

Stem Cells
The concept of the stem cell derives from the study of embryogenesis and has a strong historical basis in the field of developmental biology[1]. Adult stem cells comprise a small, highly regulated cell population characterized by their capacities for self-renewal and differentiation into a variety of mature cell types. Stem cells reside in a growing list of tissues and organs and participate to various extents in the replenishment of mature cells responsible for the specialized functional properties of the tissue in which they reside. It is because of these properties that stem cells are at the center of attention for the treatment of congenital and acquired diseases, and amelioration of the deleterious effects of aging. The degree to which the developmental potential of adult stem cells from a given tissue is restricted to the tissue in which it resides, is open to a debate that is difficult to resolve since stem cells circulate continuously in the blood and lymphatic vasculature[2]. The recent discovery that fully differentiated epithelial cells from adults can, through the (re-)initiation of expression of as few as four genes, appear to acquire most, if not all, of the attributes of embryonic stem cells with pluripotentiality, demonstrates the close affinity between at least some differentiated cells and the cells from which they ultimately arose[3, 4]. It must be emphasized that these findings have been obtained under experimental conditions not encountered in nature; nonetheless, they may enable clinical tissue regeneration for the treatment of a number of diseases. If many adult tissues and organs are continuously replenished by cells derived from stem cells, then why do they show signs of aging? One possibility is that stem cells themselves age and senesce, resulting in a decreased ability to replace worn-out progeny and/or the fact that they pass on aged phenotypes to their progeny.
Missing in this discussion until now is the effect of the cellular and molecular environment on stem cell properties, although the molecular re-programming of epithelial cells into pluripotent stem cells demonstrates the importance of the intracellular environment. Indeed, ample evidence exists showing that intrinsic and extrinsic regulators are inextricably linked in determining stem cell functional properties. Of special current interest is the extracellular stem cell environment, commonly referred to as the stem cell ‘niche’, as originally coined for hematopoietic stem cells in the bone marrow. The cells and their extracellular matrices comprising the ‘niche’ for stem cells in different tissues are likely to be different, but the signals that mediate effects on stem cells, such as maintaining them in a replicative quiescent (or active) state often involve similar if not identical pathways (see review in ref. [5]). Moreover, the cellular and molecular composition of ‘niches’ is probably dynamic and responsive to needs for stem cell renewal and the differentiation of progeny into specific lineages.
Stem cell aging
In most tissues, aging has the effect of blunting processes akin to regeneration, such as wound healing[6] and hematopoietic reconstitution after chemotherapy or following bone marrow transplant[7, 8]. Moreover, the role of stem cell aging in the extent and pace of regeneration remains to be fully explored. There is evidence in the hematopoietic system that stem cells progressively lose the breadth of their developmental potency. Serially transplanted bone marrow stem cells rapidly lose the capacity to produce the normal spectrum and proportions of blood cell lineages[9–13]. These studies suggest that stem cell plasticity, if such exists, may be higher when the organism is young and diminishes, or is lost, with age[14]. Highly purified young stem cells engraft the marrow of young recipients with high efficiency[15, 16], but old stem cells have considerably decreased ability to home to the marrow and contribute to engraftment [17–19]. In addition, age-related changes in the functional abilities of HSC are clearly demonstrated by transplantation experiments using embryonic and adult cells, e.g., HSC from old bone marrow were capable of fewer repetitive transplantations than those from young marrow[20], HSC from fetal liver engraft better than adult mouse bone marrow cells in lethally irradiated mice[21], and limiting dilution repopulation assays show that fetal liver stem cells have extensive functional advantage over young adult bone marrow[22]. Thus, what are the changes seen in stem cells during the aging process?
A number of factors may interfere with the overall potency of the stem cell population during aging. However, much of the difficulty in assessing the effect of age on the stem cell population has to do with the way in which stem cell properties are measured. Functional tests require the proliferation and differentiation of stem cells, because it is their progeny that are actually measured. In transplantation experiments, engraftment by hematopoietic stem cell progeny is measured. In secondary transfers, it is stem cells produced by self-renewing stem cells of the primary graft that in turn generate progeny responsible for engraftment in secondary hosts. Under the best conditions, it is not possible to exceed five successful passages[23]. Failure to fully regenerate the stem cell population may be due to intrinsic effects such as aging and extrinsic effects associated with the transplant procedure[23–25]. Extrinsic factors associated with the transplant procedure, such as HSC reassociation with bone marrow stromal components and their integuments, may be responsible for the failure of the transplanted HSC to fully regenerate the host hematopoietic system[26].
Stem cells numbers
If stem cells play a role in the limitation of organismal longevity, the simplest explanation of how they might do so would be a decline in their numbers in old age. Qualitative changes in stem cells and in the composition of the stem cell population with respect to its qualities may be an important additional factor in stem cell aging. The amount of active bone marrow in humans progressively diminishes from childhood to old age, during which time active marrow is restricted to the pelvis, sternum, and vertebrae, and where even in these sites the marrow cellularity is reduced[27]. Moreover, the number of CD34+ cells in the marrow and blood of the very old, including centenarians, is greatly reduced[28, 29]. Despite these quantitative changes there is little solid evidence that longevity is threatened by a lack of stem cells[30]. Rather, as discussed below, the response of old stem cell populations in many tissues is a lack of the robust response of their young counterparts to stress.
There is little question that cellular senescence is intimately involved in the aging process and we have championed the idea that the effects of age on the stem cell population contribute, perhaps to a large extent, to the declining function of tissues during aging, and perhaps to organismal longevity[14, 31, 32]. Since the regulation of stem cells is accomplished via both intrinsic and extrinsic mechanisms, senescence, at both population and cellular levels, may involve age-related changes in either or both pathways. In this review, we examine cellular senescence from the vantage point of stem cells, particularly hematopoietic stem cells (HSC) in the bone marrow in the context of aging. Stem cell populations, by necessity, must persist for the lifetime of the organism, and for this reason, elaborate mechanisms must have evolved to preserve the cardinal stem cell functions. Nonetheless, there is strong evidence despite these protective mechanisms, that stem cell populations are not spared from the ravages of aging[32, 33]. In this review, we propose that protein mis-folding and aggregation may contribute to stem cell aging in ways that parallel amyloidoses in the nervous system, such as Alzheimer’s disease.
Senescence of a stem cell population can have quantitative and qualitative dimensions. For example, we have shown in embryo-aggregated chimeric mice that stem cell populations of the two composite strains [C57BL/6 (B6) and DBA/2 (D2)] contribute to hematopoiesis in completely different patterns during aging, despite their co-existence in a common environment, and presumably exposure to the same ‘niche’ environment[31]. In old chimeras, stem cells of the D2 genotype were either completely exhausted, senescent, or otherwise quiescent, since they gave way completely to blood cell production derived from stem cells of the partner strain (B6). We subsequently harvested marrow from chimeras in which the D2 stem cells were no longer contributing to hematopoiesis and showed that they were still present and could be re-activated by bone marrow transplantation, albeit with a greatly diminished duration of activity[34]. These results affirm the importance of intrinsic stem cell regulators and show that at least in this type of experiment, senescence at the stem cell level, if it occurs, must be reversible. Since cellular senescence has come to imply a series of irreversible changes, perhaps the more broad term, quiescence, better explains the changes observed during aging of D2 HSC. Much of the work on murine HSC has been carried out with B6 mice, often because a strain congenic for the pan-hematopoietic cell surface marker CD45.1, derived from the SJL donor strain, is readily available on the B6 genetic background. B6 mice carry the CD45.2 allele and thus in bone marrow transplant studies involving the two strains, donor and recipient hematopoietic cells can be easily distinguished and quantified by flow cytometric immunophenotyping. In contrast to diminishing HSC at old age in the relatively short-lived D2 and BALB/c strains, HSC numbers increase steadily in long-lived B6 mice up to an age of at least 30 months[35]. Whether the increased numbers of HSC during aging in B6 mice contributes to their longer lifespan is not known. What is known is that qualitative changes in B6 HSC, as in all other strains studied, accompany aging. For example, HSC from old mice have a biased differentiation pattern favoring myeloid differentiation at the expense of lymphoid lineages[18, 36]. Therefore, it would be reasonable to propose that as stem cells have accumulated intracellular damage as a result of the rigors of aging, surveillance pathways leading to apoptosis, such as the p53-initiated cascade, takes care of compromised stem cells by removing them from the pool, preventing the establishment of a clone with potentially dysfunctional or tumorigenic progeny[37–39]. The importance of apoptosis in the physiological regulation of HSC population size suggest that this mechanism may be a target of aging. For example, overexpression of bcl-2 in transgenic mice not only prevents apoptosis in response to a number of genotoxic challenges, but increases by more than twofold the number of HSC in these mice under steady-state conditions and enhances their engraftment potential on a cell-by-cell basis[40]. If cells are lost through accumulated damage, could stem cell replication recover stem cell numbers?
