

Abstract
GSK-3 is one of the very few signaling molecules that regulate a truly astonishing number of critical intracellular signaling pathways. It has been implicated in a number of diseases including heart failure, bipolar disorder, diabetes, Alzheimer’s disease, aging, inflammation and cancer. Furthermore, a recent clinical trial has validated the feasibility of targeting GSK-3 with small molecule inhibitors for human diseases. In the current review we will focus on its expanding role in the heart, concentrating primarily on recent studies that have employed cardiomyocyte- and fibroblast-specific conditional gene deletion in mouse models. We will highlight the role of the GSK-3 isoforms in various pathological conditions including myocardial aging, ischemic injury, myocardial fibrosis and cardiomyocyte proliferation. We will discuss our recent findings that deletion of GSK-3α specifically in cardiomyocytes attenuates ventricular remodeling and cardiac dysfunction post-MI by limiting scar expansion and promoting cardiomyocyte proliferation. The recent emergence of GSK-3β as a regulator of myocardial fibrosis will also be discussed. We will review our very recent findings that specific deletion of GSK-3β in cardiac fibroblasts leads to fibrogenesis, left ventricular dysfunction and excessive scarring in the ischemic heart. Finally, we will examine the underlying mechanisms that drive the aberrant myocardial fibrosis in the models in which GSK-3β is specifically deleted in cardiac fibroblasts. We will summarize these recent results and offer explanations, whenever possible, and hypotheses when not. For these studies we will rely heavily on our models and those of others to reconcile some of the apparent inconsistencies in the literature.
Keywords: GSK-3, heart failure, myocardial infarction, cardiac fibroblast, fibrosis
Introduction
Glycogen synthase kinase-3 (GSK-3) is a ubiquitously expressed, serine/threonine kinase that was identified in 1980 for its role in regulating glycogen synthase, the rate limiting enzyme in glycogen synthesis. GSK-3 was first cloned in 1990, based on partial peptide sequencing.1 GSK-3 is a highly conserved protein kinase as isoenzymes from species as distant as flies and humans display more than 90% sequence homology within the protein kinase domain.2 The GSK-3 family consists of two isoforms, α and β, which are 98% identical within their kinase domains but differ substantially in their N- and C-terminal sequences which accounts for an overall sequence homology of 85%. This exceptionally high homology in the kinase domain makes the prospect of development of isoform-specific small molecule inhibitors daunting (though still possible). Although the GSK-3 isoforms have very similar structures and overlapping functions, they also have some unique properties: 1) only the beta isoform is phosphorylated at Ser389/Thr390; 2) Beta isoform has a neuronal-specific splice-site inserted at AA-13; and 3) some substrates are isoform-specific. Furthermore, the phenotype of global deletion of both isoforms are very different, as global deletion of GSK-3β is embryonic lethal but GSK-3α deficient mice born are normal and can survive up to several years. Both isoforms also have some unique regulatory signaling mechanism as cells lacking GSK-3β prevent proper activation of NF-kB (although the precise mechanism is unclear) but alpha null cells show no effect. Another example of isoform specific regulation is SMAD-3, which is strongly regulated by GSK-3β in a GSK-3α independent manner.
Unlike most protein kinases, GSK-3 is typically active in un-stimulated cells and is inhibited in response to a variety of stimuli. While GSK-3 is one of the few protein kinases that can be inactivated by phosphorylation, the molecular mechanism of GSK-3 regulation is very complex and not fully understood. GSK-3s are negatively regulated by N-terminal phosphorylation of serine residues of the enzyme (Ser21 for GSK-3α and Ser9 for GSK-3β), herein after referred to as S21 and S9 respectively. In addition to negative regulation by N-terminal phosphorylation, the activity of GSK-3 is positively regulated by tyrosine phosphorylation at Tyr279 for GSK-3α and Tyr216 for GSK-3β.2–4 p38 MAPK can also inactivate GSK-3β via phosphorylation within its C-terminal region at Ser389 and Thr390.5 The role of S21 and S9 phosphorylation in myocardial pathophysiology has been extensively studied,6–10 however, the physiological significance of tyrosine phosphorylation and p38 mediated C-terminal phosphorylation are not yet clear. The basic biology and molecular mechanisms of GSK-3 signaling have been previously reviewed and the readers are referred to a recent review for details.11
The initial in vitro studies examining the role of GSK-3 in cardiac disease processes were first published a decade ago and identified GSK-3β as a negative regulator of the hypertrophic response in cardiomyocytes.12,13 Haq et al.12 demonstrated that adenovirus-mediated gene transfer of GSK-3β with a Ser9 to Ala mutation, (a mutant that cannot be inhibited by Akt) led to a reduced hypertrophic response of cardiomyocytes following stimulation with hypertrophic agonists. This study suggested that inactivation of GSK-3β was required for cardiomyocytes to recruit hypertrophic response.12 Since then, numerous in vivo studies, utilizing a variety of genetically modified mouse models have been published and suggest an essential role of GSK-3α/β in several important aspects of cardiac biology.6,14–18 Table 1 summarizes a list of studies with genetically modified mouse models, suggesting crucial roles of GSK-3α/β in regulating cardiac homeostasis and responses to stresses in vivo.6, 7, 10, 15–28 Previously, most of the attention on the GSK-3 family has centered on GSK-3β. However, based on our recent studies in isoform-specific conditional knockouts, both isoforms appear to play overlapping, unique and even opposing functions in the heart. Herein, we will focus on recent data derived from cardiomyocyte- and fibroblast-specific conditional mouse models. We will discuss our recent findings that deletion of GSK-3α, specifically in cardiomyocytes, is protective in the setting of MI, suggesting that specifically targeting GSK-3α could be a novel strategy to limit adverse remodeling in ischemic hearts. These findings are in stark contrast with the studies of germ line global deletion of GSK-3α, which suggest that loss of GSK-3α is detrimental, leads to spontaneous hypertrophy by 6 months of age,18 aggravates MI-induced remodeling20 and shows accelerated aging-related pathologies.22 We will also discuss our recent findings that GSK-3β negatively regulates fibrotic remodeling in the ischemic heart by modulating canonical TGF-β1 signaling through direct interactions with SMAD-3, suggesting that GSK-3β–mediated negative regulation of fibrosis is essential to limit the adverse ventricular remodeling in the ischemic heart.17 Figure 1 highlights some of the important upstream activators and downstream targets of GSK-3 that have been implicated in cardiac pathology.
Table 1.
summarizes a list of studies with genetically modified mouse models, suggesting crucial roles of GSK-3α/β in regulating cardiac homeostasis and responses to stresses in vivo.6, 7, 10, 15–28
| SN | Genetic model | HF Model | Phenotype/outcome | Mechanism/characteristic | Reference No. |
|---|---|---|---|---|---|
| 1. | Cardiac specific Tg-GSK-3α | PO | Protective | GSK-3α inhibits cardiac growth and pressure overload-induced cardiac hypertrophy but increases fibrosis and apoptosis. | 19 |
| 2. | GSK-3α S21A KI | PO | Detrimental | Increased hypertrophy and heart failure. | 6 |
| 3. | GSK-3α global KO | PO | Detrimental | Born without apparent cardiac abnormalities but develops cardiac hypertrophy and LV dysfunction after 3 month of age; impaired β-adrenergic responsiveness. Increased hypertrophy and heart failure following PO. | 18 |
| 4. | GSK-3α global KO | MI | Detrimental | Deletion of GSK-3α promotes ischemic injury, increases risk of cardiac rupture, accentuate post-MI remodeling and left ventricular dysfunction. | 20 |
| 5. | Tg-GSK-3α/CA-MEK1 bigenic mice | Aging heart, PO | Detrimental | Constitutively active MEK1 rescues cardiac dysfunction caused by overexpressed GSK-3α during aging and hemodynamic pressure overload | 21 |
| 6. | GSK-3α global KO | Aging heart | Detrimental | KO mice shows premature death and acceleration of age-related pathologies including cardiac hypertrophy, contractile dysfunction, sarcomere disruption and striking sarcopenia in cardiac and skeletal muscle. Hyper-activation of mTORC1 in the KOs causes suppression of autophagy which leads to accelerated age-related pathologies. | 22 |
| 7. | Cardiac specific conditional GSK-3α KO | MI | Protective | In contrast to global KO models, this study suggest that cardiomyocyte specific deletion of GSK-3α limits ventricular remodeling and preserves cardiac function post-MI. | 28 |
| 8. | GSK-3β global KO | – | Lethal | Embryonic lethality caused by severe liver degeneration during mid-gestation. This is primarily due to inability to recruit pro-survival NF-kB signaling in setting of increased TNF-α production in response to pathogen. In the absence of pathogens, died in late gestation or at birth due to near obliteration of the ventricular cavities by proliferating cardiomyocytes. | 16, 23 |
| 9. | Cardiac specific Tg-GSK-3β-S9A | PO | Protective | Expression of activated GSK-3β diminished hypertrophy in response to chronic beta-adrenergic stimulation and pressure overload. | 7 |
| 10. | Cardiac specific Tg-GSK-3β | – | Detrimental | Dramatic impairment of normal post-natal cardiomyocyte growth, impaired contractile function and diastolic relaxation, abnormal calcium handling leading to an inability to normalize cytosolic [Ca2+] in diastole. | 24 |
| 11. | Cardiac specific tg-GSK-3β-DN | PO | Protective | Protected apoptosis and fibrosis with increased cardiac contractility after PO, Induction of compensatory hypertrophy. | 15 |
| 12. | GSK-3β S9A KI | PO | Protective | Attenuated hypertrophy and heart failure following PO. | 6 |
| 13. | GSK-3α/β-S21A-S9A-DKI | PO | Protective | GSK-3α/β DKI Mice Develop Hypertrophy with Preserved LV Function following PO | 6 |
| 14. | Cardiac specific conditional GSK-3β KO | PO, MI | Protective | Preserved LV dilation and function post MI. Increased proliferation following PO and MI. No role in PO-induced hypertrophy. | 25 |
| 15. | Cardiac specific Tg-GSK-3β-S9A | I/R | Detrimental | This study suggests that inhibition of GSK3β by postconditioning is required to prevent opening of the mitochondrial permeability transition pore during reperfusion. | 8 |
| 16. | GSK-3α/β-S21A-S9A-DKI | I/R | – | This study suggests that inhibition of GSK3β is not required for ischemic preconditioning. | 9 |
| 17. | GSK-3α/β-S21A-S9A-DKI | ISO infusion, MI | Protective | Attenuated cardiac hypertrophy and interstitial fibrosis with improved contractile function induced by chronic adrenergic stimulation. No role in MI induced remodeling. | 10, 26 |
| 18. | Tg-DnGSK-3β, GSK-3β+/−), tg-GSK-3β-KI | I/R | differential | Inhibition of GSK-3β exacerbates ischemic injury but protects against I/R injury by modulating mTOR and autophagy signaling. | 27 |
| 19. | Fibroblast specific GSK-3β KO | MI | Detrimental | Deletion of GSK-3β in cardiac fibroblast leads to fibrogenesis, left ventricular dysfunction and excessive scarring in the ischemic heart. Mechanistically, deletion of GSK-3β leads to hyper-activation of pro-fibrotic TGF-β1-SMAD-3 signaling. | 17 |
Figure 1. Signaling cascades regulated by GSK-3 in heart.
