

Abstract
Classical chemotherapeutics, such as cisplatin and its analogues, have been highly successful in the clinic, yet improvements can certainly be made, given the significant side effects associated with the killing of healthy cells. Recent advances in the field of chemotherapy include the development of targeted anticancer agents, compounds that are directed towards a specific biomarker of cancer, with the hopes that such targeted therapies might have reduced side effects given their greater selectivity. Here we discuss several transition metal complexes that are tailored towards various biomolecules associated with cancer. Most notably, the success of rhodium metalloinsertors, which specifically bind to nucleic acid base mismatches in DNA, highlight the enormous potential of this exciting new strategy.
Keywords: medicinal chemistry, bioinorganic, DNA mismatch repair
Since the discovery of the anticancer properties of cis-dichlorodiammine-platinum(II) (cisplatin), the field of medicinal inorganic chemistry has burgeoned.1–5 For many years, the field focused on the development of more potent analogues, second and third generation derivatives, leading to the FDA approval of two additional cis-platinum(II) complexes, carboplatin and oxaliplatin (Figure 1).2,3 Cisplatin and carboplatin, in particular, have been highly successful in the treatment of a variety of cancers, including testicular, ovarian, cervical, and non-small cell lung cancers.4 However, these treatments are often associated with severe side effects and a build-up of resistance. These issues have led researchers to focus more recently on the development of novel non-platinum chemotherapeutics.
Figure 1.

Chemical structures of classical platinum-based chemotherapeutics.
The rich photophysical and photochemical properties of metal complexes, in addition to their basic coordination chemistry, make them ideal scaffolds for a wide variety of biological applications. Though the pharmaceutical industry in general has shied away from “heavy metal” pharmaceuticals, with the exception of cis-platin and its derivatives, there are in fact real opportunities in the development of transition metal pharmaceuticals, given their high modularity, ease of synthesis in preparing molecules of complex shapes and symmetries, and the ability to monitor their fate within the cell using a variety of spectroscopies. In this Comment, we do not intend to carry out an exhaustive review of research in this area. Instead, we discuss here illustrative, recent efforts to develop the next generation of metal chemotherapeutics. The focus of many laboratories has been earlier in the preparation of basically more potent metal complexes that function like cis-platin in coordinating to DNA but are more effective, either because of more optimum uptake characteristics, or the inability of lesions formed to be easily detected and repaired. Much time and attention have been spent in this arena. However the goal has moved also to the design of complexes with a new strategy based upon selectivity, with the preparation of transition metal complexes that are more selective than cis-platin owing to a design strategy where the complex interacts with a specific biological target found prominently in cancer cells.
Cisplatin and its analogues were developed as “classical” chemotherapeutics. These types of drugs are meant to interfere with replication and/or the mitotic processes of tumor cells.5 In this way, they achieve potency by damaging cancer cells more than healthy cells. While this approach has been fruitful, it has been known to cause a litany of side effects, such as nephrotoxicity, neurotoxicity, leukopenia, and thrombocytopenia, as well as nausea, vomiting, and hair loss.6 For these reasons, research efforts in recent years have shifted towards the development of targeted chemotherapy. In “targeted” therapy, a drug is developed to target a specific cellular signaling pathway on which cancer cells depend for growth, metastasis, or angiogenesis.7 These types of compounds aim to damage cancer cells instead of healthy cells. Targeted therapy focuses on the development of selective therapeutics, whereas classical therapy has focused on the development of increasingly cytotoxic compounds. The next generation of chemotherapeutics has focused on targeting biomolecules, including proteins, organelles, and specific DNA lesions (Figure 2).
Figure 2.

Chemical structures of targeted chemotherapeutics discussed in this Comment: (top, left to right) The octasporine complex OS1, a potent inhibitor of the protein kinase GSK3α; General architecture of RAPTA cathepsin B inhibitors; Ruthenocene analogues of tamoxifen for the selective targeting of ERα; (bottom, left to right) The first generation rhodium metalloinsertor, [Rh(bpy)2(chrysi)]3+, selectively binds to mismatched and abasic sites in duplex DNA; Structure of mtPt, a cisplatin analogue designed to localize to the mitochondria.