Stem cell replication and the effect on telomere length
A hallmark of the population of stem cells from normal young and middle-aged mice or humans is its overwhelming quiescence[41, 42]. Bromodeoxyuridine (Brdu) administration has been used to label the stem cell compartments of young, but not old, mice. In young marrow, as expected, a short pulse of Brdu has a very small labeling index, consistent with a quiescent population. Long-term BrdU administration to young mice via drinking water showed that essentially all stem cells replicated at least once about every 2 months[43, 44]. These results are in disagreement with the concept that most stem cells are deeply quiescent, and many of which may not enter cycle during a mammal’s entire lifetime[45]. The pattern of cell cycle kinetics of stem cells may be altered during aging[35], but further studies are required in both mouse and man to resolve this issue. However, an increased proportion of stem cells in cycle might be correlated with the increased incidences of leukemia, lymphoma, and myelodysplastic syndrome at old age[35].
Interest in the importance of telomeres to mammalian aging, replicative stress, and cancer etiology in the last several years has led to the their study in stem cells[46–52]. The assumption has been that telomeres serve as a pacemaker of senescence in replicating stem cells, as in other cell types, in which the length of telomeres shortens with each round of replication until a critical short length is reached that signals induction of cell senescence[53, 54]. Despite the fact that HSC produce telomerase[55–57] telomeres of stem cells have been shown to progressively shorten during aging[49, 58– 60] and following hematopoietic stress[61], especially that following stem cell transplantation[59, 62–66]. Recent studies have shed light on the question of whether telomere length in stem cells plays a role in their function under physiological conditions. Inter-species comparisons of stem cell numbers in mice, cats and humans revealed that, despite the variation in sizes and lifespans of the three species, the total number of hematopoietic stem cells did not reflect those variations and were, in fact, remarkably similar[67]. By measuring telomere lengths in blood granulocytes, and applying stochastic simulation, Shepherd et al.[68] and Lopes et al. [69]have shown that while the absolute number of stem cells may not reflect the variation in organismal size or longevity, the rate of hematopoietic stem cell turnover does. Replication rates for HSCs in mice, cats and humans were estimated to be once in 2.5, 10, and 45 weeks, respectively[68]. Thus, the way in which stem cell populations are organized and parsed for use is a reflection of a combination of physiological parameters including the time-scale over which functional activity is required.
Organismal Aging
If aging is viewed as the cumulative wear-and-tear on cells of an organism, the rate and degree of organismal aging is a reflection of how well the organism counteracts the deleterious effects of the environment. Currently, two major theories regarding organismal aging are proposed: an evolutionary-based theory and a damage-based theory. Many evolutionary biologists suggest that genes that favor the species’ reproductive success may have negative effects in later life, thus limiting the organism’s life span. This phenomenon has been termed antagonistic pleiotropy[70]. According to this theory, organisms are effectively maintained only to achieve reproductive success. Following a species’ reproductive phase, genes selected for their beneficial effects were not selected in later life. An example of antagonistic pleiotrophy is the role of androgens in males. Androgens, the hormones responsible for the normal growth and function of the prostate in the young, and pivotal in the generation of sperm for reproduction, are the same hormones that contribute to prostate cancer in the elderly.
The replicative senescence (replicative damage) theory was first introduced in the 1960s as a process that limits the number of cell divisions that a specific cell can undergo throughout life[71], and has subsequently been linked to the loss of telomeric DNA described above. Therefore, replicative senescence, or growth arrest, was postulated to be a direct link to how many rounds of replication that cells undergo during their lifetime[72]. During each cell division DNA is lost at the ends of the chromosomes[73] because most of our somatic cells do not express telomerase, the enzyme responsible in reconstituting the structures at the ends of the chromosomes, the telomeres[74]. Joeng et al. [75] have shown that life span could be prolonged in worms that overexpressed HRP-1, a telomere binding protein that steadily increases telomere length. However, the mechanisms involved in such a process are unknown. Further studies have shown that the affected cells underwent senescence before they acquired critical short telomeres, avoiding the genomic instability leading to cancer. Therefore, replicative senescence/growth arrest did not depend exclusively on telomere erosion – it depended on other mechanisms as well.
DNA replication itself imposes stress in the proofreading and editing mechanism necessary to keep the genome free of replication errors. The DNA repair machinery plays a pivotal role in keeping the genome free from mistakes that invariably occur during replication and that are caused by environmental insults. Therefore, there appears to be a direct link between life span and DNA repair[76]. Species that have efficient DNA repair machinery live longer than those without this capacity[77]. Mutations in DNA polymerases have also been implicated in the aging process and, not surprisingly, senescent cells have faulty DNA polymerases[78].
Throughout life, molecular mechanisms necessary to counteract the deleterious effects of the environment evolve[79]. In addition to cellular senescence, apoptosis is an important evolutionarily conserved mechanism used to remove damaged cells. Unlike necrosis, it consists of the removal of unwanted and damaged cells without exposing neighboring cells to cellular proteases. Defects in the regulation of apoptosis have been linked to degenerative and hyperproliferative diseases such as cancer[80–82]. Key molecules in this process, such as p53, Bmi-1, p16ink4a, p19Arf, and bcl-2 have been identified and strongly linked to apoptosis and senescence[83–88]. Therefore, as in senescence, apoptosis is a tumor suppressor mechanism that prevents cells carrying mutations to divide and generate defective progeny. Apoptosis and senescence may have evolved together to protect complex organisms that have a mix of proliferative and postmitotic tissues.
Aging Damage
A theory based on cellular damage was first proposed by Harman in the mid-1950’s[89]. Today it has two main components: oxidative damage and cellular/replicative damage (Figure 1). Normal metabolism produces reactive oxygen species (ROS) that may oxidize and damage cell membranes, proteins, and nucleic acids. An example of the effect on the aging process of ameliorating ROS toxicity may be found in studies in which enzymes that limit exposure of cellular components to ROS, such as catalase and superoxide dismutase (SOD1), were over-expressed. Supra-normal levels of SOD1 in Drosophila caused an increase of 20–30% in life span compared to control flies[90]. Interestingly, if over-expression was targeted to neuronal cells, flies lived even longer than flies that have total body overexpression of the enzyme, thus confirming that the nervous system is susceptible to damage by ROS and that such damage affects longevity. In mammals, the link between ROS and aging is still incomplete because there is no evidence of premature aging in mice carrying loss-of-function mutations in ROS degrading enzymes[91]. In addition to ROS, damage in the mitochondrial genome has been closely linked to aging. Mutations in mitochondrial DNA cause defects in the electron transport chain that affects energy production and creates ROS. Age-dependent declines in mitochondrial function are present in many species[92], supporting a current view that aging may be directly related to cellular metabolism and decreased mitochondrial function[93],
Figure 1. Biological processes affecting stem cells.
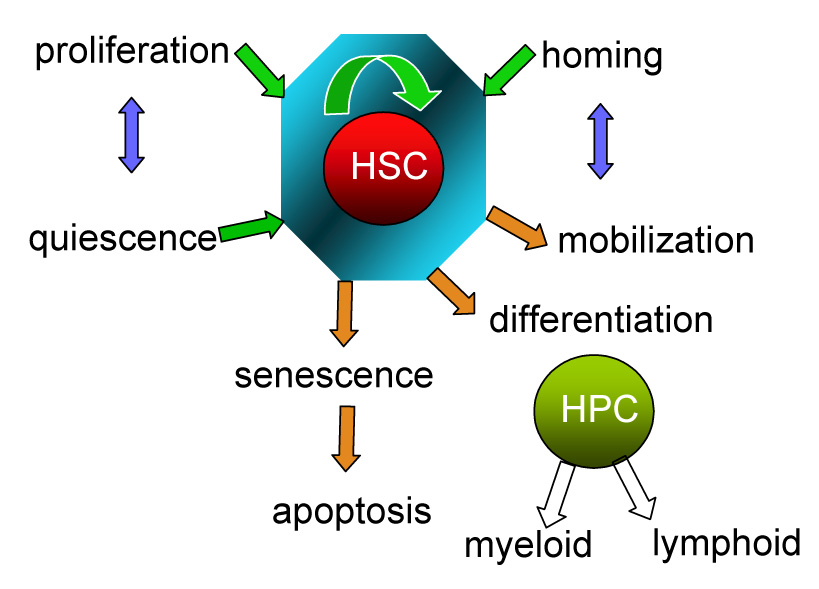
The biology of stem cells involves many different cellular processes that take place in order to maintain the appropriate number of HSCs and their functionality during variable environmental conditions throughout life. The stem cell pool is carefully maintained through a balance among proliferation vs quiescence, self-renewal vs differentiation, and the dynamic interplay between extravasation into the blood and lymph (mobilization) vs lodement in a bone marrow niche (homing). Any defect in any of the processes will apply stress on HSCs and lead them to senescence and apoptosis. The compromise of stem cell pool size and their functions will ultimately result in early “aging” involving loss of their normal potential and thus affect longevity. The green arrow indicates cellular processes favoring maintenance of HSC population, whereas brown arrows indicate those factors compromising HSCs. HSCs: hematopoietic stem cells. HPCs: hematopoietic progenitor cells.
