

Abstract
Downregulation of G-protein-coupled receptors (GPCRs) provides an important mechanism for reducing neurotransmitter signaling during sustained stimulation. Chronic stimulation of M2 muscarinic receptors (M2Rs) causes internalization of M2R and G-protein-activated inwardly rectifying potassium (GIRK) channels in neuronal PC12 cells, resulting in loss of function. Here, we show that coexpression of GABAB R2 receptors (GBR2s) rescues both surface expression and function of M2R, including M2R-induced activation of GIRKs and inhibition of cAMP production. GBR2 showed significant association with M2R at the plasma membrane but not other GPCRs (M1R, μ-opioid receptor), as detected by fluorescence resonance energy transfer measured with total internal reflection fluorescence microscopy. Unique regions of the proximal C-terminal domains of GBR2 and M2R mediate specific binding between M2R and GBR2. In the brain, GBR2, but not GBR1, biochemically coprecipitates with M2R and overlaps with M2R expression in cortical neurons. This novel heteromeric association between M2R and GBR2 provides a possible mechanism for altering muscarinic signaling in the brain and represents a previously unrecognized role for GBR2.
Introduction
Activation of G-protein-coupled receptors (GPCRs) provides an important signaling pathway for neurotransmitters in the brain. Three primary classes of neurotransmitter GPCRs have been described: rhodopsin-like (class A), secretin-like (class B), and metabotropic, glutamate/pheromone-like (class C). Although viewed initially as monomeric receptors, recent studies have shown that GPCR signaling can occur through the formation of dimers or higher order oligomers of receptors (van Koppen and Kaiser, 2003; Franco et al., 2007; Springael et al., 2007; Dalrymple et al., 2008). The best example is the GABAB receptor, which functions as an obligatory heterodimer of two subtypes, GABAB R1 receptor (GBR1) and GABAB R2 receptor (GBR2) (Jones et al., 1998; Kaupmann et al., 1998; White et al., 1998). Association of GBR2 with GBR1 shields an endoplasmic reticulum (ER) retention motif located on the C-terminal domain of GBR1, thereby allowing trafficking of the GBR1/GBR2 heterodimer to the plasma membrane (Margeta-Mitrovic et al., 2000). Subsequent studies revealed that GBR1 contains the agonist binding site, whereas GBR2 signals to G-proteins (Robbins et al., 2001).
Muscarinic receptors also have the propensity to form oligomeric complexes in heterologous cell systems (Park and Wells, 2003; Goin and Nathanson, 2006). The muscarinic receptors, which are activated by acetylcholine, have diverse roles in the CNS and have been implicated in cognition and neuropathic pain (Youdim and Buccafusco, 2005; Pan et al., 2008). Of the five different muscarinic receptors (M1R–M5R), M2R/M4R typically signal through Gαi/o G-proteins, which activate G-protein-activated inwardly rectifying potassium (GIRK or Kir3) channels, and reduce cAMP levels by inhibiting adenylyl cyclase; M1R/M3R/M5R, however, signal through Gαq G-proteins, which alter phosphatidylinositol turnover (van Koppen and Kaiser, 2003). M2R and M3R have been reported to heterodimerize in transfected cells but not with other receptors, such as opioid or V2 vasopressin receptors (van Koppen and Kaiser, 2003; Wang et al., 2005). The functional consequence of muscarinic receptor dimerization in vivo, either with itself or with other GPCRs, is not known (van Koppen and Kaiser, 2003).
We recently discovered that natively expressed M2Rs and GIRK channels coexist in a signaling complex that is downregulated in neuronal PC12 cells (Clancy et al., 2007). Surface expression of the M2R/GIRK complex is rescued by exposure to a muscarinic antagonist, suggesting that basal release of acetylcholine chronically stimulates M2R, leading to internalization of both M2R and GIRK channels (Clancy et al., 2007). Cross talk among neurotransmitter systems is common in the CNS. For example, presynaptic signaling through M2Rs can attenuate GABAergic inhibition (Koós and Tepper, 2002; Apergis-Schoute et al., 2007). Furthermore, both GABAB receptors and M2Rs are located on presynaptic afferents in the spinal cord in which they control glutamate release and mediate spinal analgesia (Iyadomi et al., 2000; Chen and Pan, 2004; Zhang et al., 2007). Since both M2R and GABAB receptors associate with GIRK channels in a signaling complex (David et al., 2006; Fowler et al., 2007), we investigated whether GABAB receptors could couple to cholinergic-dependent downregulated GIRK channels in neuronal PC12 cells. Surprisingly, we discovered the GABAB R2 subunit directly associates with M2R and rescues muscarinic signaling in neuronal PC12 cells. We report here the determination of the molecular and cellular events mediating this novel type of GABABR/M2R association. We further show M2R and GABAB R2 associate with each other in vivo.
Materials and Methods
Molecular biology and tissue culture.
The following constructs were used: GIRK2c expressed in pcDNA3.1(+), human M1 muscarinic receptor expressed in peCFP-N1, and human μ-opioid receptor (μOR) expressed in peCFP-N1. The M2 muscarinic receptor was fused to cyan fluorescent protein (CFP) (M2R-CFP) as described previously (Clancy et al., 2007). GBR1/GBR2-yellow fluorescent protein (YFP) and GBR1/GBR2-CFP were created as described previously (Fowler et al., 2007). GBR2Δ3-YFP was created by engineering a SacII site into GABABR2 after R747 and subcloning into peYFP-N1 using HindIII/SacII. GBR2Δ1 and Δ2 were made by engineering an AgeI site at amino acids 820 and 776, respectively, and subcloning into peYFP-N1 using EcoRI/AgeI. GBR1R2-YFP was created by engineering a KpnI site into a conserved region of GBR1 and GBR2, substituting a phenylalanine for a lysine (R1F883L/R2F739L), and subcloning the C terminus of GBR2 into GBR1-YFP using KpnI/AgeI. These substitutions resulted in receptors that functioned similar to wild type. GBR2R575D-YFP was created using the QuikChange XL kit from Stratagene. Chimeras swapping the C-terminal domains between M1 and M2 muscarinic receptors were created by overlap PCR (Finley et al., 2004). Glutathione S-transferase (GST) fusion constructs were created by fusing the region of interest to the 3′ end of GST using pGEX-2T. H8-fusion constructs were created using pHis8.3. Fusion constructs were expressed in BL21 (DE3) Escherichia coli and affinity purified as previously described (Lunn et al., 2007).
Neuronal PC12 cells were generated by 7 d NGF pretreatment as described previously (Clancy et al., 2007). HEK293 cells were maintained as described previously (Fowler et al., 2007). For electrophysiology, cells were plated onto 12 mm glass coverslips (Warner Instruments) coated with poly-d-lysine (20 mg/ml) and collagen (100 mg/ml) in 24-well plates. For imaging, cells were plated on 35 mm glass-bottom culture dishes (MatTek Corporation) and coated as described previously (Clancy et al., 2007). Neuronal PC12 cells were transfected using Lipofectamine 2000 (Invitrogen) at 1 μg of cDNA per construct (electrophysiology) or 2 μg of cDNA per construct [total internal reflection fluorescence (TIRF)/fluorescence resonance energy transfer (FRET)]. HEK293 cells were transfected by the calcium phosphate method as described previously (Fowler et al., 2007) using 0.2 μg of cDNA per construct (electrophysiology) or 1 μg of cDNA per construct (TIRF/FRET). Transfected cells were cultured for an additional 48 h before analysis. For experiments measuring changes in cAMP (see Fig. 1G) and functional coupling of GBR2 mutants (see Fig. 7), neuronal PC12 cells were transfected with M2R/GIRK2c cDNAs and exposed to 1 mm carbachol for 24 h to minimize variability observed previously (Clancy et al., 2007). Exogenously expressed receptors and channels are regulated in the same manner as those endogenously expressed (Clancy et al., 2007).
Figure 1.
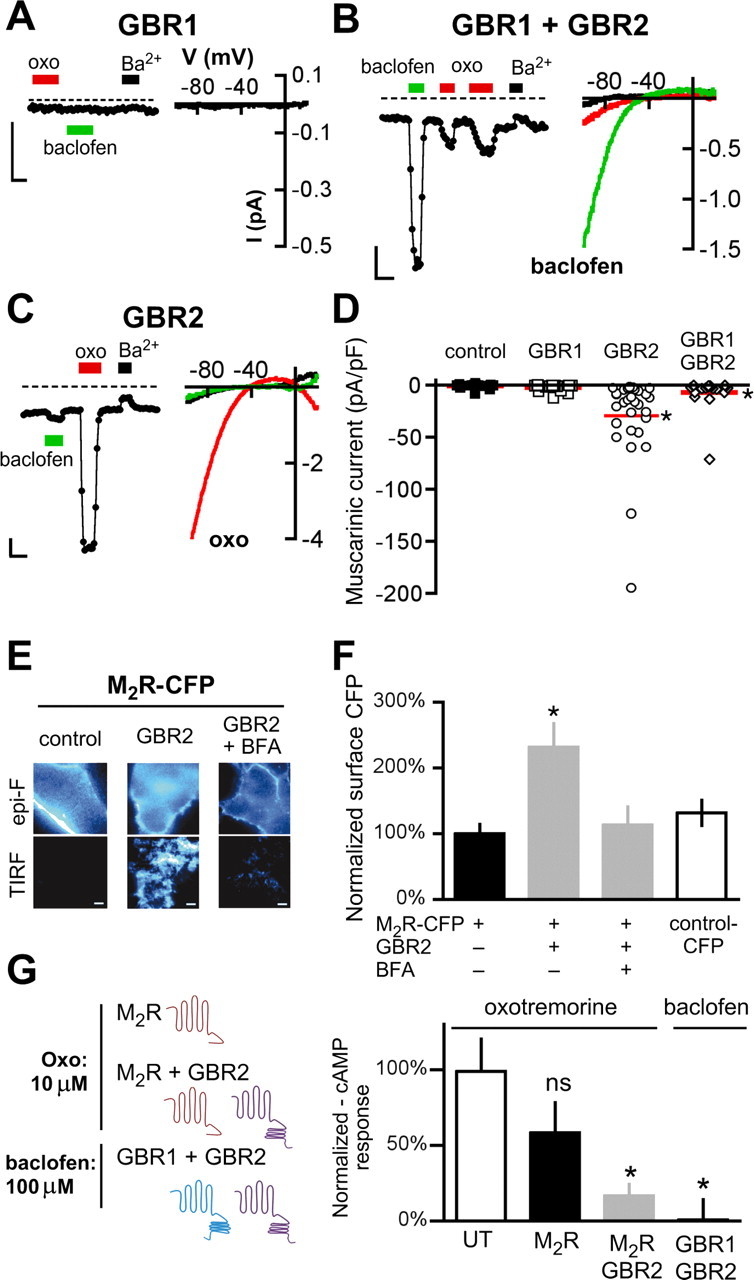
GABAB receptor expression rescues muscarinic-mediated GIRK signaling in neuronal PC12 cells. A–C, Representative current traces from neuronal PC12 cells (NGF treatment for 7 d) transfected with cDNA for GABABR1 (GBR1) (A), GABABR1 and GABABR2 (GBR1 + GBR2) (B), or GABABR2 (GBR2) (C). Whole-cell currents were recorded in 20 mm potassium (20K) in response to a voltage-ramp protocol (−120 to +50 mV; holding potential, −40 mV). The traces show the current recorded at −120 mV with 20K alone or 20K plus Ba2+ (1 mm) (black bars), oxotremorine-M (oxo) (10 μm) (red bars), or baclofen (100 μm) (green bars). The dashed lines represent zero current level. Calibration: 0.5 nA, 20 s. Insets, Current–voltage (I–V) plots for responses on left. D, Scatter plot shows muscarinic responses (+oxo) expressed as current density (in picoamperes per picofarad). The solid red lines show mean current density (N = 17–28 cells per condition with >3 transfections per condition). E, Representative images from neuronal PC12 cells expressing M2R-CFP either alone (left, control), coexpressed with GBR2-YFP (center, right), or coexpressed with GBR2-YFP and treated for 2 h with BFA (50 μm) (right). Cells were imaged under both epifluorescence (Epi-F) (top) and TIRF microscopy (bottom). Scale bar, 10 μm. F, Bar chart shows the mean surface expression for M2R-CFP measured under TIRF for cells expressing M2R-CFP alone, M2R/GBR2, or M2R/GBR2/BFA. Fluorescence was normalized to M2R-CFP. Control cells were transfected with CFP cDNA alone. The asterisk indicates statistical difference (p < 0.05) from M2R-CFP. G, Schematics show neuronal PC12 cells transfected with M2R alone or M2R/GBR2 cDNA and treated with 10 μm oxotremorine, or GBR1/GBR2 cDNA and treated with baclofen (100 μm) for 30 min, after forskolin stimulation (10 μm). The bar chart shows the effect of oxotremorine or baclofen on forskolin-stimulated cAMP. cAMP levels were normalized to untransfected (UT) cells treated with forskolin only and expressed as a percentage of forskolin-stimulated cAMP levels. The asterisk indicates statistical differences (p < 0.05 vs UT for 3 experiments). Error bars indicate SEM.
