

Abstract
Endocrine-disrupting chemicals (EDCs) represent a broad class of exogenous substances that cause adverse effects in the endocrine system by interfering with hormone biosynthesis, metabolism, or action. The molecular mechanisms of EDCs involve different pathways including interactions with nuclear hormone receptors (NHRs) which are primary targets of a large variety of environmental contaminants. Here, based on the crystal structures currently available in the Protein Data Bank, we review recent studies showing the many ways in which EDCs interact with NHRs and impact their signaling pathways. Like the estrogenic chemical diethylstilbestrol, some EDCs mimic the natural hormones through conserved protein–ligand contacts, while others, such as organotins, employ radically different binding mechanisms. Such structure-based knowledge, in addition to providing a better understanding of EDC activities, can be used to predict the endocrine-disrupting potential of environmental pollutants and may have applications in drug discovery.
Keywords: Nuclear hormone receptors, Endocrine-disrupting chemicals, Environmental pollutants, Crystal structures, Hormone
Introduction
Endocrine-disrupting chemicals (EDCs) are exogenous substances that interfere with the function of hormonal systems and produce a range of developmental, reproductive, neurological, immune, or metabolic diseases in humans and wildlife [1, 2]. Many EDCs are man-made chemicals produced by industry and released into the environment, for example phthalate or bisphenol A (BPA) plasticizers, organotins, pesticides, dioxins, polychlorinated biphenyls, flame retardants, or alkylphenols. Some naturally occurring EDCs can also be found in plants or fungi, such as the so-called phytoestrogens: genistein, daidzein, or the mycoestrogen zearalenone. The sources of exposure to EDCs are diverse and vary widely around the world. There are several historical examples of toxic pills or contamination that show a direct causal relationship between a unique chemical and the manifestation of an endocrine or reproductive dysfunction (see below). However, these types of single exposure are not representative of more common persistent exposures to a broad mix of chemicals and contaminants. Industrialized and agricultural areas are typically characterized by contamination from a wide range of chemicals that may seep into soil and groundwater. These complex mixtures enter the food chain and accumulate in animals and humans. Exposure occurs through drinking water, breathing contaminated air, ingesting food, or contacting contaminated soil. People working with pesticides, fungicides, and industrial chemicals are particularly exposed to these toxic substances and thus have a high risk for developing reproductive or endocrine abnormalities. EDCs can affect the endocrine systems of an organism in a wide variety of ways. These include mimicking natural hormones, antagonizing their action or modifying their synthesis, metabolism, and transport. Moreover, these substances can act via multiple pathways including membrane receptors, the aryl hydrocarbon receptor, or the enzymatic machineries involved in hormone biosynthesis/metabolism. However, most of the reported harmful effects of EDCs are attributed to their interference with hormonal signaling mediated by nuclear hormone receptors (NHRs) [3–6]. The first example of endocrine disruption was provided by the pharmaceutical diethylstilbestrol (DES, Fig. 1) which was used to prevent miscarriage in women with high risk pregnancies. In the 1970s, prenatal exposure to DES was linked with the development of vaginal cancer in so-called DES-daughters and DES-toxic effects were subsequently attributed to the interaction of this compound with estrogen receptors (ERs) which are members of the NHR family [7]. Thus, most of the subsequent studies have focused on NHRs involved in reproductive processes, in particular ERs and the androgen receptor (AR). More recently, studies have shown that the activity of the pregnane X receptor (PXR), the constitutive androstane receptor (CAR), the estrogen related receptors (ERRs), the thyroid hormone receptors (TRs), the retinoid X receptors (RXRs), or the peroxisome proliferator-activated receptors (PPARs) can also be affected by EDCs. Based on the conservation of structural and functional NHR features and on the large structural and chemical diversity of compounds found in the environment, one can predict that all members of the NHR family are potential targets of EDCs.
Fig. 1.
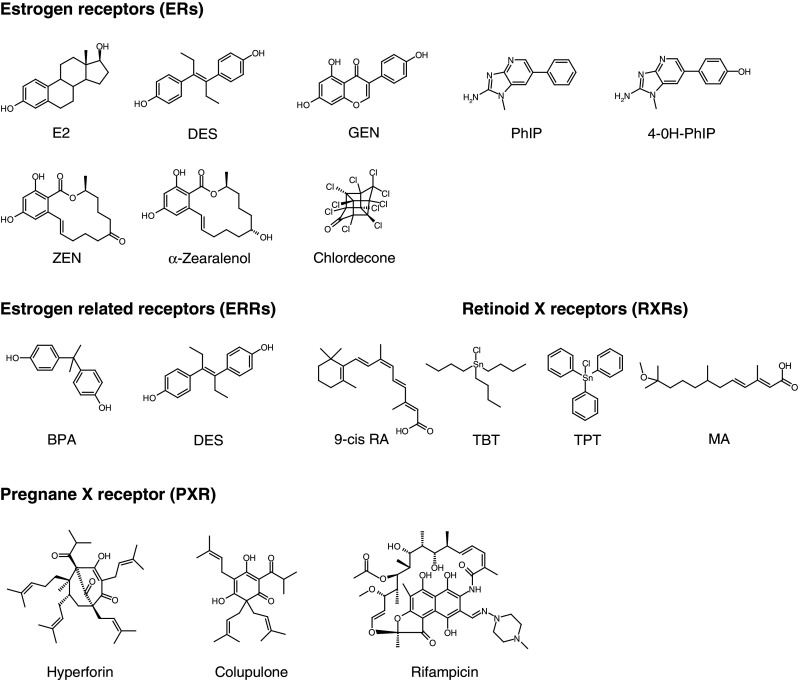
Chemical structures of representative EDCs discussed in this review. E2 17β-estradiol, DES diethylstilbestrol, GEN genistein, PhIP 2-amino-1-methyl-6-phenylimidazo [4-5-b] pyridine, 4-OH-PhIP 4-hydroxy-2-amino-1-methyl-6-phenylimidazo [4-5-b] pyridine, ZEN zearalenone, BPA bisphenol A, 9-cis RA 9-cis retinoic acid, TBT tributyltin, TPT triphenyltin, MA methoprene acid
Human NHRs (Table 1) are a family of 48 transcription factors, many of which have been shown to be activated by ligands. NHRs regulate cognate gene networks involved in key physiological functions such as cell growth and differentiation, development, homeostasis, or metabolism [8, 9]. As a consequence, dysfunctional NHR signaling (i.e., receptor mutation or inappropriate exposure to environmental pollutants) often leads to proliferative, reproductive, and metabolic diseases, including hormonal cancers, infertility, obesity, or diabetes. NHRs are modular proteins composed of several domains (Fig. 2a), most notably an N-terminal domain which harbors a ligand-independent activation function (AF-1), a central DNA binding domain (DBD), and a C-terminal ligand binding domain (LBD) hosting a ligand-dependent transcriptional activation function (AF-2) [8]. In the absence of the cognate ligand, some NHRs are located in the nucleus, bind to the DNA response elements of their target genes, and recruit corepressors, while others are located in the cytoplasm in an inactive complex with chaperones. Ligand binding induces major structural alterations of the receptor LBDs leading to (1) destabilization of corepressor or chaperone interfaces, (2) exposure of nuclear localization signals to allow nuclear translocation and DNA binding of cytoplasmic receptors, and (3) recruitment of coactivators triggering gene transcription through chromatin remodeling and activation of the general transcription machinery. The crystal structures of many NHR LBDs have been determined, revealing a conserved core of 12 α-helices (H1–H12) and a short two-stranded antiparallel β-sheet (s1 and s2) arranged into a three-layered sandwich fold (Fig. 2b). This arrangement generates a mostly hydrophobic cavity in the lower half of the domain which can accommodate the cognate ligand. In all hormone-bound LBD structures, the ligand binding pocket (LBP) is sealed by helix H12. This conformation is specifically induced by the binding of hormones or synthetic agonists and is referred to as the “active conformation” because it allows the dissociation of corepressors and favors the recruitment of transcriptional coactivators [10–12]. It is noteworthy that this conformational state can also be achieved by some constitutively active orphan receptors for which no natural ligand has been identified. In this active-form, helices H3, H4, and H12 define a hydrophobic binding groove for short LxxLL helical motifs (L stands for leucine and x for any amino acid) found within coactivators (Fig. 2b). In contrast to agonist binding, interaction with antagonists prevents the correct positioning of helix H12, thus avoiding association with the LxxLL motifs of coactivators [10–12].
Table 1.
The 48 human nuclear receptors
| NHR | Subtypes | M, D, H | Ligandsa | |
|---|---|---|---|---|
| NR1A1, A2 | TR | α, β | H | Thyroid hormones |
| NR1B1, B2, B3 | RAR | α, β, γ | H | Retinoic acid |
| NR1C1, C2, C3 | PPAR | α, β, γ | H | Fatty acids, leukotriene B4 (α), fibrates (α), prostaglandin J2 (γ), thiazolidinediones (γ) |
| NR1D1, D2 | Rev-erb | α, β | M, D | Heme |
| NR1F1, F2, F3 | ROR | α, β, γ | M | Cholesterol (α), cholesteryl sulfate (α), retinoic acid (β), Orphan (γ) |
| NR1H3, H2 | LXR | α, β | H | Oxysterols, T0901317, GW3965 |
| NR1H4, NR1H5 | FXR | α, β | H | Bile acids (α), fexaramine (α), lanosterol (β) |
| NR1I1 | VDR | H | Vitamin D, 1, 25-dihydroxyvitamin D3 | |
| NR1I2 | PXR | H | Xenobiotics, 16α-cyanopregnenolone | |
| NR1I3 | CAR | H | Xenobiotics | |
| NR2A1, A2 | HNF4 | α, γ | D | Orphans |
| NR2B1, B2, B3 | RXR | α, β, γ | D | Retinoic acids |
| NR2C1, C2 | TR | 2, 4 | D, H | Orphans |
| NR2E2 | TLL | M, D | Orphan | |
| NR2E3 | PNR | M, D | Orphan | |
| NR2, F1, F2 | COUP-TF | I, II | D, H | Orphans |
| NR2F6 | EAR-2 | M | Orphan | |
| NR3A1, A2 | ER | α, β | D | Estradiol-17β, tamoxifen (α), raloxifene (α), various synthetic compounds (β) |
| NR3B1, B2, B3 | ERR | α, β, γ | M, D | Orphan (α), DES (β, γ), 4-OH tamoxifen (β, γ) |
| NR3C1 | GR | D | Cortisol, dexamethasone, RU486 | |
| NR3C2 | MR | D | Aldosterone, spirolactone | |
| NR3C3 | PR | D | Progesterone, medroxyprogesterone acetate, RU486 | |
| NR3C2 | AR | D | Testosterone, flutamide | |
| NR4A1 | NGFI-B | M, D, H | Orphan | |
| NR4A2 | NURR1 | M, H | Orphan | |
| NR4A3 | NOR1 | M | Orphan | |
| NR5A1 | SF1 | M | Orphan | |
| NR5A2 | LRH-1 | Orphan | ||
| NR6A1 | GCNF | D | Orphan | |
| NR0B1 | DAX-1 | Orphan | ||
| NR0B2 | SHP | H | Orphan | |
Adapted from [9]
M Monomer, D dimer, H RXR-heterodimer
aFor ligand specific of a subtype, the concerned subtype is indicated in parentheses
Fig. 2.

Schematic illustration of the structural and functional organization of NHRs. a NHRs contain a well-conserved DNA binding domain (DBD), a moderately conserved ligand binding domain (LBD), and a highly divergent N-terminal A/B region. Two transcriptional activation functions have been described in these receptors: a “constitutively active” AF-1 in region A/B and an AF-2 which corresponds to a coactivator binding surface formed by helices H3, H4, and H12 of the LBD whose completion requires the presence of the hormone. b Overall structure of NHR LBDs exemplified by the ERα LBD homodimer (PDB code 1GWR) in complex with estradiol (stick representation) and a coactivator fragment (CoA). H1–H12 and s1, s2 denote α-helices and β-strands, respectively
Several receptors including ERs, RXRα, ERRα, ERRγ, and PXR have been crystallized in complex with various EDCs. This review provides an overview of the biological function of these receptors, of their associated environmental disruptors, and of the mechanisms by which some of these substances bind to NHRs and alter their signaling pathways at concentrations within the micro- to nanomolar ranges.
