An official website of the United States government
Here's how you know
Official websites use .gov
A
.gov website belongs to an official
government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you've safely
connected to the .gov website. Share sensitive
information only on official, secure websites.
As a library, NLM provides access to scientific literature. Inclusion in an NLM database does not imply endorsement of, or agreement with,
the contents by NLM or the National Institutes of Health.
Learn more:
PMC Disclaimer
|
PMC Copyright Notice
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
Parkinson's disease is one of the several neurodegenerative disorders that affects aging individuals, with approximately 1% of those over the age of 60 years developing the disorder in their lifetime. The disease has the characteristics of a progressive disorder in most people, with a common pattern of pathological change occurring in the nervous system that extends beyond the classical striatal degeneration of dopaminergic neurons. Earlier studies concluded that the disease was a disorder of alpha-synuclein, with the formation of aggregates of abnormal alpha-synuclein being characteristic. More recent studies have concluded that inflammation plays a central role in the disorder and that the characteristic findings can be accounted for by either mutation or oxidative damage to alpha-synuclein, with resulting immune reactions from surrounding microglia, astrocytes, and macrophages. What has been all but ignored in most of these studies is the role played by excitotoxicity and that the two processes are intimately linked, with inflammation triggered cell signaling enhancing the excitotoxic cascade. Further, there is growing evidence that it is the excitotoxic reactions that actually cause the neurodegeneration. I have coined the name immunoexcitotoxicity to describe this link between inflammation and excitotoxicity. It appears that the two processes are rarely, if ever, separated in neurodegenerative diseases.
Russell Blaylock has written several landmark papers for Surgical Neurology International (SNI) over the years. He introduced the concept of immunoexcitotoxicity as the cause of chronic traumatic encephalopathy in 2011.[1] The concept fundamentally changed the understanding of repeated brain trauma and explained why football players and others in head contact sports develop dementia, Alzheimer’s, and Parkinson's disease. He now explains in the following paper how immunoexcitotoxicity plays a key role in the development of Parkinson's syndrome and the disease process.
Dr. Blaylock has pioneered new concepts from which others have shied away because of controversy as a new idea. He has also written a two-part series for SNI on the immunology of a neurosurgeon.[2,3]
He writes a monthly newsletter called the Blaylock Wellness Report, which is commercially available, and discusses the value of neutaceuticals, naturally occurring substances, which have a profound effect on health and wellness. These are also pioneering, scientifically-detailed publications. Now, there is a much larger group of physicians who are exploring the value of these molecular supplements in combating major diseases.
His most recent paper deals with the pathophysiology of Parkinson's disease and the key role that immuneexcitotoxicity plays in the development of this disease over time. This concept can be extended to other neurodegenerative diseases. His paper is a major tour de force and may take some time to read, but its scientific logic is incredibly well-documented and all fits into a plausible explanation for this disease. What he has written will be a landmark paper for the 21st century about Parkinson's disease and will lead to treatments and cures of the problem. As I have written, the immune system in disease will become a major new area of medical science in the 21st century and will explain many of the diseases whose causes have eluded us. I have asked him to write a summary of the paper as an introduction for the reader who would like to know the basics of what he has found. Following that summary is his major paper on the subject.
We hope that you enjoy it. We are proud to bring this masterwork in science to the attention of our readers. Please share it with your neurologist friends and others who would be interested in this concept. You could even have a group take sections of this paper and discuss them, as the learning experience for immunology and the central nervous system will provide a basis for understanding many diseases, for which the causes are still unknown.
Publishing this paper continues SNI's long-standing concept of bringing new, creative, and innovative concepts to our readers before they are in the mainstream publications. In the coming years, you will see many more publications that echo what Russell has written.
There is growing evidence that inflammation and excitotoxicity both play a central role in Parkinson's disease, in particular the progressive aspect of neurodegeneration associated with the disorder. While the issue is not completely settled, it appears that alpha-synuclein may be playing a secondary role in this disorder rather than being the central pathological event. Several recent studies have shown there is no direct correlation between Lewy body density and clinical symptoms of Parkinsonism or with disease duration. Experimental studies using transgenic mice have likewise shown the presence of abundant alpha-synuclein in the brain accompanied by no evidence of neurodegeneration.
Among the various animal models used to study this enigmatic disorder, i.e. neurotoxic and immunological models, it is the immunological models that most closely resemble human cases of Parkinson's disease, especially its slow appearance and gradual progression with acceleration of degeneration over time.
Numerous studies, both in human cases and among animal models, demonstrate strong evidence of inflammation within the affected areas of the brain and within the cerebrospinal fluid. McGeer, almost 30 years ago, found extensive microglial activation within and around the substantia nigra in autopsied cases of Parkinson's disease. Several studies suggest that microglial activation appears to be a very early event in Parkinson's cases, and is present in the all the animal models as well, including the neurotoxic models.
The questions that most need answering is what is activating the striatal microglia and why do they remain activated for such long periods? Over the years, a multitude of factors have been associated with an increased risk of Parkinson's disease, such as exposure to certain pesticides/herbicides and fungicides, accumulation of environmental aluminum, repeated head injury, aging, exposure to high levels of manganese, exposure to high levels of polychlorinated biphenyls (PCBs) and trichloroethylene (TCE), and a genetic influence is suspected as well, one that interacts with environmental pollutants.
In a majority of such cases, one sees chronic activation of brain microglia. While such associations have gained significant attention, a more common set of conditions may be at play. It is known that most cases of Parkinson's disease are preceded by many years with chronic digestive tract problems, such as drooling, difficulty swallowing, poor gastric emptying, abdominal distention, and in a great number of cases, chronic constipation. As regards to the proposed etiology, i.e. systemic immune activation, it is interesting to note that 85% of the examined cases of Parkinson's disease were found to have dysfunctional olfaction.
Associated with these gastrointestinal-related problems, one sees the appearance of Lewy bodies and Lewy neurites in the autonomic ganglion, the vagus nerve, brainstem nuclei, and spinal cord. Simultaneously, similar Lewy bodies and Lewy neurites appear in the olfactory tract and nuclei. Based on several autopsy studies, the pathological process appears to ascend from the wall of the gastrointestinal tract and progresses along the vagus nerve and other autonomics, where the process invades brainstem nuclei and subsequently the striatum, and finally, the rest of the brain. From the olfactory tract and nuclei, one observes similar ascension of the neurodegenerative process into deeper brain structures.
Some have proposed that it is by this mechanism that the mutated alpha-synuclein enters the brain and eventually affects the neurons of the substantia nigra par compacta, i.e., by traveling along a transneural pathway. While such passage of mutated alpha-synuclein between neurons has been demonstrated experimentally, it remains to be demonstrated in human cases of Parkinson's disease.
A more likely scenario would be priming/activation of brainstem microglia via stimulation of vagal afferents by an inflamed gut or in relation to other systemic immune activation. Priming could occur from episodes of systemic immune stimulation, such as with recurrent viral or bacterial infections, or from intracranial events, such as head trauma, cerebral infections (even occult infections), accumulation of toxic metals in the brain, or exposure to agricultural chemicals that are known to affect the central nervous system (CNS).
It is known that primed microglia undergo upregulation of their immune cytokine and chemokine generation, however, without release of these elements. That is, the primed microglia are placed on high alert. A subsequent stimulus will further activate these primed microglia, triggering a much more intense immune reaction than normally occurs with microglial activation—a sort of hyperreaction. Not only do we see a massive release of proinflammatory immune elements but also several excitotoxic elements, such as glutamate, aspartate, and quinolinic acid.
It appears that the source of the immune activation of the primed microglia is some form of systemic immune response. It has been shown that when brain microglia are primed, subsequent systemic immune activation will trigger neurodegeneration with the brain, which can be prolonged and intense. In this discussion, I have focused on two sites of systemic inflammation—olfactory and gastrointestinal.
Considerable evidence has shown that there is a crosstalk between the proinflammatory pathways within affected neurons and excitotoxic receptors. The effect of this crosstalk is an increased intensity of the neurodestructive process. I coined the term immunoexcitotoxicity to describe the process, even though others described the process prior to my giving it a name. What this immunoexcitotoxic process means is that, when the brain is inflamed for any reason, one observes a magnification of excitotoxicity—so much so, that even otherwise normal concentrations of extracellular glutamate can trigger neurodegeneration.
Of particular interest is the finding that inflammatory cytokines, such as tumor necrosis factor-alpha (TNF-alpha) and IL-1ß, can alter the synaptic insertion of specific types of glutamate receptors and even GABA receptors, which ultimately magnify the excitotoxic process. TNF-alpha, for example, increases the insertion of GluR2-lacking AMPA type glutamate receptors on the synaptic membrane. These receptors, unlike the GluR2-containing AMPA receptors, are calcium permeable—thus making them more potentially neurodestructive. These inflammatory cytokines also alter NMDA-type glutamate receptor synaptic insertion and can simultaneously accelerate the removal of GABA receptors form the synaptic membrane. These series of events push the excitotoxic/GABA balance toward increased excitotoxicity.
In addition, inflammatory cytokines upregulate the enzyme glutamatase, which increases the generation of glutamate within microglia and neurons. Proinflammatory cytokines also inhibit glutamate transport proteins (excitatory amino acid transporters or EAATs), thus elevating the level of extracellular glutamate.
One of the early observations in Parkinson's disease was the presence of mitochondrial dysfunction, especially involving complex I of the electron transport chain. Several studies have shown that suppression of energy production in neurons significantly increases their sensitivity to excitotoxicity—so much so that even normal concentrations of extracellular glutamate can become excitotoxic.
It appears that the central mechanism at play in this disorder is a triggering of priming of microglia within specific brain areas that eventually progress to full microglial activation involving other areas of the brain.
Full microglial activation occurs with systemic immune stimulation. Further, compelling evidence suggest that the main process involved in neurodegeneration is excitotoxicity, with immune-induced inflammatory pathways acting to significantly enhance excitotoxic mechanisms.
Surg Neurol Int. 2017 Apr 26;8:65.
PART 1: IMMUNOEXCITOTOXICITY AS A CAUSE OF NEURODEGENERATION IN PARKINSON’S DISEASE
A. An outline of immunoexcitotoxicity in neurodegeneration
In this discussion, I will limit myself to idiopathic Parkinson's disease, a progressive neurodegenerative disorder that typically begins around the age of 60 years, with an increased incidence with aging.
a. Triggering event>Immune-induced inflammation by CNS microglia and systemic macrophages >Excitotoxicity>Progressive neurodegeneration
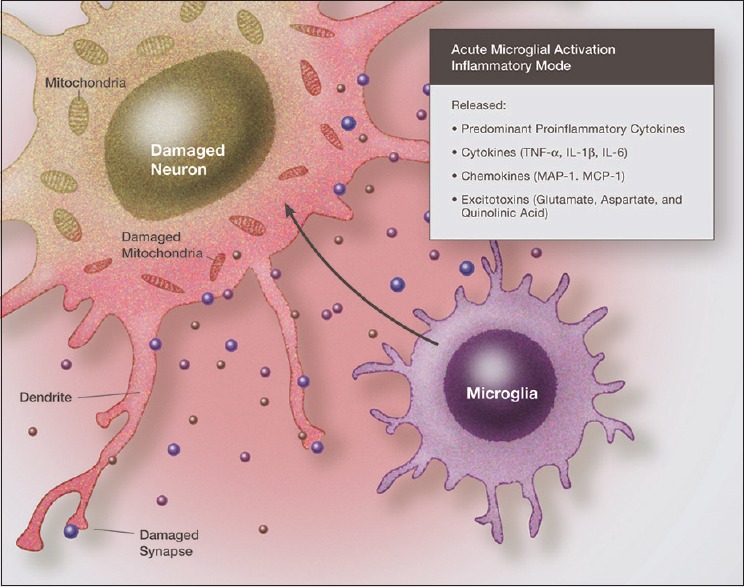
There is considerable evidence that two processes play a major role in the neurodegeneration seen in Parkinson's disease—immune-induced inflammation and excitotoxicity.[2,5,49,143,157,228,244,253] Over the past 10 years, evidence has accumulated that these two processes are intimately linked and probably never occur independently in the pathological nervous system. In the majority of cases, the source of the inflammation and excitotoxicity is the resident immune cells of the central nervous system (CNS), the microglia, and under certain conditions, invading macrophages[125,148,251] [Figure 1].
Illustration demonstrating the neurotoxic effects of proinflammatory cytokines and chemokines acting in synergy with excitatory amino acids. Immunoexcitoxicity results in damage to neuronal cell membranes, mitochondria, DNA, as well as dendrites and synapses mainly by the excitotoxic mechanisms
b. Normal resting microglia>Nurture of neurons, neurites, and synaptic structures
It has been demonstrated that microglia undergo numerous morphological and functional states depending on the microenvironment within the area of the affected brain.[118,125,136,148,251] Under normal conditions, microglia are in a resting or ramified state, which has been shown to be less of a resting state than a condition in which the microglia sends out a number of probing pseudopods to sample the surrounding microenvironment.[241] While in the resting state, microglia can also release a number of neurotrophic compounds that nourish the surrounding neurons, neurites, and the tripartite synapse (presynaptic membrane, post-synaptic bulb, and an astrocyte).
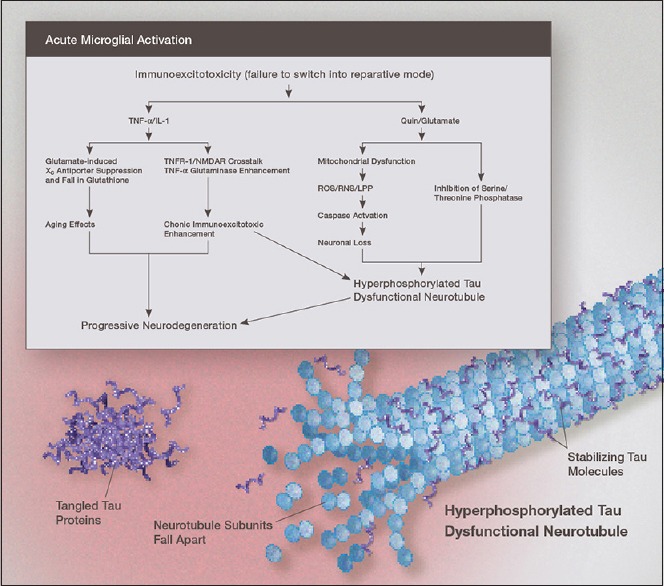
This is particularly important in recovery following microglial activation states in which varying amount of damage is done to surrounding structures that requires repair [Figure 2]. The microglia also release a number of anti-inflammatory cytokines, such as IL-10, TGF-1ß, and IL-4, to counteract inflammation induced by full microglial activation.
Illustration of the mechanism linking immunoexcitotoxicity with increased deposition of hyperphosphorylated tau in the chronically inflamed brain. Once the microglia fail to switch to the reparative or resting mode, elevated levels of IL-1ß, TNF-, and other proinflammatory cytokines and chemokines, act to enhance excitotoxicity by a number of mechanisms. High levels of glutamate and nitric oxide suppress mitochondrial energy generation. Both elevated QUIN and glutamate increase levels of oxidized and nitrated alpha-synuclein
c. Neuroprotection by resting microglia >Importance of microglial priming
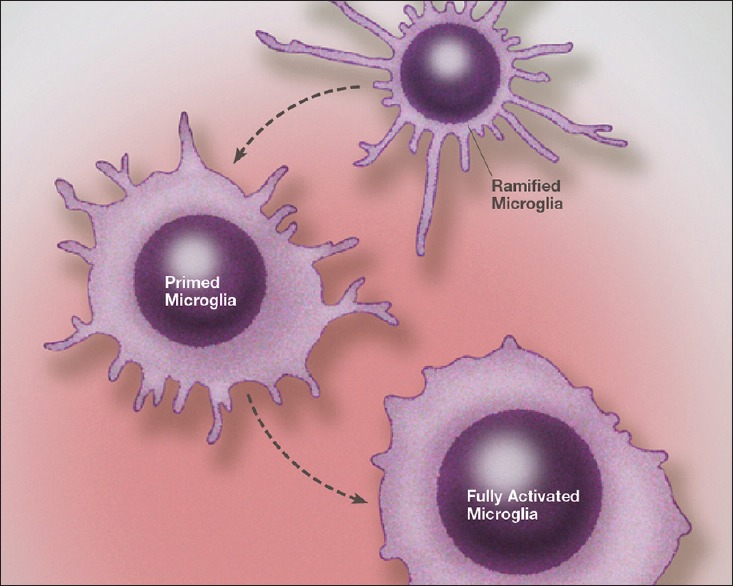
It is known that microglial activation occurs rapidly (minutes) following a pathological insult and, under specific conditions, can persist for very long periods, even a lifetime.[203] Further, there is an intimate connection between systemic inflammation and activation or priming of central microglia [Figure 3].[174] Most important is the process of microglial priming, which occurs with low-grade inflammation.[105,189] It is characterized by an increase in the production of both pro-and anti-inflammatory cytokines, chemokines, and other inflammatory molecules, as well as an increase in the production of glutamate, all of which remain within the microglia. That is, there is no release of these destructive elements from primed microglia, and hence, no damage to the surrounding neuronal or neuritic elements. However, once the microglia become fully activated, there is a hyperreaction with an intense presentation of inflammation and excitotoxicity that exceeds that normally associated with microglial activation.[175]
Illustration of microglia priming/activation transition states beginning from a resting (ramified) state. Recent studies indicate that microglia can assume a number of activation states, such as predominately phagocytic, predominately neuroprotective or predominately neurodestructive. In the primed state the mRNA for cytokines, chemokines and other reactive molecules are upregulated but active proteins are not released
This link between inflammatory mechanisms and glutamate receptors works in both directions, that is, inflammation triggers the release of excess glutamate, and likewise, excess extracellular glutamate can trigger inflammation, making this association a self-propagating pathological process. In both instances, it is the microglia that plays a central role. Not only do we see activation of microglia in the region of insult but also migration of activated microglia to other sites occurs as the process spreads.[162,246]
It is known that microglia are heterogeneously distributed in the nervous system, making some areas of the brain and spinal cord more vulnerable than others.[57,134] For example, the substantia nigra is known to have one of the highest concentrations of microglia in the brain.[124] Before discussing the immunoexcitotoxic process, I will discuss the evidence for a role of inflammation in Parkinson's disease.
