

Abstract

A new method for the synthesis of tricyclic nitrogen heterocycles from N,2-diallylaniline derivatives is described. These transformations proceed via sequential alkene aminopalladation of an intermediate LnPd(Ar)(NRR’) species followed by alkene carbopalladation of the resulting LnPd(Ar)(R) complex. Both alkene insertion steps occur in preference to C–N or C–C bond-forming reductive elimination. An unusual 1,3-palladium shift occurs when 2-Allyl-N-(2-vinylphenyl)aniline is employed as substrate, which yields a tetracyclic molecule with three contiguous stereocenters.
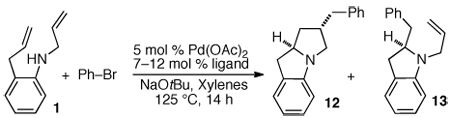
Over the past several years our group has developed a method for the construction of nitrogen heterocycles via Pd-catalyzed carboamination reactions between aryl bromides and amines bearing pendant alkenes.1 For example, treatment of an N-substituted 2-allylaniline derivative (e.g., 1) with an aryl bromide in the presence of NaOtBu and a palladium catalyst leads to the formation of a 2-benzylindoline product (4).2 These reactions proceed via intramolecular alkene aminopalladation of palladium(aryl)(amido) complex 2 to yield 3a, which undergoes reductive elimination to give 4 (Scheme 1).
Scheme 1.

Cascade Aminopalladation/Carbopalladation
It seemed plausible that this method could be extended to cascade cyclization processes that yield tricyclic products if an intermediate related to 3a could be intercepted with a pendant alkene. For example, if N,2-diallylaniline were employed as a substrate, alkene aminopalladation of 2 (R = allyl) would yield 3b, which could undergo intramolecular carbopalladation to give 5. Reductive elimination would then yield 6. Overall, this transformation would generate three bonds, two rings, and two stereocenters in a single step from a simple starting material. Importantly, this method would provide a new means to access benzo-fused 1-azabicyclo[3.3.0]octanes (6) and related 1-azabicyclo[4.3.0]nonanes. These scaffolds are a prominent feature of several natural products,3 and have also served as key intermediates in the synthesis of analogous fully saturated ring systems.4
The approach outlined in Scheme 1 sharply contrasts with related palladium-catalyzed cascade Heck reactions between polyalkene substrates bearing pendant alkenyl (or aryl) halides (Scheme 2).5,6,7 The Heck cascades occur through sequential intramolecular alkene carbopalladation reactions of R-Pd-X intermediates such as 8 or 9 (X = halide or pseudohalide), and are usually terminated by β-hydride elimination from the final R-Pd-X species (10) to generate an alkene (11). Thus, elements of molecular complexity present in 10 are removed in the terminal step, as the β-elimination leads to loss of a stereocenter and an organometallic functional group. In comparison, the final step of the aminopalladation/carbopalladation cascade shown in Scheme 1 (reductive elimination from 5 to yield 6) would produce a C–Ar bond, and the stereocenters generated in each alkene insertion step would be retained in the product.8
Scheme 2.