Estrogen receptors
Estrogen receptors and their environmental ligands (xenoestrogens)
Estrogen receptors (ERα and ERβ) are receptors for the sex hormone, 17β-estradiol (E2, Fig. 1), which play important roles in the growth and maintenance of a diverse range of tissues such as the mammary gland, uterus, bone, or the cardiovascular system. Both ERs are widely distributed throughout the body, displaying distinct but overlapping expression patterns in a variety of tissues [13]. ERα is expressed primarily in the uterus, liver, kidney, and heart, whereas ERβ is expressed primarily in the ovary, prostate, lung, gastrointestinal tract, bladder, and hematopoietic and central nervous systems [14]. However, ERα and ERβ are coexpressed in a number of tissues including the mammary gland, thyroid, adrenal, bone, and some regions of the brain. Although ERα and ERβ share similar mechanisms of action, several differences in the transcriptional abilities of each receptor as well as distinct phenotypes between gene-null animals have been identified, suggesting that these receptors may regulate distinct cellular pathways [13, 15]. Interestingly, when ERs are coexpressed, ERβ exhibits an inhibitory action on ERα-mediated gene expression [16, 17], such that ERβ has been shown to antagonize several ERα-mediated effects including fat reduction and cellular proliferation in breast, uterus, or prostate [18–20]. Breast cancer is one of the leading forms of cancer in the western world, and it is well documented that the mitogenic actions of E2 are critical in the etiology and progression of this pathology. Accordingly, current therapies of breast cancers are primarily based on estrogen antagonists that interact with ERα and completely or partially shut down the corresponding hormone-responsive pathway. Because both ERs are expressed in the normal breast, it is pertinent to ask whether EDCs with different selectivity for ERs present differential effects on breast cancer proliferation. The LBDs of ERα and ERβ share a high degree of homology in their primary amino acid sequence and are very similar in their tertiary architecture [11, 21]. It is therefore not surprising that most compounds tested so far bind to ERα and ERβ with similar affinities or have similar potencies in activation of estrogen response element (ERE)-mediated reporter gene expression [22]. However, some ERα- or ERβ-selective ligands have been identified [23].
High affinity xenoestrogens
For historical reasons, most studies on endocrine disruption have focused on sex hormone receptors, thus leading to the discovery of a large panel of substances interfering with ER signaling. Indeed, the group of molecules identified as ER endocrine disruptors is highly heterogenous and includes high affinity ligands (Kd values between 10 pM and 1 nM), which are pharmaceutical agents contained in contraceptive pills, human estrogens (E2, estriol, and estrone), and some natural compounds such as zearalenone (ZEN) and its metabolites; when available, affinities and activities of the compounds are reported in Table 2. The most notorious pharmaceutical EDC is DES (Fig. 1), an orally active synthetic nonsteroidal estrogen that, due to its teratogenic effects, was banned in 1971. In utero exposure to DES provoked abnormal cervical, uterine, and oviduct anatomy [24], vaginal adenocarcinoma [25], infertility, and ectopic pregnancy [26]. Ethynylestradiol which is present in contraceptive pills is commonly found in environmental samples such as the water of sewage treatment plants or rivers [27–29]. In spite of its structural differences with E2, ZEN (Fig. 1) is one of the most active endocrine disruptors [30]. ZEN is a mycoestrogen with non-estrogenic chemical structure which is produced by several species of Fusarium fungi, and it widely contaminates agricultural products [31] such that ZEN exists in almost all agricultural crops [32]. After consumption, ZEN is metabolized to provide two major stereo isomers, α- and β-zearalenol [33, 34], the α-metabolite displaying the highest estrogenic activity [30].
Table 2.
EC50s of ligands for human ERα, ERβ, RXRα, and PXR as reported
| ERα | ERβ | RXRα | PXR | |
|---|---|---|---|---|
| E2 | 0.02–0.05 nM [24, 47] | 0.07–0.2 nM [24, 47] | ND | 10 μM [98] |
| DES | 0.2 nM [47] | 0.4 nM [47] | ND | 10 μM [98] |
| GEN | 38 nM [24, 47] | 6–9 nM [24, 47] | ND | NA [98] |
| PhIP | 30 nM [37] | NA [37] | ND | ND |
| BPA | 4 μM [40] | 5 μM [40] | 2 μMa [124] | >10 μM [98] |
| ZEN | 1 nMb | 1 nMb | ND | 10 μM [98] |
| α-Zearalenol | 0.1 nMb | 0.1 nMb | ND | 1 μM [98] |
| Chlordecone | 12 μM [39] | Antagonist [39] | ND | ND |
| TBT | ND | ND | 3 nM [120] | ND |
| Hyperforin | ND | ND | ND | 0.11 μM [90] |
| Rifampicin | ND | ND | ND | 0.7 μM [90] |
All values are reported as EC50s in stably or transiently transfected human cell lines
ND No relevant EC50 for the ligand/receptor interaction has been reported to our knowledge, NA the ligand is inactive, Antagonist the ligand acts as an antagonist
aBPA EC50 determined by a RXR yeast two-hybrid assay
bBalaguer P., unpublished results
Medium/low affinity xenoestrogens
Many other environmental compounds interact with ERs with medium to low affinity (Kd values between 1 nM and 10 μM). Phytoestrogens are plant-derived substances that have estrogenic activity [22]. They are classified according to their chemical structure: isoflavones, flavones, flavanones, coumestans, stilbenes, and lignans. The most widely studied are the isoflavones which are present at high concentrations in soy products and red clover [35]. In recent years, efforts to implement healthier eating habits have resulted in an increased consumption of soy products and, hence, increased exposure to phytoestrogens. Genistein (GEN, Fig. 1) is the principal phytoestrogen in soy and has a wide range of biological actions. It has a structural similarity with E2 and is an agonist for both ERs, albeit with a marked preference for ERβ [22, 36]. The traditionally low breast cancer incidence rates in Asia are commonly associated with the high dietary intake of phytoestrogens, but the role of phytoestrogens as anticancer or chemopreventive agents remains under investigation. Other estrogenic substances are present in foodstuffs, including the heterocyclic amine 2-amino-1-methyl-6-phenylimidazo[4-5-b]pyridine (PhIP) contained in cooked meat. PhIP (Fig. 1) which induces tumors of the breast and prostate in mice and which has been characterized accordingly as a compound with significant estrogenic activity inducing transcription of E2-regulated genes [37, 38]. Certain insecticides and herbicides such as dichloro-diphenyl-tricholoethane (DDT), methoxychlor, hexachlorocyclohexanes, and related compounds are also suspected to act as environmental estrogenic chemicals. Chemically stable and strongly lipophilic, organochlorine compounds tend to accumulate in lipid-rich tissues and induce endocrine disruption at exposure levels measured in the environment. Interestingly, some pesticides such as chlordecone (Fig. 1) or methoxychlor display ERα agonistic, but ERβ antagonistic, activities [39], thus increasing the risk of cancer development. Finally, many industrial compounds display estrogenic activity. These include BPA (Fig. 1), alkylphenols, and benzophenones. BPA, a monomer of polycarbonate plastics, is one of the highest-volume chemicals in commerce. Polycarbonates are used in many consumer products, including food and water containers, baby bottles, medical tubing, or epoxy resin, and small amounts of BPA can migrate from polymers to food or water, especially when heated. Interestingly, BPA displays estrogen-like activities at nanomolar doses, but the mechanism by which it exerts its biological actions remains enigmatic. Although BPA binds ERs, its binding affinity is several orders of magnitude lower than that of E2 [22, 40], and it has been suggested that BPA could also act through binding to membrane ERs, G-protein coupled receptor 30 [41], or ERRγ [42]. Alkylphenols are degradation derivatives of alkylphenols ethoxylates which are widely used surfactants and detergents in domestic and industrial products. Alkylphenols are present at significant levels in samples of every environmental compartment examined, including fish muscle tissue [43], and are also generally ubiquitous in food. Benzophenones used in topical sunscreen preparations have also been shown to activate ERs with a stronger affinity for ERβ [44, 45].
Structural basis of xenoestrogen action
Natural estrogens
In the ERs, the hormone binding pockets are lined with 20 mostly hydrophobic residues that interact with the steroid scaffold [46, 47]. A few polar residues located at the two ends of the pockets form hydrogen bonds with the polar groups at the 3- and 17-positions of estrogens (Fig. 3a). Selectivity of hormone binding derives from both the shape of the hydrophobic portion of the pocket and the presence of receptor-specific hydrogen-bond networks. In contrast to other steroids which possess a 3-keto group, estrogens have a 3-hydroxyl group. Thus, part of the specificity of E2 for ERs is supported by a network of water-mediated hydrogen bonds involving a hydrogen-bond acceptor in ERs (E353 in ERα) while a hydrogen-bond donor (glutamine residue) is present in other steroid receptors. At the other end of the ligand, the 17-hydroxyl group of E2 is hydrogen-bonded to H524 (ERα).
Fig. 3.

Structural determinants of ligand recognition by ERs. a Close-up view of the E2 binding pocket in ERα (PDB code 1GWR). ERα residues involved in hormone binding as well as the E2 molecule are colored in yellow. b Superposition of ERα and ERβ LBPs in complex with E2 (yellow, PDB code 1GWR) and GEN (blue, PDB code 1QKM), respectively. Residues involved in E2 and GEN binding are colored in yellow and blue, respectively. c Close-up view of the positions of helix H12 in two GEN-ERβ complexes in the presence (blue, PDB code 1X7J) and absence (green, PDB code 1QKM) of a coactivator fragment. d Superposition of ERα LBPs in complex with PhIP (yellow, PDB code 2QXM) and 4-OH-PhIP (pink, PDB code 2QSE). Residues involved in PhIP and 4-OH-PhIP binding are colored in yellow and pink, respectively. e Superposition of ERα LBPs in complex with PhIP (blue, PDB code 2QXM) and E2 (yellow, PDB code 1GWR), respectively. f Superposition of ERβ and ERα LBPs in complex with GEN (blue, PDB code 1X7J) and DES (green, PDB code 3ERD), respectively. Residues involved in GEN and DES binding are colored in blue and green, respectively. W Water molecule. Helix and residue numbers are indicated
Xenoestrogens in food: the example of GEN and PhIP
As noted above, GEN (Fig. 1) is an isoflavonoid phytoestrogen which binds to both ER isotypes with a preference for ERβ [22, 48]. Although it is a non-steroidal compound, GEN possesses a diphenolic structure that shares structural similarity with the steroidal core of estrogens. Consequently, structures of GEN-bound ERα and ERβ reveal that it binds to both ERs nearly identically and in a manner reminiscent of that observed for the natural hormone [46, 49–51]. The phenol ring of GEN mimics the hydroxyl group of the A-ring of E2 and interacts with residues E353, R394, and a highly ordered water molecule, whereas the isoflavone portion superimposes on the C and D rings of E2 and makes a hydrogen bond with H524 (Fig. 3b). Because there are only two conservative residue substitutions in the hormone binding pockets of the two receptor subtypes (ERα L384/ERβ M336 and ERα M421/ERβ I373), the explanation of the 40-fold ERβ selectivity of GEN could not be convincingly explained by visual inspection of crystal structures. Overall, it appears that ligand selectivity arises from a combination of subtle differences involving sequence differences outside the binding pocket that alter the shape of the cavity [52] and proximal ligand–amino acid contacts [49]. Using quantum chemical calculations, Manas et al. [49] demonstrated that, although conservative, the two substitutions are capable of contributing significantly to the observed ERβ selectivity of GEN on the basis of differential electronic interactions mediated by the sulfur-containing side chain of methionines relative to purely aliphatic side chains of leucine and isoleucine residues [49]. GEN-bound ERs have been crystallized both in the presence and absence of a coactivator fragment [49, 50]. Interestingly, the activation helix H12 was observed in two different orientations in the two complexes (Fig. 3c). In the presence of the coactivator fragment, H12 adopts the canonical active conformation, whereas it is found in the antagonist-like conformation in the binary ER-GEN complex. This inactive conformation results from the unwinding of helix H11 and the consecutive lengthening of the loop connecting H11 and H12. These structural data account for the observation that GEN acts as a partial AF-2 agonist, antagonizing E2 but displaying a residual agonist activity in some cells [48] (Balaguer et al., unpublished data). As demonstrated for other partial AF-2 agonists [53] and in contrast to pure agonists, GEN appears unable to induce the receptor active conformation on its own, and the presence of a coactivator is required to fully stabilize this conformational state. Thus, the overall activity of partial AF-2 agonists like GEN is determined by the concentration of coregulators which may vary between cell types or tissues [54].