B. INFLAMMATION AND PARKINSON’S DISEASE
a. Triggering event>Inflammation induced full activation of primed microglia>Enhanced release of proinflammatory cytokines/chemokines >Generation of mutated alpha-synuclein and its release into extraneuronal space>Further enhancement of immunoexcitotoxic cascade
McGeer et al., almost 30 years ago, first identified increased microglial activation within the substantia nigra in post-mortem Parkinson's brains.[156] Most now agree that inflammation is central to the initiation and progress of Parkinson's disease as well as a number of other neurodegenerative disorders.[66,99,138,142,156,158,228,249] Further evidence comes from studies showing elevated levels of IL-1ß in the brain of Parkinson's disease patients and elevated levels of IL-1ß, TNF-α, and IL-6 in the spinal fluid of Parkinson's patients.[28,163,164] Some postmortem studies have shown extensive microglial activation within the substantia nigra, putamen, hippocampus, transenterhinal, cingulate, and temporal cortex, while others found profound microglial activation in the SN but not the putamen.[110,161] Most likely this distribution of microglial activation depends on the stage of the disease when the tissue is examined. It has also been shown that inflammatory cytokines, such as IL-1ß, promote upregulation of alpha-synuclein in neuronal cultures and in vivo, suggesting inflammation as a primary trigger for elevated alpha-synuclein accumulation in Parkinson's disease.[93]
b. Alpha-synuclein: Normal and mutated>Form solid oligomers in ECS>Toxic extrcellular aggregates>Activation of microglia; Intracellular mutated alpha-synuclein>Alters function of mitochondria, lipid membranes>Suppresses electron transport chain>Lowers energy function >Neurodegeneration
Alpha-synuclein is a 140-amino acid protein that is principally found in neurons, which under physiological conditions are localized primarily within presynaptic terminals. While abundant in the CNS, they are also abundant in erythrocytes and platelets. Within the neuronal soma, alpha-synuclein appears in a much lower concentration. While its function is poorly understood, because alpha-synuclein is found in the area of the synapse, it has been hypothesized that it plays a role in neurotransmitter release to control neuronal firing.[113]
Under pathological conditions, alpha-synuclein can undergo numerous mutational changes, such as phosphorylation, oxidation, glycation, and nitrosylation.[150,222] When mutated, alpha-synuclein can form ß-sheets, much like amyloid, which can then form soluble oligomers. These soluble oligomers can escape the neuron and enter the extracellular space, where they become much more toxic.[74,207,222] Over time, these oligomers will aggregate to form insoluble fibrils. It appears that the fibrils are much less toxic than are the oligomers. Mutation of alpha-synuclein occurs in a microenviroment of oxidative and nitrosative stress, most likely induced by inflammation and excitotoxicity (immunoexcitotoxicity).[222] Mutated alpha-synuclein can alter the function of mitochondria, lysosome, and lipid membranes.[4,107,222] Within the mitochondria, mutated alpha-synuclein has been shown to suppress complex I of the electron transport chain, which would not only lower energy production but would also increase oxidative stress, both of which, in turn, significantly enhances the neurodegenerative potential of excitotoxicity.[143,173,231]
In the extracellular space, alpha-synuclein has a high propensity for eliciting immune activation of microglia.[14,150,207,231] There is some evidence that mutated alpha-synuclein can move between neurons and neurites, thus transmitting the formation of oligomers.[4,74,207] While this has been proposed and there is some experimental evidence for this transfer, in my opinion the preponderance of evidence suggest that the progression of neurodegeneration seen in Parkinson's is secondary to microglial migration and chronic activation.
c. Inflammation, microglia, and alpha-synuclein
1. Microglial activation: Its role in the neurodegenerative process; Aggregated alpha-synuclein>Microglia response coupled with release of inflammatory cytokines
Reactive microglia are found most often around Lewy bodies and alpha-synuclein aggregations.[150] Activation of toll-like receptor-2 within the substantia nigra has also been described, however, TLR-4 seems to be critical for the inflammatory reaction of microglia.[70,76] There is growing evidence that microglial activation is occurring before one sees neuronal loss.[99,222] Neurons, the source of mutated alpha-synuclein, can escape into the extraneuronal space, where it comes into contact with microglia.[4] Roodveldt et al. found that non-aggregated mutant α-synuclein, when found outside the neuron, triggered a robust microglial immune response significantly different from nonmutated wild-type α-syn.[207] The triggered inflammatory reactions include the release of nitric oxide and an array of inflammatory cytokines, such as TNF-α, IL-Iß, IL-6, anti-inflammatory cytokine IL-10, as well as chemokines, RANTES, monocyte chemotactic protein-1 (MCP-1), CXCL-10, and macrophage inflammatory protein-1α. Other inflammatory events are also included, such as increased production of prostaglandins (PGE2), complement cascade proteins, and the humoral adaptive immune system may be involved as well.[205,231]
2. Lipopolysacchride (LPS) injection into the substantia nigra>Alpha-synuclein>Causes an immediate inflammatory reaction with rapid neurodegeneration
Lipopolysacchride, an immune stimulating substance extracted from the cell wall of Escherichia coli, is used experimentally to induce rapid and intense activation of microglia and macrophages. The intensity of the reaction is dose dependent.
There appears to exist a direct link between inflammation and the development of mutated alpha-synuclein. For instance, injection of LPS into the substantia nigra of adult wild-type mice led to widespread alpha-synuclein inclusion pathology with elevated levels of microgliosis in the SN and progressive neurodegeneration of dopaminergic neurons within the substantia nigra.[226] TNF-alpha knockout animals were not protected from neurodegeneration or behavioral parkinsonian behavior following LPS injection systemically, indicating that TNF-alpha was not necessary for dopaminergic (DA) neuron destruction. Interestingly, IL-1 knockout mice maintained normal behavior, indicating that IL-1ß was essential for microglial toxicity towards dopaminergic neurons.
3. Intraperitoneal injection (IP) of lipopolysacchride activates systemic immune stimulation and a delayed, progressive neurodegeneration of the substantia nigra
In another study, using a single intraperitoneal injection of LPS in mice (systemic immune stimulation), it was shown that delayed and progressive destruction of dopaminergic neurons occurred in the SN over a 10-month period.[199] Also, of interest, it was found in this i.p. study that TNF-α progressively increases in the SN and remained elevated at 10 months, whereas systemic TNF-α levels fell by 9 hours—indicating that the source of the TNF-α was from activated microglia within the striatum and not systemically generated and transferred into the CNS. In this study, TNF-α KO mice did not show neurodegeneration, suggesting that the mechanism for progressive neurodegeneration differs in the two models.
In the systemic immune model, TNF-α receptors were necessary for transfer of the immune signal to the CNS, however in the former direct substantia nigra LPS injection model, because the microglia were being stimulated directly, TNF-α receptors were not essential. IL-1ß is known as a major activator of microglia and was therefore necessary and essential. IL-1ß is elevated by LPS given via systemic injection and rapidly raises CNS IL-1ß levels, thus leading to neurodegeneration by activating microglia.[200] Interestingly, at 2 hours following i.p. injection of the LPS, serum IL-1 was elevated but not brain levels, but at 6 hours, IL-1 was elevated throughout the brain of the animals but was not detected in the serum. This experiment indicates that once the signal was transferred to the CNS, IL-1ß was generated by local cells, primarily microglia, and was not continuously transferred from the serum. Thus, it is accepted that IL-1ß is a major stimulus for microglial activation.[211] Microglia also release IL-1ß, leading to a cycle of self-stimulation. Indeed, activated microglial cells expressing proinflammatory cytokines, such as Il-1ß, TNF-α, IL-6, and INF-y, as well as up-regulation of iNOS have been reported in the substantia nigra of Parkinson's patients at autopsy.[104,108]
The reason for the delay in appearance of CNS neurodegeneration and its persistence is related to the effect of systemic immune activation on microglial priming and eventual full activation, which can persist for very long periods—even decades.
4. Further evidence for microglial activation and neurodegeneration in patients and animal models of Parkinson's disease: Evidence of the spread of microglial activation during disease progression
Demonstrations of progressive neurodegeneration and microglial activation have best been shown using positron emission tomography (PET) imaging techniques in both human cases of Parkinson's disease as well as in animal models. In one such study, researchers used early stage, drug naïve Parkinson patients scanned using [11C] CFT binding of the dopamine transporter, which when combined with [(11) C](R)-PK11195 demonstrated destructive microglial activation.[181,182] They found destructive microglial activation only in the midbrain area, which correlated with the loss of dopaminergic terminals in the striatum. As the disease progressed, they witnessed spreading of the microglial activation over the entire brain along with progressive destruction of dopaminergic terminals. This strongly suggests that the source of dopaminergic neuron loss was the activated microglia, which always preceded the neuronal destruction. Progression of microglial activation preceding or paralleling neurodegeneration as the disease advances has also been demonstrated using microglial activation scanning with the more accurate tracer [11C] DPA713.[230] A significant increase in microglial activation was seen in the occipital, temporal, and parietal cortex in Parkinson patients. One year later, there was a significant increase in microglial activation in the occipital and temporal areas. Another PET study of more advanced patients with a related disorder, supranuclear palsy, found microglial activation concentrated in the pons, basal ganglion, frontal, and temporal cortical regions.[85]
D. PET microglial scanning and glucose utilization in Alzheimer’s, Parkinson’s, cognitively impaired, and control patients
Another interesting study using human partcipants, which included 10 Alzheimer's patients, 10 patients with mild cognitive impairment; 10 Parkinson diseases dementia patients, and 16 controls, also performed PET scanning for microglial activation as well as glucose utilization.[75] There was a significant correlation between microglial activation and reduced glucose metabolism for all the cognitively impaired patients. Levels of microglial activation were inversely related to the results on the Mini-Mental State Examination and neuropsychometric tests, with higher levels of microglial activation associated with poorer results on the mental tests and vice versa. This study demonstrated a direct correlation between neuronal damage, microglial activation, and loss of cognitive abilities. Edison et al. found similar correlations.[73]
A second generation imaging tracer, [11C] DPA713, used for microglial activation has been shown to be more accurate in determining microglial activation than the first generation tracer [11C]®PK11195.[257]
E. Proposed mechanisms for inflammation-linked neurodegeneration: How neuronal cell death occurs
A number of mechanisms have been proposed for inflammation-linked neurodegeneration, including (1) damage by reactive oxygen/reactive nitrogen species (ROS/RNS), (2) damage by proteases, (3) TNF-linked receptor activation of death domains, (4) defects in axonal transport, and (5) mitochondrial dysfunction.[138] Most such hypotheses have at their core destruction of vital cell mechanism by ROS/RNS and lipid peroxidation products, such as 4-hydroxynonenal and acrolein.[11,115,138]
4. Alpha-synuclein in the extracellular space>Microglial activation>Inflammation>Generate reactive nitrogen and oxygen species>Effect cell components>Effects mitochondrial function; Neurotoxic cytokine TNF-alpha plays major role in neurodegeneration
Mutant or nitrated alpha-synuclein in the extracellular space has been shown to elicit immune activation (microglial activation) and trigger inflammation. The release of nitric oxide by upregulating iNOS not only increases the production of powerful radicals but also nitrates alpha-synuclein, thus making the molecule more inflammatory.[4,101]
Toll-like receptor-2 and TLR-4, PAMPS receptors located on microglia, are essential for phagocytosis and removal of alpha-synuclein.[123] TLR-4 is also essential for inflammatory activation of microglia and plays a role in phagocytosis activation.[76] The act of engulfing the mutated alpha-synuclein can activate the microglia, thus initiating a immunoexcitotoxic cascade.
Once activated, inflammatory reactions generate ROS/RNS, which damage a number of cell components, including membranes, DNA, and mitochondria. An inducible enzyme, NADPH oxidase, primarily generates the superoxide radical, which by itself is a rather weak free radical. NADPH oxidase is primarily found in microglia and can be induced when the microglia are in the proinflammatory phenotype.[39]
Superoxide reacts rapidly with nitric oxide to form the powerful radical peroxynitrite, which can nitrate alpha-synuclein. Inflammation-induced upregulation of iNOS and activation of NADPH oxidase thus creates a form of nitrated alpha-synuclein that is significantly more inflammatory than is wild-type alpha-synuclein.[231] Peroxynitrite in its own right is a very powerful radical and a major source of the lipid peroxidation product 4-hydroxynonenal, a very destructive lipid radical.[15]
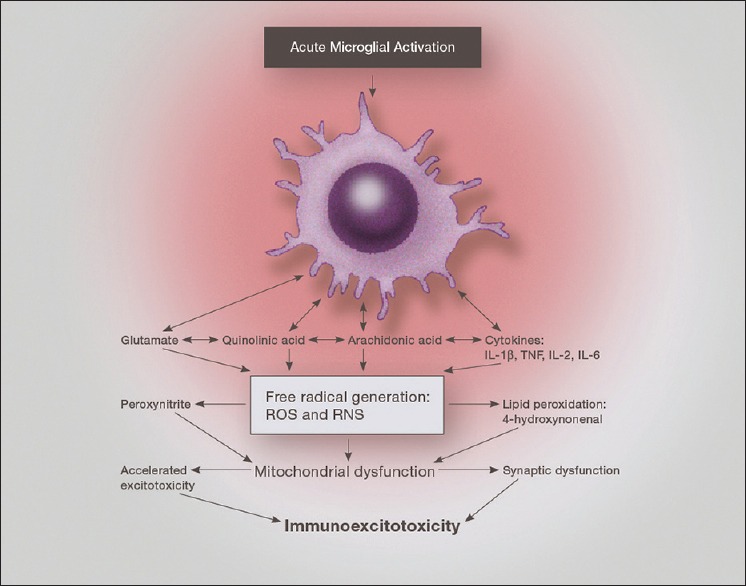
One proposed mechanism by which inflammation induces neurodegeneration is by the effect of free radicals and lipid peroxidation products (LPOs) on mitochondrial function[40,114,115,144] [Figure 4]. The rapidity of neuronal death following short-term exposure to nitric oxide suggest that neither peroxynitrite nor direct energy deficits produced by nitric oxide are responsible for neuronal death seen with inflammation.[9]
Illustration of the neurotoxic factors released from an activated microglia, demonstrating the interaction of proinflammatory cytokines, prostaglandins and excitatory amino acids. Of particular importance is the effect on mitochondrial function, which when depressed enhances excitotoxic sensitivity as well as reactive oxygen species generation
TNF-α has been proposed as a major neurotoxic cytokine in inflammatory brain disorders, including neurodegenerative diseases. It has not been proven to be directly toxic to neurons.[100] TNF-α acts through two major receptors, i.e., TNFR1 and TNFR2. TNFR1, when activated is neurotoxic and is found mostly on neurons, whereas TNFR2 is neuroprotective and is found mostly on glia, including microglia.[52,53] While activation of free radicals and lipid peroxidation products might explain the toxicity of TNF-α to neurons, there are other less direct explanations, which are discussed later. TNF-α is known to induce neuronal death by inducing microglial phagocytosis.[171]
5. Immunoexcitotoxicity and Parkinson's disease: Crosstalk between cytokines and glutamate receptors
a. What is immunoexcitotoxicity?>Proinflammatory cytokines>TNF-alpha and IL-1B; Role of glutathione in cell death
Excitotoxicity has been demonstrated in numerous human and animal studies of Parkinson's disease.[5,7,21,22,49,69,160,167,176,254] There is a considerable amount of evidence for excitotoxicity in Parkinson's disease brains, but is it a critical player in the process or is the progressive destruction primarily dependent on inflammation? In an article published in 2008, I coined the term immunoexcitotoxicity to call attention to a previously described interrelationship between inflammatory mechanisms and excitotoxicity.[23]
Proinflammatory cytokines are known to affect the excitability of specific neurons either directly or indirectly.[159,240] Indirect activation of excitatory receptors can occur by cytokine-induced release of nitric oxide, prostaglandins, or neurotropins.[3] The two most studied cytokines as regards neuronal excitability include IL-1ß and TNF-α.
In the case of TNF-α it has been shown that when used alone in organotypic hippocampal-entorhinal cortex brain slices, little neuronal damage occurs, but when submaximal glutamate concentrations were added, neurotoxicity was considerably accelerated.[264] Neurotoxicity in the brain slices was blocked by using NMDA receptor blockers in this study, and when blocked, despite the continued high level of TNF-alpha, no significant neuronal destruction was seen. It was proposed that the toxicity of TNF-α was caused by the cytokine inhibiting glutamate reuptake, which has been demonstrated several times.[179,264]
Of interest, is the finding that the glutamate transporter EAAT1 also transports glutathione precursors into dopaminergic neurons, and that inhibiting this transporter dramatically and selectively increases the sensitivity of dopaminergic neurons to excitotoxic death by reducing intracellular glutathione levels as well as raising extraneuronal glutamate levels.[167] Glutathione is a major cellular antioxidant molecule, and when exposed to high levels of free radicals, glutathione becomes oxidized and must be converted to its reduced form to have antioxidant function. Low levels of cellular glutathione are associated with increased vulnerability of cells to oxidative stress. Glutathione is mainly generated within astrocytes and is transported to neurons.
Selective loss of dopaminergic neurons in the substantia nigra was demonstrated in animal models in which a selective blocker of glutamate transporters was used.[7] In this study, the authors found that excitotoxicity was significantly enhanced in the dopaminergic neurons selectively via NMDA receptors, and that raising glutathione levels using N-acetyl-L-cysteine was protective, again emphasizing a dual role of the EAAT glutamate transporter in both glutamate reuptake and transfer of glutathione precursors into the dopaminergic neuron.
6. TNF-alpha enhances excitotoxicity by trafficking specific excitatory receptors
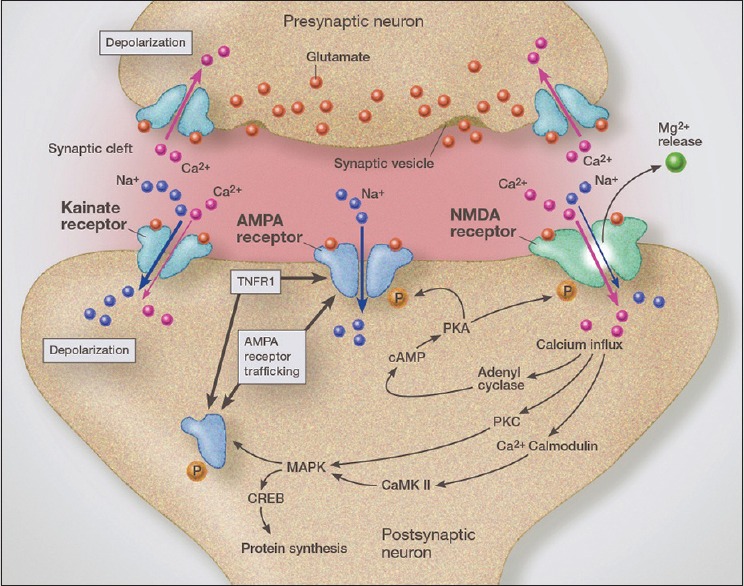
Another mechanism by which TNF-α enhanced excitotoxicity is through its receptor, primarily TNFR1.[18] The main effect of TNF-alpha is on trafficking specialized calcium-permeable AMPA glutamate receptors. AMPA glutamate receptors are fast conducting glutamate receptors that normally, unlike NMDA receptors, are calcium impermeable. It is the GLuR2 subunit that normally blocks calcium permeability by the AMPA receptor. Under pathological conditions, GLuR2-lacking AMPA receptors are constructed by the neuron's endoplasmic reticulum and transported and inserted into the synaptic plate. Stimulation of the TNFR1 receptor by TNF-alpha is a major stimulus for trafficking calcium-permeable AMPA receptors to the synapse. These specialized, calcium-permeable receptors are more excitotoxic than the calcium impermeable AMPA receptors.
Interestingly, it has been demonstrated by this study that the effect of TNF-α was dose dependent, with low doses being neuroprotective against AMPA excitotoxicity (acting on the AMPA glutamate receptor) and higher doses of TNF-alpha triggering enhanced AMPA-induced neuronal death. It appears that low dose TNF-alpha acts via the TNFR-2 receptor (neuroprotective), and the high dose of TNF-alpha acts through TNFR-1, which increases trafficking of calcium-permeable AMPA receptors to the synapse, which enhances excitotoxicity.