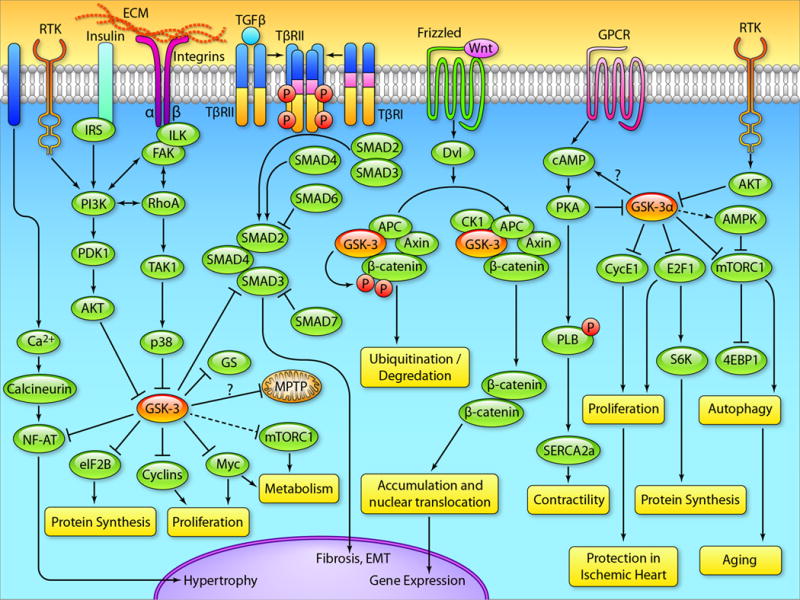
In response to growth factor binding to their receptors, the PI3K/Akt pathway is activated, leading to inhibition of GSK-3. GSK-3 negatively regulates a host of factors downstream of growth factor signaling, so the consequences of GSK-3 inhibition are activation of these factors including: 1) NF-AT-hypertrophy regulator, 2) glycogen synthase-glycogen synthesis regulator, 3) mTORC1-autophagy and metabolism regulator, 4) D- and E-type cyclins-cell cycle regulator, 3) Myc-metabolism and proliferation regulator. An alternative mechanism to inhibit GSK-3 is mediated by p38. GSK-3β directly binds with SMAD-3 to negatively regulate the profibrotic TGF-β1 signaling. GSK-3 is well known to regulate the canonical Wnt signaling. Phosphorylation of β-catenin by GSK-3 leads to the ubiquitination and degradation of β-catenin by the proteasome, preventing gene expression. In the absence of GSK-3, β-catenin is stabilized, and then translocates to the nucleus leading to gene expression. β-catenin regulates a host of processes from fibrosis to cardiac hypertrophy. GSK-3α regulates AMPK and mTOR, the master regulators of autophagy and metabolism. In the absence of GSK-3α, mTOR is dysregulated leading to impaired autophagy which accelerates the progression of aging. GSK-3α also regulates the β-adrenergic receptor responsiveness and cAMP production via unknown mechanisms. GSK-3α directly interacts and phosphorylates cyclin E1 in cardiomyocytes and its deletion promotes E2F-1 and cyclin E1 recruitment and induces the re-entry of adult cardiomyocytes into the cell cycle. (llustration Credit: Ben Smith).
Role of GSK-3α in cardiac biology
The roles of GSK-3β in cardiac biology are well recognized.4, 16, 17, 25, 29, 30 In contrast, much less is known about the roles of GSK-3α in the heart, but what is known suggests unique and overlapping functions of the two kinases.31 Germline deletion of GSK-3β is embryonic lethal (see below). In contrast, deletion of GSK-3α does not affect embryonic development. However, global deletion of GSK-3α is detrimental in the setting of MI and pressure overload (PO)-induced ventricular remodeling and heart failure.18,20 A similar detrimental phenotype of increased cardiac hypertrophy, fibrosis and heart failure has been reported with a constitutively active GSK-3α global knock-in (GSK-3αKI) mouse.6 Thus, the role of GSK-3α in cardiac pathophysiology is complex and not clear.6, 18–22 This will be addressed further below.
Role of GSK-3α in cardiac hypertrophy and fibrosis
As terminally differentiated cells, cardiomyocytes respond to various physiological (e.g. exercise) and pathological (e.g. hypertension) stresses by undergoing cellular and subsequently organ level hypertrophy. It is well established that physiological hypertrophy enhances cardiac performance via increasing stroke volume and oxygen consumption.32,33 On the other hand, pathological hypertrophy is typically associated with re-expression of the fetal gene program, increased fibrosis, cell death and cardiac dysfunction and may lead to heart failure. Henceforth, we will use the term cardiac hypertrophy to mean pathological cardiac hypertrophy.
We employed a global loss of function model to demonstrate that GSK-3α is a negative regulator of cardiac hypertrophy and fibrosis at baseline and in the stressed heart.18,20 Mechanistically, mTORC1 has been identified as a key target of GSK-3α to regulate cardiac hypertrophy. We found that mTORC1 was profoundly dysregulated as evidenced by significantly increased phosphorylation of mTORC1 targets including 4E-BP1 and ribosomal S6 kinase in the GSK-3α KO.18 Transgenic overexpression of GSK-3α in mice led to smaller hearts with increases in apoptosis and fibrosis but with preserved cardiac function at baseline. However, in response to PO, these mice showed attenuated cardiac hypertrophy, enhanced apoptosis and fibrosis, and significantly reduced ventricular function.19 Mechanistically, GSK-3α appears to inhibit physiological cardiac growth through inhibition of ERK activation.19 Maejima et al.21 employed a constitutively active MEK1 (CA-MEK1)/GSK-3α bigenic mouse model and successfully rescued most of the detrimental phenotype of the GSK-3α-tg mice. Surprisingly, increased cardiac fibrosis was not rescued. Consistently, GSK-3αKI led to markedly increased pathological cardiac hypertrophy, fibrosis and ventricular dysfunction in response to PO.6 We employed a cardiac-specific conditional GSK-3α KO (GSK-3αcKO) mouse and challenged them with MI. Consistently, GSK-3αcKO mitigates post-MI remodeling, contractile dysfunction and heart failure.28 However, our findings suggest that GSK-3α has no direct role in MI-mediated cardiac hypertrophy and fibrosis.