Figure 7.
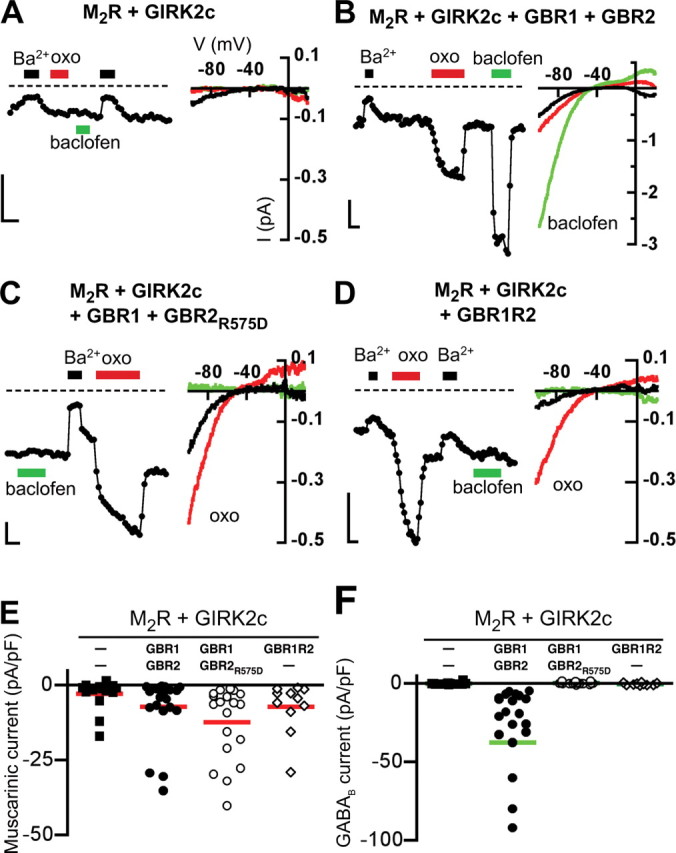
Rescue of muscarinic receptor-mediated currents is not dependent on activation of the GABAB receptor pathway. A–D, Representative current traces from neuronal PC12 cells expressing M2R and GIRK2c alone (A), or with GBR1/GBR2 (B), GBR1 and the G-protein signaling-deficient GBR2R575D (C), or the GBR1R2 chimera (D). Neuronal PC12 cells transfected with M2R and GIRK2c did not show either oxotremorine- or baclofen-induced GIRK currents. Expression of M2R with GBR1/GBR2R575D or GBR1R2 led to significant muscarinic-induced currents but no baclofen-induced GIRK currents. GBR1/GBR2 expression led to both muscarinic and baclofen-activated GIRK currents. Whole-cell currents were recorded in 20 mm potassium (20K) in response to a voltage-ramp protocol (−120 to +50 mV; holding potential, −40 mV). The traces show the current recorded at −120 mV with 20K alone or 20K plus Ba2+ (1 mm) (black bars), oxotremorine-M (oxo) (10 μm) (red bars), or baclofen (100 μm) (green bars). The dashed line represents zero current level. Calibration: 200 pA, 10 s. Current–voltage (I–V) plots are shown for different conditions. E, F, Scatter plots show summary of oxotremorine-M (muscarinic) (E) and baclofen (GABAB) (F) induced currents for different conditions. The solid lines show mean current density (N = 11–22 cells per condition with >3 transfections per condition).
TIRF microscopy and FRET measurements.
A Nikon TE2000 microscope was equipped with a 60× oil-immersion TIRF objective (1.45 numerical aperture) and a solid-state DPSS 442 nm CFP laser (Melles Griot; 85 BTL 010) and an argon 514 nm YFP laser (Melles Griot; 532-GS-A03), which could be adjusted manually for epifluorescence and TIRF. The TIRF angle was adjusted using a fixed point on the back focal plane. The Nikon filter cube contained a polychroic mirror with reflection bands at 440 and 510 nm, and bandpasses at 475/30 and 560/60 nm (z442/514rpc; Chroma Technology). CFP and YFP emission filters (470/30 and 535/50, respectively) were placed in a filter wheel (Sutter Instrument) and controlled by a Lambda 10-2 controller (Sutter Instrument). Images were acquired with a 12.5 MHz Imago CCD camera (Till Photonics). The camera, laser shutters, and filter wheel were electronically controlled by TILLvisION 4.0 software. Epifluorescent and TIRF images were acquired and analyzed as described previously (Clancy et al., 2007).
For FRET measurements, cells were fixed in ice-cold methanol on the day of the experiment. Fixation reduced background changes in CFP fluorescence with photobleaching. FRET efficiency (percentage FRET) was measured using the acceptor photobleaching (APB) method as described previously (Fowler et al., 2007). Only the change in CFP fluorescence after photobleaching YFP is used to calculate the percentage FRET (Vogel et al., 2006), in contrast to the three-cube method, which requires measuring the YFP emission with CFP excitation and correcting for bleedthrough and cross talk fluorescence (Takanishi et al., 2006; Vogel et al., 2006). Briefly, images were acquired for CFP fluorescence (100 ms exposure; 2 × 2 binning; 442 nm laser; CFPEm filter) and YFP fluorescence (30 ms exposure; 2 × 2 binning; 514 nm laser; YFPEm filter) before and after 60 s photobleaching with the 514 nm laser. Percentage FRET was measured pixel-by-pixel using NIH ImageJ plug-in FRETcalc [%FRET = 100 × (CFPEm-post − CFPEm-pre)/CFPEm-post]. Images were converted to 8-bit, background subtracted, and smoothed. Donor and acceptor thresholds were determined cell by cell to maximize colocalization between the CFP image, the YFP image, and the calculated percentage FRET image. For colocalization analysis, images were false colored red and green, and the Pearson correlation coefficient was calculated in NIH ImageJ using the Manders coefficients plug-in.
Electrophysiology.
Whole-cell patch-clamp electrophysiology was used to record macroscopic currents from neuronal PC12 cells. Borosilicate glass electrodes (P6165T; Warner Instruments) were pulled on a Narashige puller and had resistances of 5–7 MΩ. Membrane currents were recorded with an Axon Axopatch 200B (Molecular Devices) amplifier at room temperature, filtered at 2 kHz, and digitized at 5 kHz with an Axon Digidata 1320 interface (Molecular Devices). The intracellular pipette solution contained the following (in mm): 130 KCl, 20 NaCl, 5 EGTA, 2.56 K2ATP, 5.46 MgCl2, and 10 HEPES, pH 7.2 with KOH. Li2GTP (300 μm) was added fresh to the intracellular pipette solution. The external bath solution (20K) contained the following (in mm): 140 NaCl, 20 KCl, 0.5 CaCl2, 2 MgCl2, and 10 HEPES, pH 7.2 with NaOH. BaCl2 was added directly to the 20K solution. Oxotremorine was made up as a 1 mm stock and diluted on the day of experiment. A ramp voltage protocol (−120 to +50 mV) was used to study GIRK currents.
In vitro binding assay.
In vitro overlay binding assays were performed as described previously (Lunn et al., 2007). Briefly, fusion proteins were expressed and purified in BL21 (DE3) E. coli. GST-fused proteins (2 μg) were separated by SDS-PAGE, transferred to nitrocellulose, and stained with Ponceau S. Blots were placed in blocking buffer (2.5% BSA, 25 mm Tris, pH 7.4, 150 mm NaCl, 2 mm KCl, 0.05% Tween 20) and incubated overnight at 4°C. Histidine-fused protein probes (100 nm) were added to blocking buffer with 5 μm β-mercaptoethanol and incubated for 1 h on a shaker at room temperature. Western immunoblotting was performed with anti-His HRP-conjugated (Pierce; 1:2500 dilution) antibodies in TBST (0.05–0.1% Tween 20). Blots were washed, incubated with SuperSignal ECL reagents (Pierce), and exposed to BioMax XAR film (Eastman Kodak) for 30 s to 2 min.
cAMP assay.
cAMP levels were measured with a commercially available cAMP Activity Assay kit (BioVision). Briefly, cells were washed and incubated with 0.5 mm IBMX (isobutylmethylxanthine) for 15 min. Cells were then treated with 10 μm forskolin alone or with 10 μm oxotremorine or 100 μm baclofen for 30 min. Cells were lysed with 0.1 m HCl for 20 min, collected, and centrifuged. Protein quantification was determined using a standard Pierce assay kit and BSA as a standard. cAMP levels were measured following the manufacturer's instructions for equal amounts of protein from each condition and normalized to cells treated with forskolin alone. Activation of Gαi/o G-proteins, which inhibits adenylyl cyclase, is expected to lower cAMP levels.
Immunoprecipitation and immunofluorescence.
Rat and rabbit antibodies against M2R were purchased from Millipore. The specificity of guinea pig GBR1 and GBR2 antibodies has been described previously (Couve et al., 2002; Kuramoto et al., 2007). M2R antibody specificity was validated using three different techniques. First, Western analysis was performed with HEK293 cells transfected with GBR2, M2R-CFP/GBR2, M1R-CFP/GBR2 cDNAs (see Fig. 8A). Twenty micrograms of lysates were loaded on SDS-PAGE and transferred to nitrocellulose. Blots were probed with anti-M2R or anti-GBR2. Western staining of whole-brain lysates using anti-M2R antibodies revealed a single band of the predicted molecular weight for M2R (supplemental Fig. S5, available at www.jneurosci.org as supplemental material). Last, immunostaining using anti-M2R antibodies with HEK293 cells transfected with cDNA for M2R-CFP or M1R-CFP did not show significant cross-reactivity with M1R or with endogenous proteins (supplemental Fig. S6, available at www.jneurosci.org as supplemental material). For immunoprecipitation studies, brains from adult mice (∼26 g) were homogenized in 10 vol of 0.32 m sucrose using 10 strokes in a glass–Teflon homogenizer. The homogenate was centrifuged at 1400 × g for 10 min at 4°C in a benchtop centrifuge, and the supernatant was saved. The pellet was solubilized in 50 mm Tris-Cl, pH 8.0, 150 mm NaCl, and 1% NP-40, and centrifuged at 16,100 × g for 10 min. Similar results were obtained with solubilization in 50 mm Tris-Cl, pH 8.0, 150 mm NaCl, and 1% NP-40, and centrifugation at 100,000 × g for 10 min in a Beckman TLX benchtop Ultracentrifuge. The resulting supernatant was exposed to primary antibodies (5 μg) at 4°C, or control IgG coupled to protein A-Sepharose for 1 h at 4°C. Beads were centrifuged at 2000 rpm in a Microfuge, washed three times in the above buffer supplemented with 0.5 m NaCl, and then separated by SDS-PAGE. Proteins were transferred to nitrocellulose membranes and then immunoblotted with antibodies against GBR1 (1 μg/ml), GBR2 (1 μg/ml), or M2R (1 μg/ml), followed by the appropriate HRP-conjugated secondary (0.5 μg/ml) using standard techniques, and then visualized using the Fuji-LAS3000 system.
