heme is a pro-oxidant that damages vascular cells, where the iron derived from heme becomes available to produce a variety of reactive oxygen species via the Fenton reaction, [46, 101] resulting in membrane lipid peroxidation and damage to DNA and proteins [40, 47, 61]. These actions could account for some of the vascular pathology and neuronal injury or death in AD. Additionally, competition for freed Hb, outside of the normal haptoglobin and related scavenging systems, may permit or enhance vascular injury.
We present here our results of a phage display screen of a human brain cDNA library to identify proteins that interact with Aβ in which and the gamma (A)-globin subunit of fetal hemoglobin (Hb F) was identified. We also present surface plasmon resonance studies that show differential binding of adult Hb (Hb A) and Hb F, in oxidized and reduced states, to Aβ1-42. Specifically, metHb F showed reduced affinity for binding to Aβ relative to metHb A. Hb F contains two gamma chains (either A or G) in place of the two beta chains of Hb A [67]. Ten to thirty-five percent of persons in the general population have a common promoter polymorphism, C-T−158 XmnI, in the gene coding for the gamma (G)-globin chain, HBG2, that can contribute to increased levels of the usually small amount of Hb F present in normal individuals and further increases in levels when they are under hematological stress [29, 33, 39, 57, 104]. We genotyped the XmnI polymorphism in the NIMH AD cohort and present the results here. We discuss the implications of these results and how Hb may play a role in the pathogenesis of AD.
2. Methods
2.1 Phage display screening for amyloid-binding proteins
A human brain phage display human brain cDNA library (#K1006-2 & #HL6001XA, Clontech, Mountain View, CA) was used to screen for binding partners for aggregated Aß1-42 following the manufacturers' directions (user manual Clontech PT3084-1). The phage were biopanned against immobilized Aß1-42 (#H-1368, Bachem, Torrance, CA). Aß1-42 was dissolved in water (10 μg/ml) and 100 μl was add to each well of a 96-well high-capacity protein binding plate (#9502-920-00P, Labsystems, Franklin MA). Binding and aggregation of Aß1-42 in 96-well plates and preparation of negative control plates follow general methods [9, 14, 49]. A negative control plate was baited using a scrambled Aß25-35 sequence (KSGNMLGIIAG) [9]. After three rounds of biopanning, a subset of eighty-seven individual clones from the final enriched phage library pan were screened for binding to immobilized Aß1-42 and scrambled Aß25-35. The amount of specifically-bound phage was assessed by detecting pVIII coat protein in an ELISA format. Clones exhibiting absorbance ratios ≥ 2.5 were processed for plasmid DNA isolation using Wizard Plus SV Minipreps (#A1330; Promega Corporation, Madison WI). Isolated plasmids were sequenced in the Medical College of Georgia Molecular Biology Core Facility using the dye terminator method (ABI Prism Model 377) and the 5′-sequencing primer provided with the phage library.
2.2 Surface plasmon resonance protein-protein interaction assay
A BIACORE X instrument (Piscataway, NJ) with 2 microfluidic flow cells (fc) was used for analysis of unlabeled protein-protein interactions. Amyloid was allowed to aggregate and buffers were prepared as previously described [90]. Control analytes and the experimental analytes were evaluated for interaction with immobilized Aβ1-42. Each analyte was injected at a concentration of 1 uM in running/sample buffer (10 mM Na2PO4, 150 mM NaCl, pH 7.4) across the fc1 (control) and fc2 (Aβ1-42) surfaces at flow rate of 20 ul/min for 3 min, then buffer alone was flowed over the surfaces for at least an additional minute to determine the disassociation kinetics. Following each analyte injection, the control and active flow cells are regenerated with 4 M Gdn HCl (10 mM Tris HCl, pH 8.0). Binding data are corrected by subtracting the response observed in fc1 (control) from that observed in fc2 (Aβ1-42). Dissociation equilibrium constants and rate constants were determined by fitting corrected data to a 1:1 Langmuir binding model using global analysis (BIAevaluation 3.0 Software) and reported data was repeated at least twice.
For flow cell surface preparation, aggregated Aβ1-42 (#H-1368, Bachem) was immobilized in fc2 of a CM5 sensor chip by amine coupling according to the manufacturer's guidelines (amine coupling kit, Biacore Inc,). Following activation of the carboxymethylated dextran surface, 35 ul of Aβ1-42 (100 g/ml in 10 mM CH3COONa, pH 4.0) was injected across the activated surface at flow rate of 5 ul/min. Following Aβ1-42 immobilization, unreacted surface groups were blocked as usual. Non-covalently bound Aβ1-42 is removed by multiple injections (20 ul at 20 ul/min) of regeneration buffer (4 M Gdn HCl). A stable Aβ1-42 baseline of 1700 response units (RUs; 1000 RUs = 1 ng protein/mm2) was achieved following four regeneration injections.
Control analytes consisted of positive [Apolipoprotein E4 (APOE4) (#178476, Calbiochem/EMD Biosciences, San Diego, CA); anti-amyloid antibody (mouse IgG1 monoclonal antibody to beta-amyloid, #M0872, DAKO, Carpinteria, CA)] and negative controls [bovine serum albumin (BSA, #A7638, Sigma, St. Louis, MO); anti-neurofilament antibody (mouse IgG1 monoclonal antibody to phosphorylated NFH & NFM, #SMI-31, Sternberger Monoclonals Inc., Lutherville, MD)]. The same anti-beta amyloid antibody was used to determine if the amyloid peptide surface was still intact following multiple analyte and regeneration runs.
Experimental analytes consisted of adult methemoglobin (metHb A), adult oxyhemoglobin (oxyHb A), fetal methemoglobin (metHb F), fetal oxyhemoglobin (oxyHb F), and gamma (G) globin chains. Hbs were obtained by the Medical College of Georgia Sickle Cell Center from blood samples donated by patient volunteers using IRB approved informed consent forms. Hbs F were obtained from a beta thalasemia patient (>90% Hb F) or newborns using left over blood samples acquired for clinical tests under IRB approval; No patient identifiers were associated with clinical samples. MetHb was derived from fresh oxyHb that was oxidized by exposure to air at 4° C for several weeks and assayed by cooximetry as being over 95% metHb. Hbs were liberated from washed (Locke's solution, 154 mM NaCl, 5.6 mM KCl, 2.3 mM CaCl2, 1 mM MgCl2, 3.6 mM NaHCO3, 5 mM d-glucose) RBC by exposure to 4x volumes of distilled H2O. Cell debris was removed by centrifugation (12,000 × g 10 minutes) and the supernatant was used. In some cases commercially available adult human metHb was also used (#H7379, Sigma). Individual globin chains were derived by HPLC separation as previously described [54].
2.3 Study population: NIMH AD Genetic Initiative families
The family study group comprised 209 families with at least two siblings with AD with mean age of onset (MAO) 70.9 ± 7.4 (range 50-97 years) years and one unaffected sibling (used as a control). This group (candidate gene set) is a subset of a larger group of families collected as part of the NIMH AD Genetics Initiative [10] following a standardized protocol utilizing the NINCDS-ADRDA criteria for diagnosis of definite and probable AD [99]. Because parental genotypes are usually not available for this dataset, family-based association testing (FBAT) was used [55]. The candidate gene set containing 203 families with age of onset ≥50 years (non-early families) was further subdivided into two subsets by age of onset: 61 non-late onset families (age of onset between 50 and 65 years) and 142 late onset families (age of onset > 65 years). Six families with ages of onset under 50 that were part of the total set were excluded from this set (non-early). This set of 203 non-early families was also classified by APOE4 dosage, wherein 64 families were designated as APOE4E4 member (at least one member of the family is homozygous for the E4 allele) and 139 families classified as no member E4E4 (no individuals in the family possessing the E4E4 genotype). Individuals who were heterozygous for the APOE3/E4 genotype are included in families in both of these subsets. Stratifying on homozygous APOE4 status rather than carrier status separates families with strong APOE4 presence from those with no or less APOE4 presence.