Similarly, the mutagenic compound PhIP and one of its metabolites 4-OH-PhIP have been crystallized with ERα [38]. PhIP is a heterocyclic amine with no hydroxyl groups (Fig. 1) and thus differs significantly from classical ER ligands. The phenyl ring occupies a position similar to that adopted by the A- and B-rings of E2 but does not form the typical hydrogen bonds with R394 and E353 (Fig. 3d). The remaining portion of the ligand mimics the C- and D-rings of E2. Except for the NH2 moiety which interacts favorably with H524, the hydrogen-bonding potential of the other nitrogen atoms of the heterocyclic ring is not satisfied by interaction with the receptor LBP. Compared with the ERα-E2 complex, the structure with PhIP shows a substantial shift in the last three turns of helix H11 against which helix H12 docks in the active conformation (Fig. 3e). This loss of stabilizing contacts between the two secondary structural elements likely renders helix H12 more dynamic in solution [38]. Together, these structural data explain the functional properties (affinity and activity) of PhIP. Interestingly, the PhIP metabolite 4-OH-PhIP differs from its parent compound by a hydroxyl group which reproduces the interactions made by the A-ring hydroxyl of E2. This structural observation leads to the prediction that 4-OH-PhIP should display a better affinity towards ER and also a higher estrogenic activity than its parent compound.
Pharmaceuticals: the example of DES
In contrast to the partial agonists described above, the synthetic nonsteroidal DES (Fig. 1) functions as a full agonist. The interaction of DES with ERα [47] resembles that of GEN or E2 with the two conserved hydroxyl groups involved in hydrogen bonds with E353, R394, and a water molecule on one side and H524 on the other side (Fig. 3f). Nevertheless, although the DES B ring occupies the same position as the GEN C ring, the phenolic A ring and the ethyl groups of DES do not superimpose the isoflavone ring of GEN, as a rotation angle of roughly 50° is observed between the aromatic rings (Fig. 3f). Thus, DES forms additional non-polar contacts with the LBD. In particular, contacts with A350 (H3), L384 (H5), F404 (S1), or L428 (H7) may account for the higher affinity of DES for the receptor [14], whereas specific interactions made with W383 (H4) or L540 (H12) may help stabilize the active conformation and explain the full agonistic profile of this compound.
Estrogen-related receptor γ
Estrogen-related receptors and their environmental ligands
ERRγ is a member of the ERR subfamily of orphan receptors, which are closely related to ERs [55]. The ERR family includes three members, ERRα, ERRβ, and ERRγ. The three receptors are very similar, with 90% sequence identity in the DBD and more than 60% in the LBD. ERRα is highly expressed in muscle, heart, bone, and adipose tissue, as well as in the central nervous system [56]. ERRγ is expressed in a tissue-restricted manner, for example very strongly in the mammalian brain during development, and then in the brain, lung, and many others tissues during adulthood. In terms of structure, ERRs are very close to ERs. Sequence alignment reveals a 60% identity in the DBD regions and a moderate similarity (<35%) of the LBDs, consistent with the incapacity of ERRs to bind E2 [57]. Nevertheless, it has been demonstrated that ERRs can interfere with estrogen signaling [55]. Indeed, ERs and ERRs recognize the same DNA binding elements, share common target genes and are coexpressed in many tissues [58, 59]. Recent publications report that ERRα may represent a biomarker of poor prognosis in breast and ovarian cancers suggesting an involvement of the receptor in cell proliferation. In contrast, exogenous overexpression of ERRβ and ERRγ in prostate cancer cell line results in inhibition of proliferation [60]. Furthermore, treatment with an ERRβ/γ agonist has been shown to promote this antiproliferative effect, consistent with ERRγ being a favorable prognosis factor [61]. Additionally, several lines of evidence suggest that ERRs play a central role in regulating energy metabolism. ERRs are expressed in tissues associated with lipid metabolism and high energy demands, their transcriptional activity is highly dependent on the presence of coregulators implicated in the control of metabolic programs, genetic studies in mice reveal that their presence is essential for the generation of energy and related tissue-specific functions, and functional genomics/proteomics studies have associated ERRs with the control of vast metabolic gene networks, in particular those involved in mitochondrial biogenesis and function [62]. The rise in the incidence of metabolic syndromes correlates with the rise in the use and distribution of industrial chemicals that may play a role in generation of obesity [63], suggesting that EDCs and ERRs may be linked to this epidemic crisis.
To date, the ERRs have not been shown to interact with any physiologically relevant small molecules, suggesting that these receptors manifest constitutive activity [64, 65], and, indeed, crystallographic analyses of ERRs indicated that these receptors adopt the transcriptionally active conformation in the absence of any ligand [65]. The compounds screened for activity on the ERRs were known endocrine disruptors with estrogen-like activity. Two organochlorine pesticides, toxaphene and chlordane, were found to act as weak antagonists of ERRα [66]. DES inhibits the constitutive activity of all three ERRs [67, 68]. The phytoestrogen kaempferol is an ERRα and γ antagonist [69]. In contrast to the numerous reported ERR antagonists, few agonists of ERR have been identified. However, phytoestrogens (GEN, daidzein) function as non-selective and low affinity ERR agonists [70]. Finally BPA, bisphenol E (BPE), and others phenols were recently reported to bind with high affinity to ERRγ [42, 64].
Structural basis of BPA- and DES-ERR interactions
Structure of the unliganded ERRγ
The crystal structure of unliganded ERRγ LBD [65, 71] superimposes well with that of the E2-bound ERα LBD, revealing that the receptor can adopt a transcriptionally active conformation in the absence of ligand (Fig. 4a). This conformation allows an interaction with LxxLL motifs of coactivators via a charge clamp comprised of K284 (H3) and E452 (H12). The structure reveals that ERRγ exhibits a relatively small putative LBP (Fig. 4a) delimited by 22 amino acids and measuring 280 Å3 in volume compared with 480 Å3 for ERα [71]. This observation may explain why most of the exogenous compounds interacting with ERRs act as antagonists rather than agonists (see the description of the DES-bound structure bellow). The empty ERRγ pocket is formed by hydrophobic and a few polar (Y326, N346) or charged (E275, R316) residues. The substitution of several residues between ERα and ERRγ LBPs [L525/F435 (H11), G521/A431 (H11), and L540/F450 (H12)] leads to the decrease of the cavity volume for ERRγ that precludes E2 binding to the orphan receptor. Nevertheless, it was predicted that ERRγ could bind small phenol-containing ligands because the two charged residues which form a hydrogen bonding network anchoring the 3-OH group of the A-ring of estradiol in ERα (E353 in H3 and R394 in H5) are conserved in ERRγ (E275 and R316).
Fig. 4.

Structural determinants of ligand recognition by ERRs. a Overall structure of unliganded ERRγ LBD (PDB code 1KV6) in cartoon representation. The unoccupied LBP is highlighted in black. b Superposition of ERRγ LBPs in absence of ligand (yellow, PDB code 1KV6) and in complex with BPA (green, PDB code 2E2R). Residues involved in BPA binding are colored in green and equivalent residues in the unliganded receptor are colored in yellow. c Superposition of ERRγ LBP in absence of ligand (yellow, PDB code 1KV6) and of the DES-bound form (blue, PDB code 1SP9). Residues involved in DES binding are colored in blue and equivalent residues in the unliganded receptor are colored in yellow
Structure of BPA-bound ERRγ
Although a natural ligand remains to be found for ERRγ, several synthetic ligands have been identified for this receptor [42, 64]. All display a conserved phenol ring such as BPA, a small symmetric molecule with two phenol rings and two methyl groups linked to a central sp3 carbon atom (Fig. 1). The structure of ERRγ in complex with BPA (Fig. 4b) has been solved, revealing an overall protein conformation indistinguishable from that of the unliganded receptor [72–74]. A close look at the LBP shows that, as previously anticipated, one of the two phenol-hydroxyl groups of BPA forms hydrogen bonds with residues E275 (H3) and R316 (H5) while the second is hydrogen bonded to N346 in H7. A hydrogen bond between Y326 and N346 holds N346 in position to interact with the second phenol group of BPA. Interestingly, this asparagine residue is not conserved in ERRα and β, thus accounting for the specific ERRγ-BPA interaction. Moreover, comparison of ERRγ and ERRα LBPs reveals a much smaller cavity for the latter [75]. The replacement of two alanine residues (A272 and A431) in ERRγ by a phenylalanine (F328) and a valine (V491) in ERRα accounts for this size reduction and further explains why BPA does not bind to the latter. BPA binding provokes only minimal LBP rearrangements. E275, which appears disordered in the unliganded structure, adopts a unique conformation to make a hydrogen bond with one OH group of BPA, and the side chain of L345 moves away from the pocket upon BPA binding to open up the cavity and make room for the second phenol group of BPA. In summary, ERRγ possesses a LBP to which BPA can bind with high affinity and specificity while preserving the constitutively active conformation of the receptor. The limited impact of BPA binding on receptor structure is consistent with the fact that this ligand does not enhance or disrupt coactivator binding and thus appears as a functionally silent ligand [72, 73]. Thus, the classical mechanism of NHR activation involving the re-localization and stabilization of helix H12 in the active conformation does not explain how ERRγ could mediate the estrogenic effects of BPA. Rather, thermal stability studies revealed that BPA binding leads to global thermodynamic stabilization of ERRγ LBD, a phenomenon which could increase steady state levels of the receptor and impact both its cellular half-life and biological activity [71–73].
Structure of DES-bound ERRγ
In contrast to BPA, DES deactivates ERRs and acts as a so-called inverse agonist by disrupting the basal interaction between ERRs and coactivators [68]. The crystal structure of the DES-bound ERRγ (Fig. 4c) shows that DES-mediated inverse agonism is based on the rearrangement of the side chain of F435 (H11), which upon ligand binding flips out and sterically interferes with H12, thus displacing it from its agonist position [76]. Interestingly, the recently reported crystal structure of ERRγ LBD in complex with the synthetic agonist GSK4716 [71] revealed an unexpected rearrangement of the phenol binding residues E275 and R316, which allows access to an additional pocket of 390 Å3 resulting in the formation of a single combined pocket of 610 Å3. This structure reveals that ERRγ can accommodate larger ligands than previously anticipated.
Pregnane X receptor
Pregnane X receptor and its environmental ligands
PXR, also known as steroid and xenobiotic receptor (SXR) and pregnane activated receptor (PAR), is activated by xenobiotics and acts as a master regulator of phase I to III of drug metabolism. PXR is involved in the biosynthesis, distribution, and metabolism of steroids, bile acids, and xenobiotics [77]. Activated PXR binds to gene promoters as a heterodimer with RXR and induces the expression of target genes such as CYP3A. This receptor plays a prominent role as protector of the endocrine system from chemical perturbation by sensing increases in the concentration of a multitude of EDCs and inducing detoxification pathways to prevent other NHRs from interactions with these chemicals. Literature has described PXR activation as “Jekyll and Hyde” or “ying and yang” [78, 79] to illustrate both its beneficial and prejudicial effects. In fact, activation of PXR can be positive, as it accelerates the detoxification process and consequently the elimination of xenobiotics. In contrast, the premature metabolism of active compounds such as hormones or drugs means that target responses will not be activated and can lead to harmful effects or adverse interactions. The metabolism of inactive compounds can also lead to the synthesis of active metabolites, such as the transformation of methoxychlor by CYP2C11, a PXR-induced enzyme, into phenolic estrogenic compounds [80]. It has also been observed that coregulatory proteins work in concert with ligands to stabilize PXR LBD such that the ligand-induced transcriptional response would depend on which coregulator binds to PXR LBD [81]. This observation suggests that promoter context (i.e., the combination of coregulators associated with a target gene promoter) is an important parameter in determining the transcriptional activity of PXR. Support for this notion comes from greater PXR activation by pregnenolone on a CYP3A4 PXR response element (PXRE) than on a PXRE from the multidrug resistance protein 1 (MDR1) gene which correlates with recruitment of the coactivators SRC-1 and not AIB-1 [82]. Overall, it is difficult to conclude whether the xenobiotic activation of PXR is predominantly negative or positive, but clearly PXR plays an essential role in endocrine disruption. Unlike most NHRs that tend to be specialized to bind few ligands with structural homologies, PXR is able to bind a large number of structurally diverse ligands with a wide range of affinities [83–87]. PXR binds a multitude of drugs such as the antibiotic rifampicin [88–91], the anti-cancer taxol [92, 93], the anti-cholesterol SR12813 [91, 94], the barbituric phenobarbital [90], the St John’s Wort anti-depressor hyperforine (Fig. 1) [95, 96], and many more, recently reviewed in [97]. Furthermore, numerous studies have focused on its ability to bind environmental compounds, such as pesticides [91], natural and synthetic estrogens and alkylphenols [87, 98–100], polychlorinated biphenyls [98, 101], brominated flame retardants [102], or antimicrobial triclosan [98].