Figure 8.
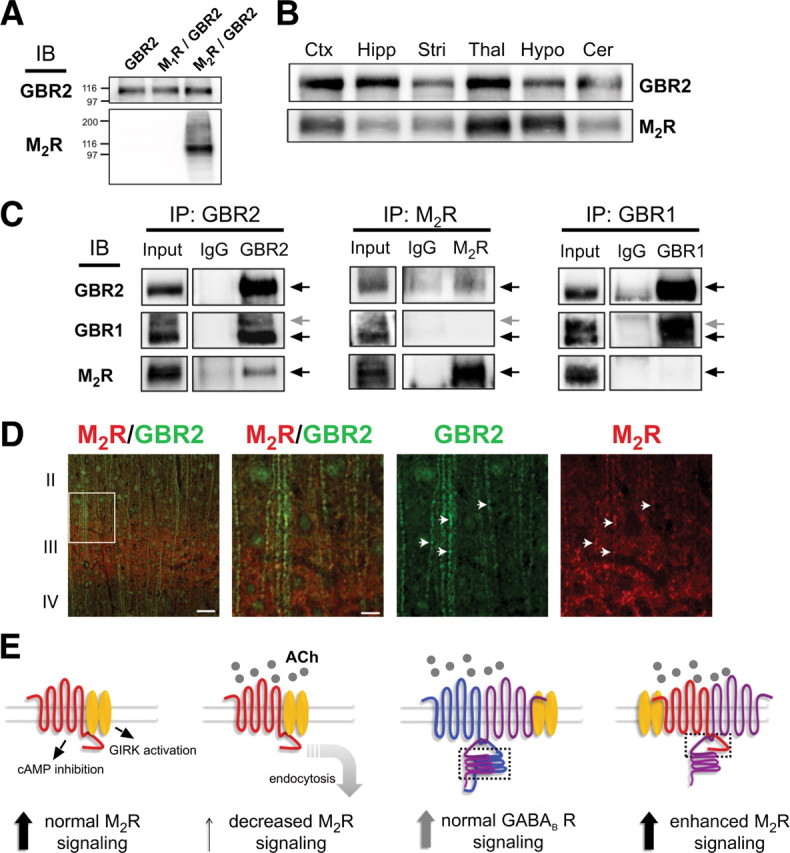
Association of M2R with GBR2 in the mouse brain: a novel model for M2 muscarinic receptor signaling. A, Validation of anti-M2R antibody specificity. Immunoblot (IB) of membranes prepared from HEK293 cells expressing GBR2, GBR2/M1R, or GBR2/M2R using anti-M2R or anti-GBR2 antibodies. B, Western analysis using anti-M2R or anti-GBR2 antibodies shows overlapping expression of M2R and GBR2 in multiple regions of mouse brain. C, Biochemical association of M2R with GBR2 in mouse brain. Immunoprecipitations (IPs) with control IgG, anti-GBR2, anti-GBR1, or anti-M2R antibodies immobilized on protein A-Sepharose were performed with detergent-solubilized mouse cortical proteins. Precipitated material was analyzed by immunoblotting with anti-GBR1, -GBR2, or -M2R antibodies and visualizing using ECL. Input represents 10% of the material used for immunoprecipitation. The arrows indicate bands of predicted molecular weight. D, Immunostaining reveals colocalization of M2R and GBR2 in somatosensory cortex. Sections were stained with anti-M2R (red) and anti-GBR2 (green) antibodies. The right-hand panels represent an enlargement of the boxed area in the extreme left-hand panel. Scale bars, 20 μm and 2 μm. The arrows highlight dendritic regions of overlapping M2R and GBR2 expression. E, Signaling models for M2R and GBR1/GBR2 in the brain (left to right). Muscarinic M2R signaling under normal conditions involves activation of GIRK channels and inhibition of cAMP production. With prolonged exposure to agonist, M2Rs downregulate via endocytosis, leading to reduced G-protein signaling. GBR1/GBR2 heterodimer signaling is unaffected by cholinergic agonist. GBR2 association with M2R restores muscarinic G-protein signaling. A direct physical interaction between the C-terminal domains (dashed box indicates binding regions) mediates the association. Note the proximal C-terminal domain of GBR2 subunit mediates binding with M2R, whereas the coiled-coil regions are involved in GBR2/GBR1 dimerization.
For immunofluorescence, mice (∼26 g) were deeply anesthetized and intracardially perfused with saline solution followed by 4% paraformaldehyde. Brains were removed, postfixed overnight, and cryoprotected in 30% sucrose. Free-floating sections were cut at 40 μm using a freezing microtome and stored at −20°C in cryoprotective solution (30% sucrose, 30% ethyleneglycol, 1% polyvinylpyrrolidone in PBS). Sections were stained in the presence of 0.05% Triton X-100 and guinea pig anti-GBR2 (1:5000) (Couve et al., 2002) and rabbit anti-mR2 (1:500) (Millipore) antibodies. Images were then collected on a Bio-Rad Radiance II microscope. All channels were first background subtracted, and the threshold value was determined for each channel that was used for all sections in each experiment.
Data analysis.
Data were analyzed using GraphPad Prism, and statistical significance was determined at p < 0.05 using one-way ANOVA followed by Dunnett's multiple comparison post hoc test or Student's t test for two groups.
Results
GABAB receptor expression rescues muscarinic/GIRK signaling in neuronal PC12 cells
NGF-differentiated PC12 cells develop neuronal-like properties, including neurite extensions, synaptic connections, and release of neurotransmitters. We reported previously that endogenous release of acetylcholine downregulates the GIRK2/M2R signaling complex (Clancy et al., 2007). Functional GIRK2/M2R complexes could be maintained either by chemically inhibiting endocytosis or by treating with a muscarinic receptor antagonist (Clancy et al., 2007). To investigate whether the downregulation of the M2R–GIRK2 signaling complex was unique to the muscarinic signaling pathway (i.e., homologous desensitization), we transiently expressed the GBR1 and/or GBR2 subunits in neuronal PC12 cells and determined the effect on GIRK channel function. Expression of GBR1 alone did not result in basal or baclofen-induced GIRK currents (Fig. 1A, Table 1), suggesting there is little or no endogenous expression of GBR2. Expression of both GBR1 and GBR2 yielded barium-sensitive basal GIRK currents and significant baclofen-induced GIRK currents (Fig. 1B,C, Table 1), suggesting that GIRK2/GBR1/R2 complexes can still form in the presence M2R-dependent desensitization.
Table 1.
Current densities for barium-inhibited basal current, oxotremorine-induced, and baclofen-induced currents for the indicated transfection conditions in neuronal PC12 cells
| Neuronal PC12 transfection conditions | Barium inhibition(pA/pF) | Oxotremorine response(pA/pF) | Baclofen response(pA/pF) | Cell number (N) | Fraction with oxotremorineresponse (%) |
|---|---|---|---|---|---|
| Untransfected | −0.65 ± 0.3 | −0.60 ± 0.5 | −0.16 ± 0.6 | 18 | 17 |
| GBR1 | −1.4 ± 0.9 | −1.8 ± 0.9 | −0.6 ± 0.7 | 17 | 29 |
| GBR2 | −5.2 ± 1.7* | −29.4 ± 7.6* | −7.0 ± 4.3* | 28 | 100 |
| GBR1 + GBR2 | −5.3 ± 1.1* | −6.9 ± 3.7* | −19.8 ± 4.0* | 19 | 68 |
| M2R + GIRK2c | −5.4 ± 2.0 | −2.8 ± 1.2 | −0.1 ± 0.16 | 16 | 19 |
| M2R + GIRK2c + GBR1 + GBR2 | −6.3 ± 2.1 | −7.1 ± 2.2** | −37.4 ± 9.6** | 22 | 41 |
| M2R + GIRK2c + GBR1 + GBR2R575D | −7.5 ± 1.8 | −12.3 ± 2.8** | −0.3 ± 0.2 | 19 | 84 |
| M2R + GIRK2c + GBR1R2 | −4.7 ± 1.7 | −7.1 ± 2.5** | −0.2 ± 0.2 | 13 | 69 |
Mean current densities ± SEM (in picoamperes per picofarad) are shown. The 75th percentile of control responses was used as the cutoff for declaring a response.
The asterisks indicate statistical significance (p < 0.05) using a one-way ANOVA followed by a post hoc Dunnett test (*vs untransfected control; **vs M2R plus GIRK2c control).
Expression of GBR1/GBR2, however, now revealed muscarinic-mediated GIRK channel signaling. Although untransfected cells failed to show muscarinic-mediated currents, small oxotremorine-induced currents were detected in cells coexpressing GBR1/GBR2 (Fig. 1B,D) (p < 0.05) (Table 1). We then examined whether muscarinic-mediated currents were detectable in cells coexpressing either GBR1 or GBR2 alone. In cells expressing GBR1, oxotremorine-mediated signaling was undetectable (Fig. 1A). In contrast, expressing GBR2 alone led to significant muscarinic-mediated GIRK currents (Fig. 1C,D) (p < 0.05). Interestingly, the oxo-induced GIRK currents for cells coexpressing GBR2 were larger than those coexpressing GBR2/GBR1. The levels of free GBR2 receptors in the two different conditions might explain this difference. We also found that surface expression of GIRK2c channels was rescued by cotransfection with GBR2 but not GBR1 (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Together, these results suggest that GBR2 promotes muscarinic signaling despite the presence of chronic acetylcholine and endogenous mechanisms for downregulating muscarinic signaling.
To investigate whether coexpression of GBR2 rescued surface expression of M2R or enhanced muscarinic signaling through some other mechanism, we transfected neuronal PC12 cells with M2R tagged with CFP at the C terminus and visualized M2R-CFP at the plasma membrane (<100 nm) using TIRF microscopy. When expressed alone in the neuronal PC12 cells, M2R-CFP showed little surface expression indistinguishable from CFP alone (Fig. 1E,F) [100 ± 15% (n = 15) vs 132 ± 19% (n = 15); p > 0.05], similar to previous studies (Clancy et al., 2007). However, coexpression of the GBR2 subunit significantly increased M2R-CFP expression (Fig. 1E,F) [232 ± 36% (n = 13); p < 0.05 vs M2R-CFP alone]. The increase in M2R-CFP with GBR2 could result from a reduction in endocytosis or an enhancement of forward trafficking. To assess the role of forward trafficking, we incubated neuronal PC12 cells in brefeldin A (BFA), a compound that inhibits forward trafficking from the endoplasmic reticulum (Chardin and McCormick, 1999). A 2 h treatment with BFA abolished the upregulation of M2R surface expression by the GBR2 subunit (Fig. 1E,F) [113 ± 28% (n = 9); p > 0.05 vs M2R-CFP alone]. These results suggest the mechanism of enhancement in muscarinic signaling is that GBR2 increases the forward trafficking of both M2R and GIRK2 channels, raising the possibility of a direct association between GBR2 and M2R (see below).
In addition to opening GIRK channels via Gβγ subunits, stimulation of muscarinic receptors also activates Gαi/o, which inhibits adenylyl cyclase and leads to a reduction in cAMP levels. To test whether rescue of muscarinic signaling by GBR2 was limited to activation of GIRK channels, we measured the cAMP levels in neuronal PC12 cells in the absence or presence of coexpressed GBR2. Stimulation of muscarinic receptors in neuronal PC12 cells had no effect on forskolin-induced levels of cAMP, suggesting endogenous muscarinic receptors do not activate Gαi/o, similar to the lack of GIRK channel activation. Neuronal PC12 cells transfected with M2R alone showed some reduction in the mean cAMP levels (58% of forskolin-stimulated control), but this was not statistically significant. In contrast, neuronal PC12 cells transfected with both M2R and GBR2 showed 83% reduction of cAMP with oxotremorine (17% of forskolin-stimulated control; p < 0.05) (Fig. 1G). This reduction is comparable with the control condition, in which baclofen stimulation of GBR1/GBR2-expressing cells reduced cAMP by 99% (p < 0.05). Together, these studies demonstrate that GBR2 can overcome muscarinic-dependent desensitization and maintain both Gβγ-dependent activation of GIRK channels and Gαi/o-dependent inhibition of cAMP synthesis.