2.4 XmnI genotyping of -158 polymorphism of HBG2 in the NIMH AD cohort
Isolation of genomic DNA and general procedures for PCR-RFLP genotyping have been described [74]. Reagents and conditions to amplify the XmnI polymorphism (C →T) at bp −158 of HBG2 were, 0.5 pmole each of left (5'-GCACTGAAACTGTTGCTTTATAGGAT-3') and right primers (5'-GCGTCTGGACTAGGAGCTTATTG-3'), 0.5 U of Taq (Promega, Madison, WI) and 37 cycles of 94°C/40 sec, 55°C/30 sec, 72°C/1 min. with a final extension of 72°C/8 min. After cycling, temperature was lowered to 12°C/30 min. to reduce condensation on the microtiter plates. Three μL of product was digested with 2 U of XmnI (N. E. Biolabs, Beverly, MA) in a 12 μL reaction at 37°C/4 hours. Digestion products were run on 1.5% agarose gel, and photographed on a Fluor-S Imager (Bio-Rad, Hercules, CA).
3. Results
3.1 Amyloid-binding proteins isolated from phage display clones
The absorbance ratios (Aß1-42 well/scrambled Aß25-35 well ) ranged from 0.85-3.14 absorbance units. We selected a subset of clones that bound to Aß1-42 coated wells, but not to scrambled Aß25-35 coated wells and with absorbance ratios greater than 2.5. These clones were isolated and sequenced. Three clones had novel sequences bearing receptor-like motifs (data not shown). One of the positive clones corresponded to a fragment (Leu81 - Val137) of the gamma (A)-globin subunit of Hb F; this peptide sequence included the heme binding site.
3.2 Surface plasmon resonance of Aβ1-42 binding
Since there is evidence that the Aβ peptide can exert its neurotoxic effects via iron-mediated oxidative damage [82, 87, 88], we further investigated and characterized the interaction and binding properties of Aβ to hemoglobin, which contains bound iron, by performing surface plasmon analysis. Figure 1 shows that the amyloid surface in fc2 binds the positive control analytes, APOE4 and the amyloid antibody, but does not bind the negative controls, BSA or a non-specific antibody (SMI-31) of the same isotype (IgG1). Analysis of the surface resonance data from the Hb analytes (Figure 2) fit a 1:1 Langmuir binding model (BIAevaluation 3.0) using the kinetic parameters listed in Table 1. This suggests that the globin chain, without an attached heme group, and both the adult and fetal metHbs bind to Aβ1-42 with a single on and off rate, indicating that there is a single binding site on the analytes and the ligand. Table 1 also shows the gamma (G)-globin chain and the metHbs have relatively fast and roughly equivalent on rates with slow off rates. However, the metHbs, particularly metHb A, have slower off rates relative to the naked globin chain. In contrast, adult or fetal oxyHbs exhibit no significant binding to Aβ1-42.
Figure 1.
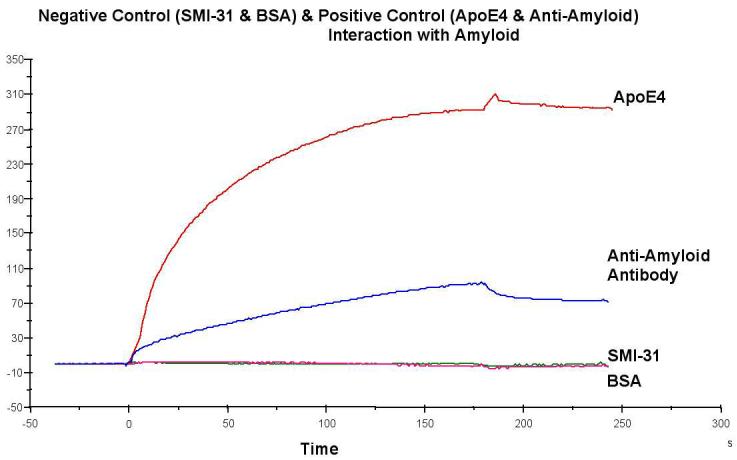
Positive controls (1M APOE4 and 7nM anti-amyloid monoclonal antibody) and negative controls (1M monoclonal antibody SMI-31 and 1M BSA). Data shown is corrected for bulk effects by subtracting the response observed in fc1 from that observed in fc2.
Figure 2.
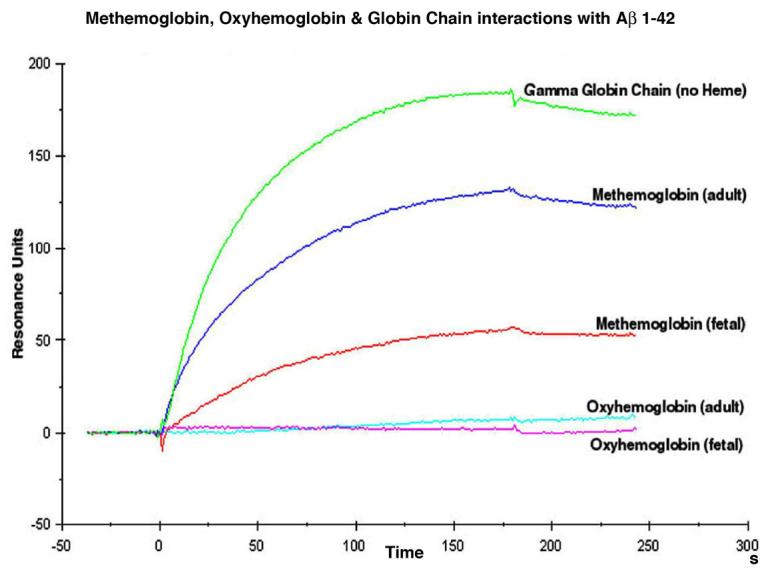
Data shown is corrected for bulk effects by subtracting the response observed in fc1 from that observed in fc2. Binding of the amyloid peptide (Aβ1-42) appears to be mediated by the redox state of the heme iron. Globin without a heme group or hemoglobin where the heme iron is oxidized to Fe+3 (metHb) is permissive for binding. In contrast, when the heme iron is in the Fe+2 reduced state (oxyHb), binding is blocked.
Table 1.
Surface Plasmon Resonance Kinetic Analysis: Fit to a 1:1 Langmuir binding model
| Analyte | ka (1/Ms) | kd (1/s) | KD (M) | Rmax (RU) | Chi2 |
|---|---|---|---|---|---|
| γ (G)-globin | 21,900 ± 99 |
1.13e-3 ± 3.58e-5 |
5.19e-8 | 196 ± 0.551 |
1.38 |
| MetHb A | 17,600 ± 144 |
2.20e-7 ± 5.22e-8 |
1.25e-11 | 130 ± 0.197 |
1.38 |
| MetHb F | 12,900 ± 237 |
5.76e-4 ± 8.30e-5 |
4.47e-8 | 63.4 ± 0.722 |
1.38 |
| OxyHb A | NF | NF | NF | NF | NF |
| OxyHb F | NF | NF | NF | NF | NF |
Kinetic data from Figure 2: Standard errors are located below the values. An association rate constant, ka, of 2e4 means that 20,000 binding events occur per second when the concentration of the analyte is 1M. A disassociation rate constant, kd, of 1e-3 means that 0.001 complexes fall apart per second when the concentration of the analyte is 1M. NF = does not fit the 1:1 Langmuir model. Rmax is maximum response unit (RU).