As an illustration, the high levels of mutagenesis in cancerous cells often lead to upregulation and overexpression of proteins, making them attractive candidates for targeting. Metal complexes, due to their modular nature and inherent chirality, are uniquely able to target selectively these chiral biomolecules. In particular, this approach has been applied toward the selective inhibition of kinase activity. Phosphorylation of proteins by kinases is a highly important regulatory activity. However, over-phosphorylation of proteins is common in many types of cancer.8 In a recent study by Meggers et al., inert metal complexes, inspired by the natural product staurosporine and termed octasporines, were designed as highly selective kinase inhibitors (Figure 2).9,10 Six complexes were synthesized, all containing a ruthenium or iridium center and a bidentate pyridocarbazole ligand designed to bind the hinge region of the ATP-binding pocket of the kinase. However, the remaining ligands on each complex were designed to make up a unique set of hydrogen-bonding interactions with the glycine-rich loop of the ATP-binding pockets of six distinct kinases (Figure 3).9 In vivo studies have revealed the anti-angiogenic properties of one of these types of compounds in zebrafish embryos, exemplifying their potential.10
Figure 3.

Design of Octasporine complexes as inhibitors of protein kinases (adapted from reference 9). The pyridocarbazole ligand, common to all complexes, binds to the hinge region (where the adenine portion of ATP binds) of the ATP-binding pocket. The remaining A, B, C, and D ligands make up a set of hydrogen-bonding interactions with the glycine-rich loop (where the ribose triphosphate portion of ATP binds) of the ATP binding pocket, each unique to a particular kinase.
Whereas the previous example utilized the structural complexity of inert metal complexes, the reactive nature of certain metal centers can also be exploited in targeted therapy. Proteases play a crucial role in tumorigenesis by suppressing cell-death pathways and promoting cell-survival pathways.11 One such protease, cathepsin B, has been targeted by ruthenium arene RAPTA compounds (Figure 2).12,13 These compounds were found to inhibit cathepsin B protease activity and exhibited selective anti-metastatic activity in vivo.13,14 Estrogen receptors such as estrogen receptor α (ERα), which is overexpressed in several types of breast cancer, have also been the subject of targeted therapy studies.15 Several organometallic analogues of tamoxifen, an antagonist of estrogen receptors, have been developed to selectively target ERα (Figure 2).16,17 These complexes have demonstrated cytotoxic activity selectively in ERα-positive breast cancer cell lines.16
In addition to protein targeting, the mitochondria can also serve as a valuable target for drug design. Mitochondria produce reactive oxygen species as a byproduct of metabolism, and they also play a crucial role in the regulation of cell death pathways.18 Targeting mitochondria and mitochondrial DNA can induce apoptosis in tumorigenic cells, as was recently demonstrated by Lippard and Kelley.19 They constructed a cis-platinum(II) complex tethered to a mitochondrial penetrating peptide, which contained alternating cationic and lipophilic residues to enhance mitochondrial uptake (Figure 2). This complex was shown to localize almost exclusively to mitochondria in several cancer cell lines. Moreover, the complex was able to induce apoptosis in cisplatin-resistant ovarian cancer cells by damaging mitochondrial DNA.
The proposed mechanism of action of classical platinum-based chemotherapeutics is the formation of covalent DNA adducts, followed by cellular processing of these lesions.4 The synthesis of new generation classical therapeutics with enhanced DNA binding properties in order to increase cytotoxicity has been extensively explored. However, the design and synthesis of therapeutics that bind to specific DNA lesions that are more prevalent in cancer cells than normal cells may represent a targeted strategy for new chemotherapy. In particular, rhodium metalloinsertors (Figure 2) are known to bind mismatches in duplex DNA in vitro with high specificity and affinity.20 These complexes, which all bear the sterically expansive 5,6-chrysene diimine (chrysi) ligand, bind 80% of DNA mismatches in all sequence contexts and preferentially target thermodynamically destabilized mismatches over matched base pairs by a factor of over 1000.21,22 The binding mode of these complexes to mismatched DNA was structurally characterized and revealed that the chrysi ligand inserts into the DNA from the minor groove and ejects both mismatched bases in a binding mode termed metalloinsertion (Figure 4).23–26 Ejection of the mismatched bases results in a large lesion that is hypothesized to have the potential to be recognized in vivo.
Figure 4.