GBR2 associates with M2 muscarinic receptors at the plasma membrane
We hypothesized that GBR2 may associate directly with M2R at the plasma membrane, altering desensitization and enhancing muscarinic signaling. To assay interactions between the GABAB and M2R receptors at the plasma membrane, we measured the possible FRET between the two receptors using TIRF microscopy. FRET efficiency was calculated using the acceptor photobleaching method (percentage FRET) (Fowler et al., 2007). One advantage of the APB method is that only the change in CFP fluorescence is used to calculate the percentage FRET, making it possible to compare the percentage FRET among different studies (Vogel et al., 2006). We examined possible percentage FRET in a PC12 cell expressing GBR2-YFP and M2R-CFP before (pre) and after (post) acceptor photobleaching (Fig. 2A). A distribution of percentage FRET calculated pixel-by-pixel was used to determine the mean percentage FRET for each cell (Fig. 2B). We measured significant percentage FRET between M2R-CFP and GBR2-YFP (Fig. 2A–C) (11.1 ± 1.2% FRET; p < 0.05 vs GBR2-CFP), but not between M2R-CFP and GBR1-YFP (2.9 ± 0.5% FRET). For comparison, we detected significant percentage FRET between GBR1-CFP and GBR2-YFP subunits compared with GBR2-CFP alone (Fig. 2C) (10.5 ± 1.3% FRET vs 0.6 ± 1.2% FRET; p < 0.05). To examine whether percentage FRET occurred from random collision or specific association (Kenworthy and Edidin, 1998), we compared the percentage FRET as a function of YFP intensity (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Note the hyperbolic increase in percentage FRET with increasing YFP concentration for the GBR1–GBR2 and M2R–GBR2 FRET pairs. Furthermore, we did not detect percentage FRET between GBR2-YFP and the μ-opioid receptor (μOR-CFP, 1.4 ± 0.7% FRET; p > 0.05), or the M1 muscarinic receptor (Fig. 2A,C) (M1R-CFP, 1.2 ± 0.4% FRET; p > 0.05), suggesting the interaction between M2R and GBR2 was specific and that random association between M2R and GBR2 was unlikely to explain the detectable percentage FRET. Thus, GBR2 and M2R are closely associated (100 Å) with each other at the plasma membrane of neuronal PC12 cells.
Figure 2.
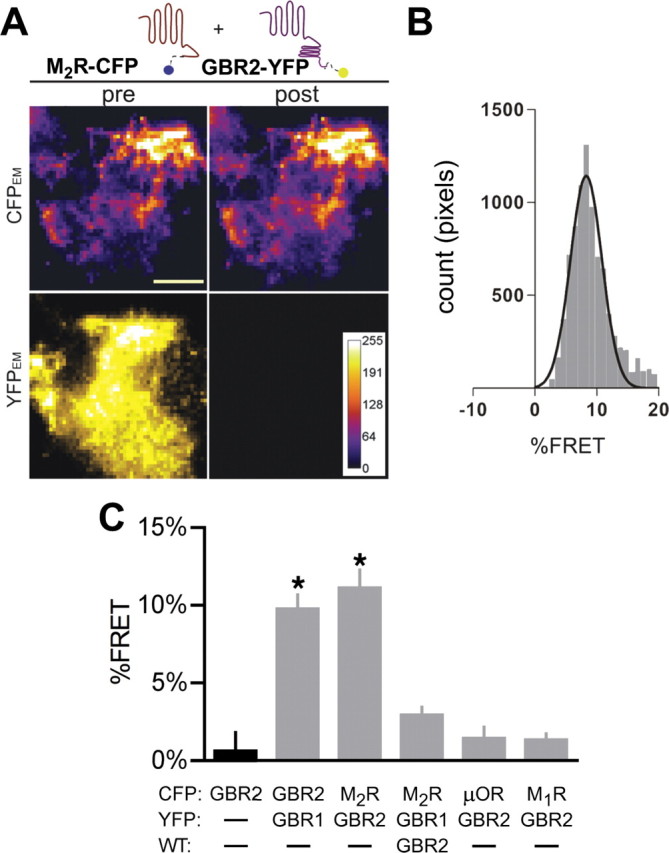
GBR2 and M2R interact at the plasma membrane in neuronal PC12 cells. A, Representative images from neuronal PC12 cells expressing M2R-CFP and GBR2-YFP. Cells were imaged using TIRF microscopy before (pre) and after (post) 60 s photobleaching YFP with a 514 nm laser. Images were collected for the CFP channel (CFPEM) and the YFP channel (YFPEM). Images are scaled to the same intensity. Scale bar, 10 μm. B, Histogram shows the distribution percentage FRET calculated per pixel using NIH ImageJ–FRETcalc plugin. The data were fit with a Gaussian distribution. C, Bar chart shows percentage FRET (mean ± SEM) for each set of transfections. Only the GBR1/GBR2 and GBR2/M2R show significant detectable percentage FRET. The asterisk indicates statistical differences [p < 0.05 vs GBR2-CFP alone (black bar); N = 8–11 cells per condition with >3 transfections per condition].
GBR2 and M2R interaction requires the GBR1 subunit in HEK293 cells
We next attempted to recapitulate the association of GBR2 with M2R in another mammalian cell line. In HEK293 cells, we observed significant percentage FRET between the GBR1-CFP and GBR2-YFP subunits of the GABAB receptor, compared with the GBR2-CFP subunit expressed alone (Fig. 3C) (6.7 ± 0.3% FRET vs 0.3 ± 0.6% FRET; p < 0.05). However, in contrast to the findings in neuronal PC12 cells, M2R-CFP did not show significant percentage FRET with GBR2-YFP (Fig. 3A,C) (0.2 ± 0.5% FRET; p > 0.05). To determine whether expression of both GABAB receptor subunits may be required for the M2R interaction, we coexpressed the GABAB heterodimer (GBR1/GBR2) with the M2R-CFP. Under these conditions, association between M2R-CFP and GBR2-YFP produced significant percentage FRET but not with GBR1-YFP, similar to neuronal PC12 cells (Fig. 3B,C) (6.6 ± 1.7% FRET, p < 0.05 vs GBR2-CFP, and 2.7 ± 0.9% FRET, respectively). Interestingly, M2R did not appear to interfere with GBR1 association with GBR2, as coexpression of M2R did not reduce percentage FRET between GBR1-CFP and GBR2-YFP (Fig. 3C) (7.5 ± 0.6% FRET). FRET was not detected between GBR2-YFP and the μOR-CFP or GBR2-YFP and muscarinic M1R-CFP (Fig. 3C). Thus, the association of M2R with GBR2 appears to be specific for these two GPCRs.
Figure 3.

GBR2 and M2R but not other GPCRs produce FRET at the plasma membrane in HEK293 cells. A, B, Representative CFPEM images from HEK293 cells expressing GBR2-YFP and M2R-CFP (A) or GBR2-YFP, M2R-CFP, and wild-type GBR1 (B) before (pre) and after (post) photobleaching. FRET between GBR2-YFP and M2R-CFP requires GBR1 in HEK293 cells. YFPEM images have been omitted for clarity. Histograms to the right show the distribution of percentage FRET per pixel for the cell shown. Scale bar, 5 μm. C, Bar chart shows mean percentage FRET for the indicated transfection conditions. The asterisk indicates statistical differences [p < 0.05 vs GBR2-CFP alone (black bar); N = 8–18 cells per condition with >3 transfections per condition]. D, Representative TIRF images from cells expressing M2R-CFP (pseudocolored red) and GBR2-YFP (green) without (left) or with (right) wild-type GBR1. The overlap in expression between M2R-CFP and GBR2-YFP increases in the presence of GBR1. Scale bars, 5 μm. E, Pearson correlation Mander's coefficient measured with NIH ImageJ for CFP and YFP channels for the given receptor combination. Statistical differences are indicated by asterisk (p < 0.05 vs coexpressed M2R and GBR2; N = 18–21 cells per condition). Error bars indicate SEM.
Thus, the detection of percentage FRET between GBR2 and M2R required coexpression of GBR1 in HEK293 cells but not in neuronal PC12 cells. To explain the requirement for GBR1 in HEK293 cells, we speculated that GBR2 was targeted to a membrane compartment different from that of M2R in HEK293 cells, in the absence of GBR1. To investigate this, we examined the colocalization of M2R-CFP and GBR2-YFP in the absence or presence of GBR1 (Fig. 3D). Indeed, GBR2-YFP and M2R-CFP showed significantly less colocalization in the absence of GBR1 (Fig. 3D,E) (p < 0.05). Calculation of the Pearson correlation coefficient (R) for GBR2-YFP and M2R-CFP (0.30 ± 0.05; n = 19) indicated significantly (p < 0.05) less colocalization than for GBR2-YFP and M2R-CFP in the presence of GBR1 (0.70 ± 0.03; n = 21). Perhaps the GBR1 subunit contains a membrane targeting motif that alters localization of the GBR1-GBR2 heterodimer in HEK293 cells. It is also possible that neurons or neuronal PC12 cells express a protein that promotes the expression of a M2R/GBR2 heterodimer on the plasma membrane (see Discussion).
C-terminal domains of M2R and GBR2 mediate GPCR interaction
If GBR2 associates directly with M2R, we hypothesized that a region in the C-terminal tail may mediate this interaction. To investigate this possibility, we created chimeras consisting of C-terminal exchanges between M2R and M1R (Fig. 4A). The chimera M2M1R-CFP, in which the M1R C-terminal domain replaces that on M2R, did not produce significant percentage FRET with GBR2-YFP, compared with the percentage FRET with M2R-CFP/GBR2-YFP/GBR1 or GBR1-CFP/GBR2-YFP in HEK293 cells (Fig. 4B,D) (p > 0.05). In contrast, M1M2R-CFP, which contains the C-terminal of M2R fused to M1R, produced detectable percentage FRET with GBR2-YFP (Fig. 4C,D) (6.0 ± 0.4% FRET, vs 0.13 ± 0.5% FRET for GBR2-CFP; p < 0.05). Thus, the C-terminal domain of M2R appears to be sufficient to associate with GBR2.
Figure 4.