In addition to mitochondria, other intracellular organelles such as lysosomes are damaged over time due to free radical-mediated modifications[94]. Lipofuscin, the age pigment first described in brain cells of the elderly, is composed of intra-lysosomal polymeric material that cannot be degraded by lysosomal hydrolases[95]. There is an inverse correlation between lipofuscin accumulation and lysosomal function, and consequently cellular life span[96]. In short-lived species, mitochondria release more electrons and peroxides than long lived species and therefore accumulate more lipofuscin granules[97]. The presence of lipofuscin in stem cells is still unknown. Only recently has the accumulation of lipofuscin been shown to interfere with cellular functions and promote age-related pathologies such as neurodegenerative diseases, heart failure, and macular degeneration[98].
Aging and protein mis-folding
Protein mis-folding is a common event in living cells, that possess a constant risk over the life of the individual[99–102]. In young and healthy cells, misfolded proteins are efficiently eliminated by an intracellular defense system[103]. The system involves production and maintenance of functional proteins, precisely regulated turnover, and the removal of damaged and aberrant proteins[104, 105]. In aging cells and in cells from individuals with specific genetic diseases, the misfolded protein load may overwhelm the system and disturb its protective function, leading to abnormal protein accumulation and subsequent self-assembly into toxic oligomers and aggregates[106, 107]. Many mechanisms may be involved in the increased risk of protein mis-folding and aggregation in senescent cells. Oxidative stress and ROS represent the major contributors[108–110]. Accumulation of ROS during biological aging ultimately leads to the widespread oxidation of biomolecules, including DNA, lipids and proteins. Oxidatively modified proteins are thermodynamically unstable and prone to structural alteration, thus promoting protein self-assembly and aggregation[111–113]. A second mechanism involves accumulation of mutated DNA with age due to increases in DNA damage and a decline in DNA repair processes[114, 115]. When non-synonymous mutations occur in coding sequences, inappropriate amino acids may be substituted, which may destabilize the native folded structure of proteins and favor the formation of mis-folded proteins[116]. When mutations occur in promoter sites of age and disease-associated genes, they may increase transcription and subsequently the concentration of aggregation-prone proteins and peptides[117–119]. The misfolded proteins and their aggregates may interfere with the normal cellular functions in a multitude of ways, all of which ultimately lead to senescence or cell death. As discussed previously, oxidative stress, DNA mutation, and perhaps other mechanisms may be involved in age-related declines in stem cell pool size and functions. Although very little is known about the role of misfolded and aggregated proteins in senescent and cancerous stem cells, it potentially represents a major determinant in defining the stem cell functions, the rate of aging, the development of age-related diseases and, yes, lifespan.
Latexin and cystatin C
The qualitative changes in stem cells and the composition of the stem cell population with respect to qualitatively distinct subclasses is an important factor in stem cell aging. We have shown that amongst mouse strains there is a strong correlation between the rate of early hematopoietic progenitor proliferation and mouse lifespan[120, 121]. Moreover, we and others have observed large strain-specific differences in the maintenance of the HSC population during aging[36, 120, 122–126], thus suggesting that genetic regulation plays an important role in the way aging affects HSCs. Using forward genetics, we recently identified a protein, latexin, whose differential expression in stem cells accounts for at least part of these differences in young murine hematopoiesis[127]. We have showed that latexin is a negative regulator of stem cell number and acts through at least two mechanisms to modulate stem cell pool size: a) it decreases HSC cell replication and b) it increases HSC apoptosis. Therefore, in the hematopoietic system, and perhaps other organs, latexin influences aging and perhaps lifespan through its action on stem cells.
Latexin was originally discovered in the lateral neocortex of rats and acts as a marker of regionality and development in both central and peripheral nervous system[128, 129]. It was also expressed in a number of other tissues, including hematopoietic and lymphoid organs[130]. It is 222 amino acids in length with a molecular weight of 29kD. Latexin is the only known carboxypeptidase inhibitor (CPI) in mammalians. It is a non-competitive, nearly irreversible, and potent inhibitor of carboxypeptidase A (CPA), but is less potent against carboxypeptidase B (CPB) and does not act on various other proteases[131– 133]. In rodents, latexin inhibits CPA1, 2 and mast cell CPA (CPA3), whereas it binds to CPA4 in humans[134]. Latexin may thus function in regulating tissue-specific protein degradation and turnover. As an endogenous carboxypeptidase inhibitor, the latexin primary sequence, however, doesn’t possess significant homology with other reported CPIs[135]. Instead, it shares high similarity in structure with a cystein protease inhibitor, cystatin C. Latexin consists of two topologically equivalent subdomains, each with a cystatin-like topology[132]. Latexin and cystatin C have structural and perhaps evolutionary ancestry, in common, but they also have their own specific characteristics as summarized in Table 1. First, functional domains and/or conformation may be different between cystatin C and latexin because they have different targets. Second, latexin is localized in the cytoplasm due to the lack of a membrane-specific signal peptide sequence, whereas most of cystatin C is secreted into biological fluids. Therefore, the inhibition of cystain C on cystein protease takes place extracellularly, whereas latexin mainly regulates cytosolic proteins. Interestingly, lateixin does not interact with its CPA target under normal conditions, because studies have shown that they are not co-localized in the same granular compartment in the cytoplasm, at least in rat peritoneal mast cells[136]. These results suggest that either latexin does not function through its inhibiton of CPA, or the inhibition occurs under certain conditions when CPA is released accidentally into the cytoplasmic space and latexin granules intercept the released CPA and inhibit it. Third, it has recently been shown that latexin and cystatin C can be induced to form amyloid-β like aggregates, although through different self-assembly mechanisms and under different induction conditions. A recent study has revealed that in vitro assembly of latexin is initiated by its conformational change under conditions where the polypeptide chain is mainly unfolded[137]. In contrast, polymerization of cystatin C takes place under conditions where the polypeptide sequence is altered because of genetic polymorphisms or mutations[138]. Cystain C variants or mutants are less stable and prone to form the folded dimer intermediates, which are the building blocks for cystatin C oligomerization. Finally, cystatin C has a broad range of biological roles because it is secreted into bodily fluids and tissues[139, 140], whereas latexin may function in specific cells depending on where it is expressed. Irrespective of venue, it is also possible they function in a similar manner due to their structural similarity.
Table 1.
Comparison of latexin and cystatin C.
| Latexin | Cystatin C | |
|---|---|---|
| Peptide length | 222 residues | 111 residues |
| Cystatin-like domain(s) | Two | One |
| Cellular location | Cytosolic | Secreted |
| Building blocks for aggregation | Unfolded polypeptide | Partially folded polypeptide |
| Aggregation condition | In vitro Mild denaturing condition (3–8M urea) | In vivo Genetic mutation-induced amino acid substitution |
| Inhibition target | Carboxypeptidase A | Cystein proteases |
| Known functions | 1) Regulation of hematopoietic stem cell pool size | 1) Regulation of embryonic and neural stem cells |
| 2) Inflammation | 2) Immune response | |
| 3) Sensory perception | 3) Tumor progression | |
| 4) Neuron marker | 4) Alzheimer’s disease | |
| 5) Bone formation | ||
| 6) Renal function marker | ||
| 7) Other functions | ||
Note: references are cited as in the main text.
Roles of latexin in stem cell aging
Despite several structure-function studies of latexin, there is still very little knowledge about its biological roles in stem cells and aging. We herein propose some potential regulatory mechanisms of latexin in stem cells, aging and age-related diseases. We have shown compelling evidence of latexin’s involvement in the regulation of HSC in young mice. Our unpublished preliminary results revealed that latexin expression in fetal hematopoiesis and early adulthood are strongly correlated with HSC numbers throughout adulthood and old age. Although latexin was originally detected in differentiated neurons, some evidence indicates it is also present in neural stem cells (NSCs)[141, 142]. According to the concept of adult stem cell plasticity and the noteworthy genetic overlap between NSCs and HSCs, our results showing regulatory roles in HSCs may also be applicable to NSCs. This hypothesis could be supported by a recent report showing cystatin C, a latexin homologue, regulated neurosphere generation either by direct induction of embryonic stem cells (ESCs) into functional NSCs, or by expansion of cells that had spontaneously differentiated into NSCs[143]. These results underscore that in the hematopoietic system, and perhaps in other organs, latexin influences aging and perhaps lifespan through its action on stem cells.
Latexin and neurological amyloidoses
The deposition and aggregation of misfolded proteins is strongly implicated in the pathogenesis of aging-associated neurologic disorders, such as Alzheimer’s disease (AD) and Parkinson’s syndrome, to name just two[144]. Latexin was found to be significantly down-regulated in an AD mouse model, and the recent findings that cystain C over-expression ameliorates this disease suggests that latexin may play a similar role[145]. Amyloid-β (Aβ) is the major constituent of the amyloid fibrils deposited in the brains of patients with AD. It is a processing product of a larger β amyloid precursor protein (βAPP)[146]. Immunohistochemical studies have revealed the colocalization of cystatin C with Aβ[147], suggesting that as a component, cystatin C either enhances amyloid fibril leading to neuronal degeneration, or inhibits fibril formation. Using an Alzheimer’s disease mouse model, two studies from Levy and Jucker laboratories have shown that overexpression of cystatin C could diminish amyloid-β deposition and thus plays a protective role in AD[148, 149]. The underlying mechanisms involve the direct binding of Cystatin C and amyloid-β rather than the effects on APP and amyloid-β expression level. The binding affinity between amyloid-β and cystatin C is high enough to prevent amyloid-β accumulation and reduce cystatin C self-association. Because of the structural similarity between latexin and cystatin C, lateixn may act as another potential protective factor that prevents the development of aging-related neurological diseases.