Deletion of GSK-3α accelerates aging in mice
Aging is a naturally occurring complex biological process which is characterized by deterioration in physiological integrity that leads to progressive functional impairment and increased mortality.34 The deterioration in cellular physiology promotes pathological conditions such as cardiovascular disorders, cancer and neurodegenerative diseases. Genetic, epigenetic, and external factors like dietary intake and intrinsic stresses such as reactive oxygen species (ROS) are known key drivers of aging.35 Understanding of the ageing process has been hampered by several issues such as gradual and heterogeneous inception in the body, diverse phenotypes, and most importantly lack of adequate molecular biomarkers to quantify the degree of ageing. Recently, we reported that GSK-3α appears to be one of the key regulators of aging.22 We found that GSK-3α actively suppresses the aging process, and global deletion of this kinase accelerates age-related pathologies, thereby reducing life span. GSK-3α KO mice showed profound aging phenotypes in most of the organs systems including the heart, gut, liver and bone. Further studies in the heart revealed vacuolar degeneration consistent with marked sarcopenia as well as significant myocyte dropout with increased fibrosis. Mechanistically, we found that insulin receptor substrate (IRS-1) expression, a direct target of GSK-3, was up-regulated and mTORC1 function was dysregulated as evidenced from increased activity of downstream targets ie., 4E-BP1, S6 kinase, and ribosomal S6 protein in the aging mice. mTORC1 dysregulation in KO hearts led to impaired autophagy as reflected by decreased LC3-II to LC3-I ratio and increased p62 expression in the GSK-3α KOs. To confirm that the observed aging phenotypes in the GSK-3α deficient mice were due to dysregulation of mTORC1, we employed a second generation inhibitor of mTORC1, everolimus, and indeed, the pathologies observed in aged KO mice were partially rescued. Thus GSK-3α is a critical regulator of mTORC1, autophagy, and aging and its deletion leads to accelerated aging in multiple tissues. This study suggests that strategies to maintain GSK-3α activity in the elderly could retard the appearance of age-related pathologies. One major limitation of this study is the exclusive dependence on an embryonic global KO model which has well known limitations of developmental and compensatory effects. Further studies with inducible tissue specific loss of function model will be useful to better understand the role of GSK-3α in aging process.
GSK-3α regulates cardiomyocyte proliferation in the adult injured heart
The neonatal heart has the capability of regeneration through cardiomyocyte proliferation.36 However, the mammalian adult heart has limited regenerative capacity and has generally been considered as a terminally differentiated post-mitotic organ.37 Various stressors, most importantly ischemic injury, can lead to progressive cardiomyocyte loss. The loss of cardiomyocytes after an MI in humans results in scar formation, loss of contractile capacity, and reduced cardiac function. Cardiomyocyte proliferation occurs at a very low level throughout life,38 but increases modestly in the heart in response to stress.39,40 The endogenous regenerative capacity of the heart appears insufficient to replenish lost myocytes, so even relatively low levels of apoptosis can have profound effects on cardiac function.41 This has necessitated employment of strategies that are required for activation of endogenous repair mechanisms of the heart.
GSK-3β is a potent regulator of cell proliferation during development (see below).16,42 However, the role of GSK-3α isoforms in cardiomyocyte proliferation has not been adequately studied. Persistent activation of GSK-3α is detrimental and abrogates cardiomyocyte proliferation.6 Moreover, this study indicated that genes involved in the cell cycle and cell proliferation were significantly down-regulated in the GSK-3αKI hearts after PO. Mechanistically, this study reported that persistent activation of GSK-3α suppresses the E2F transcription factor, likely through D-type cyclins in cardiac myocytes.28 Consistently, cardiac-specific conditional deletion of GSK-3α (GSK-3αcKO) is protective in the setting of MI.28 Furthermore; GSK-3αcKO hearts were associated with robust cardiomyocyte proliferation and protection against scar expansion and thinning in the ischemic heart. Mechanistically, GSK-3α deletion induces cardiomyocyte proliferation in the injured adult heart as evidenced by increased number of Ki67 and BrdU-positive cardiomyocytes. Deletion of GSK-3α increases E2F-1 and cyclin E1 recruitment in the heart post-MI. Further ex vivo studies show that GSK-3α regulates cyclin E1 levels in cardiomyocytes through phosphorylation. The elevated levels of E2F-1 and cyclin E1 in the GSK-3αcKO hearts appear to be the central mechanism of cardiomyocyte proliferation in vivo. These findings suggest that GSK-3α is a key regulator of cell cycle activators in the cardiomyocyte and strategies to inhibit GSK-3α could potentially be used in cardiac regeneration in patients with chronic MI. Taken together, these findings suggest that inhibition of GSK-3α limits ventricular remodeling and preserves cardiac function, post-MI. Thus, specifically targeting GSK-3α could be a novel strategy to limit adverse remodeling and heart failure.
Role of GSK-3β in ischemic injury
Numerous studies support the notion that phosphorylation (inhibition) of GSK-3β at Ser9 is required for the cardioprotection mediated by ischemic preconditioning.8,43–45 Juhaszova et al reported that inhibition of GSK-3β delays the opening of the mitochondrial permeability transition pore (MPTP) which is largely responsible for the cardioprotection. By using RNA interference, Juhaszova et al43 also showed that protective signaling is specifically mediated via the GSK-3β isoform, in a GSK-3α independent manner. Gomez et al8 used transgenic GSK-3β-S9A mice to demonstrate that serine 9 phosphorylation of GSK-3β is required for cardioprotection from ischemic postconditioning and likely acts by inhibiting opening of the MPTP in a Cyclophilin D independent mechanism. It has also been reported46,47 that GSK-3β interacts with Adenine nucleotide translocase at the inner mitochondrial membrane. However, the exact permeability transition pore–regulatory target(s) of GSK-3β is not known.
Interesting twists in the story began to appear when investigators used knock-ins (KI) of the inhibition-resistant form of GSK-3α/β, in which the phosphorylation sites on GSK-3α(Ser21) and GSK-3β(S9) are mutated to alanine.9,48 These studies questioned the obligatory role of GSK-3 isoforms in cardiac protection and suggested that the inhibition of GSK-3α/β is unlikely to be the key determinant of cardioprotective signaling.9,48 Thus, the role of GSK-3β in ischemic preconditioning is not clear and requires additional studies with conditional loss of function mouse models and isoform specific pharmacological inhibitors.
We used inducible cardiomyocyte-specific GSK-3β KO mice to demonstrate that deletion of GSK-3β specifically in cardiomyocytes is protective in the setting of permanent MI. GSK-3β knockouts displayed reduced LV remodeling, better-preserved LV function, and less dilatation post-MI.25 Importantly, this protection was not due to reduced infarct size in the GSK-3βcKO since we utilized a permanent occlusion MI model and we did not delete the gene until 5d post-MI, at a time when the infarct was completed. Surprisingly, the role of cardiomyocyte GSK-3β in heart is stress-dependent since GSK-3βcKO develops hypertrophy in response to MI but not in response to PO stress (discussed below). However, the observed hypertrophy seen in the remote myocardium of the GSK-3βcKO post-MI seems consistent with physiological, as opposed to pathological hypertrophy because these mice display better preserved heart function, reduced LV remodeling, and apoptosis with significantly increased cardiomyocyte proliferation.25
Web et al26 used double KI mice with constitutive activation of both GSK-3α and GSK-3β and demonstrated that constitutive GSK-3α/β activity has no effect on chronic stress remodeling following permanent MI, suggesting that LV remodeling after regional infarction is independent of GSK-3. These results contradict our findings in the conditional GSK-3β KO mice and are difficult to reconcile. Recently Sadoshima and colleagues27 investigated the role of GSK-3β in the heart subjected to either prolonged ischemia alone or a short period of ischemia followed by reperfusion, using genetically engineered mouse models of GSK-3β inhibition (Tg-DnGSK-3β and GSK-3β+/−) and activation (Tg-GSK-3βKI). This study suggested that inhibition of GSK-3β exacerbates ischemic injury but protects against I/R injury by modulating mTOR and autophagy. Changes in autophagy contribute to the differential effects of GSK-3β on myocardial injury during prolonged ischemia and I/R.27 Taken together, the available literature suggests that GSK-3β-mediated regulation of ventricular function in ischemic heart is complex and context dependent, as overexpression/activation is detrimental but sustained systemic inhibition could be detrimental too. Further preclinical investigation with conditional loss of function mouse models is warranted before GSK-3β inhibition could be considered as a drug therapy for ischemic heart disease.
Role of GSK-3β signaling in cardiac hypertrophy
The role of GSK-3β in cardiac hypertrophy has been extensively studied, in both in vitro culture models and in vivo with gain and loss of function mouse models.6, 7, 12, 13, 15, 24, 25, 49–53 Haq et al12 and Morisco et al13 were the first to discover that GSK-3β is a negative regulator of cardiac hypertrophy. Adenoviral gene transfer of GSK-3βS9A in cardiomyocytes was sufficient to block hypertrophic responses to α-adrenergic, β-adrenergic, and endothelin-1 stimulation. Considering the importance of this discovery, after these initial cell culture based reports, several groups created genetically modified animal models to confirm these findings in vivo.7 To determine whether activated GSK-3 can act as an antagonist of hypertrophic signaling in the adult heart in vivo, Olson and colleagues generated transgenic mice that express a constitutively active form of GSK-3β under the control of a cardiac-specific promoter. Cardiac-specific expression of activated GSK-3β diminished hypertrophy in response to chronic β-adrenergic stimulation and PO.7 These findings confirmed the role for GSK-3β as a negative regulator of hypertrophic signaling in vivo and suggest that elevation of cardiac GSK-3β activity may provide clinical benefit in the treatment of pathologic hypertrophy.
