

Abstract
The lipid A moiety of lipopolysaccharide forms the outer monolayer of the outer membrane of most Gram-negative bacteria. Escherichia coli lipid A is synthesized on the cytoplasmic surface of the inner membrane by a conserved pathway of nine constitutive enzymes. Following attachment of the core oligosaccharide, lipid A is flipped to the outer surface of the inner membrane by the ABC transporter MsbA, where the O-antigen polymer is attached. Diverse covalent modifications of lipid A may occur during its transit from the outer surface of the inner membrane to the outer membrane. Lipid A modification enzymes are therefore reporters for LPS trafficking within the bacterial cell envelope. Modification systems are highly variable and are often regulated by environmental conditions. Although not required for growth, the modification enzymes modulate the virulence of some Gram-negative pathogens. Heterologous expression of the genes encoding the lipid A modification enzymes in diverse bacteria facilitates the re-engineering of lipid A structures and should enable the development of new vaccines.
INTRODUCTION
Bacterial outer membranes
Lipid A (endotoxin), the hydrophobic anchor of lipopolysaccharide (LPS), is a glucosamine-based saccharolipid (1) that makes up the outer monolayer of the outer membranes of most Gram-negative bacteria (2-4). There are approximately 106 lipid A residues, 107 phospholipids and 105 undecaprenyl phosphate-sugar molecules in an E. coli cell (5, 6). With a few exceptions (7, 8), considered further below, the lipid A and Kdo domains of LPS (Figs. 1 and 2) are required for growth (5, 9, 10). In wild-type strains, additional core and O-antigen sugars are present (Fig. 1) (2, 3). These complex glycoforms are not needed for growth but protect bacteria from antibiotics and complement-mediated lysis. The core and O-antigen domains are required for virulence, and consequently are present in most clinical and environmental isolates (3). The structures and biosynthesis of core and O-antigen sugars are reviewed elsewhere (2, 3). Here, we focus on the biosynthesis of lipid A and its modification during transport to the outer membrane. The mechanisms of lipid A transport are covered in greater detail by Tommassen et al. in the 2007 issue of Annual Review of Microbiology.
Figure 1. Schematic structure of the E. coli K-12 cell envelope.

The structure and biosynthesis of LPS (2, 3), peptidoglycan (186), membrane-derived oligosaccharides (239, 240), lipoproteins (241) and phospholipids (242, 243) are reviewed elsewhere. Strains of E. coli K-12 normally do not make O-antigen, unless a mutation in the O-antigen operon is corrected (244). The Kdo2-lipid A region of LPS (the topic of this review) usually represent the minimal substructure required for growth of gram-negative bacteria. Exceptions include some spirochetes and strains of Sphinogmonas, in which the lipid A biosynthesis genes are absent, and Neisseria meningitidis in which lpxA knockouts lacking LPS are viable (142). If the ABC transporter MsbA (the inner membrane flippase for LPS) is over-expressed, E. coli can grow without Kdo (8). These strains still make the tetra-acylated precursor lipid IVA, which is nevertheless required for growth (8). The red phospholipids represent phosphatidylethanolamine and the yellow phosphatidylglycerol. Abbreviations: LPS, lipopolysaccharide; MDO, membrane-derived oligosaccharides; PPEtn, ethanolamine pyrophosphate.
Figure 2. The constitutive pathway for Kdo2-lipid A biosynthesis in E. coli.

Each enzyme of the constitutive lipid A pathway is encoded by a single structural gene (2, 69). The glucosamine disaccharide backbone of lipid A is blue. The Kdo disaccharide is black. LpxA, C and D are soluble cytoplasmic proteins, whereas LpxH and B are peripheral membrane proteins (2). The distal enzymes of the pathway, starting with LpxK, are integral inner membrane proteins, the active sites of which face the cytoplasm (2). The red numbers specify the glucosamine ring positions of lipid A and its precursors. The black numbers indicate the predominant fatty acid chain lengths found in E. coli lipid A. The single molecular species shown at the bottom left represents about 90 % of the total lipid A in E. coli, with most of the rest bearing a C12 secondary acyl chain at position 3′ (152). Additional minor acyl chain variants can be detected by high-resolution mass spectrometry (245).
The innate immune response to lipid A
Most Gram-negative bacteria synthesize lipid A molecules resembling those made by E. coli (Fig. 2) (2, 3). The lipid A moiety of LPS is detected by the TLR4/MD2 receptor of the mammalian innate immune system (11-17). Picomolar levels of lipid A induce macrophages to synthesize potent mediators of inflammation, like TNF-α and IL-1β (18, 19). Lipid A furthermore activates the production of co-stimulatory molecules required for adaptive immunity (20, 21). With mononuclear and endothelial cells, lipid A stimulates tissue factor production (22, 23). All these events are desirable for clearing local infections. When overproduced systemically during sepsis, however, the inflammation caused by some of these proteins damages small blood vessels and can precipitate Gram-negative septic shock (24, 25). LPS, or even synthetic E. coli lipid A by itself, causes a similar pathology when injected into animals (26-28), supporting its proposed role in sepsis. The characteristic structural features of E. coli lipid A (Fig. 2), especially its two phosphate and acyloxyacyl groups, are needed to trigger full TLR4/MD2 activation in human cells (26). However, partial activation of TLR4/MD2 by certain lipid A substructures and analogues results in the production of an altered cytokine profile that retains the beneficial adjuvant effects of endotoxin but minimizes animal toxicity (29-31). Some lipid A analogues (usually containing fewer acyl chains) are potent TLR4/MD2 antagonists (16, 32-35), with potential utility as human therapeutics (36). A crystal structure of TLR4/MD2 with a bound lipid A molecule or lipid A analogue is not yet available to clarify the mechanism of trans-membrane signaling (14). However, the crystal structure of the extra-cellular domain TLR3 (a TLR4 orthologue that is activated by double-stranded RNA) has recently been reported (37-39).
Discovery and overview of lipid A biosynthesis
The lipid A biosynthetic pathway may be viewed as having a conserved and a variable component. The conserved (constitutive) enzymes (Fig. 2) are intracellular, present in virtually all Gram-negative bacteria, and not generally subject to regulation (2, 40). In contrast, the lipid A modification enzymes, discussed below, are mostly extra-cytoplasmic and vary from organism to organism. In many instances, the lipid A modification systems are induced or repressed by growth conditions, such as changes in pH, divalent cation concentrations or the presence of anti-microbial peptides (41-45). Most modification enzymes reside either on the periplasmic surface of the inner membrane or in the outer membrane (46-54). They are excellent markers for following the translocation of nascent LPS from its initial site of biosynthesis on the inner surface of the inner membrane to the outer surface of the outer membrane (55-60) (Fig. 1).
The systematic elucidation of the constitutive pathway for lipid A biosynthesis (Fig. 2) was enabled by the discovery of 2,3-diacylglucosamine 1-phosphate (lipid X) (61, 62), a substance that had been overlooked in earlier work on E. coli lipids, because it is present at very low levels in wild-type cells (6, 63). However, it accumulates as much as 500-fold, or to about 5-10 % of the total lipid, in certain kinds of phosphatidylglycerol-deficient mutants (61, 62). The discovery of lipid X (61, 62) coincided with the correct structure determination (64, 65) and chemical synthesis of lipid A (66). Recognition of the existence of an acylated monosaccharide (62) representing a precursor to the proximal (right) subunit of lipid A (Fig. 2) greatly facilitated the development of testable hypotheses regarding the origin of lipid A from known lipids and carbohydrates present in E. coli (63, 67, 68).
THE CONSTITUTIVE ENZYMATIC PATHWAY OF LIPID A BIOSYNTHESIS
The nine enzymes of the constitutive lipid A pathway and the single-copy genes encoding them (Fig. 2) are conserved in Gram-negative bacteria like E. coli (2, 69). The only exceptions are organisms like Sphingomonas, which make bioactive sphingolipids instead of lipid A (70). The sequences of the lipid A genes are easily recognized when Gram-negative genomes are compared (71). LpxA, C and D are soluble proteins (72-74), whereas LpxB and LpxH are peripheral membrane proteins (75-77). LpxK, KdtA, LpxL and LpxM are integral inner membrane proteins (78-82). Their active sites are presumed to face the cytoplasmic surface of the inner membrane, given that their water-soluble co-substrates are cytoplasmic molecules (Fig. 2). Interestingly, higher plants like Arabidopsis thaliana encode significant orthologues of the constitutive enzymes within their nuclear genomes (2, 83), but lipid A-like molecules have yet been identified in plants by mass spectrometry or NMR spectroscopy (84).
Fatty acylation of UDP-GlcNAc
The first step of lipid A biosynthesis, the fatty acylation of UDP-GlcNAc (Fig. 2) (67, 85), is catalyzed by LpxA. E. coli LpxA requires the thioester R-3-hydroxymyristoyl acyl carrier protein (ACP) as its donor substrate (72, 85). It does not recognize R-3-hydroxymyristoyl-coenzyme A. The active site of E. coli LpxA functions as a precise hydrocarbon ruler that incorporates C14 hydroxyacyl chains two orders of magnitude faster than C12 or C16 chains (86, 87), consistent with the structure of E. coli lipid A (Fig. 2). In Pseudomonas aeruginosa, the LpxA ruler is reset to incorporate C10 chains (86, 87), while in Neisseria meningitidis and Leptospira interrogans it measures C12 chains (88, 89). Strains of E. coli in which P. aeruginosa lpxA replaces E. coli lpxA synthesize hybrid lipid A molecules in which C10 acyl chain are incorporated at positions 3 and 3′ (Fig. 2, red numbers). The rest of the lipid A molecule is unchanged (90). Single amino acid substitutions can switch P. aeruginosa LpxA to a C14- or E. coli LpxA to a C10-selective enzyme (87).
The crystal structure of LpxA (91-93) reveals that it is a homo-trimer (Fig. 3), constructed around multiple, contiguous hexad repeats. These motifs specify a unique secondary structure consisting of a left-handed helix of short parallel β-sheets. All hexad repeat-containing proteins studied to date are helical homo-trimers. Three hexads (18 amino acids) form one coil of the β-helix (91) (Fig. 3). The three identical active sites of LpxA, which were first proposed based on site-directed mutagenesis, are located at the subunit interfaces (93, 94). A recent X-ray structure of E. coli LpxA with bound UDP-3-O-(R-3-hydroxydecanoyl)-GlcNAc (Fig. 3, right panel), a slow substrate in the reverse direction (94), has recently been solved at 1.8 Å (Williams and Raetz, in preparation). In addition to validating the proposed locations of the LpxA active sites (94), these studies provide a structural explanation for the extraordinary chain length selectivity of these enzymes.
Figure 3. Structure of free LpxA and of LpxA with bound UDP-(3-O-acyl)-GlcNAc.

The LpxA homotrimer was solved at 2.6 Å (pdb 1LXA) in the absence of bound ligands (91). Each subunit has its own color. The side view (left) highlights the β-helix domain (91). The LpxA homotrimer was co-crystallized with a ∼25 fold molar excess of UDP-3-O-(R -3-hydroxydecanoyl)-GlcNAc and solved at 1.8 Å (Williams and Raetz, in preparation). The top-down view of this complex (right) reveals the location of the active site and the positioning of the acyl chain, consistent with previous proposals based on site-directed mutagenesis and NMR studies (94, 246).
An analogue of UDP-GlcNAc in which NH2 replaces the GlcNAc 3-OH group
Many bacteria, including L. interrogans and Acidithiobacillus ferroxidans, contain a dehydrogenase (GnnA) and a transaminase (GnnB) that convert UDP-GlcNAc to the analogue UDP-GlcNAc3N, in which the GlcNAc 3-OH group is replaced with an amine (Fig. 4) (89, 95). LpxA of L. interrogans, which is absolutely selective for a C12 chain (89, 95) (Fig. 4), acylates UDP-GlcNAc3N but not UDP-GlcNAc (89, 95). This remarkable selectivity accounts for the fact that L. interrogans lipid A molecules contain four N-linked hydroxyacyl chains (96), as the rest of the pathway is conserved (not shown in Fig. 4). E. coli LpxA cannot discriminate between UDP-GlcNAc and UDP-GlcNAc3N (89, 95), but the latter substrate is not available in E. coli. A. ferroxidans LpxA can utilize both UDP-GlcNAc and UDP-GlcNAc3N for lipid A biosynthesis, resulting in lipid A mixtures containing two, three of four N-linked acyl chains (89). A significant number of the Gram-negative bacteria sequenced to date contain GnnA and GnnB orthologues. The presence of additional N-linked acyl chains may increase the stability of lipid A to base hydrolysis or may prevent its degradation by lipases. A crystal structure of L. interrogans LpxA has recently been solved at 2.2 Å (Williams and Raetz, in preparation); this structural data should help elucidate the mechanism by which this enzyme differentiates sugar nucleotides.
Figure 4. Biosynthesis and acylation of UDP-GlcNAc3N in L. interrogans.

The sugar nucleotide UDP-GlcNAc3N is synthesized in two reactions from UDP-GlcNAc. The intermediate ketone has not yet been characterized, but UDP-GlcNAc3N generated in vitro by GnnA and GnnB can be isolated in mg quantities (89, 95). LpxA from L. interrogans is 41 % identical to E. coli LpxA at the protein level, and its x-ray structure has recently been determined (Williams and Raetz, in preparation). L. interrogans LpxA does not catalyze the acylation of UDP-GlcNAc and is absolutely selective for C12 hydroxyacyl chains (89, 95).
Deacetylation of UDP-3-O-(acyl)-GlcNAc
The equilibrium constant (∼0.01) for UDP-GlcNAc acylation by E. coli LpxA is unfavorable (72, 94). Thus, the deacetylation of UDP-3-O-(acyl)-GlcNAc by LpxC is the actual committed step of lipid A biosynthesis (74, 97). LpxC is a Zn2+-dependent enzyme that is highly conserved in all Gram-negative bacteria (98, 99). It displays no sequence similarity to other deacetylases or amidases. It is an excellent target for the development of novel antibiotics (10, 100, 101). Slow, tight-binding inhibitors of LpxC with low nM affinity have recently been reported (Fig. 5A). These compounds are N-aroyl-L-threonine hydroxamates (Fig. 5A). They possess antibiotic activity comparable to ciprofloxacin (102). The hydroxamate group presumably binds to the catalytic Zn2+ ion in a stereospecific manner. The recent X-ray (103-106) and NMR (107, 108) structures (Fig. 6) of LpxC with the bound substrate-mimetic hydroxamate inhibitor TU-514 (Fig. 5B), the fatty acyl chain of which occupies a hydrophobic tunnel leading away from the LpxC active site (Fig. 6), may facilitate the design of inhibitors with even greater antibiotic activity. Clinical applications would include the treatment of cystic fibrosis patients infected with multi-drug resistant P. aeruginosa.