Structural basis of PXR-xenobiotics interactions
By virtue of this low ligand selectivity and its dual response characteristics, PXR differs from the receptors previously discussed. Contrary to ER and RXR, which possess high affinity-specific physiological ligands or ERR which has no known natural ligand, PXR is activated by a variety of structurally distinct endogenous and exogenous compounds. Crystallographic studies have revealed several unique characteristics of PXR that account for its promiscuous ligand binding properties. First, PXR possesses a large LBP which can accommodate compounds with larger volumes than that of classical NHR ligands (823 Da for the macrocyclic rifampicin vs 272 for E2). Second, several loops clustering at the bottom of the LBD confer a high plasticity allowing the receptor LBP to adopt different shapes according to the bound ligands. Three of these flexible elements are found in a PXR-specific sequence of approximately 60 residues inserted between helices H1 and H3. This segment folds as a two-stranded antiparallel β-sheet (s1 and s1’, residues 210–228), a disordered loop (177–197), and two flexible loops (198–210 and 229–235) that appear to be characterized by either rather high thermal B factors (indicating structural mobility) in the unliganded and hyperforin-bound PXR [85, 94] or complete disorder in the structures with rifampicin [103] and colupulone [104] (Fig. 5a, b). Another region adjacent to the PXR LBP with high structural dynamics resides between s4 and H7 (residues 309–321). Thus, it appears that PXR displays a flexible portion of its LBP that allows the receptor to modify its shape and volume to bind ligands via an induced-fit mechanism. For example, the binding cavity of unbound PXR is 1,294 Å3 in volume but expands to 1,544 Å3 in the hyperforin-PXR complex. Third, the unliganded PXR LBP contains 28 residues including eight polar amino acids with the potential to form hydrogen bonds with ligands. Among them, three polar (S247, Q285, and H407) and three hydrophobic (M243, W299, and F420) residues are consistently involved in ligand binding. The remaining contacting residues depend on the size and the chemical nature of the bound ligands. For example, rifampicin contacts 18 residues while hyperforin interacts with 13 amino acid side chains. Rifampicin which is 40% larger than hyperforin and occupies regions not filled by the latter makes specific interactions with residues V211, L239, L308, and R410 (Fig. 5c, d). Interestingly, PXR is also able to bind smaller endogenous molecules such as steroidal hormones. The recently reported crystal structure of PXR LBD in complex with estradiol reveals that E2 fills only a very small part of the LBP, leaving 1,000 Å3 unoccupied [87]. Moreover, the structure shows that the position of the bound ligand as well as the key stabilizing interactions differ significantly from that found in the E2-bound ER complex structure. Notably, E2 in PXR binds closely adjacent to H12 in an orientation nearly perpendicular to that observed in the ER complex, and key E2-contacting residues in ER (E353, R394, and H524 in ERα) are replaced in PXR such that the 3-OH group on the A-ring forms an hydrogen bond with S247, whereas the 17 OH-group on the D-ring is hydrogen bonded to R410 and S208 (not shown). Globally, the capacity of PXR to accommodate a variety of chemical scaffolds critically involves the deformability of its cavity together with the unique distribution of hydrophobic, polar, and charged residues over the LBP. CAR is another receptor that responds to several endo- and xenobiotics [105, 106]. However, the smaller and less conformable LBP of CAR is most probably the reason why this receptor recognizes fewer EDCs and plays a lesser role than PXR as a xenosensor. Together, these structural observations contribute to the better understanding of how PXR can detect structurally and chemically different compounds. However, they also point to the fact that, due to the high plasticity of PXR LBP, modeling of the interaction between this receptor and EDCs in view of computational prediction of receptor–ligand interaction might be difficult.
Fig. 5.

Structural determinants of ligand recognition by PXR. a Close-up view of the flexible elements found specifically in PXR. They are inserted between helices H1 and H3 and comprise two-stranded antiparallel β-sheet (s1 and s1′), a disordered loop and two flexible loops. These elements are visible in the structure of unliganded PXR (green, PDB code 1ILG) whereas they are too disordered to be visible in the structure of PXR bound to rifampicin (purple, PDB code 1SKX). b The structure of PXR LBD in complex with hyperforin (PDB code 1M13) is rendered by thermal displacement parameters (B factor) ranging from blue (low) to red (high). c Close-up view of PXR LBP in complex with rifampicin (PDB code 1SKX). PXR residues involved in rifampicin binding are colored in purple and rifampicin is shown in yellow. d Close-up view of PXR LBP in complex with hyperforin (PDB code 1M13). PXR residues involved in hyperforin binding are colored in green and hyperforin is shown in yellow
Retinoid X receptors
Retinoid X receptors and their environmental ligands
RXRα, β, and γ occupy a particular position in the NHR superfamily, as they are the common heterodimerization partners for one-third of the 48 family members (Table 1). Consequently, RXRs are involved in the control of multiple signaling pathways in both ligand-dependent and -independent manners [107]. RXRs form three different types of dimers: RXR homodimer, permissive heterodimers, and non-permissive heterodimers. RXR permissive heterodimers (e.g., RXR-PPAR, RXR-LXR, or RXR-PXR) are activated upon ligand binding to RXR even in the absence of the partner receptor ligand, whereas non-permissive heterodimers (e.g., RXR-RAR, RXR-VDR, or RXR-TR) cannot be activated by the RXR ligand alone, and RXR serves as a silent partner in absence of partner ligand. Differential corepressor interaction accounts for the diverse activation profiles of permissive and non-permissive heterodimers [108]. However, in both cases, RXR ligands and ligands of the partner receptors can act synergistically to activate heterodimers [109, 110]. This regulatory control of nuclear signaling pathways by multiple RXR heterodimers allows environmental RXR ligands to potentially trigger a multitude of adverse effects on human health.
RXRs are activated by 9-cis retinoic acid (9-cis RA; Fig. 1) as well as docosahexaenoic acid [111, 112]. In addition, synthetic RXR ligands, referred to as rexinoids, are already used or are being developed for cancer therapy and treatment of metabolic diseases [113]. On the other hand, RXR can also be activated by environmental exogenous chemicals, including some organotin compounds (Fig. 1), which in so doing act as endocrine disruptors [114–116]. The RXRα/PPARγ heterodimer was reported to play a major role in mediating the deleterious effects of organotins which are ubiquitously present throughout the environment due to their widespread use since the 1960s in many industrial and agricultural processes [117]. Importantly, these studies demonstrated that the two biocides, tributyltin (TBT; Fig. 1) and triphenyltin (TPT; Fig. 1), are able to activate this heterodimer at nanomolar concentrations thereby inducing various toxicities ranging from adipogenesis in mammals [118, 119] to the development of male reproductive organs in female gastropods [120, 121]. Interestingly, a recent study revealed that in fact TBT activates all three RXR/PPARα, γ, δ heterodimers essentially through its interaction with RXR [122]. Another example of an EDC that modulates RXR function is methoprene which is an insect growth regulator in domestic and agricultural use as a pesticide. At least one metabolite of methoprene, methoprene acid (MA, Fig. 1), directly binds to and activates mammalian RXRs [123]. Finally, BPA and other phenols (4-tert-octylphenol, nonylphenol mixture) showed high induction of RXR directly or after being metabolized [124].
Structural basis of xenorexinoids action
MA binds to and activates the three subtypes of RXR with a lower affinity than 9-cis RA [123]. In contrast, TPT and TBT, two of the most active organotin compounds, bind to and activate RXRs as efficiently as 9-cis RA (Table 1). Structures of the complexes between RXR and TBT [122], TPT (described in this review) and MA [125], were obtained, and their comparison with that of the 9-cis RA-bound RXR [126] allowed the description of the binding mode of these EDCs and the explanation of their different binding affinities. In RXR, 9-cis RA is buried in an essentially hydrophobic pocket formed by residues located on helices H3, H5, H7, and H11, and the β-turn (Fig. 6a). RXR features an L-shaped pocket which requires a sharp bend or a twist of the polyene side chain in the retinoid skeleton to allow binding. The pocket is sealed by R316 which forms an ionic interaction with the carboxylate group of 9-cis RA on one side and by helices H7, H11 and H12 on the other side. Thus, the restrictive structural, chemical, and conformational features of the RXR LBP are perfectly adapted to accommodate the 9-cis isomer and not the elongated all-trans isomer of retinoic acid. Interestingly, the pesticide MA displays molecular characteristics similar to 9-cis RA which allow interaction with RXR, namely a conformable aliphatic chain and a carboxylate moiety. In the crystal structure [125], it was observed that, contrary to 9-cis RA, MA does not interact with H12 directly, an observation that could account for the poor agonist-driven H12 recruitment. MA adopts an L-shaped conformation to fit the RXR LBP by two consecutive 90° bond rotations, and thereby mimics the sharp cis-bend of 9-cis RA (Fig. 6b). In the same manner as 9-cis RA, the carboxylate is anchored by an ionic interaction with R387 from helix H5. The methoxy iso-butyl moiety is smaller than the β-ionone ring of 9-cis RA, and therefore makes fewer hydrophobic contacts with the receptor. These differences in interactions with the receptor most likely account for the lower affinity observed for MA in comparison with 9-cis RA. In contrast to MA, the two organotin compounds, TBT or TPT, interact strongly with RXRs and act as full RXR agonists whereas they neither structurally nor chemically resemble 9-cis RA (Fig. 1). Organotins form a group of more than 250 tin compounds containing a variety of mono-, di-, tri-, or tetra-substituted organic groups and, contrary to classical rexinoids (RXR-specific ligands), they do not contain an acidic head group for interaction with R316. The previously reported structure of RXR in complex with TBT [122], and the crystal structure of TPT-bound RXR presented here, reveal that, as compared with 9-cis RA, the two organotins occupy only a small part of RXR LBP (Fig. 6c). Moreover, the structures show that the high affinity of the organotins for RXR derives mainly from the formation of a covalent bond between the tin atom of organotins and the sulfur atom of a cysteine residue (C432) in helix H11. The remaining interactions involve van der Waals contacts between almost all organotin carbon atoms and RXR residues. Interestingly, although TBT and TPT interact with only a subset of binding pocket residues, they are engaged in enough essential contacts to efficiently stabilize RXRα in its active conformation. The particular position of C432 in helix H11 likely accounts for the high agonist activity of TBT and TPT. Indeed, several studies have pointed out the importance of this secondary structural element which should adopt a helical conformation for full receptor activation [38, 127, 128]. The presence of a cysteine residue at this particular H11 position is unique to RXRs and may allow for the stabilization of the helical conformation of H11 and of the active receptor form by TBT and TPT. Other NHRs including PPARγ and the glucocorticoid receptor (GR) have been identified as organotin targets [114, 118, 119, 129]. PPARγ and GR contain cysteine residues at different positions which could help to anchor the organotins in the LBPs. However, these cysteine residues could fix the tin compounds in regions of the LBPs which do not allow efficient stabilization of the active receptor conformation. Accordingly, organotins were shown to act as partial agonist or antagonist for PPARγ [122] and GR [129], respectively, indicating that tin-containing compounds could use the specific Sn-S interaction to modulate the transcriptional activity of a number of NHRs, the functional outcome being dictated by the structure of the organotin and the position of the anchoring cysteine in the LBP.
Fig. 6.