Latexin and cancer
Increased propensity to cancer is one of prominent hallmarks of aging. Latexin shares 30% sequence similarity with Tazarotene-Induced Gene 1 (TIG1), which is down-regulated or absent in an extensive list of tumor types[150]. Studies by Callahan et al. revealed that the absence of a candidate gene, although yet unidentified, whose protein product had 85% identity of the mouse latexin protein, was associated with increased incidence of ovarian cancer[151]. We have found similar expression patterns of latexin in a variety of human leukemia and lymphoma cell lines, and in primary cells from patients with these diseases (manuscript in preparation), indicating that latexin may be implicated in the regulation of tumor progression, perhaps as a suppressor. Cystatin C has demonstrated both tumor-suppressing and tumor-promoting functions. It attenuated tumor cell-mediated invasion and degradation of extracellular matrix[152].
Overexpression of cystatin C in the mouse model of glioblastoma reduced intracerebral tumor formation[153], thus demonstrating anti-tumor effects. In contrast, transplantation of a highly metastatic melanoma cell line into cystatin C-null mice significantly reduced lung translocation of malignant cells, indicating that cystatin C may enable the matastasis of malignant cells[154]. The dual role cystatin C in tumor progression could be explained by a combination of its protease inhibitor activity and cytokine-like activity[155]. As a member of the cystatin superfamily, latexin may act similarly in the malignant transformation of stem and progenitor cells and regulate tumor progression.
Many mechanisms are involved in the aging and age-related decline in tissue functions. Stem cells and their changes with age are of great interest because they contribute to life-long replenishment of functional mature cells. Genes implicated in the stem cell regulation would provide a good platform for studying the relationship among stem cells, aging and age-related diseases. Using latexin as an example, we put forth a hypothesis addressing the questions of how genetic changes affect stem cells, the nature of underlying mechanisms and how the aging phenotypes are interconnected with each other (Figure 2). Latexin initiates cellular responses through three potentially related mechanisms: 1) inhibition of carboxypeptidase A, 2) participation of intracellular signaling pathways, and 3) regulation of protein aggregation. These processes, perhaps along with other mechanisms, synergistically or independently regulate cell proliferation and apoptosis, that consequently affect stem cell numbers and function. When latexin expression increases in stem cells with age, as we have observed in human hematopoietic stem cells, the stem cell pool size will become smaller and tissue regeneration capacity in the face of stress may be impaired, a process fostering stem cell aging. On the other hand, high levels of latexin may inhibit protein aggregate-induced cellular toxicity in old stem cells and prevent their functional decline with age. As a putative tumor suppressor, elevated latexin level could reduce the increased propensity of senescent stem cells to transform into cancer stem cells and in result in tumor formation. Thus, latexin may exert an anti-aging effect at the intersection of stem cells, aging, and cancer in ways that come full circle with events in embryogenesis and tissue regeneration.
Figure 2. Potential roles of latexin in stem cell senescence.
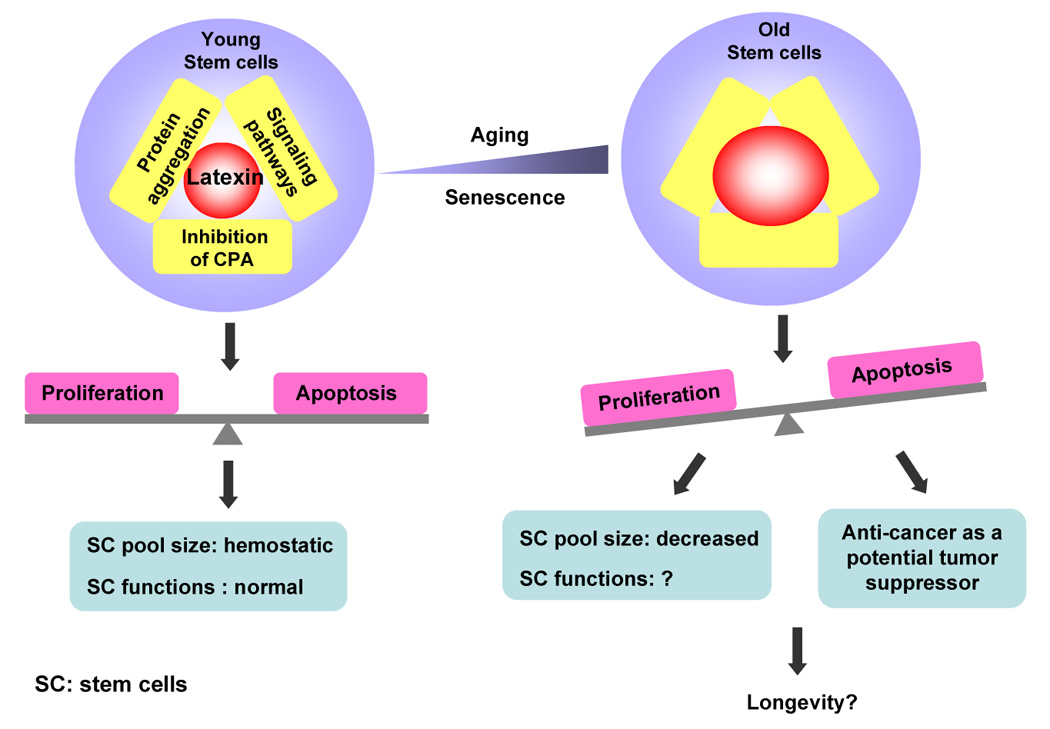
Three potential mechanisms may be involved in the regulation of stem cells by latexin: inhibition of carboxypeptidase A (CPA), involvement of intracellular signaling pathways and protein folding. In young stem cells, these mechanisms are well regulated and the biological processes affecting stem cells are balanced. In sum, these result in the homeostasis and normal functionality of stem cell population. As latexin expression increases with age, latexin-associated biological processes are affected, and may contribute to stem cell senescence, and quantitative loss. Age-associated diseases, such as amyloidoses and cancers may also be related to latexin levels in old stem cells. Therefore, latexin represents a good model to address the relationship among stem cell, the rate of aging, the development of age-related diseases, and lifespan.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Maximow A. Der Lymphozyt als gemeinsame Stammzelle der verschiedenen Blutelemente in der embryonalen Entwicklung und im postfetalen Leben der Saugetiere. Folia Haematol. 1909;8:125–134. [Google Scholar]
- 2.Massberg S, Schaerli P, Knezevic-Maramica I, Kollnberger M, Tubo N, Moseman EA, Huff IV, Junt T, Wagers AJ, Mazo IB, von Andrian UH. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell. 2007;131:994–1008. doi: 10.1016/j.cell.2007.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 4.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 5.Jones DL, Wagers AJ. No place like home: anatomy and function of the stem cell niche. Nat Rev Mol Cell Biol. 2008;9:11–21. doi: 10.1038/nrm2319. [DOI] [PubMed] [Google Scholar]
- 6.Gosain A, DiPietro LA. Aging and wound healing. World J Surg. 2004;28:321–326. doi: 10.1007/s00268-003-7397-6. [DOI] [PubMed] [Google Scholar]
- 7.Malik SM. The impact of aging on chemotherapy. Clin Lung Cancer. 2004;5:243–244. doi: 10.1016/S1525-7304(11)70345-X. [DOI] [PubMed] [Google Scholar]
- 8.Henckaerts E, Langer JC, Orenstein J, Snoeck HW. The positive regulatory effect of TGF-beta2 on primitive murine hemopoietic stem and progenitor cells is dependent on age, genetic background, and serum factors. J Immunol. 2004;173:2486–2493. doi: 10.4049/jimmunol.173.4.2486. [DOI] [PubMed] [Google Scholar]
- 9.Smith LG, Weissman IL, Heimfeld S. Clonal analysis of hematopoietic stem-cell differentiation in vivo. Proc.Natl.Acad.Sci.USA. 1991;88:2788–2792. doi: 10.1073/pnas.88.7.2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Osawa M, Hanada K-i, Hamada H, Nakauchi H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science. 1996;273:242–245. doi: 10.1126/science.273.5272.242. [DOI] [PubMed] [Google Scholar]
- 11.Spangrude GJ, Brooks DM, Tumas DB. Long-term repopulation of irradiated mice with limiting numbers of purified hematopoietic stem cells: in vivo expansion of stem cell phenotype but not function. Blood. 1995;85:1006–1016. [PubMed] [Google Scholar]
- 12.Sudo K, Ema H, Morita Y, Nakauchi H. Age-associated characteristics of murine hematopoietic stem cells. J. Exp. Med. 2000;192:1273–1280. doi: 10.1084/jem.192.9.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Offner F, Kerre T, DeSmedt M, Plum J. Bone marrow CD34+ cells generate fewer T cells in vitro with increasing age and following chemotherapy. British Journal of Haematology. 1999;104:801–808. doi: 10.1046/j.1365-2141.1999.01265.x. [DOI] [PubMed] [Google Scholar]
- 14.Liang Y, Van Zant G. Genetic control of stem-cell properties and stem cells in aging. Curr Opin Hematol. 2003;10:195–202. doi: 10.1097/00062752-200305000-00001. [DOI] [PubMed] [Google Scholar]
- 15.Matsuzaki Y, Kinjo K, Mulligan RC, Okano H. Unexpectedly efficient homing capacity of purified murine hematopoietic stem cells. Immunity. 2004;20:87–93. doi: 10.1016/s1074-7613(03)00354-6. [DOI] [PubMed] [Google Scholar]
- 16.Benveniste P, Cantin C, Hyam D, Iscove NN. Hematopoietic stem cells engraft in mice with absolute efficiency. Nat Immunol. 2003;4:708–713. doi: 10.1038/ni940. [DOI] [PubMed] [Google Scholar]