Figure 5. Structures of LpxC inhibitors CHIR-090 and TU-514.
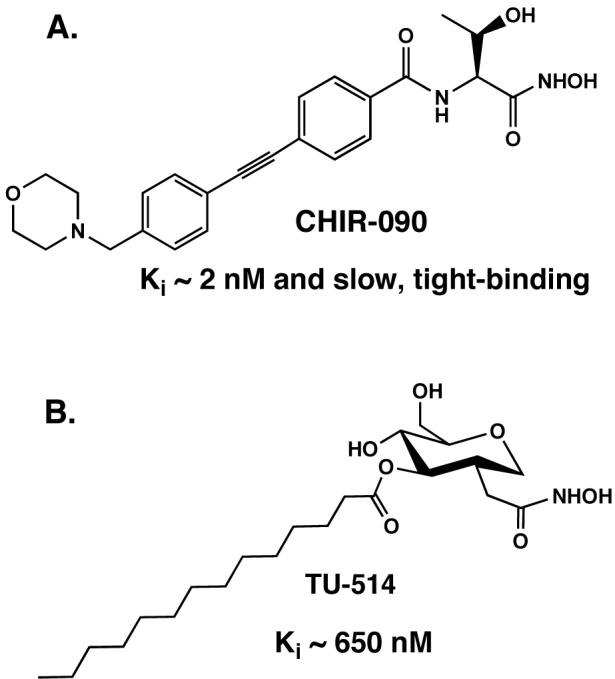
A. The slow, tight-binding inhibitor CHIR-090 inhibits diverse LpxC orthologues in the low nM range and displays potent antibiotic activity against many gram-negative bacteria (102). B. The substrate mimetic TU-514 inhibits E. coli LpxC with Ki ∼ 650 nM but has little or no antibiotic activity (100).
Figure 6. NMR structure of LpxC with bound substrate-mimetic inhibitor TU-514.
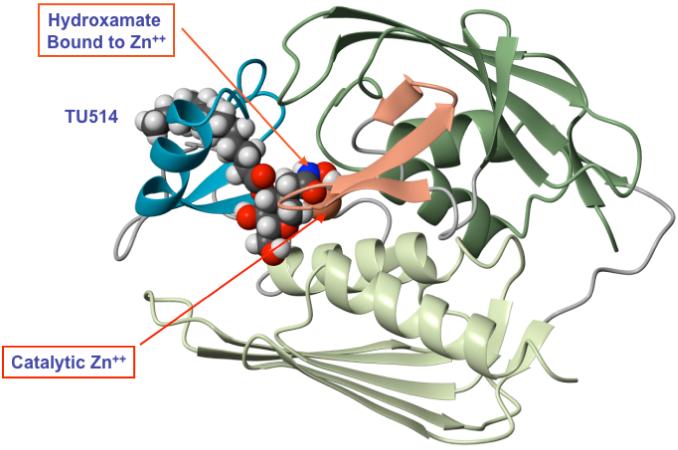
This ribbon diagram is based on the NMR studies of Coggins et al. (107, 108). The recent crystal structure of the same complex is similar, except for slight differences in the orientation of the tetrahydropyran ring (105).
LpxC levels increase five- to ten-fold in cells treated with sub-lethal doses of LpxC inhibitors (109). Induction is not associated with increased transcription (109) but may be due to reduced LpxC turnover when lipid A biosynthesis is curtailed. LpxC induction is also seen in temperature-sensitive LpxA mutants in the absence of LpxC inhibitors (109). Although the signaling mechanisms controlling LpxC induction are unknown, two amino acids at the C-terminus of LpxC are critical for this regulation (110). The FtsH protease is partially responsible for regulating LpxC turnover in vivo (111) but additional processes cannot yet be excluded.
Following deacetylation, a second R-3-hydroxymyristate chain is added by LpxD to make UDP-2,3-diacyl-GlcN (Fig. 2) (73). The X-ray structure of LpxD (W. Hunter, personal communication) shows that it, like LpxA, is a homotrimer constructed around multiple contiguous hexad repeats.
Formation of the lipid A disaccharide
The pyrophosphate linkage of UDP-2,3-diacyl-GlcN is cleaved by LpxH, which catalyzes the attack of water on the α-phosphorus atom of the UDP moiety to form 2,3-diacyl-GlcN-1-phosphate (lipid X) (76, 77) and UMP (Fig. 2). LpxH is unusual in that it is missing in about one third of the Gram-negative genomes. An alternative pyrophosphatase of this kind must exist in these strains, since all of them contain LpxD and LpxB (Fig. 2), but the relevant gene has not been identified. LpxH is a peripheral membrane protein that is stimulated in vitro by Mn2+. This enzyme is distantly related to the phosphoprotein phosphatase family. Its structure is not yet available.
The β,1′-6 linked disaccharide that is characteristic of all lipid A molecules is generated by LpxB, which condenses UDP-2,3-diacyl-GlcN with lipid X (68, 75) and releases UDP (Fig. 2). Like LpxH, LpxB is a peripheral membrane protein. It is a member of a unique family of glycosyltransferases, some of which are distantly related to MurG (112), as judged by PSI-BLAST analysis (113). LpxB is very useful for the chemo-enzymatic synthesis of lipid A analogues (114, 115). Its crystal structure has not been reported. A second LpxB orthologue of unknown function is present in strains of Legionella, where it is required for growth inside of Acanthamoeba (116).
Kdo incorporation and secondary acylation
The integral inner membrane proteins LpxK (78, 117), KdtA (WaaA) (118, 119), LpxL (HtrB) and LpxM (MsbB) (79, 80, 82, 120) catalyze the last four steps of the constitutive pathway in E. coli (Fig. 2). Each protein contains one predicted membrane-spanning segment at its N-terminus. The active sites likely face the cytoplasm. LpxK phosphorylates the 4′-position of the disaccharide 1-phosphate generated by LpxB (Fig. 2) to form lipid IVA (121). This important precursor is an excellent endotoxin antagonist in human cells, but an agonist of reduced potency in the mouse (33). This unusual pharmacology is determined by the source of the TLR4/MD2 complex (14, 122-124). Hexa-acylated lipid A dimerizes human TLR4/MD2, whereas lipid IVA does not (14, 122-124).
Next, two Kdo residues are incorporated by the bifunctional enzyme KdtA (WaaA) (118, 119, 125). The labile sugar nucleotide CMP-Kdo is the Kdo donor (40, 126, 127). The second Kdo unit is incorporated much more rapidly than the first, and therefore the intermediate with a single Kdo residue does not accumulate (Fig. 2). However, in Hemophilus influenzae, Vibrio cholerae, Bordetalla pertussis, and several other organisms, KdtA incorporates only one Kdo residue (128). A special kinase (KdkA), unique to these bacteria (128, 129), then incorporates a phosphate group at the same position where the outer Kdo residue is added by E. coli KdtA (Fig. 2). Hemophilus kdtA and kdkA in combination can rescue a heptose-deficient E. coli mutant with a deletion in its own kdtA gene (130). When the heptose region of the core is intact, however, a monofunctional Kdo transferase can rescue a KdtA deletion mutant (131). KdtA of Chlamydia trachomatis incorporates at least three Kdo residues and can also functionally substitute for E. coli KdtA (9, 132).
The last steps of E. coli lipid A biosynthesis involve the addition of the secondary lauroyl and myristoyl residues to the distal glucosamine unit (Fig. 2) (120) by LpxL and LpxM, which require the Kdo disaccharide moiety in their substrates for activity (82, 120). LpxL and LpxM prefer acyl-ACP donors but can also function with acyl-coenzyme A substrates (Six and Raetz, in preparation). LpxL and LpxM display significant sequence similarity to each other (79, 80), but not to LpxA or LpxD. LpxL and LpxM are distantly related to the lysophosphatidic acid acyltransferase family (Six and Raetz, in preparation). The lpxM gene is not required for growth in E. coli (82, 133). S. typhimurium and Shigella lpxM mutants are attenuated in their ability to cause inflammation (134-136). Both lpxL and lpxM can be deleted, provided the cells are grown on minimal medium or at low temperature (82). Tetra-acylated lipid A, which accumulates in lpxL mutants, is not rapidly transported from its site of biosynthesis on the inner surface of the inner membrane to the outer membrane (82).
The E. coli chromosome encodes an additional gene homologous to lpxL, termed lpxP, which is expressed at low temperature (12 °C) (81, 137). LpxP incorporates palmitoleate in place of laurate (not shown in Fig. 2), perhaps reflecting the need to adjust outer membrane fluidity (81, 137). In Yersinia pestis, lpxL is missing but lpxM and lpxP are present (138). Consequently, Y. pestis synthesizes tetra-acylated lipid A at 37 °C but makes hexa-acylated lipid A at 25 °C (138). As noted above, tetra-acylated lipid A is a relatively weak TLR4/MD2 agonist in the mouse and an antagonist in humans (138). Introduction of the E. coli lpxL gene into Y. pestis permits the synthesis of hexa-acylated lipid A (a potent TLR4/MD2 agonist) at all temperatures (138). Such constructs are fully attenuated in a mouse infection model and provide immunity against a subsequent challenge with wild-type Y. pestis (138).
Why lipid A is essential for growth in most Gram-negative bacteria remains uncertain. It may be required for the proper folding of some outer membrane proteins (139, 140). Recently, Nishiyama et al. have reported a lipid A-like factor required for signal recognition particle/SecYEG-dependent and -independent membrane protein integration in E. coli (141). Because mass spectrometry and NMR spectroscopy were not used for structural analysis, the chemical nature of this factor and its identification as lipid A-related remains in question (141).
N. meningitidis is unusual in that its lpxA gene can be inactivated (142); such mutants grow slowly without lipid A but nevertheless can assemble a functional outer membrane, albeit missing some lipoproteins (7).
EXPORT OF NEWLY SYNTHESIZED LPS AND PHOSPHOLIPIDS
How E. coli lipids cross the inner membrane and are transported to the outer membrane (Fig. 7) is not fully understood (55, 143). A clue to bacterial lipid transport emerged from studies of lpxL mutants (Fig. 2) and their suppression by multiple copies of msbA (144-146). LpxL is the lauroyl transferase of lipid A biosynthesis (Fig. 2) (79). LPS with tetra-acylated lipid A accumulates in inner membranes of lpxL mutants at 42°C, and growth on broth is inhibited (146). MsbA is an essential ABC transporter (Fig. 7), closely related to eucaryotic Mdr proteins (144). MsbA over-expression restores the growth of lpxL mutants at 42°C without restoring laurate addition, resulting in export of LPS with tetra-acylated lipid A to the outer membrane (146). E. coli msbA knockouts are not viable (144), but their analysis is complicated by two factors. First, long times (4-8 h) are needed to dilute out pre-existing MsbA supplied in trans from a temperature-sensitive plasmid (146). Second, the lpxK gene (Fig. 2), which is immediately downstream in an operon with msbA, is also essential for growth (117, 146).
Figure 7. Proteins implicated in O-antigen assembly and in the export of LPS.
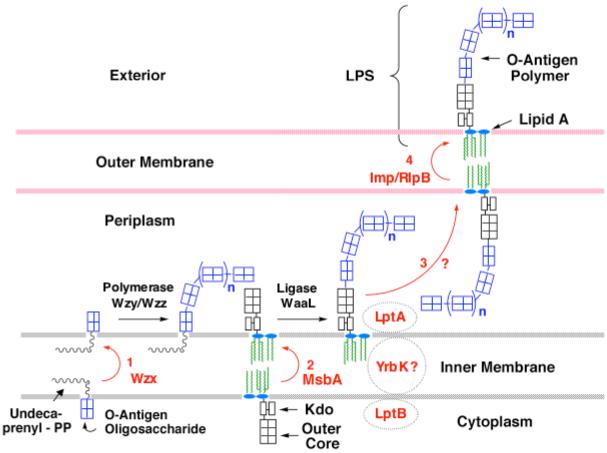
The proteins in red letters are involved in the export of LPS. The ABC transporter MsbA flips newly-synthesized core-lipid A to the outer surface of the inner membrane (55, 143). O-antigen is assembled separately on undecaprenyl phosphate and is flipped by the putative transporter Wzx (247). O-antigen oligosaccharides are polymerized on the periplasmic surface of the inner membrane by Wzy and Wzz, and transferred to nascent core-lipid A by WaaL (2). In vitro systems for the polymerase and ligase have not been reported. The periplasmic protein LptA (159) is proposed to shuttle LPS to the essential outer membrane protein complex Imp/RlpB, which is required the assembly of LPS into the outer surface of the outer membrane, as judged by accessibility to the ecto-enzymes PagL or PagP (56, 60, 197, 209). With the exception of the lipid-activated ATPase activity of MsbA (154), no in vitro assays have been developed for any of the proposed transporters. The LptA protein may function together with an additional ABC transporter protein termed LptB and the putative trans-membrane protein YrbK (159).
To gain a clearer understanding of MsbA function, a temperature-sensitive point mutant of E. coli (WD2) was isolated in which there is a single A270T substitution in the fifth predicted membrane-spanning segment (143), located near the proposed MsbA dimer interface on the periplasmic side of the inner membrane (147-149). This mutant protein is rapidly inactivated at 44 °C. Export of all major lipids (both LPS and phospholipids) to the outer membrane is inhibited by ∼90 % in WD2 after 30 minutes at 44 °C (143), as judged by pulse-labeling studies. Lipid A biosynthesis, phospholipid biosynthesis and export of major outer membrane proteins are not immediately affected (143). However, minor protein species have not been examined (143). The cells do not undergo rapid lysis, suggesting that peptidoglycan assembly is not inhibited (143). Electron microscopy reveals inner membrane invaginations in WD2 at 44°C, consistent with increased surface area secondary to a selective block of lipid export (143). However, the covalent modifications of newly-synthesized lipid A with 4-amino-4-deoxy-L-arabinose (L-Ara4N) or phosphoethanolamine moieties (Fig. 8), which occur on the outer surface of the inner membrane, are inhibited at 44 °C in temperature-sensitive MsbA mutants (55), consistent with the idea that MsbA is the flippase for LPS (Fig. 8).
Figure 8. Covalent modifications of Kdo2-lipid A in E. coli K-12 and Salmonella.