(Left) Crystal structure of [Rh(bpy)2(chrysi)]3+, the first generation metalloinsertor, bound to an AC mismatch in duplex DNA. (Right, top) Chemical structure of [Rh(chrysi)(phen)(DPE)]2+, a later generation metalloinsertor with enhanced selectivity and potency. (Right, bottom) Cell-selective cytotoxicity of [Rh(chrysi)(phen)(DPE)]2+, the complex selectively kills MMR-deficient (red) cells over MMR-proficient (green) cells.
Mismatches in genomic DNA arise naturally as a consequence of replication, but if left uncorrected can lead to mutations.27,28 The mismatch repair (MMR) pathway serves as a checkpoint to increase the fidelity of DNA replication ~1000 fold.29 Importantly, deficiencies in the mismatch repair machinery have been associated with several types of cancer, as well, notably, as increased resistance to classical chemotherapeutics such as cisplatin.30 Therefore, the development of a targeted therapy for MMR- deficient cancers would be invaluable in the clinic. Due to the unique DNA mismatch-binding properties of rhodium metalloinsertors, we sought to explore their biological properties in MMR-deficient cells. The compounds were initially found to inhibit growth in MMR-deficient colorectal cancer cells over MMR-proficient cells, as measured by antibody assays for DNA synthesis.31,32 In a follow-up study, it was discovered that metalloinsertors with accelerated uptake also exhibited preferential cytotoxicity towards MMR-deficient cells (Figure 4).33
Most recently, a structure-function study was conducted by altering the lipophilicities of the non-inserting ligands.34,35 This investigation resulted in the synthesis of a family of mismatch-binding complexes with similar binding affinities and selectivities for DNA mismatches, yet drastically different selectivities for MMR-deficient cells. It was discovered that more lipophilic complexes did not exhibit the unique cell-selective activities for which metalloinsertors are distinguished. However, complexes with more hydrophilic ancillary ligands were highly selective for the MMR-deficient cells over MMR-proficient cells. It was discovered that nuclear uptake of all metalloinsertors studied was sufficient for mismatch binding to genomic DNA. However, significant mitochondrial uptake led to an abolishment of their selective targeting of MMR-deficient cells. Most notably, simply substituting a hydroxyl group for a methyl group results in dramatic changes in cell-selective activity due to drastic changes in the subcellular localization (Figure 5).35 This study supports the notion that the unique cell-selective activities of these compounds rises from targeting of mismatches in genomic DNA. In an effort to more directly relate the biological activity of rhodium metalloinsertors to the MMR-deficiency phenotype, our laboratory has now embarked on studies to validate the biological efficacy of these compounds.
Figure 5.

Inhibitory effects of [Rh(DPAE)2chrysi]3+ (bottom, left) and [Rh(PrDPA)2chrysi]3+ (bottom, center) on cellular proliferation in MMR-deficient HCT116O (red) and MMR-proficient HCT116N (green) cells as a function of BrdU incorporation during DNA synthesis (adapted from reference 35). Percent BrdU incorporation is normalized to that of untreated cells. (Bottom, right) Subcellular localization of [Rh(DPAE)2chrysi]3+ (black) and [Rh(PrDPA)2chrysi]3+ (hashed). Mitochondrial rhodium content (left axis) has been normalized to mitochondrial protein content, and nuclear rhodium content (right axis) is expressed as the percentage of cellular rhodium in the nucleus.
All of the cell assay experiments characterizing the in cellulo effects of rhodium metalloinsertors had been undertaken on the isogenic cell lines HCT116N and HCT116O. The HCT116 parent cell line is a human colorectal carcinoma line deficient in the hMLH1 gene. This gene encodes for part of the mismatch repair (MMR) machinery; consequently this cell line is MMR-deficient. The HCT116N cell line has been transfected with human chromosome 3 (ch3), which restores MMR proficiency, while the HCT116O cell line has been transfected with human chromosome 2 (ch2), leaving it MMR-deficient.36 In this model system, however, the MMR-proficient cells and MMR-deficient cells are generated as different clones, and are distinct from the parental cell line. These differences can result in changes in chromosome stability or gene expression that are not solely due to MMR deficiency. To this end, we engineered NCI-H23 lung adenocarcinoma cells that contain a doxycycline-inducible short hairpin RNA (shRNA) that suppresses the expression of the mismatch repair gene MLH1. This provides an isogenic cell line system that can be used to directly compare MMR-proficient and MMR-deficient cells.37
It was found that these MLH1-deficient cells, which are more resistant to the DNA damaging agents doxorubicin, cisplatin, and etoposide, are indeed more sensitive to rhodium metalloinsertors (Figure 6).37 These results further validate the biological activity of rhodium metalloinsertors, as they have now been shown to exhibit selective biological effects across multiple assays and in different systems for comparing MMR deficiency to proficiency. Clearly, the strategy of targeting a specific lesion in DNA is a promising alternative to the classical approach.