Structural determinants of ligand recognition by RXRs. a Close-up view of the 9-cis-RA binding pocket of RXRα (PDB code 1FBY). RXR residues involved in 9-cis-RA binding are colored in yellow and 9-cis-RA is shown in orange. b Superposition of RXRα and RXRβ LBPs in complex with 9-cis-RA (orange, PDB code 1FBY) and methoprene acid (violet, PDB code 1UHL), respectively. Residues involved in 9-cis-RA and methoprene acid binding are colored in yellow and pink, respectively. c Superposition of RXRα LBPs in complex with 9-cis-RA (orange, PDB code 1FBY) and TPT (blue, PDB code 3KWY). Residues involved in 9-cis-RA and TPT binding are colored in yellow and blue, respectively
Concluding comments
Deregulation of NHR-mediated transcription accounts for the deleterious effects of many EDCs. Thus, characterization of the harmful interaction between receptors and environmental compounds, both at the structural and functional levels, as well as the development of robust in vivo, in vitro, and in silico screening methods, are important for assessment of the toxic potential of large numbers of chemicals [5, 130–132]. In this context, computer-aided technologies which allow activity prediction of endocrine disruptors and environmental risk assessment have been developed. Computational tools very often include the use of quantitative structure–activity relationship (QSAR) methods in which the chemical structure of a compound is quantitatively correlated with its biological activity through a number of molecular descriptors, including the molecular weight and parameters, to account for hydrophobicity, topology, or electronic properties [133–137]. The growing number of crystal structures of NHR LBDs in the presence of various ligands greatly facilitates the understanding of ligand binding and receptor modulation. This structural information can also be used with modeling and docking tools to predict the interaction of EDCs with NHRs [138–142]. However, several levels of structural complexity limit the current application of these computational approaches.
The examples reviewed in the present article indicate that EDCs can be roughly divided into three classes depending on their structural and chemical proximity with endogenous hormones. The first class contains all EDCs mimicking the natural hormones through conserved protein–ligand contacts. Representative examples for ERs and RXRs are DES, GEN, and MA (Fig. 1) which display most of the structural and chemical requirements for interaction with their respective receptors in a hormone-like fashion. Other EDCs such as α-zearalenol (Fig. 1) most probably fall into this category [143]. The second class contains compounds displaying only a fraction of the anchoring chemical groups exhibited by the endogenous hormone as exemplified by PhIP and its metabolite 4-OH-PhIP [144]. Lastly, the third class corresponds to chemicals which display molecular properties distinct from the endogenous ligands but that still bind to NHRs by employing radically different binding mechanisms. The best experimentally validated example of this concept is the case of TBT and TPT. Although they do not possess the classical features of rexinoids, namely a carboxylic head and a long aliphatic tail, these organotin compounds bind RXRs with high affinity and are able to efficiently activate these receptors through the establishment of a covalent link with a cysteine residue of the LBP. Similarly, due to a chemical structure unrelated to that of E2, the pesticide chlordecone (Fig. 1) should exert its estrogenic activity through ER via a non-estrogen-like binding mechanism.
Computational methods can be effective in recognizing the putative endocrine activity of compounds of the two-first classes of EDCs. As an example, ERRγ can be bound by a high variety of phenols [42, 69], and modeling and docking tools can provide valuable help in selecting ERRγ binding compounds from this large family of environmental contaminants [145, 146]. In contrast, prediction of the endocrine disruptive activity of the last category of compounds will likely be much less straightforward.
Another difficult case is PXR which is activated by a variety of structurally distinct endogenous and exogenous compounds. PXR ligands can be categorized into potency groups, weak, moderate, and strong [83, 84, 87, 91, 99], and modeling and docking tools could be used to predict the affinity of new chemicals for PXR. However, due to the structural flexibility of its LBP, predicting the responses of PXR to environmental chemicals remains a difficult task. One of the major challenges of computational methods could be the prediction of all the potential target receptors for a given chemical compound. Nevertheless, the example provided by the structures of E2 bound to PXR and ER revealing that the two receptors bind the same hormone in remarkably distinct manners suggests that one cannot easily take advantage of the information provided by the structure of a given receptor/ligand complex to predict the binding mode of the same ligand to another receptor. Finally, compounds that target so-called orphan receptors might also be difficult to identify owing to the significant and hardly predictable conformational changes generally associated with ligand binding.
To date, only a few EDC-bound NHRs have been crystallized as compared with the 140,000 synthetic chemicals used in consumer products. Thus, it appears that efforts in elucidating the mechanisms of NHR/EDC interactions by crystallography and other structural methods must be pursued in order to deal with difficult cases and to increase our knowledge of the structural mechanisms and molecular interactions used by different receptors and a wide range of structurally and chemically diverse compounds. As exemplified by organotin compounds, such studies can also reveal unforeseen binding modes and provide guidelines for the rational design of novel NHR modulators. Together with the improvement of computational methods, this mounting structural data should increase the effectiveness of in silico screening strategies. As the European Union’s 2006 Registration, Evaluation, Authorization, and Restriction of Chemicals (REACH) regulation aims to assess the toxicity of more than 100,000 synthetic chemicals, there is a strong demand for such computational tools which would allow reduction of the cost of the evaluation as well as animal lives [147].
Acknowledgments
This work was supported by funds from the INSERM, CNRS, Université Montpellier 1 & 2, the French National Research Agency (ANR-07-PCVI-0001-01 to W.B.), the Agence Française de Sécurité Sanitaire de l’Environnement et du Travail (AFSSET, RD-2005-007 to P.B.) and the European Union Commission (CASCADE FOOD-CT-2004-506319 to P.B.). We thank Catherine Teyssier, Pierre Germain, and Catherine A Royer for critical reading of the manuscript.
Contributor Information
William Bourguet, Phone: +33-467-417702, FAX: +33-467-417913, Email: bourguet@cbs.cnrs.fr.
Patrick Balaguer, Phone: +33-467-612409, FAX: +33-467-613787, Email: patrick.balaguer@valdorel.fnclcc.fr.
References
- 1.Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, Hauser R, Prins GS, Soto AM, Zoeller RT, Gore AC. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev. 2009;30:293–342. doi: 10.1210/er.2009-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hotchkiss AK, Rider CV, Blystone CR, Wilson VS, Hartig PC, Ankley GT, Foster PM, Gray CL, Gray LE. Fifteen years after “Wingspread”: environmental endocrine disrupters and human and wildlife health: where we are today and where we need to go. Toxicol Sci. 2008;105:235–259. doi: 10.1093/toxsci/kfn030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swedenborg E, Ruegg J, Makela S, Pongratz I. Endocrine disruptive chemicals: mechanisms of action and involvement in metabolic disorders. J Mol Endocrinol. 2009;43:1–10. doi: 10.1677/JME-08-0132. [DOI] [PubMed] [Google Scholar]
- 4.Tabb MM, Blumberg B. New modes of action for endocrine-disrupting chemicals. Mol Endocrinol. 2006;20:475–482. doi: 10.1210/me.2004-0513. [DOI] [PubMed] [Google Scholar]
- 5.Janosek J, Hilscherova K, Blaha L, Holoubek I. Environmental xenobiotics and nuclear receptors: interactions, effects and in vitro assessment. Toxicol In Vitro. 2006;20:18–37. doi: 10.1016/j.tiv.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Toppari J. Environmental endocrine disrupters. Sex Dev. 2008;2:260–267. doi: 10.1159/000152042. [DOI] [PubMed] [Google Scholar]
- 7.Herbst AL, Ulfelder H, Poskanzer DC. Adenocarcinoma of the vagina: association of maternal stilbestrol therapy with tumor appearance in young women. N Engl J Med. 1971;284:878–881. doi: 10.1056/NEJM197104222841604. [DOI] [PubMed] [Google Scholar]
- 8.Gronemeyer H, Gustafsson JA, Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov. 2004;3:950–964. doi: 10.1038/nrd1551. [DOI] [PubMed] [Google Scholar]
- 9.Germain P, Staels B, Dacquet C, Spedding M, Laudet V. Overview of nomenclature of nuclear receptors. Pharmacol Rev. 2006;58:685–704. doi: 10.1124/pr.58.4.2. [DOI] [PubMed] [Google Scholar]
- 10.Bourguet W, Germain P, Gronemeyer H. Nuclear receptor ligand-binding domains: three-dimensional structures, molecular interactions and pharmacological implications. Trends Pharmacol Sci. 2000;21:381–388. doi: 10.1016/s0165-6147(00)01548-0. [DOI] [PubMed] [Google Scholar]
- 11.Pike AC. Lessons learnt from structural studies of the oestrogen receptor. Best Pract Res Clin Endocrinol Metab. 2006;20:1–14. doi: 10.1016/j.beem.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 12.Renaud JP, Moras D. Structural studies on nuclear receptors. Cell Mol Life Sci. 2000;57:1748–1769. doi: 10.1007/PL00000656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Couse JF, Korach KS. Estrogen receptor null mice: what have we learned and where will they lead us? Endocr Rev. 1999;20:358–417. doi: 10.1210/edrv.20.3.0370. [DOI] [PubMed] [Google Scholar]
- 14.Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology. 1997;138:863–870. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- 15.Curtis SW, Washburn T, Sewall C, DiAugustine R, Lindzey J, Couse JF, Korach KS. Physiological coupling of growth factor and steroid receptor signaling pathways: estrogen receptor knockout mice lack estrogen-like response to epidermal growth factor. Proc Natl Acad Sci USA. 1996;93:12626–12630. doi: 10.1073/pnas.93.22.12626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu MM, Albanese C, Anderson CM, Hilty K, Webb P, Uht RM, Price RH, Jr, Pestell RG, Kushner PJ. Opposing action of estrogen receptors alpha and beta on cyclin D1 gene expression. J Biol Chem. 2002;277:24353–24360. doi: 10.1074/jbc.M201829200. [DOI] [PubMed] [Google Scholar]
- 17.Pettersson K, Delaunay F, Gustafsson JA. Estrogen receptor beta acts as a dominant regulator of estrogen signaling. Oncogene. 2000;19:4970–4978. doi: 10.1038/sj.onc.1203828. [DOI] [PubMed] [Google Scholar]