7. Proinflammatory cytokine IL-1ß enhances excitotoxicity by increased trafficking of NMDA glutamate receptors
Gardoni et al. demonstrated significant crosstalk between IL-1ß receptors and NMDA receptors that could also lead to enhanced excitotoxicity.[84] Of interest, they also found that stimulation of the NMDA receptor increased the expression of IL-1ß receptors on the post-synaptic membrane, indicating a two-way interaction between excitotoxicity and inflammation. Increased neuron excitability occurs not only because of cytokine modification of voltage-gated or receptor-coupled ion channels but also by promoting presynaptic changes in neurotransmission.[81]
IL-1ß application directly to neurons in the absence of microglia has little effect on neuron excitability.[81] In a mixed culture, containing microglia, the effect of IL-ß depends on the concentration. Low concentrations can decrease glutamate receptor activity and high concentrations enhance glutamate receptor activity.[45] The main influence of IL-1ß on neuronal excitability appears to be through the NMDA receptor, mainly by affecting its subunit composition. For example, a high dose of IL-1ß cause an upregulation of NR2B/A subunits that produced long-lasting alterations in subunit expression in NMDA receptors, thus increasing the excitability of affected neurons.[133,239]
8. IL-1ß and TNF-alpha enhance excitotoxicity by endocytosis of GABA receptors, exocytosis of GluR2-Lacking AMPA receptors, and NMDA receptors
Another mechanism by which IL-1ß affects neuron excitability is by modulating GABA potential. GABA receptors modulate neuron excitability by downregulating synaptic excitation. Wang et al. demonstrated a dose dependent IL-1ß suppression of GABA function that increased neuron excitability.[245]
A great number of studies have shown that elevation in TNF-alpha can produce neuronal excitability and enhance excitotoxicity. For example, using hippocampal slices and mixed cultures, researchers have shown that TNF-alpha enhances glutamate neurotransmission.[89,227] Riazi et al. also demonstrated an increase in brain excitability in relation to increasing levels of TNF-alpha induced peripherally, primarily by TNF-alpha activating microglia.[206]
Fast conduction in the CNS is mediated mostly by AMPA glutamate receptors. TNF-alpha has been shown to increase trafficking of internal AMPA receptors onto the synaptic surface, thus increasing sensitivity of the neuron to glutamate stimulation (synaptic scaling) [Figure 5].[229,235] Of critical importance is the observation that it is GluR2-lacking AMPA receptors that make up most of the trafficked receptors during inflammation[147,183] [Figure 3]. GluR2-lacking AMPA receptors, unlike GluR2-containing AMPA receptors, are calcium permeable, thus magnifying their excitotoxic potential.[140] In addition, TNF-alpha increases endocytosis of GABA receptors, thus reducing inhibitory protection against excitotoxicity.[197,220] Recent studies also indicate that higher levels of TNF-alpha, through a ceramide-dependent mechanism, increases the phosphorylation of the NR1 NMDA receptor subunit, thus increasing its surface insertion, which increases the neuron's sensitivity to glutamate.[248]
Illustration of glutamatergic synapse demonstrating rapid AMPA receptor trafficking from the endoplasmic reticulum, which is driven by activation of TNFR1. Crosstalk between the AMPA receptor and TNFR1 increase synaptic insertion of GluR2-lacking (calcium permeable) AMPA receptors, thus increasing synaptic glutamate-related sensitivity (excitotoxicity). TNFR1 activation also increases GABA receptor endocytosis, which increases synaptic sensitivity to excitotoxicity even further
9. Cytokine interaction required for neurotoxicity>Role of nitric oxide in immunoexcitotoxicity
In teasing out the actual mechanism of inflammation-induced neurodegeneration, it has been shown that IL-1ß and TNF-alpha, when either is used alone, do not cause neuron toxicity no matter the concentration, but that when combined they cause marked neuronal injury.[51] In this study, proinflammatory cytokines were linked to excessive generation of NO and blocking NO reduced the toxic effect of IL1ß and TNF-alpha on neurons by approximately 45%. Of real interest is that blocking the NMDA receptor reduced toxicity of these cytokines when used in combination by 55%. Also, of interest is the fact that during excitotoxicity NO is markedly elevated as well as reactive oxygen and nitrogen species.[65,258] It should also be appreciated that in this study only the NMDA receptor was blocked, leaving other glutamate receptors active. These unblocked glutamate receptors would include the AMPA/Kainate receptors, GluR2-lacking AMPA receptors, and metabotropic receptors. These unblocked glutamate receptors could account for an even greater contribution to the excitotoxic arm of the immunoexcitotoxic process. Further evidence of this interaction between inflammation and excitotoxicity comes from a study that found that using subtoxic concentrations of rotenone with subtoxic concentrations of the immune stimulator LPS or proinflammatory cytokines induces full neurotoxicity.[82] Rotenone is known to induce excitotoxicity via NMDA receptor potentiation.[252] Two critical mechanisms in excitotoxicity include upregulation of iNOS/nNOS and NADPH oxidase, which together provide the substrates for formation of the powerful reactive nitrogen specie, peroxyitrite.[39] Peroxynitrite is formed rapidly when NO is in the presence of superoxide.
10. IL-6 proinflammatory cytokine's role in immunoexcitotoxicity
Glutamate receptors are classified as either iontropic or metabotropic. The ionotropic glutamate receptors are further divided into NMDA, AMPA, and Kainate type receptors, each with specific characteristics. In most areas of the brain, only the NMDA receptors are calcium permeable, but under certain conditions, both physiological and pathological, AMPA receptors can be calcium permeable (by lacking the GLuR2 subunit). Kainate receptors are generally classified along with AMPA receptors but also have unique characteristics. The metabotrophic receptors operate through membrane G-protein subunits and are classified within five subgroups (mGluR1-5), each with differing effects on glutamate neurotransmission, some enhancing excitotoxicity and others suppressing excitotoxicity.
Very little is known about the effect of cytokines on neuronal excitability, however, IL-6 has been shown to reduce group-II metabotropic glutamate receptors and L-type calcium channels.[236] In fact, IL-6 has been found in some studies to offer significant neuroprotection against calcium overload by reducing NMDA receptor activity.[149] This appears to be contradictory to the finding, that of all the cytokines, IL-6 alone was most closely associated with mortality in PD patients.[72] Baseline levels of IL-6 were higher in patients who died during the 4-year follow-up period and along with age, these represent two independent factors related to the risk of mortality in PD patients. There was no correlation with the other proinflammatory cytokines, even though they were elevated. While proinflammatory cytokines progressively increase within the striatum and substantia nigra in both animal models and human cases of Parkinson disease, there is a parallel fall in neurotrophins release (BDNF and NGF).[168]
Chemokines are also linked to neuronal excitation.[170,262] Prolonged exposure to chemokine CCL3 has been shown to elicit changes in NMDA-evoked calcium currents and increased the number of NMDA receptors on synaptic membranes in cultures.[129] As chemokines have as their main function the recruitment of immune cells to the CNS, the major effect is probably related to interactions of these recruited cells with the involved neural tissues.
11. How the immune system and glutamate receptors are linked in Parkinson's disease to produce cell death
With microglial activation, we see a simultaneous release of neurotoxic levels of both proinflammatory cytokines and excitatory amino acids, including glutamate, aspartate, and quinolinic acid.[96,120] Unfortunately, most of the published articles concerning Parkinson's disease link neurodegeneration only to inflammation, with all but a few even mentioning excitotoxicity. Based on a great deal of literature research, I have concluded that the two processes are inextricably linked and occur with microglial activation. There is growing evidence that the main destructive process is the excitotoxic process. As cited previously, there is a cyclic two-way stimulation between inflammation and excitotoxicity that progresses in many neurodegenerative disorders—that is, as excitotoxicity accelerates, inflammation also worsens. In addition, there are a number of conditions that modulate excitotoxicity that are to be considered, such a cellular energy supply, synergistically and additively interacting toxic molecules, effects of aging, status of the antioxidant network, effects of stress, and genetic factors.
B. Pathophysiology of immunoexcitotoxicity: Triggering event>Activated microglia>Release of cytokines, glutamate, free radicals, nitric oxide>Increase in glutamate receptors on neuron>Inhibition of GABA inhibitory receptors>Excitation of neuron to reach death>Mitochondrial dysfunction>Neuronal cell death>Progressive neuronal degeneration>Clinical disease state
Immune-glutamatergic interaction is commonly referred to as crosstalk, which describes, in most cases, an interaction among shared cell signaling mechanisms.[24] Under normal conditions, cytokines are either undetectable or detectable only in very low concentrations in the central nervous system.[62] In such low concentrations, IL-1ß, IL-6, and TNF-alpha can all act as neurotropic hormones. Being that even very high levels of proinflammatory cytokines do not damage neurons except in the presence of microglia, it becomes evident that alone, proinflammatory cytokines have no direct deleterious effect on neurons in pure culture, rather require the presence of microglia in an activated state. The question to be answered is what is the actual destructive process that leads to neurodegeneration? As we have seen, generation of high levels of free radicals and lipid peroxidation products and NO from activated microglia are indeed destructive. Still, this does not answer our question, as both inflammation and excitotoxicity can generate the same destructive elements. In fact, clinical studies using powerful antioxidants have been rather disappointing, suggesting that much more is responsible for the progressive neurodegeneration than mere oxidative stress and lipid peroxidation.[247]
Yawata et al. demonstrated that macrophages stimulated by LPS or TNF-alpha caused robust neurotoxicity that was completely prevented by blocking NMDA receptors.[256] NMDA receptor blocker MK-801 has also been shown to completely prevent neurotoxicity by immune stimulator LPS, thus making a compelling case for excitotoxicity as the main neurotoxic process in immunoexcitotoxicity. Similar protection from LPS-triggered neurodestruction was seen when glutamate release was prevented in microglia.[217,223] Metabotropic glutamate receptors may also play a role in immunoexcitotoxicity. This has been shown by stimulating mGluR group II type metabotropic glutamate receptors, which induces microglial activation and neuron death.[229] This stimulation also triggered TNF-alpha release by the activated microglia. That excitotoxicity was the ultimate cause of neuron death, was further shown by stimulating mGluR groups III (an inhibitor of excitotoxicity), which rescued the neurons.
In another study, researchers found that inflammation alone did not produce neurodegeneration in the hippocampus, but when a glutamate receptor agonist, ibotenate, was added, extensive neurodegeneration and microglial activation occurred in the hippocampus in an in vivo rat model.[165]
AMPA trafficking plays a critical role in learning and memory by changes in AMPA receptors (GluR2-lacking AMPA receptors) that make them calcium permeable thus altering calcium homeostasis, a process called synaptic scaling—that is, changes in synaptic excitability[235] [Figure 3]. These calcium-permeable AMPA receptors also play a central role in neurodegeneration for the same reasons.[12,188]
Of critical importance was the finding that the effect of TNF-alpha on neuronal excitability was dose dependent [Figure 6]. Low concentrations were neuroprotective by preferentially stimulating TNFR2 on neuron membranes, with higher concentrations inducing increased sensitivity to excitotoxins via TNFR1.[18] Stimulating TNFR1 stimulates increases trafficking of the calcium-permeable AMPA receptors (GluR2-lacking AMPA receptors) to the synaptic membrane, this enhancing excitotoxicity. Using a specific blocker of these calcium-permeable AMPA receptors (jaro spider toxin analog NASPM) researchers demonstrated that GluR2-lacking AMPA receptors are playing a major role in TNF-alpha-enhancement of excitotoxicity and that its effects on excitotoxicity occurs very rapidly.[140]
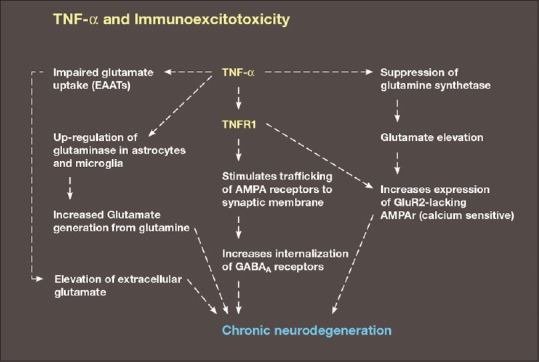
Diagram demonstrating a number of the major mechanisms of immunoexcitotoxicity, which includes the interaction of TNF- with a number of systems that enhance excitotoxicity. This includes impaired glutamate transport, upregulation of glutaminase, suppression of glutamine synthetase, increased trafficking of AMPA receptors to synaptic lipid raft and endocytosis of GABA
In-vivo evidence relating trafficking of GluR2-lacking AMPA receptors to the area of injured neural structures has also been reported using a spinal cord contusion model.[77] CNS contusions rapidly activate microglia, leading to immunoexcitotoxicity. Also, of interest is the finding that vascular endothelial growth factor (VEGF) enhances the insertion of GluR2 subunits onto motor neurons (making the calcium impermeable), thus making them more resistant to excitotoxic death.[29] In essence, what this demonstrates is that trafficking of GluR2-lacking (calcium permeable) AMPA receptors plays a major role in inflammation-generated immunoexcitotoxicity.
We have seen that TNF-alpha enhances excitotoxicity by increased trafficking of NMDA receptors as well as special calcium-permeable AMPA receptors toward the synaptic membranes. Olmos and Liabo propose three interrelated vicious cycles to explain the interrelationship between neuroinflammation and excitotoxicity.[179] The first of these cycles depends on TNF-alpha stimulating its own release from activated microglia. At the same time, glutamate is also released in increasing concentrations, which acts on microglial metabotropic glutamate receptors that in turn stimulates a greater release of TNF-alpha. The second cycle occurs when TNF-alpha inhibits glutamate reuptake, mainly by astrocytes, thus raising glutamate levels even higher in the extraneuronal space [Figure 4]. The third cycle involves all the mechanism by which TNF-alpha upsets the balance between excitotoxicity, including GABA inhibition of excitotoxicity by endocytosis of GABAA receptors.[197] As neurons and astrocytes begin to die off, they release a number of molecular substances that enhance excitotoxicity and promote microglial activation.
Bal-Price and Brown, using neuronal cultures and astrocyte/microglial cultures, demonstrated that neuronal death during inflammatory neurodegeneration was secondary to excitotoxicity and not due to inflammation, release of proteases, or peroxynitrite toxicity.[9] In the study, they found that blocking iNOS could prevent neuronal death, suggesting that nitric oxide was playing a dominate role. Blocking NMDA receptors using MK-801 also completely prevented cell death but only if done during the first 24 hours after exposure to the inflammagen. After that, only partial, but still significant, protection was afforded. Using nitric oxide donors reproduced the neurodegenerative features of inflammation activation of iNOS. It was proposed that the nitric oxide was triggering excessive glutamate release by inhibiting mitochondrial energy production.[139,172] Neuronal energy suppression is known to also considerably enhance glutamate excitotoxic injury.[173] Further, nitric oxide in higher concentrations not only enhances glutamate release but also plays a major role in excitotoxicity itself by increasing the production of peroxynitrite, activation of ADP-ribose polymerase, and/or mitochondrial permeability transition.[41] The Bal-Price/Brown study established that, because of the rapidity of the nitric oxide effect, peroxynitrite could not have been the neurotoxic principle, rather it was secondary to excitotoxicity. They found that NO inhibited mitochondrial respiration within seconds and glutamate release within one minute.
The reason glutamate blockers offered only partial protection later in the course of inflammatory stimulation most likely was secondary to the near total collapse of mitochondrial function seen at that time point. That is, by 24 hours after inflammagen exposure, a time after which glutamate blocker protection switched from full protection to only partial protection, was also the point at which neuronal ATP was almost completely depleted. At the level of glutamate release, triggered by the inflammation-induced NO elevation, full excitotoxic neuronal death would be expected to occur.[186] In essence, when mitochondrial energy generation was only partially impaired, excitotoxicity was the major damaging process, however, with later total energy loss (mitochondrial collapse) necrotic death became the major death process.
PART II
E. Animal models of Parkinson's disease: A brief review of and support for the immunoexcitotoxicity hypothesis
Several animal models of Parkinson's disease have been designed. They include rotenone, MPTP, paraquat, 6-hydroxydopamine (6-0HDA), genetic, and immune-related models.[135,194,233] Genetic models will not be considered here as this paper is more concerned with sporadic Parkinson disease.
Many of these models do not reflect human cases of the disorder.[135] This is especially so for the toxic models. For example, neither the MPTP nor 6-hydroxydopamine models induced Lewy bodies.[259] In addition, most of these toxic models cause direct toxicity to dopamine neurons and do not produce chronic progressive neurodegeneration. Regardless, these models are included as they are used to produce disorders with features of Parkinson's disease. For a more in-depth review of these models see Bove et al. and Blesa and Przedborski.[25,30]
a. MPTP model
1-methy-4-phrenyl-1,2,3,6-tetrqhydropyridine (MPTP) is a toxic analog of the narcotic meperidine, which in the early 1980s was found to produce an irreversible parkinsonian-like syndrome of rapid onset when used intravenously by drug users. One of its principle actions is to inhibit complex I of the electron transport chain. There is also evidence that MPTP (and its active component MPP+) activates microglia.[166] In general, the damage produced by MPTP is acute, however, there is some further progression of neurodegeneration later, even in human cases, and in all cases, the neuropathological damage perfectly resembles that seen in human cases of sporadic PD.[131]
Postmortem examinations of the brains of humans exposed to MPTP demonstrated that activated microglia were present up to 16 years after exposure to the drug.[132] It was concluded that a progressive neurodegenerative process was occurring that involved microglial activation.[61,132] It is also of interest that neither human autopsy studies or animal models using MPTP demonstrated Lewy bodies.[98,132] Some have made the observation that, while fully formed Lewy bodies were not seen, definite inclusions of alpha-synuclein developed in the aged animals and occurred in the exact same locations as human cases of PD.[79] It is possible that Lewy bodies are limited to humans.
Earlier reports suggested that neuronal loss in MPTP was limited to dopaminergic neurons, however, more recent studies have found additional injury outside the nigrostriatal system, which also involved other neurotransmitters, primarily norepinephrine, and less so, serotonin releasing neurons.[193] While this model is widely accepted as a workable model for Parkinson disease research, it does not represent what we are seeing in human cases, unless one can show that most cases of spontaneous PD are related to a toxin that has characteristics like MPTP.
The other chemical toxin models have similar problems, such as the 6-hydroxydopamine (6-OHDA) models, the rotenone model, and the paraquat/maneb models. In the case of the 6-hydroxydopamine model, which is extensively used, major problems exist. For example, 6-OHDA does not pass the blood–brain barrier (BBB) and must be injected directly in the brain area or given intraventricularly to produce the lesion.[30] The lesions are produced rapidly when the agent is injected into the substantia nigra, usually within hours to a few days, and only produce progressive neurodegeneration when it is injected intrastriatal and then develop much faster than is seen clinically, usually within 1–3 weeks. In addition, the toxin affects both dopaminergic and noradrenergic neurons.
Rotenone, a commonly used pesticide, reproduces many features of human cases of PD, and in some cases, generates inclusions resembling Lewy bodies.[16] Two other herbicide/fungicides, paraquat and maneb, respectively, are also used to produce animal models of PD. Paraquat also produces Lewy-like bodies in degenerating neurons.[152] Combining the two, paraquat and maneb, increases the damaging effect on dopaminergic neurons and the striatum.[232]
Another very interesting model uses a single MPTP intranasal instillation.[194] Prediger et al. have shown that this model more closely follows the progression and pathophysiology of human cases of Parkinson's disease, especially in its preclinical phases.[194,195] This model demonstrated defects in olfactory, cognitive, and motor functions with accompanying glutamatergic excitotoxicity, mitochondrial dysfunction, and neuronal death in the olfactory bulb, striatum, and prefrontal cortex.
b. Immunological models of Parkinson's disease
Some of the early immunological models of PD included the use of the powerful immune stimulant LPS injected directly into the substantia nigra, which was shown to produce selective and irreversible damage to dopaminergic neurons, while sparing serotonergic neurons.[48] The problem with these models is that they produced acute damage, rather than chronic neurodegeneration.
Injecting LPS or TNF-alpha systemically (intraperitoneal) in mice produced a rapid elevation in brain TNF-alpha levels that remained elevated for 10 months, while peripheral TNF-alpha levels fell to baseline by 9 hours.[199] The systemic LPS activated microglia in the substantia nigra and elevated proinflammatory cytokines during the entire period. Progressive destruction of the DA neurons within the substantia nigra continued for 7 months. Mice lacking TNF-alpha receptors (TNF R1/R2(-/-) experienced none of these pathological events. Gao et al. demonstrated that a low dose intranigral infusion of LPS for 2 weeks could also produce a delayed, chronically progressive loss of DA neurons.[83] In this study, microglia were rapidly activated early in the infusion and plateaued at 2 weeks. Following activation of the microglia, they found a gradual neurodegeneration that began between 4 and 6 weeks, and by 10 weeks, 70% of DA neurons in the substantia nigra were lost. Regarding the mechanism of destruction, they found a number of factors released by the microglia, but attributed most of the damage to elevations in NADPH oxidase, a major source of superoxide. Activation of NADPH oxidase is also a major reaction involved in excitotoxicity.[128]
Tanaka et al., using a cannula, injected LPS into both substantia nigra for 5 consecutive days and observed long-term activation of microglia in the SN.[226] Along with microglial activation, they observed a significant elevation in IL-1ß as well as elevations in the lipid peroxidation product 4-hydroxynonenal (4-HNE) and the powerful reactive nitrogen specie peroxynitrite. By 22 days following the last injection, they observed alpha-synuclein gene expression, and by 30 days DA neuronal death.