The known covalent modifications of Kdo2-lipid A (3) are indicated by the substituents with the dashed bonds. The glucosamine disaccharide is blue, the L-Ara4N unit green, and the phosphoethanolamine moieties red. Under some conditions, the positions of the phosphoethanolamine and L-Ara4N substituents are reversed (not shown) (248, 249). Lipid A species with two phosphoethanolamine units or two L-Ara4N moieties may also be present (44). Expression of the enzymes ArnT (47) and EptA(PmrC) is under the control of PmrA/B (50, 52). PagP and PagL are regulated by PhoP/Q (46, 47). LpxO (magenta), LpxR and EptB are not regulated by either PhoP/Q or PmrA/B (44, 53, 54). Asterisks indicate modification enzymes not found in E. coli K-12. Transfer of the Salmonella genes encoding these enzymes to E. coli results in the expected lipid A modifications.
Several recent X-ray structures of the homodimeric MsbA protein at 4.2-4.5 Å (147-149) support the proposed flippase function of MsbA (143) and suggest the existence of multiple conformational states. However, the relatively low resolution of these structures (147-149), compared with those of other ABC transporters, has hampered structural interpretation and raised serious issues regarding the published conformations of the MsbA protein (150, 151). An improved, high-resolution structure of MsbA, preferably with a well-defined, ligand such as Kdo2-lipid A (152), would greatly facilitate further mechanistic studies of MsbA.
Studies of MsbA-mediated LPS flip-flop in E. coli membrane vesicles or in purified, reconstituted systems have not been reported. Why phospholipid transport to the outer membrane is blocked in msbA mutants is also unclear (143). There is evidence that phospholipid flip-flop is not ATP-dependent in bacterial inner membrane vesicles (153). Although purified MsbA is a lipid-activated ATPase (154), phospholipid flip-flop could not be demonstrated in liposomes containing MsbA alone (155). MsbA may indeed be a specific flippase primarily for LPS, or inhibition of phospholipid export in msbA mutants (143) might be secondary to LPS accumulation. Alternatively, other proteins may be needed together with MsbA to catalyze phospholipid and LPS flip-flop. In some marine bacteria, MsbA is fused via its C-terminus to LpxK (156).
Recently, msbA has been identified as a multi-copy suppressor of Kdo-deficient mutants of E. coli (8), including strains with deletions in the Kdo transferase (Fig. 2) (9) (Reynolds and Raetz, in preparation). Additional uncharacterized suppressor mutations and multi-copy suppressors have also been reported (8); their analysis should provide exciting new insights into lipid A trafficking and function. In Kdo-deficient strains harboring the appropriate suppressor, lipid IVA is sufficient for outer membrane biogenesis and cell viability (8), but the cells are still sensitive to the LpxC inhibitor CHIR-090 (Reynolds and Raetz, in preparation). The outer membrane protein profile of Kdo-deficient strains is remarkably similar to that observed with wild-type E. coli (8). It appears that MsbA over-production can overcome the transport defect associated with both under-acylated (146) and Kdo-deficient (157) LPS precursors.
In N. meningitidis slow growth is possible without LPS (142). Consequently, one can delete the msbA gene in N. meningitidis and retain the ability to assemble an outer membrane (158), yet N. meningitidis msbA can partially complement the temperature-sensitive E. coli msbA mutant WD2 (158). It may be that E. coli MsbA is required for the rapid flipping of phospholipids, given the fast growth of E. coli versus N. meningitidis.
Additional proteins are required to assemble and attach O-antigen (2-4), and to shuttle nascent LPS across the periplasm and into the outer membrane (Fig. 7). The essential periplasmic protein YhbN(LptA) has recently been implicated in this process (159). A linked cytoplasmic ABC transporter subunit homologue, designated LptB, also plays a role (159), perhaps in conjunction with the additional trans-membrane protein YbrK (159). The LptA/B transporter complex is thought to function after MsbA catalyzed LPS flipping, possibly extracting nascent LPS from the periplasmic surface of the inner membrane on its way to the outer membrane (159). Next, the outer membrane protein Imp (56, 160) and its lipoprotein partner RlpB (60) are thought to flip nascent LPS within the outer membrane, bringing it to the exterior. Depletion of LptA/B (159) or Imp/RlpB (56, 160) causes the accumulation of aberrant, heavy membranes, apparently located within the periplasm. As with MsbA, in vitro transport assays with pure proteins have yet to be developed to validate these proposals.
Although eucaryotic Mdr proteins are thought to catalyze phospholipid flip-flop in vitro, mouse mutants lacking the three major Mdr proteins are viable and show no generalized defects in lipid trafficking (161, 162). Given the multitude of Mdr-like proteins in mammalian genomes, functional redundancy may account for the lack of phenotype. However, mouse Mdr2 knockouts display a specific lipid transport deficiency in that they cannot pump phosphatidylcholine into their bile (163). Many of the additional Mdr-like proteins present in animal cells have recently been implicated in the transport of specific lipids (164). As with MsbA, however, simple and direct in vitro assays for Mdr catalyzed lipid flip-flop are not well developed.
LIPID A MODIFICATION SYSTEMS IN GRAM-NEGATIVE BACTERIA
Overview of E. coli and Salmonella lipid A modification enzymes
E. coli K-12 and S. typhimurium contain enzymes for modifying lipid A with phosphoethanolamine (Fig 8, red) (50, 52, 53, 165), L-Ara4N (Fig. 8, green) (41, 47, 48, 165, 166) and/or palmitate (46, 167, 168) (Fig. 8, black). Two selective deacylases and a dioxygenase are additionally present in S. typhimurium (Fig. 8). Many of these enzymes are regulated in response to changes in growth conditions. For instance, the attachment of phosphoethanolamine by the enzyme EptA (50) and L-Ara4N by the enzyme ArnT (Fig. 8) (47) is induced by activation of the PmrA transcription factor, either by exposure of cells to mild acid or by pmrA constitutive mutations (44, 169, 170). The L-Ara4N group is positively charged at pH 7. It neutralizes the negative charge of the lipid A 4′-phosphate group (Fig. 8), thereby reducing bacterial susceptibility to cationic anti-microbial peptides and polymyxin (41, 169-171).
Addition of polar groups to E. coli and Salmonella lipid A
The biosynthesis of L-Ara4N and the mechanism of its attachment to lipid A have recently been elucidated. The process starts with the oxidative decarboxylation of UDP-glucuronic acid by the C-terminal domain of ArnA (Fig. 9) (172). The resulting UDP-4-ketopentose is transaminated by ArnB (173) to generate UDP-L-Ara4N, which is then formylated by the N-terminal domain of ArnA (174). X-ray structures of ArnA (175, 176) and ArnB (177) are available, as both are soluble proteins. How they interact and transfer their products between their active sites is unknown. Only the N-formyl derivative of UDP-L-Ara4N (Fig. 9, magenta modification) can be transferred to undecaprenyl phosphate by ArnC (174). The subsequent deformylation by ArnD to generate undecaprenyl phosphate L-Ara4N (Figs. 9 and 10) renders the pathway irreversible (174). After transport of undecaprenyl phosphate-L-Ara4N to the outer surface of the inner membrane, ArnT transfers the L-Ara4N residue to the 4′-phosphate group of lipid A (Fig. 9) (47). A possible inner membrane transport system for undecaprenyl phosphate-L-Ara4N, encoded by the arnE and arnF genes (Fig. 9), has recently been identified (Yan and Raetz, in preparation).
Figure 9. Biosynthesis of the L-Ara4N unit and its attachment to lipid A.

Formation of the L-Ara4N moiety begins with the oxidation of UDP-glucose to UDP-glucuronic acid (172), as first proposed by Zhou et al. (165) Next, the C-terminal domain of ArnA catalyzes an NAD+-dependent oxidative decarboxylation to yield an unusual UDP-4-ketopentose, which is converted to UDP-L-Ara4N (green moiety) by the transaminase ArnB (173). The N-terminal domain of ArnA then uses N-10-formyltetrahydrofolate to add a formyl group (magenta) to UDP-L-Ara4N (174). Next, ArnC/PmrF, a distant orthologue of dolichyl phosphate-mannose synthase, selectively transfers the formylated L-Ara4N residue to undecaprenyl phosphate (174). The ArnD-dependent deformylation of this lipid to undecaprenyl phosphate-α-L-Ara4N (which accumulates in polymyxin-resistant mutants) likely occurs on the inner leaflet of the inner membrane and may prevent reversal of the ArnC reaction (174). After transport to the outer surface of the inner membrane by a process that may involve the inner membrane proteins ArnE and ArnF (Yan and Raetz, in preparation), the polytopic membrane protein ArnT transfers the L-Ara4N moiety (shown as a green rectangle) to lipid A. Given the dual function the ArnA holoenzyme (174-176), the possibility of substrate channeling from ArnA to ArnC deserves consideration. However, ArnA and ArnB do not associate with each other in vitro.
Figure 10. Structure of undecaprenyl phosphate-α-L-Ara4N.
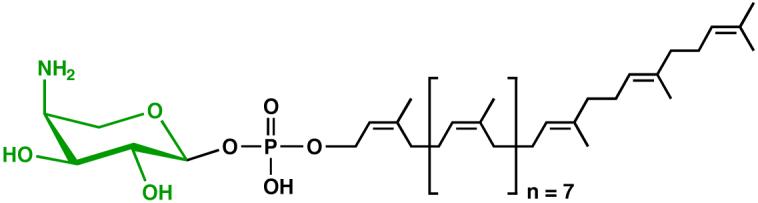
This lipid accumulates in polymyxin-resistant mutants of E. coli and Salmonella (48).
Phosphoethanolamine transfer to lipid A by the enzyme EptA (Fig. 8), predominantly to the 1 phosphate group, likewise occurs on the outer surface of the inner membrane (50, 52, 55). Under certain growth conditions or in the absence of L-Ara4N, EptA can modify the lipid A 4′ position with a second phosphoethanolamine moiety (not shown in Fig. 8) (44). Phosphatidylethanolamine serves as the phosphoethanolamine donor substrate. In contrast to the L-Ara4N group, which is critical for polymyxin resistance (41, 174, 178), the roles of the phosphoethanolamine modifications remain uncertain.
E. coli and S. typhimurium both contain a related enzyme, designated EptB, which is homologous to EptA but is not regulated by PmrA. Instead, EptB is induced by the addition of 5 mM Ca2+ to the growth medium (53, 179) and is under the control of the σE transcription factor (180). EptB transfers a phosphoethanolamine moiety from phosphatidylethanolamine to the outer Kdo residue (Fig. 8). Diacylglycerol is generated as the byproduct (53). Heptose-deficient mutants lacking EptB are killed by the presence of 5 mM Ca2+ in the growth medium (53), suggesting a function in the maintenance of outer membrane stability.
Additional Ept enzymes are likely necessary to control the incorporation of phosphoethanolamine into other cell envelope components including the first inner core heptose sugar, which is modified in a PmrA-dependent manner by an ept gene homologue in S. typhimurium (181). Phosphoethanolamine is not normally incorporated into the lipid A of E. coli K-12 until the cells are exposed to mildly acidic growth conditions (44), but pathogenic E. coli O157:H7 expresses this modification constitutively (182). The latter organism harbors an ept gene homologue in the plasmid pO157-encoded shf locus (183, 184), but the corresponding gene is replaced by virK, an unrelated gene needed for intercellular spreading in Shigella flexneri (136). The plasmid-borne shf loci of E. coli O157:H7 and S. flexneri also encode a second lpxM gene homologue, which must be inactivated together with the chromosomal copy in order to achieve virulence attenuation (136, 183). The shf loci also encode the wabB glycosyltransferase gene that controls attachment onto the third heptose sugar of GlcNAc, which is a unique feature of the R3 inner core (184).
When grown in the presence of 10 mM Mg2+ at neutral pH, E. coli and S. typhimurium synthesize a subset of lipid A molecules (20-30 %) in which a diphosphate group is present at the 1-position of lipid A (not shown in Figs. 2 or 8) (166). The diphosphate residue is generated on the periplasmic surface of the inner membrane by YeiU, an undecaprenyl diphosphate-specific phosphotransferase (185) (Trent et al., preparation). YeiU also functions as an undecaprenyl diphosphate phosphatase. Undecaprenyl diphosphate is generated on the outer surface of the inner membrane during the polymerization of peptidoglycan (Fig. 1) (186). The lipid A diphosphate groups, synthesized by YeiU and EptA, might function to stabilize and/or balance the surface electrostatics of the outer membrane depending on environmental conditions. However, YeiU is not essential for cell growth on nutrient broth.
Modification of the fatty acyl chains of E. coli and Salmonella lipid A
Modification of lipid A with palmitate by PagP (CrcA) (Fig. 8) is under control of the PhoP/PhoQ system, which is activated by low Mg2+ concentrations or cationic anti-microbial peptides (42, 44, 168). An acidic patch on the surface of the periplasmic domain of the PhoQ sensor-kinase is proposed to orient parallel to the membrane plane, which allows Mg2+ to bridge the acidic patch with anionic phospholipid polar head groups and maintain a repressed regulatory state (45, 187). The PhoP/PhoQ system is thought to be activated either by growing cells under Mg2+-limited conditions or, under Mg2+-replete conditions found during growth within macrophage phagosomal vacuoles (188), upon displacement of Mg2+ by cationic anti-microbial peptides (45). In S. typhimurium, PhoP/PhoQ activation triggers the PmrA/PmrB pathway via a post-translational mechanism using an effector known as PmrD, but the pmrD gene is non-functional in E. coli (189). Consequently, PmrA/PmrB-dependent modification of the lipid A phosphate groups can be uncoupled from the PhoP/PhoQ-dependent incorporation of palmitate only in E. coli (57).
S. typhimurium mutants unable to add palmitate to lipid A are sensitive to certain cationic anti-microbial peptides including representatives of amphipathic α-helical (C18G) and β-sheet (protegrin) structural classes, but not including polymyxin (168). Although the amphipathic α-helical peptides LL37 and C18G are both inducers of the PhoP/PhoQ system (45), PagP only provides measurable resistance to the latter (190). PagP is required for animal infections caused by Legionella pneumophila and Bordetella bronchiseptica, where it provides resistance to cationic anti-microbial peptides and antibody-mediated complement lysis, respectively (191-193). The pagP gene is distributed among a narrow group of primarily pathogenic bacteria, and its regulation often correlates with their pathogenic lifestyle (194). For example, the Bvg virulence regulator controls B. bronchiseptica PagP expression (166) and the incorporation of palmitate into Y. pseudotuberculosis lipid A predominates at the body temperature of the infected host (195).