Using a gain of function approach, we observed that transgenic expression of GSK-3β in the mouse heart leads to dramatic impairment of normal post-natal cardiomyocyte growth and results in small (hypotrophic) hearts associated with markedly abnormal cardiac contractile function due to impaired calcium handling.24 Thus, these studies confirmed that GSK-3β regulates both normal as well as pathological cardiac growth. To examine the effect of persistent inactivation of GSK-3β on cardiac phenotypes at baseline and in response to PO, transgenic mice expressing a kinase inactive (dominant negative) form of GSK-3β were employed.15 Interestingly, inhibition of GSK-3β at baseline induces well-compensated cardiac hypertrophy similar to physiological hypertrophy, and stimulates cardiac function. This report further suggests that inhibition of GSK-3β during PO stress plays a protective role by inhibiting apoptosis and fibrosis, thereby preventing cardiac decompensation. Matsuda et al6 employed GSK-3βKI mice and demonstrate that expression of GSK-3βS9A prevents cardiac hypertrophy and dysfunction in response to PO stress.
We used a cardiomyocyte-specific, conditional knockout model to define the role of GSK-3β in pathological hypertrophy.25 Based on the majority of the published data (discussed above), we expected that there would be exaggerated hypertrophy in GSK-3βcKO in response to PO. However, to our surprise, we found that GSK-3β has no role in basal or TAC-induced pathological cardiac hypertrophy.25 Furthermore, we did not find any differences in the cardiac function of WT and GSK-3βcKO at baseline and in response to TAC. In distinct contrast, GSK-3β appears to be the dominant isoform regulating hypertrophy in the ischemic heart, which seems to be physiological hypertrophy (discussed in detail in “ischemic injury” section). This study suggests that in the case of TAC-induced hypertrophy, GSK-3β is not a central regulator.
GSK-3β, but not GSK-3α regulates cardiac development and cardiomyocyte proliferation and GSK-3α is dispensable
The first set of questions to address that allowed us to begin to understand what roles were being played by the two isoforms were studies ex vivo. In these, we employed ES cells to create embryoid bodies from wild-type, GSK-3α-KO or GSK-3β-KO EBs. We found that GSK-3α KO cardiomyocytes had somewhat impaired differentiation compared to WT EBs. Most striking, however, were the GSK-3β KO EBs. These EBs exhibited marked increases in size and numbers of EBs that were proliferating based on phospo-histone H3 staining. Thus, terminal cardiomyocyte differentiation was substantially blunted in GSK-3β KO EBs. It also became apparent that abnormalities were associated with factors typically involved in promoting cell proliferation (GATA, cyclin D1, and c-Myc). Importantly, β-catenin/Wnt pathway was not affected in GSK-3β KO EBs. Similarly GATA4 and Nkx2.5 were not substantially altered in GSK-3β KO vs WT.16
Differences in GSK-3α and -3β signaling in cardiomyocyte differentiation were even more apparent in the heart development. While the GSK-3α KO embryos were born at the expected frequency and had no cardiac developmental defects, live births were not observed in GSK-3β KO mice. Most striking was the profound hypertrophic myopathy that developed, with near obliteration of the ventricular cavities due to marked myocyte proliferation. Of note, congenital heart abnormalities, including double outlet right ventricle (DORV) and ventricular septal defects (VSD), were observed but were not common, and these certainly did not account for the marked thickening of the ventricular walls in the GSK-3β KO. Equally striking, impaired cardiomyocyte differentiation in ES cells and pronounced hyperplasia of cardiomyocytes during embryonic development were blatantly obvious.
To determine the mechanism, we examined rates of proliferation of the myocytes and found highly significant increases in myocytes that were positive for phospho-histone H3, a marker of proliferation. Thus it was abundantly clear that the “hypertrophy” was virtually completely due to myocyte proliferation rather true hypertrophy. Thus GSK-3β, but not GSK-3α, regulates cardiac development, and partners with GATA4, cyclin D1, and c-Myc which appear to play key roles in the developing embryonic heart. In summary, GSK-3β is a central regulator of embryonic cardiomyocyte proliferation and differentiation, as well as outflow tract development. These finding raise the possibility that mutations in GSK-3β may contribute to cases of DORV and VSD, and possibly even some cases of idiopathic LV hypertrophy. The role of GSK-3β in congenital heart disease is still unclear to our knowledge.
Emerging role of GSK-3β signaling cascades in cardiac fibrosis
Fibrosis affects nearly every tissue in the body. It is a leading cause of morbidity and mortality, as highly aggressive fibrotic processes eventually lead to organ malfunction and death.54 In reference to myocardial diseases, virtually every form of heart disease is associated with expansion and activation of the cardiac fibroblast compartment, the primary source of ECM production and fibrosis.55 Despite its enormous impact on human health, there are currently no approved treatments that specifically target fibrosis. Although the direct evidence on the role of cardiac fibroblasts in normal cardiac function is lacking, it is believed that cardiac fibroblasts play a central role in the maintenance of ECM in the normal heart. Recent studies with fibroblast-targeted mouse models clearly demonstrate that cardiac fibroblasts could play a driving role in myocardial diseases processes e.g. pressure overload-induced hypertrophy and MI-induced remodeling and heart failure.17,56,57 In an elegant study, Takeda et al.56 for the first time, employed mouse lines in which Cre recombinase was driven by the periostin (Postn) promoter (cardiac fibroblast specific gene targeting) to clearly demonstrate that cardiac fibroblasts are essential for the adaptive response of the heart to PO. Importantly, cardiomyocyte-specific deletion of the same target did not lead to any phenotype. This pioneering work proposed the concept that cardiac fibroblasts are not mere bystanders, acting only in fibrosis, but are crucial mediators of myocardial hypertrophy and adaptive responses in the heart.56 Duan et al57 used tamoxifen-inducible Col1a2Cre(T) to delete β-catenin specifically in fibroblasts (global) and demonstrated the critical requirement of cardiac fibroblasts for preserving cardiac function after acute ischemic cardiac injury.
Several studies utilizing a variety of models have supported the roles of GSK-3β in cardiac myocyte biology and disease.6,12,16,24,25,50 However, until very recently the role of GSK-3β in myocardial fibrosis was virtually unknown. Therefore we employed both Postn and Col1a2Cre to target GSK-3β specifically in fibroblasts and demonstrated that deletion of GSK-3β leads to hyper-activation of pro-fibrotic TGF-β1-SMAD-3 signaling which results in excessive fibrosis and adverse ventricular remodeling post-MI. The GSK-3β pathway has been implicated in regulating the transition of fibroblasts into myofibroblast and fibrotic signaling.58,59 Recently, Bergmann et al58 reported that inhibition of GSK-3β induces dermal fibrosis by activation of the canonical Wnt pathway. Incubation of cultured dermal fibroblasts with specific GSK-3 inhibitor SB216763 led to increased expression and release of col1a1. Furthermore, in vivo administration of SB216763 alone was sufficient to induce progressive dermal fibrosis.58 The major caveat of the above referenced study is that the conclusion exclusively relies on non-isoform specific GSK-3 inhibitors and systemic administration. Kapoor et al60 employed fibroblast-specific knockouts of GSK-3β mice to investigate the function of GSK-3β in fibrogenic responses and wound-healing in a skin injury model. Global fibroblast-specific GSK-3β KO mice exhibited increased fibrogenesis, accelerated wound closure and excessive scarring compared with control mice. In addition, knockouts showed elevated collagen production, increased fibroblast proliferation and myofibroblast formation during wound healing. Thus this study was limited to skin tissue and the major criticism was the exclusive dependence on either a global fibroblast mouse model or non-isoform selective pharmacological inhibitors.
In our recent report17 we demonstrated that GSK-3β is phosphorylated (inhibited) in fibrotic tissues from ischemic human and mouse hearts. Using two different fibroblast-specific GSK-3β knockout mouse models, we demonstrated that deletion of GSK-3β, specifically in cardiac fibroblasts, leads to fibrogenesis and profound scarring in the post-ischemic heart. Deletion of GSK-3β also induces a pro-fibrotic, myofibroblast phenotype in isolated cardiac fibroblasts, in mouse embryonic fibroblasts (MEFs) and in post-MI hearts deleted for GSK-3β. This report was the first to study MI-induced fibrotic remodeling employing cardiac fibroblast-specific gene targeting. Furthermore, this study is the first to demonstrate the effect of cardiac fibroblast-specific gene targeting on global cardiac function and adverse remodeling post-MI.
Molecular mechanism of GSK-3β-mediated fibrosis
In the healthy heart, GSK-3β actively protects from fibrosis, and injury leads to its inhibition (phosphorylation), which allows fibrotic genes to be expressed. Two different mechanisms have been proposed by which GSK-3β exerts its inhibitory effect on fibroblast activation and fibrotic gene expression: β-catenin dependent mechanism and TGF-β1-SMAD-3- dependent mechanism.