3.3 XmnI polymorphism of the gamma (G) globin gene (HBG2)
Results of the family based association analyses (FBAT) [55] for the XmnI polymorphism (C →T) at bp −158 of HBG2 in the candidate gene set of DNA samples and the other subsets are shown in Table 2. There was a significant negative association of the XmnI promoter polymorphism with the presence of AD in the total candidate gene set and the non-early (age of onset > 50 years), late onset (age of onset > 65 years), and no member E4E4 (no individuals in the family possessing an E4E4 genotype) subsets. The T allele of the XmnI promoter polymorphism was transmitted less frequently to patients with AD than the normal (C) allele.
Table 2.
P-Values for −158 (C→T) Polymorphism of HBG2
| Subset of DNA | Z Statistic | P-Value (dominant model) |
|---|---|---|
| Total Candidate Gene Set (209 families) | −2.773 | 0.006* |
| Age of Onset ≥50 Subset (203 families) | −2.570 | 0.010* |
| Age of Onset 50-65 Subset (61 families) | −0.865 | 0.387 |
| Age if Onset > 65 Subset (142 families) | −2.471 | 0.013* |
| APOE4E4 Member Subset (64 families) | −1.454 | 0.146 |
| No E4E4 Member Subset (139 families) | −2.045 | 0.041* |
Z Statistic for T allele: A positive value of Z statistic indicates more transmission and a negative value is less transmission to affected individuals than expected under the null hypothesis
The association appears to be more prominent in the late-onset families than in the early-onset families, which is reflected in its association in the no member E4E4subset. The latter subset has a higher age of onset because the E4 allele is associated with earlier onset of disease [56]. When power analysis was performed, an effect size which resulted in 80% power in the larger groups (total, late, no member E4E4), yielded < 50% power in the smaller subsets (non-late, E4E4 member). Therefore, the non-significant results for these latter subsets could be due to insufficient power.
4. Discussion
From a phage display screen of a human brain cDNA library seeking Aβ-binding proteins, we identified the gamma (A)-globin subunit of fetal hemoglobin as a binding partner. In that only a sub-population of clones in the phage display were sequenced and identified, we did not determine if other globin chains besides the gamma (A) chain also bound to Aβ using this approach. However, the coding regions of HBG1 and HBG2, that code for the gamma (A) and gamma (G) globin chains of Hb F, respectively, exhibit a high degree of homology differing only by a single base pair[86]. Therefore, it is likely that the gamma (G)-globin chain binds Aβ with the same high avidity as the gamma (A) chain. Furthermore, additional Aβ binding proteins, such as APOE, APOJ, and alpha 2 macroglobulin, could have been present in clones not selected and sequenced by us.
Our surface plasmon resonance studies confirmed that both Hb A and Hb F specifically bind to Aβ1-42. However, binding occurs only when the iron in the heme group is in the oxidized state (metHb), and that metHb A binds to Aβ with significantly higher affinity than metHb F (Table 1). Furthermore, the globin chains lacking the heme prosthetic group avidly bind amyloid peptide. This suggests that the binding of Aβ peptides to Hb is primarily via the globin polypeptide, rather than directly to the heme group. These studies also demonstrate that the binding of the globin chain and metHbs to Aβ are in the range of efficient ligand-receptor binding, although somewhat lower than the known Aβ binding protein, APOE4, which binds strongly to Aβ.
There is in vivo and in vitro evidence of RBC binding Aβ and of Aβ mediated RBC injury [27, 34, 42, 63, 76]. Indeed, over 50 percent of Aβ in circulation within the vascular compartment may be bound to RBCs [79]. It is clear that RBCs in AD show morphological and biochemical evidence of injury that can lead to hemolysis [19, 23, 32, 41, 48, 76, 78], although it is not clear if hemolytic anemia is associated with AD [8, 26, 64, 68, 73]. Additionally, while it is not known if the Hb isoform content (e.g. adult vs. fetal) of RBCs plays a role in RBC lysis, RBC bound Aβ can cause oxidative injury to Hb within the RBC [42] and Aβ binding to Hb, in particular Hb A as we have demonstrated here, may effect the stabilization of the heme group mediating its release and potential for endothelial injury. Free or oxyHb may not only directly injure the vasculature, including endothelial cells, but may mediate blood flow through NO scavenging, resulting in hypoperfusion to the tissue [4, 35, 43, 59]. Indeed, hypoperfusion may be an early pathogenic event in AD [38, 45, 93].
The haptoglobin/hemopexin system normally binds extracellular (oxyHb) and heme [2]. However, the protective haptoglobin/hemopexin/Albumin/heme oxygenase (HO) systems can be overwhelmed in disease states, including hemolysis and inflammation, resulting in increased circulating levels of free Hb and free (or LDL bound) heme that can attack the endothelium and effect perfusion [6, 43, 69, 70, 81, 97]. Such may be the case in AD when RBC fragility and lysis are increased. Increased free Hb in human serum has recently been found associated with AD relative to controls [109]. This and several other reports indicate that haptoglobin is increased in AD plasma and CSF [44, 62, 107]. This may represent an attempt to adjust the homeostatic level in response to chronic hemolysis, although inflammation, which is also associated with AD, can lead to increased haptoglobin levels [50]. Indeed, a growing list of clinical manifestations (including hemolysis-associated smooth muscle dystonia, vasculopathy, and endothelial dysfunction) are being attributed to hemoglobin release and suggests that hemolysis and hemoglobinemia should be considered as a novel mechanism in various disease states [6, 52, 81, 97].
As mentioned above, free Hb or oxyHb spontaneously oxidizes to form metHb outside of the RBC and, in general, metHb is more toxic than oxyHb because it loses its heme group more readily [24]. [13, 16]. Heme released from metHb damages vascular endothelial cells by catalyzing oxidative injury and lipid peroxidation [15, 58, 59, 96, 106]. The strong affinity of adult metHb for binding Aβ might accelerate the normal “spontaneous” heme release when in the presence of the amyloid peptide or Aβ may preferably enhance the injury and lysis of adult Hb A containing RBCs versus Hb F containing RBCs. In addition, amyloid may interact with free heme groups or generate free radicals in the presence of iron with its tighter association to metHb A.[40, 47, 61]. Recent evidence indicates the Aβ-heme complex acts as a peroxidase, resulting in increased oxidative damage [3].
The above observations are consistent with some of the morphological and inflammatory changes associated with the vasculature in AD [20-22, 80, 95]. This may be, in part mediated by Aβ interactions with RBCs and free Hb, and, in particular, with metHb A.
Hb F expression, restricted to a subset of erythrocytes called F Cells, varies with genetic and environmental factors [12, 31]. It is estimated that eighty-nine percent of adult Hb F and F cell variance is due to heritable factors [30] and the XmnI polymorphism (C→T) at position −158 of the HBG2 promoter accounts for the greatest single share of this genetic variability [28, 94]. High F cell and Hb F levels in normal populations can be due to, at least in part, to the XmnI polymorphism [29, 57, 84, 89, 103, 108]. Additionally, with hematological stress (e.g. Sickle Cell disease, Beta Thalasemia, hemolysis, pregnancy), the XmnI polymorphism has been shown to be at least partly responsible for an even greater increase in total Hb F and F cell levels [1, 7, 18, 25, 33, 60, 66, 94, 98].