Palmitate transfer to lipid A occurs on the outer surface of the outer membrane where PagP uses phospholipids as palmitoyl donors (46). Both X-ray and NMR structures of E. coli (Fig. 11) have been reported (196, 197). The active site of PagP faces the exterior (197), suggesting that its activity is regulated by phospholipid access, as phospholipids are not always present on the outer surface of the outer membrane. PagP is an 8-stranded anti-parallel β-barrel, preceded by an N-terminal amphipathic α-helix (Fig. 11). The β-barrel axis is tilted by ∼25 degrees with respect to the membrane plane (196, 198). Like most β-barrel outer membrane proteins, the interior of the inner leaflet-exposed half of the molecule is largely polar, but the interior of the outer LPS-exposed half is decidedly hydrophobic and lined by a bound molecule of the detergent lauroyldimethylamine-N-oxide (Fig. 11). Proline residues punctuate the β-strands at two opposing sites and disrupt the continuity of β-barrel hydrogen bonding around the bound detergent, thus providing obvious routes for lateral access of lipid substrates from within the outer leaflet of the membrane.
Figure 11. The outer membrane lipid A palmitoyltransferase PagP (left) and the lipid A 3-O-deacylase PagL (right).

The disordered loop connecting the first and second β-strands of PagP (pdb 1THQ), and the bound lipid X with PagL (pdb 2ERV) were introduced subsequently and energy minimized. The authors thank Chris Neale and Régis Pomès (University of Toronto) and Lucy Rutten and Jan Tommassen (Utrecht University) for providing the coordinates of energy minimized PagP and PagL, respectively. Abbreviation: LDAO, lauroyldimethylamine-N-oxide.
The mechanism by which PagP selects palmitate in preference to other fatty acyl chains involves another “hydrocarbon ruler”, which can be reset to recognize shorter fatty acids by means of single amino acid substitutions (196). Substitution of Gly88 lining the floor of the lauroyldimethylamine-N-oxide binding pocket can make the pocket more shallow by the same length as the introduced side-chain, and this affords a corresponding shortening of the acyl-chain that is selected by the enzyme (196). The aromatic side-chains of Tyr26 and Trp66 undergo a rare exciton interaction, which can be detected by circular dichroism spectroscopy and provides a sensitive probe to gauge methylene unit resolution of acyl-chain selection (Khan et al., in preparation). The ability to modulate PagP acyl-chain selection might be important for the preparation of endotoxin antagonists and adjuvants (30, 31, 199).
The amino acid residues implicated in catalysis Asp76, His33, and Ser77 are not organized into a catalytic triad characteristic of serine esterases (196, 197), but PagP can exist in a conformational equilibrium between two dynamically distinct states (200). Although the known structure represents an inhibited R-state, an ordering of residues in and around the disordered cell-surface loop that connects the first two β-strands is thought to be necessary to afford a catalytically-competent T-state, the structural details of which remain to be elucidated. Knowledge of the T-state structure would likely reveal details of the PagP catalytic mechanism.
PagP can function as a membrane-intrinsic probe to monitor either the transport of LPS to the outer membrane (158) or the translocation of phospholipids into the outer leaflet, which occurs when LPS organization is disrupted by mutations that affect the presentation of LPS on the cell surface (60), or by EDTA that chelates Mg2+ needed to neutralize negative charge repulsions between neighboring LPS molecules (57). Phospholipid accumulation in the outer leaflet can render cells sensitive to hydrophobic antibiotics and detergents that are normally impermeable when lipid asymmetry is maintained. An outer membrane permeability defect observed in an LpxM-deficient mutant of E. coli O157:H7 was recently associated with PagP activation through a lipid perturbation mechanism, which revealed that PagP can contribute to the restoration of the permeability barrier (Kim et al., in preparation). Interestingly, PagP activation in this mutant also induced a truncation of the R3 core at the level of the first outer core glucose unit, which could be rescued by restoring the cytosolic pool of UDP-glucose. The implication that PagP activation in the outer membrane can control cytoplasmic functions is consistent with observations that LPS modifications, including palmitoylation of lipid A, can initiate signal transduction across the bacterial cell envelope to the transcription factor σE (201).
S. typhimurium contains several additional lipid A modification enzymes that are not present in wild-type E. coli K-12. PagL is an outer membrane lipase that is regulated by PhoP/PhoQ and removes the R-3-hydroxymyristoyl chain at position 3 of lipid A (Fig. 8) (49). LpxR, a distinct outer membrane lipase, cleaves the intact 3′-acyloxyacyl moiety of Kdo2-lipid A (Fig. 8) (54). In vitro studies have shown that LpxR activity is Ca2+-dependent and Kdo-activated (54). Finally, LpxO is an inner membrane enzyme that hydroxylates the 3′ secondary acyl chain of Kdo2-lipid A in the presence of O2 (Fig. 8), using Fe2+ and α-ketoglutarate as cofactors (202, 203). LpxO is not under the control of PhoP/PhoQ, and its active site faces the cytoplasm (44, 55). The crystal structures of LpxO and LpxR have not yet been determined.
Expression of LpxO, LpxR or PagL in E. coli K-12 leads to the expected lipid A modifications (Fig. 8) (49, 54, 202). Interestingly, PagL and LpxR are latent in S. typhimurium, unless over-expressed (49, 54), suggesting the presence of endogenous inhibitors such as the L-Ara4N moiety (204). The functions of these enzymes are unknown, but PagL and LpxR would likely attenuate the cytokine inducing ability of LPS (205-207).
Despite its absence from E. coli, PagL homologues are more widely distributed than PagP, although PagL is not primarily restricted to pathogenic organisms (208). The crystal structure of PagL from P. aeruginosa reveals an overall fold that is similar to PagP (209), and its active site likewise faces the outer surface of the outer membrane (Fig. 11). Both molecules are 8-stranded anti-parallel β-barrels and are strikingly tilted in the outer membrane. However, PagL differs in the presentation of its lipid substrate to a distinct catalytic triad formed by Glu140, His126, and Ser128 on the β-barrel exterior. Energy minimization of the model substrate lipid X reveals that acyl-chains likely bind into hydrophobic grooves on the β-barrel exterior (209). Although the enzyme normally encounters R-3-hydroxydecanoyl chains in its substrates in vivo, it also utilizes R-3-hydroxymyristoyl chains in vitro, which indicates that acyl-chains encountered on the β-barrel exterior cannot be measured with the same precision as performed by PagP. The potential of PagL to dimerize at an interface between active sites suggests a potential mechanism to inhibit activity in the outer membrane (209).
Modification of Francisella and Helicobacter lipid A
Francisella tularensis is the cause of tularemia, a highly contagious pulmonary disease of humans and animals (210). Francisella novicida U112, a related environmental organism, does not infect humans, and affords a practical model system for laboratory investigation (211). Strains of Francisella synthesize relatively little LPS, but do contain significant amounts of free lipid A, which can be extracted with chloroform-methanol together with the glycerophospholipids (212). The biological significance of free lipid A, which lacks Kdo and other core sugars, is unclear (212). The genomes of Francisella encode all the enzymes of the constitutive lipid A biosynthetic pathway, including the Kdo transferase and enzymes for core sugar addition (213). It may be that nascent lipid A is pumped out of the cell by MsbA more rapidly than it is glycosylated by KdtA. Alternatively, free lipid A may arise from nascent LPS by the action of an unusual Kdo hydrolase present in Francisella membranes (58). However, a Kdo hydrolase is also present in membranes of Helicobacter pylori (214), but free lipid A is not abundant. The gene encoding the Kdo hydrolase has not yet been identified.
The predominant lipid A species present in Francisella are shown in Fig. 12 (upper right). Interesting features (also seen with the portion of the lipid A that is linked to Francisella LPS) include the absence of the 4′-phosphate group and the 3′ acyl chain (212, 215). A galactosamine residue, which is a cationic sugar resembling L-Ara4N, is attached to the 1-phosphate group of Francisella lipid A (212, 215). Under some conditions, the 1-phosphate and galactosamine groups appear to be missing (Fig. 12, lower right structure) (216).
Figure 12. Extracellular covalent modifications of lipid A in F. tularensis.

The proposed modification pathway is based on the genetic characterization of the two phosphatases, LpxE and LpxF (58, 59), which are present in many bacteria that synthesize phosphate-deficient lipid A. The attachment of galactosamine to F. tularensis lipid A involves a polyisoprene phosphate donor, analogous to undecaprenyl phosphate-L-Ara4N in E. coli (Wang and Raetz, in preparation). The F. tularensis system is also unusual in that much of its lipid A is “free”, i.e. not covalently attached to LPS (212). The origin and function of free lipid A have not been established.
Some of the novel enzymatic reactions that account for these interesting lipid A modifications have been identified (Fig. 12). The lipid A phosphate groups are removed by two distinct inner membrane phosphatases, designated LpxE and LpxF (Fig. 12), the active sites of which face the periplasmic surface of the inner membrane (58, 59). Expression of LpxE in strains of E. coli or Salmonella leads to nearly quantitative dephosphorylation at the 1-position (58). Expression of LpxF in wild-type E. coli has no effect, because LpxF does not dephosphorylate lipid A species containing a secondary acyl chain at the 3′-position (59). However, LpxF expression leads to complete loss of the 4′-phosphate moiety in lpxM mutants of E. coli or Salmonella, which synthesize mainly penta-acylated lipid A species (Fig. 2) (59). Cells of Salmonella expressing lpxE synthesize the non-toxic, adjuvant form of lipid A (also known as monophospho-lipid A or MPLA) (30, 31), which is usually prepared by mild acid hydrolysis of LPS or by chemical synthesis. LpxE-expressing strains may be useful as live oral vaccines, assuming that the excessive inflammation that is normally caused by Salmonella lipid A is indeed suppressed.
Disruption of the lpxF gene in F. novicida (Fig. 12) results in the quantitative retention of the lipid A 4′ phosphate group with the accumulation of the species shown at the top center of Fig. 12 (217). When the 4′-phosphate moiety is left in place, the 3′ acyl chain is not removed (217), suggesting an obligatory order of processing. Because there is no lpxR gene in Francisella (213), it is unclear how the 3′-acyl chain is actually removed. H. pylori lipid A, like that of F. novicida, lacks both the 4′-phosphate group and the 3′-acyl chain(s). There is no lpxF gene in H. pylori (218), but an orthologue of lpxR is present. When lpxR is inactivated in H. pylori, a hexa-acylated lipid A species lacking the 4′-phosphate group accumulates (Trent et al. in preparation), suggesting that H. pylori contains a distinct 4′-phosphatase that can dephosphorylate lipid A molecules containing a secondary 3′-acyl chain.
The mutant of F. novicida lacking lpxF is hypersensitive to cationic antimicrobial peptides and is avirulent in a mouse infection model (217). Following short-term intraperitoneal injection, the lpxF mutant bacteria trigger the production of a subset of cytokines, suggestive of TLR2 activation, whereas wild-type cells do not (217). Unlike the Y. pestis construct described above (138), the lipid A of Francisella lpxF mutant bacteria does not activate TLR4 (217), and lpxF mutant cells do not trigger the production of TNFα (217). Instead, the hypersensitivity of the lpxF mutant to cationic anti-microbial peptides (217) may cause damage to the bacterial envelope and expose other ligands, such as membrane lipoproteins for TLR2 or bacterial DNA for TLR9 (17). The potential of lpxF mutants as novel vaccines for the prevention of tularemia has not yet been explored.
Deletion of the single arnT gene homologue present in F. novicida leads to the production of lipid A molecules lacking the galactosamine modification (212), consistent with the finding that undecaprenyl phosphate-galactosamine (Fig. 12) is present amongst the minor lipids of F. novicida (Wang and Raetz, in preparation). The galactosamine modification pathway in Francisella therefore appears to be analogous to the L-Ara4N pathway (Fig. 9) in E. coli. Interestingly, the live vaccine strain of F. tularensis, like the arnT mutant of F. novicida, lacks the galactosamine modification on its free lipid A (212). Whether this feature is due to the absence of ArnT in the live vaccine strain or the inability to synthesize the undecaprenyl phosphate-galactosamine donor substrate (Fig. 12) is unclear.
Lipid A processing in Rhizobium leguminosarum and Rhizobium etli
The plant endosymbionts R. leguminosarum and R. etli synthesize a complex mixture of lipid A molecules that lack the 1- and 4′-phosphate groups found in most other Gram-negative bacteria (Fig. 13) (219-221). In a subset of molecular species, the anomeric carbon atom of the proximal unit is oxidized to a carboxylic acid (Fig. 13) (219-221). Galacturonic acid residues are attached to the outer Kdo moiety (not shown) and to the 4′-position of lipid A (219-221). Both organisms synthesize mainly penta-acylated lipid A molecules (220, 221) with an unusually long secondary acyl chain at the 2′-position (Fig. 13) (220, 221); this is a characteristic structural feature of the lipid A from many Rhizobiaciae (222). A portion of the lipid A molecules are deacylated at position 3 (223), and there is considerable fatty acid chain length heterogeneity (Fig. 13), when contrasted with E. coli lipid A (152, 220, 221). The core sugars beyond the Kdo region of Rhizobium LPS are also very different from those of E. coli (224, 225) in that the heptose units are replaced by mannose, galactose and Kdo (not shown in Fig. 13).
Figure 13. Kdo2-lipid IVA versus mature lipid A of R. etli and R. leguminosarum.

These bacteria make phosphate-deficient lipid A molecules from Kdo2-lipid IVA (226, 250), using the lipid A phosphatases LpxE and LpxF, as described in the text. Lipid A molecules of Rhizobium typically contain a very long secondary acyl chain at position 2′ (222). Additional unique features include the presence of galacturonic acid (cyan) in place of phosphate at position 4′ and oxidation of the proximal glucosamine unit (blue) in a portion of the molecules to aminogluconate (magenta) (219-221). Partial substituents and micro-heterogeneity of acyl chains lengths are indicated by dashed bonds. Components C and E lack the 3-O linked hydroxyacyl chain. The schematic representations of these structures are shown below the actual chemical structures.