Figure 6.
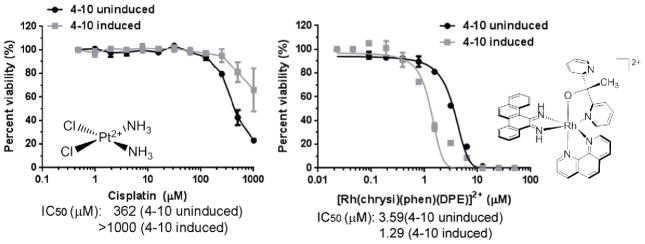
NCI-H23 subclones that were uninduced or induced for MLH1 shRNA were treated with either cisplatin (left) or the rhodium metalloinsertor [Rh(chrysi)(phen)(DPE)]2+ (right) (adapted from reference 37). Cells were treated at concentrations indicated, and cell viability assessed after 4 days using a Cell Titer-Glo assay. IC50 values are shown below the plots.
Targeted chemotherapy thus holds the potential to combat the severe side effects and acquired resistance associated with classical chemotherapeutics such as cisplatin. Many years of study have focused on achieving high potency for metal complex therapeutics. But such potency has been achieved. Just as the design of organic chemotherapeutics have shifted from potent alkylators and other inhibitors of DNA synthesis to far more tailored, subtle reagents, the design of novel metallotherapeutics now requires a targeted approach. There has really been a paradigm shift in next generation chemotherapeutic drug design that focuses on specifically tailored therapies. The unique reactivity and coordination geometry of metal complexes make them the ideal scaffold for this new tailor-made design of targeted therapeutics. The examples discussed in this Comment exemplify the enormous potential of this new strategy in transition metal chemotherapy and perhaps lay the groundwork for this burgeoning new field.
Acknowledgments
We are grateful to the NIH (GM33309) for the long time support of this work. In addition we thank NIH for a training grant to A.G.W, NSF for fellowship support for A.C.K. and the Moore Foundation for their support of the Caltech Chemistry Signaling Center.
References
- 1.Mansour VH, Rosenberg B, Vancamp L, Trosko JE. Nature. 1969;222:385–386. doi: 10.1038/222385a0. [DOI] [PubMed] [Google Scholar]
- 2.Wheate NJ, Walker S, Craig GE, Oun R. Dalton Trans. 2010;39:8113–8127. doi: 10.1039/c0dt00292e. [DOI] [PubMed] [Google Scholar]
- 3.Kelland LR, Sharp SY, O’Neill CF, Raynaud FI, Beale PJ, Judson IR. J Inorg Biochem. 1999;77:111–115. [PubMed] [Google Scholar]
- 4.Jamieson ER, Lippard SJ. Chem Rev. 1999;99:2467–2498. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- 5.Wang D, Lippard SJ. Nat Rev Drug Discovery. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 6.Decatris MP, Sundar S, O’Byrne KJ. Cancer Treat Rev. 2004;30:53–81. doi: 10.1016/S0305-7372(03)00139-7. [DOI] [PubMed] [Google Scholar]
- 7.Ang WH, Dyson PJ. Eur J Inorg Chem. 2006:4003–4018. [Google Scholar]
- 8.Beltran B, Casado P, Rodríguez-Prados JC, Cutillas PR. J Proteomics. 2012;77:492–503. doi: 10.1016/j.jprot.2012.09.029. [DOI] [PubMed] [Google Scholar]
- 9.Feng L, et al. J Am Chem Soc. 2011;133:5976–5986. doi: 10.1021/ja1112996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kunick C, Ott I. Angew Chem Int Ed. 2010;49:5226–5227. doi: 10.1002/anie.201002062. [DOI] [PubMed] [Google Scholar]
- 11.Koblinski JE, Ahram M, Sloane BF. Clin Chim Acta. 2000;291:113–135. doi: 10.1016/s0009-8981(99)00224-7. [DOI] [PubMed] [Google Scholar]
- 12.Meggers E. Chem Commun. 2009:1001–1010. doi: 10.1039/b813568a. [DOI] [PubMed] [Google Scholar]
- 13.Casini A, et al. J Med Chem. 2008;51:6773–6781. doi: 10.1021/jm8006678. [DOI] [PubMed] [Google Scholar]