- 17.Morrison SJ, Wandycz AM, Akashi K, Globerson A, Weissman IL. The aging of hematopoietic stem cells. Nat Med. 1996;2:1011–1016. doi: 10.1038/nm0996-1011. [DOI] [PubMed] [Google Scholar]
- 18.Liang Y, Van Zant G, Szilvassy SJ. Effects of aging on the homing and engraftment of murine hematopoietic stem and progenitor cells. Blood. 2005;106:1479–1487. doi: 10.1182/blood-2004-11-4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ross E, Anderson N, Micklem HS. Serial depletion and regeneration of the murine hematopoietic system. Implications for hematopoietic organization and the study of cellular aging. J.Exp.Med. 1982;155:432–444. doi: 10.1084/jem.155.2.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ogden DA, Mickliem HS. The fate of serially transplanted bone marrow cell populations from young and old donors. Transplantation. 1976;22:287–293. doi: 10.1097/00007890-197609000-00010. [DOI] [PubMed] [Google Scholar]
- 21.Micklem HS, Ford CE, Evans EP, Ogden DA, Papworth DS. Competitive in vivo proliferation of foetal and adult hematopoietic cells in lethally irradiated mice. J. Cell Physiol. 1972;79:293–298. doi: 10.1002/jcp.1040790214. [DOI] [PubMed] [Google Scholar]
- 22.Rebel VI, Miller CL, Eaves CJ, Lansdorp PM. The repopulation potential of fetal liver hematopoietic stem cells in mice exceeds that of their adult bone marrow counterparts. Blood. 1996;87:3500–3507. [PubMed] [Google Scholar]
- 23.Siminovitch L, Till JE, McCulloch EA. Decline in colony-forming ability of marrow cells subjected to serial transplantation into irradiated mice. J.Cell.Comp.Physiol. 1964;64:23–31. doi: 10.1002/jcp.1030640104. [DOI] [PubMed] [Google Scholar]
- 24.Ogden DA, Micklem HS. The fate of serially transplanted bone marrow cell populations from young and old donors. Transplantation. 1976;22:287–293. doi: 10.1097/00007890-197609000-00010. [DOI] [PubMed] [Google Scholar]
- 25.Iscove N, Nawa K. Hematopoietic stem cells expand during serial transplantation in vivo without apparent exhaustion. Curr.Biol. 1997;7:805–808. doi: 10.1016/s0960-9822(06)00341-1. [DOI] [PubMed] [Google Scholar]
- 26.Harrison DE, Astle CM, Delaittre JA. Loss of proliferative capacity in immunohemopoietic stem cells is caused by serial transplantation rather than aging. J.Exp.Med. 1978;147:1526–1531. doi: 10.1084/jem.147.5.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harstock RJ, S E, Ketter CS. Normal variation with aging of the amount of hematopoietic tissue in bone marrow from anterior iliac crest. Am J Clin Pathol. 1965;43:325–333. doi: 10.1093/ajcp/43.4.326. [DOI] [PubMed] [Google Scholar]
- 28.Bagnara GP, Bonsi L, Strippoli P, Bonifazi F, Tonelli R, D'Addato S, Paganelli R, Scala E, Fagiolo U, Monti D, Cossarizza A, Bonafe M, Franceschi C. Hemopoiesis in healthy old people and centenarians: well-maintained responsiveness of CD34+ cells to hemopoietic growth factors and remodeling of cytokine network. J Gerontol A Biol Sci Med Sci. 2000;55:B61–B66. doi: 10.1093/gerona/55.2.b61. discussion B67–70. [DOI] [PubMed] [Google Scholar]
- 29.Waterstrat A, Oakley EJ, Miller A, Swiderski C, Liang Y, Van Zant G. Mechanisms of stem cell aging. In: Rudolph KL, editor. Telomeres and Telomerase in Ageing, Disease, and Cancer. Springer-Verlag; Berlin: 2008. [Google Scholar]
- 30.Rando TA. Stem cells, ageing and the quest for immortality. Nature. 2005;441:1080–1086. doi: 10.1038/nature04958. [DOI] [PubMed] [Google Scholar]
- 31.Van Zant G, Holland BP, Eldridge PW, Chen J-J. Genotype-restricted growth and aging patterns in hematopoietic stem cell populations of allophenic mice. J.Exp.Med. 1990;171:1547–1565. doi: 10.1084/jem.171.5.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schlessinger D, Van Zant G. Does functional depletion of stem cells drive aging? Mech Ageing Dev. 2001;122:1537–1553. doi: 10.1016/s0047-6374(01)00299-8. [DOI] [PubMed] [Google Scholar]
- 33.Sharpless NE, DePinho RA. How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol. 2007;8:703–713. doi: 10.1038/nrm2241. [DOI] [PubMed] [Google Scholar]
- 34.Van Zant G, Scott-Micus K, Thompson BP, Fleischman RA, Perkins S. Stem cell quiescence/activation is reversible by serial transplantation and is independent of stromal cell genotype in mouse aggregation chimeras. Exp. Hematol. 1992;20:470–475. [PubMed] [Google Scholar]
- 35.Morrison SJ, Wandycz AM, Akashi K, Globerson A, Weissman IL. The aging of hematopoietic stem cells. Nature Med. 1996;2:1011–1016. doi: 10.1038/nm0996-1011. [DOI] [PubMed] [Google Scholar]
- 36.Kim M, Moon HB, Spangrude GJ. Major age-related changes of mouse hematopoietic stem/progenitor cells. Ann N Y Acad Sci. 2003;996:195–208. doi: 10.1111/j.1749-6632.2003.tb03247.x. [DOI] [PubMed] [Google Scholar]
- 37.Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, Hee Park S, Thompson T, Karsenty G, Bradley A, Donehower LA. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- 38.Donehower LA. Does p53 affect organismal aging? J Cell Physiol. 2002;192:23–33. doi: 10.1002/jcp.10104. [DOI] [PubMed] [Google Scholar]
- 39.Dumble M, Moore L, Chambers SM, Geiger H, Van Zant G, Goodell MA, Donehower LA. The impact of altered p53 dosage on hematopoietic stem cell dynamics during aging. Blood. 2006 doi: 10.1182/blood-2006-03-010413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Domen J, Cheshier SH, Weissman IL. The role of apoptosis in the regulation of hematopoietic stem cells: Overexpression of BCL-2 increases both their number and repopulation potential. J. Exp. Med. 2000;191:253–263. doi: 10.1084/jem.191.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weissman IL. Stem cells: Units of development, units of regeneration, and units in evolution. Cell. 2000;100:157–168. doi: 10.1016/s0092-8674(00)81692-x. [DOI] [PubMed] [Google Scholar]
- 42.Morrison SJ, Uchida N, Weissman IL. The biology of hematopoietic stem cells. Annu.Rev.Cell Dev.Biol. 1995;11:35–71. doi: 10.1146/annurev.cb.11.110195.000343. [DOI] [PubMed] [Google Scholar]
- 43.Bradford GB, Williams B, Rossi R, Bertoncello I. Quiescence, cycling, and turnover in the primitive hematopoietic stem cell compartment. Exp.Hematol. 1997;25:445–453. [PubMed] [Google Scholar]
- 44.Cheshier SH, Morrison SJ, Liao X, Weissman IL. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc. Natl. Acad. Sci. USA. 1999;96:3120–3125. doi: 10.1073/pnas.96.6.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kay HEM. How many cell-generations? Lancet. 1965;ii:418. doi: 10.1016/s0140-6736(65)90763-4. [DOI] [PubMed] [Google Scholar]
- 46.Finkel T, Serrano M, Blasco MA. The common biology of cancer and ageing. Nature. 2007;448:767–774. doi: 10.1038/nature05985. [DOI] [PubMed] [Google Scholar]
- 47.Effros RB, Globerson A. Hematopoietic cells and replicative senescence. Exp Gerontol. 2002;37:191–196. doi: 10.1016/s0531-5565(01)00183-8. [DOI] [PubMed] [Google Scholar]
- 48.Liu Y, Kha H, Ungrin M, Robinson MO, Harrington L. Preferential maintenance of critically short telomeres in mammalian cells heterozygous for mTert. Proc Natl Acad Sci U S A. 2002;99:3597–3602. doi: 10.1073/pnas.062549199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brummendorf TH, Mak J, Sabo KM, Baerlocher GM, Dietz K, Abkowitz JL, Lansdorp PM. Longitudinal studies of telomere length in feline blood cells: implications for hematopoietic stem cell turnover in vivo. Exp Hematol. 2002;30:1147–1152. doi: 10.1016/s0301-472x(02)00888-3. [DOI] [PubMed] [Google Scholar]