Comparison to Cascade Alkene Carbopalladation
Although the cascade aminopalladation/carbopalladation sequence could have considerable utility, in order to achieve our desired transformation, we would need to overcome a significant obstacle that is not present in the cascade Heck reactions. The key intermediates in the Heck cascades (8 and 9) contain only a single C–Pd bond. Thus, premature termination of the cascade via competing reductive elimination from 8 or 9 cannot occur, as C–X bond forming reductive elimination from Pd(II) is thermodynamically unfavorable.9 In contrast, intermediate 3b contains two Pd–C bonds, and can potentially undergo competing irreversible C–C bond-forming reductive elimination to afford undesired monocyclized product 4. In addition, the catalyst employed for the cascade cyclization must not only favor alkene insertion over reductive elimination from 3b, but also must allow the requisite reductive elimination from 5 to proceed.
In our initial experiments we sought to find a catalyst that would facilitate the desired cascade reaction. To this end, we examined the coupling of 1 (R = allyl) with bromobenzene using catalysts generated in situ from mixtures of palladium acetate and phosphine ligands (Table 1). As anticipated, these reactions afforded two major products: 12 and 13.10 After some exploration, we discovered that bulky triaryl phosphines 14 and 15 provided 12 in acceptable chemical yields and diastereoselectivities.
Table 1.
Optimization Studiesa
 | ||||
|---|---|---|---|---|
| entry | ligand | 12:13 | dr 12 | yield 12b |
| 1 | Dpe-phos | 2:1 | 1:1 | 24% |
| 2 | Dppf | 2:1 | 2:1 | 34% |
| 3 | Nixantphos | 0:100 | – | – |
| 4 | P(o-tol)3 | 3:1 | 4:1 | 59% |
| 5 | PCy3 | 2:1 | 2:1 | 20% |
| 6 | 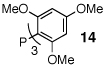 |
6:1 | 3:1 | 68% |
| 7 | 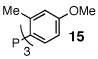 |
5:1 | 5:1 | 60% (50%)c |
Conditions: Reactions were conducted on a 0.11 mmol scale using 1.0 equiv 1, 1.5 equiv PhBr, 1.5 equiv NaOtBu, xylenes (0.2 M), 125 °C, 14 h.
Yields were determined by 1H NMR analysis of crude reaction mixtures that contained phenanthrene as an internal standard. The mass balance of these reaction mixtures was composed of products resulting from β–hydride elimination of intermediate 2, 3b, or 5.
Isolated yield.
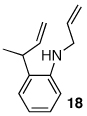
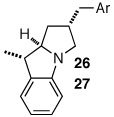
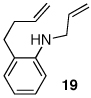
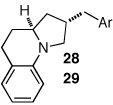
Having discovered a viable catalyst system for the cascade cyclization reaction of 1, we proceeded to examine analogous transformations of related substrates. As illustrated in Table 2, a number of N,2-diallylaniline derivatives (1, 16–17) were converted to benzo-fused-1-azabicyclo[3.3.0]octanes 20–25 in moderate yields with moderate to good diastereoselectivities. Substitution at the benzylic position was tolerated, as illustrated by the conversion of 18 to 26 and 27. The conversion of N-allyl-2-(but-3-enyl)aniline (19) to benzo-fused-1-azabicyclo[4.3.0]nonanes 28–29 was also achieved with moderate to good yields and selectivities. However, efforts to transform substrates bearing disubstituted alkenes have been unsuccessful. In addition, the coupling of 2-bromotoluene with 1 afforded monocyclic N-allyl-2-(2-methylbenzyl)indoline (4, Ar = 2-methylphenyl) as the major product, which was isolated in 72% yield. Only a small amount of the desired bicyclic compound was generated in this reaction.
Table 2.
Cascade Reactionsa
| substrate | product | R | Ar | ratio bicyclic: monocyclicb |
drc | yieldd |
|---|---|---|---|---|---|---|
 |
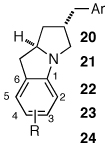 |
H | p-CF3C6H4 | 10:1 | >20:1 (5:1) | 46% |
| H | p-MeC6H4 | 3:1 | 10:1 (10:1) | 66% | ||
| H | p-MeOC6H4 | 5:1 | 13:1 (5:1) | 53% | ||
| 3-MeO | p-MeC6H4 | 2:1 | 10:1 (5:1) | 57% | ||
| 4-F | p-(Me2N)C6H4 | 2:1 | 7:1 (3:1) | 58% | ||
| 1 | 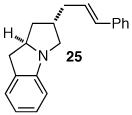 |
2:1 | 3:1 (3:1) | 42% | ||
 |
 |
Ph p-PhC(O)C6H4 |
7:1 7:1 |
18:1 (7:1) >20:1 (10:1) |
67% 68% |
|
 |
 |
Ph p-MeOC6H4 |
5:1 5:1 |
3:1 (3:1) 3:1 (3:1) |
70% 47% |
Conditions: Reactions were conducted on a 0.3–0.5 mmol scale using 1.0 equiv substrate, 1.5 equiv ArBr, 1.5 equiv NaOtBu, xylenes (0.4 M), 125 °C, 14 h.
Ratio of bicyclic product:monocyclic product observed in crude reaction mixtures.
Diastereomeric ratios are for isolated material. Numbers in parentheses represent diastereomeric ratios observed in crude reaction mixtures. In some instances the minor diastereomer was partially or entirely removed during isolation.
Isolated yield (average of two or more experiments).
Although most substrates examined afforded the anticipated products, reactions of 30 with aryl bromides provided surprising results (eq 1). The expected products 32a–c, were not obtained, but instead 31a–c were generated in moderate yield with excellent diastereoselectivity.
 |
(1) |
In order to probe the origin of this unexpected regioisomer, we conducted three experiments with deuterium labeled substrates 33a–c. As shown in eq 2, substrates 33a and 33b were converted to 34a and 34b, with no migration of the label observed in either case. In contrast, substrate 33c, bearing deuterium atoms on the terminal carbon of the allyl group were transformed to 34c with migration of a deuterium atom to the C5-methyl group.
 |
(2) |
On the basis of this data, we suggest that formation of 34c may proceed as illustrated in Scheme 3. An initial aminopalladation followed by carbopalladation could effect the conversion of 33c to key intermediate 35 as outlined above (Scheme 1). A surprising and unprecedented 1,3-palladium/hydride shift would then yield 36, which could undergo reductive elimination to afford 34c. Although through-space palladium migrations have previously been observed,11 only a single report has described Pd-migration from one sp3-hybridized carbon to another.12 The conversion of 35 to 36 is particularly surprising given the fact that 35 also contains hydrogen atoms that could potentially undergo β-elimination.
Scheme 3.