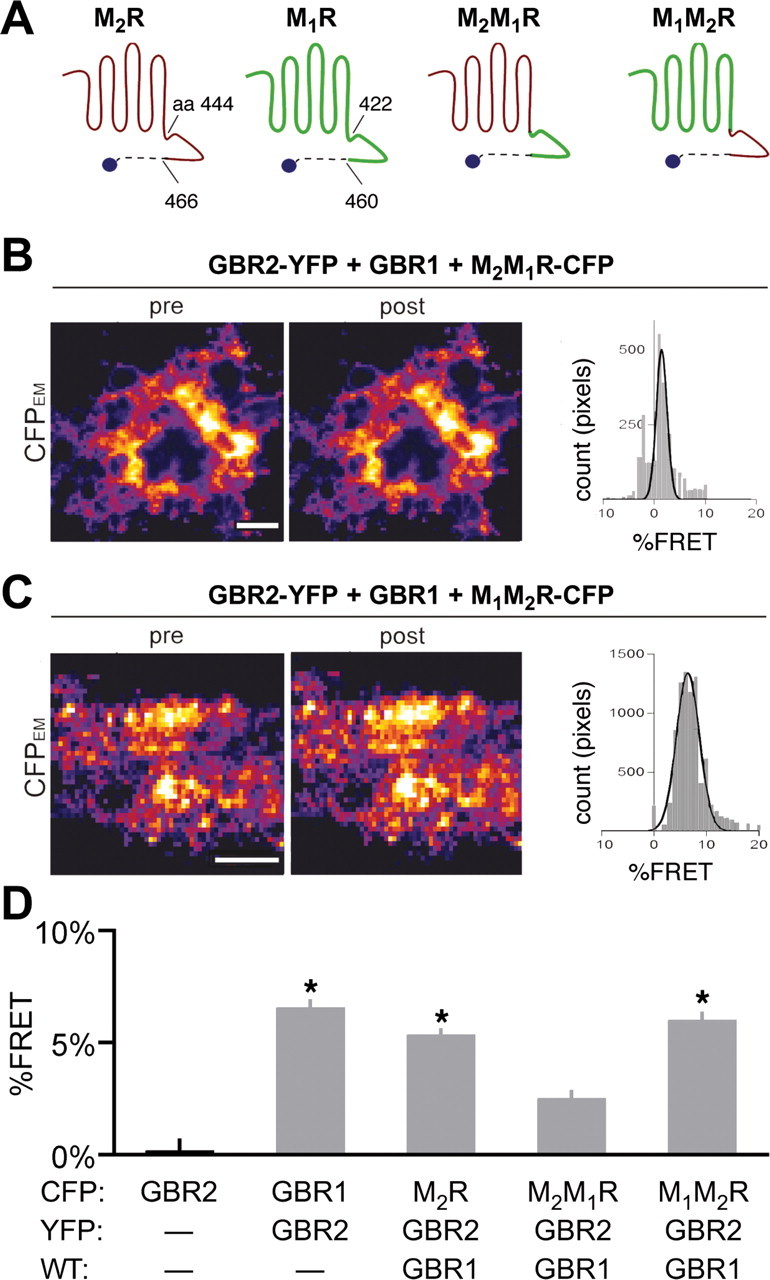
C-terminal domain of M2R required for association with GBR2. A, Schematic illustrates the amino acid regions exchanged between the M1R and M2R to generate the M2M1R and M1M2R chimeras. B, C, Representative TIRF images from HEK293 cells transfected with cDNA for GBR2-YFP, GBR1, and either M2M1R-CFP (B) or M1M2R-CFP (C). Histograms show distribution of percentage FRET per pixels for each cell shown. Scale bars, 5 μm. D, The bar chart shows percentage FRET for each receptor pair. Statistical differences are indicated by asterisk [p < 0.05 vs GBR2-CFP alone (black bar); N = 9–23 cells per condition with >3 transfections per condition]. Error bars indicate SEM.
We next localized the region of GBR2 that mediates the interaction with M2R. Previously, the coiled-coil domain in the C termini of GBR1 and GBR2 was shown to mediate dimerization (Kammerer et al., 1999). We created a series of YFP-fused C-terminal truncations of GBR2, removing the distal C terminus (at E820; GBR2Δ1), the coiled-coil domain (at V776; GBR2Δ2), or the entire C terminus (at R747; GBR2Δ3). Neither GBR2Δ2-YFP nor GBR2Δ3-YFP produced detectable percentage FRET with M2R-CFP, in the presence of GBR1 (0.25 ± 0.2% FRET, n = 12; and 1.8 ± 0.5% FRET, n = 7, respectively). GBR2Δ1-YFP, however, exhibited significant percentage FRET with M2R-CFP (4.4 ± 0.7% FRET, n = 10, vs −0.35 ± 0.4% FRET for GBR2-CFP alone, n = 13; p < 0.05). Because the coiled-coil domain mediates the interaction of GBR1 and GBR2 (Kammerer et al., 1999), we investigated whether the lack of percentage FRET with GBR2Δ2-YFP or GBR2Δ3-YFP could be attributable to the loss of association with GBR1. To address this, we created a chimeric receptor containing the N-terminal and transmembrane domains of the GBR1 and the C-terminal domain of GBR2, GBR1R2 (Fig. 5A). This chimera contains the GBR2 C-terminal domain and the putative M2R dimerization site and should rescue the interaction with M2R-CFP and generate percentage FRET. Indeed, the GBR1R2-YFP chimera showed significant percentage FRET with M2R-CFP, compared with the GBR1-YFP plus GBR2 (Fig. 5B,D) (6.0 ± 0.5 vs 1.9 ± 0.6% FRET; p < 0.05; n = 20). Interestingly, the GBR1R2-YFP chimera could now associate with M2R-CFP without requiring coexpression of GBR1 or GBR2. This finding suggests that formation of a GBR1/R2 heterodimer per se is not essential for interaction with M2R. Rather, a motif on the GBR1 N-terminal or transmembrane region is necessary for localizing the GABAB receptor to the same subcellular region as M2R in HEK293 cells. Consistent with this, the GBR1R2-YFP chimera showed a high degree of colocalization with M2R-CFP [Pearson's correlation coefficient (R), 0.61 ± 0.02; n = 14]. In contrast, GBR2Δ2-YFP, which does not contain the coiled-coil domain necessary for GBR1 interaction, showed significantly lower colocalization with M2R-CFP [Pearson's correlation coefficient (R), 0.46 ± 0.04, n = 11, vs GBR1R2-YFP, p < 0.05].
Figure 5.
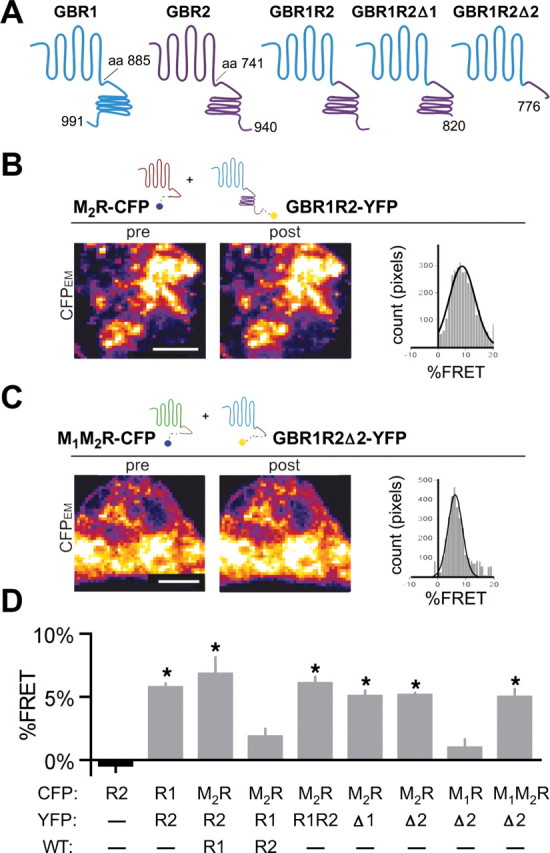
Proximal region of GBR2 C-terminal domain is required for association with M2R. A, Schematic illustrates the amino acid regions involved in constructing the GBR1R2 chimera and its truncations. B, C, Representative TIRF images from HEK293 cells expressing GBR1R2-YFP and M2R-CFP (B) or the truncated GABAB chimera, GBR1R2Δ2-YFP, and the muscarinic chimera M1M2R-CFP (C). Histograms show the percentage FRET per pixel for each cell shown. Scale bars, 5 μm. D, Bar chart shows mean percentage FRET for each transfection condition. Statistical differences are indicated by an asterisk [p < 0.05 vs GBR2-CFP alone (black bar); N = 8–20 cells per condition with >3 transfections per condition]. Error bars indicate SEM.
To precisely identify the site on GBR2 C terminus that mediates the association with M2R, the same series of C-terminal truncations were incorporated into GBR1R2-YFP (Fig. 5A). Surprisingly, deletion of the distal C-terminal tail (GBR1R2Δ1-YFP) or the coiled-coil domain (GBR1R2Δ2-YFP) did not prevent percentage FRET with M2R-CFP (Fig. 5D) (5.6 ± 0.5 and 5.4 ± 0.6% FRET, respectively, vs GBR2-CFP, −0.4 ± 0.5% FRET; p > 0.05). Furthermore, GBR1R2Δ2-YFP showed significant percentage FRET with the muscarinic M1M2R chimera [M1M2R-CFP (Fig. 5C,D), 5.0 ± 0.6, vs GBR2-CFP, p > 0.05]. Together, these spectroscopic measurements suggest that the proximal C-terminal domain of GBR2 is important for dimerization with M2R, and that the C termini of both GBR2 and M2R are sufficient for this interaction.
The proximal C terminus of GBR2 binds directly to the M2R C terminus
The finding that the C-terminal domains are required for the detection of significant percentage FRET between M2R and GBR2 suggests this association is mediated by direct protein–protein binding between to the two GPCRs. To investigate this, we used an in vitro overlay binding assay to measure direct protein–protein binding using GST fusion proteins containing the C-terminal domain of M2R (GST-M2CT), M1R (GST-M1CT), or GBR1 (GST-GBR1CT) (Fig. 6A) (Lunn et al., 2007). We examined whether a His8-tagged C-terminal domain of GBR2 could bind to the GST fusion proteins. As expected, His8-GBR2CT exhibited strong binding to the GST-GBR1CT (Fig. 6B), shown previously to involve the coiled-coil domains (Margeta-Mitrovic et al., 2000). Similar to GST-GBR1CT, His8-GBR2CT also showed binding to GST-M2RCT but importantly did not bind to GST-M1RCT (Fig. 6B). Thus, the direct binding of the C-terminal domains of GBR2 and M2R may mediate the association, bringing the receptors close enough to generate a FRET signal.
Figure 6.
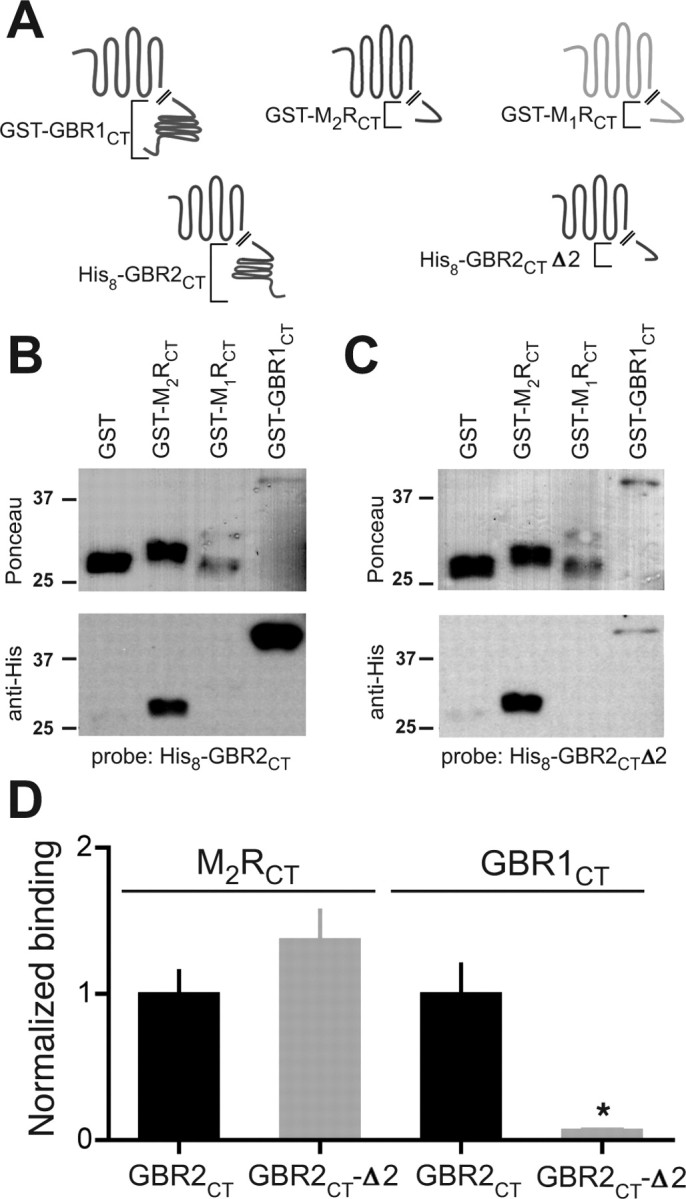
Proximal C-terminal domain of GBR2 binds directly to the M2R C-terminal domain. A, Schematic shows the design of GST- and His8-tagged fusion constructs for in vitro overlay binding assay. GST was fused to the M2R C terminus (GST-M2RCT), the M1R C terminus (GST-M1RCT), or the GBR1 C terminus (GST-GBR1CT). B, C, GBR2CT and GBR2CTΔ2 both bind to M2RCT but not to M1RCT. Representative Ponceau and Western blots. Membranes were incubated with 100 nm His8-GBR2CT (B) or His8-GBR2CTΔ2 (C). Ponceau-stained membranes (top) show size and concentration of GST-tagged fusion proteins. Note degradation for GST-M1RCT. Overlay blots were incubated with anti-His antibody to visualize bound protein (bottom). D, Bar graph shows the relative change in binding for GBR2CT and GBR2CTΔ2. OD of His8 bands were divided by the OD of the corresponding Ponceau-stained bands for GBR2CTΔ2 and normalized to GBR2CT (N = 3). Statistical difference is indicated by an asterisk (p < 0.05 by Student's t test). Error bars indicate SEM.