The decreased transmission of the T allele in the AD patients relative to the unaffecteds that we found may indicate that the presence of the T allele provides a protective mechanism due to the higher Hb F:Hb A ratio. Because metHb F does not bind Aβ1-42 as avidly as metHb A, the interaction of metHb F with Aβ may be less toxic than the interaction of metHb A with Aβ resulting in less RBC lysis and damage to the vascular epithelium. Evidence for the contribution of the XmnI polymorphism in the protective role of Hb F against vascular damage has been observed in patients with sickle cell disease [1, 66, 94]. These results suggest that further investigation into the effects of the XmnI polymorphism upon Hb F levels in AD subjects may provide additional evidence supporting this hypothesis.
We also genotyped this polymorphism in a small cohort of African American AD patients (n=126) and age-matched controls (n=93) (data not shown). Although we did not show any statistically significant association, the direction of the outcomes was the same (C allele increased in AD patients). The lack of significance could be explained by small sample size and therefore, lack of statistical power.
In conclusion, we identified the gamma subunit of Hb F from the initial screening of a normal human brain phage display library while isolating proteins that bind to Aβ1-42. Using plasmon studies, we further investigated the kinetics of this interaction and found there was much stronger binding of oxidized or met Hb (Fe+3) to Aβ1-42 when compared to reduced or oxy Hb (Fe+2), and that oxidized adult Hb (metHb A, 2α2β chains) binds with much greater affinity to Aβ1-42 than the oxidized form of fetal Hb (metHb F, 2α2γ) Becauese the XmnI polymorphism in the promoter region of the gene, HBG2, which codes for the gamma (G)-globin subunit of Hb F is the most common known genetic factor that contributes to adult Hb F expression, we genotyped this polymorphism in the NIMH cohort of AD patients and unaffected siblings. We observed a significant association of AD with a higher frequency of the ‘normal’ C allele of the HBG2 polymorphism. In contrast, unaffected individuals showed greater transmission of the T allele, which has been reported to be associated with increased adult HBG2 expression and Hb F levels. The increased percentage of Hb F in the circulating blood, if present, could be a mechanism that reduces damage to vascular epithelium due to the decreased binding affinity that metHb F has for Aβ1-42 in comparison to metHb A, or the reduced injury and lysis of Hb F containing RBCs (F Cells) versus Hb A containing RBCs. Specifially, if there is increased Hb F due to the presence of the T allele, it could result in: 1) decreased Aβ interaction with Hb A resulting in less interference with the Hb scavenging systems and/or 2) reduced interaction of Aβ with free or oxyHb resulting in reduced heme release and subsequent heme mediated injury; and/or 3) diminished Aβ mediated lysis of F Cells relative to Hb A containing RBCs resulting in reduced oxyHb and heme and their toxic effects upon the vascular cells. Further, decreased RBC lysis and Hb/heme mediated damage to the microvasculature may help reduce hypoperfusion and inflammation in the brain as well. As such, results of this study suggest further investigations into the association of the XmnI polymorphism to AD, the effects of the XmnI polymorphism upon Hb F levels in AD subjects, and the role of differential binding of Aβ1-42 with Hb isoform content of RBC are needed.
5. Acknowledgements
This research was supported by NIMH grants R01-NS045934-05, NINDS R29NS32835-01(WDH), Medical College of Georgia MCGRI grant (WDH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. Disclosure Statement:
I so state for myself and on behalf of the other authors on the manuscript, Hemoglobin binding to Aβ and HBG2 SNP association suggest a role in Alzheimer's disease, that we have no conflicts and sources of funding that influence the results of this paper, the data is not published or submitted elsewhere, and that appropriate approval and procedures were used concerning human subjects.
I also verify that all authors on this manuscript have reviewed the contents of the manuscript, approve of its contents, and validate the accuracy of the data.

Rodney T. Perry, Ph.D.- 1st and corresponding author
7. References
- 1.Adekile AD, Yacoub F, Gupta R, Sinan T, Haider MZ, Habeeb Y, Al-Bloushi M, Moosa A. Silent brain infarcts are rare in Kuwaiti children with sickle cell disease and high Hb F. Am J Hematol. 2002;70(3):228–31. doi: 10.1002/ajh.10143. [DOI] [PubMed] [Google Scholar]
- 2.Anderson GJ, Frazer DM. Hepatic iron metabolism. Semin Liver Dis. 2005;25(4):420–32. doi: 10.1055/s-2005-923314. [DOI] [PubMed] [Google Scholar]
- 3.Atamna H, Boyle K. Amyloid-{beta} peptide binds with heme to form a peroxidase: Relationship to the cytopathologies of Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:3381–6. doi: 10.1073/pnas.0600134103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Azarov I, Huang KT, Basu S, Gladwin MT, Hogg N, Kim-Shapiro DB. Nitric oxide scavenging by red blood cells as a function of hematocrit and oxygenation. J Biol Chem. 2005;280(47):39024–32. doi: 10.1074/jbc.M509045200. [DOI] [PubMed] [Google Scholar]
- 5.Balla J, Jacob HS, Balla G, Nath K, Eaton JW, Vercellotti GM. Endothelial-cell heme uptake from heme proteins: induction of sensitization and desensitization to oxidant damage. Proc Natl Acad Sci USA. 1993;90(20):9285–9. doi: 10.1073/pnas.90.20.9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balla J, Vercellotti GM, Jeney V, Yachie A, Varga Z, Eaton JW, Balla G. Heme, heme oxygenase and ferritin in vascular endothelial cell injury. Mol Nutr Food Res. 2005;49(11):1030–43. doi: 10.1002/mnfr.200500076. [DOI] [PubMed] [Google Scholar]
- 7.Bandyopadhyay S, Mondal BC, Sarkar P, Chandra S, Das MK, Dasgupta UB. Two beta-globin cluster-linked polymorphic loci in thalassemia patients of variable levels of fetal hemoglobin. Eur J Haematol. 2005;75(1):47–53. doi: 10.1111/j.1600-0609.2005.00416.x. [DOI] [PubMed] [Google Scholar]
- 8.Beard CM, Kokmen E, O'Brien PC, Ania BJ, Melton LJ., 3rd Risk of Alzheimer's disease among elderly patients with anemia: population-based investigations in Olmsted County, Minnesota. Ann Epidemiol. 1997;7(3):219–24. doi: 10.1016/s1047-2797(97)00015-x. [DOI] [PubMed] [Google Scholar]