While these immunological models seemed to reproduce more closely what one would expect in human cases of Parkinson's disease, a newer model seemed to add significantly to our understanding of the pathophysiology of Parkinson's disease. This has been referred to as the two-hit model. It was known that mice genetically engineered to produce excess human mutant A53T alpha-synuclein (Tg mice) rarely develop signs of SN neurodegeneration, even though they developed widespread neurodegeneration throughout the CNS.[64,86]
Injecting the Tg mice with normal saline (NS) did not produce detectable neuron loss and injecting the wild-type (WT) mice systemically with LPS likewise produce no neuron loss. However, injecting the Tg mice systemically with LPS reproduced many of the features of Parkinson's disease including chronic progressive and relatively selective degeneration of dopaminergic neurons within the substantia nigra pars compacta and destruction of fibers of the nigrostriatal pathways. The process was chronically progressive with persistent inflammation and also produced Lewy bodies with aggregation of alpha-synuclein. In this model, degeneration began at 2.5 months and was significant at 5 months. In a previous study, also using systemic LPS, neuronal degeneration began 7 months following systemic LPS injection.[199]
What this study demonstrated was that the presence of mutated alpha-synuclein within the brain alone was not sufficient to produce parkinsonian pathology and progressive neurodegeneration; however, systemic immune stimulation considerably accelerated the formation of insoluble nitrated alpha-synuclein aggregates within DA neurons. Interestingly, both the WT mice and the Tg mice reacted the same to systemic LPS stimulation, which implies that the magnification of the destructive process was localized within the brain itself and most likely was triggered by activated microglia. Continuous blocking of iNOS and NADPH oxidase prevented the neurodegeneration. Both of these enzymes are located within microglia.
As neurons begin to die they release a number of noxious compounds, including glutamate, which accelerates the neurodegenerative process and triggers further microglial activation. It is also known that DA neurons are uniquely sensitive to oxidative insults.[116] The substantia nigra has been shown to have the highest concentration of microglia in the brain, which may explain its unusual sensitivity to microglial activation-related neurodegeneration.[134]
c. Critical importance of microglial priming
Mild-to-moderate stimulation of microglia is known to induce a phenotype that significantly upregulates a number of pro- and anti-inflammatory cytokines (predominately proinflammatory), chemokines, and glutamate generation from glutamine.[103] In this primed state, the microglia does not release these elements to any significant extent. Subsequent immune stimulation of these primed microglia will fully activate the microglia, resulting in an exaggerated release of its inflammatory/excitotoxic elements [Figure 5]. The immunoexcitotoxic reaction elicited from primed microglia is dramatically greater than that from ordinarily activated microglia.
A number of clinical conditions are associated with microglial priming including brain ischemia, hypoxia, brain trauma, systemic infections, systemic inflammatory disorders (autoimmune disorders), stress, neurodegenerative diseases, surgical procedures (even minor procedures), and even aging itself.[78,174,175,189,198,208,210,263]
Of special importance is the observation that microglial priming occurs progressively with aging because most cases of Parkinson's occur in the elderly.[68,174] In addition, by the time one reaches the extremes of life, they are more likely to have accumulated a number of neurological insults of varying severity—strokes, trauma, glial scars, and latent infections. The studies of Cunningham et al., using a prion mouse model, demonstrate that, in the face of an intracerebral inflammatory lesion, subsequent immune stimulation, either central or systemic, dramatically increased the level of neurological damage by the then fully activated microglia.[60] It was shown in this study that the prion infectious agent triggered microglial priming and the systemic immune stimulation with LPS then fully activated these hyperreactive microglia.
Infectious agents are not required for this two-hit process, as other CNS conditions have also demonstrated microglial priming enhanced neurological injury following systemic immune stimulation, even many years after the initial priming stimulus.[190,219] It should also be appreciated that, in the real world, people are subjected to numerous systemic immune-stimulating episodes, which not only would include periodic acute infectious diseases but also systemic trauma, depression, exposure to particulate exhaust, exposure to agricultural and industrial chemicals, stress, endocrine diseases (diabetes, thyroiditis, etc.), exposure to toxic metals, and recurrent activation of latent viruses, such as herpes simplex virus and cytomegalovirus. Increasingly, Lyme borreliosis is becoming a significant cause for chronic systemic and neuroinflammation.[202] One of the more common disorders associated with microglial priming/activation is diabetes. For example, studies have shown that hyperglycemia is associated with a significant worsening of LPS-induced microglial inflammation.[261] Other studies have confirmed that hyperglycemia is associated with microglial activation in vivo in rats.[169,204]
d. Role of inflammation in excitotoxicity
Inflammation, as we have seen, significantly enhances the insertion of GluR2-lacking, calcium permeable AMPA receptors into the synaptic membrane, which significantly enhances excitotoxicity. Combined with the enhancement of excitotoxicity by TNF-alpha and IL1ß through inhibition of the glutamate transport proteins, enhancement of glutaminase, suppression of glutamate dehydrogenase, and glutamine synthetase, one can appreciate the significant link between inflammation and excitotoxicity.[42] Glutaminase, found in high concentrations in astrocytes and microglia, catalyzes the conversion of glutamine to glutamate and when enhanced can magnify excitotoxicity by raising extraneuronal glutamate levels.[106] Glutamine synthetase is an enzyme that catalyzes the conversion of glutamate to glutamine, thereby reducing the risk of excitotoxicity. Glutamate dehydrogenase in a two-way reaction interconverts glutamate and alpha-ketogluterate, which also is protective when glutamate levels are elevated. Both of these enzymes are redox sensitive and under inflammatory conditions are impaired (particularly in the presence of free iron), thus interfering with glutamate homeostasis.[6,141] Also at play is the cysteine-glutamate antiporter (system Xc-), which exchanges extracellular cystine for intracellular glutamate, which under pathological conditions can raise extraneuronal glutamate levels and induce excitotoxicity.[37] Normally, this exchange system is essential for generating astrocytic glutathione from cystine, which when assembled, is then transferred to the neuron.
e. Role of alpha-synuclein
Alpha-synuclein is normally secreted by neurons in small amounts but is also released by dying neurons in larger concentrations and has been detected in the CSF of Parkinson's disease patients.[137,151] Normal function of alpha-synuclein is not fully understood but is known to be important for folding and refolding synaptic proteins, thus protecting nerve terminals against injury.[50] When oxidized or nitrated, alpha-synuclein develops enhanced immune characteristics.[14,222] Mutant forms of alpha-synuclein also significantly increased the proinflammatory response and triggered the release of nitric oxide and proinflammatory cytokines from microglia.[4] Gu et al., using transgenic mice secreting A53T mutant alpha-synuclein, found that accumulation within the astrocytes impaired glutamate transport, thus triggering excitotoxicity and microglial activation, which further enhanced the destruction of DA neurons in the midbrain, brainstem, and spinal cord.[95] Others have found mutant alpha-synuclein to be a potent activator of astrocytes with a dramatic increase in IL-6 and ICAM-1 secretion.[126] In addition, inflammatory cytokines, such as IL-1ß, promotes the upregulation of alpha-synuclein, which would increase the accumulation of oxidized and nitrosated alpha-synuclein.[93]
f. Alpha-synuclein and microglial activation
Couch et al. compared the immune reaction to intranigral injections of alpha-synuclein and LPS, and found that, while the immune reaction to alpha-synuclein was significant, it was overall considerably weaker than with LPS.[58] In comparison, 24 hours after injection of the substantia nigra with alpha-synuclein, TNF-alpha gene activation was increased two-fold and IL-1ß was increased five-fold, whereas after the direct intranigral LPS injection TNF-alpha, gene expression increased 77-fold and IL-1ß increased 1200-fold. In their cell culture study, Couch et al. found that more TNF-alpha was released by microglia when exposed to alpha-synuclein than LPS, signifying a difference when using in-vivo models. Microglial activation was markedly increased with both substances.
In a second part of the study, they found that systemic immune stimulation with LPS dramatically enhanced the inflammatory response of the intranigral alpha-synuclein injection, almost to the levels seen with direct LPS injection of the SN, again demonstrating a two-hit process. It was concluded that the intranigral alpha-synuclein was acting as a microglial priming agent, and systemic immune stimulation by the LPS converted the primed microglia into fully, but hyperactive, microglial phenotype. It is important to note that direct injection of alpha-synuclein into the SN did not lead to significant neuron death as was seen with direct injection of the LPS, but did result in significant neuron death following systemic LPS immune stimulation. This two-hit phenomenon has been confirmed by several others independently.[55,156,214]
g. Alpha-synuclein and excitotoxicity
There also is a strong link to excitotoxicity. Yang et al. found that overexpression of alpha-synuclein simultaneously increased phosphorylation of the NMDA receptor, thus increasing the NR1 and NR2B subunits, which enhanced this glutamate receptor's sensitivity to glutamate.[255] Another study found that oligomers of alpha-synuclein were particularly toxic to DA neurons and that these oligomers rapidly enhanced synaptic transmission via AMPA type glutamate receptors, which would significantly enhance excitotoxicity.[107]
F. Mutated alpha-synuclein and its role in immunoexcitotoxicity: Role of mitochondria in excitotoxicity
In essence, normally alpha-synuclein plays a role in protecting the synapse and neuron, but when present in excess or especially when mutated or oxidized/nitrated, it becomes inflammatory and can prime the surrounding microglia. With activation of the peripheral immune system, especially if intense or repetitive, the immune reaction inside the brain becomes dramatically enhanced through a hyperreactive activation of the previously primed microglia. Once in this destructive phenotype, iNOS and nNOS systems are upregulated, resulting in high levels of extraneuronal nitric oxide. NO suppresses mitochondrial function, thus lowering neuronal energy production, which magnifies sensitivity of the neuron and its synapse to excitotoxicity.[172] NO also stimulates the release of glutamate from microglia and astrocytes, again triggering excitotoxicity.[117]
At the same time, NADPH oxidase, mainly within activated microglia, is upregulated and generates high levels of superoxide and hydrogen peroxide. Superoxide and NO react rapidly to produce the powerful reactive nitrogen specie peroxynitrite, which further damages the mitochondria as well as other neuronal structures. The reactive microglia also release an assortment of proinflammatory cytokines, chemokines, and interferons, which further stimulate excitotoxicity. Elevated extraneuronal glutamate levels suppress the normal exchange of cystine for glutamate, resulting in a fall in intraneuronal glutathione (GSH).
a. Alpha-synuclein and cognition
Despite these findings, there is some evidence that alpha-synuclein aggregations have a lesser role in the pathology, and especially, the clinical manifestations of Parkinson's disease. For instance, Burke et al. argued that a number of elderly without neurological disease are found to have extensive synucleinopathy ranging up to Braak stages 4 to 6.[43] In addition, they add, there is no correlation between the Braak stage and the clinical severity of the disease—that is, there is no correlation with a measure of the density and extensive presence of Lewy bodies throughout the CNS. In other word, there are a number of cases in which Lewy bodies are incidental inclusions. Others have argued that the lack of correlation is based on the fact that, as the neurons are destroyed, the released Lewy bodies are removed by phagocytosis.[91] Others have found that individuals can demonstrate significant cognitive disturbance with no or minimal cortical Lewy bodies, and conversely, widespread cortical Lewy body pathology can occur without significant cognitive decline.[56,88] Parkkinen et al. examined from a very large autopsy series (n = 1720) a cohort of 226 alpha-synuclein positive individuals, and found that almost half with extensive alpha-synuclein aggregates in the dorsal motor nucleus of the vagus, substantia nigra, and basal forebrain were completely free of extrapyramidal or cognitive symptoms.[185]
There are a number of possible explanations for this discrepancy, the main one being that the intensity of the microglial reaction depends on the intensity and frequency of systemic inflammation. It could also depend on the strength of the person's antioxidant network, detoxification systems, and mitochondrial health.[63]
b. Mitochondrial dysfunction and excitotoxicity: Complex I
The electron transport system in mitochondria operates through three enzyme complexes. Complex I is the entry point for electron transfer energy production. Earlier studies in untreated Parkinson's disease patients have frequently been shown to have decreased activity of complex I their platelets.[87,212] A more recent study found no inhibition of mitochondrial function in the platelets of Parkinson patients, but did find inhibition of complex I of the electron transport chain in persons exposed to a certain class of pesticides.[38] Some earlier studies also failed to find defects in respiratory chain function in Parkinson patients, and attributed earlier observations to the difficulty in measuring enzyme function in cells with known low mitochondrial function, such as blood cells.[153] Mitochondrial dysfunction is known to occur within neurons of the striatum.[27]
The mechanism involved in mitochondrial impairment in neurons involves to a large degree the upregulation of microglial and astrocytic iNOS with resulting elevated levels of NO [Figure 6]. NO inhibits mitochondrial respiration in neuronal synaptosomes even in nanomolar concentrations by inhibiting cytochrome c oxidase.[40] Another mechanism for mitochondrial inhibition is by the formation of peroxynitrite from its reaction with superoxide, also generated from microglia.[47,201]
c. Mitochondrial dysfunction and excitotoxicity
As described previously, mitochondrial energy suppression markedly increases excitotoxicity by increasing the sensitivity of glutamate receptors.[10,90,111,173] In addition, impairment of intracellular calcium buffering occurs when mitochondria are dysfunctional.[196] Endoplasmic reticulum stress, which occurs with inflammation and excitotoxicity, also impairs intracellular buffering, thus triggering deadly excitotoxic cascades.
Inflammation is also known to injure mitochondria.[59] Significant mitochondrial damage and subsequent degeneration of the nigrostriatal pathway was demonstrated by injecting LPS into the striatum of rats.[109] Excitotoxicity is also associated with mitochondrial dysfunction, and it has been shown using an animal model of excitotoxin-induced spinal cord paralysis in rats that supplying energy substrates, such as pyruvate, could completely prevent the paralysis.[209] From these observations, it is apparent that neuronal damage associated with both immune and excitotoxic (immunoexcitotoxicity) processes is significantly worsened in the face of mitochondrial energy deficits.
G. Two major pathways for systemic inflammation in Parkinson's disease: Triggering event pathways
a. The olfactory tract as an entry point for CNS microglial stimulation
The olfactory nerves are first order neurons, meaning they project their axons directly to the brain without intervening synapses, which allows toxic substances to by-pass the BBB.[71] The link between Parkinson's disease with olfactory dysfunction is strengthened by a number of clinical studies.[97,192,250] Clinically, it is known that 85% of PD patients show a loss of olfactory function and that it manifests before clinical signs of PD appear.[17,221] By age 80, approximately 75% of normal aged individuals will suffer some impairment of olfaction, but a majority of these Parkinson's patients are younger.
Using 8-month-old C57/B6 mice, unilateral (right-sided) intranasal instillation using a low dose of LPS (10 μg) was given every other day for 5 months. A number of features that closely resembled behavioral and neuropathological parkinsonism were found in the animals.[102] The test animals displayed progressive hypokinesia, chronic progressive bradykinesia, selective DA neuron loss in the striatum, accumulation of alpha-synuclein aggregates in the substantia nigra, and microglial activation. At 5 months after the instillation of the LPS, they found a progressive loss of tyrosine hydroxylase-immunoreactive neurons in the right substantia nigra, the same side showing the motor disability. As was shown in other studies using immune-based models, the DA neurons in the ventral tegmental nucleus, which is close to the substantia nigra, were spared.
The olfactory bulb contains large populations of dopaminergic neurons. With LPS intranasal instillation, a loss of the neurons and a reduced thickness of the olfactory bulb was seen. Within the substantia nigra when exposed to the LPS via the olfactory tract, they observed Lewy body-like alpha-synuclein aggregations. Using various markers, the researchers demonstrated marked microglia activation in the affected SN nucleus. Levels of TNF-alpha and IL-1ß were also elevated within the nucleus. Microglial activation was also seen in the olfactory bulb. Intranasal instillation of LPS had no effect on systemic immune activation. Dopaminergic neurons show a special sensitivity to inflammation, excitotoxicity, and oxidative stress not seen in neurons in the hippocampus or cortex.[27]
People are frequently exposed to fine particles of substances (<2.5 μM) suspended in the air, which can originate from a number of sources, including oil refineries, metal processing facilities, automotive emissions, power plants, and wild fires.[44] These particles have been shown to be composed of both organic and inorganic compounds, such as sulfates, nitrates, carbon, and ammonium, and various toxic metals, including mercury, cadmium, lead, tin, aluminum, and fluoride. Of particular importance, it has been shown that humans are frequently exposed to LPS suspended in the air as fine particles as part of house dust and dust from biosolids—a treated sewage sludge used for landscaping that contains a considerable amount of organic material, including bacterial components.[234] These biosolids, which have been shown to contain components of gram-negative bacteria (the source of LPS), are used not only in agriculture but also for landfills around parks and children's play areas.[155] When carried in the wind and inhaled they potentially can trigger the same immune reactions along the olfactory tact as seen in this study.
b. The gastrointestinal tract as an entry point for CNS microglial stimulation: Evidence for a spreading inflammatory disorder in response to environmental toxins through the GI autonomic nervous system pathways to the brain
One of the consistent observations has been the presence of widespread Parkinson disease-related lesions throughout the nervous system, including autonomic nuclei of the spinal cord, brainstem, olfactory nerves, sympathetic ganglion, cardiac and pelvic parasympathetic plexuses and vagus nerve, adrenal medulla, salivary glands, and even the skin.[26,242,243]
Consistent with this finding are the non-CNS symptoms and signs that often occur prior to the onset of typical Parkinson's symptoms. These symptoms include drooling, dysphagia, nausea, delayed gastric emptying, abdominal distention, and chronic constipation.[119] In fact, chronic constipation can occur years or even decades before the onset of PD motor symptoms.[191]
Oinas et al., in a study of 302 patients aged 85 years or older, found alpha-synuclein aggregates, some as mini-aggregates, and some as mature Lewy bodies and Lewy neurites, in 44% and 34% of the individuals, respectively.[178] Of patients with moderate-to-severe thoracic spinal cord alpha-synuclein pathology, 60% had diffuse Lewy body pathology in the brain as well. This finding increased to 89% in those with moderate or severe sacral alpha-synuclein pathology, demonstrating that spinal cord and autonomic pathology reflects CNS pathology in regard to alpha-synuclein aggregates.
Based on the examination of a great number of brains from Parkinson's patients, Braak et al. concluded that Parkinson's appears to be an ascending neuropathological disorder, in which alpha-synuclein immunopositive Lewy bodies and Lewy neurites begin to develop first in the gastrointestinal systems and then ascend through the vagus nerves and other autonomic nerves and ganglion into the dorsal motor nucleus of the vagus and glossopharyngeal nerves.[32,33] Simultaneously, Lewy bodies and neurites also appear in the olfactory tract and anterior olfactory nucleus. As the disease progresses cortical involvement ensues, beginning in the anteriomedial temporal mesocortex and progressing into the neocortex, high order association sensory cortex, and prefrontal areas.