β-catenin dependent mechanism
It is well established that GSK-3β is a central component of the Wnt/frizzled signal transduction pathways that use β-catenin as a second messenger.61–63 The components and molecular mechanisms of Wnt signaling have been extensively studied and the readers are referred to a recent review for details.64 In brief, β-catenin levels are normally kept low by a phosphorylation event that is mediated by GSK-3β, which targets β-catenin for ubiquitination and proteasomal degradation. Since GSK-3β is an essential component of this degradation complex, its deletion leads to β-catenin accumulation in the cytoplasm and enters the nucleus, where it co-activates transcription of target genes.
There is growing evidence suggesting that this accumulated β-catenin leads to fibroblast activation and fibrogenesis in multiple organ systems.65–67 The role of Wnt/β-catenin signaling in fibro-proliferative disorders was initially supported by strong correlations between β-catenin mutations and aggressive fibromatosis in human desmoids tumors.68,69 Consistently, forced activation of β-catenin employing a mutant that resists ubiquitination-dependent degradation, is sufficient to drive exuberant collagen synthesis and fibrosis.70 In a dermal fibroblast model, Bergmann et at58 applied a SiRNA approach and demonstrated that deletion of β-catenin is sufficient to abolish the profibrotic effect observed upon inactivation of GSK-3β. These data suggest that inhibition of GSK-3 induces the fibrotic phenotype by activating the canonical Wnt pathway. Subsequently it was shown that β-catenin is a central mediator of profibrotic signaling in systemic sclerosis.67,71 Using a tamoxifen-inducible model, Duan et al57 demonstrated that deletion of β-catenin in fibroblasts leads to impaired fibrogenic responses and decreased cardiac performance in an ischemia reperfusion model. This study suggests that a profibrotic β-catenin injury response is critically required for healing and thus preserving cardiac function after acute ischemia/reperfusion injury. Taken together these data suggest that inhibition/deletion of GSK-3 induces a fibrotic phenotype by activating the canonical Wnt-β-catenin pathway.
TGF-β1-SMAD-3 dependent mechanism
TGF-β1 is the most potent pro-fibrotic cytokine identified to date, is a key mediator of fibroblast activation, and drives the aberrant synthesis of ECM in fibrotic diseases. The detailed components and mechanisms of TGF-β1 signaling have been reviewed elsewhere.72–74 In brief, TGF-β1 signals through at least two independent routes: primarily through the SMAD-dependent canonical pathway and the SMAD-independent or non-canonical pathways. In the SMAD-dependent pathway, activation of TGFβ type 2 receptor (TGFBR2) activates TGF-β type I receptor (TGFBRI1) and then type 1 receptor phosphorylates the transcription factor SMAD2/3 on the two serine residues at their C-termini (Ser423/25). Phosphorylated SMAD2/3 collaborates with the common mediator SMAD-4 (CO-SMAD) for cytosol to nucleus translocation and subsequent transcriptional regulation.
Most of the literature on TGF-β signaling pathways have predominantly focused on TGF-β receptors directly phosphorylating and activating SMAD transcription factors within the C-terminal domain.72,73 However, recently there is increasing interest in alternate serine and threonine phosphorylation sites within the linker region of SMADs, which control a number of cellular responses including epithelial-mesenchymal transition and SMAD-3 transcriptional activity.75–77 The linker region is defined as the domain which lies between the MH1 and MH2 domains of a SMAD protein.78 In contrast to C-terminal domain phosphorylation (which leads to activation), linker region phosphorylation leads to inhibition of SMAD transcriptional activity.75,79
Phosphorylation and nuclear translocation of SMAD2/3 is a rate-limiting step in TGF-β signaling and is important for determining the strength and duration of the signal and biological response.80,81 Emerging evidence suggests that GSK-3β exerts its anti-fibrotic effect by negatively regulating SMAD-3 activity (Figure 2). Guo et al82 were the first to demonstrate that GSK-3β controls SMAD-3 protein stability to modulate TGF-β1 signaling. This study demonstrates that reduction in the expression or activity of GSK-3β leads to increased SMAD-3 stability and transcriptional activity without affecting TGF-β receptors or SMAD-2. Conversely, overexpression of GSK-3β promotes SMAD-3 basal degradation and desensitizes cells to TGF-beta.82 Subsequently, it was shown that GSK-3β phosphorylates SMAD-3 within the linker region at Ser204 to negatively regulate its activity.79,83 However, all of this work was done either in immortalized cell lines or in isolated primary cells. Thus until very recently the in vivo role and physiological significance of this pathway was virtually unknown. We administered a small molecule inhibitor of SMAD-3 (SIS3) in vivo to test if hyper-activation of SMAD-3 is really responsible for excessive fibrotic responses observed in cardiac fibroblast-specific GSK-3β KO mice.17 SIS3 treatment significantly blunted scar expansion in KOs hearts compared to KOs treated with vehicle. Moreover, SIS3 administration nearly abolished the detrimental phenotype of cardiac fibroblast specific GSK-3β deletion as evidenced by restored ventricular function and chamber dimensions. Together, these findings provide strong evidence that hyper-activation of SMAD-3 is largely responsible for the detrimental phenotype following selective inhibition or deletion of GSK-3β in cardiac fibroblasts. Furthermore, we demonstrated that GSK-3β directly interacts with SMAD-3 in human heart, and cardiac fibroblasts. Inhibition/deletion of GSK-3β leads to increased phosphorylation of SMAD-3 at the carboxyl terminus (S423/425) and decreased phosphorylation at the n-terminus (S204). Thus our study suggests that GSK-3β exerts its effect on TGF-β1-SMAD-3 signaling specifically by regulating both the C-terminal domain as well as the linker region (Fig. 2). Taken together the available evidence suggests that inhibition/deletion of GSK-3β induces the fibrotic phenotype by activating the SMAD-3 pathway.
Figure 2. GSK-3β mediated regulation of pro-fibrotic TGF-β1-SMAD-3 signaling in cardiac fibroblast.
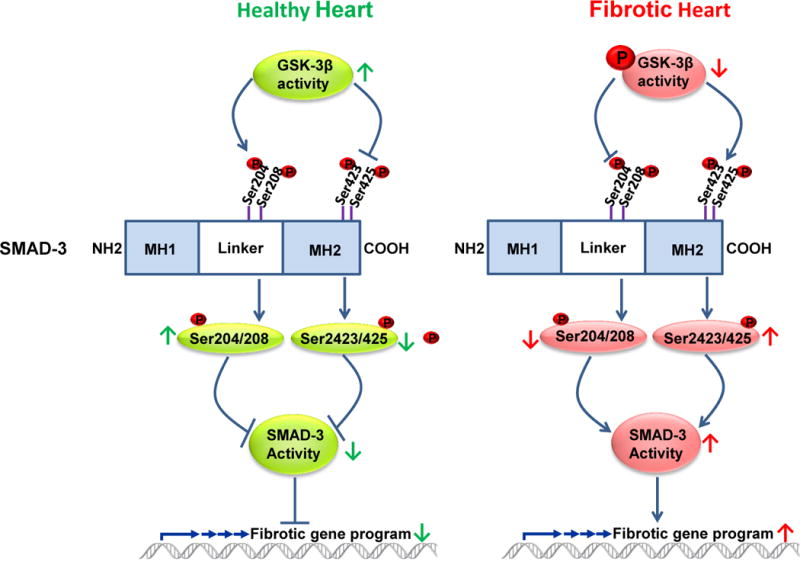
GSK-3β exerts dual control on TGF-β1-SMAD-3 signaling by regulating both the C-terminal domain as well as the linker region of SMAD3. In a healthy heart, GSK-3β interacts with, and thereby maintains, the low level activity of SMAD-3. Thus, GSK-3β is a critical regulator of canonical TGF-β1 signaling and its deletion, or inhibition, leads to aberrant hyper-activation of pro-fibrotic TGF-β1-SMAD-3 signaling.