The enzymology, genetics and topography of R. leguminosarum and R. etli lipid A biosynthesis have been investigated in considerable detail. These bacteria contain orthologues of the first seven lpx genes found in E. coli (Fig. 2) (226), and therefore they initially synthesize the tetra-acylated precursor Kdo2-lipid IVA, shown in Figs. 13 and 14. The diverse lipid A molecular species of Rhizobium (Fig. 13) are generated from Kdo2-lipid IVA by enzymes that are not present in E. coli. For instance, the long secondary acyl chain is incorporated by LpxXL, a distant orthologue of LpxL (227), requiring a special acyl carrier protein termed ACP-XL (Fig. 14) (228). R. leguminosarum and R. etli also contain the phosphatases LpxE (58, 229) and LpxF (59), which catalyze the removal of the 1- and 4′-phosphate groups respectively (Fig. 14), as in Francisella. Following removal of the 1-phosphate moiety, LpxQ (230, 231) can oxidize the proximal glucosamine of Rhizobium lipid A in the presence of O2 to form an aminogluconate unit. Removal of the 4′-phosphate group by LpxF is necessary for the incorporation of the 4′-galacturonic acid moiety (Fig. 14), which appears to involve a polyisoprene phosphate sugar donor (rather than a sugar nucleotide) (Ingram and Raetz, unpublished). Similarly, the incorporation of galacturonate into the Kdo region of Rhizobium LPS by the enzymes RgtA and RgtB (Fig. 14) requires dodecaprenyl phosphate galacturonic acid as the donor substrate (232, 233). RgtA and RgtB (Fig. 14) are distantly related in sequence and membrane topography to ArnT (232) (Fig. 9).
Figure 14. Kdo2-lipid IVA processing in R. etli and R. leguminosarum membranes.

The processing enzymes unique to the Rhizbium system can be assayed using E. coli Kdo2-lipid IVA as the model substrate (223, 225, 227-229, 232, 233, 250-252). The enzymes that have been characterized to date are labeled in red according to the genes that encode them. Other colors: glucosamine, blue; galacturonic acid, cyan; aminogluconate, magenta; Kdo, white; other core sugars, black dashed line; fatty acids, green.
The precise order of Kdo2-lipid IVA modification in Rhizobium (Fig. 14) is not fully established. In vitro, there is no obligatory order of enzymatic processing, with the exception of LpxE preceding LpxQ, and LpxF preceding RgtD (Fig. 14). However, several lines of evidence strongly support the enzymatic topography shown in Fig. 15, in which LpxE, LpxF, and the Rgt enzymes function on the outer surface of the inner membrane, and LpxQ and PagL act in the outer membrane. Heterologous expression of the inner membrane phosphatases LpxE or LpxF in the appropriate strains of E. coli results in nearly complete and selective lipid A dephosphorylation, provided that the LPS flippase MsbA (Figs. 7 and 15) is functional (58, 59). The involvement of a dodecaprenyl phosphate-linked galacturonic acid donor, instead of a sugar nucleotide, is likewise consistent with galacturonate addition to lipid A occurring on the outer surface of the inner membrane (Fig. 15) (232, 233), analogous to L-Ara4N in polymyxin-resistant E. coli (Figs. 8 and 9) (48). PagL and LpxQ are recovered in the outer membrane when expressed in E. coli (49, 230, 231). LpxQ efficiently generates lipid A containing aminogluconate when co-expressed with LpxE in E. coli (Ingram and Raetz, unpublished).
Figure 15. Topography of lipid A modifications in R. etli and R. leguminosarum.

The proposed topography of the processing enzymes is based on the finding that lipid A dephosphorylation by LpxE and LpxF requires a functional msbA gene, when foreign lpxE or lpxF genes are expressed in E. coli (51, 58, 59). The Rgt proteins require a polyisoprene donor as their co-substrate, consistent with a periplasmic localization (232, 233). PagL and LpxQ are known to be outer membrane proteins (47, 231). The x-ray structure of PagL shows that its active site is oriented towards the outside (209). The orientation of LpxQ is unknown (231). PagP is not present in Rhizobium. Enzymes are indicated in red, and putative transport proteins are shown in black. Letters in the outer membrane refer to lipid A components B, C, D and E, shown in Fig. 13. Other colors: glucosamine, blue; galacturonic acid, cyan; aminogluconate, magenta; Kdo, white; other core sugars, black dashed line; fatty acids, green.
The biological significance of the lipid A modification systems of Rhizobium has been evaluated by genetics. In each instance studied to date, the relevant enzymes were first identified by the development of in vitro biochemical assays, followed by the expression cloning of the corresponding structural genes (59, 225, 227-229, 231, 232). Subsequent deletion of these structural genes caused the accumulation of the predicted, structurally altered lipid A species, demonstrating the biological relevance of the enzymatic approach. For instance, deletion of lpxXL or acpXL results in the failure to incorporate the long secondary acyl chain (Fig. 13) (234, 235). These mutant bacteria grow slowly, and they are hypersensitive to detergents and low pH (234, 235). However, these mutants are nevertheless able to form partially functional nodules in their host plants. Deletion of Rhizobium LpxE or LpxF results in the retention of the 1- or 4′-phosphate moieties, respectively (Sohlenkamp, Ingram and Raetz, unpublished). In the lpxE mutant, the aminogluconate cannot be generated, whereas in the lpxF mutant the 4′-galacturonate residue is not incorporated. Studies of the phenotypes of the mutants should provide insights into the functions of these structural features.
Unusual lipid A modifications in other bacteria
Given the diversity of gram-negative bacteria, it is likely that many additional lipid A modification enzymes exist. Some of modification enzymes are present in only a few types of organisms. For instance, L. interrogans contains a lipid A methyl transferase (LmtA) that uses S-adenosylmethionine to methylate the 1-phosphate group (236). LmtA is distantly related to eucaryotic methyl transferases involved in the C-terminal processing of farnesylated proteins (236). LmtA can be expressed in E. coli resulting in methylation of about half the lipid A (236). The function of lipid A methylation is unknown.
Some strains of Acinetobacter make LPS containing an analogue of Kdo, known as Ko, in which the CH2 moiety at the Kdo 3-position is hydroxylated (237). Interestingly, the KdtA orthologue of this organism utilizes CMP-Kdo in vitro (238), raising the possibility Ko is formed after the addition of Kdo to lipid IVA. A Kdo-selective dioxygenase, analogous to LpxO (Fig. 8), might account for these observations. The relevant gene has not yet been identified.
FUTURE DIRECTIONS FOR LIPID A AND LPS RESERACH
The following questions and research directions will dominate lipid A research in the coming years.
1) Why is lipid A essential for growth in most gram-negative bacteria?
Careful genetic and biochemical examination of unusual systems like N. meningitidis, in which lipid A is not required for growth (142), might prove informative. The powerful approach of searching for second-site suppressor mutations in E. coli has recently provided new insights into the role of the Kdo disaccharide (8) but has not been applied systematically to all the genes of the constitutive pathway.
2) What is the relevance of the lpx gene orthologues present in higher plants?
Mutants of Arabidopsis lacking lpxA are viable but lack fine root hairs (Liu, Nikolau and Raetz, in preparation). However, despite suggestive recent immunochemical studies (84), no lipid A-like molecules have been characterized by chemical methods in plants. Such molecules might also have been overlooked, if localized to root hairs or stem cells. Arabidopsis lpxA can rescue a temperature sensitive lpxA mutant of E. coli, showing that plant LpxA can catalyze UDP-GlcNAc acylation (83).
3) Can in vitro assays for LPS flipping in membrane vesicles be developed?
The genetic and biochemical evidence for the involvement of proteins such as MsbA (55, 143) and Imp/RlpB (56, 60) in catalyzing trans-membrane movement of LPS in cells is very compelling, but it needs to be bolstered by the development of in vitro assays in reconstituted liposomes with purified components. The use of lipid A modification enzymes, like LpxE or PagL, as reporters for the trans-membrane movement of LPS in vitro should be explored, as this promising approach may facilitate development of robust in vitro systems.
4) Can crystal structures of the cytoplasmic membrane proteins involved in lipid A biosynthesis, modification, and signaling be obtained?
A great deal of progress has already been made with soluble proteins like LpxA (91, 93) and LpxC (103, 107), but the other constitutive enzymes are also of great interest. A structure of TLR4/MD2 with a bound ligand, like Kdo2-lipid A, should provide important new insights into signaling mechanisms. Methods for crystallizing cytoplasmic membrane proteins have improved in recent years. However, the problems encountered with the structural studies of MsbA show how difficult this approach can be (150, 151).
5) Will lipid A-like molecules that block the TLR4/MD2 receptor be useful for the treatment of gram-negative sepsis?
Many clinical trials have failed in the arena of sepsis (25), but the current work with the antagonist E5564 is unique with regard to potency and selectivity of this LPS antagonist in humans (36). Phase III trials are in progress.
6) Can clinically useful antibiotics be developed around the constitutive lipid A pathway?
The best available LpxC inhibitors (Fig. 5A) (102) may be effective enough for clinical trials, pending improvements in bioavailability, pharmacokinetics and toxicology.
7) Can lipid A modification systems be exploited for vaccine development?
Work with Y. pestis expressing lpxL is the most advanced in this regard (138). The opportunities afforded by attenuated lpxF mutants of F. tularensis (59, 217) or strains lacking lpxM (msbB) (135, 136) are under active investigation. The properties of Salmonella strains expressing LpxE (58), which synthesize the mono-phosphorylated, non-toxic adjuvant form of lipid A, will also be of great interest.
Answers to many of the above questions should emerge from careful studies of the unique proteins and enzymes that assemble and modify lipid A. A combination of genetics and biochemistry will be required to bridge the remaining gaps between lipid A structural diversity and biological function.
ACKNOWLEDGMENTS
Lipid A research in our laboratories is supported by the following grants: NIH GM-51310 and GM-51796 to C. R. H. R., CIHR MOP-43886 to R. E. B., and NIH AI-064184 to M. S. T.
References
- 1.Fahy E, Subramaniam S, Brown HA, Glass CK, Merrill AH, Jr., et al. J. Lipid Res. 2005;46:839–62. doi: 10.1194/jlr.E400004-JLR200. [DOI] [PubMed] [Google Scholar]
- 2.Raetz CRH, Whitfield C. Annu. Rev. Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brade H, Opal SM, Vogel SN, Morrison DC, editors. Endotoxin in Health and Disease. Marcel Dekker, Inc.; New York: 1999. p. 950. [Google Scholar]
- 4.Nikaido H. Microbiol. Mol. Biol. Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galloway SM, Raetz CRH. J. Biol. Chem. 1990;265:6394–402. [PubMed] [Google Scholar]
- 6.Guan Z, Breazeale SD, Raetz CRH. Anal. Biochem. 2005;345:336–9. doi: 10.1016/j.ab.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 7.Steeghs L, de Cock H, Evers E, Zomer B, Tommassen J, van der Ley P. Embo J. 2001;20:6937–45. doi: 10.1093/emboj/20.24.6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meredith TC, Aggarwal P, Mamat U, Lindner B, Woodard RW. ACS Chem. Biol. 2006;1:33–42. doi: 10.1021/cb0500015. [DOI] [PubMed] [Google Scholar]
- 9.Belunis CJ, Clementz T, Carty SM, Raetz CRH. J. Biol. Chem. 1995;270:27646–52. doi: 10.1074/jbc.270.46.27646. [DOI] [PubMed] [Google Scholar]