- 9.Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell. 1994;77(6):817–27. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 10.Blacker D, Bertram L, Saunders AJ, Moscarillo TJ, Albert MS, Wiener H, Perry RT, Collins JS, Harrell LE, Go RC, Mahoney A, Beaty T, Fallin MD, Avramopoulos D, Chase GA, Folstein MF, McInnis MG, Bassett SS, Doheny KJ, Pugh EW, Tanzi RE. Results of a high-resolution genome screen of 437 Alzheimer's disease families. Hum Mol Genet. 2003;12(1):23–32. doi: 10.1093/hmg/ddg007. [DOI] [PubMed] [Google Scholar]
- 11.Bosman GJ, Bartholomeus IG, de Man AJ, van Kalmthout PJ, de Grip WJ. Erythrocyte membrane characteristics indicate abnormal cellular aging in patients with Alzheimer's disease. Neurobiol Aging. 1991;12(1):13–8. doi: 10.1016/0197-4580(91)90033-g. [DOI] [PubMed] [Google Scholar]
- 12.Boyer SH, Belding TK, Margolet L, Noyes AN. Fetal hemoglobin restriction to a few erythrocytes (F cells) in normal human adults. Science. 1975;188(4186):361–3. doi: 10.1126/science.804182. [DOI] [PubMed] [Google Scholar]
- 13.Bunn HF, Jandl JH. Exchange of heme among hemoglobins and between hemoglobin and albumin. J. Biol Chem. 1968;243(3):465–75. [PubMed] [Google Scholar]
- 14.Burdick D, Soreghan B, Kwon M, Kosmoski J, Knauer M, Henschen A, Yates J, Cotman C, Glabe C. Assembly and aggregation properties of synthetic Alzheimer's A4/beta amyloid peptide analogs. J Biol Chem. 1992;267(1):546–54. [PubMed] [Google Scholar]
- 15.Chiu DT, Liu TZ. Free Radical and Oxidative Damage in Human Blood Cells. J Biomed Sci. 1997;4(5):256–259. doi: 10.1007/BF02253426. [DOI] [PubMed] [Google Scholar]
- 16.Chiu DT, van den Berg J, Kuypers FA, Hung IJ, Wei JS, Liu TZ. Correlation of membrane lipid peroxidation with oxidation of hemoglobin variants: possibly related to the rates of hemin release. Free Radic Biol Med. 1996;21(1):89–95. doi: 10.1016/0891-5849(96)00035-4. [DOI] [PubMed] [Google Scholar]
- 17.Cullen KM, Kocsi Z, Stone J. Pericapillary haem-rich deposits: evidence for microhaemorrhages in aging human cerebral cortex. J Cereb Blood Flow Metab. 2005;25(12):1656–67. doi: 10.1038/sj.jcbfm.9600155. [DOI] [PubMed] [Google Scholar]
- 18.De Angioletti M, Lacerra G, Pagano L, Alessi M, D'Avino R, Manca L, Carestia C. Beta-thalassaemia-87 C-->G: relationship of the Hb F modulation and polymorphisms in compound heterozygous patients. Br J Haematol. 2004;126(5):743–9. doi: 10.1111/j.1365-2141.2004.05089.x. [DOI] [PubMed] [Google Scholar]
- 19.De Franceschi L, Olivieri O, Corrocher R. Erythrocyte aging in neurodegenerative disorders. Cell Mol Biol (Noisy-le-grand) 2004;50(2):179–85. [PubMed] [Google Scholar]
- 20.De Jong GI, De Vos RA, Steur EN, Luiten PG. Cerebrovascular hypoperfusion: a risk factor for Alzheimer's disease? Animal model and postmortem human studies. Ann N Y Acad Sci. 1997;826:56–74. doi: 10.1111/j.1749-6632.1997.tb48461.x. [DOI] [PubMed] [Google Scholar]
- 21.de La Torre JC. Vascular Basis of Alzheimer's Disease Pathogenesis. Ann. N.Y. Acad. Sci. 2002;977:196–215. doi: 10.1111/j.1749-6632.2002.tb04817.x. [DOI] [PubMed] [Google Scholar]
- 22.de la Torre JC. Is Alzheimer's disease preceded by neurodegeneration or cerebral hypoperfusion? Ann Neurol. 2005;57(6):783–4. doi: 10.1002/ana.20516. [DOI] [PubMed] [Google Scholar]
- 23.Engstrom I, Ronquist G, Pettersson L, Waldenstrom A. Alzheimer amyloid beta-peptides exhibit ionophore-like properties in human erythrocytes. Eur J Clin Invest. 1995;25(7):471–6. doi: 10.1111/j.1365-2362.1995.tb01732.x. [DOI] [PubMed] [Google Scholar]
- 24.Everse J, Hsia N. The toxicities of native and modified hemoglobins. Free Radic Biol Med. 1997;22(6):1075–99. doi: 10.1016/s0891-5849(96)00499-6. [DOI] [PubMed] [Google Scholar]
- 25.Ferrara M, Matarese SM, Francese M, Borrelli B, Perrotta A, Meo A, La Rosa MA, Esposito L. Role of polymorphic sequences 5′ to the G(gamma) gene and 5′ to the beta gene on the homozygous beta thalassemic phenotype. Hemoglobin. 2003;27(3):167–75. doi: 10.1081/hem-120023380. [DOI] [PubMed] [Google Scholar]
- 26.Fujiwara Y, Takahashi M, Tanaka M, Hoshi T, Someya T, Shinkai S. Relationships between plasma beta-amyloid peptide 1-42 and atherosclerotic risk factors in community-based older populations. Gerontology. 2003;49(6):374–9. doi: 10.1159/000073765. [DOI] [PubMed] [Google Scholar]
- 27.Galeazzi L, Galeazzi R, Valli MB, Corder EH, Giunta S. Albumin protects human red blood cells against Abeta25-35-induced lysis more effectively than ApoE. Neuroreport. 2002;13(16):2149–54. doi: 10.1097/00001756-200211150-00032. [DOI] [PubMed] [Google Scholar]
- 28.Garner C, Tatu T, Game L, Cardon LR, Spector TD, Farrall M, Thein SL. A candidate gene study of F Cell levels in sibiling pairs using joint linkage and association analysis. Genescreen. 2000;1:9–14. [Google Scholar]
- 29.Garner C, Menzel S, Martin C, Silver N, Best S, Spector TD, Thein SL. Interaction between two quantitative trait loci affects fetal haemoglobin expression. Annals of Human Genetics. 2005;69:707–714. doi: 10.1111/j.1529-8817.2005.00188.x. [DOI] [PubMed] [Google Scholar]
- 30.Garner C, Tatu T, Reittie JE, Littlewood T, Darley J, Cervino S, Farrall M, Kelly P, Spector TD, Thein SL. Genetic influences on F cells and other hematologic variables: a twin heritability study. Blood. 2000;95(1):342–346. [PubMed] [Google Scholar]
- 31.Garner CP, Tatu T, Best S, Creary L, Thein SL. Evidence of genetic interaction between the beta-globin complex and chromosome 8q in the expression of fetal hemoglobin. American Journal of Human Genetics. 2002;70(3):793–799. doi: 10.1086/339248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gibson GE, Huang HM. Oxidative processes in the brain and non-neuronal tissues as biomarkers of Alzheimer's disease. Front Biosci. 2002;7:d1007–15. doi: 10.2741/A827. [DOI] [PubMed] [Google Scholar]
- 33.Gilman JG. Expression of G gamma and A gamma globin genes in human adults. Hemoglobin. 1988;12(56):707–16. doi: 10.3109/03630268808991664. [DOI] [PubMed] [Google Scholar]