It is during the preclinical stages that one sees involvement isolated mostly in the enteric system. Shannon et al. examined colon biopsy samples taken previously from Parkinson's patients before their symptoms appeared.[215] They found alpha-synuclein inclusion in all of the samples taken from 2 years to as long as 5 years before Parkinsonian symptom onset. Examination of 23 healthy controls found no such lesions. In a follow-up study, they performed colon biopsies on 10 untreated Parkinson's patients and 23 healthy controls and 23 patients with inflammatory bowel disease.[216] It is of interest that they found alpha-synuclein in all the colonic biopsies of Parkinson's patients but in none of the healthy controls or those with inflammatory bowel disease. With 3-nitrotyrosine being present in the biopsies of all three groups and inflammation in the inflammatory bowel disease group, they concluded that the presence of alpha-synuclein inclusion within the nerve fibers of the colonic submucosa was not related to either inflammation or oxidative stress. More recently, others have questioned the reliability of using alpha-synuclein in the colon as a biomarker for Parkinson's disease.[238]
Using a genetic model of Parkinson disease, researchers demonstrated that enteric symptoms and alpha-synuclein deposits within the ganglion of the enteric nervous system occurred before neurological symptoms.[130] To further elucidate the mechanism by which alpha-synuclein aggregation was being triggered within the neurons in the submucosa of the colon, Forsyth et al. not only used intestinal biopsies but also tested for intestinal hyperpermeability (leaky gut) in newly diagnosed cases of Parkinson's disease.[80] They found that Parkinson patients have significantly greater intestinal permeability than controls and also increased staining for E. coli, nitrotyrosine, and alpha-synuclein within the intestinal mucosa. The patients also had decrease serum staining for LPS binding protein (LBP), which indicates that LPS from the cell wall of the E. coli that had entered the blood stream from the breached intestinal barrier may have been reduce in concentration by the liver. This observation indicates that the primary immune reaction was taking place in the wall of the colon and that microglial activation in the brainstem (principally in the DMN of the vagus) was being signaled through the vagus nerve.
One must wonder why there is not a strong link between inflammatory bowel disease and Parkinson disease risk. The differences between inflammatory bowel disease and what was seen in Parkinson disease cases may be a difference in E. coli translocation and in probiotic diversity and concentrations.[13,54,94] E. coli contains within its cell wall the powerful immune stimulant LPS. Of note is a study using an ulcerative colitis rat model in which researchers found elevated levels of proinflammatory cytokines, including IL-1ß, IL-6, and ICAM-1, as well an upregulation of iNOS within the animal's substantia nigra.[237]
There is some evidence that the microbiota species can affect Parkinson disease symptoms. For example, Scheperjans et al. in examining the microbiota of 72 PD patients versus 72 healthy controls found that the Parkinson patients had a 77.6% reduced abundance of Prevotellaceae bacterial family compared with controls. In addition, Parkinson patients with a greater abundance of Enterobacteriaceae had greater postural instability and gait difficulty as opposed to Parkinson patients with tremor-dominant form of Parkinson disease.[213] Tan et al. found a high incidence of small intestine bacterial overgrowth (SIBO) in Parkinson's disease patients.[225] A quarter of 103 Parkinson patients examined had SIBO, which was independently associated with worse motor symptoms. Helicobacter pylori infection was also found to be associated with worse severity of Parkinson's disease.[224] These observations support the idea that systemic inflammation, in particular gastrointestinal inflammation, is linked to the pathological process within the CNS in Parkinson's disease.
Kelly et al. developed a Parkinson's mouse model in which they exposed mice to either a low dose of LPS or saline systemically and were sacrificed at monthly intervals over a 5-month period.[122] The LPS exposure caused an immediate and progressive increase in alpha-synuclein expression in the large intestine but not the small intestine. They also observed an increase in alpha-synuclein in a subset of myenteric neurons in the colon. There was also an abrupt increase in alpha-synuclein in the brainstem, but there was no evidence of nigrostriatal degeneration. This observation could represent the early preclinical stages of Parkinson's disease. From this study, it was concluded that a low dose exposure to LPS could induce the gastrointestinal alpha-synuclein pathology in the myenteric neurons and leaky gut without producing obvious brain pathology characteristic of Parkinson's disease.
The gradual increase in large intestine permeability seen in this study appeared secondary to the abrupt increases in myenteric alpha-synuclein, suggesting that this pathological process precedes the colon's hyperpermeability (leaky gut).
There is also evidence that dopaminergic nigrostriatal neurodegeneration can increase intestinal inflammation, oxidative stress, and enhance the motility of the intestine.[187] These events would indicate that induction of inflammation between the brain and gut is a two-way connection, most likely via the vagus nerve.[92] They did observe infiltration of mast cells and eosinophils into the submucosa and glial activation within the myenteric ganglia. It is known that the dorsal motor nucleus of the vagus, one of the first areas involved in PD, can induce inflammation in the GI tract.[154]
c. Evidence for stages of progression
Braak et al. in 1996 hypothesized a morphological staging system based on progressive pathological involvement beginning in the brainstem and eventually extending through the higher brain centers.[31] They outlined a series of six stages of progression with the first two stages being preclinical.[34] During the presymptomatic stages 1–2, the inclusion pathology was confined to the medulla oblongata/pontine tegmentum and olfactory bulb/anterior olfactory nucleus. There appears to be some extension of the pathology to the caudal raphe nuclei and gigantocellular portions of the adjacent reticular formations as well as locus coeruleus. During stages 3–4, severe pathological changes are seen in the locus coerulus and central nucleus of the amygdala, tegmental pedunculopontine nucleus, and cholinergic nuclei of the basal forebrain, including Meynart's nucleus. In stage 3, extensive Lewy bodies are seen in the substantia nigra pars compacta but without neuronal loss, which then occurs in stage four. Stages 5–6 represent the end stages where the pathology invades the temporal neocortex, limbic regions, and prefrontal cortex and is associated with varying degrees of dementia.[36]
Braak et al. also hypothesized that some unknown pathogen was able to pass from neuron to neuron, and because each of the areas involved in PD are intimately interconnected, the pathogen would be able to easily spread along these pathways.[35] No such pathogen has been discovered. It has been shown that alpha-synuclein can pass between neurons and some have proposed this passage as a mechanism of the progression and spread of pathological alpha-synuclein aggregates in Parkinson's disease.[1]
A more likely explanation would be the progressive activation of microglia, especially if these microglia had been previously primed. It is known that microglia can travel throughout the brain once they are activated.[74,177]
More recently, the Braak system of orderly progression of pathogenic alpha-synuclein from enteric nervous system to brainstem and higher cortical areas has been challenged. These studies suggest that alpha-synuclein aggregations occur simultaneously across multiple regions of the central and peripheral nervous system, arguing against a caudal-rostral progression.[67] Some studies have shown that incidental alpha-synuclein pathology may affect solely the locus coeruleus and substantia nigra without affecting the medullary nuclei.[8,113]
Kalaitzkis et al. examined 71 cases of PD from the UK Parkinson's Disease Society Tissue Bank at the Imperial College in London and found that, while 53% of cases did follow the Braak caudal-rostral staging, 47% of the cases did not and in 7% the dorsal motor nucleus of the vagus (DMV) was not involved at all.[121] In the study, 100% of cases had severe involvement of the substantia nigra and 98.5% had involvement of the nucleus basalis of Meynert.
Jellinger examined the brains of 53 cases of PD and found that all had extensive Lewy body pathology and neuronal loss in the substantia nigra, medullary and pontine nuclei, and locus coeruleus.[112] Additional lesions were also found in 24% of amygdala, 58% of allocortex, 34% of cingulate area, and 26.5% of isocortex. All of these cases were within stages 4–6 of the Braak staging system. The largest autopsy series, involving 1720 people over 40 years of age at death, found 14% had alpha-synuclein lesions, with 83% of these having a distribution pattern that followed the Braak staging system. Of these, 55% having widespread alpha-synuclein pathology (Braak stages 5–6) lacked clinical signs of dementia or extrapyramidal signs or symptoms.[185]
The earliest pathological lesions in several studies were found in the spinal cord, DMV, olfactory bulb, and basal forebrain nuclei, but did not always indicate progression to clinical PD.[127,184] In the Parkkinen et al., study of 106 autopsy cases having alpha-synuclein positive brains, only 30% were diagnosed with a neurodegenerative disorder, indicating that alpha-synuclein pathology by itself (even when substantial) was not a marker for clinical disease.[27,184] As mentioned previously, Burke et al. found no correlation between Braak stage and the clinical severity of Parkinson's disease, and they argued further that many asymptomatic individuals have brain synucleinopathies ranging up to Braak stages 4–6.[43] This is compatible with the findings in wild-type Tg transgenic mice expressing excessive wild type alpha-synuclein, in which their brains have abundant alpha-synuclein production but no neuronal degeneration or neurological impairment, and only develop neurodegeneration when they undergo systemic immune stimulation with lipopolysaccharide (LPS).[86]
The mystery as to why some human case of PD follow closely the caudal-rostral Braak staging system and others do not may depend on the source of the immune stimulation triggering the microglial activation. Cases with primarily intracerebral immune activations, as with latent viruses, exposure to pesticides/herbicides, certain autoimmune disorders, and exposure to MPTP, might have direct activation of microglia within the nuclei above the level of the lower brainstem nuclei (DMV, LC nuclei) first or even exclusively. It is also possible that cases having the main entry point of immune stimulation via the olfactory system would show a different pattern of neurodegeneration and alpha-synuclein deposition than cases in which the gastrointestinal system was the main entry point of the inflammaogen.
It has been estimated that only 50% of neurons need be intact in the striatal innervation pathways for normal function of motor control.[19] While some have tried to explain the large number of elderly having significant concentrations of alpha-synuclein lesions without having neurological symptoms by claiming a very prolonged preclinical phase (up to 20 years), in a number of these series the elderly patients were at the extremes of life and still neurologically intact.[218] This could mean that they never reached the threshold of losing more than 50% of the striatal inputs, that is, the disease in these individuals may progress so slow that they die before symptoms develop. Again, there appears to be no correlation between Lewy body density and clinical symptoms or disease duration.[88]
CONCLUSIONS
Considerable evidence suggests a link between Parkinson's disease and chronic inflammation. The immediate source of this inflammation within the affected brain areas involves mostly microglia with some involvement of astrocytes, macrophages, mast cells, and T-lymphocytes, as well as inflammatory mediators, such as cytokines, chemokines, and interferons. Most of the studies and discussions of studies have concluded that the neurodegeneration associated with this inflammation is secondary to activated cellular mechanisms involved in inflammatory cell signaling, including lipid peroxidation, reactive oxygen and reactive nitrogen species, proinflammatory prostaglandins, proteases, and nitric oxide. It has been shown that alpha-synuclein in a glial free culture produces no neuron loss—microglia are necessary for neurodegeneration.[180,260]
Several studies have shown that alone, inflammatory cytokines, such as TNF-alpha, are unable to kill neurons, and that in most cases, especially during the initial stages, cell death is triggered by excitotoxicity. That is, the inflammatory link to neuronal death is through excitotoxicity. I think that immunoexcitotoxicity is descriptive of this disease process and can fully explain all aspect of clinical Parkinson's disease.
Inflammation triggers and enhances excitotoxicity by a number of mechanisms, including trafficking of calcium-permeable AMPA receptors to the synaptic membrane, endocytosis of GABA receptors, free radical and lipid peroxidation product inhibition of glutamate transporters, oxidative inactivation of glutamine synthetase and glutamic acid decarboxylase, cytokine enhancement of glutaminase enzyme activity, and effects on NMDA glutamate receptor subtypes and trafficking.
One of the early observations concerning Parkinson's disease was an impairment in mitochondrial function, which affected not only dopaminergic neurons in the substantia nigra but also systemic cells, such as platelets.[20] A more recent study found no inhibition of mitochondrial function in the platelets of PD patients but did find inhibition of complex I of the electron transport chain in persons exposed to a certain class of pesticides.[38] The major factors leading to mitochondrial dysfunction within affected neurons included direct inhibition of complex I by nitric oxide and mitochondrial damage by peroxynitrite generated from an interaction between mitochondrial produced superoxide and up-regulation of inducible nitric oxide synthetase (iNOS). LPS can suppress mitochondrial function by increasing levels of nitric oxide.[109]
The importance of mitochondrial dysfunction in Parkinson disease is that it considerably enhances excitotoxicity by a number of mechanisms, meaning that under such conditions of low energy supply, even low concentrations of extraneuronal glutamate can become excitotoxic.
Also to be considered in evaluating risk is the observation that early exposure in life to immune insults, either in utero or soon after birth, can increase sensitivity to substantia nigral neurotoxic agents later in life.[46] In addition, these early life exposures to immune activation have been shown to reduce the number of DA neurons in the substantia nigra, thus reducing the number of neurons supporting motor function.[146] They also observed a progressive additional loss of these neurons as the animals age. Importantly, they observed a life-long elevation in proinflammatory cytokines in the striatum.
Ling et al. demonstrated that exposing animals prenatally to LPS reduced the number of dopamine neurons in the substantia nigra, and that exposing these animals later in life to subtoxic doses of rotenone led to a significant loss of DA neurons.[145] Translated to real life situations, it could mean that those exposed to early life inflammatory conditions would be much more susceptible to certain environmental toxic substances later in life—most likely because of having fewer surviving substantia nigra cells at that stage of life. This finding would help explain why some individuals are more prone to develop Parkinson's disease as they age than are others.
The future treatment of Parkinson’s, and more importantly, prevention, may depend on utilizing compounds that suppress neurodegenerative immunity, and more importantly, excitotoxicity. Because of the complexity of the immunoexcitotoxic process, a number of possibilities now exist in attaining this goal.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
1.Abounit S, Bousset L, Loria F, Zhu S, de Chaumont F, Pieri L, et al. Tunneling nanotubes spread fibrillary α-synuclein by intercellular trafficking lysosomes. EMBO J. 2016;35:2120–38. doi: 10.15252/embj.201593411. [DOI] [PMC free article] [PubMed] [Google Scholar]
2.Alam Q, Alam MZ, Mushtaq G, Damenhouri GA, Rasool M, Kamai MA, Haque A. Inflammatory process in Alzheimer's and Parkinson's diseases: Central role of cytokines. Curr Pharm Des. 2016;22:541–8. doi: 10.2174/1381612822666151125000300. [DOI] [PubMed] [Google Scholar]
4.Alvarez-Erviti L, Couch Y, Richardson J, Cooper JM, Wood MJ. Alpha-synuclein released by neurons activate the inflammatory response in microglial cell line. Neurosci Res. 2011;69:337–42. doi: 10.1016/j.neures.2010.12.020. [DOI] [PubMed] [Google Scholar]
5.Ambrosi G, Cerri S, Blandini F. A further update of excitotoxicity in the pathogenesis of Parkinson's disease. J Neural Transm. 2014;121:849–59. doi: 10.1007/s00702-013-1149-z. [DOI] [PubMed] [Google Scholar]
6.Aquirre J, Rodriguez R, Hansberg W. Oxidation of Neurospora crassa NADP-specific glutamate dehydrogenase by activated oxygen species. J Bacteriol. 1989;17:6243–50. doi: 10.1128/jb.171.11.6243-6250.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
7.Assous M, Had-Aissouni L, Gubellini P, Melon C, Nafi I, Salin P, et al. Progressive Parkinsonism by acute dysfunction of excitatory amino acid transporters in rat substantia nigra. Neurobiol Dis. 2014;65:69–81. doi: 10.1016/j.nbd.2014.01.011. [DOI] [PubMed] [Google Scholar]
8.Attems J, Jellinger KA. The dorsal motor nucleus of the vagus is not an obligatory trigger of Parkinson's disease. Neuropath Appl Neurobiol. 2008;34:466–67. doi: 10.1111/j.1365-2990.2008.00937.x. [DOI] [PubMed] [Google Scholar]
9.Bal-Price A, Brown GC. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci. 2001;21:6480–91. doi: 10.1523/JNEUROSCI.21-17-06480.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
10.Baltan S. Excitotoxicity and mitochondrial dysfunction underlie age-dependent ischemic white matter injury. Adv Neurobiol. 2014;11:151–70. doi: 10.1007/978-3-319-08894-5_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