- 10.Onishi HR, Pelak BA, Gerckens LS, Silver LL, Kahan FM, et al. Science. 1996;274:980–2. doi: 10.1126/science.274.5289.980. [DOI] [PubMed] [Google Scholar]
- 11.Poltorak A, He X, Smirnova I, Liu MY, Huffel CV, et al. Science. 1998;282:2085–8. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 12.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, et al. J. Immunol. 1999;162:3749–52. [PubMed] [Google Scholar]
- 13.Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, et al. J. Exp. Med. 1999;189:1777–82. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gangloff M, Gay NJ. Trends Biochem. Sci. 2004;29:294–300. doi: 10.1016/j.tibs.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 15.Miller SI, Ernst RK, Bader MW. Nat. Rev. Microbiol. 2005;3:36–46. doi: 10.1038/nrmicro1068. [DOI] [PubMed] [Google Scholar]
- 16.Visintin A, Halmen KA, Latz E, Monks BG, Golenbock DT. J. Immunol. 2005;175:6465–72. doi: 10.4049/jimmunol.175.10.6465. [DOI] [PubMed] [Google Scholar]
- 17.Akira S, Uematsu S, Takeuchi O. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 18.Beutler B, Cerami A. Annu. Rev. Biochem. 1988;57:505–18. doi: 10.1146/annurev.bi.57.070188.002445. [DOI] [PubMed] [Google Scholar]
- 19.Dinarello CA. Blood. 1991;77:1627–52. [PubMed] [Google Scholar]
- 20.Medzhitov R, Janeway C., Jr. N. Engl. J. Med. 2000;343:338–44. doi: 10.1056/NEJM200008033430506. [DOI] [PubMed] [Google Scholar]
- 21.van Duin D, Medzhitov R, Shaw AC. Trends Immunol. 2006;27:49–55. doi: 10.1016/j.it.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 22.Li A, Chang AC, Peer GT, Hinshaw LB, Taylor FB., Jr. Shock. 1996;5:274–9. doi: 10.1097/00024382-199604000-00007. [DOI] [PubMed] [Google Scholar]
- 23.Drake TA, Cheng J, Chang A, Taylor FB., Jr. Am. J. Pathol. 1993;142:1458–70. [PMC free article] [PubMed] [Google Scholar]
- 24.Parillo JE. N. Engl. J. Med. 1993;328:1471–7. doi: 10.1056/NEJM199305203282008. [DOI] [PubMed] [Google Scholar]
- 25.Russell JA. N. Engl. J. Med. 2006;355:1699–713. doi: 10.1056/NEJMra043632. [DOI] [PubMed] [Google Scholar]
- 26.Rietschel ET, Kirikae T, Schade FU, Mamat U, Schmidt G, et al. FASEB Journal. 1994;8:217–25. doi: 10.1096/fasebj.8.2.8119492. [DOI] [PubMed] [Google Scholar]
- 27.Prabhakar U, Conway TM, Murdock P, Mooney JL, Clark S, et al. DNA Cell Biol. 2005;24:410–31. doi: 10.1089/dna.2005.24.410. [DOI] [PubMed] [Google Scholar]
- 28.Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, et al. Nature. 2005;437:1032–7. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- 29.Qureshi N, Takayama K, Ribi E. J. Biol. Chem. 1982;257:11808–15. [PubMed] [Google Scholar]
- 30.Persing DH, Coler RN, Lacy MJ, Johnson DA, Baldridge JR, et al. Trends Microbiol. 2002;10:S32–7. doi: 10.1016/s0966-842x(02)02426-5. [DOI] [PubMed] [Google Scholar]
- 31.Baldridge JR, McGowan P, Evans JT, Cluff C, Mossman S, et al. Expert. Opin. Biol. Ther. 2004;4:1129–38. doi: 10.1517/14712598.4.7.1129. [DOI] [PubMed] [Google Scholar]
- 32.Takayama K, Qureshi N, Beutler B, Kirkland TN. Infect. Immun. 1989;57:1336–8. doi: 10.1128/iai.57.4.1336-1338.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Golenbock DT, Hampton RY, Qureshi N, Takayama K, Raetz CRH. J. Biol. Chem. 1991;266:19490–8. [PubMed] [Google Scholar]
- 34.Christ WJ, Asano O, Robidoux AL, Perez M, Wang Y, et al. Science. 1995;265:80–3. doi: 10.1126/science.7701344. [DOI] [PubMed] [Google Scholar]
- 35.Hawkins LD, Christ WJ, Rossignol DP. Curr. Top. Med. Chem. 2004;4:1147–71. doi: 10.2174/1568026043388123. [DOI] [PubMed] [Google Scholar]
- 36.Lynn M, Rossignol DP, Wheeler JL, Kao RJ, Perdomo CA, et al. J. Infect. Dis. 2003;187:631–9. doi: 10.1086/367990. [DOI] [PubMed] [Google Scholar]
- 37.Bell JK, Botos I, Hall PR, Askins J, Shiloach J, et al. Proc Natl Acad Sci U S A. 2005;102:10976–80. doi: 10.1073/pnas.0505077102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choe J, Kelker MS, Wilson IA. Science. 2005;309:581–5. doi: 10.1126/science.1115253. [DOI] [PubMed] [Google Scholar]
- 39.Bell JK, Askins J, Hall PR, Davies DR, Segal DM. Proc. Natl. Acad. Sci. U S A. 2006;103:8792–7. doi: 10.1073/pnas.0603245103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raetz CRH. Annu. Rev. Biochem. 1990;59:129–70. doi: 10.1146/annurev.bi.59.070190.001021. [DOI] [PubMed] [Google Scholar]
- 41.Gunn JS, Lim KB, Krueger J, Kim K, Guo L, et al. Mol. Microbiol. 1998;27:1171–82. doi: 10.1046/j.1365-2958.1998.00757.x. [DOI] [PubMed] [Google Scholar]
- 42.Guo L, Lim KB, Gunn JS, Bainbridge B, Darveau RP, et al. Science. 1997;276:250–3. doi: 10.1126/science.276.5310.250. [DOI] [PubMed] [Google Scholar]
- 43.Groisman EA. J. Bacteriol. 2001;183:1835–42. doi: 10.1128/JB.183.6.1835-1842.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gibbons HS, Kalb SR, Cotter RJ, Raetz CRH. Mol. Microbiol. 2005;55:425–40. doi: 10.1111/j.1365-2958.2004.04409.x. [DOI] [PubMed] [Google Scholar]
- 45.Bader MW, Sanowar S, Daley ME, Schneider AR, Cho U, et al. Cell. 2005;122:461–72. doi: 10.1016/j.cell.2005.05.030. [DOI] [PubMed] [Google Scholar]
- 46.Bishop RE, Gibbons HS, Guina T, Trent MS, Miller SI, Raetz CRH. Embo J. 2000;19:5071–80. doi: 10.1093/emboj/cdd507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trent MS, Ribeiro AA, Lin S, Cotter RJ, Raetz CRH. J. Biol. Chem. 2001;276:43122–31. doi: 10.1074/jbc.M106961200. [DOI] [PubMed] [Google Scholar]
- 48.Trent MS, Ribeiro AA, Doerrler WT, Lin S, Cotter RJ, Raetz CRH. J. Biol. Chem. 2001;276:43132–44. doi: 10.1074/jbc.M106962200. [DOI] [PubMed] [Google Scholar]
- 49.Trent MS, Pabich W, Raetz CRH, Miller SI. J. Biol. Chem. 2001;276:9083–92. doi: 10.1074/jbc.M010730200. [DOI] [PubMed] [Google Scholar]
- 50.Trent MS, Raetz CRH. J. Endotoxin Res. 2002;8:158. [Google Scholar]
- 51.Tran AX, Karbarz MJ, Wang X, Raetz CRH, McGrath SC, et al. J. Biol. Chem. 2004;279:55780–91. doi: 10.1074/jbc.M406480200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee H, Hsu FF, Turk J, Groisman EA. J. Bacteriol. 2004;186:4124–33. doi: 10.1128/JB.186.13.4124-4133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reynolds CM, Kalb SR, Cotter RJ, Raetz CRH. J. Biol. Chem. 2005;280:21202–11. doi: 10.1074/jbc.M500964200. [DOI] [PubMed] [Google Scholar]
- 54.Reynolds CM, Ribeiro AA, McGrath SC, Cotter RJ, Raetz CRH, Trent MS. J. Biol. Chem. 2006;281:21974–87. doi: 10.1074/jbc.M603527200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Doerrler WT, Gibbons HS, Raetz CRH. J. Biol. Chem. 2004;279:45102–9. doi: 10.1074/jbc.M408106200. [DOI] [PubMed] [Google Scholar]
- 56.Bos MP, Tefsen B, Geurtsen J, Tommassen J. Proc. Natl. Acad. Sci. U S A. 2004;101:9417–22. doi: 10.1073/pnas.0402340101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jia W, Zoeiby AE, Petruzziello TN, Jayabalasingham B, Seyedirashti S, Bishop RE. J. Biol. Chem. 2004;279:44966–75. doi: 10.1074/jbc.M404963200. [DOI] [PubMed] [Google Scholar]
- 58.Wang X, Karbarz MJ, McGrath SC, Cotter RJ, Raetz CRH. J. Biol. Chem. 2004;279:49470–8. doi: 10.1074/jbc.M409078200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang X, McGrath SC, Cotter RJ, Raetz CRH. J. Biol. Chem. 2006;281:9321–30. doi: 10.1074/jbc.M600435200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu T, McCandlish AC, Gronenberg LS, Chng SS, Silhavy TJ, Kahne D. Proc. Natl. Acad. Sci. U S A. 2006;103:11754–9. doi: 10.1073/pnas.0604744103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nishijima M, Raetz CRH. J. Biol. Chem. 1979;254:7837–44. [PubMed] [Google Scholar]
- 62.Takayama K, Qureshi N, Mascagni P, Nashed MA, Anderson L, Raetz CRH. J. Biol. Chem. 1983;258:7379–85. [PubMed] [Google Scholar]
- 63.Bulawa CE, Raetz CRH. J. Biol. Chem. 1984;259:4846–51. [PubMed] [Google Scholar]
- 64.Imoto M, Kusumoto S, Shiba T, Naoki H, Iwashita T, et al. Tetrahedron Lett. 1983;24:4017–20. [Google Scholar]
- 65.Strain SM, Fesik SW, Armitage IM. J. Biol. Chem. 1983;258:13466–77. [PubMed] [Google Scholar]
- 66.Galanos C, Lüderitz O, Rietschel ET, Westphal O, Brade H, et al. Eur. J. Biochem. 1985;148:1–5. doi: 10.1111/j.1432-1033.1985.tb08798.x. [DOI] [PubMed] [Google Scholar]
- 67.Anderson MS, Bulawa CE, Raetz CRH. J. Biol. Chem. 1985;260:15536–41. [PubMed] [Google Scholar]
- 68.Ray BL, Painter G, Raetz CRH. J. Biol. Chem. 1984;259:4852–9. [PubMed] [Google Scholar]
- 69.Riley M, Abe T, Arnaud MB, Berlyn MK, Blattner FR, et al. Nucleic Acids Res. 2006;34:1–9. doi: 10.1093/nar/gkj405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Krziwon C, Zähringer U, Kawahara K, Weidemann B, Kusumoto S, et al. Infect. Immun. 1995;63:2899–905. doi: 10.1128/iai.63.8.2899-2905.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wheeler DL, Church DM, Federhen S, Lash AE, Madden TL, et al. Nucleic Acids Research. 2003;31:28–33. doi: 10.1093/nar/gkg033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Anderson MS, Bull HS, Galloway SM, Kelly TM, Mohan S, et al. J. Biol. Chem. 1993;268:19858–65. [PubMed] [Google Scholar]
- 73.Kelly TM, Stachula SA, Raetz CRH, Anderson MS. J. Biol. Chem. 1993;268:19866–74. [PubMed] [Google Scholar]
- 74.Young K, Silver LL, Bramhill D, Cameron P, Eveland SS, et al. J. Biol. Chem. 1995;270:30384–91. doi: 10.1074/jbc.270.51.30384. [DOI] [PubMed] [Google Scholar]
- 75.Radika K, Raetz CRH. J. Biol. Chem. 1988;263:14859–67. [PubMed] [Google Scholar]
- 76.Babinski KJ, Kanjilal SJ, Raetz CRH. J. Biol. Chem. 2002;277:25947–56. doi: 10.1074/jbc.M204068200. [DOI] [PubMed] [Google Scholar]
- 77.Babinski KJ, Ribeiro AA, Raetz CRH. J. Biol. Chem. 2002;277:25937–46. doi: 10.1074/jbc.M204067200. [DOI] [PubMed] [Google Scholar]
- 78.Garrett TA, Kadrmas JL, Raetz CRH. J. Biol. Chem. 1997;272:21855–64. doi: 10.1074/jbc.272.35.21855. [DOI] [PubMed] [Google Scholar]
- 79.Clementz T, Bednarski JJ, Raetz CRH. J. Biol. Chem. 1996;271:12095–102. doi: 10.1074/jbc.271.20.12095. [DOI] [PubMed] [Google Scholar]
- 80.Clementz T, Zhou Z, Raetz CRH. J. Biol. Chem. 1997;272:10353–60. doi: 10.1074/jbc.272.16.10353. [DOI] [PubMed] [Google Scholar]
- 81.Carty SM, Sreekumar KR, Raetz CRH. J. Biol. Chem. 1999;274:9677–85. doi: 10.1074/jbc.274.14.9677. [DOI] [PubMed] [Google Scholar]
- 82.Vorachek-Warren MK, Ramirez S, Cotter RJ, Raetz CRH. J. Biol. Chem. 2002;277:14194–205. doi: 10.1074/jbc.M200409200. [DOI] [PubMed] [Google Scholar]
- 83.Liu D, Sun TP, Raetz CRH. FASEB J. 2003;17(Suppl S):A579–A. Part1. [Google Scholar]
- 84.Armstrong MT, Theg SM, Braun N, Wainwright N, Pardy RL, Armstrong PB. Faseb. J. 2006;20:2145–6. doi: 10.1096/fj.05-5484fje. [DOI] [PubMed] [Google Scholar]
- 85.Anderson MS, Raetz CRH. J. Biol. Chem. 1987;262:5159–69. [PubMed] [Google Scholar]
- 86.Williamson JM, Anderson MS, Raetz CRH. J. Bacteriol. 1991;173:3591–6. doi: 10.1128/jb.173.11.3591-3596.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wyckoff TJO, Lin S, Cotter RJ, Dotson GD, Raetz CRH. J. Biol. Chem. 1998;273:32369–72. doi: 10.1074/jbc.273.49.32369. [DOI] [PubMed] [Google Scholar]
- 88.Odegaard TJ, Kaltashov IA, Cotter RJ, Steeghs L, van der Ley P, et al. J. Biol. Chem. 1997;272:19688–96. doi: 10.1074/jbc.272.32.19688. [DOI] [PubMed] [Google Scholar]
- 89.Sweet CR, Williams AH, Karbarz MJ, Werts C, Kalb SR, et al. J. Biol. Chem. 2004;279:25411–9. doi: 10.1074/jbc.M400597200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dotson GD, Kaltashov IA, Cotter RJ, Raetz CRH. J. Bacteriol. 1998;180:330–7. doi: 10.1128/jb.180.2.330-337.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Raetz CRH, Roderick SL. Science. 1995;270:997–1000. doi: 10.1126/science.270.5238.997. [DOI] [PubMed] [Google Scholar]
- 92.Lee BI, Suh SW. Proteins. 2003;53:772–4. doi: 10.1002/prot.10436. [DOI] [PubMed] [Google Scholar]
- 93.Williams AH, Immormino RM, Gewirth DT, Raetz CRH. Proc. Natl. Acad. Sci. U S A. 2006:10877–82. doi: 10.1073/pnas.0604465103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wyckoff TJ, Raetz CRH. J. Biol. Chem. 1999;274:27047–55. doi: 10.1074/jbc.274.38.27047. [DOI] [PubMed] [Google Scholar]