- 50.Campisi J, Kim S, Lim CS, Rubio M. Cellular senescence, cancer and aging: the telomere connection. Exp Gerontol. 2001;36:1619–1637. doi: 10.1016/s0531-5565(01)00160-7. [DOI] [PubMed] [Google Scholar]
- 51.Shay JW, Wright WE. When do telomeres matter? Science. 2001;291:839–840. doi: 10.1126/science.1058546. [DOI] [PubMed] [Google Scholar]
- 52.Satyanarayana A, Wiemann SU, Buer J, Lauber J, Dittmar KE, Wustefeld T, Blasco MA, Manns MP, Rudolph KL. Telomere shortening impairs organ regeneration by inhibiting cell cycle re-entry of a subpopulation of cells. Embo J. 2003;22:4003–4013. doi: 10.1093/emboj/cdg367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Campisi J. Cancer and ageing: rival demons? Nat Rev Cancer. 2003;3:339–349. doi: 10.1038/nrc1073. [DOI] [PubMed] [Google Scholar]
- 54.Vaziri H, Dragowska W, Allsopp RC, Thomas TE, Harley CB, Lansdorp PM. Evidence for a mitotic clock in human hematopoietic stem cells: loss of telomeric DNA with age. Proc.Natl.Acad.Sci.USA. 1994;91:9857–9860. doi: 10.1073/pnas.91.21.9857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morrison SJ, Prowse KR, Ho P, Weissman IL. Telomerase activity in hematopoietic cells is associated with self-renewal potential. Immunity. 1996;5:207–216. doi: 10.1016/s1074-7613(00)80316-7. [DOI] [PubMed] [Google Scholar]
- 56.Yui J, Chiu CP, Lansdorp PM. Telomerase activity in candidate stem cells from fetal liver and adult bone marrow. Blood. 1998;91:3255–3262. [PubMed] [Google Scholar]
- 57.Chiu CP, Dragowska W, Kim NW, Vaziri H, Yui J, Thomas TE, Harley CB, Lansdorp PM. Differential expression of telomerase activity in hematopoietic progenitors from adult human bone marrow. Stem Cells. 1996;14:239–248. doi: 10.1002/stem.140239. [DOI] [PubMed] [Google Scholar]
- 58.Lansdorp PM. Developmental changes in the function of hematopoietic stem cells. Exp.Hematol. 1995;23:187–191. [PubMed] [Google Scholar]
- 59.Rufer N, Brummendorf TH, Chapuis B, Heig C, Lansdorp PM, Roosnek E. Accelerated telomere shortening in hematological lineages is limited to the first year following stem cell transplantation. Blood. 2001;97:575–577. doi: 10.1182/blood.v97.2.575. [DOI] [PubMed] [Google Scholar]
- 60.Rufer N, Dragowska W, Thornbury G, Roosnek E, Lansdorp PM. Telomere length dynamics in human lymphocyte subpopulations measured by flow cytometry. Nat.Biotechnol. 1998;16:743–747. doi: 10.1038/nbt0898-743. [DOI] [PubMed] [Google Scholar]
- 61.Ball SE, Gibson FM, Rizzo S, Tooze JA, Marsh JCW, Gordonsmith EC. Progressive telomere shortening in aplastic anemia. Blood. 1998;91:3582–3592. [PubMed] [Google Scholar]
- 62.Notaro R, Cimmino A, Tabarini D, Rotoli B, Luzzatto L. In vivo telomere dynamics of human hematopoietic stem cells. Proc.Natl.Acad.Sci.USA. 1997;94:13782–13785. doi: 10.1073/pnas.94.25.13782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Akiyama M, Asai O, Kuraishi Y, Urashima M, Hoshi Y, Sakamaki H, Yabe H, Furukawa T, Yamada O, Mizoguchi H, Yamada H. Shortening of telomeres in recipients of both autologous and allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplantation. 2000;25:441–447. doi: 10.1038/sj.bmt.1702144. [DOI] [PubMed] [Google Scholar]
- 64.Wynn R, Cross M, Hatton C, Will A, Lashford L, Dexter T, Testa N. Accelerated telomere shortening in young recipients of allogeneic bone-marrow transplants. Lancet. 1998;351:178–181. doi: 10.1016/S0140-6736(97)08256-1. [DOI] [PubMed] [Google Scholar]
- 65.Wynn R, Thornley I, Freedman M, Saunders EF. Telomere shortening in leucocyte subsets of long-term survivors of allogeneic bone marrow transplantation. British Journal of Haematology. 1999;105:997–1001. doi: 10.1046/j.1365-2141.1999.01450.x. [DOI] [PubMed] [Google Scholar]
- 66.Allsopp RC, Cheshier S, Weissman IL. Telomere shortening accompanies increased cell cycle activity during serial transplantation of hematopoietic stem cells. J. Exp. Med. 2001;193:917–924. doi: 10.1084/jem.193.8.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Abkowitz JL, Catlin SN, McCallie MT, Guttorp P. Evidence that the number of hematopoietic stem cells per animal is conserved in mammals. Blood. 2002;100:2665–2667. doi: 10.1182/blood-2002-03-0822. [DOI] [PubMed] [Google Scholar]
- 68.Shepherd BE, Guttorp P, Lansdorp PM, Abkowitz JL. Estimating human hematopoietic stem cell kinetics using granulocyte telomere lengths. Exp Hematol. 2004;32:1040–1050. doi: 10.1016/j.exphem.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 69.Lopes, JV, Pacheco JM, Dingli D. Acquired hematopoietic stem-cell disorders and mammalian size. Blood. 2007;110:4120–4122. doi: 10.1182/blood-2007-05-089805. [DOI] [PubMed] [Google Scholar]
- 70.Kirkwood TB, Austad SN. Why do we age? Nature. 2000;408:233–238. doi: 10.1038/35041682. [DOI] [PubMed] [Google Scholar]
- 71.Hayflick L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 72.Campisi J. Replicative senescence: An old lives' tale? Cell. 1996;84:497–500. doi: 10.1016/s0092-8674(00)81023-5. [DOI] [PubMed] [Google Scholar]
- 73.Blackburn EH. Telomeres and telomerase. Keio J Med. 2000;49:59–65. doi: 10.2302/kjm.49.59. [DOI] [PubMed] [Google Scholar]
- 74.Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 75.Joeng KS, Song EJ, Lee KJ, Lee J. Long lifespan in worms with long telomeric DNA. Nat Genet. 2004;36:607–611. doi: 10.1038/ng1356. [DOI] [PubMed] [Google Scholar]
- 76.Nijnik A, Woodbine L, Marchetti C, Dawson S, Lambe T, Liu C, Rodrigues NP, Crockford TL, Cabuy E, Vindigni A, Enver T, Bell JI, Slijepcevic P, Goodnow CC, Jeggo PA, Cornall RJ. DNA repair is limiting for haematopoietic stem cells during ageing. Nature. 2007;447:686–690. doi: 10.1038/nature05875. [DOI] [PubMed] [Google Scholar]
- 77.Hart RW, Setlow RB. Correlation between deoxyribonucleic acid excision-repair and life-span in a number of mammalian species. Proc Natl Acad Sci U S A. 1974;71:2169–2173. doi: 10.1073/pnas.71.6.2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Murray V, Holliday R. Increased error frequency of DNA polymerases from senescent human fibroblasts. J Mol Biol. 1981;146:55–76. doi: 10.1016/0022-2836(81)90366-1. [DOI] [PubMed] [Google Scholar]
- 79.Campisi J. Cellular senescence and apoptosis: how cellular responses might influence aging phenotypes. Exp Gerontol. 2003;38:5–11. doi: 10.1016/s0531-5565(02)00152-3. [DOI] [PubMed] [Google Scholar]
- 80.Beausejour CM, Campisi J. Ageing: balancing regeneration and cancer. Nature. 2006;443:404–405. doi: 10.1038/nature05221. [DOI] [PubMed] [Google Scholar]
- 81.Martin GM. Frontiers of aging. Science. 2001;2944:13. doi: 10.1126/science.294.5540.13. [DOI] [PubMed] [Google Scholar]
- 82.Sharpless NE, DePinho RA. Telomeres, stem cells, senescence, and cancer. J Clin Invest. 2004;113:160–168. doi: 10.1172/JCI20761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Park IK, Morrison SJ, Clarke MF. Bmi1, stem cells, and senescence regulation. J Clin Invest. 2004;113:175–179. doi: 10.1172/JCI20800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pelicci PG. Do tumor-suppressive mechanisms contribute to organism aging by inducing stem cell senescence? J Clin Invest. 2004;113:4–7. doi: 10.1172/JCI200420750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- 86.Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- 87.Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443:448–452. doi: 10.1038/nature05091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–275. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 89.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 90.Sun J, Tower J. FLP recombinase-mediated induction of Cu/Zn-superoxide dismutase transgene expression can extend the life span of adult Drosophila melanogaster flies. Mol Cell Biol. 1999;19:216–228. doi: 10.1128/mcb.19.1.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wallace DC, Melov S. Radicals r'aging. Nat Genet. 1998;19:105–106. doi: 10.1038/448. [DOI] [PubMed] [Google Scholar]