Perspectives and future directions
The pivotal functions of GSK-3α/β in cardiac repair and, to a significant but lesser degree, regeneration, combined with the fibrosis and degeneration seen, are fascinating issues in cardiac biology that clearly deserve much more attention. These dichotomy’s of GSK-3 signaling raise serious problems in terms of pharmacological targeting of GSK-3s. It is therefore essential to acquire much more knowledge on isoform-specific functions in various cardiac model systems. This should allow better prediction as to pre-clinical outcomes. Recent findings that cardiomyocyte-specific deletion of GSK-3α is protective in the ischemic heart, places GSK-3α as a prime candidate to focus efforts on patients with MI, particularly those with incomplete revascularization or larger infarcts. Given the fact that small molecule inhibitors targeting GSK-3 are available and have been used in clinical trials in patients, one could envision going forward into large animal models with the possibility of eventually going into the clinic. The caveat is that all GSK-3 targeted drugs to date target both isoforms of GSK-3 and as discussed above, long term, non-isoform specific inhibition of GSK-3α/β could be detrimental. However, it is reasonable to hypothesize that partial or short term inhibition could be beneficial in the setting of ischemia-reperfusion injury. Furthermore, as pharmacological strategies are enhanced, and are not so reliant on standard approaches for drug discovery, it is clearly feasible that specific compounds could be generated that would specifically target GSK-3α, thereby leading to better cardioprotection in the setting of MI. In order to achieve isoform specific selectivity, It is possible that small molecule inhibitors could be generated that are not targeted to the active site (which is highly conserved). There are specific regions in alpha and beta that could be targeted. Alternatively, RNAi approaches or monoclonal antibodies could be utilized in some circumstances. Furthermore, finding the appropriate delivery route and timing of the treatment will be key determinants to achieve the therapeutic outcome while avoiding or limiting any possible adverse effects. As discussed, most of the knowledge about the role of GSK-3 isoforms in failing heart has been generated by employing various genetic mouse models. Although these mouse models are excellent tools to understand the basic biology of the targeted molecule, they all have some limitations. In fact, degree of inhibition achieved by pharmacological agents and a complete loss of function by genetic knockout cannot be directly compared. We believe that more preclinical studies with small molecule inhibitors will be warranted to take the GSK-3 inhibitors into the clinic for the management of cardiovascular diseases. Indeed, in searching Clinicaltrials.gov, we could not find any registered clinical trials in cardiovascular disease with GSK-3 inhibitors. However, there is one trial employing an agent other than the traditional inhibitors of GSK-3 (lithium and valproic acid) for Alzheimer disease. (ClinicalTrials.gov Identifier: NCT00948259). This pilot, double-blind, placebo-controlled, randomized, escalating-dose trial explored the safety and efficacy of NP031112 (tideglusib) (Noscira SA) (an inhibitor of glycogen synthase kinase-3), in Alzheimer’s disease (AD) patients. Treatment was reasonably well-tolerated except for some moderate increases of serum transaminases, which were fully reversible. This small pilot study provides valuable safety data for the use of GSK-3 inhibitors. Importantly there was no indication of any adverse consequences in the heart. Furthermore, many patients have been treated with the GSK-3 inhibitor lithium for years without any noticeable side effect on the cardiac system further validates the feasibility of targeting GSK-3 for cardiac diseases. In any case, we need isoform-selective small molecule inhibitors as long-term non-isoform selective inhibition could lead to myocardial fibrotic remodeling.17 Furthermore, our findings in the developing heart raise concerns about the long-term use of more potent GSK-3 inhibitors in women of child-baring potential (these subjects were excluded from the Noscira trials). We believe that the greatest promise for drug development may lie in the development of isoform selective small molecule GSK-3 inhibitors. We believe that such precision drugs, while much harder to develop, will allow chronic treatment with GSK-3 inhibitors in various disorders.
Acknowledgments
We apologize to those whose original work could not be cited owing to space limitations.
Sources of funding: This work was supported by grants from the NHLBI to T. Force (HL061688, HL091799) and American Heart Association Scientist Development Grant (13SDG16930103) to H.L.
Non-standard Abbreviations and Acronyms
- 4E-BP1
eIF4E binding protein
- DORV
double outlet right ventricle
- ERK
Extracellular signal-regulated kinase
- GSK-3
glycogen synthase kinase-3
- GSK-3αcKO
cardiac-specific conditional GSK-3α KO
- GSK-3αKI
GSK-3α knock-in
- GSK-3βcKO
cardiac-specific conditional GSK-3β KO
- GSK-3βKI
GSK-3β knock-in
- IRS-1
insulin receptor substrate
- MEK1
MAPK/Erk kinase 1
- MI
myocardial infarction
- MPTP
mitochondrial permeability transition pore
- mTORC1
Mammalian target of rapamycin
- NF-kB
Nuclear factor κ B
- PO
pressure overload
- SMAD
Contraction of Sma and Mad (Mothers against decapentaplegic)
- TAC
Transverse aortic constriction
- Tg-DnGSK-3β
transgenic dominant negative GSK-3β
- TGF-β1
Transforming growth factor β1
- Tg-GSK-3βKI
transgenic GSK-3β knock-in
- VSD
ventricular septal defects
Footnotes
Disclosure:
None
References
- 1.Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990;9:2431–8. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hughes K, Nikolakaki E, Plyte SE, Totty NF, Woodgett JR. Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation. EMBO J. 1993;12:803–8. doi: 10.1002/j.1460-2075.1993.tb05715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lochhead PA, Kinstrie R, Sibbet G, Rawjee T, Morrice N, Cleghon V. A chaperone-dependent GSK3beta transitional intermediate mediates activation-loop autophosphorylation. Mol Cell. 2006;24:627–33. doi: 10.1016/j.molcel.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 4.Kirk JA, Holewinski RJ, Kooij V, Agnetti G, Tunin RS, Witayavanitkul N, de Tombe PP, Gao WD, Van Eyk J, Kass DA. Cardiac resynchronization sensitizes the sarcomere to calcium by reactivating GSK-3beta. J Clin Invest. 2014;124:129–38. doi: 10.1172/JCI69253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thornton TM, Pedraza-Alva G, Deng B, Wood CD, Aronshtam A, Clements JL, Sabio G, Davis RJ, Matthews DE, Doble B, Rincon M. Phosphorylation by p38 MAPK as an alternative pathway for GSK3beta inactivation. Science. 2008;320:667–70. doi: 10.1126/science.1156037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsuda T, Zhai P, Maejima Y, Hong C, Gao S, Tian B, Goto K, Takagi H, Tamamori-Adachi M, Kitajima S, Sadoshima J. Distinct roles of GSK-3alpha and GSK-3beta phosphorylation in the heart under pressure overload. Proc Natl Acad Sci U S A. 2008;105:20900–5. doi: 10.1073/pnas.0808315106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antos CL, McKinsey TA, Frey N, Kutschke W, McAnally J, Shelton JM, Richardson JA, Hill JA, Olson EN. Activated glycogen synthase-3 beta suppresses cardiac hypertrophy in vivo. Proc Natl Acad Sci U S A. 2002;99:907–12. doi: 10.1073/pnas.231619298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gomez L, Paillard M, Thibault H, Derumeaux G, Ovize M. Inhibition of GSK3beta by postconditioning is required to prevent opening of the mitochondrial permeability transition pore during reperfusion. Circulation. 2008;117:2761–8. doi: 10.1161/CIRCULATIONAHA.107.755066. [DOI] [PubMed] [Google Scholar]
- 9.Nishino Y, Webb IG, Davidson SM, Ahmed AI, Clark JE, Jacquet S, Shah AM, Miura T, Yellon DM, Avkiran M, Marber MS. Glycogen synthase kinase-3 inactivation is not required for ischemic preconditioning or postconditioning in the mouse. Circ Res. 2008;103:307–14. doi: 10.1161/CIRCRESAHA.107.169953. [DOI] [PubMed] [Google Scholar]
- 10.Webb IG, Nishino Y, Clark JE, Murdoch C, Walker SJ, Makowski MR, Botnar RM, Redwood SR, Shah AM, Marber MS. Constitutive glycogen synthase kinase-3alpha/beta activity protects against chronic beta-adrenergic remodelling of the heart. Cardiovasc Res. 2010;87:494–503. doi: 10.1093/cvr/cvq061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaidanovich-Beilin O, Woodgett JR. GSK-3: Functional Insights from Cell Biology and Animal Models. Front Mol Neurosci. 2011;4:1–25. doi: 10.3389/fnmol.2011.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haq S, Choukroun G, Kang ZB, Ranu H, Matsui T, Rosenzweig A, Molkentin JD, Alessandrini A, Woodgett J, Hajjar R, Michael A, Force T. Glycogen synthase kinase-3beta is a negative regulator of cardiomyocyte hypertrophy. J Cell Biol. 2000;151:117–30. doi: 10.1083/jcb.151.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morisco C, Zebrowski D, Condorelli G, Tsichlis P, Vatner SF, Sadoshima J. The Akt-glycogen synthase kinase 3beta pathway regulates transcription of atrial natriuretic factor induced by beta-adrenergic receptor stimulation in cardiac myocytes. J Biol Chem. 2000;275:14466–75. doi: 10.1074/jbc.275.19.14466. [DOI] [PubMed] [Google Scholar]