11.Beal MF. Mitochondria take center stage in aging and neurodegeneration. Ann Neurol. 2005;58:495–505. doi: 10.1002/ana.20624. [DOI] [PubMed] [Google Scholar]
15.Bentz M, Zaouter C, Shi Q, Fatmi H, Mildovan F, Fermandes JC, Benderdour M. Inhibition of inducible nitric oxide synthase prevents lipid peroxidation in osteoarthritic chrondrocytes. J Cell Biochem. 2012;113:2256–67. doi: 10.1002/jcb.24096. [DOI] [PubMed] [Google Scholar]
16.Berbarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–6. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
17.Berendse HW, Ponsen MM. Detection of preclinical Parkinson's disease along the olfactory tract. J Neural Transm Suppl. 2006;70:321–5. doi: 10.1007/978-3-211-45295-0_48. [DOI] [PubMed] [Google Scholar]
18.Bernardino L, Xapelli S, Silva AP, Jakobsen B, Poulsen FR, Oliverira CR, et al. Modulator effects of interleukin-1beta and tumor necrosis factor-alpha on AMPA-induced excitotoxicity in mouse organotypic hippocampal slice cultures. J Neurosci. 2005;25:6734–44. doi: 10.1523/JNEUROSCI.1510-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
19.Bernheimer H, Birkmayer W, Hornykiewicz O, Jelliner K, Seitelberger F. Brain dopamine and the syndrome of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neural Sci. 1973;20:415–55. doi: 10.1016/0022-510x(73)90175-5. [DOI] [PubMed] [Google Scholar]
20.Blake CI, Spitz E, Leehey M, Hoffer BJ, Boyson SJ. Platelet mitochondrial respiratory chain function in Parkinson's disease. Mov Disord. 1997;12:3–8. doi: 10.1002/mds.870120103. [DOI] [PubMed] [Google Scholar]
21.Blandini F. An update on the potential role of excitotoxicity in the pathogenesis of Parkinson's disease. Funct Neurol. 2010;25:65–71. [PubMed] [Google Scholar]
22.Blandini F, Porter RH, Greenamyre JT. Glutamate and Parkinson's disease. Mol Neurobiol. 1996;12:73–94. doi: 10.1007/BF02740748. [DOI] [PubMed] [Google Scholar]
23.Blaylock RL. A possible central mechanism in autism spectrum disorders, part 1. Altern Ther Health Med. 2008;14:46–53. [PubMed] [Google Scholar]
24.Blaylock RL, Strunecka A. Immune-glutamatergic dysfunction as a central mechanism of the autism spectrum disorders. Curr Med Chem. 2009;16:157–70. doi: 10.2174/092986709787002745. [DOI] [PubMed] [Google Scholar]
25.Blesa J, Prezedborski S. Parkinson's disease: Animal models and dopaminergic cell vulnerability. Front Neuroanat. 2014;8:155. doi: 10.3389/fnana.2014.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
26.Bloch A, Probst A, Bissig H, Adams H, Tolnay M. Alpha-synuclein pathology of the spinal cord and peripheral autonomic nervous system in neurologically unimpaired elderly subjects. Neuropath Appl Neurobiol. 2006;32:284–95. doi: 10.1111/j.1365-2990.2006.00727.x. [DOI] [PubMed] [Google Scholar]
28.Blum-Degen D, Muller T, Kuhn W, Gerlach M, Przuntek H, Riederer P. Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer's and de novo parkinsonian disease patients. Neurosci Lett. 1995;202:17–20. doi: 10.1016/0304-3940(95)12192-7. [DOI] [PubMed] [Google Scholar]
29.Bogaert E, Van Damme P, Poesen K, Dhondt J, Heramus N, Kiraly D, et al. VEGF protects motor neurons against excitotoxicity by upregulation of GluR2. Neurobiol Aging. 2010;31:2185–91. doi: 10.1016/j.neurobiolaging.2008.12.007. [DOI] [PubMed] [Google Scholar]
30.Bove J, Prou D, Perier C, Prezedborski S. Toxin-induced models of Parkinson's disease. J Am Soc Exp Neurother. 2005;2:484–94. doi: 10.1602/neurorx.2.3.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
31.Braak H, Braak E, Yilmazer D, de Vos RA, Jansen EN, Bohl J. Pattern of brain destruction in Parkinson's and Alzheimer's diseases. J Neural Transm. 1996;103:455–90. doi: 10.1007/BF01276421. [DOI] [PubMed] [Google Scholar]
32.Braak H, Del Tredici K, Bratzke H, Hamm-Clement J, Sandmann-Keil D, Rub U. Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson's disease (Preclinical and clinical stages) J Neurol. 2002;249(Suppl 3):III/1–5. doi: 10.1007/s00415-002-1301-4. [DOI] [PubMed] [Google Scholar]
33.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporatic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
34.Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K. Stages in the development of Parkinson's disease-related pathology. Cell Tissue Res. 2004;318:121–34. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
35.Braak H, Rub U, Gai WP, Del Tredici K. Idiopathic Parkinson's disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm. 2003;110:517–36. doi: 10.1007/s00702-002-0808-2. [DOI] [PubMed] [Google Scholar]
36.Braak H, Rub U, Jansen Steur EN, Del Tredici K, de Vos RA. Cognitive status correlates with neuropathological stage in Parkinson's disease. Neurology. 2005;64:1404–10. doi: 10.1212/01.WNL.0000158422.41380.82. [DOI] [PubMed] [Google Scholar]
37.Bridges R, Lutgen V, Lobner D, Baker DA, Daws LC. Thinking outside the cleft to understand synaptic activity: Contribution of the cystine-glutamate antiporter (system Xc-) to normal and pathological glutamatergic signaling. Pharmacol Rev. 2012;64:780–802. doi: 10.1124/pr.110.003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
38.Bronstein JM, Paul K, Yang L, Haas RH, Shults CW, Le T, et al. Platelet mitochondrial activity and pesticide exposure in early Parkinson's disease. Mov Disord. 2015;30:862–6. doi: 10.1002/mds.26164. [DOI] [PMC free article] [PubMed] [Google Scholar]
39.Brown GC. Mechanisms of inflammatory neurodegeneration: iNOS and NADPH oxidase. Biochem Soc Trans. 2007;35(pt5):1119–21. doi: 10.1042/BST0351119. [DOI] [PubMed] [Google Scholar]
40.Brown GC, Cooper CE. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 1994;345:50–4. doi: 10.1016/0014-5793(94)01290-3. [DOI] [PubMed] [Google Scholar]
42.Brown GC, Vilalta A. How microglia kill neurons. Brain Res. 2015;1628(PtB):288–97. doi: 10.1016/j.brainres.2015.08.031. [DOI] [PubMed] [Google Scholar]
43.Burke RE, Dauer WT, Vonsattel JP. A critical analysis of the Braak staging scheme for Parkinson's disease. Ann Neurol. 2008;64:485–91. doi: 10.1002/ana.21541. [DOI] [PMC free article] [PubMed] [Google Scholar]
44.Calderon C, Garciduenas L, Leray E, Heydapour P, Torres-Jardon R, Reis J. Air pollution, a rising environmental risk factor for cognition, neuroinflammation and neurodegeneration: The clinical impact on children and beyond. Rev Neurol. 2016:69–80. doi: 10.1016/j.neurol.2015.10.008. [DOI] [PubMed] [Google Scholar]
45.Campbell V, Lynch MA. Biphasic modulation of intracellular Ca+2 concentrations by interleukin-1beta in cortical synaptosomes: Involvement of a pertussis toxin-sensitive G-protein and mitogen-activated protein kinase. NeuroReport. 1998;9:1923–7. doi: 10.1097/00001756-199806220-00002. [DOI] [PubMed] [Google Scholar]
46.Carvey PM, Chang Q, Lipton JW, Ling Z. Prenatal exposure to the bacteriotoxin lipopolysaccharide leads to long-term losses of dopamine neurons in offspring: A potential new model for Parkinson's disease. Front Biosci. 2003;8:s826–37. doi: 10.2741/1158. [DOI] [PubMed] [Google Scholar]
47.Cassina A, Radi R. Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch Biochem Biophys. 1996;328:309–16. doi: 10.1006/abbi.1996.0178. [DOI] [PubMed] [Google Scholar]
48.Castano A, Herra AJ, Cano J, Machado A. Lipopolysacchride intranigral injection induces inflammatory reaction and damage in nigrostriatal dopamine system. J Neurochem. 1998;70:584–92. doi: 10.1046/j.1471-4159.1998.70041584.x. [DOI] [PubMed] [Google Scholar]
49.Caudle WM, Zhang J. Glutamate, excitotoxicity, and programmed cell death in Parkinson's disease. Exp Neurol. 2009;220:230–3. doi: 10.1016/j.expneurol.2009.09.027. [DOI] [PubMed] [Google Scholar]
50.Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–96. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
51.Chao CC, Hu S, Ehrlich L, Peterson PK. Interleukin-1 and tumor necrosis factor-alpha synergistically mediate neurotoxicity: Involvement of nitric oxide and N-methy-D-aspartate receptors. Brain Behav Immun. 1995;9:355–65. doi: 10.1006/brbi.1995.1033. [DOI] [PubMed] [Google Scholar]
52.Chen Z, Palmer TD. Differential roles of TNFR1 and TNFR2 signaling in adult hippocampal neurogenesis. Brain Behav Immun. 2013;30:45–53. doi: 10.1016/j.bbi.2013.01.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
53.Chertoff M, Di Paolo N, Schoeneberg A, Depino A, Ferrari C, Wurst W, et al. Neuroprotective and neurodegenerative effects of the chronic expression of tumor necrosis factor α in the nigrostriatal dopaminergic circuit of adult mice. Exp Neurol. 2011;227:237–51. doi: 10.1016/j.expneurol.2010.11.010. [DOI] [PubMed] [Google Scholar]
54.Clairembault T, Leclair-Visonneau L, Coron E, Bourreille A, Le Dily S, Vavasseur F, et al. Structural alterations of the intestinal barrier in Parkinson's disease. Acta Neuropathol Commun. 2015;3:12. doi: 10.1186/s40478-015-0196-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
55.Codoy MC, Tarelli R, Ferrari CC, Sarchi MI, Pitossi FJ. Central and systemic IL-1 exacerbates neurodegeneration and motor symptoms in a model of Parkinson's disease. Brain. 2008;13((pt7) 1):1880–94. doi: 10.1093/brain/awn101. [DOI] [PMC free article] [PubMed] [Google Scholar]
56.Coloslmo C, Hughes AJ, Kilford L, Lees AJ. Lewy body cortical involvement may not always predict dementia in Parkinson's disease. J Neurol Neurosurg Psychiology. 2003;74:852–6. doi: 10.1136/jnnp.74.7.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
57.Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4:399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
58.Couch Y, Alvarez-Erviti L, Sibson NR, Wood MJA, Anthony DC. The acute inflammatory response to intranigral α-synuclein differs significantly from intranigral lipo polysaccharide and is exacerbated by peripheral inflammation. J Neuroinflammation. 2011;8:166. doi: 10.1186/1742-2094-8-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
59.Csaba S. The pathophysiological role of peroxynitrite in shock, inflammation, and ischemia reperfusion injury. Shock. 1996;6:79–88. doi: 10.1097/00024382-199608000-00001. [DOI] [PubMed] [Google Scholar]
60.Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci. 2005;25:9275–84. doi: 10.1523/JNEUROSCI.2614-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
61.Czlonkowska A, Kohuknika M, Kurowska-Jatrzebska I, Czlonkowski A. Microglial reaction in MPTP (1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine.) induced Parkinson's disease mice model. Neurodegeneration. 1996;5:137–43. doi: 10.1006/neur.1996.0020. [DOI] [PubMed] [Google Scholar]
62.Danzer R. Cytokine-induced sickness behavior: Mechanisms and implications. Ann NY Acad Sci. 2001;933:222–34. doi: 10.1111/j.1749-6632.2001.tb05827.x. [DOI] [PubMed] [Google Scholar]
66.De Virgilo A, Greco A, Fabbrini G, Inghilleri M, Rizzo MI, Galio A, et al. Parkinson's disease: Autoimmunity and neurodegeneration. Autoimmun Rev. 2016;15:1005–11. doi: 10.1016/j.autrev.2016.07.022. [DOI] [PubMed] [Google Scholar]
67.Dickson DW, Fujishiro H, DelloDonne A, Menke J, Ahmed Z, Klos J, et al. Evidence that incidental Lewy body disease is pre-symptomatic Parkinson's disease. Acat Neuropathol. 2008;115:437–44. doi: 10.1007/s00401-008-0345-7. [DOI] [PubMed] [Google Scholar]
68.Dilger RN, Johnson RW. Aging, microglial priming, and the discordant central inflammatory response to signals from the peripheral immune system. J Leukoc Biol. 2008;84:932–9. doi: 10.1189/jlb.0208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
69.Doble A. The role of excitotoxicity in neurodegenerative disease: Implications for therapy. Pharmacol Ther. 1999;81:163–221. doi: 10.1016/s0163-7258(98)00042-4. [DOI] [PubMed] [Google Scholar]
70.Doorn KJ, Moors T, Druckarch B, van de Berg W, Lucassen PJ, van Dam AM. Microglial phenotypes and toll-like receptor-2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson's disease patients. Acta Neuropathol Commun. 2014;2:90. doi: 10.1186/s40478-014-0090-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
71.Doty RL. The olfactory vector hypothesis of neurodegenerative diseases: Is it viable? Ann Neurol. 2008;63:7–15. doi: 10.1002/ana.21327. [DOI] [PubMed] [Google Scholar]
72.Dufek M, Rektorova I, Thon V, Lokaj J, Rektor I. Interleukin-6 may contribute to mortality in Parkinson's disease patients: A 4-year prospective study. Parkinsons Dis 2015. 2015 doi: 10.1155/2015/898192. 898192. [DOI] [PMC free article] [PubMed] [Google Scholar]
73.Edison P, Ahmed I, Fan Z, Hinz R, Gelosa G, Ray Chaudhuri K, et al. Microglia, amyloid, and glucose metabolism in Parkinson's disease with and without dementia. Neuropsychopharmacology. 2013;38:938–49. doi: 10.1038/npp.2012.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
74.Emmanouilidou E, Vekrellis K. Exocytosis and spreading of normal and aberrant α-synuclein. Brain Path. 2016;26:398–403. doi: 10.1111/bpa.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
75.Fan Z, Aman Y, Ahmed L, Chetelat G, Landeau B, Ray Chaudhuri K, et al. Influence of microglial activation on neuronal function in Alzheimer's and Parkinson's disease dementia. Alzheimer Dement. 2015;11:608–21. doi: 10.1016/j.jalz.2014.06.016. [DOI] [PubMed] [Google Scholar]
76.Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, et al. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia. 2013;61:349–60. doi: 10.1002/glia.22437. [DOI] [PMC free article] [PubMed] [Google Scholar]
77.Ferguson AR, Christensen RN, Gensel JC, Miller BA, Sun F, Beattie EC, et al. Cell death after spinal cord injury is exacerbated by rapid TNF alpha-induced trafficking of GluR2-lacking AMPARs to the plasma membrane. J Neuorsci. 2008;28:11391–400. doi: 10.1523/JNEUROSCI.3708-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
78.Fonken LK, Weber MD, Daut RA, Kitt MM, Frank MG, Watkins LR, Maier SF. Stress-induced neuroinflammatory priming is time of day dependent. Psychoneuroendocrinology. 2016;66:82–90. doi: 10.1016/j.psyneuen.2016.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
79.Forno LS, Langston JW, DeLanney LE, Irwin I, Ricaurte GA. Locus ceruleus lesions and eosinophilic inclusions in MPTP-treated monkeys. Ann Neurol. 1986;20:449–55. doi: 10.1002/ana.410200403. [DOI] [PubMed] [Google Scholar]
80.Forsyth CB, Shannon KM, Kordower JH, Voight RM, Shaikh M, Jaglin JA, et al. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson's disease. PLoS One. 2011;6:e28032. doi: 10.1371/journal.pone.0028032. [DOI] [PMC free article] [PubMed] [Google Scholar]
82.Gao HM, Hong JS, Zhang W, Liu B. Synergestic dopaminergic neurotoxicity of the pesticide rotenone and inflammogen lipopolysaccharide: Relevance to the etiology of Parkinson's disease. J Neurosci. 2003;23:1228–36. doi: 10.1523/JNEUROSCI.23-04-01228.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
83.Gao HM, Jiang J, Wilson B, Zhang W, Hong JS, Liu B. Microglial activation-mediated delayed and progressive degeneration of rat dopaminergic neurons: Relevance to Parkinson's disease. J Neurochem. 2002;81:1285–97. doi: 10.1046/j.1471-4159.2002.00928.x. [DOI] [PubMed] [Google Scholar]
84.Gardoni F, Boraso M, Zianni E, Corsini E, Galli CL, Cattabeni F, et al. Distribution of interleukin-1 receptor complex at the synaptic membrane driven by interleukin-1ß and NMDA stimulation. J Neuroinflammation. 2011;8:14. doi: 10.1186/1742-2094-8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
85.Gerhard A, Trender-Gerhard I, Turkheimer F, Quinn NP, Bhatia KP, Brooks DJ. In vivo imaging of microglial activation with [11c]-PK11195 in progressive supranuclear palsy. Mov Disorder. 2006;21:89–93. doi: 10.1002/mds.20668. [DOI] [PubMed] [Google Scholar]
86.Glasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM. Neuronal α-synucleinopathy with severe movement disorder in mice expressing A53T human α-synuclein. Neuron. 2002;34:521–33. doi: 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
88.Gomez-Tortosa E, Newell K, Irizarry MC, Albert M, Crowdon JH, Hyman BT. Clinical and quantitative pathological correlates of dementia with Lewy bodies. Neurology. 1999;53:1284–91. doi: 10.1212/wnl.53.6.1284. [DOI] [PubMed] [Google Scholar]
89.Grassi F, Mileo AM, Monaco L, Punturieri A, Santoni A, Eusebi F. TNF-alpha increases frequency of spontaneous miniature synaptic currents in cultured rat hippocampal neurons. Brain Res. 1994;659:226–30. doi: 10.1016/0006-8993(94)90883-4. [DOI] [PubMed] [Google Scholar]
90.Nicholis DG, Budd SL. Mitochondria and neuronal glutamate excitotoxicity. Biochim Biophys Bioenergetics. 1998;1366:97–112. doi: 10.1016/s0005-2728(98)00123-6. [DOI] [PubMed] [Google Scholar]
91.Greffard S, Verny M, Bonnet AM, Seilhean D, Hauw JJ, Duyckaerts C. A stable proportion of Lewy body bearing neurons in the substantia nigra suggest a model in which the Lewy body causes neuronal death. Neurobiol Aging. 2010;31:99–103. doi: 10.1016/j.neurobiolaging.2008.03.015. [DOI] [PubMed] [Google Scholar]
92.Grenham S, Clarke G, Cryan JF, Dinan TG. Brain-gut-microbe communication in health and disease. Front Physiol. 2011;2:94. doi: 10.3389/fphys.2011.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
93.Griffin WS, Liu L, Li Y, Mrak RE, Barger SW. Interleukin-1 mediates Alzheimer's and Lewy body pathologies. J Neuroinflammation. 2006;3:5. doi: 10.1186/1742-2094-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
95.Gu XL, Long CX, Sun L, Xie C, Lin X, Cai H. Astrocytic expression of Parkinson's disease-related A53T alpha-synuclein causes neurodegeneration in mice. Mol Brain. 2010;3:2. doi: 10.1186/1756-6606-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
96.Guillemin GJ. Quinolenic acid, the inescapable neurotoxin. FEB J. 2012;279:1356–65. doi: 10.1111/j.1742-4658.2012.08485.x. [DOI] [PubMed] [Google Scholar]
97.Haehner A, Hummel T, Reichmann H. Olfactory dysfunction as a diagnostic marker for Parkinson's disease. Expert Rev Neurother. 2009;9:1773–9. doi: 10.1586/ern.09.115. [DOI] [PubMed] [Google Scholar]
98.Halliday G, Herrero MT, Murphy K, McCann H, Ros-Bernal F, Barcia C, et al. No Lewy pathology in monkeys with over 10 years of severe MPTP Parkinsonism. Mov Disord. 2009;24:1519–23. doi: 10.1002/mds.22481. [DOI] [PubMed] [Google Scholar]
99.Halliday GM, Stevens CH. Glia initiator and progressor of pathology in Parkinson's disease. Mov Disord. 2011;26:6–17. doi: 10.1002/mds.23455. [DOI] [PubMed] [Google Scholar]
100.Hamano H, Noguchi M, Fukui H, Issiki A, Watanabe Y. Regulation of brain cell environment on neuronal protection: Role of TNF alpha in glia cells. Life Sci. 2002;72:565–74. doi: 10.1016/s0024-3205(02)02252-x. [DOI] [PubMed] [Google Scholar]
101.Harms AS, Cao S, Rowse AL, Thome AD, Li X, Manieri LR, et al. MHCH is required for alpha-synuclein-induced activation of microglia. J Neurosci. 2013;33:95–6. doi: 10.1523/JNEUROSCI.5610-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