- 18.Weihua Z, Saji S, Makinen S, Cheng G, Jensen EV, Warner M, Gustafsson JA. Estrogen receptor (ER) beta, a modulator of ERalpha in the uterus. Proc Natl Acad Sci USA. 2000;97:5936–5941. doi: 10.1073/pnas.97.11.5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindberg MK, Moverare S, Skrtic S, Gao H, Dahlman-Wright K, Gustafsson JA, Ohlsson C. Estrogen receptor (ER)-beta reduces ERalpha-regulated gene transcription, supporting a “ying yang” relationship between ERalpha and ERbeta in mice. Mol Endocrinol. 2003;17:203–208. doi: 10.1210/me.2002-0206. [DOI] [PubMed] [Google Scholar]
- 20.Ogawa S, Eng V, Taylor J, Lubahn DB, Korach KS, Pfaff DW. Roles of estrogen receptor-alpha gene expression in reproduction-related behaviors in female mice. Endocrinology. 1998;139:5070–5081. doi: 10.1210/endo.139.12.6357. [DOI] [PubMed] [Google Scholar]
- 21.Nahoum V, Bourguet W. Androgen and estrogen receptors: potential of crystallography in the fight against cancer. Int J Biochem Cell Biol. 2007;39:1280–1287. doi: 10.1016/j.biocel.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der Burg B, Gustafsson JA. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology. 1998;139:4252–4263. doi: 10.1210/endo.139.10.6216. [DOI] [PubMed] [Google Scholar]
- 23.Veeneman GH. Non-steroidal subtype selective estrogens. Curr Med Chem. 2005;12:1077–1136. doi: 10.2174/0929867053764662. [DOI] [PubMed] [Google Scholar]
- 24.Kaufman RH, Korhonen MO, Strama T, Adam E, Kaplan A. Development of clear cell adenocarcinoma in DES-exposed offspring under observation. Obstet Gynecol. 1982;59:68S–72S. [PubMed] [Google Scholar]
- 25.Herbst AL. The current status of the DES-exposed population. Obstet Gynecol Annu. 1981;10:267–278. [PubMed] [Google Scholar]
- 26.Newbold R. Cellular and molecular effects of developmental exposure to diethylstilbestrol: implications for other environmental estrogens. Environ Health Perspect. 1995;103(Suppl 7):7–83. doi: 10.1289/ehp.95103s783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cargouet M, Perdiz D, Mouatassim-Souali A, Tamisier-Karolak S, Levi Y. Assessment of river contamination by estrogenic compounds in Paris area (France) Sci Total Environ. 2004;324:55–66. doi: 10.1016/j.scitotenv.2003.10.035. [DOI] [PubMed] [Google Scholar]
- 28.Ribeiro C, Tiritan ME, Rocha E, Rocha MJ. Seasonal and spatial distribution of several endocrine-disrupting compounds in the Douro River Estuary, Portugal. Arch Environ Contam Toxicol. 2009;56:1–11. doi: 10.1007/s00244-008-9158-x. [DOI] [PubMed] [Google Scholar]
- 29.Ying GG, Kookana RS, Kumar A, Mortimer M. Occurrence and implications of estrogens and xenoestrogens in sewage effluents and receiving waters from South East Queensland. Sci Total Environ. 2009;407:5147–5155. doi: 10.1016/j.scitotenv.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 30.Takemura H, Shim JY, Sayama K, Tsubura A, Zhu BT, Shimoi K. Characterization of the estrogenic activities of zearalenone and zeranol in vivo and in vitro. J Steroid Biochem Mol Biol. 2007;103:170–177. doi: 10.1016/j.jsbmb.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 31.Trenholm HL, Warner RM, Fitzpatrick DW. Rapid, sensitive liquid chromatographic method for determination of zearalenone and alpha- and beta-zearalenol in wheat. J Assoc Off Anal Chem. 1984;67:968–972. [PubMed] [Google Scholar]
- 32.Veldman A, Borggreve GJ, Mulders EJ, van de Lagemaat D. Occurrence of the mycotoxins ochratoxin A, zearalenone and deoxynivalenol in feed components. Food Addit Contam. 1992;9:647–655. doi: 10.1080/02652039209374120. [DOI] [PubMed] [Google Scholar]
- 33.Kiessling KH, Pettersson H. Metabolism of zearalenone in rat liver. Acta Pharmacol Toxicol (Copenh) 1978;43:285–290. doi: 10.1111/j.1600-0773.1978.tb02267.x. [DOI] [PubMed] [Google Scholar]
- 34.Kuiper-Goodman T, Scott PM, Watanabe H. Risk assessment of the mycotoxin zearalenone. Regul Toxicol Pharmacol. 1987;7:253–306. doi: 10.1016/0273-2300(87)90037-7. [DOI] [PubMed] [Google Scholar]
- 35.Messina M, Kucuk O, Lampe JW. An overview of the health effects of isoflavones with an emphasis on prostate cancer risk and prostate-specific antigen levels. J AOAC Int. 2006;89:1121–1134. [PubMed] [Google Scholar]
- 36.Escande A, Pillon A, Servant N, Cravedi JP, Larrea F, Muhn P, Nicolas JC, Cavailles V, Balaguer P. Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta. Biochem Pharmacol. 2006;71:1459–1469. doi: 10.1016/j.bcp.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 37.Lauber SN, Ali S, Gooderham NJ. The cooked food derived carcinogen 2-amino-1-methyl-6-phenylimidazo[4, 5-b] pyridine is a potent oestrogen: a mechanistic basis for its tissue-specific carcinogenicity. Carcinogenesis. 2004;25:2509–2517. doi: 10.1093/carcin/bgh268. [DOI] [PubMed] [Google Scholar]
- 38.Nettles KW, Bruning JB, Gil G, Nowak J, Sharma SK, Hahm JB, Kulp K, Hochberg RB, Zhou H, Katzenellenbogen JA, Katzenellenbogen BS, Kim Y, Joachmiak A, Greene GL. NFkappaB selectivity of estrogen receptor ligands revealed by comparative crystallographic analyses. Nat Chem Biol. 2008;4:241–247. doi: 10.1038/nchembio.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lemaire G, Mnif W, Mauvais P, Balaguer P, Rahmani R. Activation of alpha- and beta-estrogen receptors by persistent pesticides in reporter cell lines. Life Sci. 2006;79:1160–1169. doi: 10.1016/j.lfs.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 40.Paris F, Balaguer P, Terouanne B, Servant N, Lacoste C, Cravedi JP, Nicolas JC, Sultan C. Phenylphenols, biphenols, bisphenol-A and 4-tert-octylphenol exhibit alpha and beta estrogen activities and antiandrogen activity in reporter cell lines. Mol Cell Endocrinol. 2002;193:43–49. doi: 10.1016/s0303-7207(02)00094-1. [DOI] [PubMed] [Google Scholar]
- 41.Bouskine A, Nebout M, Brucker-Davis F, Benahmed M, Fenichel P. Low doses of bisphenol A promote human seminoma cell proliferation by activating PKA and PKG via a membrane G-protein-coupled estrogen receptor. Environ Health Perspect. 2009;117:1053–1058. doi: 10.1289/ehp.0800367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okada H, Tokunaga T, Liu X, Takayanagi S, Matsushima A, Shimohigashi Y. Direct evidence revealing structural elements essential for the high binding ability of bisphenol A to human estrogen-related receptor-gamma. Environ Health Perspect. 2008;116:32–38. doi: 10.1289/ehp.10587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ying GG, Williams B, Kookana R. Environmental fate of alkylphenols and alkylphenol ethoxylates: a review. Environ Int. 2002;28:215–226. doi: 10.1016/s0160-4120(02)00017-x. [DOI] [PubMed] [Google Scholar]
- 44.Kunz PY, Galicia HF, Fent K. Comparison of in vitro and in vivo estrogenic activity of UV filters in fish. Toxicol Sci. 2006;90:349–361. doi: 10.1093/toxsci/kfj082. [DOI] [PubMed] [Google Scholar]
- 45.Molina-Molina JM, Escande A, Pillon A, Gomez E, Pakdel F, Cavailles V, Olea N, Ait-Aissa S, Balaguer P. Profiling of benzophenone derivatives using fish and human estrogen receptor-specific in vitro bioassays. Toxicol Appl Pharmacol. 2008;232:384–395. doi: 10.1016/j.taap.2008.07.017. [DOI] [PubMed] [Google Scholar]
- 46.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 47.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 48.Barkhem T, Carlsson B, Nilsson Y, Enmark E, Gustafsson J, Nilsson S. Differential response of estrogen receptor alpha and estrogen receptor beta to partial estrogen agonists/antagonists. Mol Pharmacol. 1998;54:105–112. doi: 10.1124/mol.54.1.105. [DOI] [PubMed] [Google Scholar]
- 49.Manas ES, Xu ZB, Unwalla RJ, Somers WS. Understanding the selectivity of genistein for human estrogen receptor-beta using X-ray crystallography and computational methods. Structure. 2004;12:2197–2207. doi: 10.1016/j.str.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 50.Pike AC, Brzozowski AM, Hubbard RE, Bonn T, Thorsell AG, Engstrom O, Ljunggren J, Gustafsson JA, Carlquist M. Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. EMBO J. 1999;18:4608–4618. doi: 10.1093/emboj/18.17.4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vivat V, Gofflo D, Garcia T, Wurtz JM, Bourguet W, Philibert D, Gronemeyer H. Sequences in the ligand-binding domains of the human androgen and progesterone receptors which determine their distinct ligand identities. J Mol Endocrinol. 1997;18:147–160. doi: 10.1677/jme.0.0180147. [DOI] [PubMed] [Google Scholar]
- 52.Nettles KW, Sun J, Radek JT, Sheng S, Rodriguez AL, Katzenellenbogen JA, Katzenellenbogen BS, Greene GL. Allosteric control of ligand selectivity between estrogen receptors alpha and beta: implications for other nuclear receptors. Mol Cell. 2004;13:317–327. doi: 10.1016/s1097-2765(04)00054-1. [DOI] [PubMed] [Google Scholar]
- 53.Nahoum V, Perez E, Germain P, Rodriguez-Barrios F, Manzo F, Kammerer S, Lemaire G, Hirsch O, Royer CA, Gronemeyer H, de Lera AR, Bourguet W. Modulators of the structural dynamics of the retinoid X receptor to reveal receptor function. Proc Natl Acad Sci USA. 2007;104:17323–17328. doi: 10.1073/pnas.0705356104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith CL, O’Malley BW. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr Rev. 2004;25:45–71. doi: 10.1210/er.2003-0023. [DOI] [PubMed] [Google Scholar]
- 55.Giguere V. To ERR in the estrogen pathway. Trends Endocrinol Metab. 2002;13:220–225. doi: 10.1016/s1043-2760(02)00592-1. [DOI] [PubMed] [Google Scholar]
- 56.Bonnelye E, Vanacker JM, Dittmar T, Begue A, Desbiens X, Denhardt DT, Aubin JE, Laudet V, Fournier B. The ERR-1 orphan receptor is a transcriptional activator expressed during bone development. Mol Endocrinol. 1997;11:905–916. doi: 10.1210/mend.11.7.9948. [DOI] [PubMed] [Google Scholar]
- 57.Horard B, Vanacker JM. Estrogen receptor-related receptors: orphan receptors desperately seeking a ligand. J Mol Endocrinol. 2003;31:349–357. doi: 10.1677/jme.0.0310349. [DOI] [PubMed] [Google Scholar]
- 58.Vanacker JM, Pettersson K, Gustafsson JA, Laudet V. Transcriptional targets shared by estrogen receptor- related receptors (ERRs) and estrogen receptor (ER) alpha, but not by ERbeta. EMBO J. 1999;18:4270–4279. doi: 10.1093/emboj/18.15.4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lu D, Kiriyama Y, Lee KY, Giguere V. Transcriptional regulation of the estrogen-inducible pS2 breast cancer marker gene by the ERR family of orphan nuclear receptors. Cancer Res. 2001;61:6755–6761. [PubMed] [Google Scholar]
- 60.Yu S, Wang X, Ng CF, Chen S, Chan FL. ERRgamma suppresses cell proliferation and tumor growth of androgen-sensitive and androgen-insensitive prostate cancer cells and its implication as a therapeutic target for prostate cancer. Cancer Res. 2007;67:4904–4914. doi: 10.1158/0008-5472.CAN-06-3855. [DOI] [PubMed] [Google Scholar]
- 61.Ariazi EA, Jordan VC. Estrogen-related receptors as emerging targets in cancer and metabolic disorders. Curr Top Med Chem. 2006;6:203–215. doi: 10.2174/1568026610606030203. [DOI] [PubMed] [Google Scholar]
- 62.Giguere V. Transcriptional control of energy homeostasis by the estrogen-related receptors. Endocr Rev. 2008;29:677–696. doi: 10.1210/er.2008-0017. [DOI] [PubMed] [Google Scholar]