- 34.Giunta S, Valli MB, Galeazzi R, Fattoretti P, Corder EH, Galeazzi L. Transthyretin inhibition of amyloid beta aggregation and toxicity. Clin Biochem. 2005;38(12):1112–9. doi: 10.1016/j.clinbiochem.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 35.Gladwin MT, Crawford JH, Patel RP. The biochemistry of nitric oxide, nitrite, and hemoglobin: role in blood flow regulation. Free Radic Biol Med. 2004;36(6):707–17. doi: 10.1016/j.freeradbiomed.2003.11.032. [DOI] [PubMed] [Google Scholar]
- 36.Goodall HB, Reid AH, Findlay DJ, Hind C, Kay J, Coghill G. Irregular distortion of the erythrocytes (acanthocytes, spur cells) in senile dementia. Dis Markers. 1994;12(1):23–41. doi: 10.1155/1994/493810. [DOI] [PubMed] [Google Scholar]
- 37.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 38.Hirao K, Ohnishi T, Hirata Y, Yamashita F, Mori T, Moriguchi Y, Matsuda H, Nemoto K, Imabayashi E, Yamada M, Iwamoto T, Arima K, Asada T. The prediction of rapid conversion to Alzheimer's disease in mild cognitive impairment using regional cerebral blood flow SPECT. Neuroimage. 2005 doi: 10.1016/j.neuroimage.2005.06.066. [DOI] [PubMed] [Google Scholar]
- 39.Ho PJ, Hall GW, Luo LY, Weatherall DJ, Thein SL. Beta-thalassaemia intermedia: is it possible consistently to predict phenotype from genotype? Br J Haematol. 1998;100(1):70–8. doi: 10.1046/j.1365-2141.1998.00519.x. [DOI] [PubMed] [Google Scholar]
- 40.Howlett D, Cutler P, Heales S, Camilleri P. Hemin and related porphyrins inhibit beta-amyloid aggregation. FEBS Lett. 1997;417(2):249–51. doi: 10.1016/s0014-5793(97)01290-8. [DOI] [PubMed] [Google Scholar]
- 41.Janoshazi A, Sellal F, Marescaux C, Danion JM, Warter JM, de Barry J. Alteration of protein kinase C conformation in red blood cells: a potential marker for Alzheimer's disease but not for Parkinson's disease. Neurobiol Aging. 2006;27(2):245–51. doi: 10.1016/j.neurobiolaging.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 42.Jayakumar R, Kusiak JW, Chrest FJ, Demehin AA, Murali J, Wersto RP, Nagababu E, Ravi L, Rifkind JM. Red cell perturbations by amyloid beta-protein. Biochim Biophys Acta. 2003;1622(1):20–8. doi: 10.1016/s0304-4165(03)00101-6. [DOI] [PubMed] [Google Scholar]
- 43.Jeney V, Balla J, Yachie A, Varga Z, Vercellotti GM, Eaton JW, Balla G. Pro-oxidant and cytotoxic effects of circulating heme. Blood. 2002;100(3):879–87. doi: 10.1182/blood.v100.3.879. [DOI] [PubMed] [Google Scholar]
- 44.Johnson G, Brane D, Block W, van Kammen DP, Gurklis J, Peters JL, Wyatt RJ, Kirch DG, Ghanbari HA, Merril CR. Cerebrospinal fluid protein variations in common to Alzheimer's disease and schizophrenia. Appl Theor Electrophor. 1992;3(2):47–53. [PubMed] [Google Scholar]
- 45.Johnson KA, Jones K, Holman BL, Becker JA, Spiers PA, Satlin A, Albert MS. Preclinical prediction of Alzheimer's disease using SPECT. Neurology. 1998;50(6):1563–71. doi: 10.1212/wnl.50.6.1563. [DOI] [PubMed] [Google Scholar]
- 46.Juckett M, Zheng Y, Yuan H, Pastor T, Antholine W, Weber M, Vercellotti G. Heme and the endothelium. Effects of nitric oxide on catalytic iron and heme degradation by heme oxygenase. J Biol Chem. 1998;273(36):23388–97. doi: 10.1074/jbc.273.36.23388. [DOI] [PubMed] [Google Scholar]
- 47.Kalaria RN. Cerebrovascular degeneration is related to amyloid-beta protein deposition in Alzheimer's disease. Ann N Y Acad Sci. 1997;826:263–71. doi: 10.1111/j.1749-6632.1997.tb48478.x. [DOI] [PubMed] [Google Scholar]
- 48.Kawamoto EM, Munhoz CD, Glezer I, Bahia VS, Caramelli P, Nitrini R, Gorjao R, Curi R, Scavone C, Marcourakis T. Oxidative state in platelets and erythrocytes in aging and Alzheimer's disease. Neurobiol Aging. 2005;26(6):857–64. doi: 10.1016/j.neurobiolaging.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 49.Kay BK, Winter J, McCafferty J. Phage display of peptides and proteins: a laboratory manual. Academic Press, Inc.; San Diego: 1996. [Google Scholar]
- 50.Kormoczi GF, Saemann MD, Buchta C, Peck-Radosavljevic M, Mayr WR, Schwartz DW, Dunkler D, Spitzauer S, Panzer S. Influence of clinical factors on the haemolysis marker haptoglobin. Eur J Clin Invest. 2006;36(3):202–9. doi: 10.1111/j.1365-2362.2006.01617.x. [DOI] [PubMed] [Google Scholar]
- 51.Krizanac-Bengez L, Mayberg MR, Janigro D. The cerebral vasculature as a therapeutic target for neurological disorders and the role of shear stress in vascular homeostatis and pathophysiology. Neurol Res. 2004;26(8):846–53. doi: 10.1179/016164104X3789. [DOI] [PubMed] [Google Scholar]
- 52.Kumar S, Bandyopadhyay U. Free heme toxicity and its detoxification systems in human. Toxicology Letters. 2005;157(3):175–188. doi: 10.1016/j.toxlet.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 53.Kuo YM, Kokjohn TA, Kalback W, Luehrs D, Galasko DR, Chevallier N, Koo EH, Emmerling MR, Roher AE. Amyloid-B Peptides interact with plasma proteins and Erythrocytes: Implications for their quantitation in plasma. Biochem Biophys Res Commun. 2000;268(3):750–6. doi: 10.1006/bbrc.2000.2222. [DOI] [PubMed] [Google Scholar]
- 54.Kutlar F, Huisman TH. New ultra-micro high performance liquid chromatographic method for determining the gamma chain composition of hemoglobin F in normal adults. J Chromatogr. 1993;620(2):183–9. doi: 10.1016/0378-4347(93)80002-l. [DOI] [PubMed] [Google Scholar]
- 55.Lake SL, Blacker D, Laird NM. Family-based tests of association in the presence of linkage. Am J Hum Genet. 2000;67(6):1515–25. doi: 10.1086/316895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lange C, Laird NM. Power calculations for a general class of family-based association tests: dichotomous traits. Am J Hum Genet. 2002;71(3):575–84. doi: 10.1086/342406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leonova J, Kazanetz EG, Smetanina NS, Adekile AD, Efremov GD, Huisman TH. Variability in the fetal hemoglobin level of the normal adult. Am J Hematol. 1996;53(2):59–65. doi: 10.1002/(SICI)1096-8652(199610)53:2<59::AID-AJH1>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 58.Lin G, Macdonald RL, Marton LS, Kowalczuk A, Solenski NJ, Weir BK. Hemoglobin Increases Endothelin-1 in Endothelial Cells by Decreasing Nitric Oxide. Biochemical and Biophysical Research Communications. 2001;280(3):824–830. doi: 10.1006/bbrc.2000.4167. [DOI] [PubMed] [Google Scholar]
- 59.Liu X, Spolarics Z. Methemoglobin is a potent activator of endothelial cells by stimulating IL-6 and IL-8 production and E-selectin membrane expression. Am. J. Physiol Cell Physiol. 2003;285:C1036–C1046. doi: 10.1152/ajpcell.00164.2003. [DOI] [PubMed] [Google Scholar]