Proposed Mechanism for Formation of 34c
In conclusion we have developed a new cascade reaction for the synthesis of polycyclic nitrogen heterocycles that proceeds by way of sequential aminopalladation and carbopalladation. These transformations illustrate that catalyst tuning can allow alkene insertion processes to occur in preference to C–N or C–C bond-forming reductive elimination in LnPd(Ar)(NR2) or LnPd(Ar)(R) complexes. In addition, we have observed the first occurrence of 1,3-palladium migration of an alkylpalladium intermediate. Studies on the scope of this method and the application of these concepts to the development of new catalytic reactions are underway.
Supplementary Material
Acknowledgment
The authors acknowledge the NIH-NIGMS for financial support of this work (GM-071650). Additional funding was provided by GlaxoSmithKline, 3M, Amgen, and Eli Lilly. The authors thank prior Wolfe group member Dr. Josephine S. Nakhla (currently at Sigma-Aldrich) for conducting initial experiments in this area.
Footnotes
Supporting Information Available. Experimental procedures, characterization data for all new compounds, descriptions of stereochemical assignments with supporting crystallographic structural data, and copies of 1H and 13C NMR spectra for all new compounds reported in the text (102 pages). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Reviews: Wolfe JP. Eur. J. Org. Chem. 2007:571. Wolfe JP. Synlett. 2008:2913.
- 2.(a) Lira R, Wolfe JP. J. Am. Chem. Soc. 2004;126:13906. doi: 10.1021/ja0460920. [DOI] [PubMed] [Google Scholar]; (b) Bertrand MB, Wolfe JP. Tetrahedron. 2005;61:6447. [Google Scholar]
- 3.(a) Liu J-F, Jiang Z-Y, Wang R-R, Zheng Y-T, Chen J-J, Zhang X-M, Ma Y-B. Org. Lett. 2007;9:4127. doi: 10.1021/ol701540y. [DOI] [PubMed] [Google Scholar]; (b) Kariba RM, Houghton PJ, Yenesew A. J. Nat. Prod. 2002;65:566. doi: 10.1021/np010298m. [DOI] [PubMed] [Google Scholar]
- 4.(a) Ito Y, Nakajo E, Natatsuka M, Saegusa T. Tetrahedron Lett. 1983;24:2881. [Google Scholar]; (b) Pearson WH, Fang W-K. J. Org. Chem. 2000;65:7158. doi: 10.1021/jo0011383. [DOI] [PubMed] [Google Scholar]
- 5.(a) Negishi E-i, Wang G, Zhu G. Top. Organomet. Chem. 2006;19:1. [Google Scholar]; (b) Maddaford SP, Andersen NG, Cristofoli WA, Keay BA. J. Am. Chem. Soc. 1996;118:10766. [Google Scholar]; (c) de Meijere A, Meyer FE. Angew. Chem. Int. Ed. Engl. 1994;33:2379. [Google Scholar]; (d) Zeni G, Larock RC. Chem. Rev. 2006;106:4644. doi: 10.1021/cr0683966. [DOI] [PubMed] [Google Scholar]
- 6.For cascade reactions of enynes that proceed through similar intermediates, see: Trost BM, Shi YJ. Am. Chem. Soc. 1993;115:9421. Meyer FE, Parsons PJ, de Meijere A. J. Org. Chem. 1991;56:6487.
- 7.For Pd(II)-catalyzed oxidative cascade reactions that generate heterocycles through aminopalladation of LnPdX2 alkene complexes followed by alkene carbopalladation, see: Yip K-T, Zhu N-Y, Yang D. Org. Lett. 2009;11:1911. doi: 10.1021/ol900355h. and references cited therein
- 8.To the best of our knowledge, only two reports of intermolecular cascade Heck reactions between aryl or alkenyl halides and 1,n-dienes have appeared in the literature. In these cases the transformations were terminated by aryl C-H activation followed by C-C bond formation; all alkene insertion steps proceed through R-Pd-X intermediates. See: Hu Y, Ouyang Y, Qu Y, Hu Q, Yao H. Chem. Commun. 2009:4575. doi: 10.1039/b900770a. Hu Y, Song F, Wu F, Cheng D, Wang S. Chem. Eur. J. 2008;14:3110. doi: 10.1002/chem.200702035.
- 9.Roy AH, Hartwig JF. J. Am. Chem. Soc. 2001;123:1232. doi: 10.1021/ja0034592. [DOI] [PubMed] [Google Scholar]
- 10.The formation of N-(prop-1-enyl)-2-methylindoline was observed in some instances. The origin of this side product is not entirely clear, but it may result from reduction or protonolysis of intermediate 3b.
- 11. Huang Q, Fazio A, Dai G, Campo MA, Larock RC. J Am. Chem. Soc. 2004;126:7460. doi: 10.1021/ja047980y. Kesharwani T, Verma AK, Emrich D, Ward JA, Larock RC. Org. Lett. 2009;11:2591. doi: 10.1021/ol900940k. and references cited therein Ma S, Gu Z. Angew. Chem. Int. Ed. 2005;44:7512. doi: 10.1002/anie.200501298.
- 12.For rare examples of 1,5-Pd migration of alkylpalladium intermediates, which occur under oxidative conditions, see: Heumann A, Bäckvall JE. Angew. Chem. Int. Ed. 1985;24:207.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.