- 92.Boffoli D, Scacco SC, Vergari R, Solarino G, Santacroce G, Papa S. Decline with age of the respiratory chain activity in human skeletal muscle. Biochim Biophys Acta. 1994;1226:73–82. doi: 10.1016/0925-4439(94)90061-2. [DOI] [PubMed] [Google Scholar]
- 93.Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 94.Cuervo AM, Dice JF. How do intracellular proteolytic systems change with age? Front Biosci. 1998;3:D25–D43. doi: 10.2741/a264. [DOI] [PubMed] [Google Scholar]
- 95.Terman A, Brunk UT. Lipofuscin. Int J Biochem Cell Biol. 2004;36:1400–1404. doi: 10.1016/j.biocel.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 96.Cuervo AM, Dice JF. When lysosomes get old. Exp Gerontol. 2000;35:119–131. doi: 10.1016/s0531-5565(00)00075-9. [DOI] [PubMed] [Google Scholar]
- 97.Barja G, Herrero A. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. Faseb J. 2000;14:312–318. doi: 10.1096/fasebj.14.2.312. [DOI] [PubMed] [Google Scholar]
- 98.Brunk UT, Terman A. The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur J Biochem. 2002;269:1996–2002. doi: 10.1046/j.1432-1033.2002.02869.x. [DOI] [PubMed] [Google Scholar]
- 99.Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 100.Chan HS, Dill KA. Protein folding in the landscape perspective: chevron plots and non-Arrhenius kinetics. Proteins. 1998;30:2–33. doi: 10.1002/(sici)1097-0134(19980101)30:1<2::aid-prot2>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 101.Dill KA, Chan HS. From Levinthal to pathways to funnels. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 102.Dill KA, Phillips AT, Rosen JB. Protein structure and energy landscape dependence on sequence using a continuous energy function. J Comput Biol. 1997;4:227–239. doi: 10.1089/cmb.1997.4.227. [DOI] [PubMed] [Google Scholar]
- 103.Ellis RJ. The molecular chaperone concept. Semin Cell Biol. 1990;1:1–9. [PubMed] [Google Scholar]
- 104.Frydman J. Folding of newly translated proteins in vivo: the role of molecular chaperones. Annu Rev Biochem. 2001;70:603–647. doi: 10.1146/annurev.biochem.70.1.603. [DOI] [PubMed] [Google Scholar]
- 105.McClellan AJ, Frydman J. Molecular chaperones and the art of recognizing a lost cause. Nat Cell Biol. 2001;3:E51–E53. doi: 10.1038/35055162. [DOI] [PubMed] [Google Scholar]
- 106.Grune T, Jung T, Merker K, Davies KJ. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and 'aggresomes' during oxidative stress, aging, and disease. Int J Biochem Cell Biol. 2004;36:2519–2530. doi: 10.1016/j.biocel.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 107.Poppek D, Grune T. Protein oxidation and proteolysis during cellular senescence. Z Gerontol Geriatr. 2004;37:175–183. doi: 10.1007/s00391-004-0228-z. [DOI] [PubMed] [Google Scholar]
- 108.Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem. 1997;272:20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- 109.Stadtman ER, Berlett BS. Reactive oxygen-mediated protein oxidation in aging and disease. Chem Res Toxicol. 1997;10:485–494. doi: 10.1021/tx960133r. [DOI] [PubMed] [Google Scholar]
- 110.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 111.Gafni A. Altered protein metabolism in aging. Annu Rev Gerontol Geriatr. 1990;10:117–131. doi: 10.1007/978-3-662-38445-9_7. [DOI] [PubMed] [Google Scholar]
- 112.Wisser KC, Schauerte JA, Burke DT, Galecki A, Chen S, Miller RA, Gafni A. Mapping tissue-specific genes correlated with age-dependent changes in protein stability and function. Arch Biochem Biophys. 2004;432:58–70. doi: 10.1016/j.abb.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 113.Gafni A. Structural modifications of proteins during aging. J Am Geriatr Soc. 1997;45:871–880. doi: 10.1111/j.1532-5415.1997.tb01518.x. [DOI] [PubMed] [Google Scholar]
- 114.Butler JS, Loh SN. Kinetic partitioning during folding of the p53 DNA binding domain. J Mol Biol. 2005;350:906–918. doi: 10.1016/j.jmb.2005.05.060. [DOI] [PubMed] [Google Scholar]
- 115.Welch WJ. Role of quality control pathways in human diseases involving protein misfolding. Semin Cell Dev Biol. 2004;15:31–38. doi: 10.1016/j.semcdb.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 116.Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol. 2002;320:369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 117.Mizuta I, Satake W, Nakabayashi Y, Ito C, Suzuki S, Momose Y, Nagai Y, Oka A, Inoko H, Fukae J, Saito Y, Sawabe M, Murayama S, Yamamoto M, Hattori N, Murata M, Toda T. Multiple candidate gene analysis identifies alpha-synuclein as a susceptibility gene for sporadic Parkinson's disease. Hum Mol Genet. 2006;15:1151–1158. doi: 10.1093/hmg/ddl030. [DOI] [PubMed] [Google Scholar]
- 118.Holzmann C, Kruger R, Saecker AM, Schmitt I, Schols L, Berger K, Riess O. Polymorphisms of the alpha-synuclein promoter: expression analyses and association studies in Parkinson's disease. J Neural Transm. 2003;110:67–76. doi: 10.1007/s00702-002-0769-5. [DOI] [PubMed] [Google Scholar]
- 119.Tanzi RE, Kovacs DM, Kim TW, Moir RD, Guenette SY, Wasco W. The gene defects responsible for familial Alzheimer's disease. Neurobiol Dis. 1996;3:159–168. doi: 10.1006/nbdi.1996.0016. [DOI] [PubMed] [Google Scholar]
- 120.de Haan G, Nijhof W, Van Zant G. Mouse strain-dependent changes in frequency and proliferation of hematopoietic stem cells during aging: correlation between lifespan and cycling activity. Blood. 1997;89:1543–1550. [PubMed] [Google Scholar]
- 121.de Haan G, Van Zant G. Genetic analysis of hemopoietic cell cycling in mice suggests its involvement in organismal life span. FASEB J. 1999;13:707–713. doi: 10.1096/fasebj.13.6.707. [DOI] [PubMed] [Google Scholar]
- 122.Geiger H, Rennebeck G, Van Zant G. Regulation of hematopoietic stem cell aging in vivo by a distinct genetic element. Proc Natl Acad Sci U S A. 2005;102:5102–5107. doi: 10.1073/pnas.0408654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Geiger H, True JM, de Haan G, Van Zant G. Age- and stage-specific regulation patterns in the hematopoietic stem cell hierarchy. Blood. 2001;98:2966–2972. doi: 10.1182/blood.v98.10.2966. [DOI] [PubMed] [Google Scholar]
- 124.KnaanShanzer S, Verlinden SFF, vanBeusechem VW, VanBekkum DW, Valerio D. Intrinsic potential of phenotypically defined human hemopoietic stem cells to self-renew in short-term in vitro cultures. Experimental Hematology. 1999 SEP;27:1440–1450. doi: 10.1016/s0301-472x(99)00074-0. [DOI] [PubMed] [Google Scholar]
- 125.Morrison SJ, Qian D, Jerabek L, Thiel BA, Park IK, Ford PS, Kiel MJ, Schork NJ, Weissman IL, Clarke MF. A genetic determinant that specifically regulates the frequency of hematopoietic stem cells. J Immunol. 2002;168:635–642. doi: 10.4049/jimmunol.168.2.635. [DOI] [PubMed] [Google Scholar]
- 126.Yuan R, Astle CM, Chen J, Harrison DE. Genetic regulation of hematopoietic stem cell exhaustion during development and growth. Exp Hematol. 2005;33:243–250. doi: 10.1016/j.exphem.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 127.Liang Y, Jansen M, Aronow B, Geiger H, Van Zant G. The quantitative trait gene latexin influences the size of the hematopoietic stem cell population in mice. Nat Genet. 2007;39:178–188. doi: 10.1038/ng1938. [DOI] [PubMed] [Google Scholar]