- 60.Lolis D, Georgiou I, Loizou P, Makrydimas G. High Hbf in Pregnancy Is Associated with the Xmn-I Polymorphism at the −158bp of the G-Gamma-Globin Gene. European Journal of Obstetrics Gynecology and Reproductive Biology. 1995;60(2):153–156. doi: 10.1016/0028-2243(95)02105-2. [DOI] [PubMed] [Google Scholar]
- 61.Markesbery WR. The role of oxidative stress in Alzheimer disease. Arch Neurol. 1999;56(12):1449–52. doi: 10.1001/archneur.56.12.1449. [DOI] [PubMed] [Google Scholar]
- 62.Mattila KM, Pirttila T, Blennow K, Wallin A, Viitanen M, Frey H. Altered blood-brain-barrier function in Alzheimer's disease? Acta Neurol Scand. 1994;89(3):192–8. doi: 10.1111/j.1600-0404.1994.tb01660.x. [DOI] [PubMed] [Google Scholar]
- 63.Mattson MP, Begley JG, Mark RJ, Furukawa K. Abeta25-35 induces rapid lysis of red blood cells: contrast with Abeta1-42 and examination of underlying mechanisms. Brain Res. 1997;771(1):147–53. doi: 10.1016/s0006-8993(97)00824-x. [DOI] [PubMed] [Google Scholar]
- 64.McCaddon A, Tandy S, Hudson P, Gray R, Davies G, Hill D, Duguid J. Absence of macrocytic anaemia in Alzheimer's disease. Clin Lab Haematol. 2004;26(4):259–63. doi: 10.1111/j.1365-2257.2004.00618.x. [DOI] [PubMed] [Google Scholar]
- 65.Miao J, Vitek MP, Xu F, Previti ML, Davis J, Van Nostrand WE. Reducing cerebral microvascular amyloid-beta protein deposition diminishes regional neuroinflammation in vasculotropic mutant amyloid precursor protein transgenic mice. J Neurosci. 2005;25(27):6271–7. doi: 10.1523/JNEUROSCI.1306-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miller BA, Olivieri N, Salameh M, Ahmed M, Antognetti G, Huisman TH, Nathan DG, Orkin SH. Molecular analysis of the high-hemoglobin-F phenotype in Saudi Arabian sickle cell anemia. N Engl J Med. 1987;316(5):244–50. doi: 10.1056/NEJM198701293160504. [DOI] [PubMed] [Google Scholar]
- 67.Miller JL. Signaled expression of fetal hemoglobin during development. Transfusion. 2005;45(7):1229–32. doi: 10.1111/j.1537-2995.2005.00182.x. [DOI] [PubMed] [Google Scholar]
- 68.Milward EA, Grayson DA, Creasey H, Janu MR, Brooks WS, Broe GA. Evidence for association of anaemia with vascular dementia. Neuroreport. 1999;10(11):2377–81. doi: 10.1097/00001756-199908020-00029. [DOI] [PubMed] [Google Scholar]
- 69.Minneci PC, Deans KJ, Zhi H, Yuen PS, Star RA, Banks SM, Schechter AN, Natanson C, Gladwin MT, Solomon SB. Hemolysis-associated endothelial dysfunction mediated by accelerated NO inactivation by decompartmentalized oxyhemoglobin. J Clin Invest. 2005;115(12):3409–17. doi: 10.1172/JCI25040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Na N, Ouyang J, Taes YE, Delanghe JR. Serum free hemoglobin concentrations in healthy individuals are related to haptoglobin type. Clin Chem. 2005;51(9):1754–5. doi: 10.1373/clinchem.2005.055657. [DOI] [PubMed] [Google Scholar]
- 71.Nicoll JA, Yamada M, Frackowiak J, Mazur-Kolecka B, Weller RO. Cerebral amyloid angiopathy plays a direct role in the pathogenesis of Alzheimer's disease. Pro-CAA position statement. Neurobiol Aging. 2004;25(5):589–97. doi: 10.1016/j.neurobiolaging.2004.02.003. discussion 603-4. [DOI] [PubMed] [Google Scholar]
- 72.Oyama R, Yamamoto H, Titani K. Glutamine synthetase, hemoglobin alpha-chain, and macrophage migration inhibitory factor binding to amyloid beta-protein: their identification in rat brain by a novel affinity chromatography and in Alzheimer's disease brain by immunoprecipitation. Biochim Biophys Acta. 2000;1479(12):91–102. doi: 10.1016/s0167-4838(00)00057-1. [DOI] [PubMed] [Google Scholar]
- 73.Pandav RS, Chandra V, Dodge HH, DeKosky ST, Ganguli M. Hemoglobin levels and Alzheimer disease: an epidemiologic study in India. Am J Geriatr Psychiatry. 2004;12(5):523–6. doi: 10.1176/appi.ajgp.12.5.523. [DOI] [PubMed] [Google Scholar]
- 74.Perry RT, Collins JS, Harrell LE, Acton RT, Go RCP. Investigation of association of 13 polymorphisms in eight genes in southeastern African American Alzheimer disease patients as compared to age-matched controls. Am J Med Genet. 2001;105(4):332–42. doi: 10.1002/ajmg.1371. [DOI] [PubMed] [Google Scholar]
- 75.Poljak A, McLean CA, Sachdev P, Brodaty H, Smythe GA. Quantification of hemorphins in Alzheimer's disease brains. J Neurosci Res. 2004;75(5):704–14. doi: 10.1002/jnr.20020. [DOI] [PubMed] [Google Scholar]
- 76.Ravi LB, Poosala S, Ahn D, Chrest FJ, Spangler EL, Jayakumar R, Nagababu E, Mohanty JG, Talan M, Ingram DK, Rifkind JM. Red cell interactions with amyloid-beta(1-40) fibrils in a murine model. Neurobiol Dis. 2005;19(12):28–37. doi: 10.1016/j.nbd.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 77.Regan RF, Panter SS. Hemoglobin potentiates excitotoxic injury in cortical cell culture. J Neurotrauma. 1996;13(4):223–31. doi: 10.1089/neu.1996.13.223. [DOI] [PubMed] [Google Scholar]
- 78.Repetto MG, Reides CG, Evelson P, Kohan S, de Lustig ES, Llesuy SF. Peripheral markers of oxidative stress in probable Alzheimer patients. Eur J Clin Invest. 1999;29(7):643–9. doi: 10.1046/j.1365-2362.1999.00506.x. [DOI] [PubMed] [Google Scholar]
- 79.Rogers J, Li R, Mastroeni D, Grover A, Leonard B, Ahern G, Cao P, Kolody H, Vedders L, Kolb WP, Sabbagh M. Peripheral clearance of amyloid beta peptide by complement C3-dependent adherence to erythrocytes. Neurobiol Aging. 2005 doi: 10.1016/j.neurobiolaging.2005.09.043. [DOI] [PubMed] [Google Scholar]
- 80.Roher AE, Kokjohn TA, Beach TG. An association with great implications: vascular pathology and Alzheimer disease. Alzheimer Dis Assoc Disord. 2006;20(1):73–5. doi: 10.1097/01.wad.0000201855.39246.2d. [DOI] [PubMed] [Google Scholar]
- 81.Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. Jama. 2005;293(13):1653–62. doi: 10.1001/jama.293.13.1653. [DOI] [PubMed] [Google Scholar]
- 82.Rottkamp CA, Raina AK, Zhu X, Gaier E, Bush AI, Atwood CS, Chevion M, Perry G, Smith MA. Redox-active iron mediates amyloid-b toxicity. Free Radical Biology and Medicine. 2001;30(4):447–450. doi: 10.1016/s0891-5849(00)00494-9. [DOI] [PubMed] [Google Scholar]
- 83.Sadrzadeh SM, Anderson DK, Panter SS, Hallaway PE, Eaton JW. Hemoglobin potentiates central nervous system damage. J Clin Invest. 1987;79(2):662–4. doi: 10.1172/JCI112865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sampietro M, Thein SL, Contreras M, Pazmany L. Variation of HbF and F-cell number with the G-gamma Xmn I (C-T) polymorphism in normal individuals. Blood. 1992;79(3):832–3. [PubMed] [Google Scholar]