- 63.Baillie-Hamilton PF. Chemical toxins: a hypothesis to explain the global obesity epidemic. J Altern Complement Med. 2002;8:185–192. doi: 10.1089/107555302317371479. [DOI] [PubMed] [Google Scholar]
- 64.Li J, Ma M, Wang Z (2009). In vitro profiling of endocrine disrupting effects of phenols. Toxicol In Vitro (in press) [DOI] [PubMed]
- 65.Greschik H, Wurtz JM, Sanglier S, Bourguet W, van Dorsselaer A, Moras D, Renaud JP. Structural and functional evidence for ligand-independent transcriptional activation by the estrogen-related receptor 3. Mol Cell. 2002;9:303–313. doi: 10.1016/s1097-2765(02)00444-6. [DOI] [PubMed] [Google Scholar]
- 66.Yang C, Chen S. Two organochlorine pesticides, toxaphene and chlordane, are antagonists for estrogen-related receptor alpha-1 orphan receptor. Cancer Res. 1999;59:4519–4524. [PubMed] [Google Scholar]
- 67.Coward P, Lee D, Hull MV, Lehmann JM. 4-Hydroxytamoxifen binds to and deactivates the estrogen-related receptor gamma. Proc Natl Acad Sci USA. 2001;98:8880–8884. doi: 10.1073/pnas.151244398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tremblay GB, Kunath T, Bergeron D, Lapointe L, Champigny C, Bader JA, Rossant J, Giguere V. Diethylstilbestrol regulates trophoblast stem cell differentiation as a ligand of orphan nuclear receptor ERR beta. Genes Dev. 2001;15:833–838. doi: 10.1101/gad.873401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang J, Fang F, Huang Z, Wang Y, Wong C. Kaempferol is an estrogen-related receptor alpha and gamma inverse agonist. FEBS Lett. 2009;583:643–647. doi: 10.1016/j.febslet.2009.01.030. [DOI] [PubMed] [Google Scholar]
- 70.Suetsugi M, Su L, Karlsberg K, Yuan YC, Chen S. Flavone and isoflavone phytoestrogens are agonists of estrogen-related receptors. Mol Cancer Res. 2003;1:981–991. [PubMed] [Google Scholar]
- 71.Wang L, Zuercher WJ, Consler TG, Lambert MH, Miller AB, Orband-Miller LA, McKee DD, Willson TM, Nolte RT. X-ray crystal structures of the estrogen-related receptor-gamma ligand binding domain in three functional states reveal the molecular basis of small molecule regulation. J Biol Chem. 2006;281:37773–37781. doi: 10.1074/jbc.M608410200. [DOI] [PubMed] [Google Scholar]
- 72.Abad MC, Askari H, O’Neill J, Klinger AL, Milligan C, Lewandowski F, Springer B, Spurlino J, Rentzeperis D. Structural determination of estrogen-related receptor gamma in the presence of phenol derivative compounds. J Steroid Biochem Mol Biol. 2008;108:44–54. doi: 10.1016/j.jsbmb.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 73.Matsushima A, Kakuta Y, Teramoto T, Koshiba T, Liu X, Okada H, Tokunaga T, Kawabata S, Kimura M, Shimohigashi Y. Structural evidence for endocrine disruptor bisphenol A binding to human nuclear receptor ERR gamma. J Biochem. 2007;142:517–524. doi: 10.1093/jb/mvm158. [DOI] [PubMed] [Google Scholar]
- 74.Matsushima A, Teramoto T, Okada H, Liu X, Tokunaga T, Kakuta Y, Shimohigashi Y. ERRgamma tethers strongly bisphenol A and 4-alpha-cumylphenol in an induced-fit manner. Biochem Biophys Res Commun. 2008;373:408–413. doi: 10.1016/j.bbrc.2008.06.050. [DOI] [PubMed] [Google Scholar]
- 75.Kallen J, Schlaeppi JM, Bitsch F, Filipuzzi I, Schilb A, Riou V, Graham A, Strauss A, Geiser M, Fournier B. Evidence for ligand-independent transcriptional activation of the human estrogen-related receptor alpha (ERRalpha): crystal structure of ERRalpha ligand binding domain in complex with peroxisome proliferator-activated receptor coactivator-1alpha. J Biol Chem. 2004;279:49330–49337. doi: 10.1074/jbc.M407999200. [DOI] [PubMed] [Google Scholar]
- 76.Greschik H, Flaig R, Renaud JP, Moras D. Structural basis for the deactivation of the estrogen-related receptor gamma by diethylstilbestrol or 4-hydroxytamoxifen and determinants of selectivity. J Biol Chem. 2004;279:33639–33646. doi: 10.1074/jbc.M402195200. [DOI] [PubMed] [Google Scholar]
- 77.Orans J, Teotico DG, Redinbo MR. The nuclear xenobiotic receptor pregnane X receptor: recent insights and new challenges. Mol Endocrinol. 2005;19:2891–2900. doi: 10.1210/me.2005-0156. [DOI] [PubMed] [Google Scholar]
- 78.Biswas A, Mani S, Redinbo MR, Krasowski MD, Li H, Ekins S. Elucidating the ‘Jekyll and Hyde’ nature of PXR: the case for discovering antagonists or allosteric antagonists. Pharm Res. 2009;26:1807–1815. doi: 10.1007/s11095-009-9901-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Moreau A, Vilarem MJ, Maurel P, Pascussi JM. Xenoreceptors CAR and PXR activation and consequences on lipid metabolism, glucose homeostasis, and inflammatory response. Mol Pharm. 2008;5:35–41. doi: 10.1021/mp700103m. [DOI] [PubMed] [Google Scholar]
- 80.Mikamo E, Harada S, Nishikawa J, Nishihara T. Endocrine disruptors induce cytochrome P450 by affecting transcriptional regulation via pregnane X receptor. Toxicol Appl Pharmacol. 2003;193:66–72. doi: 10.1016/j.taap.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 81.Watkins RE, Davis-Searles PR, Lambert MH, Redinbo MR. Coactivator binding promotes the specific interaction between ligand and the pregnane X receptor. J Mol Biol. 2003;331:815–828. doi: 10.1016/s0022-2836(03)00795-2. [DOI] [PubMed] [Google Scholar]
- 82.Masuyama H, Suwaki N, Tateishi Y, Nakatsukasa H, Segawa T, Hiramatsu Y. The pregnane X receptor regulates gene expression in a ligand- and promoter-selective fashion. Mol Endocrinol. 2005;19:1170–1180. doi: 10.1210/me.2004-0434. [DOI] [PubMed] [Google Scholar]
- 83.Benod C, Subra G, Nahoum V, Mallavialle A, Guichou JF, Milhau J, Roblés S, Bourguet W, Pascussi JM, Balaguer P, Chavanieu A. N-1H-Benzimidazol-5-ylbenzenesulfonamide derivatives as potent hPXR agonists. Bioorg Med Chem. 2008;16:3537–3549. doi: 10.1016/j.bmc.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 84.Lemaire G, Benod C, Nahoum V, Pillon A, Boussioux AM, Guichou JF, Subra G, Pascussi JM, Bourguet W, Chavanieu A, Balaguer P. Discovery of a highly active ligand of human pregnane x receptor: a case study from pharmacophore modeling and virtual screening to “in vivo” biological activity. Mol Pharmacol. 2007;72:572–581. doi: 10.1124/mol.106.033415. [DOI] [PubMed] [Google Scholar]
- 85.Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, Willson TM, Kliewer SA, Redinbo MR. The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science. 2001;292:2329–2333. doi: 10.1126/science.1060762. [DOI] [PubMed] [Google Scholar]
- 86.Xue Y, Chao E, Zuercher WJ, Willson TM, Collins JL, Redinbo MR. Crystal structure of the PXR-T1317 complex provides a scaffold to examine the potential for receptor antagonism. Bioorg Med Chem. 2007;15:2156–2166. doi: 10.1016/j.bmc.2006.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xue Y, Moore LB, Orans J, Peng L, Bencharit S, Kliewer SA, Redinbo MR. Crystal structure of the pregnane X receptor-estradiol complex provides insights into endobiotic recognition. Mol Endocrinol. 2007;21:1028–1038. doi: 10.1210/me.2006-0323. [DOI] [PubMed] [Google Scholar]
- 88.Bertilsson G, Berkenstam A, Blomquist P. Functionally conserved xenobiotic responsive enhancer in cytochrome P450 3A7. Biochem Biophys Res Commun. 2001;280:139–144. doi: 10.1006/bbrc.2000.4066. [DOI] [PubMed] [Google Scholar]
- 89.Blumberg B, Sabbagh W, Jr, Juguilon H, Bolado J, Jr, van Meter CM, Ong ES, Evans RM. SXR, a novel steroid and xenobiotic-sensing nuclear receptor. Genes Dev. 1998;12:3195–3205. doi: 10.1101/gad.12.20.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest. 1998;102:1016–1023. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lemaire G, Mnif W, Pascussi JM, Pillon A, Rabenoelina F, Fenet H, Gomez E, Casellas C, Nicolas JC, Cavailles V, Duchesne MJ, Balaguer P. Identification of new human pregnane X receptor ligands among pesticides using a stable reporter cell system. Toxicol Sci. 2006;91:501–509. doi: 10.1093/toxsci/kfj173. [DOI] [PubMed] [Google Scholar]
- 92.Jones SA, Moore LB, Shenk JL, Wisely GB, Hamilton GA, McKee DD, Tomkinson NC, LeCluyse EL, Lambert MH, Willson TM, Kliewer SA, Moore JT. The pregnane X receptor: a promiscuous xenobiotic receptor that has diverged during evolution. Mol Endocrinol. 2000;14:27–39. doi: 10.1210/mend.14.1.0409. [DOI] [PubMed] [Google Scholar]
- 93.Synold TW, Dussault I, Forman BM. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat Med. 2001;7:584–590. doi: 10.1038/87912. [DOI] [PubMed] [Google Scholar]
- 94.Watkins RE, Maglich JM, Moore LB, Wisely GB, Noble SM, Davis-Searles PR, Lambert MH, Kliewer SA, Redinbo MR. 2.1 Å crystal structure of human PXR in complex with the St. John’s wort compound hyperforin. Biochemistry. 2003;42:1430–1438. doi: 10.1021/bi0268753. [DOI] [PubMed] [Google Scholar]
- 95.Moore JT, Kliewer SA. Use of the nuclear receptor PXR to predict drug interactions. Toxicology. 2000;153:1–10. doi: 10.1016/s0300-483x(00)00300-0. [DOI] [PubMed] [Google Scholar]
- 96.Wentworth JM, Agostini M, Love J, Schwabe JW, Chatterjee VK. St John’s wort, a herbal antidepressant, activates the steroid X receptor. J Endocrinol. 2000;166:R11–R16. doi: 10.1677/joe.0.166r011. [DOI] [PubMed] [Google Scholar]
- 97.di Masi A, Marinis ED, Ascenzi P, Marino M (2009). Nuclear receptors CAR and PXR: molecular, functional, and biomedical aspects. Mol Aspects Med (in press) [DOI] [PubMed]
- 98.Jacobs MN, Nolan GT, Hood SR. Lignans, bacteriocides and organochlorine compounds activate the human pregnane X receptor (PXR) Toxicol Appl Pharmacol. 2005;209:123–133. doi: 10.1016/j.taap.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 99.Mnif W, Pascussi JM, Pillon A, Escande A, Bartegi A, Nicolas JC, Cavailles V, Duchesne MJ, Balaguer P. Estrogens and antiestrogens activate hPXR. Toxicol Lett. 2007;170:19–29. doi: 10.1016/j.toxlet.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 100.Kinani S, Bouchonnet S, Creusot N, Bourcier S, Balaguer P, Porcher JM, Ait-Aissa S. Multiple endocrine activities in sediments from small rivers in North of France assessed by the combined use of in vitro bioassays and chemical analysis. Environ Pollut. 2010;158:74–83. doi: 10.1016/j.envpol.2009.07.041. [DOI] [PubMed] [Google Scholar]
- 101.Tabb MM, Kholodovych V, Grun F, Zhou C, Welsh WJ, Blumberg B. Highly chlorinated PCBs inhibit the human xenobiotic response mediated by the steroid and xenobiotic receptor (SXR) Environ Health Perspect. 2004;112:163–169. doi: 10.1289/ehp.6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pacyniak EK, Cheng X, Cunningham ML, Crofton K, Klaassen CD, Guo GL. The flame retardants, polybrominated diphenyl ethers, are pregnane X receptor activators. Toxicol Sci. 2007;97:94–102. doi: 10.1093/toxsci/kfm025. [DOI] [PubMed] [Google Scholar]
- 103.Chrencik JE, Orans J, Moore LB, Xue Y, Peng L, Collins JL, Wisely GB, Lambert MH, Kliewer SA, Redinbo MR. Structural disorder in the complex of human pregnane X receptor and the macrolide antibiotic rifampicin. Mol Endocrinol. 2005;19:1125–1134. doi: 10.1210/me.2004-0346. [DOI] [PubMed] [Google Scholar]