- 14.Guidi F, et al. J Inorg Biochem. 2013;118:94–99. doi: 10.1016/j.jinorgbio.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 15.Holst F, et al. Nat Genet. 2007;39:655–660. doi: 10.1038/ng2006. [DOI] [PubMed] [Google Scholar]
- 16.Vessières A, Top S, Beck W, Hillard E, Jaouen G. Dalton Trans. 2006:529–541. doi: 10.1039/b509984f. [DOI] [PubMed] [Google Scholar]
- 17.Pigeon P, Top S, Vessières A, Huché M, Hillard E, Salomon E, Jaouen G. J Med Chem. 2005;48:2814–2821. doi: 10.1021/jm049268h. [DOI] [PubMed] [Google Scholar]
- 18.Gogvadze V, Orrenius S, Zhivotovsky B. Trends Cell Biol. 2008;18:166–173. doi: 10.1016/j.tcb.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 19.Wisnovsky SP, Wilson JJ, Radford RJ, Pereira MP, Laposa RR, Lippard SJ, Kelley SO. Chem Biol. 2013;20:1–6. doi: 10.1016/j.chembiol.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jackson BA, Barton JK. J Am Chem Soc. 1997;119:12986–12987. [Google Scholar]
- 21.Jackson BA, Barton JK. Biochemistry. 2000;39:6176–6182. doi: 10.1021/bi9927033. [DOI] [PubMed] [Google Scholar]
- 22.Jackson BA, Alekseyev VY, Barton JK. Biochemistry. 1999;38:4655–4662. doi: 10.1021/bi990255t. [DOI] [PubMed] [Google Scholar]
- 23.Pierre VC, Kaiser JT, Barton JK. Proc Natl Acad Sci USA. 2007;104:429–434. doi: 10.1073/pnas.0610170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cordier C, Pierre VC, Barton JK. J Am Chem Soc. 2007;129:12287–12295. doi: 10.1021/ja0739436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeglis BM, Pierre VC, Kaiser JT, Barton JK. Biochemistry. 2009;48:4247–4253. doi: 10.1021/bi900194e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song H, Kaiser JT, Barton JK. Nature Chem. 2012;4:615–620. doi: 10.1038/nchem.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loeb LA. Cancer Res. 2001;61:3230–3239. [PubMed] [Google Scholar]
- 28.Bhattacharya NP, Skandalis A, Ganesh A, Groden J, Meuth M. Proc Natl Acad Sci USA. 1994;91:6319–6323. doi: 10.1073/pnas.91.14.6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iyer RR, Pluciennik A, Burdett V, Modrich PL. Chem Rev. 2006;106:302–323. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- 30.Carethers JM, Hawn MT, Chauhan DP, Luce MC, Marra G, Koi M, Boland CR. J Clin Invest. 1996;98:199–206. doi: 10.1172/JCI118767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hart JR, Glebov O, Ernst RJ, Kirsch IR, Barton JK. Proc Natl Acad Sci USA. 2006;103:15359–15363. doi: 10.1073/pnas.0607576103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ernst RJ, Song H, Barton JK. J Am Chem Soc. 2009;131:2359–2366. doi: 10.1021/ja8081044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ernst RJ, Komor AC, Barton JK. Biochemistry. 2011;50:10919–10928. doi: 10.1021/bi2015822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Komor AC, Schneider CJ, Weidmann AG, Barton JK. J Am Chem Soc. 2012;134:19223–19233. doi: 10.1021/ja3090687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weidmann AG, Komor AC, Barton JK. Philos Trans R Soc A. 2013;371:20120117. doi: 10.1098/rsta.2012.0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koi M, Umar A, Chauhan DP, Cherian SP, Carethers JM, Kunkel TA, Boland CR. Cancer Res. 1994;54:4308–4312. [PubMed] [Google Scholar]
- 37.Bailis JM, Gordon ML, Gurgel JL, Komor AC, Barton JK, Kirsch IR. PLoS One. 2013;8:e78726. doi: 10.1371/journal.pone.0078726. [DOI] [PMC free article] [PubMed] [Google Scholar]