- 95.Sweet CR, Ribeiro AA, Raetz CRH. J. Biol. Chem. 2004;279:25400–10. doi: 10.1074/jbc.M400596200. [DOI] [PubMed] [Google Scholar]
- 96.Que-Gewirth NLS, Ribeiro AA, Kalb SR, Cotter RJ, Bulach DM, et al. J. Biol. Chem. 2004;279:25420–9. doi: 10.1074/jbc.M400598200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Anderson MS, Robertson AD, Macher I, Raetz CRH. Biochemistry. 1988;27:1908–17. doi: 10.1021/bi00406a017. [DOI] [PubMed] [Google Scholar]
- 98.Jackman JE, Raetz CRH, Fierke CA. Biochemistry. 1999;38:1902–11. doi: 10.1021/bi982339s. [DOI] [PubMed] [Google Scholar]
- 99.Jackman JE, Raetz CRH, Fierke CA. Biochemistry. 2001;40:514–23. doi: 10.1021/bi001872g. [DOI] [PubMed] [Google Scholar]
- 100.Jackman JE, Fierke CA, Tumey LN, Pirrung M, Uchiyama T, et al. J. Biol.Chem. 2000;275:11002–9. doi: 10.1074/jbc.275.15.11002. [DOI] [PubMed] [Google Scholar]
- 101.Clements JM, Coignard F, Johnson I, Chandler S, Palan S, et al. Antimicrob. Agents Chemother. 2002;46:1793–9. doi: 10.1128/AAC.46.6.1793-1799.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.McClerren AL, Endsley S, Bowman JL, Andersen NH, Guan Z, et al. Biochemistry. 2005;44:16574–83. doi: 10.1021/bi0518186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Whittington DA, Rusche KM, Shin H, Fierke CA, Christianson DW. Proc. Natl. Acad. Sci. U S A. 2003;100:8146–50. doi: 10.1073/pnas.1432990100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hernick M, Gennadios HA, Whittington DA, Rusche KM, Christianson DW, Fierke CA. J. Biol. Chem. 2005;280:16969–78. doi: 10.1074/jbc.M413560200. [DOI] [PubMed] [Google Scholar]
- 105.Gennadios HA, Whittington DA, Li X, Fierke CA, Christianson DW. Biochemistry. 2006;45:7940–8. doi: 10.1021/bi060823m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gennadios HA, Christianson DW. Biochemistry. 2006;45:15216–23. doi: 10.1021/bi0619021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Coggins BE, Li X, McClerren AL, Hindsgaul O, Raetz CRH, Zhou P. Nat. Struct. Biol. 2003;10:645–51. doi: 10.1038/nsb948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Coggins BE, McClerren AL, Jiang L, Li X, Rudolph J, et al. Biochemistry. 2005;44:1114–11126. doi: 10.1021/bi047820z. [DOI] [PubMed] [Google Scholar]
- 109.Sorensen PG, Lutkenhaus J, Young K, Eveland SS, Anderson MS, Raetz CRH. J. Biol. Chem. 1996;271:25898–905. doi: 10.1074/jbc.271.42.25898. [DOI] [PubMed] [Google Scholar]
- 110.Fuhrer F, Langklotz S, Narberhaus F. Mol. Microbiol. 2006;59:1025–36. doi: 10.1111/j.1365-2958.2005.04994.x. [DOI] [PubMed] [Google Scholar]
- 111.Ogura T, Inoue K, Tatsuta T, Suzaki T, Karata K, et al. Mol. Microbiol. 1999;31:833–44. doi: 10.1046/j.1365-2958.1999.01221.x. [DOI] [PubMed] [Google Scholar]
- 112.Hu Y, Chen L, Ha S, Gross B, Falcone B, et al. Proc. Natl. Acad. Sci. U S A. 2003;100:845–9. doi: 10.1073/pnas.0235749100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, et al. NucleicAcids Research. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Haselberger A, Hildebrandt J, Lam C, Liehl E, Loibner H, et al. Triangle. 1987;26:33–49. [Google Scholar]
- 115.Scholz D, Bednarik K, Ehn G, Neruda W, Janzek E, et al. J. Med. Chem. 1992;35:2070–4. doi: 10.1021/jm00089a019. [DOI] [PubMed] [Google Scholar]
- 116.Albers U, Reus K, Shuman HA, Hilbi H. Microbiology. 2005;151:167–82. doi: 10.1099/mic.0.27563-0. [DOI] [PubMed] [Google Scholar]
- 117.Garrett TA, Que NL, Raetz CRH. J. Biol. Chem. 1998;273:12457–65. doi: 10.1074/jbc.273.20.12457. [DOI] [PubMed] [Google Scholar]
- 118.Belunis CJ, Raetz CRH. J. Biol. Chem. 1992;267:9988–97. [PubMed] [Google Scholar]
- 119.Clementz T, Raetz CRH. J. Biol. Chem. 1991;266:9687–96. [PubMed] [Google Scholar]
- 120.Brozek KA, Raetz CRH. J. Biol. Chem. 1990;265:15410–7. [PubMed] [Google Scholar]
- 121.Ray BL, Raetz CRH. J. Biol. Chem. 1987;262:1122–8. [PubMed] [Google Scholar]
- 122.Lien E, Means TK, Heine H, Yoshimura A, Kusumoto S, et al. J. Clin. Invest. 2000;105:497–504. doi: 10.1172/JCI8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Poltorak A, Ricciardi-Castagnoli P, Citterio S, Beutler B. Proc. Natl. Acad. Sci. U S A. 2000;97:2163–7. doi: 10.1073/pnas.040565397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Saitoh S, Akashi S, Yamada T, Tanimura N, Kobayashi M, et al. Int. Immunol. 2004;16:961–9. doi: 10.1093/intimm/dxh097. [DOI] [PubMed] [Google Scholar]
- 125.Brozek KA, Hosaka K, Robertson AD, Raetz CRH. J. Biol. Chem. 1989;264:6956–66. [PubMed] [Google Scholar]
- 126.Meredith TC, Woodard RW. J Biol Chem. 2003;278:32771–7. doi: 10.1074/jbc.M303661200. [DOI] [PubMed] [Google Scholar]
- 127.Sperandeo P, Pozzi C, Deho G, Polissi A. Res. Microbiol. 2006;157:547–58. doi: 10.1016/j.resmic.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 128.White KA, Kaltashov IA, Cotter RJ, Raetz CRH. J. Biol. Chem. 1997;272:16555–63. doi: 10.1074/jbc.272.26.16555. [DOI] [PubMed] [Google Scholar]
- 129.White KA, Lin S, Cotter RJ, Raetz CR. J. Biol. Chem. 1999;274:31391–400. doi: 10.1074/jbc.274.44.31391. [DOI] [PubMed] [Google Scholar]
- 130.Brabetz W, Muller-Loennies S, Brade H. J. Biol. Chem. 2000;275:34954–62. doi: 10.1074/jbc.M005204200. [DOI] [PubMed] [Google Scholar]
- 131.Isobe T, White KA, Allen AG, Peacock M, Raetz CRH, Maskell DJ. J. Bacteriol. 1999;181:2648–51. doi: 10.1128/jb.181.8.2648-2651.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Belunis CJ, Mdluli KE, Raetz CRH, Nano FE. J. Biol. Chem. 1992;267:18702–7. [PubMed] [Google Scholar]
- 133.Karow M, Georgopoulos C. J. Bacteriol. 1992;174:702–10. doi: 10.1128/jb.174.3.702-710.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Somerville JE, Jr., Cassiano L, Bainbridge B, Cunningham MD, Darveau RP. J. Clin. Invest. 1996;97:359–65. doi: 10.1172/JCI118423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Low KB, Ittensohn M, Le T, Platt J, Sodi S, et al. Nat. Biotechnol. 1999;17:37–41. doi: 10.1038/5205. [DOI] [PubMed] [Google Scholar]
- 136.D’Hauteville H, Khan S, Maskell DJ, Kussak A, Weintraub A, et al. J. Immunol. 2002;168:5240–51. doi: 10.4049/jimmunol.168.10.5240. [DOI] [PubMed] [Google Scholar]
- 137.Vorachek-Warren MK, Carty SM, Lin S, Cotter RJ, Raetz CRH. J. Biol. Chem. 2002;277:14186–93. doi: 10.1074/jbc.M200408200. [DOI] [PubMed] [Google Scholar]
- 138.Montminy SW, Khan N, McGrath S, Walkowicz MJ, Sharp F, et al. Nat. Immunol. 2006;7:1066–73. doi: 10.1038/ni1386. [DOI] [PubMed] [Google Scholar]
- 139.Ferguson AD, Hofmann E, Coulton JW, Diederichs K, Welte W. Science. 1998;282:2215–20. doi: 10.1126/science.282.5397.2215. [DOI] [PubMed] [Google Scholar]
- 140.Ferguson AD, Welte W, Hofmann E, Lindner B, Holst O, et al. Structure Fold. Des. 2000;8:585–92. doi: 10.1016/s0969-2126(00)00143-x. [DOI] [PubMed] [Google Scholar]
- 141.Nishiyama K, Ikegami A, Moser M, Schiltz E, Tokuda H, Muller M. J. Biol.Chem. 2006;281:35667–76. doi: 10.1074/jbc.M608228200. [DOI] [PubMed] [Google Scholar]
- 142.Steeghs L, den Hartog R, den Boer A, Zomer B, Roholl P, van der Ley P. Nature. 1998;392:449–50. doi: 10.1038/33046. [DOI] [PubMed] [Google Scholar]
- 143.Doerrler WT, Reedy MC, Raetz CRH. J. Biol. Chem. 2001;276:11461–4. doi: 10.1074/jbc.C100091200. [DOI] [PubMed] [Google Scholar]
- 144.Karow M, Georgopoulos C. Mol. Microbiol. 1993;7:69–79. doi: 10.1111/j.1365-2958.1993.tb01098.x. [DOI] [PubMed] [Google Scholar]
- 145.Polissi A, Georgopoulos C. Mol. Microbiol. 1996;20:1221–33. doi: 10.1111/j.1365-2958.1996.tb02642.x. [DOI] [PubMed] [Google Scholar]
- 146.Zhou Z, White KA, Polissi A, Georgopoulos C, Raetz CRH. J. Biol. Chem. 1998;273:12466–75. doi: 10.1074/jbc.273.20.12466. [DOI] [PubMed] [Google Scholar]
- 147.Chang G, Roth CB. Science. 2001;293:1793–800. doi: 10.1126/science.293.5536.1793. [DOI] [PubMed] [Google Scholar]
- 148.Chang G. J. Mol. Biol. 2003;330:419–30. [Google Scholar]
- 149.Reyes CL, Chang G. Science. 2005;308:1028–31. doi: 10.1126/science.1107733. [DOI] [PubMed] [Google Scholar]
- 150.Dawson RJ, Locher KP. Nature. 2006;443:180–5. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- 151.Chang G, Roth CB, Reyes CL, Pornillos O, Chen Y-J, Chen AP. Science. 2006;314:1875. doi: 10.1126/science.314.5807.1875b. [DOI] [PubMed] [Google Scholar]
- 152.Raetz CRH, Garrett TA, Reynolds CM, Shaw WA, Moore JD, et al. J. Lipid Res. 2006;47:1097–111. doi: 10.1194/jlr.M600027-JLR200. [DOI] [PubMed] [Google Scholar]
- 153.Huijbregts RP, de Kroon AI, de Kruijff B. J. Biol. Chem. 1998;273:18936–42. doi: 10.1074/jbc.273.30.18936. [DOI] [PubMed] [Google Scholar]
- 154.Doerrler WT, Raetz CRH. J. Biol. Chem. 2002;277:36697–705. doi: 10.1074/jbc.M205857200. [DOI] [PubMed] [Google Scholar]
- 155.Kol MA, van Dalen A, de Kroon AI, de Kruijff B. J. Biol. Chem. 2003;278:24586–93. doi: 10.1074/jbc.M301875200. [DOI] [PubMed] [Google Scholar]
- 156.Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, et al. Science. 2004;304:66–74. doi: 10.1126/science.1093857. [DOI] [PubMed] [Google Scholar]
- 157.Osborn MJ, Rick PD, Rasmussen NS. J. Biol. Chem. 1980;255:4246–51. [PubMed] [Google Scholar]
- 158.Tefsen B, Bos MP, Beckers F, Tommassen J, de Cock H. J. Biol. Chem. 2005;280:35961–6. doi: 10.1074/jbc.M509026200. [DOI] [PubMed] [Google Scholar]
- 159.Sperandeo P, Cescutti R, Villa R, Di Benedetto C, Candia D, et al. J. Bacteriol. 2007;189:244–53. doi: 10.1128/JB.01126-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Braun M, Silhavy TJ. Mol. Microbiol. 2002;45:1289–302. doi: 10.1046/j.1365-2958.2002.03091.x. [DOI] [PubMed] [Google Scholar]
- 161.Borst P, Zelcer N, van Helvoort A. Biochim. Biophys. Acta. 2000;1486:128–44. doi: 10.1016/s1388-1981(00)00053-6. [DOI] [PubMed] [Google Scholar]
- 162.Borst P, Elferink RO. Annu. Rev. Biochem. 2002;71:537–92. doi: 10.1146/annurev.biochem.71.102301.093055. [DOI] [PubMed] [Google Scholar]
- 163.Smit JJM, Schinkel AH, Oude Elferink RPJ, Groen AK, Wagenaar E, et al. Cell. 1993;75:451–62. doi: 10.1016/0092-8674(93)90380-9. [DOI] [PubMed] [Google Scholar]
- 164.Schmitz G, Liebisch G, Langmann T. FEBS Lett. 2006;580:5597–610. doi: 10.1016/j.febslet.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 165.Zhou Z, Lin S, Cotter RJ, Raetz CRH. J. Biol. Chem. 1999;274:18503–14. doi: 10.1074/jbc.274.26.18503. [DOI] [PubMed] [Google Scholar]
- 166.Zhou Z, Ribeiro AA, Lin S, Cotter RJ, Miller SI, Raetz CRH. J. Biol. Chem. 2001;276:43111–21. doi: 10.1074/jbc.M106960200. [DOI] [PubMed] [Google Scholar]
- 167.Brozek KA, Bulawa CE, Raetz CRH. J. Biol. Chem. 1987;262:5170–9. [PubMed] [Google Scholar]
- 168.Guo L, Lim KB, Poduje CM, Daniel M, Gunn JS, et al. Cell. 1998;95:189–98. doi: 10.1016/s0092-8674(00)81750-x. [DOI] [PubMed] [Google Scholar]
- 169.Helander IM, Kilpeläinen I, Vaara M. Mol. Microbiol. 1994;11:481–7. doi: 10.1111/j.1365-2958.1994.tb00329.x. [DOI] [PubMed] [Google Scholar]
- 170.Nummila K, Kilpeläinen I, Zähringer U, Vaara M, Helander IM. Mol. Microbiol. 1995;16:271–8. doi: 10.1111/j.1365-2958.1995.tb02299.x. [DOI] [PubMed] [Google Scholar]
- 171.Helander IM, Kato Y, Kilpelainen I, Kostiainen R, Lindner B, et al. Eur. J. Biochem. 1996;237:272–8. doi: 10.1111/j.1432-1033.1996.0272n.x. [DOI] [PubMed] [Google Scholar]
- 172.Breazeale SD, Ribeiro AA, Raetz CRH. J. Biol. Chem. 2002;277:2886–96. doi: 10.1074/jbc.M109377200. [DOI] [PubMed] [Google Scholar]
- 173.Breazeale SD, Ribeiro AA, Raetz CRH. J. Biol. Chem. 2003;279:24731–9. doi: 10.1074/jbc.M304043200. [DOI] [PubMed] [Google Scholar]
- 174.Breazeale SD, Ribeiro AA, McClerren AL, Raetz CRH. J. Biol. Chem. 2005;280:14154–67. doi: 10.1074/jbc.M414265200. [DOI] [PubMed] [Google Scholar]