To localize the M2R binding site in GBR2, we created a truncated fusion protein, His8-GBR2CTΔ2, which is similar to the truncated receptor used in the FRET experiments. His8-GBR2CTΔ2 exhibited ∼80% less binding to GST-GBR1CT (Fig. 6C,D) compared with His8-GBR2CT, confirming the importance of the coiled-coil domain in the association of GBR2 with GBR1. In contrast, His8-GBR2CTΔ2 binding to GST-M2RCT was indistinguishable from that of His8-GBR2CT (Fig. 6C,D). Together, these results suggest the proximal C terminus of GBR2 mediates the association with M2R and does not involve the coiled-coil domain required for GBR1/GBR2 dimerization.
G-protein signaling via GBR2 is not required for M2R rescue
The involvement of the GABAB receptor signaling pathway in the functional rescue of the GIRK channel/M2R complex could be explained by agonist binding to the M2R and GBR2 signaling to the G-proteins. Indeed, the GBR2 signals to G-proteins in the GBR1/R2 heterodimer (Robbins et al., 2001). To examine this possibility, we created a GBR2 mutant (GBR2R575D) that was shown previously to suppress GABAB receptor G-protein activity (Binet et al., 2007). In agreement with this finding, R575D mutation rendered the GBR2 subunit incapable of signaling to G-proteins when coexpressed with GBR1 and GIRK2c channels in HEK293 cells (−1.1 ± 0.3 pA/pF, compared with −20.8 ± 9.0 pA/pF for wild-type GBR2, p < 0.05; n = 4). Importantly, this mutation did not affect basal GIRK current levels or surface expression of GBR2-YFP as measured by TIRF microscopy (data not shown).
We next coexpressed GBR1 and GBR2R575D in neuronal PC12 cells. Previous studies suggested that exogenously expressed M2Rs and GIRK channels undergo the same regulation as their endogenously expressed counterparts (Clancy et al., 2007). To minimize heterogeneity with endogenous GIRK2/M2R expression, neuronal PC12 cells were transfected with the cDNA for M2R and GIRK2c. Control cells expressing only M2R and GIRK2c exhibited little or no muscarinic-induced GIRK currents (Fig. 7A,E) (−2.8 ± 1.2 pA/pF) (Table 1), similar to untransfected neuronal PC12 cells. In contrast, coexpression of wild-type GBR2 or GBR2R575D rescued muscarinic receptor-induced (“oxo”) currents to a similar degree (Fig. 7B,C,E, Table 1) (−7.1 ± 2.2 and −12.3 ± 2.8 pA/pF, respectively, vs control, p < 0.05). Although only GBR1/R2 wild-type transfected neuronal PC12 cells showed baclofen-mediated signaling (Fig. 7F), expression of both GBR2 and GBR2R575D increased the amplitude of muscarinic receptor-induced currents, compared with control cells (Fig. 7E, Table 1). Notably, the GBR1R2 chimera lacked baclofen-mediated currents, consistent with studies suggesting that the first and second intracellular loops of the GBR2 subunit are necessary for G-protein signaling (Robbins et al., 2001; Duthey et al., 2002) Like GBR2R575D, the GBR1R2 chimera rescued muscarinic-induced currents (Fig. 7D,E).
Because a small number of control cells showed small muscarinic-induced currents, we analyzed the percentage of responders for each condition (Table 1). Setting the cutoff for a response at the 75th percentile of control responses revealed that 3 of 16 (19%) control cells showed measurable oxotremorine induced currents above background. In contrast, 9 of 22 (41%) of GBR1/R2 wild-type, 16 of 19 (84%) of GBR1/R2R575D, and 9 of 13 (69%) of GBR1R2 transfected cells showed responses, which were significantly different from M2R plus GIRK2c control (binomial test, p < 0.05). In PC12 cells expressing GABAB receptors, there is some variability in the percentage of cells with a muscarinic response, which could be attributable to slight variations in the amount of cDNAs transfected into each cell. Last, we investigated whether the association with GBR2 altered muscarinic coupling to GIRKs. The EC50 value for muscarinic activation of GIRK channels expressing M2R/GIRK2c/GBR1/GBR2 was indistinguishable from that of cells expressing M2R/GIRK2c in HEK293 (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Together, these data suggest that GBR2-dependent enhancement of muscarinic signaling occurs through association with M2R but does not require G-protein signaling through the GABAB receptor or alterations in the coupling efficiency.
Association of GBR2 with M2R in the brain
The association of M2R and GBR2 in neuronal PC12 cells leads to the prediction that GBR2 associates with M2R in the brain. This interaction has not been investigated previously. We therefore performed several biochemical and histochemical experiments to investigate the possible in vivo association of M2R and GBR2. Using specific M2R and GBR2 antibodies, we examined the relative distribution of GBR2 and M2R protein in several different brain regions using Western analysis (Fig. 8B). Notably, cerebral cortex, thalamus, and hypothalamus displayed significant levels of both GBR2 and M2R. To determine whether GBR2 and M2R could be coimmunoprecipitated from brain, we prepared detergent solubilized cortical membranes. Anti-GBR2 antibodies but not control IgG coimmunoprecipitated GBR1 (Fig. 8C), similar to previous studies with native receptors (Kaupmann et al., 1998). We found that anti-GBR2 antibodies coimmunoprecipitated M2R (Fig. 8C, left). Importantly, the reciprocal immunoprecipitation using anti-M2R antibodies pulled down GBR2 but not GBR1 (Fig. 8C, middle). Interestingly, anti-GBR1 antibodies coimmunoprecipitated GBR2 but not M2R (Fig. 8C, right), suggesting that GBR1 may not be present in the higher order oligomer. These coimmunoprecipitation findings suggest that the association of GBR2 with M2R is specific and is not attributable to the formation of a large aggregate. Based on the optical density of the protein bands, we estimate that ∼20% of M2R would be complexed with GBR2, indicating that not all of the M2Rsare bound to GBR2s. In comparison, Ramírez et al. (2009) also found that a large fraction of GBR1 and GBR2 are not associated with each other in intracellular compartments.
To corroborate the findings of association between GBR2 and M2R, we performed immunofluorescence for GBR2 and M2R in cortical neurons (Fig. 8D). Consistent with the coimmunoprecipitation data, a high degree of colocalization for GBR1 and M2R is evident along dendrites of somatosensory cortical neurons (Fig. 8D). Together, the coimmunoprecipitation and immunostaining experiments support the conclusion that GPCR signaling complexes of M2R and GBR2 exist in the brain.
Discussion
An emerging concept in the GPCR signaling field is that the formation of dimeric receptors can generate new signaling properties for GPCRs in the brain (Franco et al., 2007). In the current study, we demonstrate a novel association of the GBR2 with the M2R that leads to rescue of muscarinic receptor-mediated signaling in neuronal PC12 cells despite chronic cholinergic receptor stimulation. Detection of FRET between GBR2 and M2R and the demonstration of direct binding between two proximal C-terminal sequences of M2R and GBR2 suggest that GBR2 forms a heteromeric complex with M2R. The GBR2/M2R association appears specific since GBR2 did not associate closely with other related muscarinic receptors (M1R) or with a different GPCR (μOR). The functional rescue of cholinergic signaling did not appear to require G-protein signaling via the GBR2, since rescue was competent with a G-protein-coupling deficient mutant of GBR2 or the GBR1R2 chimera. The findings that M2R and GBR2 coimmunoprecipitate from cortex and that M2R and GBR2 colocalize in cortical neurons, which overlap with regions of the brain that receive cholinergic projections, suggests the association of GBR2 with M2R could provide a novel mechanism for enhancing cholinergic signaling in the brain.
Dimerization of different types of GPCRs has been shown to alter G-protein signaling specificity, receptor trafficking, and/or desensitization (Dalrymple et al., 2008). Heterodimerization of opioid μOR and δOR, for example, enables signaling through pertussis toxin (PTX)-insensitive G-proteins, in contrast to the normal G-protein pathway through PTX-sensitive G-proteins (George et al., 2000). Similarly, heterodimerization of μOR and α2A adrenergic receptors alters norepinephrine signaling through G-proteins (Vilardaga et al., 2008). Dimerization of dopamine D1R and D3R receptors, however, reduces D1R internalization on stimulation with a D1R-specific agonist, but promotes internalization with coapplication of D1R and D3R specific agonists (Fiorentini et al., 2008). Similarly, β2 adrenergic receptors interaction with both κORs and δORs can also affect internalization (Jordan et al., 2001). β2R interaction with δOR promotes δOR internalization in response to adrenergic stimulation but β2R interaction with κOR prevents β2R internalization in response to adrenergic stimulation (Jordan et al., 2001). One potential limitation to these studies is that heterologous expression systems are used where levels of GPCRs can be high on the cell membrane surface as well as in intracellular compartments.
Recent studies have begun to investigate the functional consequence of GPCR dimerization in vivo, however. For example, dimerization of the 5-HT2AR with the metabotropic glutamate mGluR2 greatly enhances serotonin stimulation of Gαi (González-Maeso et al., 2008). Interestingly, this association correlates with a change in signaling found in patients with schizophrenia (González-Maeso et al., 2008). Similarly, opioid receptor heterodimers have been shown to be functionally relevant in vivo. A selective agonist for δOR/κOR heterodimers exhibits analgesic properties when injected into the spinal cord of mice (Waldhoer et al., 2005). In the current study, we provide evidence for the functional association of GBR2 with M2R in vivo, using a combination of FRET/TIRF microscopy for imaging proteins on the membrane surface, functional analyses using whole-cell patch-clamp electrophysiology of natively expressed M2R/GIRK signaling complexes in neuronal PC12 cells, immunoprecipitation of M2R and GBR2, and colocalization of M2R and GBR2 in somatosensory cortex.