- 128.Arimatsu Y. Latexin: a molecular marker for regional specification in the neocortex. Neurosci Res. 1994;20:131–135. doi: 10.1016/0168-0102(94)90030-2. [DOI] [PubMed] [Google Scholar]
- 129.Hatanaka Y, Uratani Y, Takiguchi-Hayashi K, Omori A, Sato K, Miyamoto M, Arimatsu Y. Intracortical regionality represented by specific transcription for a novel protein, latexin. Eur J Neurosci. 1994;6:973–982. doi: 10.1111/j.1460-9568.1994.tb00592.x. [DOI] [PubMed] [Google Scholar]
- 130.Liu Q, Yu L, Gao J, Fu Q, Zhang J, Zhang P, Chen J, Zhao S. Cloning, tissue expression pattern and genomic organization of latexin, a human homologue of rat carboxypeptidase A inhibitor. Mol Biol Rep. 2000;27:241–246. doi: 10.1023/a:1010971219806. [DOI] [PubMed] [Google Scholar]
- 131.Normant E, Martres MP, Schwartz JC, Gros C. Purification, cDNA cloning, functional expression, and characterization of a 26-kDa endogenous mammalian carboxypeptidase inhibitor. Proc Natl Acad Sci U S A. 1995;92:12225–12229. doi: 10.1073/pnas.92.26.12225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Aagaard A, Listwan P, Cowieson N, Huber T, Ravasi T, Wells CA, Flanagan JU, Kellie S, Hume DA, Kobe B, Martin JL. An inflammatory role for the mammalian carboxypeptidase inhibitor latexin: relationship to cystatins and the tumor suppressor TIG1. Structure (Camb) 2005;13:309–317. doi: 10.1016/j.str.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 133.Garcia-Castellanos R, Bonet-Figueredo R, Pallares I, Ventura S, Aviles FX, Vendrell J, Gomis-Rutha FX. Detailed molecular comparison between the inhibition mode of A/B-type carboxypeptidases in the zymogen state and by the endogenous inhibitor latexin. Cell Mol Life Sci. 2005;62:1996–2014. doi: 10.1007/s00018-005-5174-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pallares I, Bonet R, Garcia-Castellanos R, Ventura S, Aviles FX, Vendrell J, Gomis-Ruth FX. Structure of human carboxypeptidase A4 with its endogenous protein inhibitor, latexin. Proc Natl Acad Sci U S A. 2005;102:3978–3983. doi: 10.1073/pnas.0500678102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Reverter D, Fernandez-Catalan C, Baumgartner R, Pfander R, Huber R, Bode W, Vendrell J, Holak TA, Aviles FX. Structure of a novel leech carboxypeptidase inhibitor determined free in solution and in complex with human carboxypeptidase A2. Nat Struct Biol. 2000;7:322–328. doi: 10.1038/74092. [DOI] [PubMed] [Google Scholar]
- 136.Uratani Y, Takiguchi-Hayashi K, Miyasaka N, Sato M, Jin M, Arimatsu Y. Latexin, a carboxypeptidase A inhibitor, is expressed in rat peritoneal mast cells and is associated with granular structures distinct from secretory granules and lysosomes. Biochem J. 2000;346(Pt 3):817–826. [PMC free article] [PubMed] [Google Scholar]
- 137.Pallares I, Berenguer C, Aviles FX, Vendrell J, Ventura S. Self-assembly of human latexin into amyloid-like oligomers. BMC Struct Biol. 2007;7:75. doi: 10.1186/1472-6807-7-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rodziewicz-Motowidlo S, Wahlbom M, Wang X, Lagiewka J, Janowski R, Jaskolski M, Grubb A, Grzonka Z. Checking the conformational stability of cystatin C and its L68Q variant by molecular dynamics studies: why is the L68Q variant amyloidogenic? J Struct Biol. 2006;154:68–78. doi: 10.1016/j.jsb.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 139.Mussap M, Plebani M. Biochemistry and clinical role of human cystatin C. Crit Rev Clin Lab Sci. 2004;41:467–550. doi: 10.1080/10408360490504934. [DOI] [PubMed] [Google Scholar]
- 140.Newman DJ. Cystatin C. Ann Clin Biochem. 2002;39:89–104. doi: 10.1258/0004563021901847. [DOI] [PubMed] [Google Scholar]
- 141.Fortunel NO, Otu HH, Ng HH, Chen J, Mu X, Chevassut T, Li X, Joseph M, Bailey C, Hatzfeld JA, Hatzfeld A, Usta F, Vega VB, Long PM, Libermann TA, Lim B. Comment on " 'Stemness': transcriptional profiling of embryonic and adult stem cells" and "a stem cell molecular signature". Science. 2003;302:393. doi: 10.1126/science.1086384. author reply 393. [DOI] [PubMed] [Google Scholar]
- 142.Ramalho-Santos M, Yoon S, Matsuzaki Y, Mulligan RC, Melton DA. "Stemness": transcriptional profiling of embryonic and adult stem cells. Science. 2002;298:597–600. doi: 10.1126/science.1072530. [DOI] [PubMed] [Google Scholar]
- 143.Kato T, Heike T, Okawa K, Haruyama M, Shiraishi K, Yoshimoto M, Nagato M, Shibata M, Kumada T, Yamanaka Y, Hattori H, Nakahata T. A neurosphere-derived factor, cystatin C, supports differentiation of ES cells into neural stem cells. Proc Natl Acad Sci U S A. 2006;103:6019–6024. doi: 10.1073/pnas.0509789103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Glabe CG. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol Aging. 2006;27:570–575. doi: 10.1016/j.neurobiolaging.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 145.Mirnics ZK, Mirnics K, Terrano D, Lewis DA, Sisodia SS, Schor NF. DNA microarray profiling of developing PS1-deficient mouse brain reveals complex and coregulated expression changes. Mol Psychiatry. 2003;8:863–878. doi: 10.1038/sj.mp.4001389. [DOI] [PubMed] [Google Scholar]
- 146.Selkoe DJ. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biol. 1998;8:447–453. doi: 10.1016/s0962-8924(98)01363-4. [DOI] [PubMed] [Google Scholar]
- 147.Nagai A, Kobayashi S, Shimode K, Imaoka K, Umegae N, Fujihara S, Nakamura M. No mutations in cystatin C gene in cerebral amyloid angiopathy with cystatin C deposition. Mol Chem Neuropathol. 1998;33:63–78. doi: 10.1007/BF02815860. [DOI] [PubMed] [Google Scholar]
- 148.Kaeser SA, Herzig MC, Coomaraswamy J, Kilger E, Selenica ML, Winkler DT, Staufenbiel M, Levy E, Grubb A, Jucker M. Cystatin C modulates cerebral beta-amyloidosis. Nat Genet. 2007;39:1437–1439. doi: 10.1038/ng.2007.23. [DOI] [PubMed] [Google Scholar]
- 149.Mi W, Pawlik M, Sastre M, Jung SS, Radvinsky DS, Klein AM, Sommer J, Schmidt SD, Nixon RA, Mathews PM, Levy E. Cystatin C inhibits amyloid-beta deposition in Alzheimer's disease mouse models. Nat Genet. 2007;39:1440–1442. doi: 10.1038/ng.2007.29. [DOI] [PubMed] [Google Scholar]
- 150.Youssef EM, Chen XQ, Higuchi E, Kondo Y, Garcia-Manero G, Lotan R, Issa JP. Hypermethylation and silencing of the putative tumor suppressor Tazarotene-induced gene 1 in human cancers. Cancer Res. 2004;64:2411–2417. doi: 10.1158/0008-5472.can-03-0164. [DOI] [PubMed] [Google Scholar]
- 151.Callahan G, S V, Hartmann L, Smith D. Characterization of a carboxypeptidase-A inhibitor identified by DD-PCR in primary ovarian tumors and cell lines. Faseb J. 1999;f637 [Google Scholar]
- 152.Corticchiato O, Cajot JF, Abrahamson M, Chan SJ, Keppler D, Sordat B. Cystatin C and cathepsin B in human colon carcinoma: expression by cell lines and matrix degradation. Int J Cancer. 1992;52:645–652. doi: 10.1002/ijc.2910520425. [DOI] [PubMed] [Google Scholar]
- 153.Konduri SD, Yanamandra N, Siddique K, Joseph A, Dinh DH, Olivero WC, Gujrati M, Kouraklis G, Swaroop A, Kyritsisq AP, Rao JS. Modulation of cystatin C expression impairs the invasive and tumorigenic potential of human glioblastoma cells. Oncogene. 2002;21:8705–8712. doi: 10.1038/sj.onc.1205949. [DOI] [PubMed] [Google Scholar]
- 154.Huh CG, Hakansson K, Nathanson CM, Thorgeirsson UP, Jonsson N, Grubb A, Abrahamson M, Karlsson S. Decreased metastatic spread in mice homozygous for a null allele of the cystatin C protease inhibitor gene. Mol Pathol. 1999;52:332–340. doi: 10.1136/mp.52.6.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sokol JP, Schiemann WP. Cystatin C antagonizes transforming growth factor beta signaling in normal and cancer cells. Mol Cancer Res. 2004;2:183–195. [PubMed] [Google Scholar]