- 14.Cho J, Rameshwar P, Sadoshima J. Distinct roles of glycogen synthase kinase (GSK)-3alpha and GSK-3beta in mediating cardiomyocyte differentiation in murine bone marrow-derived mesenchymal stem cells. J Biol Chem. 2009;284:36647–58. doi: 10.1074/jbc.M109.019109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirotani S, Zhai P, Tomita H, Galeotti J, Marquez JP, Gao S, Hong C, Yatani A, Avila J, Sadoshima J. Inhibition of glycogen synthase kinase 3beta during heart failure is protective. Circ Res. 2007;101:1164–74. doi: 10.1161/CIRCRESAHA.107.160614. [DOI] [PubMed] [Google Scholar]
- 16.Kerkela R, Kockeritz L, Macaulay K, Zhou J, Doble BW, Beahm C, Greytak S, Woulfe K, Trivedi CM, Woodgett JR, Epstein JA, Force T, Huggins GS. Deletion of GSK-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J Clin Invest. 2008;118:3609–18. doi: 10.1172/JCI36245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lal H, Ahmad F, Zhou J, Yu JE, Vagnozzi RJ, Guo Y, Yu D, Tsai EJ, Woodgett J, Gao E, Force T. Cardiac Fibroblast GSK-3beta Regulates Ventricular Remodeling and Dysfunction in Ischemic Heart. Circulation. 2014;130:419–30. doi: 10.1161/CIRCULATIONAHA.113.008364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou J, Lal H, Chen X, Shang X, Song J, Li Y, Kerkela R, Doble BW, MacAulay K, DeCaul M, Koch WJ, Farber J, Woodgett J, Gao E, Force T. GSK-3alpha directly regulates beta-adrenergic signaling and the response of the heart to hemodynamic stress in mice. J Clin Invest. 2010;120:2280–91. doi: 10.1172/JCI41407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhai P, Gao S, Holle E, Yu X, Yatani A, Wagner T, Sadoshima J. Glycogen synthase kinase-3alpha reduces cardiac growth and pressure overload-induced cardiac hypertrophy by inhibition of extracellular signal-regulated kinases. J Biol Chem. 2007;282:33181–91. doi: 10.1074/jbc.M705133200. [DOI] [PubMed] [Google Scholar]
- 20.Lal H, Zhou J, Ahmad F, Zaka R, Vagnozzi RJ, Decaul M, Woodgett J, Gao E, Force T. Glycogen synthase kinase-3alpha limits ischemic injury, cardiac rupture, post-myocardial infarction remodeling and death. Circulation. 2012;125:65–75. doi: 10.1161/CIRCULATIONAHA.111.050666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maejima Y, Galeotti J, Molkentin JD, Sadoshima J, Zhai P. Constitutively active MEK1 rescues cardiac dysfunction caused by overexpressed GSK-3alpha during aging and hemodynamic pressure overload. Am J Physiol Heart Circ Physiol. 2012;303:H979–88. doi: 10.1152/ajpheart.00415.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou J, Freeman TA, Ahmad F, Shang X, Mangano E, Gao E, Farber J, Wang Y, Ma XL, Woodgett J, Vagnozzi RJ, Lal H, Force T. GSK-3alpha is a central regulator of age-related pathologies in mice. J Clin Invest. 2013;123:1821–32. doi: 10.1172/JCI64398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- 24.Michael A, Haq S, Chen X, Hsich E, Cui L, Walters B, Shao Z, Bhattacharya K, Kilter H, Huggins G, Andreucci M, Periasamy M, Solomon RN, Liao R, Patten R, Molkentin JD, Force T. Glycogen synthase kinase-3beta regulates growth, calcium homeostasis, and diastolic function in the heart. J Biol Chem. 2004;279:21383–93. doi: 10.1074/jbc.M401413200. [DOI] [PubMed] [Google Scholar]
- 25.Woulfe KC, Gao E, Lal H, Harris D, Fan Q, Vagnozzi R, DeCaul M, Shang X, Patel S, Woodgett JR, Force T, Zhou J. Glycogen synthase kinase-3beta regulates post-myocardial infarction remodeling and stress-induced cardiomyocyte proliferation in vivo. Circ Res. 2010;106:1635–45. doi: 10.1161/CIRCRESAHA.109.211482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Webb IG, Sicard P, Clark JE, Redwood S, Marber MS. Myocardial stress remodelling after regional infarction is independent of glycogen synthase kinase-3 inactivation. J Mol Cell Cardiol. 2010;49:897–900. doi: 10.1016/j.yjmcc.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhai P, Sciarretta S, Galeotti J, Volpe M, Sadoshima J. Differential roles of GSK-3beta during myocardial ischemia and ischemia/reperfusion. Circ Res. 2011;109:502–11. doi: 10.1161/CIRCRESAHA.111.249532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahmad F, Lal H, Zhou J, Vagnozzi R, Yu JE, Shang X, Woodgett J, Gao E, Force T. Cardiomyocyte-specific Deletion of Gsk3α Mitigates Post-myocardial Infarction Remodeling, Contractile Dysfunction, and Heart Failure. J Am Coll Cardiol. 2014;64:696–706. doi: 10.1016/j.jacc.2014.04.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuster DW, Sequeira V, Najafi A, Boontje NM, Wijnker PJ, Witjas-Paalberends ER, Marston SB, Dos Remedios CG, Carrier L, Demmers JA, Redwood C, Sadayappan S, van der Velden J. GSK3beta phosphorylates newly identified site in the proline-alanine-rich region of cardiac myosin-binding protein C and alters cross-bridge cycling kinetics in human: short communication. Circ Res. 2013;112:633–9. doi: 10.1161/CIRCRESAHA.112.275602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cho J, Zhai P, Maejima Y, Sadoshima J. Myocardial injection with GSK-3beta-overexpressing bone marrow-derived mesenchymal stem cells attenuates cardiac dysfunction after myocardial infarction. Circ Res. 2011;108:478–89. doi: 10.1161/CIRCRESAHA.110.229658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Force T, Woodgett JR. Unique and overlapping functions of GSK-3 isoforms in cell differentiation and proliferation and cardiovascular development. J Biol Chem. 2009;284:9643–7. doi: 10.1074/jbc.R800077200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernardo BC, Weeks KL, Pretorius L, McMullen JR. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther. 2010;128:191–227. doi: 10.1016/j.pharmthera.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 33.Dorn GW., 2nd The fuzzy logic of physiological cardiac hypertrophy. Hypertension. 2007;49:962–70. doi: 10.1161/HYPERTENSIONAHA.106.079426. [DOI] [PubMed] [Google Scholar]
- 34.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sahin E, Depinho RA. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature. 2010;464:520–8. doi: 10.1038/nature08982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078–80. doi: 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leri A, Kajstura J, Anversa P. Mechanisms of myocardial regeneration. Trends Cardiovasc Med. 2011;21:52–8. doi: 10.1016/j.tcm.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisen J. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kajstura J, Urbanek K, Rota M, Bearzi C, Hosoda T, Bolli R, Anversa P, Leri A. Cardiac stem cells and myocardial disease. J Mol Cell Cardiol. 2008;45:505–13. doi: 10.1016/j.yjmcc.2008.05.025. [DOI] [PubMed] [Google Scholar]
- 40.Laflamme MA, Murry CE. Regenerating the heart. Nat Biotechnol. 2005;23:845–56. doi: 10.1038/nbt1117. [DOI] [PubMed] [Google Scholar]
- 41.Chiong M, Wang ZV, Pedrozo Z, Cao DJ, Troncoso R, Ibacache M, Criollo A, Nemchenko A, Hill JA, Lavandero S. Cardiomyocyte death: mechanisms and translational implications. Cell Death Dis. 2011;2:e244. doi: 10.1038/cddis.2011.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Doble BW, Patel S, Wood GA, Kockeritz LK, Woodgett JR. Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev Cell. 2007;12:957–71. doi: 10.1016/j.devcel.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–49. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miura T, Tanno M. Mitochondria and GSK-3beta in cardioprotection against ischemia/reperfusion injury. Cardiovasc Drugs Ther. 2010;24:255–63. doi: 10.1007/s10557-010-6234-z. [DOI] [PubMed] [Google Scholar]
- 45.Tong H, Imahashi K, Steenbergen C, Murphy E. Phosphorylation of glycogen synthase kinase-3beta during preconditioning through a phosphatidylinositol-3-kinase–dependent pathway is cardioprotective. Circ Res. 2002;90:377–9. doi: 10.1161/01.res.0000012567.95445.55. [DOI] [PubMed] [Google Scholar]