Before cloning of the GABAB receptors, it was speculated that there would be numerous types of GABAB receptor subtypes. However, only two subunits (GBR1/GBR2) were discovered and these form an obligatory heterodimer (Kammerer et al., 1999; Margeta-Mitrovic et al., 2000). Our FRET and biochemical experiments implicate a unique and specific association between GBR2 and M2R. There are a few examples of GABAB receptors associating with other receptors. In one case, both GBR1 and GBR2 subunits were shown to independently interact with an extracellular calcium sensing receptor (ECaR), leading to increased surface expression of the ECaR (Chang et al., 2007). However, this study did not address the mechanism by which the GABAB receptor alters ECaR expression, nor the region of the GABAB subunits involved in the interaction. Furthermore, the interaction between GBR1/GBR2 subunits and ECaR was competitive, such that if GBR1 and GBR2 were both present, the formation of GBR1/GBR2 heterodimer would preclude formation of ECaR/GBR2 dimer (Chang et al., 2007). In PC12 cells, the association of M2R with GBR2, as detected by FRET, did not appear to be affected by coexpression of GBR1. Both biochemical studies and FRET measurements of truncated and chimeric receptors implicate a 36 aa sequence in the proximal C-terminal domain of GBR2 (P741 to V776) for interaction with the last 23 aa of the C-terminal domain of M2R. This sequence length is similar to that shown previously to be involved in GPCR dimerization. For example, deleting the last 15 aa of the C-terminal domain disrupts dimerization of δ-opioid receptors (Cvejic and Devi, 1997) and truncating the C-terminal domains of μ- and δ-opioid receptors reduces dimerization (Fan et al., 2005). In contrast, GBR1 and GBR2 dimerize via a coiled-coil domain in the C terminus of both subunits (Kammerer et al., 1999; Margeta-Mitrovic et al., 2000). We found that the association of GBR2 and M2R does not require the coiled-coil domain of GBR2, indicating a different mechanism of assembly than that for GBR1/GBR2 (Fig. 8E). Since the GBR1 and GBR2 interaction is situated within the coiled-coil domains of the C-terminal domains, whereas the M2R and the GBR2 subunit associate via the proximal GBR2 C-terminal domain, it raises the possibility of a complex forming between M2R and the GBR1/R2. Although we detected FRET between only GBR2 and M2R, and measured direct biochemical binding between their respective C-terminal domains, the lack of FRET cannot rule out an association, such as between GBR1 and M2R, since two GPCRs could be physically close but orthogonal orientation of the fluorophore dipoles does not produce a FRET signal (Takanishi et al., 2006). Thus, it remains a possibility that GBR1 associates with M2R/GBR2 in a higher-order multimeric complex.
We observed an important difference between neuronal PC12 cells and HEK293 cells. Coexpression of GBR2 was sufficient to rescue muscarinic signaling and enable detectable FRET between M2R and GBR2 in neuronal PC12 cells. In contrast, coexpression of GBR1 was required to measure FRET between GBR2 and M2R in HEK293 cells. One plausible explanation is that trafficking pathways in neuronal PC12 and kidney HEK293 cells are different. For example, differences in endocytosis of the GABAB receptor have been described previously among different cell lines. In HEK293 cells, GBR1/R2 shows rapid constitutive endocytosis but no agonist-induced acceleration (Grampp et al., 2007), whereas in cortical neurons GBR1/R2 shows neither constitutive nor agonist-induced endocytosis (Fairfax et al., 2004), and in CHO cells GBR1/R2 shows rapid agonist-induced internalization (González-Maeso et al., 2003). Therefore, it seems reasonable that similar differences may exist for forwarding trafficking as well. We found the GBR1 receptor chimera containing the C-terminal domain of GBR2 (GBR1R2) traffics properly and produces significant percentage FRET with M2R. These findings suggest the C-terminal domain of GBR2 is sufficient to associate with M2R and that the GBR1 N-terminal domain and/or transmembrane domains provide the appropriate information for targeting in HEK293 cells. GBR1 contains two sushi domain repeats in the N-terminal domain that bind extracellular matrix proteins and could be important for targeting (Blein et al., 2004). In neuronal PC12 cells, it is also possible that a neuron-specific protein promotes targeting of GBR2-M2R, such as through a related GABAB receptor (Calver et al., 2003), a GISP (GPCR interacting scaffolding protein) (Kantamneni et al., 2007), or a modulator of GABAB receptors [e.g., RAMPs (receptor activity-modifying proteins)] (Parameswaran and Spielman, 2006). Alternatively, neuronal PC12 cells may express a low level of endogenous GBR1 that is sufficient to promote M2R-GBR2 expression on the plasma membrane. Indeed, we could detect small baclofen-activated currents in some neuronal PC12 cells transfected with GBR2 (−7.0 ± 4.3 pA/pF). Future studies are needed to reveal whether a higher-order GPCR complex of M2R/GBR2 and associated proteins or complex of M2R/GBR1/GBR2 exists in native tissues.
What is the mechanism underlying GBR2 enhancement of cholinergic M2R signaling in neuronal PC12 cells? Brefeldin A, a metabolite of the fungus Eupenicillium brefeldianum, specifically blocks protein transport from the ER to the Golgi apparatus and can lead to inhibition of protein secretion (Chardin and McCormick, 1999). Brefeldin A treatment abrogated the GBR2-dependent rescue of M2R signaling. One interpretation of this result is that the association of GBR2 with M2R involves forward trafficking of the receptor complex, perhaps from the ER or endosomal compartments, to the plasma membrane. Consistent with this, previous studies have shown that GBR2 promotes forward trafficking of GBR1 to produce GABAB heterodimers on the plasma membrane (Margeta-Mitrovic et al., 2000). The association of GBR2 and M2R does not appear to involve a cross-conformational switch, as described for μOR and α2A adrenergic receptors (Vilardaga et al., 2008). In fact, disrupting the G-protein signaling of GBR2 did not interfere with the ability of GBR2 to rescue M2R signaling, and the dose–response for cholinergic activation did not change in the presence of GBR2 receptors. Together, these data suggest GBR2 serves as a trafficking protein specific for M2R that efficiently interferes with the mechanism of M2R receptor-dependent downregulation.
The interaction of GBR2 with M2R could have important functional consequences for muscarinic signaling in the brain. Expression of GBR2 in M2R-expressing neurons would afford some neurons with the ability to maintain muscarinic signaling during elevated or chronic agonist exposure. The interplay between classical mechanisms of muscarinic receptor desensitization (e.g., GRKs, phosphorylation, etc.) and a “resensitization” pathway described in the current study could determine the strength of muscarinic signaling in the brain. Notably, cholinergic neurons in the nucleus basalis of Meynert project to the cerebral cortex, in which neuroanatomical studies have shown that small interneurons contain M2R, GABA, calcium binding proteins, and several inhibitory neuropeptides (Mufson et al., 2003). We find that M2R coimmunoprecipitates with GBR2 from mouse cortical tissue and exhibits overlapping expression with GBR2 in cortical neurons, highlighting one region in which GBR2 could enhance M2R signaling. Previous studies have demonstrated functional interaction between cholinergic and GABAergic signaling systems. Cholinergic activation of muscarinic receptors inhibits the release of GABA in the cortex (Sugita et al., 1991). In fact, modulation of GABAergic transmission by muscarinic receptors is impaired in a mouse transgenic model of Alzheimer's disease (Zhong et al., 2003). Upregulating GBR2 expression in these neurons might provide a mechanism for enhancing cholinergic signaling and possibly lead to novel therapeutic strategies for treating Alzheimer's disease.
Footnotes
This work was supported by the Chapman Charitable Trust at The Salk Institute (S.B.B.); National Institute of Neurological Disorders and Stroke (NINDS) Grant NS37682 (P.A.S.) and Grants NS047478, NS048045, NS051195, NS056359, and NS054900 (S.J.M.); and National Institute on Drug Abuse (NIDA) Grant DA019022 (P.A.S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIDA, NINDS, or National Institutes of Health. We thank Dr. Montminy for PC12 cells, Dr. Yu for the human μ-opioid receptor cDNA, and Dr. Jan for the GABAB receptor cDNAs.
References
- Apergis-Schoute J, Pinto A, Paré D. Muscarinic control of long-range GABAergic inhibition within the rhinal cortices. J Neurosci. 2007;27:4061–4071. doi: 10.1523/JNEUROSCI.0068-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binet V, Duthey B, Lecaillon J, Vol C, Quoyer J, Labesse G, Pin JP, Prézeau L. Common structural requirements for heptahelical domain function in class A and class C G protein-coupled receptors. J Biol Chem. 2007;282:12154–12163. doi: 10.1074/jbc.M611071200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blein S, Ginham R, Uhrin D, Smith BO, Soares DC, Veltel S, McIlhinney RA, White JH, Barlow PN. Structural analysis of the complement control protein (CCP) modules of GABA(B) receptor 1a: only one of the two CCP modules is compactly folded. J Biol Chem. 2004;279:48292–48306. doi: 10.1074/jbc.M406540200. [DOI] [PubMed] [Google Scholar]
- Calver AR, Michalovich D, Testa TT, Robbins MJ, Jaillard C, Hill J, Szekeres PG, Charles KJ, Jourdain S, Holbrook JD, Boyfield I, Patel N, Medhurst AD, Pangalos MN. Molecular cloning and characterisation of a novel GABAB-related G-protein coupled receptor. Brain Res Mol Brain Res. 2003;110:305–317. doi: 10.1016/s0169-328x(02)00662-9. [DOI] [PubMed] [Google Scholar]
- Chang W, Tu C, Cheng Z, Rodriguez L, Chen TH, Gassmann M, Bettler B, Margeta M, Jan LY, Shoback D. Complex formation with the type B gamma-aminobutyric acid receptor affects the expression and signal transduction of the extracellular calcium-sensing receptor. Studies with HEK-293 cells and neurons. J Biol Chem. 2007;282:25030–25040. doi: 10.1074/jbc.M700924200. [DOI] [PubMed] [Google Scholar]
- Chardin P, McCormick F. Brefeldin A: the advantage of being uncompetitive. Cell. 1999;97:153–155. doi: 10.1016/s0092-8674(00)80724-2. [DOI] [PubMed] [Google Scholar]
- Chen SR, Pan HL. Activation of muscarinic receptors inhibits spinal dorsal horn projection neurons: role of GABAB receptors. Neuroscience. 2004;125:141–148. doi: 10.1016/j.neuroscience.2004.01.015. [DOI] [PubMed] [Google Scholar]
- Clancy SM, Boyer SB, Slesinger PA. Coregulation of natively expressed pertussis toxin-sensitive muscarinic receptors with G-protein-activated potassium channels. J Neurosci. 2007;27:6388–6399. doi: 10.1523/JNEUROSCI.1190-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couve A, Thomas P, Calver AR, Hirst WD, Pangalos MN, Walsh FS, Smart TG, Moss SJ. Cyclic AMP-dependent protein kinase phosphorylation facilitates GABAB receptor-effector coupling. Nat Neurosci. 2002;5:415–424. doi: 10.1038/nn833. [DOI] [PubMed] [Google Scholar]
- Cvejic S, Devi LA. Dimerization of the δ opioid receptor. J Biol Chem. 1997;272:26959–26964. doi: 10.1074/jbc.272.43.26959. [DOI] [PubMed] [Google Scholar]