102.He Q, Yu W, Wu J, Chen C, Lou Z, Zhang Q, et al. Intranasal LPS-mediated Parkinson's model challenges the pathogenesis of nasal cavity and environmental toxins. PLoS One. 2013;8:e78418. doi: 10.1371/journal.pone.0078418. [DOI] [PMC free article] [PubMed] [Google Scholar]
103.Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopoplysacchride (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav Immun. 2009;23:309–17. doi: 10.1016/j.bbi.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
104.Hirsh EC, Huno S, Damier P, Faucheaux B. Glial cells and inflammation in Parkinson's disease: A role in neurodegeneration? Ann Neurol. 1998;44(3 Suppl 1):S115–20. doi: 10.1002/ana.410440717. [DOI] [PubMed] [Google Scholar]
105.Hoeijmakers L, Heinen Y, van Dam AM, Lucassen PJ, Korisi A. Microglial priming and Alzheimer's disease: A possible role for (early) immune challenges and epigenetics. Front Hum Neurosci. 2016;10:398. doi: 10.3389/fnhum.2016.00398. [DOI] [PMC free article] [PubMed] [Google Scholar]
106.Hoffman EM, Zhang Z, Schechter R, Miller KE. Glutaminase increases in rat dorsal root ganglion neurons after unilateral adjuvant-induced hind paw inflammation. Biomolecules. 2016;6:10. doi: 10.3390/biom6010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
107.Huls S, Hogen T, Vassallo N, Danzer KM, Hengerer B, Giese A, et al. AMPA-receptor-mediated excitatory synaptic transmission is enhanced by iron-induced alpha-synuclein oligomers. J Neurochem. 2011;117:868–78. doi: 10.1111/j.1471-4159.2011.07254.x. [DOI] [PubMed] [Google Scholar]
108.Hunot S, Boissiere F, Faucheux B, Brugg B, Mouatt-Prigent A, Agid Y. Nitric oxide synthase and neuronal vulnerability in Parkinson's disease. Neuroscience. 1996;72:355–63. doi: 10.1016/0306-4522(95)00578-1. [DOI] [PubMed] [Google Scholar]
109.Hunter R, Ojha U, Bhurtel S, Bing G, Choi DY. Lipopolysacchride-induced functional and structural injury of the mitochondria in the nigrostriatal pathway. Neurosci Res. 2017;114:62–9. doi: 10.1016/j.neures.2016.09.007. [DOI] [PubMed] [Google Scholar]
110.Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson's disease brains. Acta Neuropathol. 2003;106:518–26. doi: 10.1007/s00401-003-0766-2. [DOI] [PubMed] [Google Scholar]
111.Izumi Y, Zorumski CF. Neuroprotective effects of pyruvate following NMDA-mediated excitotoxic insults in hippocampal slices. Neurosci Lett. 2010;478:131–5. doi: 10.1016/j.neulet.2010.04.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
112.Jellinger KA. Alpha-synuclein pathology in Parkinson's and Alzheimer's disease brain: Incidence and topographic distribution—a pilot study. Acta Neuropath. 2003;106:191–201. doi: 10.1007/s00401-003-0725-y. [DOI] [PubMed] [Google Scholar]
113.Jellinger KA. Lewy body-related alpha-synucleinopathy in the aged human brain. J Neural Transm. 2004;111:1219–35. doi: 10.1007/s00702-004-0138-7. [DOI] [PubMed] [Google Scholar]
114.Jenner P. Oxidative stress and Parkinson's disease. Handb Clin Neurol. 2007;83:507–20. doi: 10.1016/S0072-9752(07)83024-7. [DOI] [PubMed] [Google Scholar]
115.Jenner P, Olanow CW. Oxidative stress and the pathogenesis of Parkinson's disease. Neurology. 1996;47:S161–70. doi: 10.1212/wnl.47.6_suppl_3.161s. [DOI] [PubMed] [Google Scholar]
116.Jenner P, Olanow CW. Understanding cell death in Parkinson's disease. Ann Neurol. 1998;44(Suppl 1):S72–84. doi: 10.1002/ana.410440712. [DOI] [PubMed] [Google Scholar]
117.Jimenez-Jimenez FJ, Alonso-Navarro H, Herrero MT, Garcia-Martin E, Agundez JA. An update on the role of nitric oxide in the neurodegenerative process of Parkinson's disease. Curr Med Chem. 2016;23:2666–79. doi: 10.2174/0929867323666160812151356. [DOI] [PubMed] [Google Scholar]
118.Joers V, Tansey MG, Mulas G, Carta AR. Microglial phenotypes in Parkinson's disease and animal models of the disease. Prog Neurobiol. 2016 doi: 10.1016/j.pneurobio.2016.04.006. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
119.Jost WH. Gastrointestinal dysfunction in Parkinson's disease. J Neurol Sci. 2010;289:69–73. doi: 10.1016/j.jns.2009.08.020. Edwards LL, Quigley EM, Pfeiffer RF. Gastrointestinal dysfunction in Parkinson's disease: Frequency and pathophysiology Neurology 1992;42:26.32. [DOI] [PubMed] [Google Scholar]
120.Kaindl AM, Degos V, Peineau S, Gouadon E, Chhor V, Lorin G, et al. Activation of microglial N-methyl-D-aspartate receptors triggers inflammation and neuronal cell death in the developing and mature brain. Ann Neurol. 2012;72:536–49. doi: 10.1002/ana.23626. [DOI] [PubMed] [Google Scholar]
121.Kalaitzkis ME, Graeber MB, Gentleman SM, Pearce RK. The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson's disease: A critical analysis of alpha-synuclein staging. Neuropath Appl Neurobiol. 2008;34:284–95. doi: 10.1111/j.1365-2990.2007.00923.x. [DOI] [PubMed] [Google Scholar]
122.Kelly LP, Carvey PM, Keshavarzian A, Shanon KM, Shaikh M, Bakay RA, et al. Progression of intestinal permeability changes and alpha-synuclein expression in a mouse model of Parkinson's disease. Mov Disord. 2014;29:999–1009. doi: 10.1002/mds.25736. [DOI] [PMC free article] [PubMed] [Google Scholar]
123.Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR-2 for paracrine activation of microglia. Nat Commun. 2013;4:1562. doi: 10.1038/ncomms2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
124.Kim WG, Mohoney RP, Wilson B, Jeohn GH, Liu B, Hong JS. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: Role of microglia. J Neurosci. 2000;20:6309–16. doi: 10.1523/JNEUROSCI.20-16-06309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
125.Kim YS, Joh TH. Microglia, major player in the brain inflammation: Their roles in the pathogenesis of Parkinson's disease. Exp Mol Med. 2006;38:333–47. doi: 10.1038/emm.2006.40. [DOI] [PubMed] [Google Scholar]
126.Klegeris A, Pelech S, Giasson BI, Maguire J, Zhang H, McGeer EG, et al. Alpha-synuclein activates stress signaling protein kinases in THD-2 cells and microglia. Neurobiol Aging. 2006;20:2000–8. doi: 10.1016/j.neurobiolaging.2006.11.013. [DOI] [PubMed] [Google Scholar]
127.Klos KJ, Ahiskog JE, Josephs KA, Apaydin H, Parisi JE, Boeve BF, et al. Alpha-synuclein pathology in the spinal cords of neurologically asymptomatic aged individuals. Neurology. 2006;66:1100–2. doi: 10.1212/01.wnl.0000204179.88955.fa. [DOI] [PubMed] [Google Scholar]
128.Koriauli S, Natsvlishvili N, Barbakadze T, Mikeladze D. Knockdown of interleukin-10 induces the redistribution of sigma 1-receptor and increases the glutamate-dependent NADPH oxidase activity in mouse brain neurons. Biol Res. 2015;48:55. doi: 10.1186/s40659-015-0048-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
129.Kuijpers M, van Gassen KL, de Graan PN, Kushikata T, Gruol D. Chronic exposure to the chemokine CCL3 enhances neuronal network activity in rat hippocampal cultures. J Neuroimmunol. 2010;229:73–80. doi: 10.1016/j.jneuroim.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
130.Kuo YM, Li Z, Jiao Y, Gaborit N, Pani AK, Orrison BM, et al. Extensive enteric nervous system abnormalities in mice transgenic for artificial chromosomes containing Parkinson disease-associated alpha-synuclein gene mutations precede central nervous system changes. Hum Mol Genet. 2010;19:1633–50. doi: 10.1093/hmg/ddq038. [DOI] [PMC free article] [PubMed] [Google Scholar]
131.Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of merperidine-analog synthesis. Science. 1983;219:979–80. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
132.Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, Karluk D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrdine exposure. Ann Neurol. 1999;46:598–605. doi: 10.1002/1531-8249(199910)46:4<598::aid-ana7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
133.Larre EM, Galic MA, Monuihate A. Neonatal inflammation produces selective behavioral deficits and alters N-methyl-D-aspartate receptor subunit mRNA in the adult rat brain. Eur J Neurosci. 2008;27:644–53. doi: 10.1111/j.1460-9568.2008.06031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
134.Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39:151–70. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
135.Leal PC, Lins LC, de Gois AM, Marchioro M, Santos JR. Commentary: Evaluation of models of Parkinson's disease. Front Neurosci. 2016;10:283. doi: 10.3389/fnins.2016.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
136.Le W, Wu J, Tang Y. Protective microglia and their regulation in Parkinson's disease. Front Mol Neurosci. 2016;9:89. doi: 10.3389/fnmol.2016.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
137.Lee HJ, Patel S, Lee SJ. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci. 2005;25:6016–24. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
139.Leist M, Fara E, Montecucco C, Nicotera P. Peroxynitrite and nitric oxide donors induce neuronal apoptosis by eliciting autocrine excitotoxicity. Eur J Neurosci. 1997;9:1488–98. doi: 10.1111/j.1460-9568.1997.tb01503.x. [DOI] [PubMed] [Google Scholar]
141.Levine RL. Oxidative modification of glutamine synthetase. I Inactivation is due to loss of one histidine residue. J Biol Chem. 1983;258:11828–33. [PubMed] [Google Scholar]
142.Li Z, Zheng Z, Ruan J, Li Z, Tzeng CM. Chronic inflammation links cancer and Parkinson's disease. Front Aging Neurosci. 2916;8:126. doi: 10.3389/fnagi.2016.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
143.Lim S, Chun Y, Lee JS, Lee SJ. Neuroinflammation in synucleinopathies. Brain Path. 2016;26:404–9. doi: 10.1111/bpa.12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
144.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–95. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
145.Ling Z, Chang QA, Tong CW, Leurgans SE, Lipton JW, Carvey PM. Rotenone potentiates dopamine neuron loss in animals exposed to lipopolysacchride prenatally. Exp Neurol. 2004;190:373–83. doi: 10.1016/j.expneurol.2004.08.006. [DOI] [PubMed] [Google Scholar]
146.Ling Z, Gayle DA, Ma SY, Lipton JW, Tong CW, Hong JS, et al. In utero bacterial endotoxin exposure causes loss of tyrosine hydroxylase neurons in the postnatal rat midbrain. Mov Disord. 2002;17:116–24. doi: 10.1002/mds.10078. [DOI] [PubMed] [Google Scholar]
148.Long-Smith CM, Sullivan AM, Nolan YM. The influence of microglia on the pathogenesis of Parkinson's disease. Prog Neurobiol. 2009;89:277–87. doi: 10.1016/j.pneurobio.2009.08.001. [DOI] [PubMed] [Google Scholar]
150.MacKenzie IR. Activated microglia in dementia with Lewy bodies. Neurology. 2000;55:132–4. doi: 10.1212/wnl.55.1.132. [DOI] [PubMed] [Google Scholar]
151.Majbour NK, Vaikath NN, Eusebi P, Chiasserini D, Ardah M, Varghese S, et al. Longitudinal changes in CSF alpha-synuclein species reflect Parkinson's disease progression. Mov Disord. 2016;31:1535–42. doi: 10.1002/mds.26754. [DOI] [PubMed] [Google Scholar]
152.Manning-Bog AB, McCormack AL, Li J, Uversky VN, Fink AL, Di Monte DA. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: Paraquat and alpha-synuclein. J Biol Chem. 2002;277:1641–4. doi: 10.1074/jbc.C100560200. [DOI] [PubMed] [Google Scholar]
153.Martin MA, Mlina JA, Jimenez-Jimenez FJ, Benito-Leon J, Orti-Pareja M, Campos Y, Arenas J. Respiratory-chain enzyme activities in isolated mitochondria of lymphocytes from untreated Parkinson's disease patients. Grupo-Centro de Trastornos del Movimiento. Neurology. 1996;46:1343–6. doi: 10.1212/wnl.46.5.1343. [DOI] [PubMed] [Google Scholar]
154.Matteoli G, Boeckxstaens GE. The vagal innervation of the gut and immune homeostasis. Gut. 2013;62:1214–22. doi: 10.1136/gutjnl-2012-302550. [DOI] [PMC free article] [PubMed] [Google Scholar]
155.McCall CA, Jordan KS, Habash MB, Dunfield KE. Monitoring Bacteroides spp, markers, nutrients, metals and Escherichia coli in soil and leachate after land application of three types of municipal biosolids. Water Res. 2015;70:255–65. doi: 10.1016/j.watres.2014.12.004. [DOI] [PubMed] [Google Scholar]
156.McColl BW, Rothwell NJ, Allan SM. Systemic inflammatory stimulus potentiates the acute phase and CXC chemokine responses to experimental stroke and exacerbates brain damage via interleukin-1 and neutrophil-dependent mechanisms. J Neurosci. 2007;27:4403–12. doi: 10.1523/JNEUROSCI.5376-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
157.McGeer PL, Itagaki S, Akiyama H, McGeer EG. Rate of cell death in parkinsonism indicates active neuropathological process. Ann Neurol. 1988;24:574–6. doi: 10.1002/ana.410240415. [DOI] [PubMed] [Google Scholar]
159.Marin I, Kipnis J. Learning and memory…and the immune system. Learn Mem. 2013;20:601–6. doi: 10.1101/lm.028357.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
160.Metha A, Prabhakar M, Kumar P, Deshmukh R, Sharma PL. Excitotoxicity: Bridge to various triggers in neurodegenerative disorders. Eur J Pharmacol. 2013;698:6–18. doi: 10.1016/j.ejphar.2012.10.032. [DOI] [PubMed] [Google Scholar]
161.Mirza B, Hadberg H, Thomson P, Moos T. The absence of reactive astrocytes is indicative of a unique inflammatory process in Parkinson's disease. Neuroscience. 2000;95:425–32. doi: 10.1016/s0306-4522(99)00455-8. [DOI] [PubMed] [Google Scholar]
162.Miyake T, Shirakawa H, Nakagawa T, Kaneko S. Activation of mitochondrial transient receptor potential vanilloid 1 channel contributes to microglial migration. Glia. 2015;63:1870–82. doi: 10.1002/glia.22854. [DOI] [PubMed] [Google Scholar]
163.Mogi M, Harada M, Riederer P, Narabyashi H, Fujta J, Nagatsu T. Interleukin-1 beta growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett. 1994;180:147–50. doi: 10.1016/0304-3940(94)90508-8. [DOI] [PubMed] [Google Scholar]
164.Morgi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett. 1994;165:208–10. doi: 10.1016/0304-3940(94)90746-3. [DOI] [PubMed] [Google Scholar]
165.Morimoto K, Murasugi T, Oda T. Acute neuroinflammation exacerbates excitotoxicity in rat hippocampus in vivo. Exp Neurol. 2002;177:95–104. doi: 10.1006/exnr.2002.7991. [DOI] [PubMed] [Google Scholar]
166.Munoz-Manchado AB, Villadiego J, Romo-Madero S, Suarez-Luna N, Bermejo-Navas A, Rodriguez-Gomez JA, et al. Chronic and progressive Parkinson's disease MPTP model in adult and aged mice. J Neurochem. 2016;136:373–87. doi: 10.1111/jnc.13409. [DOI] [PubMed] [Google Scholar]
167.Nafia I, Re DB, Masmejean F, Melon C, Kachidian P, Kererian-Le Goff L, et al. Preferential vulnerability of mesencephalic dopamine neurons to glutamate transporter dysfunction. J Neurochem. 2008;105:484–96. doi: 10.1111/j.1471-4159.2007.05146.x. [DOI] [PubMed] [Google Scholar]
168.Nagatsu T, Mogi M, Ichinose H, Togari A. Changes in cytokines and neurotrophins in Parkinson's disease. J Neural Transm Suppl. 2000;(60):277–90. doi: 10.1007/978-3-7091-6301-6_19. [DOI] [PubMed] [Google Scholar]
169.Nagayach A, Patro N, Patro I. Astrocytic and microglial responses in experimentally induced diabetic rat brain. Metab Brain Dis. 2014;29:747–61. doi: 10.1007/s11011-014-9562-z. [DOI] [PubMed] [Google Scholar]
170.Nelson TE, Gruol DL. The chemokine CXCL10 modulates excitatory activity and intracellular calcium signaling in cultured hippocampal neurons. J Neuroimmunol. 2004;156:74–87. doi: 10.1016/j.jneuroim.2004.07.009. [DOI] [PubMed] [Google Scholar]
171.Neniskyte U, Vilalta A, Brown GC. Tumor necrosis factor alpha-induced neuronal loss is mediated by microglial phagocytosis. FEBS Lett. 2014;588:2952–56. doi: 10.1016/j.febslet.2014.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
172.Netzahualcoyotzi C, Tapia R. Degeneration of spinal motor neurons by chronic AMPA-induced excitotoxicity in vivo and protection by energy substrates. Acta Neuropathol Commun. 2015;3:27. doi: 10.1186/s40478-015-0205-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
173.Netzahualcoyotzi C, Tapia R. Energy substrates protect hippocampus against endogenous glutamate-mediated neurodegeneration in awake rats. Neurochem Res. 2014;39:1346–54. doi: 10.1007/s11064-014-1318-y. [DOI] [PubMed] [Google Scholar]
174.Norden DM, Godbout JP. Review: Microglia of the aged brain: Primed to be activated and resistant to regulation. Neuropathol Appl Neurobiol. 2013;39:19–34. doi: 10.1111/j.1365-2990.2012.01306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
175.Norden DM, Muccigrosso MM, Godbout JP. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology. 2015;96(Pt A):29–41. doi: 10.1016/j.neuropharm.2014.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
176.Obal I, Majlath Z, Toldi J, Vescsei L. Mental disturbances in Parkinson's disease and related disorders: The role of excitotoxins. J Parkinson Dis. 2014;4:139–50. doi: 10.3233/JPD-130294. [DOI] [PubMed] [Google Scholar]
177.Ohsawa K, Kohsaka S. Dynamic motility of microglia: purinergic modulation of microglial movement in the normal and pathological brain. Glia. 2011;59:1793–9. doi: 10.1002/glia.21238. [DOI] [PubMed] [Google Scholar]
178.Oinas M, Paetau A, Myllkagamgas L, Notkola IL, Kalimo H, Polvikoski T. Alpha-synuclein pathology in the spinal cord autonomic nuclei associates with alpha-synuclein pathology in the brain: A population-based Vantaa 85+study. Acta Neuropathol. 2010;119:715–22. doi: 10.1007/s00401-009-0629-6. [DOI] [PubMed] [Google Scholar]
179.Olamos G, Llado J. Tumor necrosis alpha: A link between neuroinflammation and excitotoxicity. Mediators Inflam 2014. 2014 doi: 10.1155/2014/861231. 861231. [DOI] [PMC free article] [PubMed] [Google Scholar]
180.Olmos G, Conde I, Arenas I, Del Peso L, Castellanos C, Landazuri MO, et al. Accumulation of hypoxia-inducable factor-1 alpha through a novel electrolytic, thiol antioxidant-sensitive mechanism. Cell Signal. 2007;19:2098. doi: 10.1016/j.cellsig.2007.06.004. [DOI] [PubMed] [Google Scholar]
181.Ouchi Y, Yagi S, Yokokura M, Sakamoto M. Neuroinflammation in the living brain of Parkinson's disease. Parkinsonism Relat Disord. 2009;(Suppl 3):S200–4. doi: 10.1016/S1353-8020(09)70814-4. [DOI] [PubMed] [Google Scholar]
182.Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, et al. Microglial activation and dopamine terminal loss in early Parkinson's disease. Ann Neurol. 2005;57:168–75. doi: 10.1002/ana.20338. [DOI] [PubMed] [Google Scholar]
184.Parkkinen L, Kauppinen T, Pirttila T, Autere JM, Alafuzoff I. Alpha-synuclein pathology does not predict extrapyramidal symptoms or dementia. Ann Neurol. 2005;57:82–91. doi: 10.1002/ana.20321. [DOI] [PubMed] [Google Scholar]
185.Parkkinen L, Pirttula T, Alafuzoff I. Applicability of current staging/categorization of alpha-synuclein pathology and their clinical relevance. Acta Neuropath. 2008;115:399–407. doi: 10.1007/s00401-008-0346-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