- 175.Williams GJ, Breazeale SD, Raetz CRH, Naismith JH. J. Biol. Chem. 2005;280:23000–8. doi: 10.1074/jbc.M501534200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Gatzeva-Topalova PZ, May AP, Sousa MC. Structure (Camb) 2005;13:929–42. doi: 10.1016/j.str.2005.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Noland BW, Newman JM, Hendle J, Badger J, Christopher JA, et al. Structure (Camb) 2002;10:1569–80. doi: 10.1016/s0969-2126(02)00879-1. [DOI] [PubMed] [Google Scholar]
- 178.Gunn JS, Ryan SS, Van Velkinburgh JC, Ernst RK, Miller SI. Infect. Immun. 2000;68:6139–46. doi: 10.1128/iai.68.11.6139-6146.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kanipes MI, Lin S, Cotter RJ, Raetz CRH. J. Biol. Chem. 2001;276:1156–63. doi: 10.1074/jbc.M009019200. [DOI] [PubMed] [Google Scholar]
- 180.Figueroa-Bossi N, Lemire S, Maloriol D, Balbontin R, Casadesus J, Bossi L. Mol. Microbiol. 2006;62:838–52. doi: 10.1111/j.1365-2958.2006.05413.x. [DOI] [PubMed] [Google Scholar]
- 181.Tamayo R, Choudhury B, Septer A, Merighi M, Carlson R, Gunn JS. J. Bacteriol. 2005;187:3391–9. doi: 10.1128/JB.187.10.3391-3399.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kim SH, Jia W, Parreira VR, Bishop RE, Gyles CL. Microbiology. 2006;152:657–66. doi: 10.1099/mic.0.28692-0. [DOI] [PubMed] [Google Scholar]
- 183.Kim SH, Jia W, Bishop RE, Gyles C. Infect. Immun. 2004;72:1174–80. doi: 10.1128/IAI.72.2.1174-1180.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kaniuk NA, Vinogradov E, Li J, Monteiro MA, Whitfield C. J. Biol. Chem. 2004;279:31237–50. doi: 10.1074/jbc.M401879200. [DOI] [PubMed] [Google Scholar]
- 185.El Ghachi M, Derbise A, Bouhss A, Mengin-Lecreulx D. J. Biol. Chem. 2005;280:18689–95. doi: 10.1074/jbc.M412277200. [DOI] [PubMed] [Google Scholar]
- 186.Lazar K, Walker S. Curr. Opin. Chem. Biol. 2002;6:786–93. doi: 10.1016/s1367-5931(02)00355-1. [DOI] [PubMed] [Google Scholar]
- 187.Cho US, Bader MW, Amaya MF, Daley ME, Klevit RE, et al. J. Mol. Biol. 2006;356:1193–206. doi: 10.1016/j.jmb.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 188.Martin-Orozco N, Touret N, Zaharik ML, Park E, Kopelman R, et al. Mol. Biol. Cell. 2006;17:498–510. doi: 10.1091/mbc.E04-12-1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Winfield MD, Groisman EA. Proc. Natl. Acad. Sci. U S A. 2004;101:17162–7. doi: 10.1073/pnas.0406038101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Guina T, Yi EC, Wang H, Hackett M, Miller SI. J. Bacteriol. 2000;182:4077–86. doi: 10.1128/jb.182.14.4077-4086.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Robey M, O’Connell W, Cianciotto NP. Infect. Immun. 2001;69:4276–86. doi: 10.1128/IAI.69.7.4276-4286.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Preston A, Maxim E, Toland E, Pishko EJ, Harvill ET, et al. Mol. Microbiol. 2003;48:725–36. doi: 10.1046/j.1365-2958.2003.03484.x. [DOI] [PubMed] [Google Scholar]
- 193.Pilione MR, Pishko EJ, Preston A, Maskell DJ, Harvill ET. Infect. Immun. 2004;72:2837–42. doi: 10.1128/IAI.72.5.2837-2842.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Bishop RE. Mol. Microbiol. 2005;57:900–12. doi: 10.1111/j.1365-2958.2005.04711.x. [DOI] [PubMed] [Google Scholar]
- 195.Rebeil R, Ernst RK, Gowen BB, Miller SI, Hinnebusch BJ. Mol. Microbiol. 2004;52:1363–73. doi: 10.1111/j.1365-2958.2004.04059.x. [DOI] [PubMed] [Google Scholar]
- 196.Ahn VE, Lo EI, Engel CK, Chen L, Hwang PM, et al. Embo. J. 2004;23:2931–41. doi: 10.1038/sj.emboj.7600320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Hwang PM, Choy WY, Lo EI, Chen L, Forman-Kay JD, et al. Proc. Natl. Acad. Sci. U S A. 2002;99:13560–5. doi: 10.1073/pnas.212344499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Evanics F, Hwang PM, Cheng Y, Kay LE, Prosser RS. J. Am. Chem. Soc. 2006;128:8256–64. doi: 10.1021/ja0610075. [DOI] [PubMed] [Google Scholar]
- 199.Stover AG, Da Silva Correia J, Evans JT, Cluff CW, Elliott MW, et al. J. Biol. Chem. 2004;279:4440–09. doi: 10.1074/jbc.M310760200. [DOI] [PubMed] [Google Scholar]
- 200.Hwang PM, Bishop RE, Kay LE. Proc. Natl. Acad. Sci. U S A. 2004;101:9618–23. doi: 10.1073/pnas.0402324101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Tam C, Missiakas D. Mol. Microbiol. 2005;55:1403–12. doi: 10.1111/j.1365-2958.2005.04497.x. [DOI] [PubMed] [Google Scholar]
- 202.Gibbons HS, Lin S, Cotter RJ, Raetz CRH. J. Biol. Chem. 2000;275:32940–9. doi: 10.1074/jbc.M005779200. [DOI] [PubMed] [Google Scholar]
- 203.Raetz CRH. J. Endotoxin Res. 2001;7:73–8. [PubMed] [Google Scholar]
- 204.Kawasaki K, Ernst RK, Miller SI. J. Bacteriol. 2005;187:2448–57. doi: 10.1128/JB.187.7.2448-2457.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Geurtsen J, Steeghs L, Hamstra HJ, Ten Hove J, de Haan A, et al. Infect. Immun. 2006;74:5574–85. doi: 10.1128/IAI.00834-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kawasaki K, Ernst RK, Miller SI. J. Biol. Chem. 2004;279:20044–8. doi: 10.1074/jbc.M401275200. [DOI] [PubMed] [Google Scholar]
- 207.Ernst RK, Adams KN, Moskowitz SM, Kraig GM, Kawasaki K, et al. J. Bacteriol. 2006;188:191–201. doi: 10.1128/JB.188.1.191-201.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Geurtsen J, Steeghs L, Hove JT, van der Ley P, Tommassen J. J. Biol. Chem. 2005;280:8248–59. doi: 10.1074/jbc.M414235200. [DOI] [PubMed] [Google Scholar]
- 209.Rutten L, Geurtsen J, Lambert W, Smolenaers JJ, Bonvin AM, et al. Proc. Natl. Acad. Sci. U S A. 2006;103:7071–6. doi: 10.1073/pnas.0509392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Ellis J, Oyston PC, Green M, Titball RW. Clin. Microbiol. Rev. 2002;15:631–46. doi: 10.1128/CMR.15.4.631-646.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kieffer TL, Cowley S, Nano FE, Elkins KL. Microbes Infect. 2003;5:397–403. doi: 10.1016/s1286-4579(03)00052-2. [DOI] [PubMed] [Google Scholar]
- 212.Wang X, Ribeiro AA, Guan Z, McGrath S, Cotter R, Raetz CRH. Biochemistry. 2006;45:14427–40. doi: 10.1021/bi061767s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Larsson P, Oyston PC, Chain P, Chu MC, Duffield M, et al. Nat. Genet. 2005;37:153–9. doi: 10.1038/ng1499. [DOI] [PubMed] [Google Scholar]
- 214.Stead C, Tran A, Ferguson D, Jr., McGrath S, Cotter R, Trent S. J. Bacteriol. 2005;187:3374–83. doi: 10.1128/JB.187.10.3374-3383.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Phillips NJ, Schilling B, McLendon MK, Apicella MA, Gibson BW. Infect. Immun. 2004;72:5340–8. doi: 10.1128/IAI.72.9.5340-5348.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Vinogradov E, Perry MB, Conlan JW. Eur. J. Biochem. 2002;269:6112–8. doi: 10.1046/j.1432-1033.2002.03321.x. [DOI] [PubMed] [Google Scholar]
- 217.Wang X, Ribeiro AA, Guan Z, Abraham SN, Raetz CRH. Proc. Natl. Acad. Sci. U S A. 2007 doi: 10.1073/pnas.0611606104. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, et al. Nature. 1997;388:539–47. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 219.Bhat UR, Forsberg LS, Carlson RW. J. Biol. Chem. 1994;269:14402–10. [PubMed] [Google Scholar]
- 220.Que NLS, Lin S, Cotter RJ, Raetz CRH. J. Biol. Chem. 2000;275:28006–16. doi: 10.1074/jbc.M004008200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Que NLS, Ribeiro AA, Raetz CRH. J. Biol. Chem. 2000;275:28017–27. doi: 10.1074/jbc.M004009200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Bhat UR, Mayer H, Yokota A, Hollingsworth RI, Carlson R. J. Bacteriol. 1991;173:2155–9. doi: 10.1128/jb.173.7.2155-2159.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Basu SS, White KA, Que NL, Raetz CRH. J. Biol. Chem. 1999;274:11150–8. doi: 10.1074/jbc.274.16.11150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Carlson RW, Reuhs B, Chen TB, Bhat UR, Noel KD. J. Biol. Chem. 1995;270:11783–8. doi: 10.1074/jbc.270.20.11783. [DOI] [PubMed] [Google Scholar]
- 225.Brozek KA, Kadrmas JL, Raetz CRH. J. Biol. Chem. 1996;271:32112–8. [PubMed] [Google Scholar]
- 226.Price NPJ, Kelly TM, Raetz CRH, Carlson RW. J. Bacteriol. 1994;176:4646–55. doi: 10.1128/jb.176.15.4646-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Basu SS, Karbarz MJ, Raetz CRH. J. Biol. Chem. 2002;277:28959–71. doi: 10.1074/jbc.M204525200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Brozek KA, Carlson RW, Raetz CRH. J. Biol. Chem. 1996;271:32126–36. doi: 10.1074/jbc.271.50.32126. [DOI] [PubMed] [Google Scholar]
- 229.Karbarz MJ, Kalb SR, Cotter RJ, Raetz CRH. J. Biol. Chem. 2003;278:39269–79. doi: 10.1074/jbc.M305830200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Que-Gewirth NLS, Lin S, Cotter RJ, Raetz CRH. J. Biol. Chem. 2003;278:12109–19. doi: 10.1074/jbc.M300378200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Que-Gewirth NLS, Karbarz MJ, Kalb SR, Cotter RJ, Raetz CRH. J. Biol. Chem. 2003;278:12120–9. doi: 10.1074/jbc.M300379200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Kanjilal-Kolar S, Basu SS, Kanipes MI, Guan Z, Garrett TA, Raetz CRH. J. Biol. Chem. 2006;281:12865–78. doi: 10.1074/jbc.M513864200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Kanjilal-Kolar S, Raetz CRH. J. Biol. Chem. 2006;281:12879–87. doi: 10.1074/jbc.M513865200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Ferguson GP, Datta A, Carlson RW, Walker GC. Mol. Microbiol. 2005;56:68–80. doi: 10.1111/j.1365-2958.2005.04536.x. [DOI] [PubMed] [Google Scholar]
- 235.Vedam V, Kannenberg EL, Haynes JG, Sherrier DJ, Datta A, Carlson RW. J. Bacteriol. 2003;185:1841–50. doi: 10.1128/JB.185.6.1841-1850.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Boon Hinckley M, Reynolds CM, Ribeiro AA, McGrath SC, Cotter RJ, et al. J. Biol. Chem. 2005;280:30214–24. doi: 10.1074/jbc.M506103200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Vinogradov EV, Muller-Loennies S, Petersen BO, Meshkov S, Thomas-Oates JE, et al. Eur. J. Biochem. 1997;247:82–90. doi: 10.1111/j.1432-1033.1997.00082.x. [DOI] [PubMed] [Google Scholar]
- 238.Bode CE, Brabetz W, Brade H. Eur. J. Biochem. 1998;254:404–12. doi: 10.1046/j.1432-1327.1998.2540404.x. [DOI] [PubMed] [Google Scholar]
- 239.Schneider JE, Reinhold V, Rumley MK, Kennedy EP. J. Biol. Chem. 1979;254:10135–8. [PubMed] [Google Scholar]
- 240.Kennedy EP. In: Escherichia coli and Salmonella typhimurium. Neidhardt FC, editor. I. ASM Publications; Washington, D. C: 1987. pp. 672–9. [Google Scholar]
- 241.Wu HC. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt FC, editor. American Society for Microbiology; Washington, D. C.: 1996. pp. 1005–14. [Google Scholar]
- 242.Raetz CRH. Annu. Rev. Genet. 1986;20:253–95. doi: 10.1146/annurev.ge.20.120186.001345. [DOI] [PubMed] [Google Scholar]
- 243.Cronan JE. Annu. Rev. Microbiol. 2003;57:203–24. doi: 10.1146/annurev.micro.57.030502.090851. [DOI] [PubMed] [Google Scholar]
- 244.Stevenson G, Neal B, Liu D, Hobbs M, Packer NH, et al. J. Bacteriol. 1994;176:4144–56. doi: 10.1128/jb.176.13.4144-4156.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Murphy RC, Raetz CRH, Reynolds CM, Barkley RM. Prostaglandins Other Lipid Mediat. 2005;77:131–40. doi: 10.1016/j.prostaglandins.2004.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Jain NU, Wyckoff TJ, Raetz CRH, Prestegard JH. J. Mol. Biol. 2004;343:1379–89. doi: 10.1016/j.jmb.2004.08.103. [DOI] [PubMed] [Google Scholar]
- 247.Feldman MF, Marolda CL, Monteiro MA, Perry MB, Parodi AJ, Valvano MA. J. Biol. Chem. 1999;274:35129–38. doi: 10.1074/jbc.274.49.35129. [DOI] [PubMed] [Google Scholar]
- 248.Strain SM, Armitage IM, Anderson L, Takayama K, Qureshi N, Raetz CRH. J. Biol. Chem. 1985;260:16089–98. [PubMed] [Google Scholar]
- 249.Zhou Z, Ribeiro AA, Raetz CRH. J. Biol. Chem. 2000;275:13542–51. doi: 10.1074/jbc.275.18.13542. [DOI] [PubMed] [Google Scholar]
- 250.Price NJP, Jeyaretnam B, Carlson RW, Kadrmas JL, Raetz CRH, Brozek KA. Proc. Natl. Acad. Sci. U. S. A. 1995;92:7352–6. doi: 10.1073/pnas.92.16.7352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Kadrmas JL, Brozek KA, Raetz CRH. J. Biol. Chem. 1996;271:32119–25. [PubMed] [Google Scholar]
- 252.Kadrmas JL, Allaway D, Studholme RE, Sullivan JT, Ronson CW, et al. J. Biol. Chem. 1998;273:26432–40. doi: 10.1074/jbc.273.41.26432. [DOI] [PubMed] [Google Scholar]