- Dalrymple MB, Pfleger KD, Eidne KA. G protein-coupled receptor dimers: functional consequences, disease states and drug targets. Pharmacol Ther. 2008;118:359–371. doi: 10.1016/j.pharmthera.2008.03.004. [DOI] [PubMed] [Google Scholar]
- David M, Richer M, Mamarbachi AM, Villeneuve LR, Dupré DJ, Hebert TE. Interactions between GABA-B1 receptors and Kir 3 inwardly rectifying potassium channels. Cell Signal. 2006;18:2172–2181. doi: 10.1016/j.cellsig.2006.05.014. [DOI] [PubMed] [Google Scholar]
- Duthey B, Caudron S, Perroy J, Bettler B, Fagni L, Pin JP, Prézeau L. A single subunit (GB2) is required for G-protein activation by the heterodimeric GABA(B) receptor. J Biol Chem. 2002;277:3236–3241. doi: 10.1074/jbc.M108900200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairfax BP, Pitcher JA, Scott MG, Calver AR, Pangalos MN, Moss SJ, Couve A. Phosphorylation and chronic agonist treatment atypically modulate GABAB receptor cell surface stability. J Biol Chem. 2004;279:12565–12573. doi: 10.1074/jbc.M311389200. [DOI] [PubMed] [Google Scholar]
- Fan T, Varghese G, Nguyen T, Tse R, O'Dowd BF, George SR. A role for the distal carboxyl tails in generating the novel pharmacology and G protein activation profile of mu and delta opioid receptor hetero-oligomers. J Biol Chem. 2005;280:38478–38488. doi: 10.1074/jbc.M505644200. [DOI] [PubMed] [Google Scholar]
- Finley M, Arrabit C, Fowler C, Suen KF, Slesinger PA. βL-βM loop in the C-terminal domain of GIRK channels is important for Gβγ activation. J Physiol. 2004;555:643–657. doi: 10.1113/jphysiol.2003.056101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentini C, Busi C, Gorruso E, Gotti C, Spano P, Missale C. Reciprocal regulation of dopamine D1 and D3 receptor function and trafficking by heterodimerization. Mol Pharmacol. 2008;74:59–69. doi: 10.1124/mol.107.043885. [DOI] [PubMed] [Google Scholar]
- Fowler CE, Aryal P, Suen KF, Slesinger PA. Evidence for association of GABAB receptors with Kir3 channels and RGS4 proteins. J Physiol. 2007;580:51–65. doi: 10.1113/jphysiol.2006.123216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, Casadó V, Cortés A, Ferrada C, Mallol J, Woods A, Lluis C, Canela EI, Ferré S. Basic concepts in G-protein-coupled receptor homo- and heterodimerization. ScientificWorldJournal. 2007;7:48–57. doi: 10.1100/tsw.2007.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George SR, Fan T, Xie Z, Tse R, Tam V, Varghese G, O'Dowd BF. Oligomerization of mu- and delta-opioid receptors. Generation of novel functional properties. J Biol Chem. 2000;275:26128–26135. doi: 10.1074/jbc.M000345200. [DOI] [PubMed] [Google Scholar]
- Goin JC, Nathanson NM. Quantitative analysis of muscarinic acetylcholine receptor homo- and heterodimerization in live cells: regulation of receptor down-regulation by heterodimerization. J Biol Chem. 2006;281:5416–5425. doi: 10.1074/jbc.M507476200. [DOI] [PubMed] [Google Scholar]
- González-Maeso J, Wise A, Green A, Koenig JA. Agonist-induced desensitization and endocytosis of heterodimeric GABAB receptors in CHO-K1 cells. Eur J Pharmacol. 2003;481:15–23. doi: 10.1016/j.ejphar.2003.09.002. [DOI] [PubMed] [Google Scholar]
- González-Maeso J, Ang RL, Yuen T, Chan P, Weisstaub NV, López-Giménez JF, Zhou M, Okawa Y, Callado LF, Milligan G, Gingrich JA, Filizola M, Meana JJ, Sealfon SC. Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature. 2008;452:93–97. doi: 10.1038/nature06612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grampp T, Sauter K, Markovic B, Benke D. Gamma-aminobutyric acid type B receptors are constitutively internalized via the clathrin-dependent pathway and targeted to lysosomes for degradation. J Biol Chem. 2007;282:24157–24165. doi: 10.1074/jbc.M702626200. [DOI] [PubMed] [Google Scholar]
- Iyadomi M, Iyadomi I, Kumamoto E, Tomokuni K, Yoshimura M. Presynaptic inhibition by baclofen of miniature EPSCs and IPSCs in substantia gelatinosa neurons of the adult rat spinal dorsal horn. Pain. 2000;85:385–393. doi: 10.1016/S0304-3959(99)00285-7. [DOI] [PubMed] [Google Scholar]
- Jones KA, Borowsky B, Tamm JA, Craig DA, Durkin MM, Dai M, Yao WJ, Johnson M, Gunwaldsen C, Huang LY, Tang C, Shen Q, Salon JA, Morse K, Laz T, Smith KE, Nagarathnam D, Noble SA, Branchek TA, Gerald C. GABAB receptors function as a heteromeric assembly of the subunits GABABR1 and GABABR2. Nature. 1998;396:674–679. doi: 10.1038/25348. [DOI] [PubMed] [Google Scholar]
- Jordan BA, Trapaidze N, Gomes I, Nivarthi R, Devi LA. Oligomerization of opioid receptors with beta 2-adrenergic receptors: a role in trafficking and mitogen-activated protein kinase activation. Proc Natl Acad Sci U S A. 2001;98:343–348. doi: 10.1073/pnas.011384898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammerer RA, Frank S, Schulthess T, Landwehr R, Lustig A, Engel J. Heterodimerization of a functional GABAB receptor is mediated by parallel coiled-coil alpha-helices. Biochemistry. 1999;38:13263–13269. doi: 10.1021/bi991018t. [DOI] [PubMed] [Google Scholar]
- Kantamneni S, Corrêa SA, Hodgkinson GK, Meyer G, Vinh NN, Henley JM, Nishimune A. GISP: a novel brain-specific protein that promotes surface expression and function of GABA(B) receptors. J Neurochem. 2007;100:1003–1017. doi: 10.1111/j.1471-4159.2006.04271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaupmann K, Malitschek B, Schuler V, Heid J, Froestl W, Beck P, Mosbacher J, Bischoff S, Kulik A, Shigemoto R, Karschin A, Bettler B. GABAB-receptor subtypes assemble into functional heterometic complexes. Nature. 1998;396:683–687. doi: 10.1038/25360. [DOI] [PubMed] [Google Scholar]
- Kenworthy AK, Edidin M. Distribution of a glycosylphosphatidylinositol-anchored protein at the apical surface of MDCK cells examined at a resolution of <100 Å using imaging fluorescence resonance energy transfer. J Cell Biol. 1998;142:69–84. doi: 10.1083/jcb.142.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koós T, Tepper JM. Dual cholinergic control of fast-spiking interneurons in the neostriatum. J Neurosci. 2002;22:529–535. doi: 10.1523/JNEUROSCI.22-02-00529.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramoto N, Wilkins ME, Fairfax BP, Revilla-Sanchez R, Terunuma M, Tamaki K, Iemata M, Warren N, Couve A, Calver A, Horvath Z, Freeman K, Carling D, Huang L, Gonzales C, Cooper E, Smart TG, Pangalos MN, Moss SJ. Phospho-dependent functional modulation of GABA(B) receptors by the metabolic sensor AMP-dependent protein kinase. Neuron. 2007;53:233–247. doi: 10.1016/j.neuron.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn ML, Nassirpour R, Arrabit C, Tan J, McLeod I, Arias CM, Sawchenko PE, Yates JR, 3rd, Slesinger PA. A unique sorting nexin regulates trafficking of potassium channels via a PDZ domain interaction. Nat Neurosci. 2007;10:1249–1259. doi: 10.1038/nn1953. [DOI] [PubMed] [Google Scholar]
- Margeta-Mitrovic M, Jan YN, Jan LY. A trafficking checkpoint controls GABA(B) receptor heterodimerization. Neuron. 2000;27:97–106. doi: 10.1016/s0896-6273(00)00012-x. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Ginsberg SD, Ikonomovic MD, DeKosky ST. Human cholinergic basal forebrain: chemoanatomy and neurologic dysfunction. J Chem Neuroanat. 2003;26:233–242. doi: 10.1016/s0891-0618(03)00068-1. [DOI] [PubMed] [Google Scholar]
- Pan HL, Wu ZZ, Zhou HY, Chen SR, Zhang HM, Li DP. Modulation of pain transmission by G-protein-coupled receptors. Pharmacol Ther. 2008;117:141–161. doi: 10.1016/j.pharmthera.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parameswaran N, Spielman WS. RAMPs: the past, present and future. Trends Biochem Sci. 2006;31:631–638. doi: 10.1016/j.tibs.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Park PS, Wells JW. Monomers and oligomers of the M2 muscarinic cholinergic receptor purified from Sf9 cells. Biochemistry. 2003;42:12960–12971. doi: 10.1021/bi034491m. [DOI] [PubMed] [Google Scholar]
- Ramírez OA, Vidal RL, Tello JA, Vargas KJ, Kindler S, Härtel S, Couve A. Dendritic assembly of heteromeric gamma-aminobutyric acid type B receptor subunits in hippocampal neurons. J Biol Chem. 2009;284:13077–13085. doi: 10.1074/jbc.M900575200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins MJ, Calver AR, Filippov AK, Hirst WD, Russell RB, Wood MD, Nasir S, Couve A, Brown DA, Moss SJ, Pangalos MN. GABAB2 is essential for G-protein coupling of the GABAB receptor heterodimer. J Neurosci. 2001;21:8043–8052. doi: 10.1523/JNEUROSCI.21-20-08043.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springael J-Y, Urizar E, Costagliola S, Vassart G, Parmentier M. Allosteric properties of G protein-coupled receptor oligomers. Pharmacol Ther. 2007;115:410–418. doi: 10.1016/j.pharmthera.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Sugita S, Uchimura N, Jiang ZG, North RA. Distinct muscarinic receptors inhibit release of gamma-aminobutyric acid and excitatory amino acids in mammalian brain. Proc Natl Acad Sci U S A. 1991;88:2608–2611. doi: 10.1073/pnas.88.6.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takanishi CL, Bykova EA, Cheng W, Zheng J. GFP-based FRET analysis in live cells. Brain Res. 2006;1091:132–139. doi: 10.1016/j.brainres.2006.01.119. [DOI] [PubMed] [Google Scholar]
- van Koppen CJ, Kaiser B. Regulation of muscarinic acetylcholine receptor signaling. Pharmacol Ther. 2003;98:197–220. doi: 10.1016/s0163-7258(03)00032-9. [DOI] [PubMed] [Google Scholar]
- Vilardaga JP, Nikolaev VO, Lorenz K, Ferrandon S, Zhuang Z, Lohse MJ. Conformational cross-talk between alpha2A-adrenergic and mu-opioid receptors controls cell signaling. Nat Chem Biol. 2008;4:126–131. doi: 10.1038/nchembio.64. [DOI] [PubMed] [Google Scholar]
- Vogel SS, Thaler C, Koushik SV. Fanciful FRET. Sci STKE. 2006;2006:re2. doi: 10.1126/stke.3312006re2. [DOI] [PubMed] [Google Scholar]
- Waldhoer M, Fong J, Jones RM, Lunzer MM, Sharma SK, Kostenis E, Portoghese PS, Whistler JL. A heterodimer-selective agonist shows in vivo relevance of G protein-coupled receptor dimers. Proc Natl Acad Sci U S A. 2005;102:9050–9055. doi: 10.1073/pnas.0501112102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Sun X, Bohn LM, Sadée W. Opioid receptor homo- and heterodimerization in living cells by quantitative bioluminescence resonance energy transfer. Mol Pharmacol. 2005;67:2173–2184. doi: 10.1124/mol.104.010272. [DOI] [PubMed] [Google Scholar]
- White JH, Wise A, Main MJ, Green A, Fraser NJ, Disney GH, Barnes AA, Emson P, Foord SM, Marshall FH. Heterodimerization is required for the formation of a functional GABAB receptor. Nature. 1998;396:679–682. doi: 10.1038/25354. [DOI] [PubMed] [Google Scholar]
- Youdim MB, Buccafusco JJ. CNS Targets for multi-functional drugs in the treatment of Alzheimer's and Parkinson's diseases. J Neural Transm. 2005;112:519–537. doi: 10.1007/s00702-004-0214-z. [DOI] [PubMed] [Google Scholar]
- Zhang HM, Zhou HY, Chen SR, Gautam D, Wess J, Pan HL. Control of glycinergic input to spinal dorsal horn neurons by distinct muscarinic receptor subtypes revealed using knockout mice. J Pharmacol Exp Ther. 2007;323:963–971. doi: 10.1124/jpet.107.127795. [DOI] [PubMed] [Google Scholar]
- Zhong P, Gu Z, Wang X, Jiang H, Feng J, Yan Z. Impaired modulation of GABAergic transmission by muscarinic receptors in a mouse transgenic model of Alzheimer's disease. J Biol Chem. 2003;278:26888–26896. doi: 10.1074/jbc.M302789200. [DOI] [PubMed] [Google Scholar]