186.Patneau DK, Mayer ML. Structure-activity relationship for amino acid transmitter candidates acting at N-methyl-D aspartate and quisqualate receptors. J Neurosci. 1990;10:2385–97. doi: 10.1523/JNEUROSCI.10-07-02385.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
187.Pellegrini C, Forai M, Colucci R, Tiritta E, Blandini F, Levandis G, et al. Alteration of colonic excitatory tachykininergic motility and enteric inflammation following dopaminergic nigrostriatal neurodegeneration. J Neuroinflammation. 2016;13:146. doi: 10.1186/s12974-016-0608-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
188.Pellegrini-Giampletro DE, Gorter JA, Bennett MV, Zukin RS. The GluR2 (GluR-B) hypothesis: Ca2+-permeable AMPA receptors in neurological disorders. Trends Neurosci. 1997;20:464–70. doi: 10.1016/s0166-2236(97)01100-4. [DOI] [PubMed] [Google Scholar]
189.Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. 2014;10:217–24. doi: 10.1038/nrneurol.2014.38. [DOI] [PubMed] [Google Scholar]
190.Perry VH, Teeling J. Microglia and macrophages of the central nervous system: The contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin Immunopathol. 2013;35:601–12. doi: 10.1007/s00281-013-0382-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
192.Ponsen MM, Stoffers D, Booij J, van Eck-Smit BL, Wolters ECh, Berserndse HW. Idiopathic hyposmia as a preclinical sign of Parkinson's disease. Ann Neurol. 2004;56:173–81. doi: 10.1002/ana.20160. [DOI] [PubMed] [Google Scholar]
193.Porras G, Li Q, Bezard E. Modeling Parkinson's disease in primates: The MPTP model. Cold Spring Harb Perspect Med. 2012;2:a009308. doi: 10.1101/cshperspect.a009308. [DOI] [PMC free article] [PubMed] [Google Scholar]
194.Prediger RD, Aguiar AS, Jr, Moreira EL, Matheus FC, Castro AA, Walz R, et al. The intranasal administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): A new rodent model to test palliatve and neuroprotective agents for Parkinson's disease. Curr Pharm Des. 2011;17:489–507. doi: 10.2174/138161211795164095. [DOI] [PubMed] [Google Scholar]
195.Prediger RD, Aguar AS, Jr, Rojas-Mayorquin AE, Figueiredo CP, Matheus FC, Ginesete L, et al. Single intranasal administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in C57BL/6 mice models early preclinical phase of Parkinson's disease. Neurotox Res. 2010;17:114–29. doi: 10.1007/s12640-009-9087-0. [DOI] [PubMed] [Google Scholar]
196.Prentice H, Modi JP, Wu JY. Mechanisms of neuronal protection against excitotoxicity, endoplasmic reticulum stress, and mitochondrial dysfunction in stroke and neurodegenerative diseases. Oxid Med Cell Longev. 2015 doi: 10.1155/2015/964518. 964518. [DOI] [PMC free article] [PubMed] [Google Scholar]
197.Pribiag H, Stellwagen D. TNF-α downregulates inhibitory neurotransmission through protein phosphatase 1-dependent trafficking of GABA (A) receptors. J Neurosci. 2013;33:15879–93. doi: 10.1523/JNEUROSCI.0530-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
198.Putener U, Booth SG, Perry VH, Teeling JL. Long-term impact of systemic bacterial infection on the cerebral vasculature and microglia. J Neuroinflammation. 2012;9:146. doi: 10.1186/1742-2094-9-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
199.Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–62. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
200.Quan N, Sundar SK, Weiss JM. Induction of interleukin-1 in various brain regions after peripheral and central injections of lipopolysaccharide. J Neuroimmunol. 1994;49:125–34. doi: 10.1016/0165-5728(94)90188-0. [DOI] [PubMed] [Google Scholar]
201.Radi R, Rodriguez M, Castro L, Telleri R. Inhibition of mitochondrial electron transport by peroxynitrite. Arch Biochem Biophys. 1994;308:89–95. doi: 10.1006/abbi.1994.1013. [DOI] [PubMed] [Google Scholar]
202.Ramesh G, MacLean AG, Philipp MT. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediators Inflamm. 2013;2013:480739. doi: 10.1155/2013/480739. [DOI] [PMC free article] [PubMed] [Google Scholar]
203.Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, et al. Inflammation after trauma: Microglial activation and traumatic brain injury. Ann Neurol. 2011;70:374–83. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
204.Rana I, Badoer E, Alahmadi E, Leo CH, Woodman OL, Stebbing MJ. Microglia are selectively activated in endocrine and cardiovascular control centres in streptozocin-induced diabetic rats. J Neuroendocrinol. 2014;26:413–25. doi: 10.1111/jne.12161. [DOI] [PubMed] [Google Scholar]
205.Ransohoff RM, Perry VH. Microglial physiology: Unique stimuli, specialized responses. Ann Rev Immunol. 2009;27:119–45. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
206.Riazi K, Galic MA, Kuzmiski JB, Sharkey KA, Pittman QJ. Microglial activation and TNF-alpha production mediate altered CNS excitability following peripheral inflammation. Proc Natl Acad Sci USA. 2008;105:17151–6. doi: 10.1073/pnas.0806682105. [DOI] [PMC free article] [PubMed] [Google Scholar]
207.Roodeveldt C, Labrador-Garrido A, Gonzales-Rey E, Fernendez-Montesinos R, Caro M, Nachaud CC, et al. Glial innate immunity generated by non-aggregated alpha-synuclein in mouse: Difference between wild-type and Parkinson's disease-linked mutants. PloS One. 2010;5:e13481. doi: 10.1371/journal.pone.0013481. [DOI] [PMC free article] [PubMed] [Google Scholar]
208.Rosczyk HA, Sparkman NL, Johnson RW. Neuroinflammation and cognitive function in aged mice following minor surgery. Exp Gerontol. 2008;43:840–6. doi: 10.1016/j.exger.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
209.Santa-Cruz LD, Guerrero-Castillo S, Uribe-Carvajal S, Tapia R. Mitochondrial dysfunction during the early stages of excitotoxic spinal motor neuron degeneration in vivo. ACS Chem Neurosci. 2016;7:886–96. doi: 10.1021/acschemneuro.6b00032. [DOI] [PubMed] [Google Scholar]
210.Sapin E, Peyron C, Roche F, Gay N, Carcenac C, Savasta M, Levy P, Dematteis M. Chronic intermittent hypoxia induces chronic low-grade neuroinflammation in the dorsal hippocampus of mice. Sleep. 2015;38:1537–46. doi: 10.5665/sleep.5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
211.Sato A, Ohtaki H, Tsumuraya T, Song D, Ohara K, Asano M, et al. Interleukin-1 participates in the classical and alternative activation of microglia/macrophages after spinal cord injury. J Neuroinflam. 2012;9:65. doi: 10.1186/1742-2094-9-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
213.Scheperjans F, Aho V, Pererira PA, Koskinen K, Paulin L, Pekkonen E, et al. Gut microbiota are related to Parkinson's disease and clinical phenotype. Mov Disord. 2015;30:350–8. doi: 10.1002/mds.26069. [DOI] [PubMed] [Google Scholar]
214.Serres S, Anthony DC, Jiang Y, Broom KA, Campbell SJ, Tyler DJ, et al. Systemic inflammation response reactivates immune-mediated lesions in the rat brain. J Neurosci. 2009;29:4820–8. doi: 10.1523/JNEUROSCI.0406-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
215.Shannon KM, Keshavarzian A, Dodiya HB, Jakate S, Kordower JH. Is alpha-synuclein in the colon a biomarker for premotor Parkinson's disease? Evidence from 3 cases. Mov Disord. 2012;27:716–9. doi: 10.1002/mds.25020. [DOI] [PubMed] [Google Scholar]
216.Shannon KM, Keshavazian A, Mutlu E, Dodiya HB, Daian D, Jaglin JA, et al. Alpha-synuclein in colonic submucosa in early untreated Parkinson's disease. Mov Disord. 2012;27:709–15. doi: 10.1002/mds.23838. [DOI] [PubMed] [Google Scholar]
217.Shijle J, Takeuchi H, Yawata I, Harada Y, Sonobe Y, Doi Y, et al. Blockade of glutamate release from microglia attenuates experimental autoimmune encephalomyelitis in mice. Tohoku J Exp Med. 2009;217:87–92. doi: 10.1620/tjem.217.87. [DOI] [PubMed] [Google Scholar]
218.Siderowf A, Stern MB. Preclinical diagnosis of Parkinson's disease: Are we there yet? Curr Neurol Neurosci. 2006;6:295–301. doi: 10.1007/s11910-006-0021-z. [DOI] [PubMed] [Google Scholar]
219.Somera-Molina KC, Robin B, Somera CA, Anderson C, Stine C, Koh S, et al. Glial activation links early-life seizures and long-term neurologic dysfunction: Evidence using a small molecule inhibitor of proinflammatory cytokine regulation. Epilepsia. 2007;48:1785–800. doi: 10.1111/j.1528-1167.2007.01135.x. [DOI] [PubMed] [Google Scholar]
220.Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci. 2006;25:3119–28. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
221.Stern MB, Doty RL, Dotti M, Cororan P, Crowford D, McKeown DA, et al. Olfactory function in Parkinson's disease subtypes. Neurology. 1994;44:266–8. doi: 10.1212/wnl.44.2.266. [DOI] [PubMed] [Google Scholar]
223.Takeuchi H, Jin S, Wang G, Kawanokuchi J, Kuno R, Sonobe Y, et al. Tumor necrosis factor-alpha induces neurotoxicity via release from hemichannels of activated microglia in an autokine manner. J Biol Chem. 2006;281:31362–8. doi: 10.1074/jbc.M600504200. [DOI] [PubMed] [Google Scholar]
224.Tan AH, Mahadeva S, Marras C, Thalha AM, Kiew CK, Yeat CM, et al. Helicobacter pylori infection is associated with worse severity of Parkinson's disease. Parkinsonism Relat Disord. 2015;321:221–5. doi: 10.1016/j.parkreldis.2014.12.009. [DOI] [PubMed] [Google Scholar]
225.Tan AH, Mahadeva S, Thalha AM, Gibson PR, Kiew CK, Yeat CM, et al. Small intestinal bacterial overgrowth in Parkinson's disease. Parkinsonism Relat Disord. 2014;20:535–40. doi: 10.1016/j.parkreldis.2014.02.019. [DOI] [PubMed] [Google Scholar]
226.Tanaka S, Ishii A, Chtaki H, Yoshida T, Numazawa S. Activation of microglia induces symptoms of Parkinson's disease in wild-type, but not in IL-1 knockout mice. J Neuroinflammation. 2013;10:143. doi: 10.1186/1742-2094-10-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
227.Tancredi V, D’Arcangelo G, Grassi F, Tarroni P, Palmieri G, Santoni A, et al. Tumor necrosis factor alters synaptic transmission in rat hippocampal slices. Neuroci Lett. 1992;146:176–8. doi: 10.1016/0304-3940(92)90071-e. [DOI] [PubMed] [Google Scholar]
228.Tansey MG, Goldberg MS. Neuroinflammation in Parkinson's disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis. 2010;37:510–8. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
229.Taylor DL, Jones F, Kubota ES, Pocock JM. Stimulation of microglial metabotropic glutamate receptor mGlu2 triggers tumor necrosis factor alpha-induced neurotoxicity in concert with microglial-derived Fas ligand. J Neurosci. 2005;25:2952–64. doi: 10.1523/JNEUROSCI.4456-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
230.Terada T, Yokokura M, Yoshikawa E, Fustatsubashi M, Kono S, Konishi T, et al. Extrastriatal spreading of microglial activation in Parkinson's disease: A positron emission tomography study. Ann Nucl Med. 2016;30:579–87. doi: 10.1007/s12149-016-1099-2. [DOI] [PubMed] [Google Scholar]
231.Theodore S, Cao S, McLean PJ, Standaert DG. Targeted overexpression of human alpha-synuclein triggers microglial activation and an adoptive immune response in a mouse model of Parkinson's disease. J Neuropathol Exp Neurol. 2008;67:1149–58. doi: 10.1097/NEN.0b013e31818e5e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
232.Thiuchelvam M, McCormack A, Richfield EK, Baggs RB, Tank AW, Di Monte DA, et al. Age-related irreversible progressive nigrostriatal dopaminergic neurotoxicity in the paraquat and maneb model of the Parkinson's disease phenotype. Eur J Neurosci. 2003;18:589–600. doi: 10.1046/j.1460-9568.2003.02781.x. [DOI] [PubMed] [Google Scholar]
233.Tolwani RJ, Jakowec MW, Petzinger GM, Green S, Waggie K. Experimental models of Parkinson's disease: Insights from many models. Lab Anim Sci. 1999;49:363–71. [PubMed] [Google Scholar]
234.Tozzoli R, Di Bartolo I, Gigliucci F, Btamnilia G, Monini M, Vignolo E, et al. Pathogenic Escherichia and enteric viruses in biosolids and related top soil improvers in Italy. J Appl Microbiol. 2017;122:239–47. doi: 10.1111/jam.13308. [DOI] [PubMed] [Google Scholar]
236.Vereyken EJ, Bajova H, Chow S, de Graan PN, Gruol DL. Chronic interleukin-6 alters the level of synaptic proteins in hippocampus in culture and in vivo. Eur J Neurosci. 2007;25:3605–16. doi: 10.1111/j.1460-9568.2007.05615.x. [DOI] [PubMed] [Google Scholar]
237.Villaran RF, Espinosa-Oliva AM, Sarmiento M, De Pablos RM, Arguelles S, Delgado-Cortes MJ, et al. Ulcerative colitis exacerbates lipopolysaccharide-induced damage to the nigral dopaminergic system: Potential risk factor in Parkinson's disease. J Neurochem. 2010;114:1687–700. doi: 10.1111/j.1471-4159.2010.06879.x. [DOI] [PubMed] [Google Scholar]
238.Visanji NP, Marras C, Hazrati LN, Liu LW, Lang AE. Alimentary, my dear Watson? The challenges of enteric α-synuclein as a Parkinson's disease biomarker. Mov Disord. 2014;29:444–50. doi: 10.1002/mds.25789. [DOI] [PubMed] [Google Scholar]
239.Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, et al. Interleukin-1beta enhances NMDA receptor mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. 2003;23:8692–700. doi: 10.1523/JNEUROSCI.23-25-08692.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
240.Viviani B, Gardoni F, Marinovich M. Cytokines and neuronal ion channels in health and disease. In Rev Neurobiol. 2007;82:247–83. doi: 10.1016/S0074-7742(07)82013-7. [DOI] [PubMed] [Google Scholar]
241.Von Bernhardi R, Heredia F, Salgado N, Munoz P. Microglia function in the normal brain. Adv Exp Med Biol. 2016;949:67–92. doi: 10.1007/978-3-319-40764-7_4. [DOI] [PubMed] [Google Scholar]
242.Wakabayashi K, Takahashi H. Neuropathology of autonomic nervous system in Parkinson's disease. Eur Neurol. 1997;38(Suppl 2):2–7. doi: 10.1159/000113469. [DOI] [PubMed] [Google Scholar]
243.Wakabayashi K, Takahashi H, Takeda S, Ohana E, Ikuta F. Parkinson's disease: The presence of Lewy bodies in Auerbach's and Meissner's plexuses. Acta Neuropath. 1988;76:217–21. doi: 10.1007/BF00687767. [DOI] [PubMed] [Google Scholar]
244.Wang Q, Liu Y, Zhou J. Neuroinflammation in Parkinson's disease and its potential as therapeutic target. Transl Neurodegener. 2015;4:19. doi: 10.1186/s40035-015-0042-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
245.Wang S, Cheng Q, Malik S, Yang J. Interleukin-1beta inhibits gamma-aminobutyric acid type A (GABA (a)) receptor current in hippocampal neurons. J Pharmacol Exp Ther. 2000;292:497–504. [PubMed] [Google Scholar]
246.Wang S, Chu CH, Stewart T, Ginghina C, Wang Y, Nie H, et al. α-synuclein, a chemoattractant, directs microglial migration via H202-dependent Lyn phosphorylation. Proc Natl Acad Sci USA. 2015;112:E1926–35. doi: 10.1073/pnas.1417883112. [DOI] [PMC free article] [PubMed] [Google Scholar]
247.Weber CA, Ernst ME. Antioxidants, Supplements, and Parkinson's Disease. Ann Pharmacother. 2006;40:935–8. doi: 10.1345/aph.1G551. [DOI] [PubMed] [Google Scholar]
248.Wheeler D, Knapp E, Bandaru VV, Wang Y, Knorr D, Poitier C, et al. Tumor necrosis factor-alpha induced neuronal sphingomyelinase-2 modulates synaptic plasticity by controlling the membrane insertion of NMDA receptors. J Neurochem. 2009;109:1237–49. doi: 10.1111/j.1471-4159.2009.06038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
249.Wilms H, Zecca L, Rosenthiel P, Sievers J, Deuschi G, Lucius R. Inflammation in Parkinson's diseases and other neurodegenerative diseases: Cause and therapeutic implications. Curr Pharm Des. 2007;13:1925–8. doi: 10.2174/138161207780858429. [DOI] [PubMed] [Google Scholar]
250.Wszolek ZK, Markopoulou K. Olfactory dysfunction in Parkinson's disease. Clin Neurosci. 1998;5:94–101. [PubMed] [Google Scholar]
251.Wu DC, Tieu K, Cohen O, Choi DK, Vila M, Jackson-Lewis V, et al. Glial cell response: A pathogenic factor in Parkinson's disease. J Neurovirol. 2002;8:551–8. doi: 10.1080/13550280290100905. [DOI] [PubMed] [Google Scholar]
252.Wu YN, Johnson SW. Rotenone potentiates NMDA currents in substantia nigra dopamine neurons. Neurosci Lett. 2007;42:96–100. doi: 10.1016/j.neulet.2007.05.030. [DOI] [PubMed] [Google Scholar]
253.Xia N, Assous M, Had-Aissouni L, Gubellini P, Melon C, Nafi I, Salin P, et al. Progressive Parkinsonism by acute dysfunction of excitatory amino acid transporters in rat substantia nigra. Neurobiol Dis. 2014;65:69–81. doi: 10.1016/j.nbd.2014.01.011. [DOI] [PubMed] [Google Scholar]
254.Xia N, Zhang Q, Wang ST, Gu L, Yang HM, Liu L, et al. Blockade of metabotrophic glutamate receptor 5 protects against DNA damage in a rotenone-induced Parkinson's disease model. Free Radic Biol Med. 2015;89:567–80. doi: 10.1016/j.freeradbiomed.2015.09.017. [DOI] [PubMed] [Google Scholar]
255.Yang J, Hertz E, Zhang X, Leinartaite L, Lundius EG, Li J, Svenningsson P. Overexpression of α-synuclein simultaneously increases glutamate NMDA receptor phosphorylation and reduces glucoserebrosidase activity. Neurosci Lett. 2016;611:51–8. doi: 10.1016/j.neulet.2015.11.023. [DOI] [PubMed] [Google Scholar]
256.Yawata I, Takeuchi H, Doi Y, Liang J, Mizuno T, Suzumura A. Macrophage induced neurotoxicity is mediated by glutamate and attenuated by glutaminase inhibitors and gap junction inhibitors. Life Sci. 2008;82:1111–6. doi: 10.1016/j.lfs.2008.03.010. [DOI] [PubMed] [Google Scholar]
257.Yokokawa M, Terada T, Bunai T, Nakaizumi K, Takebayashi K, Iwata Y, et al. Depiction of microglial activation in gaining and dementia: Positron emission tomography with [11C] DPA713 versus [11C](R) PK11195. J Cereb Blood Flow Metab. 2016 doi: 10.1177/0271678X16646788. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
259.Zeng BY, Dass B, Owen A, Rose S, Cannizzaro C, Tel BC, Jenner P. 6-hydroxydopamine lesioning differentially affects alpha-synuclein mRNA expression in the nucleus accumbens, striatum and substantia nigra of adults rats. Neurosci Lett. 2002;322:33–6. doi: 10.1016/s0304-3940(02)00083-6. [DOI] [PubMed] [Google Scholar]
260.Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, et al. Aggregated alpha-synclein activates microglia: A process leading to disease progression in Parkinson's disease. FASEB J. 2005;19:533042. doi: 10.1096/fj.04-2751com. [DOI] [PubMed] [Google Scholar]
261.Zhang X, Dong H, Zhang S, Lu S, Sun J, Qian Y. Enhancement of LPS-induced microglial inflammation response via TLR4 under high glucose conditions. Cell Physiol Biochem. 2015;35:1571–81. doi: 10.1159/000373972. [DOI] [PubMed] [Google Scholar]
262.Zhou Y, Tang H, Liu J, Xiong H. Chemokine CCL2 modulation of neuronal excitability and synaptic transmission in rat hippocampal slices. J Neurochem. 2011;116:406–14. doi: 10.1111/j.1471-4159.2010.07121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
263.Ziebell JM, Rowe RK, Muccigrosso MM, Reddaway JT, Adelson PD, Godbout JP, et al. Aging with a traumatic injury: Could behavioral morbidities and endocrine symptoms be influenced by microglial priming? Brain Behav Immun. 2017;59:1–7. doi: 10.1016/j.bbi.2016.03.008. [DOI] [PubMed] [Google Scholar]
264.Zou JY, Crews FT. TNF alpha potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: Neuroprotection by NF kappa B inhibition. Brain Res. 2005;1034:11–24. doi: 10.1016/j.brainres.2004.11.014. [DOI] [PubMed] [Google Scholar]