- 85.Schonberger SJ, Edgar PF, Kydd R, Faull RL, Cooper GJ. Proteomic analysis of the brain in Alzheimer's disease: molecular phenotype of a complex disease process. Proteomics. 2001;1(12):1519–28. doi: 10.1002/1615-9861(200111)1:12<1519::aid-prot1519>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 86.Schroeder WA, Huisman TH, Shelton JR, Shelton JB, Kleihauer EF, Dozy AM, Robberson B. Evidence for multiple structural genes for the gamma chain of human fetal hemoglobin. Proc Natl Acad Sci U S A. 1968;60(2):537–44. doi: 10.1073/pnas.60.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schubert D, Chevion M. The role of iron in beta amyloid toxicity. Biochem Biophys Res Commun. 1995;216(2):702–7. doi: 10.1006/bbrc.1995.2678. [DOI] [PubMed] [Google Scholar]
- 88.Schubert D, Behl C, Lesley R, Brack A, Dargusch R, Sagara Y, Kimura H. Amyloid peptides are toxic via a common oxidative mechanism. Proc Natl Acad Sci U S A. 1995;92(6):1989–93. doi: 10.1073/pnas.92.6.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shimizu K, Park KS, Enoki Y. The Xmnl Site 5′ to the G-Gamma-Globin Gene Polymorphism and Its Relationship to Percent-Hb-F and Percent-G-Gamma in Normal Japanese and Korean Adults. Human Heredity. 1992;42(4):253–258. doi: 10.1159/000154078. [DOI] [PubMed] [Google Scholar]
- 90.Shuvaev VV, Siest G. Interaction between human amphipathic apolipoproteins and amyloid beta-peptide: surface plasmon resonance studies. FEBS Lett. 1996;383(12):9–12. doi: 10.1016/0014-5793(96)00206-2. [DOI] [PubMed] [Google Scholar]
- 91.Slemmon JR, Hughes CM, Campbell GA, Flood DG. Increased levels of hemoglobin-derived and other peptides in Alzheimer's disease cerebellum. J Neurosci. 1994;14(4):2225–35. doi: 10.1523/JNEUROSCI.14-04-02225.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Solerte SB, Ceresini G, Ferrari E, Fioravanti M. Hemorheological changes and overproduction of cytokines from immune cells in mild to moderate dementia of the Alzheimer's type: adverse effects on cerebromicrovascular system. Neurobiol Aging. 2000;21(2):271–281. doi: 10.1016/s0197-4580(00)00105-6. [DOI] [PubMed] [Google Scholar]
- 93.Spilt A, Weverling-Rijnsburger AW, Middelkoop HA, van Der Flier WM, Gussekloo J, de Craen AJ, Bollen EL, Blauw GJ, van Buchem MA, Westendorp RG. Late-onset dementia: structural brain damage and total cerebral blood flow. Radiology. 2005;236(3):990–5. doi: 10.1148/radiol.2363041454. [DOI] [PubMed] [Google Scholar]
- 94.Steinberg MH. Predicting clinical severity in sickle cell anaemia. Br J Haematol. 2005;129(4):465–81. doi: 10.1111/j.1365-2141.2005.05411.x. [DOI] [PubMed] [Google Scholar]
- 95.Suo Z, Humphrey J, Kundtz A, Sethi F, Placzek A, Crawford F, Mullan M. Soluble Alzheimers beta-amyloid constricts the cerebral vasculature in vivo. Neurosci Lett. 1998;257(2):77–80. doi: 10.1016/s0304-3940(98)00814-3. [DOI] [PubMed] [Google Scholar]
- 96.Szebeni J, Winterbourn CC, Carrell RW. Oxidative interactions between haemoglobin and membrane lipid. A liposome model. Biochem J. 1984;220(3):685–92. doi: 10.1042/bj2200685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tabbara IA. Hemolytic anemias. Diagnosis and management. Med Clin North Am. 1992;76(3):649–68. doi: 10.1016/s0025-7125(16)30345-5. [DOI] [PubMed] [Google Scholar]
- 98.Thein SL, Wainscoat JS, Sampietro M, Old JM, Cappellini D, Fiorelli G, Modell B, Weatherall DJ. Association of thalassaemia intermedia with a beta-globin gene haplotype. Br J Haematol. 1987;65(3):367–73. doi: 10.1111/j.1365-2141.1987.tb06870.x. [DOI] [PubMed] [Google Scholar]
- 99.Tierney MC, Fisher RH, Lewis AJ, Zorzitto ML, Snow WG, Reid DW, Nieuwstraten P. The NINCDS-ADRDA Work Group criteria for the clinical diagnosis of probable Alzheimer's disease: a clinicopathologic study of 57 cases. Neurology. 1988;38(3):359–64. doi: 10.1212/wnl.38.3.359. [DOI] [PubMed] [Google Scholar]
- 100.Vinters HV, Vonsattel JP. Neuropathologic features and grading of Alzheimer-related and sporadic CAA. Kluwer Academic Publishers; Dordrecht: 2000. [Google Scholar]
- 101.Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF. Heme and iron metabolism: role in cerebral hemorrhage. J Cereb Blood Flow Metab. 2003;23(6):629–52. doi: 10.1097/01.WCB.0000073905.87928.6D. [DOI] [PubMed] [Google Scholar]
- 102.Wisniewski HM, Wegiel J, Vorbrodt AW, Mazur-Kolecka B, Frackowiak J. Role of perivascular cells and myocytes in vascular amyloidosis. Ann N Y Acad Sci. 2000;903:6–18. doi: 10.1111/j.1749-6632.2000.tb06344.x. [DOI] [PubMed] [Google Scholar]
- 103.Wood WG. Increased HbF in adult life. Baillière Tindall; London: 1993. [DOI] [PubMed] [Google Scholar]
- 104.Wood WG. Hereditary Persistance of Fetal Hemoglobin and Delta Beta Thalassemia. Cambridge University Press; Cambridge, UK: 2001. [Google Scholar]
- 105.Wu CW, Liao PC, Yu L, Wang ST, Chen ST, Wu CM, Kuo YM. Hemoglobin promotes Abeta oligomer formation and localizes in neurons and amyloid deposits. Neurobiol Dis. 2004;17(3):367–77. doi: 10.1016/j.nbd.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 106.Yeh LH, Alayash AI. Effects of cell-free hemoglobin on hypoxia-induced factor (HIF-1alpha) and heme oxygenase 1(HO1) expression in endothelial cells subjected to hypoxia. Antioxid. Redox Signal. 2004;6(6):944–53. doi: 10.1089/ars.2004.6.944. [DOI] [PubMed] [Google Scholar]
- 107.Yu HL, Chertkow HM, Bergman H, Schipper HM. Aberrant profiles of native and oxidized glycoproteins in Alzheimer plasma. Proteomics. 2003;3(11):2240–8. doi: 10.1002/pmic.200300475. [DOI] [PubMed] [Google Scholar]
- 108.Zertal-Zidani S, Ducrocq R, Sahbatou M, Satta D, Krishnamoorthy R. Foetal haemoglobin in normal healthy adults: relationship with polymorphic sequences cis to the beta globin gene. European Journal of Human Genetics. 2002;10(5):320–326. doi: 10.1038/sj.ejhg.5200809. [DOI] [PubMed] [Google Scholar]
- 109.Zhang R, Barker L, Pinchev D, Marshall J, Rasamoelisolo M, Smith C, Kupchak P, Kireeva I, Ingratta L, Jackowski G. Mining biomarkers in human sera using proteomic tools. Proteomics. 2004;4(1):244–56. doi: 10.1002/pmic.200300495. [DOI] [PubMed] [Google Scholar]