- 46.Nishihara M, Miura T, Miki T, Tanno M, Yano T, Naitoh K, Ohori K, Hotta H, Terashima Y, Shimamoto K. Modulation of the mitochondrial permeability transition pore complex in GSK-3beta-mediated myocardial protection. J Mol Cell Cardiol. 2007;43:564–70. doi: 10.1016/j.yjmcc.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 47.Das S, Wong R, Rajapakse N, Murphy E, Steenbergen C. Glycogen synthase kinase 3 inhibition slows mitochondrial adenine nucleotide transport and regulates voltage-dependent anion channel phosphorylation. Circ Res. 2008;103:983–91. doi: 10.1161/CIRCRESAHA.108.178970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Murphy E, Steenbergen C. Does inhibition of glycogen synthase kinase protect in mice? Circ Res. 2008;103:226–8. doi: 10.1161/CIRCRESAHA.108.181602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng H, Woodgett J, Maamari M, Force T. Targeting GSK-3 family members in the heart: a very sharp double-edged sword. J Mol Cell Cardiol. 2011;51:607–13. doi: 10.1016/j.yjmcc.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hardt SE, Sadoshima J. Glycogen synthase kinase-3beta: a novel regulator of cardiac hypertrophy and development. Circ Res. 2002;90:1055–63. doi: 10.1161/01.res.0000018952.70505.f1. [DOI] [PubMed] [Google Scholar]
- 51.Kerkela R, Woulfe K, Force T. Glycogen synthase kinase-3beta – actively inhibiting hypertrophy. Trends Cardiovasc Med. 2007;17:91–6. doi: 10.1016/j.tcm.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 52.Sugden PH, Fuller SJ, Weiss SC, Clerk A. Glycogen synthase kinase 3 (GSK3) in the heart: a point of integration in hypertrophic signalling and a therapeutic target? A critical analysis. Br J Pharmacol. 2008;153(Suppl 1):S137–53. doi: 10.1038/sj.bjp.0707659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blankesteijn WM, van de Schans VA, ter Horst P, Smits JF. The Wnt/frizzled/GSK-3 beta pathway: a novel therapeutic target for cardiac hypertrophy. Trends Pharmacol Sci. 2008;29:175–80. doi: 10.1016/j.tips.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 54.Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest. 2007;117:524–9. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen W, Frangogiannis NG. Fibroblasts in post-infarction inflammation and cardiac repair. Biochim Biophys Acta. 2013;1833:945–53. doi: 10.1016/j.bbamcr.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takeda N, Manabe I, Uchino Y, Eguchi K, Matsumoto S, Nishimura S, Shindo T, Sano M, Otsu K, Snider P, Conway SJ, Nagai R. Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. J Clin Invest. 2010;120:254–65. doi: 10.1172/JCI40295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Duan J, Gherghe C, Liu D, Hamlett E, Srikantha L, Rodgers L, Regan JN, Rojas M, Willis M, Leask A, Majesky M, Deb A. Wnt1/betacatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J. 2012;31:429–42. doi: 10.1038/emboj.2011.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bergmann C, Akhmetshina A, Dees C, Palumbo K, Zerr P, Beyer C, Zwerina J, Distler O, Schett G, Distler JH. Inhibition of glycogen synthase kinase 3beta induces dermal fibrosis by activation of the canonical Wnt pathway. Ann Rheum Dis. 2011;70:2191–8. doi: 10.1136/ard.2010.147140. [DOI] [PubMed] [Google Scholar]
- 59.Caraci F, Gili E, Calafiore M, Failla M, La Rosa C, Crimi N, Sortino MA, Nicoletti F, Copani A, Vancheri C. TGF-beta1 targets the GSK-3beta/beta-catenin pathway via ERK activation in the transition of human lung fibroblasts into myofibroblasts. Pharmacol Res. 2008;57:274–82. doi: 10.1016/j.phrs.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 60.Kapoor M, Liu S, Shi-wen X, Huh K, McCann M, Denton CP, Woodgett JR, Abraham DJ, Leask A. GSK-3beta in mouse fibroblasts controls wound healing and fibrosis through an endothelin-1-dependent mechanism. J Clin Invest. 2008;118:3279–90. doi: 10.1172/JCI35381. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 61.Rao TP, Kuhl M. An updated overview on Wnt signaling pathways: a prelude for more. Circ Res. 2010;106:1798–806. doi: 10.1161/CIRCRESAHA.110.219840. [DOI] [PubMed] [Google Scholar]
- 62.Metcalfe C, Bienz M. Inhibition of GSK3 by Wnt signalling–two contrasting models. J Cell Sci. 2011;124:3537–44. doi: 10.1242/jcs.091991. [DOI] [PubMed] [Google Scholar]
- 63.Wu D, Pan W. GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem Sci. 2010;35:161–8. doi: 10.1016/j.tibs.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim W, Kim M, Jho EH. Wnt/beta-catenin signalling: from plasma membrane to nucleus. Biochem J. 2013;450:9–21. doi: 10.1042/BJ20121284. [DOI] [PubMed] [Google Scholar]
- 65.Lam AP, Gottardi CJ. beta-catenin signaling: a novel mediator of fibrosis and potential therapeutic target. Curr Opin Rheumatol. 2011;23:562–7. doi: 10.1097/BOR.0b013e32834b3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guo Y, Xiao L, Sun L, Liu F. Wnt/beta-catenin signaling: a promising new target for fibrosis diseases. Physiol Res. 2012;61:337–46. doi: 10.33549/physiolres.932289. [DOI] [PubMed] [Google Scholar]
- 67.Akhmetshina A, Palumbo K, Dees C, Bergmann C, Venalis P, Zerr P, Horn A, Kireva T, Beyer C, Zwerina J, Schneider H, Sadowski A, Riener MO, MacDougald OA, Distler O, Schett G, Distler JH. Activation of canonical Wnt signalling is required for TGF-beta-mediated fibrosis. Nat Commun. 2012;3:735. doi: 10.1038/ncomms1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tejpar S, Nollet F, Li C, Wunder JS, Michils G, dal Cin P, Van Cutsem E, Bapat B, van Roy F, Cassiman JJ, Alman BA. Predominance of beta-catenin mutations and beta-catenin dysregulation in sporadic aggressive fibromatosis (desmoid tumor) Oncogene. 1999;18:6615–20. doi: 10.1038/sj.onc.1203041. [DOI] [PubMed] [Google Scholar]
- 69.Alman BA, Li C, Pajerski ME, Diaz-Cano S, Wolfe HJ. Increased beta-catenin protein and somatic APC mutations in sporadic aggressive fibromatoses (desmoid tumors) Am J Pathol. 1997;151:329–34. [PMC free article] [PubMed] [Google Scholar]
- 70.Cheon SS, Cheah AY, Turley S, Nadesan P, Poon R, Clevers H, Alman BA. beta-Catenin stabilization dysregulates mesenchymal cell proliferation, motility, and invasiveness and causes aggressive fibromatosis and hyperplastic cutaneous wounds. Proc Natl Acad Sci U S A. 2002;99:6973–8. doi: 10.1073/pnas.102657399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Beyer C, Schramm A, Akhmetshina A, Dees C, Kireva T, Gelse K, Sonnylal S, de Crombrugghe B, Taketo MM, Distler O, Schett G, Distler JH. beta-catenin is a central mediator of pro-fibrotic Wnt signaling in systemic sclerosis. Ann Rheum Dis. 2012;71:761–7. doi: 10.1136/annrheumdis-2011-200568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov. 2012;11:790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–30. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moustakas A, Heldin CH. The regulation of TGFbeta signal transduction. Development. 2009;136:3699–714. doi: 10.1242/dev.030338. [DOI] [PubMed] [Google Scholar]
- 75.Bae E, Kim SJ, Hong S, Liu F, Ooshima A. Smad3 linker phosphorylation attenuates Smad3 transcriptional activity and TGF-beta1/Smad3-induced epithelial-mesenchymal transition in renal epithelial cells. Biochem Biophys Res Commun. 2012;427:593–9. doi: 10.1016/j.bbrc.2012.09.103. [DOI] [PubMed] [Google Scholar]
- 76.Cohen-Solal KA, Merrigan KT, Chan JL, Goydos JS, Chen W, Foran DJ, Liu F, Lasfar A, Reiss M. Constitutive Smad linker phosphorylation in melanoma: a mechanism of resistance to transforming growth factor-beta-mediated growth inhibition. Pigment Cell Melanoma Res. 2011;24:512–24. doi: 10.1111/j.1755-148X.2011.00858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Velden JL, Alcorn JF, Guala AS, Badura EC, Janssen-Heininger YM. c-Jun N-terminal kinase 1 promotes transforming growth factor-beta1-induced epithelial-to-mesenchymal transition via control of linker phosphorylation and transcriptional activity of Smad3. Am J Respir Cell Mol Biol. 2011;44:571–81. doi: 10.1165/rcmb.2009-0282OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kamato D, Burch ML, Piva TJ, Rezaei HB, Rostam MA, Xu S, Zheng W, Little PJ, Osman N. Transforming growth factor-beta signalling: role and consequences of Smad linker region phosphorylation. Cell Signal. 2013;25:2017–24. doi: 10.1016/j.cellsig.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 79.Wang G, Matsuura I, He D, Liu F. Transforming growth factor-{beta}-inducible phosphorylation of Smad3. J Biol Chem. 2009;284:9663–73. doi: 10.1074/jbc.M809281200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen X, Xu L. Mechanism and regulation of nucleocytoplasmic trafficking of smad. Cell Biosci. 2011;1:40. doi: 10.1186/2045-3701-1-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schmierer B, Tournier AL, Bates PA, Hill CS. Mathematical modeling identifies Smad nucleocytoplasmic shuttling as a dynamic signal-interpreting system. Proc Natl Acad Sci U S A. 2008;105:6608–13. doi: 10.1073/pnas.0710134105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guo X, Ramirez A, Waddell DS, Li Z, Liu X, Wang XF. Axin and GSK3- control Smad3 protein stability and modulate TGF- signaling. Genes Dev. 2008;22:106–20. doi: 10.1101/gad.1590908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Millet C, Yamashita M, Heller M, Yu LR, Veenstra TD, Zhang YE. A negative feedback control of transforming growth factor-beta signaling by glycogen synthase kinase 3-mediated Smad3 linker phosphorylation at Ser-204. J Biol Chem. 2009;284:19808–16. doi: 10.1074/jbc.M109.016667. [DOI] [PMC free article] [PubMed] [Google Scholar]