- 104.Teotico DG, Bischof JJ, Peng L, Kliewer SA, Redinbo MR. Structural basis of human pregnane X receptor activation by the hops constituent colupulone. Mol Pharmacol. 2008;74:1512–1520. doi: 10.1124/mol.108.050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kretschmer XC, Baldwin WS. CAR and PXR: xenosensors of endocrine disrupters? Chem Biol Interact. 2005;155:111–128. doi: 10.1016/j.cbi.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 106.di Masi A, Marinis ED, Ascenzi P, Marino M. Nuclear receptors CAR and PXR: molecular, functional, and biomedical aspects. Mol Aspects Med. 2009;30:297–343. doi: 10.1016/j.mam.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 107.Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 108.Germain P, Iyer J, Zechel C, Gronemeyer H. Co-regulator recruitment and the mechanism of retinoic acid receptor synergy. Nature. 2002;415:187–192. doi: 10.1038/415187a. [DOI] [PubMed] [Google Scholar]
- 109.Chen JY, Clifford J, Zusi C, Starrett J, Tortolani D, Ostrowski J, Reczek PR, Chambon P, Gronemeyer H. Two distinct actions of retinoid-receptor ligands. Nature. 1996;382:819–822. doi: 10.1038/382819a0. [DOI] [PubMed] [Google Scholar]
- 110.Botling J, Castro DS, Oberg F, Nilsson K, Perlmann T. Retinoic acid receptor/retinoid X receptor heterodimers can be activated through both subunits providing a basis for synergistic transactivation and cellular differentiation. J Biol Chem. 1997;272:9443–9449. doi: 10.1074/jbc.272.14.9443. [DOI] [PubMed] [Google Scholar]
- 111.Heyman RA, Mangelsdorf DJ, Dyck JA, Stein RB, Eichele G, Evans RM, Thaller C. 9-cis retinoic acid is a high affinity ligand for the retinoid X receptor. Cell. 1992;68:397–406. doi: 10.1016/0092-8674(92)90479-v. [DOI] [PubMed] [Google Scholar]
- 112.de Urquiza AM, Liu S, Sjoberg M, Zetterstrom RH, Griffiths W, Sjovall J, Perlmann T. Docosahexaenoic acid, a ligand for the retinoid X receptor in mouse brain. Science. 2000;290:2140–2144. doi: 10.1126/science.290.5499.2140. [DOI] [PubMed] [Google Scholar]
- 113.de Lera AR, Bourguet W, Altucci L, Gronemeyer H. Design of selective nuclear receptor modulators: RAR and RXR as a case study. Nat Rev Drug Discov. 2007;6:811–820. doi: 10.1038/nrd2398. [DOI] [PubMed] [Google Scholar]
- 114.Nakanishi T. Endocrine disruption induced by organotin compounds; organotins function as a powerful agonist for nuclear receptors rather than an aromatase inhibitor. J Toxicol Sci. 2008;33:269–276. doi: 10.2131/jts.33.269. [DOI] [PubMed] [Google Scholar]
- 115.Grun F, Blumberg B. Environmental obesogens: organotins and endocrine disruption via nuclear receptor signaling. Endocrinology. 2006;147:S50–S55. doi: 10.1210/en.2005-1129. [DOI] [PubMed] [Google Scholar]
- 116.Grun F, Blumberg B. Endocrine disrupters as obesogens. Mol Cell Endocrinol. 2009;304:19–29. doi: 10.1016/j.mce.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Antizar-Ladislao B. Environmental levels, toxicity and human exposure to tributyltin (TBT)-contaminated marine environment: a review. Environ Int. 2008;34:292–308. doi: 10.1016/j.envint.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 118.Grun F, Watanabe H, Zamanian Z, Maeda L, Arima K, Cubacha R, Gardiner DM, Kanno J, Iguchi T, Blumberg B. Endocrine-disrupting organotin compounds are potent inducers of adipogenesis in vertebrates. Mol Endocrinol. 2006;20:2141–2155. doi: 10.1210/me.2005-0367. [DOI] [PubMed] [Google Scholar]
- 119.Kanayama T, Kobayashi N, Mamiya S, Nakanishi T, Nishikawa J. Organotin compounds promote adipocyte differentiation as agonists of the peroxisome proliferator-activated receptor gamma/retinoid X receptor pathway. Mol Pharmacol. 2005;67:766–774. doi: 10.1124/mol.104.008409. [DOI] [PubMed] [Google Scholar]
- 120.Castro LF, Lima D, Machado A, Melo C, Hiromori Y, Nishikawa J, Nakanishi T, Reis-Henriques MA, Santos MM. Imposex induction is mediated through the Retinoid X Receptor signalling pathway in the neogastropod Nucella lapillus. Aquat Toxicol. 2007;85:57–66. doi: 10.1016/j.aquatox.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 121.Nishikawa J, Mamiya S, Kanayama T, Nishikawa T, Shiraishi F, Horiguchi T. Involvement of the retinoid X receptor in the development of imposex caused by organotins in gastropods. Environ Sci Technol. 2004;38:6271–6276. doi: 10.1021/es049593u. [DOI] [PubMed] [Google Scholar]
- 122.le Maire A, Grimaldi M, Roecklin D, Dagnino S, Vivat-Hannah V, Balaguer P, Bourguet W. Activation of RXR-PPAR heterodimers by organotin environmental endocrine disruptors. EMBO Rep. 2009;10:367–373. doi: 10.1038/embor.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Harmon MA, Boehm MF, Heyman RA, Mangelsdorf DJ. Activation of mammalian retinoid X receptors by the insect growth regulator methoprene. Proc Natl Acad Sci USA. 1995;92:6157–6160. doi: 10.1073/pnas.92.13.6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li J, Ma M, Wang Z. A two-hybrid yeast assay to quantify the effects of xenobiotics on retinoid X receptor-mediated gene expression. Toxicol Lett. 2008;176:198–206. doi: 10.1016/j.toxlet.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 125.Svensson S, Ostberg T, Jacobsson M, Norstrom C, Stefansson K, Hallen D, Johansson IC, Zachrisson K, Ogg D, Jendeberg L. Crystal structure of the heterodimeric complex of LXRalpha and RXRbeta ligand-binding domains in a fully agonistic conformation. EMBO J. 2003;22:4625–4633. doi: 10.1093/emboj/cdg456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Egea PF, Mitschler A, Rochel N, Ruff M, Chambon P, Moras D. Crystal structure of the human RXRalpha ligand-binding domain bound to its natural ligand: 9-cis retinoic acid. EMBO J. 2000;19:2592–2601. doi: 10.1093/emboj/19.11.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pogenberg V, Guichou JF, Vivat-Hannah V, Kammerer S, Perez E, Germain P, de Lera AR, Gronemeyer H, Royer CA, Bourguet W. Characterization of the interaction between retinoic acid receptor/retinoid X receptor (RAR/RXR) heterodimers and transcriptional coactivators through structural and fluorescence anisotropy studies. J Biol Chem. 2005;280:1625–1633. doi: 10.1074/jbc.M409302200. [DOI] [PubMed] [Google Scholar]
- 128.Nettles KW, Bruning JB, Gil G, O’Neill EE, Nowak J, Guo Y, Kim Y, DeSombre ER, Dilis R, Hanson RN, Joachimiak A, Greene GL. Structural plasticity in the oestrogen receptor ligand-binding domain. EMBO Rep. 2007;8:563–568. doi: 10.1038/sj.embor.7400963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gumy C, Chandsawangbhuwana C, Dzyakanchuk AA, Kratschmar DV, Baker ME, Odermatt A. Dibutyltin disrupts glucocorticoid receptor function and impairs glucocorticoid-induced suppression of cytokine production. PLoS One. 2008;3:e3545. doi: 10.1371/journal.pone.0003545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Witorsch RJ. Endocrine disruptors: can biological effects and environmental risks be predicted? Regul Toxicol Pharmacol. 2002;36:118–130. doi: 10.1006/rtph.2002.1564. [DOI] [PubMed] [Google Scholar]
- 131.de Fur PL. Use and role of invertebrate models in endocrine disruptor research and testing. ILAR J. 2004;45:484–493. doi: 10.1093/ilar.45.4.484. [DOI] [PubMed] [Google Scholar]
- 132.Roncaglioni A, Benfenati E. In silico-aided prediction of biological properties of chemicals: oestrogen receptor-mediated effects. Chem Soc Rev. 2008;37:441–450. doi: 10.1039/b616276m. [DOI] [PubMed] [Google Scholar]
- 133.Waller CL, Juma BW, Gray LE, Jr, Kelce WR. Three-dimensional quantitative structure–activity relationships for androgen receptor ligands. Toxicol Appl Pharmacol. 1996;137:219–227. doi: 10.1006/taap.1996.0075. [DOI] [PubMed] [Google Scholar]
- 134.Jacobs MN. In silico tools to aid risk assessment of endocrine disrupting chemicals. Toxicology. 2004;205:43–53. doi: 10.1016/j.tox.2004.06.036. [DOI] [PubMed] [Google Scholar]
- 135.Devillers J, Marchand-Geneste N, Carpy A, Porcher JM. SAR and QSAR modeling of endocrine disruptors. SAR QSAR Environ Res. 2006;17:393–412. doi: 10.1080/10629360600884397. [DOI] [PubMed] [Google Scholar]
- 136.Fang H, Tong W, Branham WS, Moland CL, Dial SL, Hong H, Xie Q, Perkins R, Owens W, Sheehan DM. Study of 202 natural, synthetic, and environmental chemicals for binding to the androgen receptor. Chem Res Toxicol. 2003;16:1338–1358. doi: 10.1021/tx030011g. [DOI] [PubMed] [Google Scholar]
- 137.Ekins S, Chang C, Mani S, Krasowski MD, Reschly EJ, Iyer M, Kholodovych V, Ai N, Welsh WJ, Sinz M, Swaan PW, Patel R, Bachmann K. Human pregnane X receptor antagonists and agonists define molecular requirements for different binding sites. Mol Pharmacol. 2007;72:592–603. doi: 10.1124/mol.107.038398. [DOI] [PubMed] [Google Scholar]
- 138.D’Ursi P, Salvi E, Fossa P, Milanesi L, Rovida E. Modelling the interaction of steroid receptors with endocrine disrupting chemicals. BMC Bioinformatics. 2005;6(Suppl 4):S10. doi: 10.1186/1471-2105-6-S4-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Celik L, Davey J, Lund D, Schiott B. Exploring interactions of endocrine-disrupting compounds with different conformations of the human estrogen receptor alpha ligand binding domain: a molecular docking study. Chem Res Toxicol. 2008;21:2195–2206. doi: 10.1021/tx800278d. [DOI] [PubMed] [Google Scholar]
- 140.Marchand-Geneste N, Cazaunau M, Carpy AJ, Laguerre M, Porcher JM, Devillers J. Homology model of the rainbow trout estrogen receptor (rtERalpha) and docking of endocrine disrupting chemicals (EDCs) SAR QSAR Environ Res. 2006;17:93–105. doi: 10.1080/10659360600562137. [DOI] [PubMed] [Google Scholar]
- 141.Lill MA, Winiger F, Vedani A, Ernst B. Impact of induced fit on ligand binding to the androgen receptor: a multidimensional QSAR study to predict endocrine-disrupting effects of environmental chemicals. J Med Chem. 2005;48:5666–5674. doi: 10.1021/jm050403f. [DOI] [PubMed] [Google Scholar]
- 142.Sivanesan D, Rajnarayanan RV, Doherty J, Pattabiraman N. In-silico screening using flexible ligand binding pockets: a molecular dynamics-based approach. J Comput Aided Mol Des. 2005;19:213–228. doi: 10.1007/s10822-005-4788-9. [DOI] [PubMed] [Google Scholar]
- 143.Zavodszky MI, Lei M, Thorpe MF, Day AR, Kuhn LA. Modeling correlated main-chain motions in proteins for flexible molecular recognition. Proteins. 2004;57:243–261. doi: 10.1002/prot.20179. [DOI] [PubMed] [Google Scholar]
- 144.Bennion BJ, Cosman M, Lightstone FC, Knize MG, Montgomery JL, Bennett LM, Felton JS, Kulp KS. PhIP carcinogenicity in breast cancer: computational and experimental evidence for competitive interactions with human estrogen receptor. Chem Res Toxicol. 2005;18:1528–1536. doi: 10.1021/tx0501031. [DOI] [PubMed] [Google Scholar]
- 145.Kim Y, Koh M, Kim DK, Choi HS, Park SB. Efficient discovery of selective small molecule agonists of estrogen-related receptor gamma using combinatorial approach. J Comb Chem. 2009;11:928–937. doi: 10.1021/cc900081j. [DOI] [PubMed] [Google Scholar]
- 146.Nose T, Shimohigashi Y. A docking modelling rationally predicts strong binding of bisphenol A to estrogen-related receptor gamma. Protein Pept Lett. 2008;15:290–296. doi: 10.2174/092986608783744261. [DOI] [PubMed] [Google Scholar]
- 147.Hartung T, Rovida C. Chemical regulators have overreached. Nature. 2009;460:1080–1081. doi: 10.1038/4601080a. [DOI] [PubMed] [Google Scholar]